

Abstract
Relationships between novel phenotypic behaviors and specific genetic alterations are often discovered using target‐specific, directed mutagenesis or phenotypic selection following chemical mutagenesis. An alternative approach is to exploit deficiencies in DNA repair pathways that maintain genetic integrity in response to spontaneously induced damage. Mice deficient in the DNA glycosylase NEIL1 show elevated spontaneous mutations, which arise from translesion DNA synthesis past oxidatively induced base damage. Several litters of Neil1 knockout mice included animals that were distinguished by their backwards‐walking behavior in open‐field environments, while maintaining frantic forward movements in their home cage environment. Other phenotypic manifestations included swim test failures, head tilting and circling. Mapping of the mutation that conferred these behaviors showed the introduction of a stop codon at amino acid 4 of the Ush1g gene. Ush1g bw/bw null mice displayed auditory and vestibular defects that are commonly seen with mutations affecting inner‐ear hair‐cell function, including a complete lack of auditory brainstem responses and vestibular‐evoked potentials. As in other Usher syndrome type I mutant mouse lines, hair cell phenotypes included disorganized and split hair bundles, as well as altered distribution of proteins for stereocilia that localize to the tips of row 1 or row 2. Disruption to the bundle and kinocilium displacement suggested that USH1G is essential for forming the hair cell's kinocilial links. Consistent with other Usher type 1 models, Ush1g bw/bw mice had no substantial retinal degeneration compared with Ush1g bw /+ controls. In contrast to previously described Ush1g alleles, this new allele provides the first knockout model for this gene.
Keywords: backward walking mouse, cochlea, DNA repair, hair cell, hyperactivity, retina, stereocilia
We identified a novel allele of Ush1g from mice deficient in the DNA glycosylase NEIL1, which show elevated spontaneous mutations arising from translesion DNA synthesis past oxidatively induced base damage. The Ush1g bw mice have no visual phenotype, consistent with other mouse models of Usher I syndrome, but severe defects in auditory and vestibular function.
Abbreviations
- ABR
auditory brainstem response
- GCL
ganglion cell layer
- INL
inner nuclear layer
- ONL
outer nuclear layer
- RPE
retinal pigmented epithelium
- SEM
standard error of the mean
- SEM
scanning electron microscopy
- USH1
Usher I
- VsEP
vestibular evoked potential
1. INTRODUCTION
Strategies to identify and establish the functional significance of genes and their products has been pioneered through manipulation of genomes via increasingly complex experimental designs. 1 Many of these advances have been reduced to practice using the mouse genome as an experimental and screening platform. Insights gained into establishing the functional significance of individual genes has provided both the foundation from which to understand specific structure–function relationships and the framework to dissect complex pathways and processes. Although not a perfect surrogate for analyses of human genes or therapeutic design and validation, the mouse genome shares an 80% synteny with the organization of the human genome and a high percentage of readily identifiable orthologs. Thus, analyses of the phenotypic consequences of genotypic manipulation in the mouse serves as a strong foundational point from which to understand human disease. 1 The International Mouse Genotyping Consortium, whose primary objective is to coordinate the cataloging, design, production and documentation of all human gene orthologs found within the mouse genome is close to achieving this goal, with documentation of nearly 17,000 gene knockouts and covering nearly 80% of the target goal. 2
Along this discovery pathway, significant advances were made via the observation and collection of mouse phenotypes arising from not only spontaneous mutations, but also large‐scale, random mutagenesis induced by exposures to ionizing radiation and chemical mutagenesis. 1 For strategies organized around chemical mutagenesis, several large international consortia utilized DNA alkylating agents such as ethylnitrosourea and ethylmethanesulfonate to create widespread base damage, located primarily on purines. The expectation was that these base modifications would ultimately lead to randomized mutagenesis of the entire genome. However, to mitigate the potentially deleterious effects of these base damages, cells possess a variety of DNA repair pathways, with small molecule DNA alkylation damage primarily removed by the base excision repair pathway (BER). BER consists of a series of coordinated sequential steps that are initiated by DNA glycosylases, which scan the genome for altered bases, flip the damaged nucleotide into a catalytic pocket, and initiate chemistry for base release via glycosyl bond cleavage, with the potential cleavage of the phosphodiester backbone. Following base release, the remainder of the pathway is devoted to the creation and filling in of short gaps in the strand from which the base was released followed by sealing the nick. 3 This is a generally high‐fidelity pathway that relies on an undamaged complementary strand as a template for high‐fidelity gap filling. However, if the base damage is not removed prior to DNA replication by high‐fidelity DNA polymerases (α, δ and ε), blocked replication can be rescued following translesion DNA synthesis that is catalyzed by low‐fidelity, low‐processivity DNA polymerases. 4 This damage‐tolerance mechanism is the primary mechanism for introducing point mutations and short deletions and insertions.
Thus, random chemical mutagenesis strategies to create saturation mutagenesis for phenotypic selection have relied on overwhelming DNA‐repair pathways without inducing cytotoxicity. To the best of our knowledge, mice deficient in DNA‐repair mechanisms have not been used in conjunction with large‐scale chemical mutagenesis and subsequent selection. However, it would be anticipated that deficiencies in repair of spontaneously induced base damage, including depurination, deamination, alkylation and oxidation, as well as damage produced by exogenous sources, would continuously generate point mutations and short deletions. In this regard, base modification by exposure to reactive oxygen species is not only a by‐product of normal biochemical reactions, but also from ionizing radiation, ultraviolet irradiation, heavy metal exposures and inflammation. 5 Repair of the plethora of oxidatively induced damages is initiated by a series of DNA glycosylases including NEIL1, NEIL2, NEIL3, NTH1 and OGG1. 6 NEIL1 is responsible for the removal of several saturated pyrimidines, both ring‐fragmented purines, alkylated ring‐fragmented purines, psoralen‐induced DNA crosslinks, and secondary oxidation products of 8‐oxoGua. 7 Given the very broad substrate specificity of NEIL1, it would be anticipated that mice which are deficient in Neil1 would show elevated levels of spontaneous mutagenesis. For over 20 years, the Lloyd laboratory has maintained Neil −/− mice, which are frequently bred as knockouts for metabolic and carcinogenesis studies. As part of routine colony expansion, mice are systematically monitored for unusual phenotypes. The current investigation arose by the observation of mice exhibiting a backwards‐walking phenotype in an open‐field environment, as well as exhibiting a variety of other abnormal behaviors. As the literature reviews did not initially yield matches to a backwards‐walking phenotype, studies were initiated to identify the genetic alteration that produced this phenotype.
We mapped the backwards‐walking trait to the Ush1g gene, and found that it was caused by a nonsense mutation that truncated the protein after the third amino acid. We found that Ush1g bw/bw mice had auditory and vestibular defects, as well as disruption of the sensory hair bundle of cochlear hair cells, much like that seen with other mouse lines mutant for Usher syndrome type I (USH1) genes. Examination of protein distribution by immunocytochemistry showed that loss of Ush1g affects row 1 and row 2 tip proteins similarly to loss of Cdh23 or Pcdh15. We noted defects in kinocilial location that support the hypothesis that USH1G assists in stabilizing CDH23‐PCDH15 kinocilial links. Finally, like other mouse USH1 models, we detected no visual phenotypes in these mice. We conclude that Ush1g bw is a null Ush1g allele that will be useful for investigation of auditory and vestibular function.
2. MATERIALS AND METHODS
2.1. Animal models
All animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at Oregon Health & Science University (IP00000145 for RSL, IP00000610 for RR and IP00000714 for PGBG). Neil1 −/− mice have been described previously. 8 , 9 Mice arising from the original mutagenic event creating Ush1g bw were backcrossed to C57BL/6J (B6) mice (RRID:IMSR_JAX:000664, Jackson Laboratories, Bar Harbor, ME) and were maintained on a B6 background. Mouse pups were assumed to be born at midnight, so the animal age on the first day is referred to as P0.5. Both female and male pups were used for all experiments. Except for locomotor experiments, experiments were carried out during the light phase. Mice were group‐housed (maximum five animals), except for Ush1g bw/bw males; because they often became aggressive toward other male cagemates at 4–6 weeks of age, these mice were sometimes single‐housed as per the Barr‐Gillespie and Ryals lab IACUC protocols.
The Ush1g genotype was determined either in‐house using forward primer CATGGCGTTTAGGACCCAGGG and reverse primer CGCAGAGACTCCAGGTTGCC or by Transnetyx (Cordova, TN, United States) using real‐time PCR and proprietary primers.
2.2. Statistical analysis
Unless otherwise stated, statistical comparisons between two sets of data used the Student's t test with unpaired data and the untested assumption of equal variance. In figures, asterisks indicate: *p < 0.05; **p < 0.01; ***p < 0.001.
2.3. Body composition
Body composition was assessed in age‐matched backwards‐walking (bw) and normal (Norm) mice by low‐resolution nuclear magnetic resonance (NMR; EchoMRI 3‐in‐1 system, Echo Medical Systems, Houston, TX, United States) as previously described. 10 , 11 Four Norm males, three Norm females, four bw males and four bw females were used.
2.4. Locomotor activity
For these experiments, three male bw and three male Norm mice were individually housed in Plexiglass cages placed in an environmental chamber (Percival Scientific, Perry, IA) maintained at 20–21°C, with food and water available ad lib. Housing light intensity was approximately 300 lux during the 12‐h light phase of the 24‐h light: dark cycle. Zeitgeber Time 12 (ZT12) was defined as lights‐on. General locomotor activity was measured by passive infrared sensors (Vitalview; Minimitter, Bend, OR) using 10 min bins. Time‐series data were imported into Clocklab (Actimetrics, Wilmette, IL), and actograms were double‐plotted for analysis of behavioral activity. The activity measures were compared using linear mixed models with REML estimation to account for the correlation between repeated measures within a mouse. To evaluate the difference between bw and Norm mice at different light phases, significance (p < 0.05) of the interaction term between genotype and phase was interpreted first. Then pairwise comparisons of the estimated marginal means of genotypes within phase from the mixed model were computed. To compare bw and Norm mice during constant darkness, the significance of the fixed effect of genotype was interpreted. Both models included days into protocol as a covariate and mouse as a random effect. Mixed model analyses were conducted with the lme4 and emmeans packages using R (version 4.2.2; https://www.r-project.org).
2.5. Optokinetic tracking
Optokinetic tracking (OKT) thresholds were used to identify spatial frequencies of gratings (cycles/degree), which define visual performance for animals (OptoMotry; CerebralMechanics, Lethbridge, Alberta, Canada). Briefly, mice (Ush1g+/+, n = 5–8, depending on age; Ush1g bw /+, n = 8–14; Ush1g bw/bw , n = 14) were placed on the pedestal in the OptoMotry system and given 5 min to acclimate to the new environment. A simple staircase method at 100% contrast in normal lighting conditions was used for testing. Right and left eyes were tested separately and averaged together to obtain one spatial frequency per animal.
2.6. Electroretinography
Mice were dark‐adapted overnight. Under dim red light, mice (Ush1g bw /+, n = 10–11; Ush1g bw/bw , n = 10–11) were anesthetized with ketamine (100 mg/kg) and xylazine (10 mg/kg). Mouse pupils were dilated with 1% tropicamide and 2.5% phenylephrine and lubricated during the procedure with hypromellose 2.5% ophthalmic lubricant. Bilateral platinum electrodes were placed on the corneal surface to record the light‐induced electroretinography (ERG) potentials. The reference and ground electrodes placed subcutaneously in the forehead and tail, respectively. Flashes with increasing light intensity from −4 to 3.39 log cd·s/m2 were used for the recordings. A two‐way analysis of variance (ANOVA) with Šídák's multiple comparisons test was used to compare amplitudes between the groups.
2.7. Retinal spectral domain‐optical coherence tomography
At P180, mouse retinas (Ush1g+/+, n = 5; Ush1g bw /+, n = 7; Ush1g bw/bw , n = 7) were imaged with a Heidelberg Spectralis Multimodal Imaging Platform (Heidelberg Engineering Inc., Franklin, MA, United States) using spectral domain‐optical coherence tomography (SD‐OCT). Images were segmented using the Heidelberg Eye Explorer software included with the imaging platform. To measure photoreceptor thickness, REC+ was defined as the thickness from the base of the RPE to the interface of the inner nuclear layer (INL) and outer plexiform layer. REC+ values were taken from 10 locations spanning superior, inferior, nasal and temporal retina. REC+ values from both eyes were averaged for each animal. A one‐way ANOVA was used to compare photoreceptor thickness between groups.
2.8. Retina histology
At P180, enucleated eyes were placed immediately in 4% formaldehyde and incubated overnight at 4°C. Eyes (Ush1g+/+, n = 5; Ush1g bw /+, n = 5; Ush1g bw/bw , n = 5) were processed and embedded in paraffin for sectioning (Tissue‐Tek VIP 6, Tissue‐Tek TEC 5; Sakura Finetek USA, Inc., Torrance, CA, United States). Sections were cut with a microtome to a thickness of 4 μm, stained with hematoxylin–eosin, and viewed on a Leica DMI3000 B microscope (Leica Microsystems GmbH, Wetzlar, Germany). A total of 12 images per eye encompassing the nasal, temporal, superior and inferior retinal quadrants were captured with a 40× objective. The number of photoreceptor nuclei in a row were counted and averaged from these 12 images, to obtain an outer nuclear layer (ONL) cell count per animal. A one‐way ANOVA was used to compare ONL cell counts between groups.
2.9. Retina immunofluorescence
At P180, WT, Het and KO eyes were harvested and placed in 4% formaldehyde. At 24 h after fixation, whole eyes were placed in a 30% sucrose solution for 4 h, and then frozen in Scigen Tissue‐Plus O.C.T. Compound (Thermo Fisher Scientific, Waltham, MA, United States) at −78°C. Eyes were sectioned at 12 μm on a Leica CM1860 cryostat. Cross‐sections were incubated at 4°C in primary antibody consisting of anti‐rhodopsin (1D4) (1:200, Cat# NBP1‐30046; RRID:AB_1968611; Novus Biologicals, Centennial, CO, United States), 0.3% Triton X‐100, and 1% bovine serum albumin overnight. Tissues were rinsed and incubated in secondary antibody containing goat anti‐rabbit Alexa 488 (1:500, Thermo Fisher Scientific Cat# A11008; RRID:AB_143165), 1% bovine serum albumin, and PBS (1×) for 2 h at 20°C. After incubation in secondary antibody, retinas were rinsed again and placed in a DAPI solution (1:2400, Thermo Fisher Scientific Cat# D1306) for 5 min. Retinas were imaged with a white light laser confocal microscope (Leica Microsystems TCS SP8 X). Z‐stacks (spanning 14–23 μm in 1 μm intervals) were collected using a 63× objective. Confocal images were processed using the maximum intensity tool in ImageJ software (version 1.49; National Institutes of Health, Bethesda, MD, United States).
2.10. Functional tests for hearing and balance
Auditory brainstem response (ABR) experiments were carried out with seven Ush1g bw /+ animals (all male) and seven Ush1g bw/bw animals (four female and three male). Animals were anesthetized with xylazine (10 mg/kg, i.m.; Animal Health Inc., Greeley, CO, United States) and ketamine (40 mg/kg, i.m.; Hospira, Inc., Lake Forest, IL, United States), and placed on a heating pad in a sound‐isolated chamber. ABR recording procedures followed methods previously published by Jones and colleagues. 12 , 13 Briefly, pure tone burst stimuli were generated and controlled using National Instruments (Austin, TX) data acquisition system and custom software. Tone bursts at 8, 16 and 32 kHz had 1.0 ms rise‐fall times with 1.0 ms plateau (3 ms total duration). Stimuli for ABR testing were calibrated using a Bruel & Kjaar 1/8″ microphone and Nexus amplifier. Stimuli were calibrated in dB peSPL (peak‐equivalent sound‐pressure level) and were presented via high frequency transducers (SA1 driver, MF1 speakers, Tucker‐Davis Technologies, Alachua, FL) coupled at the left ear via a modified commercial eartip (ER 10D‐T03, Etymotic Research, Inc., Elk Grove Village, IL). Auditory stimuli were presented at a rate of 17 stimuli/s. ABR intensity series were collected by reducing the stimulus in 10 dB steps at higher stimulus levels and 5 dB steps closer to threshold. Threshold was defined as the stimulus level midway between the minimum stimulus level that produced a discernible response and the maximum level where no response was detectable.
Animals were tested for ABRs, followed by a few minutes' transition, then VsEPs. One Ush1g bw /+ died between ABR and DPOAE, so only six animals were used for Ush1g bw /+ VsEPs.
Vestibular evoked potential (VsEP) experiments were carried out as described previously 14 with six Ush1g bw /+ animals and seven Ush1g bw/bw animals. Mice were anesthetized by intraperitoneal injection of ketamine and xylazine (18 and 2 mg/mL; 5–7 μL/g body weight), followed by maintenance doses as needed to maintain adequate anesthesia. Core body temperature was maintained at 37.0 ± 0.2°C using a homeothermic heating pad (FHC Inc., Bowdoin, ME, United States). For VsEP testing, linear acceleration ramps producing rectangular jerk pulses were generated and controlled using a National Instruments (Austin, TX, United States) data acquisition system and custom software. Mice were placed supine on a stationary platform and the head was secured within a spring clip coupled to a voltage‐controlled mechanical shaker (Model 132‐2; Labworks, Costa Mesa, CA, United States). The head was oriented with nose up and linear translation stimuli were presented in the naso‐occipital axis parallel to the Earth‐vertical axis. Vestibular stimuli consisted of 2 ms linear jerk pulses, delivered to the head using two stimulus polarities—normal, with an initial upward jerk, and inverted, with an initial downward jerk—at a rate of 17 pulses per second. Stimulus amplitudes ranged from +6 to −18 dB re: 1.0 g/ms (where 1 g = 9.8 m/s2), adjusted in 3 dB steps. A broadband forward masker (50–50,000 Hz, 94 dB SPL) was presented during VsEP measurements to confirm the absence of auditory components. 15 Signal averaging was used to extract the VsEP responses from the background electrophysiological activity. Ongoing electroencephalographic (EEG) activity was amplified (200,000×), filtered (300–3000 Hz, −6 dB points) and digitized beginning at the onset of each jerk stimulus (1024 points, 10 μs/point) to produce one primary response trace. For each stimulus intensity and polarity, 128 primary responses were averaged to produce an averaged response waveform. Four averaged response waveforms were recorded for each stimulus intensity (two waveforms recorded for normal stimulus polarity and two for inverted polarity). Final individual response traces were produced by summing one averaged response to each stimulus polarity and dividing the result by two, thus producing two response traces for each stimulus intensity for each animal. As with the ABR analysis, threshold was defined as the stimulus level midway between the minimum stimulus level that produced a discernible response and the maximum level where no response was detectable.
2.11. Scanning electron microscopy
Periotic bones with cochleas were dissected in Leibovitz's L‐15 medium (Thermo Fisher Scientific) from P8.5 or P21.5 littermates from Ush1g bw crosses. After isolating the periotic bone, several small holes were made to provide access for fixative solutions; encapsulated cochleas were fixed for an hour in 2.5% glutaraldehyde in 0.1 M cacodylate buffer supplemented with 2 mM CaCl2. Next, cochleas were washed with distilled water and the cochlear sensory epithelium was dissected out; the tectorial membrane was manually removed. The cochlear tissues were then transferred to scintillation vials and dehydrated in a series of ethanol and critical‐point dried using liquid CO2. Samples were immobilized on aluminum specimen holders using a carbon tape and sputter coated with 3–4 nm of platinum. Samples were imaged using the Helios scanning electron microscope (SEM).
2.12. Immunofluorescence sample preparation for cochlea
Inner ears from Ush1g bw littermates were dissected at the indicated ages in cold Hank's balanced salt solution (Thermo Fisher Scientific Cat# 14025076), supplemented with 5 mM HEPES, pH 7.4 (dissection buffer). Small openings were made within the periotic bones to allow perfusion of the fixative. Cochleas were fixed in 4% formaldehyde (Cat# 1570; Electron Microscopy Sciences, Hatfield, PA, United States) in dissection buffer for 20–60 min at room temperature. In experiments using row 2 protein antibodies (EPS8L2, CAPZB), fixation was for 20 min; row 1 protein antibodies were not sensitive to fixative duration and were usually fixed for 60 min. Ears were washed in PBS, then cochleas were dissected out from the periotic bone and the lateral wall was removed. Cochleas were permeabilized in 0.2% Triton X‐100 diluted in 1× PBS for 10 min and blocked in 5% normal donkey serum (Cat# 017‐000‐121; Jackson ImmunoResearch, West Grove, PA, United States) for 1 h at room temperature. For staining against USH1C, cochleas were permeabilized and blocked for 1 h in 10% normal donkey serum (Cat# 017‐000‐121; Jackson ImmunoResearch, West Grove, PA, United States) and 0.2% Triton X‐100 diluted in 1× PBS. Organs were incubated overnight at 4°C with primary antibodies in blocking buffer (5% normal donkey serum diluted in 1× PBS) and then washed three times in 1× PBS. Dilutions were 1:250 for anti‐acetylated tubulin; 1:250 for anti‐GPSM2, 1:500 for anti‐GNAI3; 1:250 for anti‐EPS8, 1:200 for USH1C; 1:250 for anti‐EPS8L2, 1:250 for anti‐CAPZB. Tissue was then incubated with secondary antibodies, which were 2 μg/mL donkey anti‐rabbit Alexa Fluor 488 (Thermo Fisher Scientific Cat# A21206; RRID:AB_2535792) and 2 μg/mL donkey anti‐mouse Alexa Fluor 568 (Thermo Fisher Scientific Cat# A10037; RRID:AB_2534013); 1 U/mL CF405 phalloidin (Cat# 00034; Biotium, Fremont, CA, United States) was also included for the 3–4 h room treatment. Cochleas were washed three times in PBS and mounted on a glass slides in ~50 μL of Vectashield and covered with a #1.5 thickness 22 × 22 mm cover glass (Cat# 2850‐22, Corning) or a #1.5 thickness 22 × 40 mm cover glass (Thermo Fisher Scientific Cat# CLS‐1764‐2240).
Primary antibodies were as follows. Mouse anti‐acetylated tubulin (clone 6‐11B‐1) was Cat# T6793 (RRID:AB_477585) from Sigma‐Aldrich (St. Louis, MO). Rabbit anti‐GPSM2 Cat# HPA007327 (RRID: AB_1849941) from Sigma‐Aldrich. Rabbit anti‐GNAI3, directed against a C‐terminal mouse GNAI3 peptide (KNNLKECGLY), was Cat# G4040 (RRID: AB_259896) from Sigma‐Aldrich. Mouse monoclonal anti‐EPS8 (clone 15), against mouse EPS8 amino acids 628‐821, was Cat# 610143 (RRID: AB_397544) from BD Bioscience (San Jose, CA). Mouse monoclonal anti‐CAPZB2, against a C‐terminal peptide of human CAPZB2, was Cat# AB6017 (RRID: AB_10806205) from EMD Millipore (Burlington, MA). Mouse monoclonal anti‐EPS8L2, against human EPS8L2 amino acids 615‐715, was Cat# ab57571 (RRID: AB_941458) from Abcam (Cambridge, United Kingdom). Rabbit anti‐pan‐MYO15A was antibody PB48 from the Thomas Friedman lab. 16 We used Genemed Biosynthesis (San Antonio, TX) to generate a rabbit anti‐USH1C antiserum using recombinantly expressed PDZ3 of mouse Ush1c. For attempted immunolocalization of USH1G, we used anti‐sans J63 rabbit 73 (a gift from Amel El Bahloul‐Jaziri).
2.13. Fluorescence microscopy of stereocilia proteins
Organs were imaged using a 63×, 1.4 NA Plan‐Apochromat objective on a Zeiss (Oberkochen, Germany) Elyra PS.1/LSM710 system equipped with an Airyscan detector and ZEN 2012 (black edition, 64‐bit software) acquisition software. Settings for x–y pixel resolution, z‐spacing, as well as pinhole diameter and grid selection, were set according to software‐suggested settings for optimal Nyquist‐based resolution. Raw data processing for Airyscan‐acquired images was performed using manufacturer‐implemented automated settings. Settings for x–y pixel resolution, z‐spacing, as well as pinhole diameter and grid selection, were set according to software‐suggested settings for optimal Nyquist‐based resolution. Raw data processing for Airyscan‐acquired images was performed using manufacturer‐implemented automated settings. Display adjustments in brightness and contrast and reslices and/or maximum intensity Z‐projections were made in Fiji software (https://imagej.net/software/fiji/).
3. RESULTS
3.1. Discovery and identification of Ush1g bw mutant allele
As part of ongoing investigations into the role of the NEIL1 DNA glycosylase, routine expansion and health monitoring of the Neil1 −/− colony showed an unexpected phenotype. Because NEIL1 plays a major role in the initiation of base‐excision repair of oxidatively induced DNA damage and alkylated ring‐fragmented purines, 7 throughout the maintenance of this strain, spontaneous mutations arise. Offspring of Neil1 −/− crosses have generated mice with a variety of readily observable phenotypes, including but not limited to the generation of petite mice (<20 g at maturity), alopecia, microphthalmia, staggering and seizures (unpublished observations).
As part of routine colony expansion of phenotypically normal Neil1 −/− mice, 9 we observed a hyperactive male mouse. This mouse was mated with Neil1 −/− females, generating phenotypically normal F1 offspring. When these offspring were set up in pair‐wise mating, a portion of this F2 generation, representing both males and females, exhibited hyperactivity, head‐tilting and unresponsiveness to auditory stimuli. Further observation of these mice showed that when picked up by the tail, they displayed tremors and rapid spinning. If they were placed in an open space context, such as a hood or a tabletop, they immediately stopped all forward motion and only walked backwards (Video S1). Backward walking was also associated with backward circling, raising their heads to a vertical position, and head tilting. Affected mice could not perceive the edge of a surface and would routinely fall off the table top, mandating vigilance during behavioral monitoring. In their home cage, affected mice only walk or run forward and boxes containing only affected mice show rapid and sustained forward running in circular patterns within the cage. Affected mice were also unable to function in a swim test, rapidly swimming to the bottom of the tank and requiring rescue.
Subsequent mating in which both genders were phenotypically affected, or mating that used an affected female with a phenotypically normal male, resulted in successful pregnancies, albeit with small litters. All pups died within days after birth, however, presumably because of maternal disinterest or from being trampled during frantic, in‐cage running. Analyses of mice generated from the mating of affected males with wild‐type C57BL/6J females, followed by subsequent generation of F2 pups, showed that the acquisition of these phenotypes was independent of the Neil1 genotype.
3.2. Body composition and locomotor activity of backwards‐walking mice
Initial characterization of the backwards‐walking mice took place prior to identification of Ush1g as the causative gene; later experiments indicated that backwards‐walking and hyperactive phenotype was correlated completely with the Ush1g bw/bw genotype. Nevertheless, we refer to mice in these early experiments as being backwards‐walking (bw) or normal (Norm). Body weights were measured and body composition was assessed at 48–49 weeks of age. Body weights were reduced by 26% in male bw mice (t test, t(6) = 4.513, p = 0.004) and 15% in female bw mice (t test, t(5) = 3.271, p = 0.022) relative to Norm mice (Figure 1A). This significant reduction in total body weight was driven largely by reductions in fat mass. Fat mass was reduced by a remarkable 82% in male bw mice (t test, t(6) = 8.317, p = 0.0002) and 54% of body weight in female bw mice (t test, t(5) = 13.82, p = <0.0001) (Figure 1B). Correspondingly, lean body mass was increased (Figure 1C) in male bw mice (t test, t(6) = 7.673, p = 0.0003) and in female bw mice (t test, t(5) = 8.229, p = 0.0004), indicating that the reductions in fat mass and body weight likely stemmed from metabolic alterations in bw mice. Future studies should investigate potential changes in food intake and energy expenditure in these mice, ideally prior to the onset of significant divergence in body weights or body composition.
FIGURE 1.
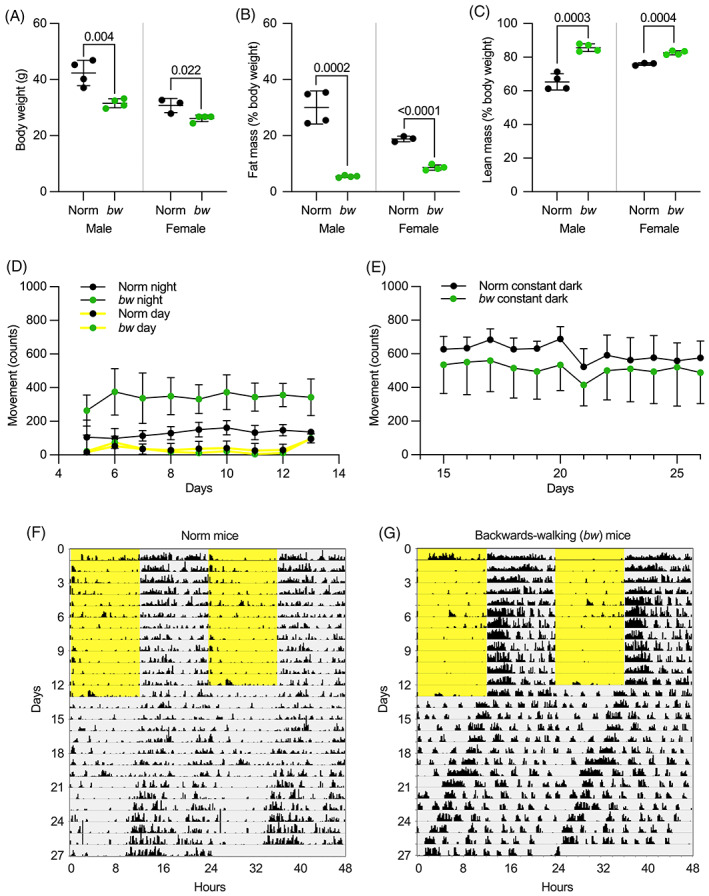
Body composition and activity of backwards‐walking mice. (A) Body weights for Norm and bw mice. (B) Fat mass as % of total mass, measured by nuclear magnetic resonance (NMR), for Norm and bw mice. (C) Lean mass as % of total mass, measured by NMR, for Norm and bw mice. (D–G) Locomotor activity of Norm and bw mice. (D) Daily activity locomotor activity during light and dark phases for mice maintained in a light: dark cycle. Each point represents the mean ± SEM of the activity during the light or dark phase. (E) The amount of activity of the non‐hyperactive and hyperactive mice during constant darkness. (F, G) Example actograms from Norm and bw mice were recorded during light (yellow) and dark (gray) periods.
All mice showed greater locomotor activity during the dark phase than during the light phase (Figure 1D,F–G). There was a significant interaction effect detected between genotype and phase in the linear mixed model (p < 0.001). During the light phase, bw and Norm mice had similar levels of activity (bw 33 ± 40 vs. Norm 40 ± 40, estimated marginal mean ± SEM, p = 0.91). During the dark phase, the bw mice showed a significantly higher activity level than Norm mice (341 ± 40 vs. 130 ± 40, estimated marginal mean ± SEM, p = 0.016). When placed in constant darkness, the two groups of mice showed similar activity levels (bw 591 ± 90 vs. Norm 491 ± 90, estimated marginal mean ± SEM, p = 0.456). The circadian period was measured from the activity onsets during the complete darkness. The mean period was 23.7 ± 0.5 h in bw mice and 23.9 ± 0.1 in Norm mice and was not statistically significantly different (p = 0.594, unpaired t test).
3.3. Mapping mutation to Ush1g
To map the mutation, affected C57BL/6 males were mated with BALB/c females; the F1 generation mice were all phenotypically normal (Figure 2). Offspring resulting from matings within this F1 generation produced a total of nine open‐field, backwards‐walking mice out of a total of 37 pups. Tails from a total of these nine mice and two additional backwards‐walking mice and nine phenotypically normal mice were sent to The Jackson Laboratory for differential single nucleotide polymorphic (SNP) analyses. In this analysis, the approximate chromosomal location of the mutated gene was anticipated to map to inherited regions that were homozygous for C57BL/6 SNPs in the 11 affected mice and either heterozygotic or homozygotic for BALB/c SNPs in unaffected mice. Analyses of a total of 126 SNPs for each mouse showed a single chromosomal location, extreme distal 11q that mapped to the mutated site (Figure 2). The linkage was evident from SNP #11‐117818566‐N, marker RS3023766 at 116,784,04 bp. The boundaries of the gene of interest were established by SNP #11‐105306632‐N, marker RS3024066 at 104,384,279 bp and the end of chromosome 11 as the lower limit. Genes mapping to this general region were analyzed for potential functional interest, with Ush1g being the leading candidate. Sequence analyses showed that the DNA encoding codon 4 of Ush1g (CAG, encoding glutamine) had been mutated to a stop codon (TAG). The mutant USH1G protein thus should be truncated after the third amino acid. Because of the previously unreported backwards‐walking phenotype of this allele, we designated it as Ush1g bw (MGI:7432804). Given the location of the introduced stop codon, we presume that the allele is a complete null.
FIGURE 2.
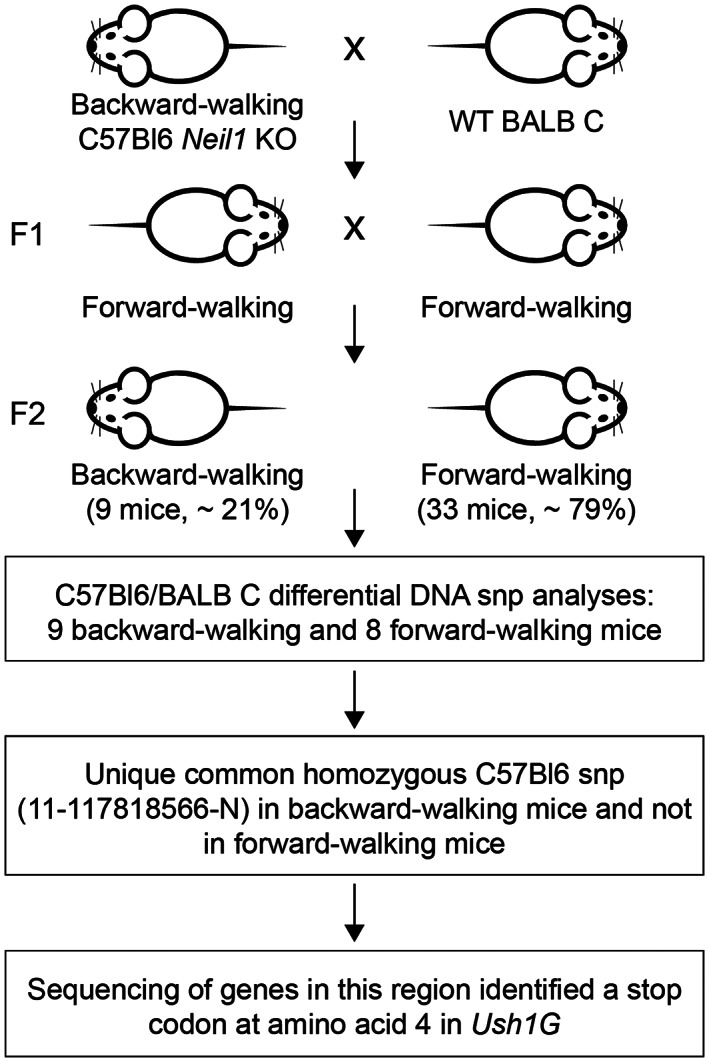
Scheme for identification of gene responsible for backwards walking. The mapping scheme used to identify Ush1g bw is shown. A backwards‐walking Neil1 −/− mouse was mated with a female BALB/c mouse; the F1 generation (all phenotypically wild type) was intercrossed to generate F2 mice. Roughly a quarter of the F2 generation had the backwards‐walking phenotype. SNP analysis showed that SNP #11‐117818566 was present in backwards‐walking mice but not their phenotypically normal littermates. A nonsense mutation was identified in the Ush1g gene that terminates the protein after the third amino acid.
3.4. Visual phenotype
USH1G causes progressive retinal degeneration in humans. 17 In Ush1g bw mice, visual behavior was measured in normal lighting conditions at P30, P90 and P180 using OKT (Figure 3A); there were no significant differences between wild type and heterozygous Ush1g animals at these time points. A two‐way repeated‐measures ANOVA indicated: based on genotype F(1, 25) = 2.430, p = 0.1316, not significant (ns); based on time and genotype F(2, 20) = 2.049, p = 0.1551, ns. By contrast, these measurements highlighted the Ush1g bw/bw phenotype; these animals were unable to balance on the platform, and measurements could not be acquired (Figure 3A).
FIGURE 3.
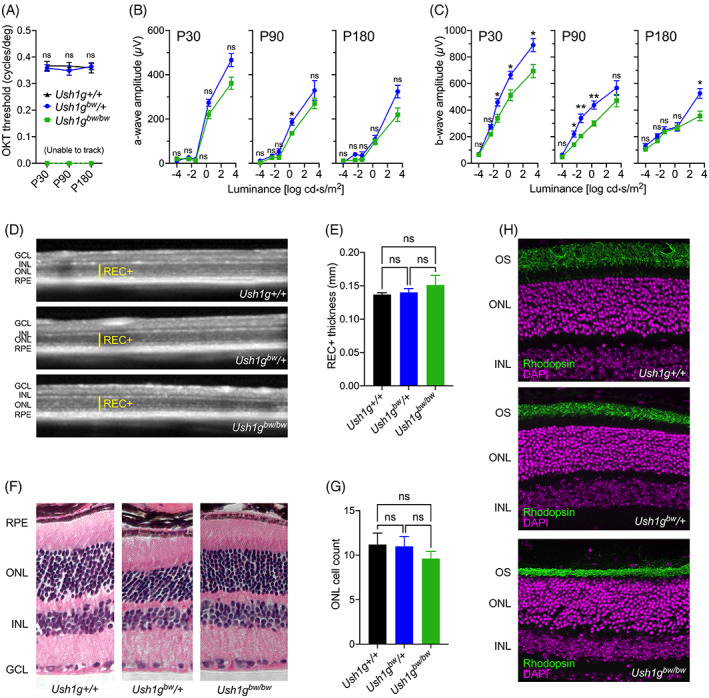
Retinal phenotype associated with the Ush1g bw mice. (A) Visual performance measured by optokinetic tracking (OKT). (B, C) Amplitudes of a‐wave (B) and b‐wave (C) generated from scotopic ERGs at range of light intensities. In each case, the three panels show results at P30, P90 and P180. Legend in A applies to panels B and C as well. Two‐way ANOVA analyses were used to compare heterozygote and homozygote responses. (D) Representative SD‐OCT images of the retina from Ush1g+/+, Ush1g bw /+ and Ush1g bw/bw mice. Yellow lines indicate photoreceptor layer thickness (REC+). (E) REC+ values were compared between groups. (F) Representative images of retinal histology from each of the genotypes at P180. (G) Quantitation of photoreceptor nuclei number in a row in the ONL. (H) Representative confocal images of retinal cross‐sections stained with anti‐rhodopsin (green) and DAPI (magenta). Key: GCL, ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer; RPE, retinal pigmented epithelium. Panel widths: (D) 4 mm; (F) 297 μm (Ush1g+/+), 230 μm (Ush1g bw /+), 365 μm (Ush1g bw/bw ); (H) 150 μm.
Scotopic ERGs were recorded at these time points; while Ush1g bw/bw mice had significantly lower b‐wave amplitudes than Ush1g bw /+ controls (Figure 3C), a‐wave amplitudes (corresponding to photoreceptor function) were essentially unchanged between the genotypes (Figure 3B). A two‐way repeated‐measures ANOVA indicated: P30 column factor (two groups), F(1, 40) = 10.66, p = 0.0023; P90 column factor (two groups), F(1, 36) = 6.869, p = 0.0128; P180 column factor (two groups), F(1, 24) = 5.522, p = 0.0273. By contrast, b‐wave amplitudes were significantly reduced at multiple luminance levels (Figure 3C). A two‐way repeated‐measures ANOVA indicated: P30 column factor (two groups), F(1, 40) = 14.01, p = 0.0006; P90 column factor (two groups), F(1, 36) = 11.85, p = 0.0015; P180 column factor (two groups), F(1, 24) = 4.071, p = 0.0549.
At P180, prior to eye harvest for histology and immunofluorescence, SD‐OCT was used to collect in vivo retinal images (Figure 3D). Photoreceptor thickness calculated from SD‐OCT images showed no difference between Ush1g+/+, Ush1g bw /+ and Ush1g bw/bw animals (Figure 3E). An ordinary one‐way ANOVA gave F(2, 16) = 3.783, p = 0.0452; the multiple comparisons showed no difference between the groups. These results were confirmed with photoreceptor nuclei counts from histology images (Figure 3F,G). For Figure 3G, an ordinary one‐way ANOVA gave F(2, 12) = 3.091, p = 0.0827. Immunofluorescence of eyes harvested at P180 showed that rhodopsin levels in the photoreceptor outer segments were consistent between groups (Figure 3H).
3.5. Auditory and vestibular functional phenotype
USH1G mutations also cause profound deafness and constant vestibular dysfunction in humans at birth. 18 The behaviors that led to the initial isolation of the Ush1g bw allele are largely similar to those seen in other mouse lines with mutations affecting the vestibular system; the allele descriptors “circler,” “shaker” and “waltzer,” commonly used for these lines, 19 all indicate hyperactivity and stereotyped motor behavior. Because most mouse lines with vestibular dysfunction also lack auditory function, we predicted that Ush1g bw/bw mice would have disruptions in hearing and balance. Indeed, Ush1g bw/bw mice had profound hearing loss, objectively shown with ABR measurements. 20 None of the Ush1g bw/bw mice tested had an ABR response at 8, 16, or 32 kHz, while heterozygote mice had normal ABR thresholds (Figure 4A). Likewise, Ush1g bw/bw mice had profound vestibular dysfunction, objectively shown by threshold elevation in VsEP measurements. 21 None of the Ush1g bw/bw mice tested had a VsEP response, while heterozygote mice had normal VsEP thresholds (Figure 4B). Comparing Ush1g bw /+ and Ush1g bw/bw animals that either showed any response or no response, for both ABR and VsEP results, the Fisher exact test statistic values were 0.0006 and the results were significant at p < 0.01.
FIGURE 4.
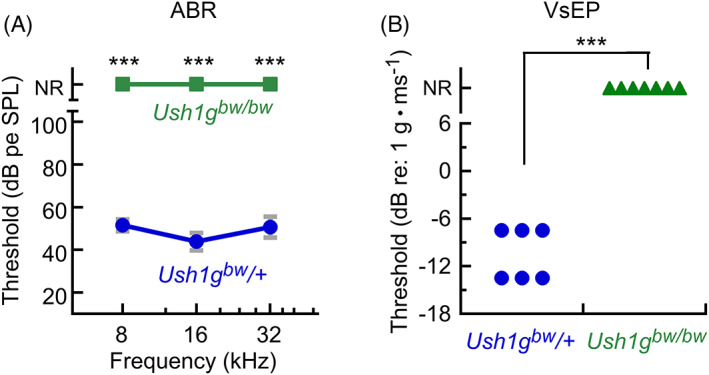
Auditory and vestibular function in Ush1g bw mice. (A) ABR measurements. None of the seven Ush1g bw/bw mice had an ABR response; ABRs in the seven Ush1g bw /+ control mice were normal. (B) VsEP measurements. None of the seven Ush1g bw/bw mice had a VsEP response; VsEPs in the six Ush1g bw /+ control mice were normal. NR, no response. Mean ± SEM plotted.
3.6. Morphology of inner‐ear hair bundles
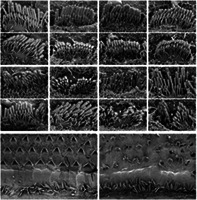
The disruption to auditory and vestibular function and the known role of USH1G in hair cells suggested examination of their sensory hair bundles would be illuminating. Mutations in other genes that cause USH1 have severe disruption of their bundles, in large part because of loss of tip links, transient lateral links and kinocilial links, all of which are formed by CDH23 and PCDH15. 22 SEM showed significant disorganization of P8.5 Ush1g bw/bw bundles (Figure 5A,E), although the disruption was not as severe as seen with homozygotes from USH1 mutant lines, including Pcdh15 av3J/av3J , Cdh23 v2J/v2J and Myo7a 8J/8J (Figure 5B–D,F–H). Stereocilia were more coherent in Ush1g bw/bw bundles than in bundles of homozygous mice from the other strains. The most notable disruption in Ush1g bw/bw bundles was the loss of connection of the stereocilia with the kinocilium, the axonemal structure that orients a bundle. While the kinocilium degenerates after P10, 23 it still can be seen in the scanning electron micrographs of Ush1g KO /+ control bundles (Figure 5, arrows). In Ush1g bw/bw hair cells, the kinocilium was usually detached from the bundle (Figure 5, arrows); in addition, there was a striking notch in most bundles, where the central stereocilia of each row were displaced medially (Figure 5, arrowheads). The notch phenotype was also seen in hair cells of the other homozygous USH1 mutant lines (Figure 5C,D,G,H), with the strongest effect seen in Myo7a 8J/8J bundles (Figure 5H).
FIGURE 5.
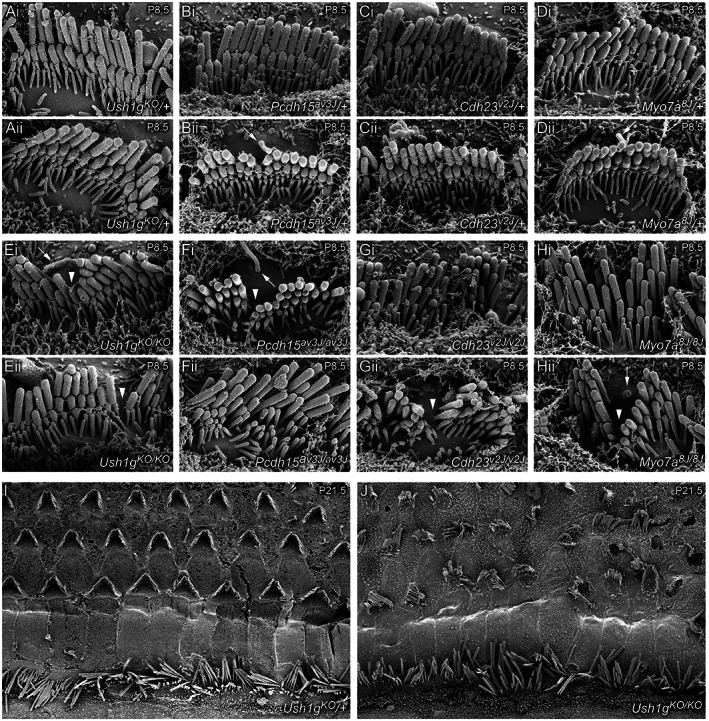
Scanning electron microscopy of hair bundles from Usher 1 mouse models. (A–H) Scanning electron micrographs of single IHC hair bundles from P8.5 cochleas of indicated genotypes. For each genotype, two examples (i and ii) are provided. Arrows indicate kinocilia; arrowheads indicate notch in bundles of mutant hair cells. (I–J) Scanning electron micrographs of P21.5 cochleas of indicated genotypes. Panel widths: (A–H) 6 μm; (I–J) 50 μm.
Many row 2 stereocilia in USH1 heterozygotes had beveled tips (Figure 4A–D), indicative of force production via the tip link and resulting actin remodeling. 24 , 25 All stereocilia tips in Ush1g bw/bw hair bundles were rounded, which suggests that functional tip links were not present (Figure 5E); this observation was consistent with the extreme elevation of ABR thresholds. A similar rounded stereocilia tip phenotype was seen in Pcdh15 av3J/av3J , Cdh23 v2J/v2J and Myo7a 8J/8J stereocilia (Figure 5F–H). Other links were still present in Ush1g bw/bw bundles (Figure 5E), which may account for the increased bundle coherence compared with the other USH1 mutants (Figure 5F–H).
At P21.5, Ush1g bw/bw OHC hair bundles were particularly disrupted compared with controls (Figure 5I,J). Adjacent stereocilia in P21.5 Ush1g bw/bw IHC bundles were not as coordinated in length as those in Ush1g KO /+ controls (Figure 5I,J).
Using phalloidin labeling to highlight the actin cytoskeleton, we noted that utricle hair bundles were disrupted in Ush1g bw/bw mice; their stereocilia were not coherent, and projected in multiple directions (Figure 6A). Cochlear bundles remained relatively coherent in Ush1g bw/bw mice (Figure 6B,C), although a notch in the bundle was usually obvious (Figure 6C, arrow).
FIGURE 6.
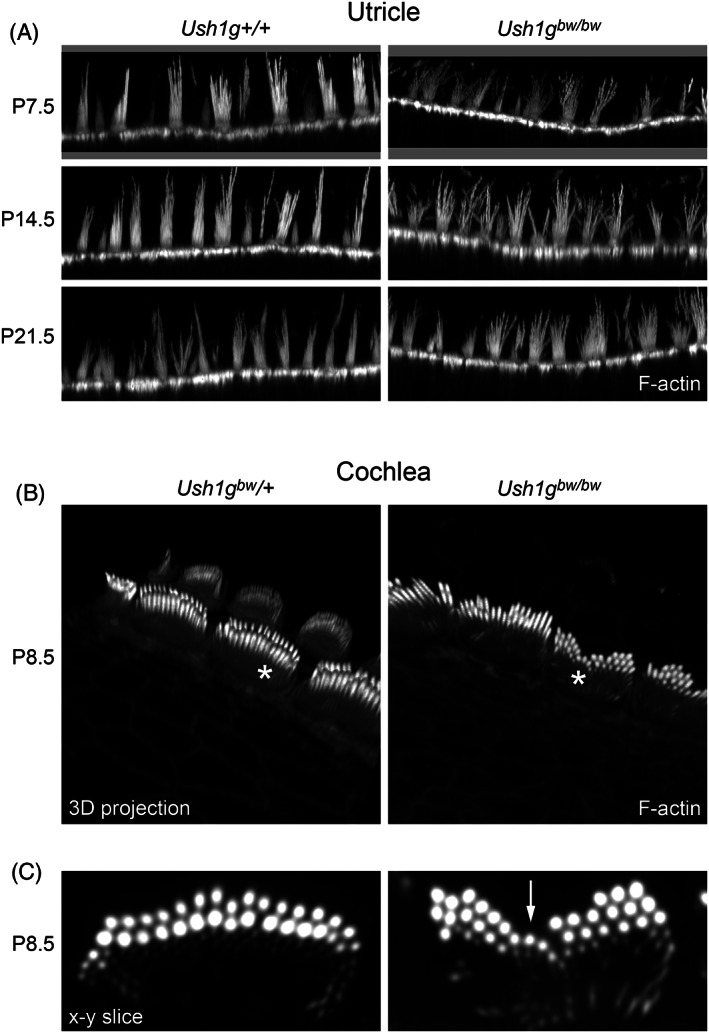
Phalloidin‐labeled hair bundles from utricle and cochlea Ush1g bw hair cells. (A) Utricle from P7.5 through P21.5. (B, C) Cochlea. Panels in B are 3D projections of an x–y–z stack. Panels in (C) show single x–y slices through IHCs indicated with asterisks in (B). Arrow in right panel of (C) indicates prominent notch in the hair bundle. Panel widths: (A) 84 μm; (C) 10 μm.
3.7. Distribution of row‐specific proteins in cochlear hair cells
In our hands, immunolocalization with an antibody reported to be specific for USH1G 26 showed similar patterns in Ush1g bw /+ controls and Ush1g bw/bw mutants (not shown), that is, it was not specific. We also used antibodies against acetylated tubulin to mark the tip of the kinocilium. 27 Similar to results seen when the kinocilium was localized using SEM, acetylated tubulin was associated with the central stereocilia in Ush1g bw /+ hair bundles (arrows in Figure 7A,B), while it was less consistently found adjacent to stereocilia in Ush1g bw/bw bundles (Figure 7A,B). The immunolocalization and SEM results indicate that USH1G is essential for function of the kinocilial links.
FIGURE 7.
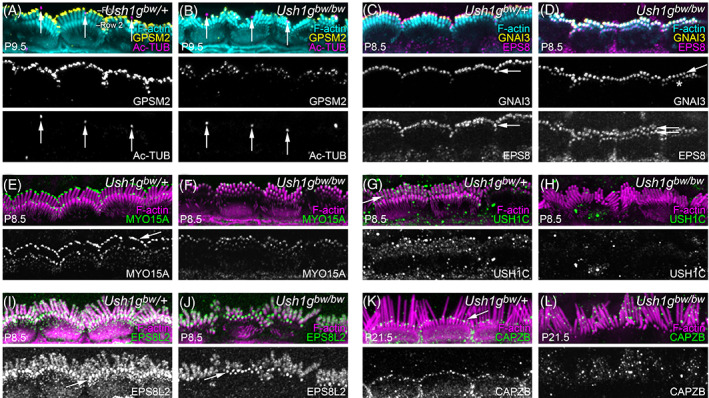
Localization of row‐specific proteins and other proteins in Ush1g bw hair cells. (A, B) GPSM2 and acetylated tubulin (Ac‐TUB) at P9.5. GPSM2 levels decrease in Ush1g bw/bw hair bundles but the row 1 concentration is unchanged. Arrows indicate kinocilia tips. (C, D) GNAI3 and EPS8 at P8.5. GNAI3 is largely still row 1‐specific (arrows) in Ush1g bw/bw bundles but some row 2 labeling is apparent (asterisk). EPS8 shifts from highly row 1‐specific in Ush1g bw /+ bundles (arrow) to equally distributed in rows 1 and 2 in Ush1g bw/bw bundles (double arrows). (E, F) MYO15A at P8.5. MYO15A is abundant in row 1 tips in Ush1g bw /+ bundles (arrow) but reduced there in Ush1g bw/bw bundles. (G, H) USH1C at P8.5. Punctate USH1C labeling in Ush1g bw /+ bundles (arrow) is missing in Ush1g bw/bw bundles. (I, J) EPS8L2 at P8.5. EPS8L2 is normally concentrated at row 2 tips but is found throughout stereocilia membranes at low levels; this pattern is unchanged in Ush1g bw/bw bundles. (K, L) CAPZB at P21.5. CAPZB labeling is present at row 2 tips in Ush1g bw /+ bundles (arrow) but is found throughout Ush1g bw/bw bundles in a punctate pattern. Panel widths, 30 μm.
A protein complex made of GPSM2, GNAI3, WHRN, MYO15A and EPS8 concentrates at the tips of row 1 stereocilia in cochlear inner hair cells 28 , 29 , 30 ; each of these proteins is required for normal lengthening of row 1 stereocilia. 30 We used immunocytochemistry to localize proteins of the row 1 complex in Ush1g bw IHCs (Figure 7). GPSM2 is normally exclusively found at row 1 tips; in Ush1g bw/bw hair bundles, it was also concentrated only in row 1 (Figure 7A). GNAI3 had a similar distribution (Figure 7C,D), although more GNAI3 was found at tips of row 2 stereocilia in Ush1g bw/bw bundles (asterisk in Figure 7D). By contrast, EPS8 shifted from having most labeling at row 1 tips in Ush1g bw /+ bundles (arrow in Figure 7C) to nearly identical levels at both row 1 and 2 tips in Ush1g bw/bw bundles (double arrow in Figure 7D). This broadened distribution of EPS8 resembled that seen in Pcdh15 av3J/av3J and Cdh23 v2J/v2J mutants. 31 MYO15A showed a similar altered distribution; while most MYO15A was found at row 1 tips in Ush1g bw /+ bundles (Figure 7E), its overall level was reduced in Ush1g bw/bw bundles and it was evenly distributed between rows 1 and 2 (Figure 7F).
USH1C (harmonin) is a binding partner for USH1G at the upper insertion of the tip link. 32 In Ush1g bw /+ bundles, USH1C was distributed throughout the hair bundle, but appeared to concentrate in a line on row 1 stereocilia that could correspond to the tip‐link upper insertions (arrow in Figure 7G). USH1C labeling disappeared in Ush1g bw/bw bundles (Figure 7H), consistent with a requirement of USH1G for proper localization of USH1C.
Several proteins concentrate at tips of row 2 stereocilia in developing hair bundles, including EPS8L2, ESPNL, BAIAP2L2, CAPZB, TWF2, DSTN and CFL1, and the locations of most of these are regulated by mechanotransduction. 33 , 34 , 35 , 36 , 37 , 38 We examined the location of two of these, EPS8L2 and CAPZB, in Ush1g bw hair cells. In Ush1g bw /+ bundles, EPS8L2 was found along the shafts of all stereocilia, but was also strongly concentrated at row 2 tips (Figure 7I); the distribution of EPS8L2 was similar in Ush1g bw/bw bundles (Figure 7J). By contrast, CAPZB no longer was found at row 2 tips in Ush1g bw/bw bundles (Figure 7L), unlike its robust tip localization in row 2 of Ush1g bw /+ controls (Figure 7K).
4. DISCUSSION
The Jackson shaker (js) allele of Ush1g was originally identified in 1967. 39 The js allele was mapped to the distal part of chromosome 11, allowing identification of the Ush1g gene. 40 Simultaneously, human Usher syndrome type 1G (USH1G) was mapped to the USH1G gene. 17 The protein product was termed sans (scaffold protein containing ankyrin repeats and SAM domain), although we refer it to here by the protein symbol derived from its gene symbol (USH1G).
As with other USH1 genes, including Cdh23, Pcdh15, Ush1c and Myo7a, Ush1g is primarily expressed in the brain, testis, eye and inner ear. 40 , 41 USH1G is particularly well characterized in the inner ear. There, CDH23 and PCDH15 form the hair cell's tip links, and USH1G, USH1C and MYO7A anchor the CDH23 end of the tip link. 42 MYO7A, which interacts with USH1C, also forms a strong complex with USH1G 43 , 44 ; moreover, USH1G and USH1C bind tightly together. 43 , 45 Finally, MYO7A, USH1C and USH1G form a liquid‐phase biomolecular condensate in vitro, which suggests that these three proteins form a multivalent, stable complex. 46 Thus in the inner ear, and probably in the retina too, MYO7A, USH1C and USH1G form a stable protein complex that can anchor CDH23 and transmit force generated by MYO7A's motor activity.
We identified a new allele for Ush1g, called Ush1g bw , which occurred unexpectedly when Neil1 −/− mice developed spontaneous behavioral changes, presumably because of the lack of base‐excision repair with the loss of NEIL1 protein. The Ush1g bw allele may be useful for dissection of auditory and vestibular function; this allele replicates the constellation of behavioral and morphological phenotypes seen in mouse lines with disruptive mutations in other USH1 genes, including Cdh23, Pcdh15 and Myo7a. Results with the new allele confirmed the essential role USH1G plays in establishing and operating key links within the hair bundle, including tip links, transient lateral links and kinocilial links.
4.1. Backwards walking phenotype
Backwards walking has not been reported as a phenotype for other Jackson shaker alleles of Ush1g (https://www.informatics.jax.org/marker/MGI:2450757). The original js allele has a frameshift mutation that leads to a truncated protein at amino acid 245, which deletes the C‐terminal SAM domain but leaves intact the ankyrin repeats, sterile α‐motif (SAM) domain and 95‐aa central region. The js‐2J, js‐3J and js‐seal alleles all introduce mutations that may produce truncated proteins but not fully eliminate expression. Given polypeptide chain termination following the third amino acid, Ush1g bw is surely a true null; consequentially, it may have more disruptive phenotypes compared with the other Jackson shaker alleles. Indeed, it is not uncommon for phenotypes of mouse alleles with naturally occurring mutations to be less profound than those of nulls, so the Ush1g bw mouse line may be particularly useful for further characterization of auditory and vestibular function.
Backwards walking has also been reported as a phenotype in other auditory and vestibular mutant mouse lines, including tailchaser (Myo6) 47 and spinner (Tmie). 48 Whether backwards walking also is present in other mutant lines that show hyperactivity, circling and other connected vestibular phenotypes is not clear. All evidence suggests that this phenotype is because of auditory and vestibular disruption, however, not because of a new role for USH1G in other tissues.
4.2. Retinal phenotype
Mouse models of USH1 are congenitally deaf but show minimal signs of retinal degeneration. 49 , 50 , 51 Although some models showed modest reductions in ERG, these changes could either be because of a phenotype resulting from the loss of an USH1 protein or be a result of variation. 49 Our model was consistent with previous USH1 models in that there was no measurable retinal degeneration in Ush1g bw/bw mice by P180. The USH1 proteins co‐localize at the calyceal processes in the retina, which is a membrane–membrane connection site between a photoreceptor's outer segment and its inner segment. Mouse photoreceptors lack calyceal processes, however, and USH1 proteins cannot be detected in the outer‐segment basal region. 50 Without the proper cellular structure, mouse models cannot recapitulate the retinal phenotype associated with USH1. To overcome this limitation, CRISPR/Cas9 editing of the MYO7A gene has recently been used to generate an USH1B rhesus macaque model, 52 while the USH1C gene was edited to produce an USH1C pig model 53 ; each model will be useful in characterizing retinal degeneration.
4.3. Hair‐cell phenotype
USH1G interacts with both CDH23 and PCDH15, 26 and we found that the morphological phenotype of Ush1g bw strongly resembled those of Cdh23 v2J and Pcdh15 av3J . One of the more striking alterations seen in hair bundles from these three mutant lines was the loss of attachment of the kinocilium to the bundle and a resulting notch in the bundle. As CDH23‐PCDH15 kinocilial links anchor the tallest stereocilia to the kinocilium, 54 , 55 these results suggest that USH1G is an essential component of the intracellular anchors for these links. USH1G is well known for its critical role in forming the motor complex at the bundle's upper tip‐link density, 26 which controls tension in tip links; while the bundle fragmentation and kinocilial displacement has been noted previously in outer hair cells using a late‐knockout Ush1g model, 26 a role of USH1G role in kinocilia links themselves has not been previously proposed.
As previously noted, 22 USH1C labeling is absent in hair bundles from mice with disruptions in Ush1g. Given that USH1C interacts strongly with USH1G, 43 , 45 we agree that USH1C is incapable of being delivered to stereocilia in the absence of USH1G. 56 The USH1G PDZ binding motif (PBM) and SAM domain form a highly stable complex (K D ≈ 2 nM) with the combined N‐terminus/PDZ1 domain (NPDZ1) of USH1C. 56 In this tight complex, USH1C is still capable of binding to CDH23. 56 USH1G is also essential for tight interaction of USH1C with MYO7A. 56
Interaction of USH1G with PCDH15 is less well understood. Double heterozygous Pcdh15 av3J /+; Ush1g js /+ mice showed elevated ABR thresholds, however, showing a genetic interaction that may reflect a physical association. 57 In support of a direct USH1G‐PCDH15 interaction, tagged USH1G was recruited to the plasma membrane of COS‐7 cells when PCDH15 was co‐expressed but not when it was expressed on its own. 26 MYO7A interacts with PCDH15, 58 and as MYO7A and USH1G interact strongly, 44 it is likely that the functional complex of PCDH15 and USH1G also includes MYO7A. Indeed, PCDH15 is mislocalized in Myo7a sh1/sh1 mutants. 58
Homozygous null mutations in Cdh23 and Pcdh15 lead to altered distribution of proteins that concentrate at the tips of row 1 stereocilia in postnatal hair bundle development. 31 While localization of GPSM2 and GNAI3 was only modestly affected, EPS8 shifted from being primarily found at row 1 tips to being equally distributed between row 1 and 2 tips. We also showed that CDH23 and PCDH15 play an underappreciated role in coordinating the lengths of adjacent stereocilia. 31 Row 1 proteins showed similar altered distribution in Ush1g bw/bw , and adjacent stereocilia lengths were less coordinated, albeit not as profoundly disrupted as in Cdh23 v2J/v2J and Pcdh15 av3J/av3J hair cells. The protein redistribution seen in Ush1g bw/bw hair cells highlights the importance of CDH23‐PCDH15 links and their intracellular partners—like USH1G—in controlling the location of proteins important for control of actin polymerization, like the row 1 complex.
Distribution of the row 2 protein EPS8L2 was relatively unaffected in Ush1g bw/bw , Cdh23 v2J/v2J and Pcdh15 av3J/av3J hair cells, but the concentration of CAPZB at row 2 tips was eliminated in all three mutant lines. Because functional transduction is required for CAPZB localization at row 2 tips, 59 these results are consistent with there being no mechanotransduction in Ush1g bw/bw hair cells, as also suggested by the profound elevation of both ABR and VsEP thresholds.
5. CONCLUSIONS
The Ush1g bw mouse line is a null mutation of Ush1g, which will be of utility for characterizing USH1G in tissues that express the protein, including the retina, cochlea and vestibular system. As with other mouse models of Usher I syndrome, however, the lack of retinal phenotype renders this model of limited utility for understanding the USH1G phenotype in humans, which includes severe retinal degeneration. By contrast, the Ush1g bw model described here should serve as an excellent model for deafness seen with USH1G mutations in humans. Alterations in hair‐bundle structure in Ush1g bw/bw mice suggest that USH1G contributes to anchoring kinocilia links, not just tip links. In cochlear hair cells, the Ush1g bw/bw phenotype is similar to those seen in Cdh23 v2J/v2J and Pcdh15 av3J/av3J mice, suggesting that USH1G is a necessary subunit in complexes with these two large cadherins.
Supporting information
VIDEO S1. Backwards‐walking and hyperactivity phenotypes in Ush1g bw mice.
ACKNOWLEDGMENTS
We thank Jennifer Goldsmith for mouse husbandry and Michael Bateschell for genotyping. We also thank Dan Austin, Oregon Institute of Occupational Health Sciences, for filming and editing of the movie documenting Ush1g bw/bw mice behavior. We carried out confocal microscopy in the OHSU Advanced Light Microscopy Core @ The Jungers Center and electron microscopy in the OHSU Multiscale Microscopy Core. PGBG was supported by National Institutes of Health grant R01DC002368. RSL was supported by National Institute of Environmental Health Sciences grant R01ES031086, as well as the Oregon Institute of Occupational Health Sciences at Oregon Health & Science University (via funds from the Division of Consumer and Business Services of the State of Oregon; ORS 656.630). RR was supported by Casey Eye Institute Core Grant P30 EY010572 from the National Institutes of Health and unrestricted departmental funding from Research to Prevent Blindness.
Vartanian V, Krey JF, Chatterjee P, et al. Spontaneous allelic variant in deafness–blindness gene Ush1g resulting in an expanded phenotype. Genes, Brain and Behavior. 2023;22(4):e12849. doi: 10.1111/gbb.12849
R. Stephen Lloyd and Peter G. Barr‐Gillespie are co‐senior authors.
Contributor Information
R. Stephen Lloyd, Email: lloydst@ohsu.edu.
Peter G. Barr‐Gillespie, Email: gillespp@ohsu.edu.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Gridley T, Murray SA. Mouse mutagenesis and phenotyping to generate models of development and disease. Curr Top Dev Biol. 2022;148:1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Peterson KA, Murray SA. Progress towards completing the mutant mouse null resource. Mamm Genome. 2022;33:123‐134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sampath H, McCullough AK, Lloyd RS. Regulation of DNA glycosylases and their role in limiting disease. Free Radic Res. 2012;46:460‐478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ling JA, Frevert Z, Washington MT. Recent advances in understanding the structures of translesion synthesis DNA polymerases. Genes (Basel). 2022;13:915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dizdaroglu M, Coskun E, Jaruga P. Measurement of oxidatively induced DNA damage and its repair, by mass spectrometric techniques. Free Radic Res. 2015;49:525‐548. [DOI] [PubMed] [Google Scholar]
- 6. Dizdaroglu M, Coskun E, Jaruga P. Repair of oxidatively induced DNA damage by DNA glycosylases: mechanisms of action, substrate specificities and excision kinetics. Mutat Res Rev Mutat Res. 2017;771:99‐127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lloyd RS. Complex roles of NEIL1 and OGG1: insights gained from murine knockouts and human polymorphic variants. DNA. 2022;2:279‐301. [Google Scholar]
- 8. Calkins MJ, Vartanian V, Owen N, et al. Enhanced sensitivity of Neil1−/− mice to chronic UVB exposure. DNA Repair (Amst). 2016;48:43‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vartanian V, Minko IG, Chawanthayatham S, et al. NEIL1 protects against aflatoxin‐induced hepatocellular carcinoma in mice. Proc Natl Acad Sci U S A. 2017;114:4207‐4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Komakula SSB, Tumova J, Kumaraswamy D, et al. The DNA repair protein OGG1 protects against obesity by altering mitochondrial energetics in white adipose tissue. Sci Rep. 2018;8:14886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Burchat N, Akal T, Ntambi JM, Trivedi N, Suresh R, Sampath H. SCD1 is nutritionally and spatially regulated in the intestine and influences systemic postprandial lipid homeostasis and gut‐liver crosstalk. Biochim Biophys Acta Mol Cell Biol Lipids. 2022;1867:159195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Braude JP, Vijayakumar S, Baumgarner K, et al. Deletion of Shank1 has minimal effects on the molecular composition and function of glutamatergic afferent postsynapses in the mouse inner ear. Hear Res. 2015;321:52‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mock B, Jones TA, Jones SM. Gravity receptor aging in the CBA/CaJ strain: a comparison to auditory aging. J Assoc Res Otolaryngol. 2011;12:173‐183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Krey JF, Krystofiak ES, Dumont RA, et al. Plastin 1 widens stereocilia by transforming actin filament packing from hexagonal to liquid. J Cell Biol. 2016;215:467‐482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jones TA, Jones SM. Short latency compound action potentials from mammalian gravity receptor organs. Hear Res. 1999;136:75‐85. [DOI] [PubMed] [Google Scholar]
- 16. Liang Y, Wang A, Belyantseva IA, et al. Characterization of the human and mouse unconventional myosin XV genes responsible for hereditary deafness DFNB3 and shaker 2. Genomics. 1999;61:243‐258. [DOI] [PubMed] [Google Scholar]
- 17. Weil D, El‐Amraoui A, Masmoudi S, et al. Usher syndrome type I G (USH1G) is caused by mutations in the gene encoding SANS, a protein that associates with the USH1C protein, harmonin. Hum Mol Genet. 2003;12:463‐471. [DOI] [PubMed] [Google Scholar]
- 18. Mustapha M, Chouery E, Torchard‐Pagnez D, et al. A novel locus for Usher syndrome type I, USH1G, maps to chromosome 17q24‐25. Hum Genet. 2002;110:348‐350. [DOI] [PubMed] [Google Scholar]
- 19. Brown SD, Hardisty‐Hughes RE, Mburu P. Quiet as a mouse: dissecting the molecular and genetic basis of hearing. Nat Rev Genet. 2008;9:277‐290. [DOI] [PubMed] [Google Scholar]
- 20. Willott JF. Overview of methods for assessing the mouse auditory system. Curr Protoc Neurosci. 2006. [DOI] [PubMed] [Google Scholar]
- 21. Jones SM, Jones TA. Genetics of peripheral vestibular dysfunction: lessons from mutant mouse strains. J Am Acad Audiol. 2014;25:289‐301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lefevre G, Michel V, Weil D, et al. A core cochlear phenotype in USH1 mouse mutants implicates fibrous links of the hair bundle in its cohesion, orientation and differential growth. Development. 2008;135:1427‐1437. [DOI] [PubMed] [Google Scholar]
- 23. Kaltenbach JA, Falzarano PR, Simpson TH. Postnatal development of the hamster cochlea. II. Growth and differentiation of stereocilia bundles. J Comp Neurol. 1994;350:187‐198. [DOI] [PubMed] [Google Scholar]
- 24. Kachar B, Parakkal M, Kurc M, Zhao Y, Gillespie PG. High‐resolution structure of hair‐cell tip links. Proc Natl Acad Sci U S A. 2000;97:13336‐13341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vélez‐Ortega AC, Freeman MJ, Indzhykulian AA, Grossheim JM, Frolenkov GI. Mechanotransduction current is essential for stability of the transducing stereocilia in mammalian auditory hair cells. Elife. 2017;6:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Caberlotto E, Michel V, Foucher I, et al. Usher type 1G protein sans is a critical component of the tip‐link complex, a structure controlling actin polymerization in stereocilia. Proc Natl Acad Sci U S A. 2011;108:5825‐5830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lu X, Sipe CW. Developmental regulation of planar cell polarity and hair‐bundle morphogenesis in auditory hair cells: lessons from human and mouse genetics. Wiley Interdiscip Rev Dev Biol. 2016;5:85‐101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tarchini B, Tadenev AL, Devanney N, Cayouette M. A link between planar polarity and staircase‐like bundle architecture in hair cells. Development. 2016;143:3926‐3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mauriac SA, Hien YE, Bird JE, et al. Defective Gpsm2/Gαi3 signalling disrupts stereocilia development and growth cone actin dynamics in Chudley‐McCullough syndrome. Nat Commun. 2017;8:14907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tadenev ALD, Akturk A, Devanney N, et al. GPSM2‐GNAI specifies the tallest stereocilia and defines hair bundle row identity. Curr Biol. 2019;29:921‐934.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Krey JF, Chatterjee P, Halford J, Cunningham CL, Perrin BJ, Barr‐Gillespie PG. Control of stereocilia length during development of hair bundles. bioRxiv. 2023; 520962. [DOI] [PMC free article] [PubMed]
- 32. Grati M, Kachar B. Myosin VIIa and sans localization at stereocilia upper tip‐link density implicates these Usher syndrome proteins in mechanotransduction. Proc Natl Acad Sci U S A. 2011;108:11476‐11481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Furness DN, Johnson SL, Manor U, et al. Progressive hearing loss and gradual deterioration of sensory hair bundles in the ears of mice lacking the actin‐binding protein Eps8L2. Proc Natl Acad Sci U S A. 2013;110:13898‐13903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ebrahim S, Avenarius MR, Grati M, et al. Stereocilia‐staircase spacing is influenced by myosin III motors and their cargos espin‐1 and espin‐like. Nat Commun. 2016;7:10833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Halford J, Bateschell M, Barr‐Gillespie PG. Ca2+ entry through mechanotransduction channels localizes BAIAP2L2 to stereocilia tips. Mol Biol Cell. 2022;33:br6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Peng AW, Belyantseva IA, Hsu PD, Friedman TB, Heller S. Twinfilin 2 regulates actin filament lengths in cochlear stereocilia. J Neurosci. 2009;29:15083‐15088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Avenarius MR, Krey JF, Dumont RA, et al. Heterodimeric capping protein is required for stereocilia length and width regulation. J Cell Biol. 2017;216:3861‐3881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. McGrath J, Tung CY, Liao X, et al. Actin at stereocilia tips is regulated by mechanotransduction and ADF/cofilin. Curr Biol. 2021;31:1141‐1153.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Deol MS, Dickie MM. Rotating, a new gene affecting behavior and the inner ear in the mouse. J Hered. 1967;58:69‐72. [Google Scholar]
- 40. Kikkawa Y, Shitara H, Wakana S, et al. Mutations in a new scaffold protein Sans cause deafness in Jackson shaker mice. Hum Mol Genet. 2003;12:453‐461. [DOI] [PubMed] [Google Scholar]
- 41. Overlack N, Maerker T, Latz M, Nagel‐Wolfrum K, Wolfrum U. SANS (USH1G) expression in developing and mature mammalian retina. Vision Res. 2008;48:400‐412. [DOI] [PubMed] [Google Scholar]
- 42. Michalski N, Petit C. Genetics of auditory mechano‐electrical transduction. Pflugers Arch. 2015;467:49‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Adato A, Michel V, Kikkawa Y, et al. Interactions in the network of Usher syndrome type 1 proteins. Hum Mol Genet. 2005;14:347‐356. [DOI] [PubMed] [Google Scholar]
- 44. Wu L, Pan L, Wei Z, Zhang M. Structure of MyTH4‐FERM domains in myosin VIIa tail bound to cargo. Science. 2011;331:757‐760. [DOI] [PubMed] [Google Scholar]
- 45. Yan J, Pan L, Chen X, Wu L, Zhang M. The structure of the harmonin/sans complex reveals an unexpected interaction mode of the two Usher syndrome proteins. Proc Natl Acad Sci U S A. 2010;107:4040‐4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. He Y, Li J, Zhang M. Myosin VII, USH1C, and ANKS4B or USH1G together form condensed molecular assembly via liquid‐liquid phase separation. Cell Rep. 2019;29:974‐986.e4. [DOI] [PubMed] [Google Scholar]
- 47. Kiernan AE, Zalzman M, Fuchs H, et al. Tailchaser (Tlc): a new mouse mutation affecting hair bundle differentiation and hair cell survival. J Neurocytol. 1999;28:969‐985. [DOI] [PubMed] [Google Scholar]
- 48. D'Amico E, Stygelbout V, Schurmans S. The Spinner‐IBMM mouse is a new spontaneous mutant in the Tmie gene. Genes Genom. 2012;34:107‐110. [Google Scholar]
- 49. Williams DS. Usher syndrome: animal models, retinal function of Usher proteins, and prospects for gene therapy. Vision Res. 2008;48:433‐441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sahly I, Dufour E, Schietroma C, et al. Localization of Usher 1 proteins to the photoreceptor calyceal processes, which are absent from mice. J Cell Biol. 2012;199:381‐399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zallocchi M, Binley K, Lad Y, et al. EIAV‐based retinal gene therapy in the shaker1 mouse model for usher syndrome type 1B: development of UshStat. PLoS One. 2014;9:e94272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ryu J, Statz JP, Chan W, et al. CRISPR/Cas9 editing of the MYO7A gene in rhesus macaque embryos to generate a primate model of Usher syndrome type 1B. Sci Rep. 2022;12:10036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Grotz S, Schäfer J, Wunderlich KA, et al. Early disruption of photoreceptor cell architecture and loss of vision in a humanized pig model of usher syndromes. EMBO mol Med. 2022;14:e14817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Goodyear RJ, Forge A, Legan PK, Richardson GP. Asymmetric distribution of cadherin 23 and protocadherin 15 in the kinocilial links of avian sensory hair cells. J Comp Neurol. 2010;518:4288‐4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Webb SW, Grillet N, Andrade LR, et al. Regulation of PCDH15 function in mechanosensory hair cells by alternative splicing of the cytoplasmic domain. Development. 2011;138:1607‐1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pan L, Zhang M. Structures of usher syndrome 1 proteins and their complexes. Physiology (Bethesda). 2012;27:25‐42. [DOI] [PubMed] [Google Scholar]
- 57. Zheng QY, Scarborough JD, Zheng Y, Yu H, Choi D, Gillespie PG. Digenic inheritance of deafness caused by 8J allele of myosin‐VIIA and mutations in other Usher I genes. Hum Mol Genet. 2012;21:2588‐2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Senften M, Schwander M, Kazmierczak P, et al. Physical and functional interaction between protocadherin 15 and myosin VIIa in mechanosensory hair cells. J Neurosci. 2006;26:2060‐2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Krey JF, Chatterjee P, Dumont RA, et al. Mechanotransduction‐dependent control of stereocilia dimensions and row identity in inner hair cells. Curr Biol. 2020;30:442‐454.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
VIDEO S1. Backwards‐walking and hyperactivity phenotypes in Ush1g bw mice.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.