

Abstract
Congenital pulmonary airway malformation (CPAM) is a rare congenital dysplastic malformation characterized by failure of bronchial development and localized glandular overgrowth. Previously known as Congenital Cystic Adenoid Malformation (CCAM), CPAM is classified into 5 types, from type 0 to type IV, depending upon the origin of pulmonary areas of the lung, cyst size, and cyst appearance. CPAM is a rare congenital anomaly typically diagnosed prenatally in ultrasound. However, few cases are diagnosed in childhood and even fewer in adulthood. CPAM can be differentiated from pulmonary sequestration based on the origin of the arterial supply; the former has its arterial supply from the pulmonary artery, whereas pulmonary sequestration has its arterial supply from the systemic circulation. Another differential diagnosis of CPAM includes congenital bronchogenic cyst, congenital lobar emphysema, pleuropulmonary blastoma, congenital cystic bronchiectasis, and congenital diaphragmatic hernia. The most common presentation is recurrent chest infection and chest pain, whereas other presentation includes pneumothorax and hemoptysis. Here, we present a case of a 6-year-old child with recurrent episodes of fever and cough diagnosed as a type II CPAM based on computed tomography findings.
Keywords: Child, Congenital pulmonary airway malformation, Computed tomography
Background
Congenital lung malformation is a heterogeneous group of embryologically related lung malformations. It includes congenital pulmonary airway malformation (CPAM), pulmonary sequestrations, bronchial atresia, lobar agenesis, bronchogenic cyst, congenital airway malformation, and poly-alveolar lobe [1]. CPAM is the most common type of congenital lung malformation that presents in 0.004% of all pregnancies and constitutes approximately 25% of all congenital pulmonary anomalies. The estimated incidence is 1 in 25,000 to 1 in 35,000 live births [2], [3], [4]. It is characterized by failure of bronchial development and overgrowth of terminal bronchopulmonary tissue. Typically, it is diagnosed prenatally in ultrasound, with only a few cases diagnosed in children and adults, the most common presentation being recurrent and resistant infections [2]. CPAM is now classified into 5 different types according to the origin of the tracheobronchial tree and the presence of cystic components, radiologic appearance, and dimensions [5]. Stocker's criteria classify CPAM from type 0 to type IV. Type I CPAM is the most common and is associated with a favorable prognosis [6]. Bilateral CPAM is rare and is associated with poor prognosis [7]. While the prenatally diagnosed CPAM has typical sonographic features, those diagnosed in childhood and adults may have recurrent infection and inflammation leading to altered radiological and histological appearance [5,8].
Case presentation
A 6-year-old child presented with recurrent episodes of cough and fever for 6 months. During the last presentation, he had a history of fever and right-sided chest pain for the past 7 days that did not resolve with oral antibiotics. On examination, the temperature was 102.4ºF, and the child was in moderate respiratory distress. Laboratory examination showed marked leukocytosis and chest X-ray showed the right lower lobe consolidation with some areas of cystic bronchiectasis.
CT scan with IV contrast was done, which revealed a multiseptated cystic mass in the postero-basal and mediobasal segments of the right lower lobe (Fig.1, Fig. 2, Fig. 3). No surrounding consolidations or ground glass opacities were seen. No communication was noted with the segmental bronchi. No separate systemic vascular supply to the segments was seen. Therefore, the case was diagnosed as CPAM radiologically.
Fig.1.

Axial HRCT image shows air-filled cystic lesion with multiple septations seen in the basal segment of the right lower lobe (shown by red arrow).
Fig. 2.

Axial HRCT image shows air-filled cystic lesion with multiple septations seen in the basal segment of the right lower lobe along with a peripheral area of consolidation (red arrow).
Fig. 3.
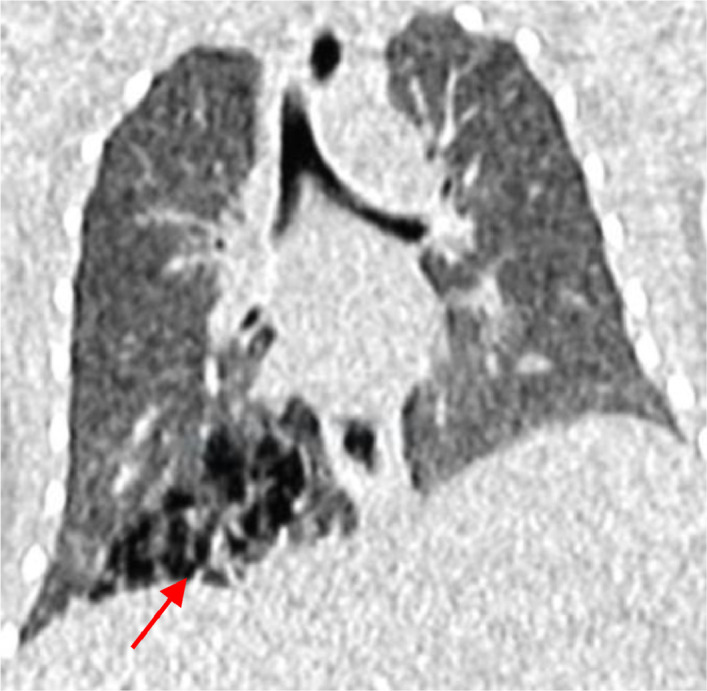
Coronal HRCT image shows a multiseptated air-filled cystic lesion in the right lower lobe (red arrow).
The child was managed with intravenous antibiotics and was stable after the treatment. He is under close observation and follow-up now. The patient remained asymptomatic till we contacted him for follow-up at the last time.
Discussion
CPAM is a rare congenital dysplastic malformation resulting from the disruptive morphogenesis of the tracheobronchial tree [9]. It is a rare congenital anomaly and accounts for 25% of congenital lung lesions. The exact pathogenesis of CPAM is not clear. However, studies in rats showed the role of overexpressed mesenchymal growth factors in lung branching embryogenesis [10,11]. Some authors have observed the germline mutations in the genes responsible for lung cancer, suggesting a predisposition to malignancy [12]. Typically, CPAM is diagnosed prenatally via ultrasound. However, some cases are reported in children and adults. When presented postnatally, CPAM commonly occurs as recurrent chest infections. However, some cases are asymptomatic and detected incidentally [8,13].
CPAM was first described as a new disease entity by Ch'in and Tang in 1949 [3]. It was classified into 3 types in 1977 [14] but was expanded into 5 types by Stocker et al. in 2002. The new name, CPAM, was assigned by Stocker [6]. About 80%-85% of the cases are diagnosed in the first 2 years of life, and adults with CPAM are rarely diagnosed [15]. Usually, CPAM involves 1 lobe and presents commonly with infection or abscess [13,16].
CPAM, initially called CCAM, was first classified into 3 types [14]. Intrinsically, all the types of CPAM are similar. However, CPAM varies in radiological presentation. Type I CPAM is the most prevalent CPAM and accounts for 60%-70% of all cases. It is characterized by single or multiple cysts more than 3 cm in diameter. These patients have the most favorable prognosis. Type II CPAM arises from terminal bronchioles and accounts for approximately 15%-30% of the cases. The lesion has cysts that are smaller in size (< 2cm) and also have solid areas [6]. The abnormality most associated with other fetal anomalies is type II-CPAM; most commonly, other intrathoracic, abdominal, or skeletal malformations are associated with this type of CPAM [14,17]. Type III CPAM arises from distal bronchiolar and alveolar regions and is associated with large lung lesions and subcentimeter cysts. This type is uncommon and occurs only in about 5%-10% of cases.
The lesions are usually solid because the cysts are very small in size. Stocker added type 0 and type IV to CPAM in 2002 [6]. Type 0 arises from the trachea and bronchus of the lungs and is the least common form of CPAM, occurring in 1%-3% of all cases. It is challenging to differentiate type 0 CPAM from the bronchogenic cyst and is almost always lethal. So, Type 0 CPAM is not frequently discussed in the literature. Finally, type IV is associated with the most distal acinar region of the lung and contains multiple large cysts similar to type I [13]. This type accounts for 5%-15% of CPAM cases. Type 4-CPAM has also been associated with malignancy [12]. Our case is CPAM-type II based on radiological findings, as the cysts are uniform in size. Although type II CPAM is mostly associated with other malformations, our case did not have any associated malformations other than CPAM.
Patients with CPAM have different clinical presentations. The most common presentation is recurrent and resistant infections. However, some of these lesions are incidentally found on chest imaging in asymptomatic patients. Other manifestations include pneumothorax, dyspnea, and hemoptysis [13]. A few rare cases of aspergilloma in CPAM have also been reported [8]. About 80%-85% of CPAM are diagnosed by the first 2 years of life [15]. In instances where CPAM is diagnosed later in life, chronic inflammation from recurrent infections may cause alteration in the radiological and histological presentation [5]. Malignant cases from CPAM have also been reported. The chronic inflammatory process is thought to be a stimulus for malignant transformation. A few cases have been reported where bronchoalveolar carcinoma developed in an area of CPAM, with subsequent progression to metastatic adenocarcinoma [18,19].
Pathologically, CPAMs are hamartomatous lesions that are comprised of cystic and adenomatous elements arising from tracheal, bronchial, bronchiolar, or alveolar tissue. Large lesions can compromise alveolar growth and development by compressing adjacent normal tissue. Histologically, these cysts are made up of the same cell types as the region from which they were derived, from pseudostratified ciliated columnar in type I, transitioning to the more cuboidal epithelium in types II and III, and finally squamous cells in type IV [4,6,14].
CPAM should be differentiated from various other conditions. The most important one is bronchopulmonary sequestration, a nonfunctional lung tissue supplied by systemic vasculature. Bronchopulmonary sequestration is frequently present in the lower lobe of the lung and typically appears as a homogenous or heterogeneous solid mass or consolidation on imaging. Less commonly, it appears as multiple cystic masses with air-fluid levels, cavitary lesions, or a single cystic mass. The most crucial distinguishing feature of pulmonary sequestration from CPAM is its blood supply from the systemic arteries, the thoracic aorta being the most common site, while CPAM has its vascular supply from the pulmonary artery. Type III CPAM is the most difficult to differentiate from pulmonary sequestration. Another important differential diagnosis is a bronchogenic cyst. In a bronchogenic cyst, abnormal budding occurs during the development of the tracheobronchial tree and can occur in the mediastinum or intrapulmonary [1,20]. Also, type IV CPAM is very difficult to distinguish from pleuropulmonary blastoma [13].
Treatment depends on the clinical presentation and the extent of the disease. Most of the symptomatic cases with recurrent pulmonary infections or respiratory distress require surgical excision. Small asymptomatic cases are followed up. There are evidences of spontaneous regression in the cases diagnosed with prenatal ultrasound [21].
Conclusion
In conclusion, CPAM is a rare congenital lung malformation characterized by bronchial development failure and localized glandular overgrowth. It is typically diagnosed prenatally but can also occur in childhood and adulthood. CPAM has various types and presents with recurrent respiratory symptoms.
Differential diagnoses include pulmonary sequestration and bronchogenic cyst. Treatment involves appropriate management of symptoms and may also require surgery. Long-term follow-up is important due to potential complications, including malignancy. Further research is needed to enhance our understanding of CPAM and improve diagnostic and management approaches.
Patient consent
The authors claim that there is no personal information in this report that might be used to identify the patient. Since the patient is a minor, informed, written, consent was obtained from his parent.
Footnotes
Competing Interests: The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1.Seear M, Townsend J, Hoepker A, Jamieson D, McFadden D, Daigneault P, et al. A review of congenital lung malformations with a simplified classification system for clinical and research use. Pediatr Surg Int. 2017;33(6):657–664. doi: 10.1007/s00383-017-4062-y. [DOI] [PubMed] [Google Scholar]
- 2.McDonough RJ, Niven AS, Havenstrite KA. Congenital pulmonary airway malformation: a case report and review of the literature. Respir Care. 2012;57(2):302–306. doi: 10.4187/respcare.00727. [DOI] [PubMed] [Google Scholar]
- 3.Ch'in KY, Tang MY. Congenital adenomatoid malformation of one lobe of a lung with general anasarca. Arch Pathol. 1949;48(3):221–229. [PubMed] [Google Scholar]
- 4.Stocker JT. Cystic lung disease in infants and children. Fetal Pediatr Pathol. 2009;28(4):155–184. doi: 10.1080/15513810902984095. [DOI] [PubMed] [Google Scholar]
- 5.Il'ina NA, Alekseeva AL. [Computed tomography in the diagnosis of congenital cystic adenomatoid malformation of the lung in children] Vestn Rentgenol Radiol. 2014;(1):33–38. [PubMed] [Google Scholar]
- 6.Stocker J. Congenital pulmonary airway malformation: a new name and expanded classification of congenital cystic adenomatoid malformation of the lung. Histopathology. 2002;41(2):424–431. [Google Scholar]
- 7.Sabbagha RE. HarperCollins Publishers; Philadelphia, Pensylvinnea: 1980. Diagnostic ultrasound applied to obstetrics and gynecology. [Google Scholar]
- 8.Feng A, Cai H, Sun Q, Zhang Y, Chen L, Meng F. Congenital cystic adenomatoid malformation of lung in adults: 2 rare cases report and review of the literature. Diagn Pathol. 2012;7:37. doi: 10.1186/1746-1596-7-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilson RD, Hedrick HL, Liechty KW, Flake AW, Johnson MP, Bebbington M, et al. Cystic adenomatoid malformation of the lung: review of genetics, prenatal diagnosis, and in utero treatment. Am J Med Genet A. 2006;140(2):151–155. doi: 10.1002/ajmg.a.31031. [DOI] [PubMed] [Google Scholar]
- 10.Lezmi G, Verkarre V, Khen-Dunlop N, Vibhushan S, Hadchouel A, Rambaud C, et al. FGF10 Signaling differences between type I pleuropulmonary blastoma and congenital cystic adenomatoid malformation. Orphanet J Rare Dis. 2013;8(1):130. doi: 10.1186/1750-1172-8-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.David M, Lamas-Pinheiro R, Henriques-Coelho T. Prenatal and postnatal management of congenital pulmonary airway malformation. Neonatology. 2016;110(2):101–115. doi: 10.1159/000440894. [DOI] [PubMed] [Google Scholar]
- 12.Hsu JS, Zhang R, Yeung F, Tang CS, Wong JK, So MT, et al. Cancer gene mutations in congenital pulmonary airway malformation patients. ERJ Open Res. 2019;5(1):2–7. doi: 10.1183/23120541.00196-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Collins AM, Ridgway PF, Killeen RP, Dodd JD, Tolan M. Congenital cystic adenomatoid malformation of the lung: hazards of delayed diagnosis. Respirol Carlton Vic. 2009;14(7):1058–1060. doi: 10.1111/j.1440-1843.2009.01603.x. [DOI] [PubMed] [Google Scholar]
- 14.Stocker JT, Madewell JE, Drake RM. Congenital cystic adenomatoid malformation of the lung. Classification and morphologic spectrum. Hum Pathol. 1977;8(2):155–171. doi: 10.1016/s0046-8177(77)80078-6. [DOI] [PubMed] [Google Scholar]
- 15.Rosado-de-Christenson ML, Stocker JT. Congenital cystic adenomatoid malformation. Radiogr Rev Publ Radiol Soc N Am Inc. 1991;11(5):865–886. doi: 10.1148/radiographics.11.5.1947321. [DOI] [PubMed] [Google Scholar]
- 16.Herrero Y, Pinilla I, Torres I, Nistal M, Pardo M, Gómez N. Cystic adenomatoid malformation of the lung presenting in adulthood. Ann Thorac Surg. 2005;79(1):326–329. doi: 10.1016/S0003-4975(03)01655-2. [DOI] [PubMed] [Google Scholar]
- 17.Hellmund A, Berg C, Geipel A, Bludau M, Heydweiller A, Bachour H, et al. Prenatal diagnosis and evaluation of sonographic predictors for intervention and adverse outcome in congenital pulmonary airway malformation. PloS One. 2016;11(3) doi: 10.1371/journal.pone.0150474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Benouaich V, Marcheix B, Begueret H, Brouchet L, Velly JF, Jougon J. Malignancy of congenital cystic adenomatoid malformation of lung in aged. Asian Cardiovasc Thorac Ann. 2009;17(6):634–636. doi: 10.1177/0218492309349810. [DOI] [PubMed] [Google Scholar]
- 19.Ioachimescu OC, Mehta AC. From cystic pulmonary airway malformation, to bronchioloalveolar carcinoma and adenocarcinoma of the lung. Eur Respir J. 2005;26(6):1181–1187. doi: 10.1183/09031936.05.00011705. [DOI] [PubMed] [Google Scholar]
- 20.Sfakianaki AK, Copel JA. Congenital cystic lesions of the lung: congenital cystic adenomatoid malformation and bronchopulmonary sequestration. Rev Obstet Gynecol. 2012;5(2):85–93. [PMC free article] [PubMed] [Google Scholar]
- 21.Tran H, Fink MA, Crameri J. Congenital cystic adenomatoid malformation: monitoring the antenatal and short-term neonatal outcome. Aust N Z J Obstet Gynaecol. 2008;48(5):462–466. doi: 10.1111/j.1479-828X.2008.00887.x. [DOI] [PubMed] [Google Scholar]