

Abstract
Epigenetic alterations marked by DNA methylation are frequent events during the early development of nasopharyngeal carcinoma (NPC). We identified that TRIM29 is hypomethylated and overexpressed in NPC cell lines and tissues. TRIM29 silencing not only limited the growth of NPC cells in vitro and in vivo, but also induced cellular senescence, along with reactive oxygen species (ROS) accumulation. Mechanistically, we found that TRIM29 interacted with voltage‐dependent anion‐selective channel 1 (VDAC1) to activate mitophagy clearing up damaged mitochondria, which are the major source of ROS. In patients with NPC, high levels of TRIM29 expression are associated with an advanced clinical stage. Moreover, we detected hypomethylation of TRIM29 in patient nasopharyngeal swab DNA. Our findings indicate that TRIM29 depends on VDAC1 to induce mitophagy and prevents cellular senescence by decreasing ROS. Detection of aberrantly methylated TRIM29 in the nasopharyngeal swab DNA could be a promising strategy for the early detection of NPC.
Keywords: hypomethylation, ROS, senescence, TRIM29, VDAC1
TRIM29 is hypomethylated and overexpressed in nasopharyngeal carcinoma. TRIM29 combines with VDAC1 to regulate cellular senescence through reactive oxygen species. Detection of aberrantly methylated TRIM29 in the nasopharyngeal swab DNA could be a promising strategy for the early detection of nasopharyngeal carcinoma.
Abbreviations
- CCCP
carbonyl cyanide m‐chlorophenylhydrazone
- Co‐IP
co‐immunoprecipitation
- CXCL
CXC chemokine ligand
- IL
interleukin
- MSP
methylation‐specific PCR
- NPC
nasopharyngeal carcinoma
- PARP
poly(ADP‐ribose) polymerase
- Rb
retinoblastoma protein
- ROS
reactive oxygen species
- SA‐β‐gal
senescence‐associated β‐galactosidase
- SASP
senescence‐associated secretory phenotype
- SILAC
stable isotope labeling with amino acids in cell culture
- TRIM
tripartite motif
- VDAC1
voltage‐dependent anion‐selective channel 1
1. INTRODUCTION
Aberrant epigenetic events, such as DNA hypo‐ and hypermethylation, play important roles during the early stages of tumor development. 1 , 2 Furthermore, changes in methylation patterns are also detectable, being of clinical value in tumor detection and diagnosis. 3 In NPC, which is a human malignancy endemic in Southeast Asia and southern China, 4 , 5 , 6 aberrant DNA methylation is an important mechanism for inactivation of cancer‐associated genes, such as CDKN2A/B, RASSF1, and LMP1. 7 , 8 Early diagnosis of NPC will greatly improve overall survival. Diagnosis of NPC at early stages (stage I or II) is associated with a high rate of survival (on average 95%). However, most NPC patients are diagnosed at an advanced stage, when treatment results are not satisfactory. 9 , 10 Thus, there is a need to develop new methylation markers for NPC detection and to identify their potential function in carcinogenesis.
TRIM29, also known as ataxia‐telangiectasia group D complementing gene (ATDC), is a member of the TRIM protein family. 11 TRIM29 has been reported to be highly expressed in lung, bladder, pancreatic, gastric, prostate, and nasopharyngeal carcinomas. 12 , 13 , 14 , 15 , 16 , 17 In these cancer cell types, TRIM29 promotes cell proliferation and enhances cell invasion. Mechanistically, TRIM29 interacts with p53 and restricts its activities by its sequestration outside of the nucleus. 18 TRIM29 binds to Dvl2 and suppresses the activity of glycogen synthase kinase 3β, with resultant stabilization of β‐catenin. 14 TRIM29 also exerts its oncogenic effects through silencing of PTEN, and regulation of the p63‐mediated pathway. 19 , 20 Recent studies have also shown that in a genetically engineered mouse model, physiologic TRIM29 overexpression is sufficient to accelerate KRAS‐mediated tumorigenesis early in the course of pancreatic ductal adenocarcinoma. 21 However, the methylation status of TRIM29 has been reported rarely in NPC.
Cellular senescence, an irreversible form of growth arrest, can be induced in response to shortened telomeres, accumulation of oxidative stress, DNA damage, and activated oncogenes (such as oncogenic RAS). 22 Senescent cells show several distinctive characteristics, including a large flat cellular morphology, SA‐β‐gal activity, metabolically active, altered gene expression, and a robust secretory phenotype (also known as SASP). 23 Cellular senescence acts as a critical tumor suppressor mechanism. In addition, recent findings suggest that DNA methylation that modulates the expression of key genes during senescence could be crucial events in oncogenesis. 1 , 24
In this study, we found that TRIM29 was hypomethylated and overexpressed in NPC cells and tissues. TRIM29 silencing limited the growth of NPC cells, triggered an elevation in ROS, and induced cellular senescence. We also detected TRIM29 hypomethylation in patient nasopharyngeal swab DNA.
2. MATERIALS AND METHODS
2.1. Tissue samples
All clinical specimens used for pyrosequencing, western blot, and immunohistochemical analyses were collected from NPC patients at Sun Yat‐sen University Cancer Center. Patients' consent and approval from Sun Yat‐sen University Cancer Center Institute Research Ethics Committee were obtained for the use of these clinical materials.
2.2. Cell culture
CNE2, CNE1, SUNE1, and HK‐1 cell lines were obtained from Dr Chaonan Qian (Sun Yat‐sen University). HONE1 cells were obtained from Professor Qian Tao (The Chinese University of Hong Kong). The NP69 cell line was obtained from Professor Wenlin Huang (Sun Yat‐sen University). CNE2, HONE1, CNE1, HK‐1, and SUNE1 cell lines were maintained in RPMI‐1640 (Gibco) supplemented with 10% heat‐inactivated FBS (Hyclone). The NP69 cell line was maintained in keratinocyte/serum‐free medium (Invitrogen). The cells were incubated at 37°C in 5% CO2 humidified air.
2.3. RNA interference
The shTRIM29#1 (TRC number: TRCN0000016348), shTRIM29#2 (TRCN0000016350), and sh‐control (SHC002) plasmids were purchased from Sigma‐Aldrich. Inducible knockdown of the TRIM29 gene was carried out with the specific shRNAs delivered by a pLKO‐Tet‐On system (Sigma‐Aldrich) according to the instruction manual. The target sequences were: pLKO‐Tet‐On shGFP sense, CAAGCTGACCCTGAAGTTCAT; and antisense, ATGAACTTCAGGGTCAGCTTGC. pLKO‐Rb‐shRNA and pLKO‐p53‐shRNA were purchased from Addgene. The shRNA sequences against VDAC1 (sense, 5′‐GCTTGGTCTAGGACTGGAATT‐3′ and antisense, 5′‐AATTCCAGTCCTAGACCAAGC‐3′; and sense, 5′‐GCAGTTGGCTACAAGACTGAT‐3′ and antisense, 5′‐ATCAGTCTTGTAGCCAACTGC‐3′) were cloned into pLKO‐Tet‐On.
2.4. Plasmid construction and cell transfection
The plasmids expressing human TRIM29 were generated by PCR amplification with Myc‐tag and subcloned into pLVX‐DsRed‐N1‐Monomer expression vector (Invitrogen) and pCDNA6B (Invitrogen). The WT TRIM29 expression plasmid was mutagenized by PCR to generate TRIM29 (shTRIM29#1‐Res) plasmids that contain six silent mutations introduced in the TRIM29‐shRNA#1 target sequence. VDAC1 was amplified from HONE1 cells with FLAG‐tag and inserted into pCDNA6B to construct pCDNA6B‐VDAC1‐FLAG, pET‐28a to construct pET‐28a‐GST‐VDAC1. Expression plasmids transfection as well as lentivirus preparation and infection were carried out as described previously. 25
2.5. Antibodies and chemicals
The following Abs were used: TRIM29 (mouse, sc‐166,718, Santa Cruz Biotechnology; rabbit, 222,070, Abcam), GAPDH, actin, and HA tag (60,004‐1‐Ig, 60,008‐1‐Ig, and 66,006‐1‐lg; Proteintech), Ki‐67, PARP1, and VDAC1 (sc‐23,900, 8007, and 390,996; Santa Cruz Biotechnology), p53 (1026‐1; Epitomics), GST, Rb, p‐Rb (Ser780), cleaved caspase‐3, p62, Beclin1, p21, and Myc tag (2624 s, 9309, 9307, 9664, 28,359, 3738, 2947, and 2278S; Cell Signaling Technology), FLAG tag (F1804; Sigma‐Aldrich), LC3B (2775 s; Novus Biologicals Inc.), HRP‐conjugated goat anti‐mouse and goat anti‐rabbit IgG (31,430 and 31,460; Pierce), and Alexa‐488 and Alexa‐546 (A11008 and A10036; Invitrogen Molecular Probes). 5‐Aza‐2′‐deoxycytidine, S‐adenosyl‐L‐methionine, doxycycline, n‐acetyl‐l‐cysteine, and puromycin were purchased from Sigma‐Aldrich.
2.6. Quantitative real‐time PCR
Total RNA was collected by using TRIzol reagent (Invitrogen) according to the manufacturer's protocol. To avoid amplification of genomic DNA, RNA was pretreated with DNase (DNA‐Free Kit; Takara). cDNA was synthesized from total RNA using M‐MLV Reverse Transcriptase (Invitrogen) and quantitative RT‐PCR was carried out using Platinum SYBR Green qPCR SuperMix (Invitrogen) according to the manufacturer's instructions. Gene‐specific primers are listed in Table S1.
2.7. Western blot analysis
Cells and tumor samples were subjected to lysis using RIPA buffer. Protein concentration was determined by using the Bradford dye method. The extracts were concentrated and separated by SDS‐PAGE, and then transferred to nitrocellulose membranes (Millipore). Membranes were probed with specific primary Abs and then with peroxidase‐conjugated secondary Abs. The bands were visualized with chemiluminescence.
2.8. MitoTracker staining
MitoTracker Deep Red (M22426; Invitrogen) and MitoTracker Green (M7514; Invitrogen) was used. Cells were stained for 25 min with MitoTracker (200 nM) in growth media at 37°C in an incubator.
2.9. Mitophagy assay
Mitophagy in live cells was monitored by a mitophagy detection kit (MD01; Dojindo). Carbonyl cyanide m‐chlorophenylhydrazone was used to trigger mitophagy. The level of mitophagy was defined by the area of Mtphagy Dye per cell. At least 50 cells were qualified in each group. The level of colocalization of Mtphagy Dye and Lyso Dye (to monitor lysosomes) was also analyzed. Quantification analysis was carried out using ImageJ software (NIH).
2.10. Immunofluorescence staining
Cells were fixed in 4% paraformaldehyde at room temperature for 15 min and permeabilized in 0.25% Triton X‐100 in PBS for 10 min. Slides were incubated with the primary Ab overnight. The immune complexes were stained with secondary Ab conjugated to Alexa‐488 or Alexa‐546 (Molecular Probes). Nuclei were stained with DAPI (Sigma‐Aldrich) and viewed with a confocal microscope (A1R; Nikon). To determine colocalization, the correlation coefficient values were calculated using ImageJ software.
2.11. DNA methylation analysis
Bisulphite DNA modification was carried out according to the manufacturer's instructions (EpiTect Bisulfite Kit; Qiagen). The MSP analysis was undertaken using standard protocols. 26 A segment of the TRIM29 gene was amplified from bisulfite‐converted DNA using primers outlined in Table S1 and pyrosequencing was carried out using the indicated sequencing primer.
2.12. Soft agar assay
Anchorage‐independent cell growth in soft agar was undertaken by seeding cells in complete growth media with 0.3% agar on top of a 0.6% agar layer in 6‐well plates. The plates were incubated at 37°C, 5% CO2 and checked every 2–3 days for colony formation. After 2–3 weeks, individual colonies were counted in 10 random fields.
2.13. Senescence‐associated β‐galactosidase staining
The SA‐β‐gal activity was assessed using a Senescence Detection Kit (Sigma‐Aldrich). Briefly, cultured cells were washed with PBS and fixed for 15 min (room temperature) in a fixation solution. Cells were then washed three times with PBS and incubated 4 h to overnight at 37°C with a fresh staining solution. The percentage of SA‐β‐Gal‐positive cells was calculated on the sum of total and SA‐β‐gal‐positive cells counted in six independent fields.
2.14. Measurement of ROS and mitochondrial membrane potential
To measure intracellular production of ROS, cells were collected and labeled in PBS containing 5 μM CM‐H2DCFDA (Sigma‐Aldrich) for 30 min. Cells were analyzed in a Becton Dickinson FACScan flow cytometer (Oxford) analysis. To measure superoxide level of mitochondria, cells were incubated with 5 μM MitoSOX (Invitrogen) for 30 min and analyzed by flow cytometry or confocal microscopy (FV1000; Olympus). Flow cytometric analysis of mitochondrial membrane potential was carried out by using a JC‐1 assay kit (Molecular Probes). Cells were incubated with 10 μg/mL JC‐1 for 10 min at 37°C.
2.15. Mouse xenograft assay
A single‐cell suspension of lentivirally transduced cells (3 × 105) was injected s.c. into BALB/c nude mice (4–6 weeks old). Tumor size was determined as (L × W2)/2; measurements of tumor length (L) and width (W) were taken with a caliper. 27 Tumors were removed for further analysis. Animal procedures were carried out with the approval of the Institutional Animal Care and Use Committee at Sun Yat‐sen University Cancer Center.
2.16. Immunoprecipitation
Immunoprecipitation was carried out as previously described, 25 with A/G agarose beads coated with anti‐Myc tag, anti‐FLAG tag, anti‐VDAC1, and anti‐GST Abs. The proteins were detected by western blot analysis using Abs against anti‐Myc tag, anti‐FLAG tag, and anti‐TRIM29. Mouse/rabbit IgG was used as a negative control.
2.17. Identification of interacting candidates of TRIM29
Cells expressing shTRIM29‐Tet‐On were grown in RPMI‐1640 medium lacking arginine, lysine, and methionine (a custom preparation from Gibco) supplemented with L‐methionine (15 mg/L) and 5% dialyzed FBS (Hyclone). “Heavy” and “light” media were prepared by adding 84 mg/L 13C6 15N4 L‐arginine and 40 mg/L 13C6 15N2 L‐lysine (Sigma Isotec) or the corresponding nonlabelled amino acids, respectively. The cells were cultivated for eight passages. In order to induce shRNA expression, 1 μM doxycycline (Sigma‐Aldrich) was added to 2 × 107 “light cells” directly after transfer to fresh tissue culture dishes, while the “heavy cells” were transferred without further treatment. Cells were grown for 2 days and solubilized with modified RIPA buffer containing 150 mM NaCl, 50 mM Tris (pH 7.5), 1 mM EDTA, 1% Nonidet P‐40 (NP‐40), 0.25% sodium deoxycholate, and protease inhibitors (Complete Tablet; Roche). A mixture of proteins from “heavy” and “light” labeled cells was immunoprecipitated using anti‐TRIM29 Ab. The eluted proteins were separated by SDS‐PAGE and subjected to liquid chromatography–mass spectrometry analysis as previously described. 25
2.18. Statistical analysis
Statistical values were expressed as the mean of three biological replicates ± SEM and performed by Student's t‐test or one‐way ANOVA. The probability values were noted as follows: *p < 0.05, **p < 0.01, ***p < 0.001. Relationships between TRIM29 levels and clinicopathological factors were analyzed by Spearman's rank correlation coefficients or the χ2‐test. All p values (two‐sided) less than 0.05 indicated a statistically significant difference. Statistical analysis was undertaken using GraphPad Prism software or IBM SPSS version 26.
3. RESULTS
3.1. TRIM29 is hypomethylated and its expression is elevated in NPC
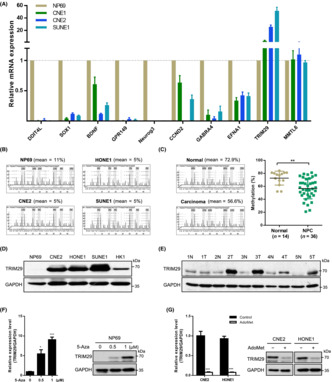
To explore the epigenetic changes mediated by DNA methylation in NPC, our group performed a Methyl‐Capture high‐throughput sequencing (MethylCap‐seq) assay previously in NPC cell lines CNE1, CNE2, and SUNE1, the untransformed nasopharyngeal epithelial cell line NP69, and a nontumor nasopharyngeal epithelial tissue sample. The candidate genes shown in Figure 1A were selected with significantly different methylation levels and we detected the mRNA expression of these genes. Among these genes, we observed the expression level of TRIM29 was enriched in several NPC cell lines compared with the noncancerous nasopharyngeal epithelial cell line NP69. Therefore, subsequent experiments were focused on TRIM29 in the present study, while other interesting candidates such as SOX1 were studied elsewhere. 28
FIGURE 1.
TRIM29 is hypomethylated and its expression is elevated in nasopharyngeal carcinoma (NPC). (A) mRNA levels of candidate genes in nasopharyngeal epithelial cell line NP69 and NPC cell lines were verified with real‐time PCR. (B) The mean percentage of methylation (%) and representative pyrograms in the TRIM29 gene region analyzed by pyrosequencing in NP69 and NPC cell lines. (C) The mean percentage of methylation (%) by pyrosequencing and representative pyrograms in the TRIM29 gene region in nasopharyngeal epithelial (normal) and nasopharyngeal carcinoma tissues (NPC); graph shows the combined data. (D) Expression of TRIM29 in NPC cell lines was measured by western blot (WB) analysis. (E) Expression of TRIM29 in normal nasopharyngeal tissues (N) and NPC tissues (T) was detected by WB analysis. (F) Real‐time PCR and WB analysis showed that treatment with 5‐aza‐deoxycytidine (5‐Aza) induced TRIM29 mRNA and protein expression in NP69 cells. (G) Real‐time PCR and WB analyses showed that treatment with S‐adenosylmethionine (AdoMet, 200 μM) reduced TRIM29 mRNA and protein expression in CNE2 and HONE1 cells. *p < 0.05**p < 0.01; ***p < 0.001.
The methylation status of CpG sites of the TRIM29 gene in nasopharyngeal epithelial cell line NP69 and human NPC cell lines CNE2, SUNE1, and HONE1 was analyzed by MSP and pyrosequencing (Figures 1B and S1A,B). The NPC cell lines contained lower levels of CpG methylation (mean = 5%) compared to the NP69 cell line (mean = 11%). We also examined TRIM29 gene methylation in normal nasopharyngeal epithelium tissues and primary NPC tissues. Pyrosequencing of bisulfite‐modified DNA showed that TRIM29 was significantly hypomethylated in primary NPC tissues (Figure 1C, p < 0.01).
Next, the expression level of TRIM29 was determined in NPC. As shown in Figure S1C,D, TRIM29 mRNA levels were enriched in NPC cell lines and tissues. Moreover, immunoblotting analysis confirmed that TRIM29 protein expression was also elevated in NPC (Figure 1D,E). To further explore whether high expression of TRIM29 is due to hypomethylation, we treated NP69 cells with an inhibitor of DNA methyltransferases, 5‐aza‐deoxycytidine. TRIM29 expression was restored at both the mRNA and protein levels (Figure 1F). Conversely, CNE2 and HONE1 cells were treated with the methyl donor S‐adenosylmethionine, an inhibitor of demethylase activity. 29 The treatment caused suppression of TRIM29 expression (Figure 1G), indicating that hypomethylation is responsible for gene activation.
3.2. TRIM29 silencing inhibits tumor growth in NPC
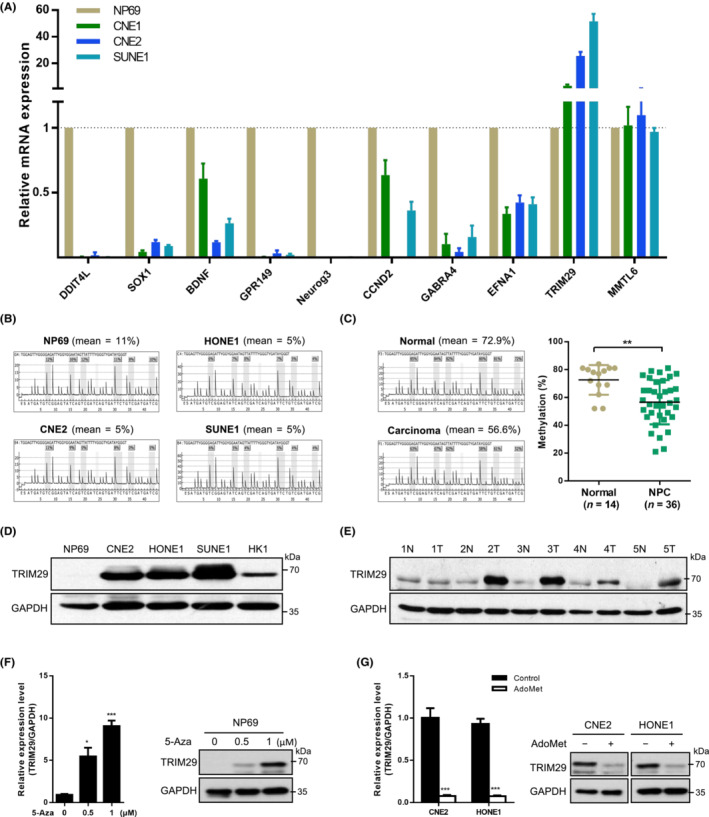
As TRIM29 levels were consistently higher in NPC cell lines, we used short‐hairpin RNA (shRNA) to generate knockdown cell model to study the role of TRIM29 in cancer cell growth. As shown in Figure 2A,B, endogenous TRIM29 expression was silenced by stable transfection with two different shRNA vectors (shT#1 and shT#2) compared to control (shNC). TRIM29 silencing remarkably inhibited the growth of CNE2 and HONE1 cells as measured by plate colony formation assay (Figure 2C,D). The soft agar assay also showed 3‐ to 10‐fold reduction of colony numbers and sizes in TRIM29‐shRNA infected cells (Figures 2E,F and S2A). In order to verify the observed phenotype was due to suppressed TRIM29 expression, we generated cell lines harboring the doxycycline‐inducible TRIM29 shRNA, and then transfected an shRNA‐resistant TRIM29 vector containing mutations in the region that is targeted by shRNA (Figures 2G and S2B). As expected, the proliferation defects of HONE1 cells induced by knockdown of endogenous TRIM29 were rescued by expression of shRNA‐resistant TRIM29 (Figure 2H). In addition, knockdown of TRIM29 reduced the percentage of Ki‐67‐positive cells (Figure S2C). We also tested the overexpression of TRIM29 in noncancerous NP69 cells, and found that TRIM29 could promote cell proliferation (Figure S2D).
FIGURE 2.
TRIM29 silencing inhibits tumor growth in nasopharyngeal carcinoma (NPC). (A, B) CNE2 and HONE1 cells were transfected with shRNA vector (sh‐control [shNC], shTRIM29#1 [shT#1], or shTRIM29#2 [shT#2]) using lentivirus‐mediated gene transfer. Knockdown efficiency was monitored by (A) quantitative RT‐PCR analysis after 48 h and (B) western blot analysis. (C, D) Cells were subjected to colony formation assay in plates. The colonies were photographed and counted. (E, F) Cells were subjected to colony formation assay in soft agar. The colonies were photographed and counted when the diameter was bigger than 60 μm. (G, H) HONE1 cells were cotransfected with doxycycline (Dox)‐inducible TRIM29 shRNA vector and the overexpression vector (vector or shRNA‐resistant TRIM29 [shT#1‐Res] containing mutations in the region that is targeted by the shRNA). Cells were treated with or without 1 μM Dox for 96 h and subjected to western blot analysis and colony formation assay in plates. (I, J) Lentivirally transduced CNE2 cells (shT#1, shT#2, or shNC) were injected s.c. (3 × 105 cells per mouse) into nude mice. Tumor volume was determined (n = 6); scale bar, 0.5 cm. (K) Tumor weight was determined (n = 6). (L) Representative photograph of immunohistochemistry staining of TRIM29, Ki‐67, and p‐Rb (S780) expression in tumor sections from the shT#1 and control xenografts. *p < 0.05; ***p < 0.001. Scale bar, 100 μm.
To investigate whether TRIM29 silencing impairs tumorigenesis in vivo, we s.c. injected CNE2 cells expressing a control shRNA or TRIM29 shRNA1 into the nude mice and measured the tumor size postinoculation. The mean volume of tumor was significantly smaller in the group of mice injected with cells expressing shTRIM29 compared to those injected with cells expressing shNC (Figure 2I,J, p < 0.05). The tumor weight was also decreased in different groups (Figure 2K). Immunohistochemical analysis revealed that the tumors of mice injected with CNE2 cells expressing TRIM29 shRNA showed lower staining of Ki‐67 and Rb phosphorylation (Ser780), markers of cell proliferation 30 (Figure 2L). These data suggest that TRIM29 silencing inhibits tumor growth in NPC.
3.3. TRIM29 silencing induces cancer cell into senescence
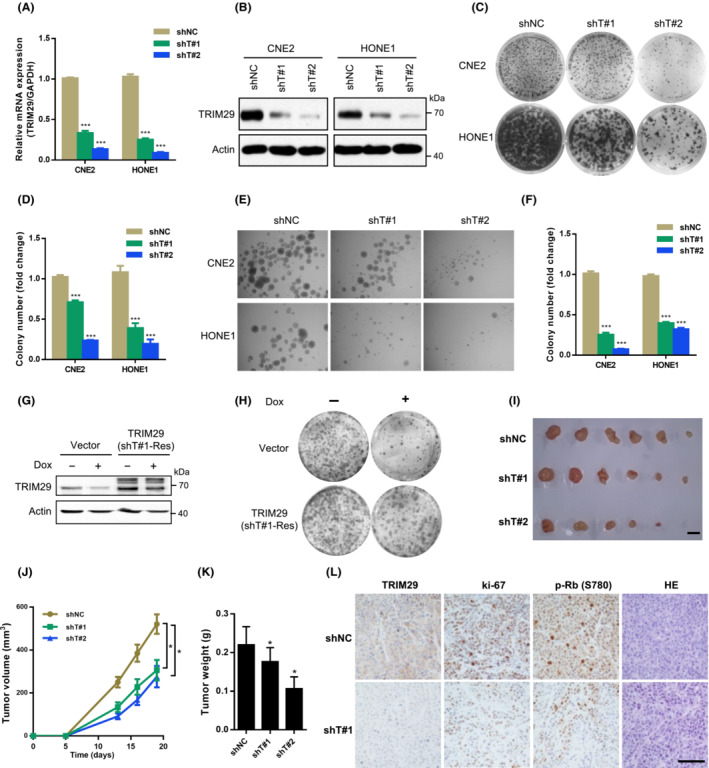
Next, we observed that TRIM29‐knockdown CNE2 and HONE1 cells showed enlarged cell size and flat vacuolated morphology (Figure 3A). The staining of SA‐β‐Gal was positive in approximately 20% of TRIM29 shRNA‐infected cells, but in less than 2% of the control cells (Figure 3B,C). The SA‐β‐gal phenotypes of TRIM29 knockdown were rescued by expression of an RNAi‐resistant form of TRIM29 (Figure S3A). Other markers of senescence were also detected, such as a dramatic increase in overall cellular granularity (Figure 3D), and elevated expression of SASP factors (Figure 3E), such as IL‐6, IL‐8, CXCL1, CXCL2, and insulin‐like growth factor binding protein 7 (IGFBP7). 31 In addition, we evaluated different molecular events of apoptosis, but we could not observe significant effects of TRIM29 silencing on the number of apoptotic cells as judged by the levels of cleaved PARP1 and cleaved caspase 3 (Figure S3B), nor TUNEL assay (Figure S3C).
FIGURE 3.
TRIM29 silencing induces cancer cell into senescence. (A‐C) CNE2 and HONE1 cells were transfected with shRNA vector (shNC, shT#1, or shT#2). (A) Morphology of cells is shown in phase contrast photomicrographs. Scale bar, 100 μm. (B) Photographs (magnification × 100) and (C) statistical graphs of senescence‐associated β‐galactosidase (β‐gal) staining. (D) Side scatter (SSC) of the cells was analyzed by flow cytometry. (E) Total RNA was extracted for quantitative PCR analysis of the indicated genes, which were normalized to GAPDH mRNA. (F) Expression of indicated proteins was analyzed by western blotting. **p < 0.01.
The senescence response in human cells relies mostly on the activation of p53 and/or Rb pathways, 32 so we examined whether p53 and/or Rb are involved in the cellular senescence induced by TRIM29 silencing. As shown in Figure 3F, the expression of p53 and p21 were upregulated in TRIM29 knockdown CNE2 and HONE1 cells, while the expression of p‐pRb (Ser780) was downregulated.
3.4. TRIM29 silencing triggers ROS accumulation and mitochondrial dysfunction
Because intracellular ROS are important mediators of senescence, 33 we next tested whether ROS play a role in the activation of senescence induced by TRIM29 silencing. As shown in Figure 4A,B, the ROS level was significantly increased in TRIM29 knockdown cells compared to control cells, as measured by DCFH‐DA, a fluorescent indicator for cellular oxidant production. TRIM29 knockdown also resulted in elevated mitochondrial ROS production (Figure 4C) and depolarization of mitochondrial membrane (Figure 4D). When ROS were scavenged by treatment of shTRIM29‐expressing HONE1 cells with an antioxidant, n‐acetyl‐cysteine, the percentage of SA‐β‐gal‐positive cells reduced with increasing doses (Figure 4E,F). These data suggest that TRIM29 silencing increases ROS accumulation and mitochondrial dysfunction.
FIGURE 4.
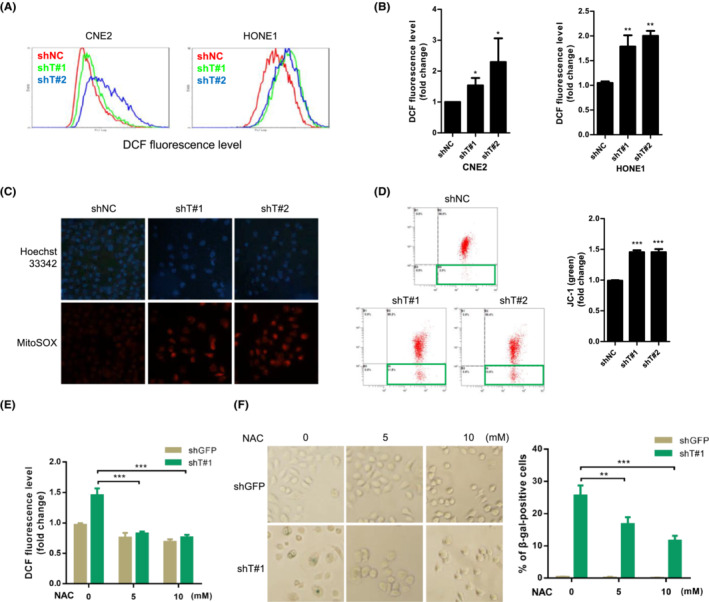
TRIM29 mediates senescence by a mechanism involving reactive oxygen species (ROS) accumulation. (A) The intracellular level of ROS was measured in CNE2 and HONE1 cells by 2′,7′‐dichlorodihydrofluorescein (DCF) fluorescence using flow cytometry. (B) Graphs show the quantification of fluorescence mean. (C) Mitochondrial superoxide of cells was measured by MitoSOX (5 μM, 30 min) and stained with Hoechst 33342. Representative immunofluorescent images are shown. (D) JC‐1 red and green fluorescence were measured by flow cytometry in HONE1 cells expressing control (shNC), shT#1, or shT#2. JC‐1 Red / JC‐1 Green ratios are plotted in the right panel. (E) HONE1 shGFP and shT#1‐Tet‐On cells were pretreated with indicated concentrations of n‐acetyl‐cysteine (NAC) (2 h), followed by cotreatment with doxycycline (1 μM) for 48 h. ROS generation was measured by DCF fluorescence using flow cytometry. (F) Senescence‐associated β‐galactosidase (β‐gal) staining was carried out in cells treated as described in (E). Photographs (left) (original magnification, ×100) and statistical graphs (right) are shown. *p < 0.05; **p < 0.01; ***p < 0.001.
3.5. TRIM29 combines with VDAC1
We next determined the mediator proteins that were critical for TRIM29 promoted accumulation of ROS and induction of cellular senescence. We investigated the proteins interacting with TRIM29 by SILAC combined with knockdown (Table S2). 34 As we observed that TRIM29 was localized to mitochondria (Figures 5A and S4A), we searched for proteins that are localized to mitochondria and regulators of ROS, and identified VDAC1, a protein of outer mitochondrial membrane, as a candidate (Figures 5B and S4B). The interaction between TRIM29 and VDAC1 was validated by Co‐IP of ectopically expressed (Figure 5C,D) and endogenous (Figure 5E) proteins. Figure 5F shows the colocalization of TRIM29 and VDAC1 by immunofluorescence. Together these data suggest that TRIM29 interacts with VDAC1.
FIGURE 5.
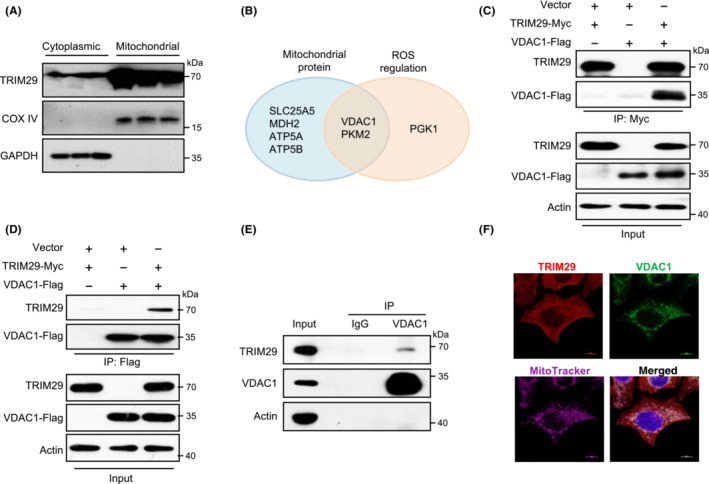
TRIM29 combines with voltage‐dependent anion‐selective channel 1 (VDAC1). (A) Mitochondrial protein fractions were subjected to western blotting (WB) using Abs as indicated. COX IV and GAPDH were used as loading controls. (B) TRIM29‐interacting proteins identified using stable isotope labeling with amino acids in cell culture (SILAC) assay. Proteins presented in both categories of mitochondrial proteins and reactive oxygen species (ROS) regulation proteins were selected for further analysis. (C, D) Co‐immunoprecipitation (IP) of TRIM29 and VDAC1 expressed in 293 T cells. Lysates from 293 T cells transfected with the indicated constructs were used for IP with anti‐Myc (left panel) or anti‐FLAG (right panel) Abs. Samples were analyzed by WB using Abs against FLAG‐tag or Myc‐tag. (E) Co‐IP of endogenous TRIM29 with VDAC1. HONE1 cell lysates were subjected to IP and WB using Abs as indicated. IP with nonspecific IgG was done as control. (F) Representative images are shown. Confocal microscopy analysis TRIM29, VDAC1, and MitoTracker in HONE1 cells. Scale bar, 10 μm.
3.6. TRIM29 mediates mitophagy through VDAC1
Because one of the early responses to excessive ROS is the induction of mitophagy, and VDAC1 is the most abundant components of the outer mitochondrial membrane and has been reported as a key player in the mitophagy process, 35 we hypothesized whether TRIM29 mediates mitophagy through VDAC1 and is involved in clearance of dysfunctional mitochondria. To investigate this possibility, we stained cells expressing TRIM29 or vector with a commercial mitophagy. In the treatment with CCCP, which triggers mitophagy, the number of puncta stained with Mtphagy Dye were significantly increased by ectopic expression of TRIM29 (Figure 6A–C), indicating an increased mitophagy flux. To further verify autophagy induction, we also used Lyso Dye, an acidic pH‐sensitive fluorescence dye staining lysosomes. Fluorescence images showed significant increase in the number of Lyso Dye‐positive lysosomes in TRIM29 overexpression cells, indicating increased acidification of lysosomes (Figure 6A–C). Mitophagy was also detected by confocal imaging based on colocalization of mitochondria with lysosomes. Confocal assays revealed ectopic expression of TRIM29 markedly increased mitochondria–lysosome colocalization (Figure 6D,E). Meanwhile, to ascertain whether TRIM29 overexpression‐induced mitophagy occurs in a VADC1‐dependent manner, VDAC1 expression was knocked down in 293 T cells by using lentivirus‐mediated delivery of shRNAs targeting VDAC1 (shVDAC1). Compared to cells infected with lentivirus expressing a nontargeting shRNA sequence (shNC), shVDAC1‐transfected cells had lower VDAC1 levels (Figure 6B). The effect of overexpression in TRIM29 on the induction of mitophagy was abolished by VDAC1 knockdown (Figure 6C,E).
FIGURE 6.
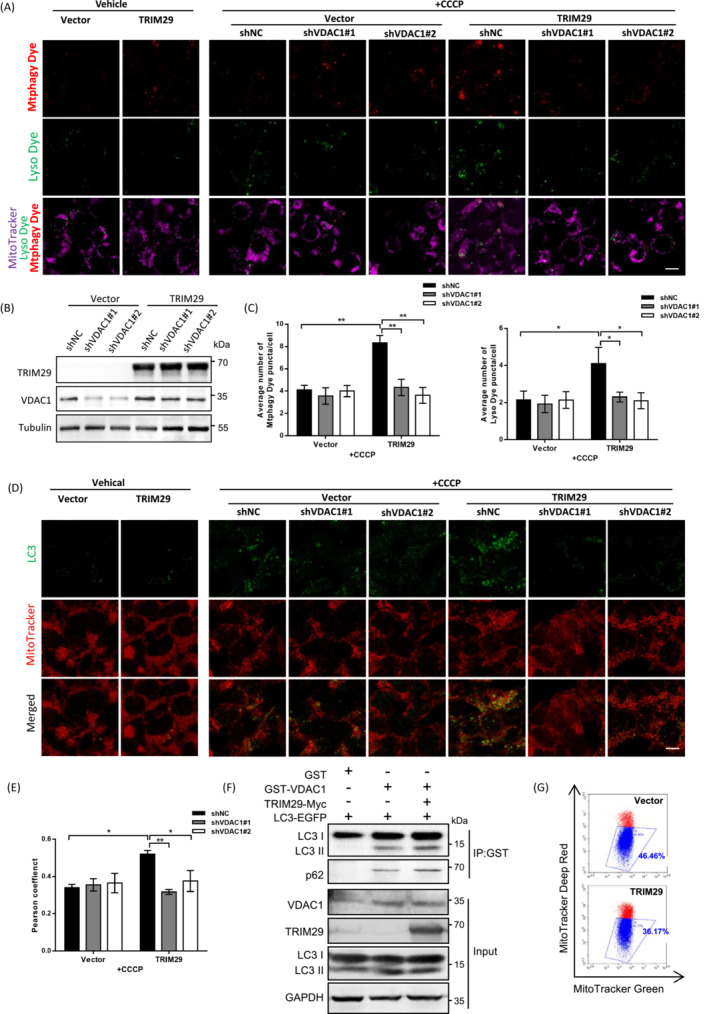
TRIM29 mediates mitophagy through voltage‐dependent anion‐selective channel 1 (VDAC1). (A) 293 T cells were infected with TRIM29 or vector in combination with shVDAC1 (shVDAC1#1 or shVDAC1#2), and treated with doxycycline (1 μM) for 48 h. Representative confocal images indicating Mtphagy Dye (red) and Lyso Dye (green) after vehicle or carbonyl cyanide m‐chlorophenylhydrazone (CCCP) exposure (6 μM, 6 h). Scale bar, 10 μm. (B) Expression of TRIM29 and VDAC1 was measured by western blot analysis. (C) Average number of Mtphagy Dye and Lyso Dye puncta per cell were counted, (D, E) Representative micrographs and quantification of colocalization for LC3 (green) and MitoTracker (red) staining in cells of (A) with or without CCCP (n > 50 cells). Scale bar, 10 μm. (F) GST‐VDAC1, TRIM29‐Myc, and LC3‐EGFP were overexpressed in 293 T cells. Cell lysates were subjected to co‐immunoprecipitation (IP) and western blot using Abs as indicated. (G) Mitochondria were stained with MitoTracker Green (Ex/Em = 490 nm/516 nm) and MitoTracker Deep Red (Ex/Em = 640 nm/665 nm) for 15 min, then analyzed by flow cytometry. Gates represent cells with damaged mitochondria. *p < 0.05, **p < 0.01.
TRIM29 overexpression also enhanced the level of LC3‐II and p62 bound to VDAC1 (Figure 6F). In extracted mitochondria fractions, we carried out immunoprecipitation with anti‐GST Abs, and the level of LC3‐II and p62 binding with VDAC1 was also elevated in cells coexpressed with TRIM29 (Figure S5A). Moreover, the Co‐IP results revealed a decreased interaction between VDAC1 and LC3‐II or p62 in shTRIM29‐expressing HONE1 cells with CCCP treatment (Figure S5B). Additionally, we investigated whether knockdown of TRIM29 suppressed VDAC1 ubiquitination and the Co‐IP results showed that TRIM29 silencing decreased the ubiquitination level of VDAC1 after CCCP exposure (Figure S5C). Furthermore, we undertook flow cytometry assays with fluorescence markers to quantitate mitochondrial damage. Consistent with the above results, TRIM29 overexpression decreased the dysfunctional mitochondria (Figure 6G). Taken together, these data support the notion that TRIM29 mediates mitophagy through VDAC1 and enhances the clearance of damaged mitochondria.
3.7. TRIM29 expression is correlated with clinical stage and TRIM29 is hypomethylated in patient nasopharyngeal swab DNA
Next, we determined the expression of TRIM29 in human NPC samples by immunohistochemistry. Staining intensity of TRIM29 is shown in Figure 7A. Clinical features are listed in Table S3 in relation to TRIM29 staining level. No correlations in staining intensity of TRIM29 with patient age, sex, lymph node status, or Epstein–Barr virus‐related serologic status were observed. However, as shown in Figure 7B, TRIM29 expression was significantly associated with clinical stage (p = 0.043). To explore whether hypomethylation of TRIM29 in nasopharyngeal swab DNA is a potential marker for detection of NPC, we detected TRIM29 methylation status of nasopharyngeal swab DNA obtained from NPC patients and normal controls by quantitative pyrosequencing analysis. The results revealed that NPC samples contained extremely low levels of CpG methylation (mean = 37.7% vs. 59.2%, p < 0.01, Figure 7C; diagnostic accuracy: area under the curve = 0.8609, 95% confidence interval, 0.7175–1.0004, p < 0.005, Figure 7D).
FIGURE 7.
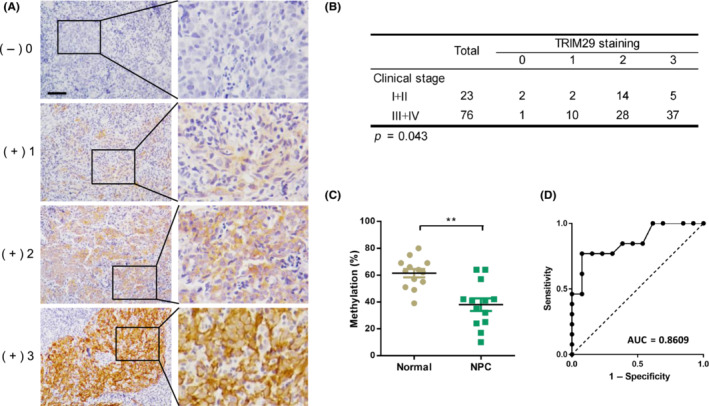
TRIM29 expression is correlated with clinical stage and TRIM29 is hypomethylated in patient nasopharyngeal swab DNA. (A) Representative images from immunohistochemistry analysis of TRIM29 expression in nasopharyngeal carcinoma (NPC) tissues. The brown granules in cytoplasm were considered as TRIM29‐positive staining. The staining intensity was scored as follows: 0, no staining; 1+, mild staining; 2+, moderate staining; and 3+, intense staining. Scale bar, 50 μm. (B) Spearman's rank correlation test was used to determine the correlation between TRIM29 staining and clinical stages. (C) Pyrosequencing of methylation status of CpG island of TRIM29 in DNAs from NPC and control swab samples; graph shows the combined data (normal, n = 13; NPC, n = 13). (D) Receiver operating characteristic (ROC) curve of TRIM29 methylation for the discrimination between NPC and normal control. **p < 0.01. AUC, area under the ROC curve.
4. DISCUSSION
In this study, we identified TRIM29 as a hypomethylation‐activated gene in NPC and high expression of TRIM29 was associated with advanced clinical stages. Suppression of TRIM29 inhibited tumor growth and triggered NPC cells into cellular senescence. The mechanism involves TRIM29‐mediated mitophagy through binding with VDAC1 and regulation of ROS accumulation. Finally, we determined that in NPC patients the TRIM29 gene was hypomethylated in DNA prepared from nasopharyngeal swabs.
In NPC, DNA hypermethylation was the most frequently reported mechanism for inactivation of cancer‐associated genes, 7 , 36 but DNA hypomethylation of the promoter CpG islands receives little attention. In this study, we detected the CpG sites located upstream of the first exon of the TRIM29 gene and found that TRIM29 was hypomethylated in NPC. As expected, elevated expression of TRIM29 is associated with hypomethylation. Our results are in agreement with earlier observations that TRIM29 is overexpressed in many cancers and may explain how TRIM29 might be activated. TRIM29 is differentially regulated in the respective cancers that are cancer‐type dependent. Reduced expression of TRIM29 was noted in breast cancer, in which TRIM29 is silenced by promoter hypermethylation. 37 Interestingly, here we undertook quantitative pyrosequencing analysis to detect the methylation status of TRIM29 using nasopharyngeal swab DNA obtained from NPC patients and found that NPC samples are hypomethylated, suggesting the possibilities for noninvasive early detection of NPC by studying TRIM29 methylation.
TRIM29 is required for NPC cell survival as its depletion inhibits the proliferation of the cells and induces senescence. Senescence is a cellular response to stress signals that is characterized by stable cell cycle arrest and major changes in cell morphology and physiology. Although most studies on senescence have been carried out on noncancer cells, it is evident that cancer cells can also mount a senescence response. 38 TRIM29 was shown to suppress expression of p53 and p21, which is consistent with previous observations that TRIM29 inhibits p53 nuclear activities and expression of p21. 18
Reactive oxygen species are well known as critical mediators of cellular senescence. 33 Reactive oxygen species are mainly generated by mitochondria, and mitochondrial dysfunction is associated with increased ROS production. We show here that ROS accumulation in response to TRIM29 knockdown is responsible for inducing senescence. One of the early responses to excessive ROS is the induction of mitophagy, and mitophagy degrades dysfunctional or excessive mitochondria through the process of the autophagosome–lysosome system. Furthermore, our results suggest that TRIM29 was localized to mitochondria and interacted with VDAC1, and TRIM29 mediates mitophagy through VDAC1 and enhances the clearance of damaged mitochondria. VDAC1 is the most abundant adaptor protein on the outer mitochondrial membrane and a regulator of mitophagy. Ubiquitination of VDAC1 induced by Parkin and PINK1 was responsible for the activation of mitophagy. 39 Our data indicates that TRIM29 silencing inhibits VDAC1 ubiquitination. The detailed molecular mechanism of the regulation of VDAC1 by TRIM29 need to be further studied in future.
In summary, our data suggest that TRIM29 is hypomethylated and overexpressed in NPC, and provide insights into the molecular mechanism underlying the role of TRIM29 in tumorigenesis. Detection of aberrantly methylated TRIM29 in nasopharyngeal swab DNA samples could be a promising strategy for the early detection of NPC.
AUTHOR CONTRIBUTIONS
QL, MY, and CY designed the experiments. CY, YQ, CW, and JC performed the experiments. CY, YQ, MY, and QL analyzed the data. CY, MY and QL wrote the manuscript. The other authors provided reagents, designed or performed specific experiments, and helped revise the paper.
FUNDING INFORMATION
This work was supported by National Key R&D Program of China (2019YFA0110300, 2022YFA1104002), National Natural Science Foundation of China (81602416, 81972594, 82341020, 82173367), Natural Science Foundation of Guangdong (2020A1515010608, 2022A1515010915, 2023A1515030063), Shenzhen Bay Laboratory Research Funds (SZBL2021080601001).
CONFLICT OF INTEREST STATEMENT
The authors declare no conflicts of interest.
ETHICS STATEMENT
Approval of the research protocol by an institutional review board: This study was approved by the Ethical Committee of Sun Yat‐sen University Cancer Center.
Informed consent: Informed consent was obtained from all patients.
Registry and registration no. of the study: All animal studies and human subject research were approved by the ethical committee of Sun Yat‐sen University Cancer Center (IRB approval number GZR2013‐110).
Animal studies: Animal experiments were approved by the Animal Care and Use Committee at Sun Yat‐sen University Cancer Center and performed according to established guidelines.
Supporting information
Appendix S1
ACKNOWLEDGMENTS
We thank all members of the Liu laboratory for their critical comments and technical support.
Yue C, Qian Y, Wang C, et al. TRIM29 acts as a potential senescence suppressor with epigenetic activation in nasopharyngeal carcinoma. Cancer Sci. 2023;114:3176‐3189. doi: 10.1111/cas.15852
Caifeng Yue, Yuanmin Qian, and Chang Wang contributed equally to this work.
Contributor Information
Min Yan, Email: yanmin@sysucc.org.cn.
Quentin Liu, Email: liuq9@mail.sysu.edu.cn.
REFERENCES
- 1. Baylin SB, Ohm JE. Epigenetic gene silencing in cancer – a mechanism for early oncogenic pathway addiction? Nat Rev Cancer. 2006;6:107‐116. [DOI] [PubMed] [Google Scholar]
- 2. Esteller M. Cancer epigenomics: DNA methylomes and histone‐modification maps. Nat Rev Genet. 2007;8:286‐298. [DOI] [PubMed] [Google Scholar]
- 3. Heyn H, Esteller M. DNA methylation profiling in the clinic: applications and challenges. Nat Rev Genet. 2012;13:679‐692. [DOI] [PubMed] [Google Scholar]
- 4. Zhang L, Chen QY, Liu H, Tang LQ, Mai HQ. Emerging treatment options for nasopharyngeal carcinoma. Drug des Devel Ther. 2013;7:37‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chua ML, Wee JT, Hui EP, Chan AT. Nasopharyngeal carcinoma. Lancet. 2016;387:1012‐1024. [DOI] [PubMed] [Google Scholar]
- 6. Zhu QY, Zhao GX, Li Y, et al. Advances in pathogenesis and precision medicine for nasopharyngeal carcinoma. MedComm. 2020;2021(2):175‐206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lo KW, Chung GT, To KF. Deciphering the molecular genetic basis of NPC through molecular, cytogenetic, and epigenetic approaches. Semin Cancer Biol. 2012;22:79‐86. [DOI] [PubMed] [Google Scholar]
- 8. Bruce JP, Yip K, Bratman SV, Ito E, Liu FF. Nasopharyngeal cancer: molecular landscape. J Clin Oncol. 2015;33:3346‐3355. [DOI] [PubMed] [Google Scholar]
- 9. Chi KH, Chang YC, Guo WY, et al. A phase III study of adjuvant chemotherapy in advanced nasopharyngeal carcinoma patients. Int J Radiat Oncol Biol Phys. 2002;52:1238‐1244. [DOI] [PubMed] [Google Scholar]
- 10. Nawaz I, Moumad K, Martorelli D, et al. Detection of nasopharyngeal carcinoma in Morocco (North Africa) using a multiplex methylation‐specific PCR biomarker assay. Clin Epigenetics. 2015;7:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hatakeyama S. TRIM proteins and cancer. Nat Rev Cancer. 2011;11:792‐804. [DOI] [PubMed] [Google Scholar]
- 12. Hawthorn L, Stein L, Panzarella J, Loewen GM, Baumann H. Characterization of cell‐type specific profiles in tissues and isolated cells from squamous cell carcinomas of the lung. Lung Cancer. 2006;53:129‐142. [DOI] [PubMed] [Google Scholar]
- 13. Dyrskjot L, Kruhoffer M, Thykjaer T, et al. Gene expression in the urinary bladder: a common carcinoma in situ gene expression signature exists disregarding histopathological classification. Cancer Res. 2004;64:4040‐4048. [DOI] [PubMed] [Google Scholar]
- 14. Wang L, Heidt DG, Lee CJ, et al. Oncogenic function of ATDC in pancreatic cancer through Wnt pathway activation and beta‐catenin stabilization. Cancer Cell. 2009;15:207‐219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kosaka Y, Inoue H, Ohmachi T, et al. Tripartite motif‐containing 29 (TRIM29) is a novel marker for lymph node metastasis in gastric cancer. Ann Surg Oncol. 2007;14:2543‐2549. [DOI] [PubMed] [Google Scholar]
- 16. Kanno Y, Watanabe M, Kimura T, Nonomura K, Tanaka S, Hatakeyama S. TRIM29 as a novel prostate basal cell marker for diagnosis of prostate cancer. Acta Histochem. 2014;116:708‐712. [DOI] [PubMed] [Google Scholar]
- 17. Santin AD, Zhan F, Bellone S, et al. Gene expression profiles in primary ovarian serous papillary tumors and normal ovarian epithelium: identification of candidate molecular markers for ovarian cancer diagnosis and therapy. Int J Cancer. 2004;112:14‐25. [DOI] [PubMed] [Google Scholar]
- 18. Yuan Z, Villagra A, Peng L, et al. The ATDC (TRIM29) protein binds p53 and antagonizes p53‐mediated functions. Mol Cell Biol. 2010;30:3004‐3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Masuda Y, Takahashi H, Hatakeyama S. TRIM29 regulates the p63‐mediated pathway in cervical cancer cells. Biochim Biophys Acta. 2015;1853:2296‐2305. [DOI] [PubMed] [Google Scholar]
- 20. Palmbos PL, Wang L, Yang H, et al. ATDC/TRIM29 drives invasive bladder cancer formation through microRNA‐mediated and epigenetic mechanisms. Cancer Res. 2015;75:5155‐5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang L, Yang H, Abel EV, et al. ATDC induces an invasive switch in KRAS‐induced pancreatic tumorigenesis. Genes Dev. 2015;29:171‐183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ben‐Porath I, Weinberg RA. The signals and pathways activating cellular senescence. Int J Biochem Cell Biol. 2005;37:961‐976. [DOI] [PubMed] [Google Scholar]
- 23. Sharpless NE, Sherr CJ. Forging a signature of in vivo senescence. Nat Rev Cancer. 2015;15:397‐408. [DOI] [PubMed] [Google Scholar]
- 24. Tommasi S, Zheng A, Weninger A, et al. Mammalian cells acquire epigenetic hallmarks of human cancer during immortalization. Nucleic Acids Res. 2013;41:182‐195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zheng F, Yue C, Li G, et al. Nuclear AURKA acquires kinase‐independent transactivating function to enhance breast cancer stem cell phenotype. Nat Commun. 2016;7:10180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation‐specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci U S A. 1996;93:9821‐9826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hallahan AR, Pritchard JI, Chandraratna RA, et al. BMP‐2 mediates retinoid‐induced apoptosis in medulloblastoma cells through a paracrine effect. Nat Med. 2003;9:1033‐1038. [DOI] [PubMed] [Google Scholar]
- 28. Guan Z, Zhang J, Wang J, et al. SOX1 down‐regulates beta‐catenin and reverses malignant phenotype in nasopharyngeal carcinoma. Mol Cancer. 2014;13:257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Detich N, Hamm S, Just G, Knox JD, Szyf M. The methyl donor S‐adenosylmethionine inhibits active demethylation of DNA: a candidate novel mechanism for the pharmacological effects of S‐adenosylmethionine. J Biol Chem. 2003;278:20812‐20820. [DOI] [PubMed] [Google Scholar]
- 30. Fry DW, Harvey PJ, Keller PR, et al. Specific inhibition of cyclin‐dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. 2004;3:1427‐1438. [PubMed] [Google Scholar]
- 31. Kuilman T, Michaloglou C, Vredeveld LC, et al. Oncogene‐induced senescence relayed by an interleukin‐dependent inflammatory network. Cell. 2008;133:1019‐1031. [DOI] [PubMed] [Google Scholar]
- 32. Collado M, Serrano M. Senescence in tumours: evidence from mice and humans. Nat Rev Cancer. 2010;10:51‐57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen Q, Fischer A, Reagan JD, Yan LJ, Ames BN. Oxidative DNA damage and senescence of human diploid fibroblast cells. Proc Natl Acad Sci U S A. 1995;92:4337‐4341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Selbach M, Mann M. Protein interaction screening by quantitative immunoprecipitation combined with knockdown (QUICK). Nat Methods. 2006;3:981‐983. [DOI] [PubMed] [Google Scholar]
- 35. Geisler S, Holmstrom KM, Skujat D, et al. PINK1/parkin‐mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12:119‐131. [DOI] [PubMed] [Google Scholar]
- 36. Yan M, Zhang Y, He B, et al. IKKalpha restoration via EZH2 suppression induces nasopharyngeal carcinoma differentiation. Nat Commun. 2014;5:3661. [DOI] [PubMed] [Google Scholar]
- 37. Ai L, Kim WJ, Alpay M, et al. TRIM29 suppresses TWIST1 and invasive breast cancer behavior. Cancer Res. 2014;74:4875‐4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang L, Lankhorst L, Bernards R. Exploiting senescence for the treatment of cancer. Nat Rev Cancer. 2022;22:340‐355. [DOI] [PubMed] [Google Scholar]
- 39. Ham S, Lee D, Yoo H, et al. Decision between mitophagy and apoptosis by parkin via VDAC1 ubiquitination. Proc Natl Acad Sci U S A. 2020;117:4281‐4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1