

Abstract
Alzheimer’s disease and related disorders feature neurofibrillary tangles and other neuropathological lesions composed of detergent-insoluble tau protein. In recent structural biology studies of tau proteinopathy, aggregated tau forms a distinct set of conformational variants specific to the different types of tauopathy disorders. However, the constituents driving the formation of distinct pathological tau conformations on pathway to tau-mediated neurodegeneration remain unknown. Previous work demonstrated RNA can serve as a driver of tau aggregation, and RNA associates with tau containing lesions, but tools for evaluating tau/RNA interactions remain limited. Here, we employed molecular interaction studies to measure the impact of tau/RNA binding on tau microtubule binding and aggregation. To investigate the importance of tau/RNA complexes (TRCs) in neurodegenerative disease, we raised a monoclonal antibody (TRC35) against aggregated tau/RNA complexes. We showed that native tau binds RNA with high affinity but low specificity, and tau binding to RNA competes with tau-mediated microtubule assembly functions. Tau/RNA interaction in vitro promotes the formation of higher molecular weight tau/RNA complexes, which represent an oligomeric tau species. Coexpression of tau and poly(A)45 RNA transgenes in Caenorhabditis elegans exacerbates tau-related phenotypes including neuronal dysfunction and pathological tau accumulation. TRC35 exhibits specificity for Alzheimer’s disease-derived detergent-insoluble tau relative to soluble recombinant tau. Immunostaining with TRC35 labels a wide variety of pathological tau lesions in animal models of tauopathy, which are reduced in mice lacking the RNA binding protein MSUT2. TRC-positive lesions are evident in many human tauopathies including Alzheimer’s disease, progressive supranuclear palsy, corticobasal degeneration and Pick’s disease. We also identified ocular pharyngeal muscular dystrophy as a novel tauopathy disorder, where loss of function in the poly(A) RNA binding protein (PABPN1) causes accumulation of pathological tau in tissue from post-mortem human brain. Tau/RNA binding drives tau conformational change and aggregation inhibiting tau-mediated microtubule assembly. Our findings implicate cellular tau/RNA interactions as modulators of both normal tau function and pathological tau toxicity in tauopathy disorders and suggest feasibility for novel therapeutic approaches targeting TRCs.
Keywords: tau, RNA, neurodegeneration, RNA binding protein, Alzheimer’s disease, OPMD
McMillan et al. show that native tau binds RNA with high affinity but low specificity. Tau/RNA binding drives tau conformational change, inhibits microtubule assembly, promotes pathological tau accumulation and exacerbates tau-mediated neuronal dysfunction.
Graphical abstract
Graphical abstract.

Introduction
Pathological tau deposits in the form of neurofibrillary tangles (NFTs) have long been appreciated as a potential driver of neurodegeneration in Alzheimer’s disease. NFTs have been shown to correlate strongly with cognitive impairment in Alzheimer’s disease, in contrast to amyloid-β plaques, which do not consistently correlate with dementia.1-3 In other types of dementia disorders, termed tauopathies, tau is the primary neuropathological lesion. Likewise, imaging studies support a causal relationship between abnormal tau and dementia.4 However, our understanding of the genesis of abnormal tau and the molecular mechanisms by which pathological tau contributes to neurodegeneration remain incompletely understood.5 The canonical biochemical function of tau is that of a microtubule (MT) binding protein, as it was first identified to both bind to MTs and promote their polymerization.6 More recently, tau has been shown to interact with a wide range of macromolecules, perhaps due to its unusual natively unfolded structure (reviewed by Limorenko and Lashuel7). Indeed, the conversion of natively unfolded tau into abnormal misfolded fibrillar structures has been an intense area of inquiry, with the recent determination of the fibril core structure for many tauopathies by cryoelectron microscopy (reviewed by Goedert8, and Scheres et al.9). The structural determination of the distinct conformers of abnormal tau fibrils has validated the tauopathy seeding/strain hypothesis by demonstrating that differing diseases exhibit structurally distinct tau aggregates contributing to disease.10-12 How structurally distinct tau aggregated species arise remains an open question, but different tau interacting macromolecules have been suggested to play a pivotal role.13-19
A wide variety of polyanions stimulate tau aggregation in vitro, including heparin, RNA, polyglutamate and fatty acids; tau also binds the highly negatively charged c-terminal tail of alpha tubulin (helix 12) to promote MT polymerization and stabilization.20 A previous study suggested that RNA is the most potent polyanion trigger of tau aggregation in vitro.16 Of the tau aggregation stimulatory polyanions mentioned above, RNA appears to be the most abundant in the neuronal cytoplasm. Further, neuropathological and macromolecular characterization of Alzheimer’s disease brain suggests NFTs contain RNA; analysis of Alzheimer’s disease-derived NFTs by microarray-based transcriptomic profiling showed certain mRNAs preferentially become trapped in tau aggregates in human disease.21-23 Next-generation sequencing–based transcriptomic analysis has confirmed these findings and extended them to cellular and transgenic animal models of tau aggregation, as well as progressive supranuclear palsy (PSP) and corticobasal degeneration (CBD).24,25 Surprisingly, tau’s MT-stabilizing activity has also been shown to be impacted by RNA in experiments where RNA homopolymers blocked tau-mediated MT assembly.26
In previous work, we identified RNA binding protein modulators of tauopathy that reside in nuclear speckles, including aly-1,2,3/ALYREF, pabp-2/PABPN1, sut-1, sut-2/MSUT2, and parn-2/TOE1.27-32 Among these RNA-binding proteins, we have shown that MSUT2 controls tauopathy-related phenotypes in brain neurons in mammals. MSUT2 knockout mice exhibit reduced accumulation of pathological tau, cognitive impairment and neurodegeneration.29 The molecular mechanism of MSUT2 modulation of tauopathy involves the nuclear RNA binding functions of MSUT2 as it binds both poly(A) RNA and the nuclear poly(A) RNA binding protein, PABPN1. We showed that MSUT2 and PABPN1 co-localize with poly(A) RNA to nuclear speckles, forming a macromolecular complex. In the brains of post-mortem Alzheimer’s disease cases both MSUT2 and PABPN1 become co-depleted. Work of others showed PABPN1 and MSUT2 have opposing effects on mRNA poly(A) tail length.33,34 Likewise, we observe that MSUT2 and PABPN1 function together in a reciprocal fashion to influence tauopathy; normal MSUT2 function drives tau aggregation while normal PAPBN1 function promotes tau proteostasis. Taken together, these findings support an RNA metabolism-related mechanism of tauopathy in need of further investigation.
While the rapid progression of tauopathy research has answered many questions about tau aggregation, seeding and pathological spread, we still lack a fundamental understanding of the initial triggers of pathological tau aggregation in vivo. Here, we further investigate tau’s interaction with RNA, a polyanion known to potently seed tau aggregation.16,17 We have developed a series of reagents and assays to detect and measure tau activity as an RNA binding protein and herein employ these tools to begin to address the impact of tau/RNA binding activity on tau normal function and tau neuropathology.
Materials and methods
Preparation of recombinant tau
Recombinant 6xHis-tagged human tau protein (isoforms 1N4R and 1N3R) were expressed in Escherichia coli and purified using a boiling lysis preparation and native nickel resin chromatography essentially as previously described.35 Recombinant untagged human tau (isoform 1N4R) was expressed in E. coli and purified using a boiling lysis preparation essentially as previously described.36,37
Microtubule assembly assays
Microtubule assembly assays were performed essentially as previously described in a low 30 µl reaction volume format in flat bottom 384-well plates.38 Porcine tubulin (20 µM final concentration, #T240, Cytoskeleton Inc.) was mixed with 2 µM recombinant 1N4R tau and varying concentrations of Poly(A) RNA (P9403, Sigma Chemical) in BRB80 (80 mM PIPES, 1 mM EGTA, 1 mM MgSO4, pH 6.8), 2 mM GTP and 1 mM DTT. Reactions were assembled on ice and Poly(A), GTP and DTT were diluted to appropriate working stock concentrations in BRB80 prior to reaction assembly. Tubulin with 20 µM taxol was utilized as a positive control for assay conditions and tubulin quality. Once all reaction components were added, the plate was allowed to incubate at room temperature for 2 min, the bottom wiped with a Kimwipe and placed in a Perkin Elmer Envision plate reader maintained at 37°C. Absorbance readings (340 nm) were taken every 60 s for 3 h. Absorbance read data were normalized by subtracting the mean of the first five reads from each absorbance read for each condition.
Analysis of tau/RNA interactions
Biotinylated RNAs were custom synthesized by Integrated DNA Technologies (IDT). Tau/RNA interactions were analysed using surface plasmon resonance (SPR; Ametek Reichert Technologies) as recommended by the manufacturer for RNA/protein interactions using H2T as SPR running buffer (50 mM HEPES, 25 mM NaCl, 0.1% Tween-20, pH 7.4) and a streptavidin SPR chip (Catalogue No. 13206071, Reichert SPR).
Generation of tau/RNA complexes
Recombinant untagged human tau was incubated with poly(A) RNA in buffer H2 for 16 h at 20°C with shaking at 400 RPM.
Analysis of tau/RNA complexes by size-exclusion chromatography
To evaluate tau aggregation or complex formation, equivalent mass of tau protein in the form of uncomplexed purified tau and tau/RNA complexes were analysed using size-exclusion chromatography using a GE AKTA purifier running a SUPERDEX 200 increase column at 1 ml/min essentially as previously described39 with H2 (50 mM HEPES, 25 mM NaCl, pH 7.4) as the chromatography running buffer. Fractions were immunoblotted for tau using the Simple Western capillary-electrophoresis system, Peggy Sue (ProteinSimple), and following the manufacturer’s recommendations. Briefly, fractions were diluted with 5× Fluorescent Master Mix, boiled for 5 min at 95°C, cooled to 4°C and loaded into a 384-well plate (12–230 kDa Size Separation module, Catalogue No. SM-S001, ProteinSimple). Primary antibody to tau (1:10,000, Catalogue No. A0024, Dako, Agilent Technologies) and goat anti-rabbit HRP (1:100, Jackson ImmunoResearch) were diluted in Antibody Diluent 2 (No Secondary Detection Module, Catalogue No. DM-003, ProteinSimple). Samples were run and analysed with the default assay in Compass for SW Version 4.0.0 (ProteinSimple).
Caenorhabdytis elegans husbandry and generation of poly(A) transgenic C. elegans
C. elegans strains were maintained as previously described40 at 20°C. The canonical wild-type (WT) C. elegans strain, Bristol N2, was used as the genetic background for transgenesis and as the WT C. elegans control strain. A transgene encoding poly(A)45 RNA was driven by the C. elegans U6 RNA polymerase III promoter and universal TTTT RNA polymerase III terminate.41 The transgene was injected into N2 at a concentration of 100 ng/µl along with Pmyo-3::mCherry (20 ng/µl) as a co-injection marker to produce worms carrying extra-chromosomal arrays. These generated lines were irradiated with UV to integrate the extra-chromosomal array into the genome as previously described.42 Successfully integrated lines were identified by isolating individual worms with 100% transmission of the Pmyo-3::mCherry marker and outcrossed with N2 males three times. The poly(A)45 expressing strain described is CK2362. The tau transgenic strain used, CK144, expresses WT 1N4R tau from the aex-3 promoter and has been previously described.41 Bigenic animals expressing both poly(A)45 and WT tau are CK2408 and were generated by crossing CK144 with CK2362 and assessed for the impact of poly(A) on tauopathy-related phenotypes essentially as previously described.27
C. elegans immunoblotting
Staged populations of C. elegans for the above-described strains were grown to Day 1 of adulthood and harvested as previously described.27 Frozen samples were resuspended in 5× Sample Buffer (5% SDS, 200 mM DTT, 50 mM Tris pH 6.8, 5 mM EDTA, 50% sucrose, 0.05% Bromophenol Blue), with volume equivalent to four times the pellet weight (4 µl/mg of packed animals). Samples were sonicated on ice for three bursts of 15 s at 70% power. Sonicated lysates were denatured for 10 min in a 95°C heat block then cooled and briefly centrifuged. For each sample, 10 µl of supernatant was loaded and run on a 4–15% Tris–HCl sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS-PAGE) gel with appropriate protein standards. The proteins were transferred to PVDF in Tris–glycine–methanol transfer buffer as per manufacturer recommendations (Bio-Rad). PVDF membrane was blocked in 5% w/v dry milk in phosphate-buffered saline (PBS) solution for 1 h, then treated overnight at 4°C in primary antibody for total tau (1:500,000, Catalogue No. A0024, Dako, Agilent Technologies), pTau pS202 (CP13, 1:1000) or pTau pS396/404 tau (PHF1, 1:1000). Tubulin [Developmental Studies Hybridoma Bank (DSHB) anti-β Tubulin E7-s] was used as a load control at 1:5000. The following day, primary antibody was removed and the membrane washed three times in 1× PBS + 1% Tween20 for 10 min each wash. Secondary antibody was then applied in 5% w/v dry milk in PBS solution and allowed to blot for 2 h, followed by an additional three 1× PBS + 1% Tween20 washes. Immunoreactivity was visualized using ECL (BioRad Clarity Western ECL Substrate 170-5061) with a LI-COR Odyssey Fc system (LI-COR Biosciences) and quantified using LI-COR Image Studio Software.
Immunization of mice with tau/RNA complexes
Tau/RNA complexes (TRCs) as described above were used to immunize mice to produce monoclonal antibodies in the Washington State University Monoclonal Antibody Center using standard operating procedures.43 To produce tau antibodies efficiently by limiting self-antigen recognition, previously described tau knockout mice were used for immunization.44 Monoclonal antibodies were produced at WSU as a fee for service project (IACUC protocol #6317) using well-established standard operating protocols for antibody production.43 Briefly, TRCs were mixed in Sigma Adjuvant System at 50% (w/v) as an emulsion prior to i.p. immunization as recommended by the manufacturer (Sigma Chemical, Catalogue No. S6322). Mice were immunized with 200 µg of tau/RNA complex and received an identical boost immunization 3 weeks after initial immunization. Spleens were harvested 3 days after boosting for hybridoma production. Clonal hybridoma lines were screened based on TRC binding activity and the TRC35-expressing hybridoma monoclonal line was isolated and cloned. TRC35 monoclonal hybridoma lysate was isotyped using Thermo Rapid Elisa monoclonal antibody (mAb) isotyping reagents (ThermoFisher, Catalogue No. 37503).
Brain specimens
Samples of post-mortem brain tissue were obtained from research participants in the University of Washington (UW) Alzheimer’s Disease Research Center and the Kaiser Permanente Washington Health Research Institute Adult Changes in Thought (ACT) Study via the UW BioRepository and Integrated Neuropathology (BRaIN) Laboratory. Informed consent for research brain donation was obtained according to protocols approved by the UW and KPWHRI Institutional Review Boards. Alzheimer’s disease brain donors (n = 19) were chosen based upon the clinical diagnosis of dementia and neuropathologically confirmed Alzheimer’s disease neuropathologic change (ADNC) sufficient to explain dementia (intermediate or high). Brain tissues used as controls for this study (n = 7) were derived from age-matched cognitively normal research participants with neuropathologically confirmed not-low levels of ADNC. Fixation of donor brains occurred by immersion in 10% neutral buffered formalin for at least 2 weeks, followed by coronal slicing and sampling that included dorsolateral prefrontal cortex, hippocampus, amygdala and cerebellum, which were processed and embedded in paraffin and sectioned at 5-μm thickness according to routine protocols for neuropathological analysis as described below.
Purification of fibrillar insoluble tau from Alzheimer’s disease post-mortem brain
Detergent-insoluble fibrillar tau protein was purified from Alzheimer’s disease brain donors with neuropathologically confirmed high pathological tau burden (Braak stage VI). Temporal lobe cortex brain specimens frozen at autopsy from Alzheimer’s disease donors were homogenized, repeatedly extracted with excess 1% sarkosyl until insoluble tau fibrils were highly enriched and soluble tau was depleted, with detergent-insoluble tau representing the major protein species present in these Alzheimer’s disease tau extracts.45
Characterization of TRC35 monoclonal antibodies
TRC35 monoclonal antibodies were used to dot blot equivalent amounts (20 ng) of E. coli-expressed native recombinant tau protein and Alzheimer’s disease tau (ADtau) fibrillar material. ADtau was standardized by quantitative immunoblot against purified recombinant tau with pan-tau anti-tau antibody (Catalogue No. A0024, Dako, Agilent Technologies) such that 20 ng of E. coli-expressed native recombinant tau protein and ADtau fibrillar material exhibited equivalent immunoreactivity. Protein preparations were not denatured prior to blotting to preserve native conformations expected of a conformation-dependent mAb. Dot blots were performed using the Bio-Dot Microfiltration Apparatus as recommended by the manufacturer (Bio-Rad, Catalogue No. 170-6545). Membranes were blocked in blocking solution [20 mM Tris, 500 mM NaCl, 1% bovine serum albumin (w/v)] and probed with TRC35 at 1:50,000 as primary antibody in TTBS buffer [20 mM Tris, 500 mM NaCl, 1% bovine serum albumin (w/v), 0.5% (w/v) Tween20]. Blots were washed three times in TTBS to remove excess primary antibody. Goat anti-rabbit IgG secondary antibodies were used at 1:10,000 in TTBS. Blots were washed three times in TTBS to remove excess secondary antibody. Immunoreactivity was measured using Clarity Western ECL reagents (Bio-Rad). Images were scanned and quantitated using LI-COR Image Studio software. Epitope profile experiments were conducted as a fee for service project at JPT Peptide Technologies, Inc. Epitope profiling employed an array of 101 overlapping peptides tiling over the entire coding sequence of tau protein. The profiling experiment was performed with antibodies diluted to 1 μg/ml and 0.1 μg/ml with TBS blocking buffer (Pierce). Subsequent to sample incubation, secondary fluorescently labelled anti-mouse-IgG at 1 μg/ml was added into the corresponding wells and left to react for 1 h. After washing and drying, the slide was scanned with a high-resolution laser scanner at 635 nm to obtain fluorescence intensity profiles.
Transgenic animal modelling
Experiments using transgenic mouse models of tauopathy were reviewed and approved by the VA Puget Sound Health Care System Institutional Animal Care and Use Committee (IACUC) and conducted in an American Association for Accreditation of Laboratory Animal Care (AAALAC)-accredited animal research facility. C57B6/J was used as the control strain of mice and all tau transgenes were maintained in a congenic state on this background. For all mouse analyses, both sexes were used and no animals were excluded. The PS19 tau transgenic mouse model expressing human P301S mutant human tau was used in this study. This mouse model is well characterized and has a highly progressive tauopathy-related phenotype. In addition, a milder mouse model of tauopathy driven by WT human tau expression (Tau4RTg2652) was also examined. This mouse line exhibits early-stage tau pathology, including phosphorylated tau, but not neurofibrillary degeneration.46 PS19 mice (n = 12 at 9 months, n = 4 at 3 months), Tau4RTg2652 mice (n = 7 at 3 months) and WT mice (n = 6 at 3 months of age) were anaesthetized and fixed by transcardial perfusion with 4% paraformaldehyde. Brains were removed and paraffin embedded for sectioning. Coronal sections (9 μm) were prepared and stored at 4°C until use.
Neuropathological evaluation
Human and mouse brain sections were deparaffinized, rehydrated through alcohols and processed through antigen retrieval steps consisting of heat pretreatment in citrate buffer by either microwave or autoclave per antibody-specific protocols. Sections were treated for endogenous peroxidases with 3% hydrogen peroxide in PBS (pH 7.4), blocked in 5% non-fat milk in PBS and incubated with primary antibodies overnight at 4°C. Biotinylated secondary goat anti-mouse or goat anti-rabbit antibody was applied for 45 min at room temperature. Finally, sections were incubated in an avidin–biotin complex with streptavidin–HRP (Vector’s Vectastain Elite ABC-HRP kit) and the reaction product was visualized with 0.05% diaminobenzidine (DAB)/0.01% hydrogen peroxide in PBS. Negative controls consisted of full protocol except primary antibody. For double label immunofluorescence experiments, AlexaFluor 488 nm goat anti-mouse and AlexaFluor 594 nm goat anti-rabbit secondary antibodies (Molecular Probes) were used and autofluorescence was quenched with 0.1% Sudan Black. Digital images were obtained using a Leica DM6 microscope with a DFC 7000 digital camera (Leica Microsystems) and imported into Adobe Photoshop.
Quantitative analysis of immunohistochemistry
HALO digital image software (Indica Labs) was used to quantify TRC35 and AT180 immunoreactivity in mouse and human brain. Brain sections were manually annotated around the regions of interest, average staining intensity for each antibody was determined to allow quantification without contribution of background staining and a common threshold was then applied to all sections for that assay. Data represent the area of positive immunoreactivity within the region of interest divided by the total annotated area. This value was then multiplied by the average optical density (OD) of immunoreactivity (IR) to yield the final normalized IR area × OD. Data are displayed as the mean±SEM. A two-tailed Student’s t-test was used to assess differences in immunoreactivity between experimental groups. Statistical analysis and graphing were performed using the Prism V8.3 software package (GraphPad).
Cell culture
ReNcell VM cells (Millipore) were cultured in DMEM/F12 media (Sigma) supplemented with 2% B-27 (ThermoFisher), 1% Glutamax (ThermoFisher), 1% penecillin/streptomycin (1000 IU/ml), 10 units/ml heparin (Sigma), 50 µg/ml gentamicin (ThermoFisher), 40 ng EGF (Sigma), and 40 ng bFGF (Sigma).47,48 Cells were grown on flasks pre-coated with laminin (Sigma) and split every 3–4 days. Cells were differentiated in the above media without growth factors for 10 or more days, replacing media every 3–4 days.
Fluorescent immunohistochemistry
ReNcell VM cells were cultured on 12 mm round coverslips coated with poly-d-lysine then laminin. Samples were fixed in a 4% formaldehyde solution, washed 3 × 5 min in PBS/Ca2+/Mg2+, then blocked in antibody buffer [PBS, 0.5% Triton X-100, 1 mM EDTA, 0.1% bovine serum albumin (BSA), 0.05% NaN3] with 10% normal goat serum. Primary antibodies were applied and incubated overnight (TRC35 1:100; SP70 1:500, Invitrogen, catalogue MA5-16404). Cells were washed three times for 5 min in PBS/Ca2+/Mg2+, then re-blocked for 10 min. Appropriate Alexa dye-labelled secondary antibodies (1:1000, Invitrogen) were applied and incubated for 20 min at room temperature. Cells were again washed three times for 5 min in PBS/Ca2+/Mg2+, counterstained with 300 nM DAPI and mounted with ProLong Gold antifade (Molecular Probes). Microscopy was performed on a Delta Vision microscope (GE Inc.) using a 100× oil immersion objective, a sCMOS camera, and 2 × 2 binning. Image analysis was performed using softWoRx 6.0 Beta software (GE Inc.).
Data availability
Upon reasonable request, the data described in this study are available from the corresponding author.
Results
Tau preferentially binds to RNA inhibiting microtubule assembly
Given that poly(A) RNA has been shown to block the canonical tau function of stimulating MT polymerization,26 we examined whether RNA might compete with tau-mediated microtubule assembly and found poly(A) RNA inhibits tau-mediated MT assembly in a dose-dependent manner (Fig. 1A). Furthermore, we tested the hypothesis that tau binds to RNA directly and may compete with tubulin dimer binding. To measure tau’s affinity for tubulin and different types of RNA molecules, we employed a highly sensitive SPR assay for RNA binding49 and adapted it to tau. SPR has been suggested to be superior to electrophoretic mobility shift assay for characterization of RNA binding proteins.50 We used purified recombinant human tau isoform 1N4R (the most abundant 4R brain isoform) to measure binding to a series of biotinylated RNA probes and observed high-affinity interactions between tau and RNA (Fig. 1B and Supplementary Table 1). We observe that human tau prefers single stranded RNAs [poly(A), poly(U), poly(N)] over highly structured tRNA (tRNALYS KD > 30 nm, Supplementary Table 1). Control experiments also showed 3R tau binding poly(A) RNA with a similar affinity to 4R tau (8.3 nM versus 7.2 nM, respectively). However, tubulin does not detectably bind poly(A) RNA. We analysed the affinity of tau for tubulin using a similar SPR assay with biotinylated α/β tubulin dimers in place of the RNA probe and observed that tau binds tubulin with high affinity as expected (KD = 39.8 nM, Fig. 1C, Butner and Kirschner51 and Kadavath et al.52). Taken together, these data suggest tau has a ∼5-fold greater affinity for linear homopolymeric RNA relative to tubulin.
Figure 1.

Tau binds to poly(A) RNA, inhibiting MT assembly and promoting tau oligomerization. (A) In vitro MT assembly detected by light scattering measured by absorbance at 340 nm expressed in absorbance units (AU). Tau initiates MT assembly under standard conditions (20 µM tubulin dimer in 1 × BRB80 with 2 µM recombinant tau). Poly(A) RNA delays and attenuates MT assembly at equivalent tau concentrations in a poly(A) RNA concentration-dependent manner (fold poly(A) levels relative to tau levels shown). Dashed lines represent SEM for n = 5 replicates for each condition. (B) Representative data trace for a biotin poly(A)20 RNA probe. SPR assay can detect tau/RNA interactions including association and dissociation to derive the parameters of binding kinetics (lines represent decreasing tau concentrations SPR in binding assay shown in descending order. Top: 250 nM tau, 200 nM tau, 150 nM tau, 100 nM tau, 50 nM tau; bottom:0 nM tau). (C) Incubation of recombinant purified tau protein and poly(A) RNA produces RNA/protein complexes detected by size-exclusion chromatography (SEC). An equivalent mass of tau protein was fractionated by SEC on a Superdex 200 increase column. (D) Bigenic poly(A)45/tau transgenic animals (strain CK2408) exhibit more severe neuronal dysfunction as detected by swimming reflex measurements when compared to the parental tau transgenic strain (CK144) alone [mean ± SEM: tau Tg = 0.098 ± 0.006, poly(A)45/tau Tg = 0.040 ± 0.007, Student’s t-test P < 0.0001]. (E) The poly(A)45 RNA transgene increases pathological tau protein accumulation commensurate with behavioural dysfunction. (F) Quantification of expression of total tau (DAKO) relative to tubulin load control, pS202 (CP13) relative to total tau, and pS396/404 (PHF1) relative to total in parental tau transgenic strain (CK144) as compared to tau/poly(A) coexpressing animals (error bars show SEM of three replicate blots. Student’s t-test P = 0.0294, 0.77 and 0.27, respectively).
Tau binding to RNA promotes formation of oligomeric tau complexes and exacerbates tauopathy phenotypes
Previously published work has demonstrated RNA induces the formation of tau aggregates in vitro and exhibits greater potency than other polyanions.16 We examined the composition of tau–poly(A) RNA complexes chromatographically and observed that the majority of tau assembles with RNA into chromatography stable complexes in vitro under standard physiologically relevant ionic and pH conditions. The assembled tau–poly(A) RNA complexes formed overnight and were resolved by native size-exclusion chromatography followed by denaturing capillary electrophoresis yielding a range in approximate molecular weight from 330 kDa to 1250 kDa (Fig. 1C). This size range is consistent with the tau/RNA complexes being composed of medium N tau oligomers consisting of ∼3 to 24 tau monomers bound to poly(A) RNA. Because RNA is by far the most abundant tau-binding macromolecule within the cytoplasm, the observation that tau/RNA interaction promotes oligomerization of tau supports a role for RNA in the initial phases of pathological tau aggregation in vivo. To further explore the relevance of poly(A) RNA-induced tau oligomerization in animal models of tauopathy, we generated transgenic C. elegans that stably express a poly(A)45 RNA transcript with the absence of any attached coding sequence in all cells. These poly(A) RNA-expressing transgenic animals are viable, normal and healthy. When crossed to pan-neuronal tau transgenic C. elegans the poly(A) RNA transgene significantly exacerbates neuronal dysfunction indicated by behavioural deficits (Fig. 1D). The poly(A)45 transgene also exacerbated accumulation of pathological tau species (Fig. 1E and F).
Mouse immunization with tau/RNA complexes to produce a monoclonal antibody recognizing a disease-specific conformational epitope of tau
To create a specific reagent for detecting TRCs in disease states and model systems, we began an antibody development project employing in vitro assembled TRCs as the immunogen to produce mouse monoclonal antibodies. Hybridoma cell lines were screened and a single hybridoma clone, #35, expressing a mAb of the IgG1 isotype was identified with selectivity for pathological tau assemblies and named TRC35 (Fig. 2A). Given the aggregated state of TRC antigen, theTRC35 mAb is expected to prefer tau assemblies over monomeric tau; we employed a native protein dot immunoblot analysis to validate the specificity of TRC35. Recombinant tau lacks pathological tau species such as pTau, but loading controls with a total tau antibody reveal similar levels of total tau (Supplementary Fig. 1). We showed TRC35 recognizes detergent-insoluble pathological tau purified from Alzheimer’s disease brain tissue, but not monomeric recombinant human tau, with ∼8-fold selectivity (Fig. 2B and C). Further TRC35 reacts strongly with Alzheimer’s disease brain lysate but not age-matched control brain lysate (Supplementary Fig. 2A and B). In addition, the TRC35 reactive conformation of tau is most strongly induced by incubation with poly(A) RNA, but heparin, a polyanion with similar charge density, can also promote tau to adopt a TRC35 reactive conformation (Supplementary Fig. 3A and B). In addition, we examined the accumulation of TRC35+ tau in cultured human ReNcell VM neural progenitors differentiated into neurons as described.48 ReNcell neurons exhibit TRC35+ accumulation throughout both the soma and nucleus, while SP70 tau mAb detecting monomeric soluble tau is confined to the soma. Thus, TRC35 and soluble tau appear to partially overlap in cultured human neuronal cells (colocalization coefficient = 0.6; Supplementary Fig. 4). To map the specific tau epitope recognized by TRC35, we conducted a peptide mapping analysis and identified a discontinuous region just outside the proline rich domain consisting of amino acids 166–176 and 225–228 (see Supplementary Table 2 and Supplementary Fig. 5 peptide epitope sequence ANATRIPAKTP and KVAV). Note that the epitope mapping of TRC35 revealed a discontinuous epitope consistent with a conformation-dependent antibody. TRC35/tau binding did not require RNA or other polyanions, demonstrating the TRC35 epitope appears to be a purely conformational peptide epitope. The c-terminal KVAV peptide component of the epitope appears immediately adjacent to the TOC1 epitope, but distinct (Ward et al.53,54 and Supplementary Table 2). Taken together these observations suggest tau binding to poly(A) RNA or perhaps other polyanions exposes the TRC35 epitope on tau when promoting tau aggregation, but RNA does not itself constitute part of the TRC35 epitope.
Figure 2.

Generation and validation of mAb TRC35 recognizing tau/RNA complexes. (A) Workflow for generating mAb TRC35. (B) mAb TRC35 recognizes a tau epitope enriched in Alzheimer’s disease. Shown is a native protein dot blot loaded with equivalent mass of E. coli-derived recombinant tau (top row, 20 ng) or Alzheimer’s disease-derived tau (ADtau) (bottom row, 20 ng). (C) Densitometry analysis of the dot blots from panel B reveals a statistically significant 8-fold selectivity of TRC35 for ADtau over native tau (P < 0.0001, statistical comparison made by two-tailed Student’s t-test). (D) Immunostaining of human tau transgenic mouse models of tauopathy shows prominent phospho-tau (AT180) and TRC35+ labelling in both 3-month-old PS19 mice, which exhibit neurofibrillary degeneration as the disease progresses, and 3-month-old Tg2652 mice, which do not exhibit any fibrillary tau deposits. C57BL/6 (B6) mouse immunostaining included as controls. Insets depict TRC35 immunoreactivity at higher magnification in the CA1. Scale bar = 500 um at low-power magnification and 50 µm in the insets.
TRC35 recognizes a pre-tangle tau epitope in mouse models of tauopathy
To ascertain whether the TRC35 epitope becomes exposed on authentic neuronal fibrillary tau deposits, we investigated two distinct transgenic mouse models of tauopathy, one exhibiting frank NFT accumulation and another that never exhibits fibrillar tau. We examined the perfused brains for P301S mutant human tau transgenic PS19 mice, a well-characterized mouse tauopathy model exhibiting progressive accumulation of Gallyas silver positive NFTs.46 We also utilized another transgenic mouse line driving high-level pan-neuronal WT human tau, Tau4RTg2652, which lacks any fibrillar tau and are Gallyas silver negative even at advanced age.55 Immunostaining for phosphorylated tau (pTau Thr231–AT180) in the mouse hippocampus reveals similar levels of pTau in both tau transgenic strains (Fig. 2D). Immunostaining with TRC35 showed that this tau conformation is not identified in WT mice but is present in both PS19 and Tau4RTg2652 animals. These findings demonstrate that TRC35 labels an epitope exposed by the accumulation of tau conformations occurring relatively early in the tauopathy cascade as it occurs in Tau4RTg2652 animals that will never exhibit frank NFT deposition, but do exhibit prominent pTau and pretangle tau accumulations.
Perturbing poly(A) regulation decreases accumulation of TRC35 immunoreactivity
We previously identified sut-2 as a suppressor of tau-induced neurodegenerative defects in C. elegans.31 The sut-2 gene encodes a zinc finger protein with a single conserved homologue in diverse species ranging from yeast to humans and is thought to regulate poly(A) RNA tail lengths on mRNAs.33,34,56,57MSUT2 is the mammalian homologue of the C. elegans sut-2 gene; post-mortem tissue studies suggest that human MSUT2 protein levels may influence neuronal vulnerability to tau toxicity and aggregation.29,30 Further, MSUT2 gene knockout ameliorates tau neurodegeneration, tau pathology and cognitive deficits in mouse models of tauopathy.29 To test whether the TRC35+ pathological tau lesions are influenced by MSUT2 function, we compared TRC35+ immunoreactivity in the stratum lacunosum moleculare (SLM) of PS19 versus PS19/MSUT2 KO mice. MSUT2 KO significantly decreases accumulation of TRC35 immunoreactivity in the hippocampus of PS19 mice by ∼4-fold (Fig. 3). These findings replicate previous observations for hyperphosphorylated and fibrillar tau in PS19/MSUT2 KO mice.29 Taken together these findings suggest that mAb TRC35 labels pathological tau lesions in mice and that modulating poly(A) RNA availability influences tau/RNA complex formation in tauopathy mouse and worm models.
Figure 3.

MSUT2 influences TRC35 immunoreactivity in PS19 tauopathy mice. (A) Nine-month-old human tau transgenic PS19 tauopathy mice exhibit abundant tau lesions labelled with TRC35 accumulating in the stratum lacunosum moleculare (SLM; arrow). (B) PS19/MSUT2 KO mice exhibit decreased accumulation of TRC35 immunoreactivity in the SLM (arrow). Insets depict TRC35 immunoreactivity at higher magnification in the SLM. (C) Measurement of TRC35+ immunoreactivity in the SLM by quantitative digital microscopy demonstrates ∼4-fold decrease in TRC35+ immunoreactivity in MSUT2 KO mice (***P < 0.0001, statistical comparison made by two-tailed Student’s t-test). Scale bar = 50 µm at low-power magnification and 100 µm in the insets.
TRC35 labels abundant tau species in Alzheimer’s disease but not controls
To further investigate the potential contribution of TRC35 immunoreactivity to the neurodegenerative cascade in tauopathy, we immunostained brain sections from Alzheimer’s disease and control brain tissues with the TRC35 mAb. Immunoreactivity for TRC35 is low but detectable in normal controls with a diffuse neuronal soma staining pattern noticeably lacking detectable neuritic or fibrillary neuropathology (Fig. 4A and D). In the frontal cortex from Alzheimer’s disease brain donors, we observe varying degrees of TRC35 immunoreactivity (Fig. 4B, C, E and F), with some cases exhibiting extensive fibrillar immunoreactivity in cortical neurons, dystrophic neurites and neuropil threads (Fig. 4C and F), while others demonstrated more modest cytoplasmic staining with regions of neuritic pathology (Fig. 4B and E). The abundance of detectable TRC35 immunoreactivity is significantly higher in Alzheimer’s disease as compared to cognitively normal age-matched controls (Fig. 4G). The distribution of TRC35-containing lesions extend throughout the brain of Alzheimer’s disease donors in regions with expected high levels of pathological tau, including the hippocampus and amygdala (Fig. 5A and B). However, unlike in the frontal cortex, lesions in the hippocampus and amygdala were uniformly abundant and robustly TRC35+ across Alzheimer’s disease cases. These brain regions are known to accumulate abundant tau pathology at an earlier stage in the disease process, suggesting TRC35 positive lesions may spread to the frontal cortex at later stages of the disease. TRC35 lesions were not observed in the cerebellum, a region typically spared from tau pathology in Alzheimer’s disease (Fig. 5C). We also examined other tauopathy disorders (Fig. 6). Brain tissue from progressive supranuclear palsy donors exhibited both NFT-like lesions and tufted astrocytes in grey matter (Fig. 6A) and TRC35+ oligodendroglial coils in subcortical white matter (Fig. 6B). Tissues from corticobasal degeneration donors exhibited staining in both TRC35+ neuropil threads and neuronal soma in grey matter and abundant dense TRC35+ neuropil threads in white matter (Fig. 6C and D). Tissues from donors with Pick’s disease exhibited globe-like TRC35+ Pick bodies (Fig. 6E). Taken together, the pattern of neurons exhibiting TRC35 positivity parallels the pattern of pathological pre-tangle and pTau deposition in Alzheimer’s disease and related tauopathy disorders.
Figure 4.

TRC35 labels abundant tau species in Alzheimer’s disease. (A and D) TRC35 immunostaining of normal control autopsy brain tissue reveals diffuse neuronal soma staining in the frontal cortex, with no neuritic or fibrillary neuropathology. (B, C, E and F) TRC35 immunostaining in the frontal cortex of Alzheimer’s disease cases revealed two distinct pathological groups: cases with modest cytoplasmic staining and sporadic regions of neuritic pathology (B and E) and cases with extensive fibrillar immunoreactivity within neuronal soma as well as dystrophic neurites and neuropil threads (C and F). (D–F) Higher magnifications of cortical staining shown in A–C. Scale bar = 100 μm A–C and 50 µm D–F. (G) Measurement of TRC35 immunoreactivity in cases (n = 19) and controls (n = 5) (statistical comparison made by Student’s t-test, P = 0.0124).
Figure 5.
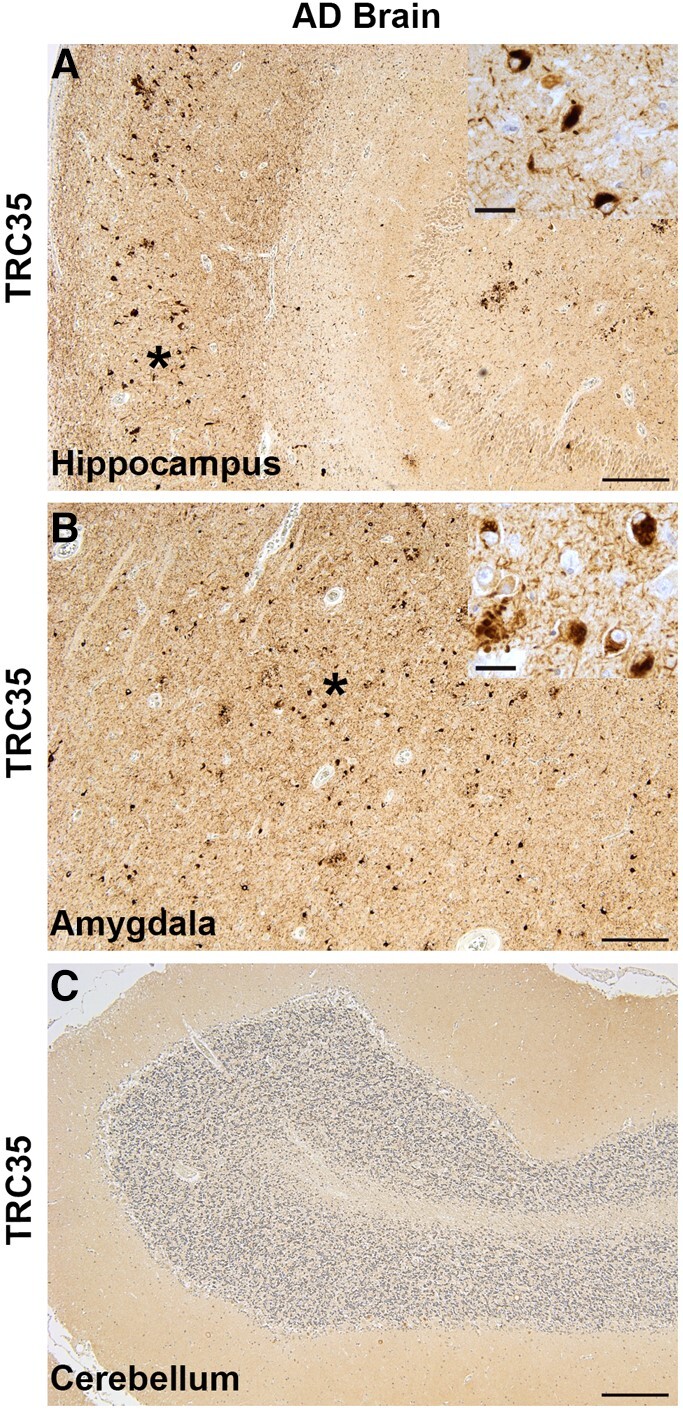
Regional distribution of TRC35 immunoreactivity in Alzheimer’s disease. (A) TRC35 immunostaining of hippocampus sections from Alzheimer’s disease autopsy brain reveal abundant TRC35 immunoreactivity. (B) TRC35 immunostaining of amygdala sections from Alzheimer’s disease autopsy brain reveal abundant TRC35 immunoreactivity. Asterisks indicate the region depicted at higher magnification in the insets in A and B. (C) TRC35 immunostaining of cerebellum sections from Alzheimer’s disease autopsy brain reveal an absence of TRC35 immunoreactivity. Scale bar = 500 μm in low-power images and 50 µm in insets.
Figure 6.

Diverse tauopathy disorders exhibit TRC35 immunoreactivity. (A) Progressive supranuclear palsy cases exhibit TRC35+ NFTs (arrow) and tufted astrocytes (arrowhead) in grey matter. (B) Progressive supranuclear palsy cases also have TRC35+ oliogdendrogial coils (arrowhead) in white matter. (C) Corticobasal degeneration cases exhibited TRC35+ neuronal soma (arrowhead) and neuropil threads in grey matter. (D) Corticobasal degeneration cases also exhibit abundant white matter pathology. (E) Pick’s disease cases exhibit spherical TRC35+ labelled Pick bodies (arrow). (F) Frontal cortex section from brain autopsy of an oculopharyngeal muscular dystrophy case exhibits TRC35 pathological tau deposition including neuropil threads and NFTs (arrowhead). Scale bar = 50 µm.
TRC35 was raised against the tau poly(A)/RNA complex. The nuclear poly(A) binding protein PABPN1 normally functions to protect and extend the length of the poly(A) tail on mRNAs.34,58 We previously showed that PABPN1 also functions to protect against tauopathy in human cells where PABPN1 knockdown exacerbates tau accumulation.25,29 PABPN1 also exhibits a reciprocal pattern of control over tau pathology relative to MSUT2. To assess whether PABPN1 impacts RNA-mediated tau oligomerization we conducted size-exclusion chromatography on TRCs made from recombinant tau and poly(A) RNA. The presence of PABPN1 disrupted oligomeric high molecular weight tau species under size-exclusion fractionation (Supplementary Fig. 6). In human disease, PABPN1 loss of function occurs in oculopharyngeal muscular dystrophy (OPMD) caused by a repeat expansion in the first coding exon of the PABPN1 gene. Neuropathological investigation of rare brain autopsy cases of OPMD revealed clear evidence of tau neuropathology in striatum, substantia nigra, hippocampus, temporal cortex and frontal cortex, while cerebellum was negative (Supplementary Fig. 7); OPMD cases also exhibited prominent TRC35+ tau inclusions in these same regions (as demonstrated in frontal cortex; Fig. 6F). To further assess the interaction between TRC35 and PABPN1, we immunostained the frontal cortex of a collection of Alzheimer’s disease cases for PABPN1. Alzheimer’s disease cases with PABPN1 depletion in the frontal cortex (n = 8; Fig. 7B) exhibited more severe accumulation of pathological tau as measured by TRC35 immunostaining, which included abundant apparent NFTs and dystrophic neurite profiles (Fig. 7D and E). In contrast, cases with normal cortical PABPN1 staining (n = 11; Fig. 7A) exhibited more modest TRC35 immunoreactivity characterized by sparse neuritic immunoreactivity and sporadic NFTs (Fig. 7C and E). Further, dual-label immunofluorescence staining for PABPN1 and TRC35 showed that tangle-bearing TRC35+ neurons appear to exhibit diminished nuclear speckle PABPN1, although this could also be consistent with neurodegenerative changes (Fig. 7F). To explore whether TRC35 immunoreactivity varied in Alzheimer’s disease with age at onset, we examined TRC35+ immunostaining and age of disease onset in post-mortem brain tissue from our collection of Alzheimer’s disease cases; the intensity of TRC35 immunoreactivity in the frontal cortex correlated with the age at disease onset in Alzheimer’s disease (Fig. 7G; Pearson correlation coefficient = −0.59, P = 0.007). Taken together, these findings demonstrate a relationship between TRC35 immunoreactivity and disease severity.
Figure 7.

Interaction between TRC35 and PABPN1. Alzheimer’s disease cases with normal cortical PABPN1 immunoreactivity (A) exhibited more modest TRC35 immunoreactivity characterized by sparse neuritic immunoreactivity and sporadic NFTs (C). In contrast, Alzheimer’s disease cases with PABPN1 depletion in the frontal cortex (B) exhibited more severe accumulation of pathological tau as measured by TRC35 immunostaining that included abundant apparent NFTs and dystrophic neurite profiles (D). (E) Densitometry analysis of TRC35 positive immunoreactivity in PABPN1-positive Alzheimer’s disease cases (n = 11) compared to PABPN1-depleted Alzheimer’s disease cases (n = 8) (**P = 0.001 by two-tailed Student’s t-test). Scale bar = 100 µm. (F) Dual-label immunofluorescence for PABPN1 (red) and TRC35 (green) in Alzheimer’s disease cases. (G) TRC35 immunostaining was quantitated and graphed with the age of onset for each Alzheimer’s disease case analysed (n = 19). TRC35 immunoreactivity correlated with the age of disease onset in Alzheimer’s disease (Pearson correlation coefficient = 0.59, P = 0.007).
Discussion
The genesis of pathological tau in normal ageing, Alzheimer’s disease and other tauopathy disorders remains incompletely understood. Pathological assembly of tau represents a gain of toxic function that disrupts cellular activities, contributing to neuronal demise in many ageing-related neurodegenerative disorders. Thorough study in Alzheimer’s disease has demonstrated a clear connection between accumulation of tau-containing pathological lesions, cognitive decline and neuronal loss.2,59 Early work in the field of tau biology identified RNA binding as a potential modulator of tau microtubule polymerization activity.26,60 Subsequently, a variety of distinct RNA binding proteins have been implicated as modifiers of tau accumulation and neurotoxicity.24,28,29,61-67 Among these RNA-binding proteins, MSUT2, PABPN1, TIA, PAP and ALYREF directly interact with either poly(A) RNA or known poly(A) RNA binding proteins, and MSUT2 and PABPN1 can influence tauopathy-related phenotypes and poly(A) tail lengths on mRNAs.29,56,58 Taken together, these previous observations led us to test the hypothesis that tau binding to RNA leads to pathological consequences because tau/RNA binding precludes tau MT dimer binding, impacting MT assembly (Fig. 1). Furthermore, coexpression of tau and poly(A)45 in C. elegans neurons exacerbates tauopathy-related behavioural phenotypes (Fig. 1D) and also exacerbates accumulation of pathological tau species including total and phosphorylated tau (Fig. 1E and F). We hypothesize the molecular mechanism of this tauopathy exacerbation occurs because poly(A) RNA abundance exceeds poly(A) RNABP capacity thus exposing tau to naked RNA driving tau/RNA binding, subsequent tau oligomerization and impaired tau proteostasis. Our future work will explore this idea further through the working hypothesis outlined in Fig. 8, but initial evidence shown in Fig. 1E and F suggest reduced tau turnover drives accumulation of a broad range of pathological tau species including pTau. However, mechanistic studies in mammalian model systems will be needed to fully establish the disease-relevant mechanisms.
Figure 8.

The role of RNA binding in tau neuropathology and MT function. Illustration of hypothesis relating tau/RNA interaction with MT function and tau neuropathology. Previous work has shown tau binds poly(A) RNA.26 Under normal conditions, RNA binding proteins shield poly(A) RNA and there is little exposed poly(A) RNA for tau to bind. Under pathological conditions, poly(A) RNA binding proteins are lost and poly(A) RNA becomes exposed. Poly(A) RNA has a higher affinity for tau than do tubulin dimers and RNA seeds tau aggregation leading to both tau loss of function from MTs and tau TRC formation.
We demonstrated that tau binds RNA with high affinity, but low specificity, although it exhibits some preference for unstructured RNA over structured tRNA. Tau binds RNA with higher affinity than tubulin dimers, and RNA binding precludes tubulin binding to tau. Thus, RNA competes with tubulin for tau. We recapitulated previous observations that poly(A) RNA inhibits tau activity in promoting microtubule polymerization, consistent with our binding studies. Further, when bound to poly(A) RNA, tau readily assembles into medium-N oligomers, suggesting the formation of tau/RNA complexes may be on pathway to pathological aggregation. Coexpression of poly(A) RNA and tau drives stronger tauopathy-related phenotypes including neuronal dysfunction and pathological tau accumulation (Fig. 1). To develop affinity reagents for probing the potential disease relevance of TRCs, we produced TRCs in vitro and employed them as an immunogen. From TRC-immunized mice, we isolated hybridoma lines expressing TRC mAbs exhibiting strong preference for aggregated human Alzheimer’s disease-derived pathological tau over recombinant soluble tau (Fig. 2).
We characterized the TRC35 mAb by immunohistology and observed it specifically stains tau lesions in transgenic mouse brains from both the Tau4RTg2652 and PS19 models of tauopathy, but not non-transgenic mice. The neuropathological characterization of TRC35 immunoreactivity in PS19 and Tau4RTg2652 animals demonstrates that tau/RNA complexes occur prior to tau fibrillization in neurons. We also examined the consequence of MSUT2 knockout in the PS19 tauopathy model, which results in dramatic decreases in other pathological tau species and observed MSUT2 KO mice exhibited reduced accumulation of TRC35 immunoreactivity in the hippocampus.
To explore the potential disease relevance of the accumulation of TRCs, we examined post-mortem brain tissue from tauopathy donors. In Alzheimer’s disease, we observed TRC35 positive somatodendritic staining, dystrophic neurites, neuropil threads and frank tangles consistent with TRC depositing with pathological tau. Characterization of Alzheimer’s disease and other tauopathy cases revealed that TRC35 labels pathological tau deposits in both 4R tauopathies (PSP and CBD) and 3R tauopathies (Pick’s disease). Further, both 3R and 4R tau show high affinity for poly(A) RNA (Fig. 1). Examination of OPMD, a neuromuscular disease with onset of dementia caused by mutations in PABPN1, revealed evidence for tauopathy including pTau and TRC35 positive lesions. To explore the relationship between TRC35 and RNA binding proteins, we characterized our cohort of Alzheimer’s disease cases with respect to PABPN1. We determined a subset of Alzheimer’s disease cases exhibited depletion of PABPN1 from the nucleus and robust accumulation of TRC35 lesions. Our working hypothesis is that tau binds RNA26 and it may have greater access to RNA in these Alzheimer’s disease cases in part because of reduced PABPN1 abundance competing for poly(A) RNA binding (Fig. 8). This idea can also be extended to other disorders including Alzheimer’s disease and related disorders where depletion of RNA binding proteins occurs and may expose cytoplasmic RNAs to tau, thereby promoting tauopathy through increased RNA-mediated tau oligomerization. Our findings support this hypothesis as TRC35 levels correlate with the age of Alzheimer’s disease onset (Fig. 7G). Taken together, these observations support the hypothesis that tau protein access to poly(A) RNA may be constrained by RNA-binding proteins.
Emerging evidence has shown that changes in RNA metabolism/processing through the actions of RNA-binding proteins modulate pathological tau accumulation. Our previous work modelling tauopathy in C. elegans has uncovered genes encoding RNA binding proteins as translationally relevant modifiers of tauopathy, including sut-1, sut-2/MSUT2, parn-2/TOE1, aly-1, aly-2 and aly-3.28-32,68 Parallel work has identified other poly(A) RNA-binding proteins like T-cell intracellular antigen 1 (TIA1), which co-localizes with phase-separated tau in stress granules and promotes fibrillary deposits of pathological tau.63,69,70 Independently, the RNA-binding protein Musashi (MSI) was found to associate with pathological tau oligomers and drive nuclear dysfunction.64,71 Multiple studies have demonstrated that tau neuropathology drives neurodegeneration by causing dysfunction of nuclear RNA processing events (reviewed by Diez and Wegmann72). For instance, spliceosome abnormalities have been hypothesized to cause cryptic RNA splicing leading to neurodegeneration in Alzheimer’s disease and related disorders.73 In addition, pathological tau can impair multiple nuclear functions including recruiting the nuclear speckle resident splicing protein SRRM2 into cytoplasmic aggregates24,25 and impairing nuclear pore complex function via co-aggregation of tau with the disordered region of Nup98.14 Other defects have been found with the U1–70 K, which disrupts U1 snRNP function in splicing.74-76 Another alternative splicing factor, TDP-43, has been shown to synergize with pathological tau in the context of Alzheimer’s disease, perhaps through an RNA-binding mechanism.77-79
Previous work has implicated poly(A) RNA both as a strong influencer of tau MT stabilizing activity26 and a potent driver of tau aggregation.16 Consistent with this, we show above that tau binds poly(A) RNA with much greater affinity than tubulin dimers (Supplementary Table 1) or polymerized MTs51,52 and disrupts tau-dependent MT assembly (Fig. 1A). In HeLa cells, cytoplasmic RNA appears approximately 3-fold more abundant than tubulin (∼27 pg RNA in HeLa cell cytoplasm80,81 versus ∼7 pg tubulin82,83). The relative abundance of tubulin versus RNA remains unknown in human brain neurons, but the above approximation suggests tubulin/RNA competition for tau binding should be considered as a possible factor relevant to tau aggregation. Thus, superficially, tau may bind RNA preferentially when other RNA binding proteins fail to shield cytoplasmic RNA from access to tau. Clearly many other RNA binding proteins play a role in modulating tauopathy,24,25,63,64,69-71,74-79 which, in sum, supports the proposed hypothetical mechanism relating tau/RNA binding with tau MT function and tau neuropathology. Here we demonstrate that PABPN1 loss of function in OPMD provokes pathological tau accumulation (Fig. 6). Further support for this hypothesis comes from the observation that RNA binds to tau fibrils and is required to sustain multiple rounds of seeding competent tau using in vitro propagation of tau-derived seeds to drive fibril formation.84
Prior study of tau aggregation yielded a diverse group of monoclonal antibodies with high utility that define conformation specific epitopes including Alz50 (pre-tangle tau85), MC1 (pretangle tau86,87), TOC1 (oligomer/dimer53), TOMA (oligomer88) and GT38 (fibrillar tau89). To this robust collection of conformation-dependent selective tau monoclonal antibodies, we now propose the addition of TRC35 for detecting pathological tau oligomers seeded by RNA or perhaps other polyanions. Note that for the majority of these conformation-dependent antibodies, the epitope has not been mapped, but in the case of TRC35 the complex epitope consists of two discontinuous peptide motifs flanking the tau proline-rich domain, which is predicted to be an unstructured region in tau fibril cores for all tauopathies solved to date (reviewed by Goedert8). We hypothesize pathological RNA access to cytoplasmic tau through RNA-binding protein deficiency or ribostatic derangement drives the accumulation of tau oligomers exposing the TRC35 epitope on tau-positive lesions in Alzheimer’s disease and related dementia disorders. However, it remains possible that other polyanions could promote TRC35 reactivity in specific circumstances (for instance with extracellular tau). We propose that greater understanding of ribostasis in human neurons will illuminate new molecular mechanisms of disease by which TRC35 accumulation leads to tauopathy.
Conclusions
Our work highlights the need for the field to further consider the previous literature implicating RNA binding in tau function and dysfunction (reviewed by Limorenko and Lashuel7). Here we present evidence supporting the disease relevance of TRCs. The observation that RNA seeded tau aggregates occur in disease suggests a renewed focus on the RNA binding activity of tau in pathology should be considered both from a basic tauopathy disease mechanisms standpoint and from the standpoint of therapeutic targeting of pathological tau for Alzheimer’s disease and related disorders. In particular, therapeutic development should take into account the possibility that targeting tau may differentially influence tau/RNA binding and MT binding activities.
Supplementary Material
Acknowledgements
We thank Elaine Loomis, Brandon Henderson and Jade Stair for outstanding research assistance. We thank Erica Melief and Aimee Schantz for outstanding administrative assistance and Lisa Keene, Katelyn Kern and Amanda Keen for support in tissue collection. We thank Victoria Hulubei for assistance with mAb purification and production. We thank Peter Davies and Virginia Lee for sharing anti-tau antibodies.
Contributor Information
Pamela J McMillan, Department of Psychiatry and Behavioral Sciences, University of Washington, Seattle, WA 98195, USA; Geriatrics Research Education and Clinical Center, Veterans Affairs Puget Sound Health Care System, Seattle, WA 98108, USA.
Sarah J Benbow, Geriatrics Research Education and Clinical Center, Veterans Affairs Puget Sound Health Care System, Seattle, WA 98108, USA; Division of Gerontology and Geriatric Medicine, Department of Medicine, University of Washington, Seattle, WA 98104, USA.
Rikki Uhrich, Geriatrics Research Education and Clinical Center, Veterans Affairs Puget Sound Health Care System, Seattle, WA 98108, USA.
Aleen Saxton, Geriatrics Research Education and Clinical Center, Veterans Affairs Puget Sound Health Care System, Seattle, WA 98108, USA.
Misa Baum, Geriatrics Research Education and Clinical Center, Veterans Affairs Puget Sound Health Care System, Seattle, WA 98108, USA.
Timothy Strovas, Geriatrics Research Education and Clinical Center, Veterans Affairs Puget Sound Health Care System, Seattle, WA 98108, USA.
Jeanna M Wheeler, Geriatrics Research Education and Clinical Center, Veterans Affairs Puget Sound Health Care System, Seattle, WA 98108, USA.
Jeremy Baker, Geriatrics Research Education and Clinical Center, Veterans Affairs Puget Sound Health Care System, Seattle, WA 98108, USA; Division of Gerontology and Geriatric Medicine, Department of Medicine, University of Washington, Seattle, WA 98104, USA.
Nicole F Liachko, Geriatrics Research Education and Clinical Center, Veterans Affairs Puget Sound Health Care System, Seattle, WA 98108, USA; Division of Gerontology and Geriatric Medicine, Department of Medicine, University of Washington, Seattle, WA 98104, USA.
C Dirk Keene, Department of Laboratory Medicine and Pathology, University of Washington, Seattle, WA 98195, USA.
Caitlin S Latimer, Department of Laboratory Medicine and Pathology, University of Washington, Seattle, WA 98195, USA.
Brian C Kraemer, Department of Psychiatry and Behavioral Sciences, University of Washington, Seattle, WA 98195, USA; Geriatrics Research Education and Clinical Center, Veterans Affairs Puget Sound Health Care System, Seattle, WA 98108, USA; Division of Gerontology and Geriatric Medicine, Department of Medicine, University of Washington, Seattle, WA 98104, USA; Department of Laboratory Medicine and Pathology, University of Washington, Seattle, WA 98195, USA.
Funding
This work was supported by grants from the Department of Veterans Affairs (Merit Review Grant #I01BX002619 to B.K. and #I01BX004044 N.L.); National Institutes of Health (# RF1AG055474 and # R01NS064131 to B.K., R01AG066729 to N.L., K08 AG065426 to C.L., P30 AG066509 and U19 AG066567 to C.D.K. for the UW ADRC and Adult Changes in Thought study, respectively) as well as the Nancy and Buster Alvord endowment (C.D.K.).
Competing interests
The authors report no competing interests.
Supplementary material
Supplementary material is available at Brain online.
References
- 1. Hyman BT, Gomez-Isla T. The natural history of Alzheimer neurofibrillary tangles and amyloid deposits. Neurobiol Aging. 1997;18:386–387; discussion 389-92. [DOI] [PubMed] [Google Scholar]
- 2. Gómez-Isla T, Hollister R, West H, et al. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann Neurol. 1997;41:17–24. [DOI] [PubMed] [Google Scholar]
- 3. Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology. 1992;42(3 Pt 1):631–639. [DOI] [PubMed] [Google Scholar]
- 4. Holtzman DM, Carrillo MC, Hendrix JA, et al. Tau: From research to clinical development. Alzheimers Dement. 2016;12:1033–1039. [DOI] [PubMed] [Google Scholar]
- 5. Ballatore C, Lee VM, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat Rev Neurosci. 2007;8:663–672. [DOI] [PubMed] [Google Scholar]
- 6. Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW. A protein factor essential for microtubule assembly. Proc Natl Acad Sci USA. 1975;72:1858–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Limorenko G, Lashuel HA. Revisiting the grammar of tau aggregation and pathology formation: How new insights from brain pathology are shaping how we study and target tauopathies. Chem Soc Rev. 2022;51:513–565. [DOI] [PubMed] [Google Scholar]
- 8. Goedert M. Cryo-EM structures of tau filaments from human brain. Essays Biochem. 2021;65:949–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Scheres SH, Zhang W, Falcon B, Goedert M. Cryo-EM structures of tau filaments. Curr Opin Struct Biol. 2020;64:17–25. [DOI] [PubMed] [Google Scholar]
- 10. Zhang W, Tarutani A, Newell KL, et al. Novel tau filament fold in corticobasal degeneration. Nature. 2020;580:283–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Falcon B, Zhang W, Murzin AG, et al. Structures of filaments from Pick’s disease reveal a novel tau protein fold. Nature. 2018;561:137–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fitzpatrick AWP, Falcon B, He S, et al. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature. 2017;547:185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wegmann S, Eftekharzadeh B, Tepper K, et al. Tau protein liquid–liquid phase separation can initiate tau aggregation. EMBO J. 2018;37:e98049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Eftekharzadeh B, Daigle JG, Kapinos LE, et al. Tau protein disrupts nucleocytoplasmic transport in Alzheimer’s disease. Neuron. 2018;99:925–940.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang Y, Mandelkow E. Tau in physiology and pathology. Nat Rev Neurosci. 2016;17:5–21. [DOI] [PubMed] [Google Scholar]
- 16. Kampers T, Friedhoff P, Biernat J, Mandelkow EM, Mandelkow E. RNA stimulates aggregation of microtubule-associated protein tau into Alzheimer-like paired helical filaments. FEBS Lett. 1996;399:344–349. [DOI] [PubMed] [Google Scholar]
- 17. Zhang X, Lin Y, Eschmann NA, et al. RNA stores tau reversibly in complex coacervates. PLoS Biol. 2017;15:e2002183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Crowe A, Ballatore C, Hyde E, Trojanowski JQ, Lee VM. High throughput screening for small molecule inhibitors of heparin-induced tau fibril formation. Biochem Biophys Res Commun. 2007;358:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Barghorn S, Mandelkow E. Toward a unified scheme for the aggregation of tau into Alzheimer paired helical filaments. Biochemistry. 2002;41:14885–14896. [DOI] [PubMed] [Google Scholar]
- 20. Al-Bassam J, Ozer RS, Safer D, Halpain S, Milligan RA. MAP2 and tau bind longitudinally along the outer ridges of microtubule protofilaments. J Cell Biol. 2002;157:1187–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ginsberg SD, Crino PB, Lee VM, Eberwine JH, Trojanowski JQ. Sequestration of RNA in Alzheimer’s disease neurofibrillary tangles and senile plaques. Ann Neurol. 1997;41:200–209. [DOI] [PubMed] [Google Scholar]
- 22. Ginsberg SD, Galvin JE, Chiu TS, Lee VMY, Masliah E, Trojanowski JQ. RNA sequestration to pathological lesions of neurodegenerative diseases. Acta Neuropathol. 1998;96:487–494. [DOI] [PubMed] [Google Scholar]
- 23. Ginsberg SD, Hemby SE, Lee VM, Eberwine JH, Trojanowski JQ. Expression profile of transcripts in Alzheimer’s disease tangle-bearing CA1 neurons. Ann Neurol. 2000;48:77–87. [PubMed] [Google Scholar]
- 24. Lester E, Ooi FK, Bakkar N, et al. Tau aggregates are RNA–protein assemblies that mislocalize multiple nuclear speckle components. Neuron. 2021;109:1675–1691.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McMillan PJ, Strovas TJ, Baum M, et al. Pathological tau drives ectopic nuclear speckle scaffold protein SRRM2 accumulation in neuron cytoplasm in Alzheimer’s disease. Acta Neuropathol Commun. 2021;9:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bryan JB, Nagle BW, Doenges KH. Inhibition of tubulin assembly by RNA and other polyanions: Evidence for a required protein. Proc Natl Acad Sci USA. 1975;72:3570–3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kow RL, Black AH, Saxton AD, Liachko NF, Kraemer BC. Loss of aly/ALYREF suppresses toxicity in both tau and TDP-43 models of neurodegeneration. Geroscience. 2022;44:747–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kow RL, Strovas TJ, McMillan PJ, et al. Distinct poly(A) nucleases have differential impact on sut-2 dependent tauopathy phenotypes. Neurobiol Dis. 2021;147:105148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wheeler JM, McMillan P, Strovas TJ, et al. Activity of the poly(A) binding protein MSUT2 determines susceptibility to pathological tau in the mammalian brain. Sci Transl Med. 2019;11:eaao6545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Guthrie CR, Greenup L, Leverenz JB, Kraemer BC. MSUT2 is a determinant of susceptibility to tau neurotoxicity. Hum Mol Genet. 2011;20:1989–1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Guthrie CR, Schellenberg GD, Kraemer BC. SUT-2 potentiates tau-induced neurotoxicity in Caenorhabditis elegans. Hum Mol Genet. 2009;18:1825–1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kraemer BC, Schellenberg GD. SUT-1 enables tau-induced neurotoxicity in C. elegans. Hum Mol Genet. 2007;16:1959–1971. [DOI] [PubMed] [Google Scholar]
- 33. Kelly SM, Bienkowski R, Banerjee A, et al. The Drosophila ortholog of the Zc3h14 RNA binding protein acts within neurons to pattern axon projection in the developing brain. Dev Neurobiol. 2016;76:93–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kelly SM, Leung SW, Pak C, Banerjee A, Moberg KH, Corbett AH. A conserved role for the zinc finger polyadenosine RNA binding protein, ZC3H14, in control of poly(A) tail length. RNA. 2014;20:681–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Baker JD, Shelton LB, Zheng D, et al. Human cyclophilin 40 unravels neurotoxic amyloids. PLoS Biol. 2017;15:e2001336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hong M, Zhukareva V, Vogelsberg-Ragaglia V, et al. Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science. 1998;282:1914–1917. [DOI] [PubMed] [Google Scholar]
- 37. Barghorn S, Davies P, Mandelkow E. Tau paired helical filaments from Alzheimer’s disease brain and assembled in vitro are based on beta-structure in the core domain. Biochemistry. 2004;43:1694–1703. [DOI] [PubMed] [Google Scholar]
- 38. Kiris E, Ventimiglia D, Sargin ME, et al. Combinatorial tau pseudophosphorylation: markedly different regulatory effects on microtubule assembly and dynamic instability than the sum of the individual parts. J Biol Chem. 2011;286:14257–14270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dujardin S, Commins C, Lathuiliere A, et al. Tau molecular diversity contributes to clinical heterogeneity in Alzheimer’s disease. Nat Med. 2020;26(8):1256–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Taylor LM, McMillan PJ, Liachko NF, et al. Pathological phosphorylation of tau and TDP-43 by TTBK1 and TTBK2 drives neurodegeneration. Mol Neurodegener. 2018;13:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mariol MC, Walter L, Bellemin S, Gieseler K. A rapid protocol for integrating extrachromosomal arrays with high transmission rate into the C. elegans genome. J Vis Exp. 2013;82:e50773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Elnaggar MM, Abdellrazeq GS, Venn-Watson SK, et al. Identification of monoclonal antibodies cross-reactive with bottlenose dolphin orthologues of the major histocompatibility complex and leukocyte differentiation molecules. Vet Immunol Immunopathol. 2017;192:54–59. [DOI] [PubMed] [Google Scholar]
- 44. Dawson HN, Ferreira A, Eyster MV, Ghoshal N, Binder LI, Vitek MP. Inhibition of neuronal maturation in primary hippocampal neurons from tau deficient mice. J Cell Sci. 2001;114:1179–1187. [DOI] [PubMed] [Google Scholar]
- 45. Guo JL, Narasimhan S, Changolkar L, et al. Unique pathological tau conformers from Alzheimer’s brains transmit tau pathology in nontransgenic mice. J Exp Med. 2016;213:2635–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yoshiyama Y, Higuchi M, Zhang B, et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–351. [DOI] [PubMed] [Google Scholar]
- 47. Chaudhuri AD, Kabaria S, Choi DC, Mouradian MM, Junn E. MicroRNA-7 promotes glycolysis to protect against 1-methyl-4-phenylpyridinium-induced cell death. J Biol Chem. 2015;290:12425–12434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Choi SH, Kim YH, Hebisch M, et al. A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature. 2014;515:274–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Amano R, Sakamoto T. Kinetic and thermodynamic analyses of RNA–protein interactions. Methods Mol Biol. 2020;2106:137–150. [DOI] [PubMed] [Google Scholar]
- 50. Matos RG, Barbas A, Arraiano CM. Comparison of EMSA and SPR for the characterization of RNA–RNase II complexes. Protein J. 2010;29:394–397. [DOI] [PubMed] [Google Scholar]
- 51. Butner KA, Kirschner MW. Tau protein binds to microtubules through a flexible array of distributed weak sites. J Cell Biol. 1991;115:717–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kadavath H, Hofele RV, Biernat J, et al. Tau stabilizes microtubules by binding at the interface between tubulin heterodimers. Proc Natl Acad Sci USA. 2015;112:7501–7506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ward SM, Himmelstein DS, Lancia JK, Fu Y, Patterson KR, Binder LI. TOC1: Characterization of a selective oligomeric tau antibody. J Alzheimers Dis. 2013;37:593–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ward SM, Himmelstein DS, Lancia JK, Binder LI. Tau oligomers and tau toxicity in neurodegenerative disease. Biochem Soc Trans. 2012;40:667–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wheeler JM, McMillan PJ, Hawk M, et al. High copy wildtype human 1N4R tau expression promotes early pathological tauopathy accompanied by cognitive deficits without progressive neurofibrillary degeneration. Acta Neuropathol Commun. 2015;3:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rha J, Jones SK, Fidler J, et al. The RNA-binding protein, ZC3H14, is required for proper poly(A) tail length control, expression of synaptic proteins, and brain function in mice. Hum Mol Genet. 2017;26:3663–3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Soucek S, Corbett AH, Fasken MB. The long and the short of it: The role of the zinc finger polyadenosine RNA binding protein, Nab2, in control of poly(A) tail length. Biochim Biophys Acta. 2012;1819:546–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Leung SW, Apponi LH, Cornejo OE, et al. Splice variants of the human ZC3H14 gene generate multiple isoforms of a zinc finger polyadenosine RNA binding protein. Gene. 2009;439:71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Nelson PT, Alafuzoff I, Bigio EH, et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: A review of the literature. J Neuropathol Exp Neurol. 2012;71:362–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Schroder HC, Bernd A, Zahn RK, Müller WE. Binding of polyribonucleotides and polydeoxyribonucleotides to bovine brain microtubule protein: Age-dependent modulation via phosphorylation of high-molecular-weight microtubule-associated proteins and tau proteins. Mech Ageing Dev. 1984;24:101–117. [DOI] [PubMed] [Google Scholar]
- 61. Vanderweyde T, Apicco DJ, Youmans-Kidder K, et al. Interaction of tau with the RNA-binding protein TIA1 regulates tau pathophysiology and toxicity. Cell Rep. 2016;15:1455–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Apicco DJ, Ash PEA, Maziuk B, et al. Reducing the RNA binding protein TIA1 protects against tau-mediated neurodegeneration in vivo. Nat Neurosci. 2018;21:72–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ash PEA, Lei S, Shattuck J, et al. TIA1 potentiates tau phase separation and promotes generation of toxic oligomeric tau. Proc Natl Acad Sci USA. 2021;118:e2014188118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Montalbano M, McAllen S, Puangmalai N, et al. RNA-binding proteins Musashi and tau soluble aggregates initiate nuclear dysfunction. Nat Commun. 2020;11:4305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tanaka H, Kondo K, Chen X, et al. The intellectual disability gene PQBP1 rescues Alzheimer’s disease pathology. Mol Psychiatry. 2018;23:2090–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kosik KS, Han S. Tau condensates. Adv Exp Med Biol. 2019;1184:327–339. [DOI] [PubMed] [Google Scholar]
- 67. Taylor LM, McMillan PJ, Kraemer BC, Liachko NF. Tau tubulin kinases in proteinopathy. FEBS J. 2019;286:2434–2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. MacMorris M, Kumar M, Lasda E, Larsen A, Kraemer B, Blumenthal T. A novel family of C. elegans snRNPs contains proteins associated with trans-splicing. RNA. 2007;13:511–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Jiang L, Ash PEA, Maziuk BF, et al. TIA1 regulates the generation and response to toxic tau oligomers. Acta Neuropathol. 2019;137:259–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Maziuk BF, Apicco DJ, Cruz AL, et al. RNA binding proteins co-localize with small tau inclusions in tauopathy. Acta Neuropathol Commun. 2018;6:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sengupta U, Montalbano M, McAllen S, Minuesa G, Kharas M, Kayed R. Formation of toxic oligomeric assemblies of RNA-binding protein: Musashi in Alzheimer’s disease. Acta Neuropathol Commun. 2018;6:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Diez L, Wegmann S. Nuclear transport deficits in tau-related neurodegenerative diseases. Front Neurol. 2020;11:1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hsieh YC, Guo C, Yalamanchili HK, et al. Tau-mediated disruption of the spliceosome triggers cryptic RNA splicing and neurodegeneration in Alzheimer’s disease. Cell Rep. 2019;29:301–316.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hales CM, Dammer EB, Diner I, et al. Aggregates of small nuclear ribonucleic acids (snRNAs) in Alzheimer’s disease. Brain Pathol. 2014;24:344–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Bishof I, Dammer EB, Duong DM, et al. RNA-binding proteins with basic–acidic dipeptide (BAD) domains self-assemble and aggregate in Alzheimer’s disease. J Biol Chem. 2018;293:11047–11066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Hales CM, Dammer EB, Deng Q, et al. Changes in the detergent-insoluble brain proteome linked to amyloid and tau in Alzheimer’s disease progression. Proteomics. 2016;16:3042–3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Latimer CS, Liachko NF. Tau and TDP-43 synergy: A novel therapeutic target for sporadic late-onset Alzheimer’s disease. Geroscience. 2021;43:1627–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Latimer CS, Burke BT, Liachko NF, et al. Resistance and resilience to Alzheimer’s disease pathology are associated with reduced cortical pTau and absence of limbic-predominant age-related TDP-43 encephalopathy in a community-based cohort. Acta Neuropathol Commun. 2019;7:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Tomé SO, Gomes LA, Li X, Vandenberghe R, Tousseyn T, Thal DR. TDP-43 interacts with pathological tau protein in Alzheimer’s disease. Acta Neuropathol. 2021;141:795–799. [DOI] [PubMed] [Google Scholar]
- 80. ThermoFisher Scientific . RNA Yields. https://www.thermofisher.com/us/en/home/references/ambion-tech-support/rna-tools-and-calculators/macromolecular-components-of-e.html.
- 81. Piwnicka M, Darzynkiewicz Z, Melamed MR. RNA and DNA content of isolated cell nuclei measured by multiparameter flow cytometry. Cytometry. 1983;3:269–275. [DOI] [PubMed] [Google Scholar]
- 82. Finka A, Goloubinoff P. Proteomic data from human cell cultures refine mechanisms of chaperone-mediated protein homeostasis. Cell Stress Chaperones. 2013;18:591–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Bulinski JC, Borisy GG. Self-assembly of microtubules in extracts of cultured HeLa cells and the identification of HeLa microtubule-associated proteins. Proc Natl Acad Sci USA. 1979;76:293–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Dinkel PD, Holden MR, Matin N, Margittai M. RNA binds to tau fibrils and sustains template-assisted growth. Biochemistry. 2015;54:4731–4740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Wolozin BL, Pruchnicki A, Dickson DW, Davies P. A neuronal antigen in the brains of Alzheimer patients. Science. 1986;232:648–650. [DOI] [PubMed] [Google Scholar]
- 86. Jicha GA, Bowser R, Kazam IG, Davies P. Alz-50 and MC-1, a new monoclonal antibody raised to paired helical filaments, recognize conformational epitopes on recombinant tau. J Neurosci Res. 1997;48:128–132. [DOI] [PubMed] [Google Scholar]
- 87. Weaver CL, Espinoza M, Kress Y, Davies P. Conformational change as one of the earliest alterations of tau in Alzheimer’s disease. Neurobiol Aging. 2000;21:719–727. [DOI] [PubMed] [Google Scholar]
- 88. Castillo-Carranza DL, Gerson JE, Sengupta U, Guerrero-Munoz MJ, Lasagna-Reeves CA, Kayed R. Specific targeting of tau oligomers in Htau mice prevents cognitive impairment and tau toxicity following injection with brain-derived tau oligomeric seeds. J Alzheimers Dis. 2014;40(Suppl 1):S97–S111. [DOI] [PubMed] [Google Scholar]
- 89. Gibbons GS, Banks RA, Kim B, et al. Detection of Alzheimer disease (AD)-specific tau pathology in AD and NonAD tauopathies by immunohistochemistry with novel conformation-selective tau antibodies. J Neuropathol Exp Neurol. 2018;77:216–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Upon reasonable request, the data described in this study are available from the corresponding author.