

Abstract
Experimental techniques for patient‐derived cancer stem‐cell organoids/spheroids can be powerful diagnostic tools for personalized chemotherapy. However, establishing their cultures from gastric cancer remains challenging due to low culture efficiency and cumbersome methods. To propagate gastric cancer cells as highly proliferative stem‐cell spheroids in vitro, we initially used a similar method to that for colorectal cancer stem cells, which, unfortunately, resulted in a low success rate (25%, 18 of 71 cases). We scrutinized the protocol and found that the unsuccessful cases were largely caused by the paucity of cancer stem cells in the sampled tissues as well as insufficient culture media. To overcome these obstacles, we extensively revised our sample collection protocol and culture conditions. We then investigated the following second cohort and, consequently, achieved a significantly higher success rate (88%, 29 of 33 cases). One of the key improvements included new sampling procedures for tumor tissues from wider and deeper areas of gastric cancer specimens, which allowed securing cancer stem cells more reproducibly. Additionally, we embedded tumor epithelial pieces separately in both Matrigel and collagen type‐I as their preference to the extracellular matrix was different depending on the tumors. We also added a low concentration of Wnt ligands to the culture, which helped the growth of occasional Wnt‐responsive gastric cancer stem‐cell spheroids without allowing proliferation of the normal gastric epithelial stem cells. This newly improved spheroid culture method may facilitate further studies, including personalized drug‐sensitivity tests prior to drug therapy.
Keywords: extracellular matrix, gastric cancer, spheroid, stem cell, Wnt
We developed a simple and efficient method to propagate patient‐derived gastric cancer stem‐cell spheroids by improving our conventional sample collection protocol and culture conditions. These spheroids can be utilized to investigate new molecular targeted therapies and their companion diagnostics for patient selection.
Abbreviations
- CI
confidence interval
- CM
conditioned medium
- CRC
colorectal cancer
- EGF
epidermal growth factor
- GC
gastric cancer
- GEI
growth effect index
- NGE‐SC
normal gastric epithelial stem cell
- PD
patient‐derived
- RNA‐seq
RNA sequencing
- SC
stem cell
- WHO
World Health Organization
1. INTRODUCTION
Gastric cancer (GC) is the fifth most common cancer in the world and fourth leading cause of cancer death even with significant improvements in surgical techniques and chemotherapy. 1 , 2 Histopathologically, GC comprises intestinal and diffuse types according to Lauren's classification, 3 which are further subdivided according to the World Health Organization (WHO) classification. 4 Recently, The Cancer Genome Atlas 5 and Asian Cancer Research Group 6 proposed molecular classifications based on the gene expression profiles. However, these classifications are of limited help in determining the most efficacious treatments, necessitating a personalized strategy. Currently, a few diagnostic markers are available to select suitable GC patients for treatment with therapeutic antibodies, such as those against HER2 7 and PD‐1/PD‐L1. 8 , 9 Since only a small proportion of patients can benefit from each therapy, more diagnostic tools are needed to stratify patients for current and upcoming therapies so that specific GC subpopulations can be effectively targeted.
Among possibly promising strategies for personalized cancer treatments, a more direct approach is to test the drug sensitivity of patient‐derived (PD) cancer stem cells (SCs) in vitro and/or in mouse xenografts. Recently, testing PD cancer stem‐cell organoids have become feasible as a clinically relevant tool for investigating personalized therapeutics, 10 , 11 as exemplified by those derived from colorectal cancer (CRC). 12 When it comes to GC, however, the success rates for establishing GC‐SC lines are substantially lower than those for CRC‐SC, with cumbersome culture methods owing to various supplementary factors and selection drugs needed for specific subtypes of GC. 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20
Recently, we have reported an efficient method for culturing PD–CRC‐SCs 21 based on the method for normal intestinal epithelial stem cells. 22 , 23 , 24 These cells embedded in Matrigel form nearly spherical structures, termed spheroids, that are comprised of nearly all mitotic stem/progenitor cells, in contrast to intestinal organoids with the budding structures that comprise mixed populations of mitotic and post‐mitotic cells. 25 In the present study, we have modified this conventional culture method for propagating PD–GC‐SC spheroids so that we can apply it for personalized clinical diagnosis and treatment.
2. MATERIALS AND METHODS
2.1. Human samples
Tumor samples were collected from GC patients who underwent primary resections at the Kyoto University Hospital (KUHP, Kyoto, Japan) and Medical Research Institute Kitano Hospital (Osaka, Japan) from January 2016 to November 2022. Their diagnosis was confirmed through histopathological examinations by board‐certified diagnostic pathologists.
2.2. L‐WRN conditioned medium
The L‐WRN cells expressing mouse Wnt3a, R‐spondin 3, and Noggin were obtained from Dr. Thaddeus S. Stappenbeck (Cleveland Clinic). Conditioned medium (CM) from L‐WRN cells was prepared according to a previous protocol. 22 Quality control testing of L‐WRN CM was conducted according to the validation procedures and guidelines reported previously. 26 A commercial L‐WRN CM was purchased from Sigma‐Aldrich.
2.3. Spheroid culture of human gastric cancer and normal gastric epithelial cells
Immediately after surgical resection, the excised stomach by operation was opened longitudinally, wrapped in gauze moistened with saline to prevent drying, and kept at room temperature. Sample specimens were collected within 1 h after the resection operation. From each stomach, one to four tumor pieces (100–1000 mm3 each) and one to two pieces of normal mucosa (500–2000 mm3) were collected in separate 15‐mL conical tubes containing 5–10 mL ice‐cold washing medium (Table S1). Sample tubes were kept on ice during transportation to the laboratory, and the isolation of epithelial cells and preparation of stem cell culture were performed within 6 h after sample collection (i.e., 7 h after the resection operation) according to a step‐by‐step protocol. 22 Specifically, the specimen pieces were minced in a 60‐mm Petri dish, digested with 1–3 mL collagenase solution (Table S1) at 37°C for 40–60 min and dissociated by pipetting. Epithelial cell clusters were filtered through a 100‐μm cell strainer (Corning), collected in a 1.5‐mL tube, and resuspended in Matrigel (Corning) or collagen type‐I matrix (Cellmatrix, Nitta Gelatin). The cell‐matrix mixture was placed at the center of each well of the 12‐well cell‐culture plate (30 μL/well; TPP). After polymerization of matrix materials at 37°C, GC and normal gastric epithelial (NGE) cells were cultured with the cancer medium and eL‐WRN medium (epidermal growth factor [EGF]‐containing 50% L‐WRN CM), respectively (Table S1). The medium was changed every other day. To passage, we collected Matrigel‐embedded spheroids and treated them with 2.5 g/L trypsin solution (Nacalai Tesque) at 37°C for 2–5 min. Collagen type‐I–embedded spheroids were treated with collagenase solution at 37°C for 30 min, followed by trypsinization. Spheroids were dissociated into small cell aggregates by pipetting, and they were resuspended in Matrigel or collagen type‐I. Dilution (based on the volume of matrix materials) was adjusted to one to six times depending on the growth rate and spheroid density. It should be noted that too much trypsinization and pipetting caused poor cell survival when spheroids grew poorly in early passages. The spheroid culture was considered successful when spheroids were expanded to 12 wells of a 12‐well cell‐culture plate.
2.4. Growth monitoring in spheroid culture using a cell imager
To monitor cell growth, we resuspended trypsinized spheroids in Matrigel or collagen type‐I at a density of approximately 150 cell aggregates/μL. Subsequently, 3 μL cell‐matrix mixture was distributed in each well of the 96‐well cell‐culture plate (TPP). After polymerization of matrix materials, cells were cultured in 100 μL of media. High‐resolution cell images were obtained using a cell imager (Cell3iMager duos, SCREEN) every 3–4 days (Figure S1A). The area of each spheroid in each well was outlined using image processing software (Figure S1B). The volume of each spheroid was estimated using the following formula: spheroid volume (μm3) = 4/3 × {[spheroid area (μm2)]3/π}1/2. The cell growth rate for each well was estimated as the proportion of total spheroid volume to that on initial measurement, and the growth effect index (GEI) was defined as the relative growth rate of an experimental group to that of its control group. At least three independent experiments were performed for each analysis.
2.5. Mutational analysis
The exonic regions of 409 cancer‐related genes in GC‐SC spheroids were sequenced using the Ion AmpliSeq Comprehensive Cancer Panel (Thermo Fisher), and the sequence alignment to the reference genome (hg19) and variant calling were performed at Macrogen Japan. We omitted the analyses of the primary tumors because we and others had shown homogeneity of driver‐gene mutations in cancer and their stability during ex vivo culture. 14 , 27 , 28 Detection of cancer‐specific mutations was performed as we described previously with modifications. 27 Specifically, polymorphic alleles were removed from the called variants using the VCFtools program (V.0.1.13) 29 by referring to the GEM Japan Whole Genome Aggregation (GEM‐J WGA) panel (https://togovar.biosciencedbc.jp/doc/datasets/gem_j_wga) or the profiles of NGE‐SC spheroids from the same patients (when available). The selected variants were annotated using the ANNOVAR program, 30 and polymorphic alleles were removed again by referring to the Human Genetic Variation Database. 31 , 32 Subsequently, they were filtered to select nonsynonymous, frameshift, and splicing mutations with more than 20% frequency. Variant calls that appeared in more than two lines were eliminated as false‐positive except for those identified in the COSMIC database. Other erroneous mutations were eliminated by surveying their coverage tracks on the Integrative Genomics Viewer software (V.2.12.3, Broad Institute).
2.6. Mutation detection from RNA sequencing (RNA‐seq) data
To save time and cost, we took advantage of our transcriptome analysis data that we completed in most GC‐SC spheroid lines. Namely, mutations in cancer‐related genes were determined by deducing from the sequences of the RNA‐seq data. Spheroid RNA samples were purified using the NucleoSpin RNA II kit (Takara Bio), and RNA‐seq analysis was performed at Macrogen Japan. The sequence alignment to the reference genome (hg19) and variant calling were performed using the Subio Platform software (V.1.24.5853, Subio). Cancer‐specific mutations in the exonic regions of expressed genes were detected with the same workflow as for the cancer panel.
Additional Materials and Methods can be found in Appendix S1.
3. RESULTS
3.1. Improvement of patient‐derived gastric cancer stem‐cell spheroid culture efficiency using a revised protocol
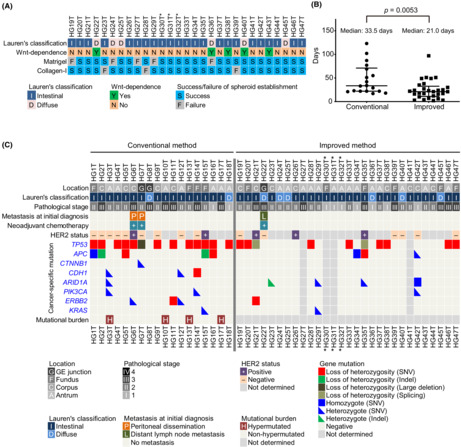
To culture GC‐SC spheroids, we conducted two sets of experiments in which we collected tumor samples from 71 patients of the first cohort, followed by those from 33 patients of the second. To the first cohort samples, we applied our conventional method originally developed for CRC‐SC spheroids (Table 1). Namely, we cultured tumor epithelial cells in a serum‐containing cancer medium (Table S1) to propagate GC‐SC spheroids. 21 In contrast, NGE‐SC spheroids were also established from normal mucosa of the same patients using the eL‐WRN medium (Table S1) containing mouse Wnt3a, R‐spondin 3, and Noggin. 21 , 22 The success rate for establishing GC‐SC spheroids was 25% (18 of 71 cases; 95% CI, 15%–35%), whereas that for NGE‐SC spheroids was 94% (67 of 71 cases; 95% CI, 89%–100%; Table 1; Table S2). To improve the low success rate, we revised our protocol in the following three points and tested its feasibility with fresh GC samples of the second patient cohort (Table 1). First and foremost, we scrutinized the sample collection maneuver from cancer tissues. One of the major reasons for our earlier failure in GC‐SC spheroid establishment by our conventional method was likely the paucity of cancer stem cells in the sampled tumor pieces as estimated histopathologically in a retrospective manner (47% with 95% CI, 30%–64%; in 16 of the 34 failed cases; Figure 1A). Another minor cause was fungal contamination (9% with 95% CI, 2%–17%; in five of the 53 failed cases), particularly, of those samples from necrotic lesions that tended to accumulate fungi and/or hyphae (Figure 1B). Therefore, we collected more tumor pieces from wider and deeper areas, avoiding necrotic lesions to harvest cancer stem cells more reproducibly (Figure 1C,D). Importantly, the revised protocol reviewed by board‐certified diagnostic pathologists of the collaborating hospitals did not affect pathological and molecular pathological assessment. Second, we embedded tumor epithelial pieces of each patient in both Matrigel and collagen type‐I separately. This was because the different extracellular matrix (ECM) was preferred in some minority cases. Third, we added 5% L‐WRN CM (containing Wnt ligands) to the cancer medium to help propagate Wnt‐responsive GC‐SCs, as the extent of dependence of GC‐SC organoids on Wnt ligands has been variable. 13 , 33 Owing to these changes, we achieved a significantly higher success rate (88% with 95% CI, 77%–99%; 29 of 33 cases) as compared to that (25% with 95% CI, 15%–35%; 18 of 71 cases) with the first patient cohort (Table 1; Table S3). We failed in four of 33 cases because of heavy contamination with yeasts (two cases) or poor cell growth in early passages (two cases). Notably, five of 29 lines (17%) were established only when embedded in collagen type‐I with a statistically significant difference (p = 0.008, Fisher's exact test), whereas three lines (10%) were only in Matrigel (Figure 2A). Regarding Wnt dependency, five GC‐SC lines required L‐WRN CM to maintain spheroid lines (Figure 2A). Our revised method also improved the culture efficiency in terms of the time needed for spheroid culture establishment, as the median time of the second cohort (21 days) was significantly shorter than that of the first cohort (33.5 days; Figure 2B).
TABLE 1.
Summary of culture methods.
| Our conventional method | Our improved method | Nanki et al. 13 | Yan et al. 14 | |
|---|---|---|---|---|
| Sampling method | ||||
| Site | Inside the tumor boundary | Both sides of the tumor boundary | NS | NS |
| Number of tissue pieces | 1–2 | 3–4 | NS | NS |
| Area (mm2)/Depth (mm) | 50–150/2–3 | 100–200/3–5 | NS | NS |
| Matrix material | Matrigel | Matrigel and collagen‐I, separately | Matrigel | Matrigel |
| Medium composition | ||||
| Growth factor | EGF, FGF2, FBS | EGF, FGF2, FBS | EGF, FGF10 | EGF, FGF10, FBS (as CM) |
| Stem cell niche factor | – | L‐WRN CM | Afamin‐Wnt3a CM, RSPO1, Noggin | Wnt3a CM, RSPO1 CM, Noggin CM |
| Inhibitor | SB431542, Y27632 | SB431542, Y27632 | A83‐01 | A83‐01, Y27632 |
| Other supplements | B27, NECA | B27, NECA | B27, Gastrin, NAC | B27, Gastrin, NAC |
| Selection procedure for cancer cell enrichment | No selection | No selection | +Nutlin‐3, –A83‐01/+TGF‐β, –EGF/–FGF10, or single‐cell dissociation | Manual picking or +Nutlin‐3 |
| Success rate | 25% (18/71) (95% CI, 15%–35%) | 88% (29/33) (95% CI, 77%–99%) | 75% (44/59) | >50% |
Note: Two representative methods reported previously are also shown as references.
Abbreviations: −, no or withdrawal from the culture medium; +, addition to the culture medium; CI, confidence interval; CM, conditioned medium; EGF, epidermal growth factor; FBS, fetal bovine serum; FGF, fibroblast growth factor; NAC, N‐Acetyl‐l‐cysteine; NECA, 5’‐N‐ethylcarboxamine adenosine; NS, not specified; RSPO1, R‐spondin 1; TGF‐β, transforming growth factor beta.
FIGURE 1.
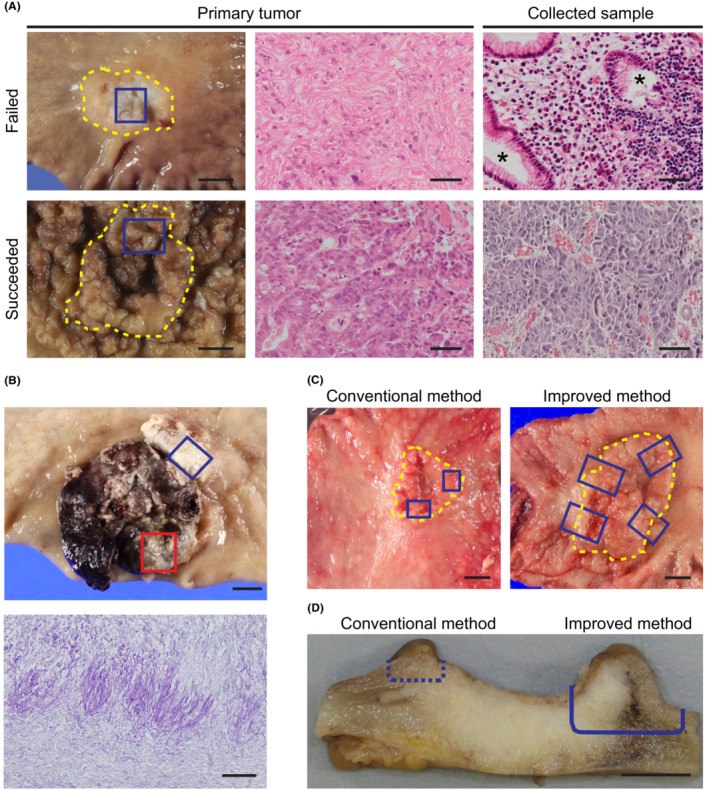
Possible reasons for unsuccessful gastric cancer stem cell (GC‐SC) spheroid culture. (A) Macroscopic luminal views of the resected specimens (left) and H&E‐stained sections of the primary tumors (center) and collected tissue samples (right) in a failed (top) and a succeeded (HG6T, bottom) case. Yellow dotted lines outline the tumor area. Blue boxes show the regions of sample collection. Note that a collected sample of the failed case contains non‐neoplastic glandular epithelial cells (asterisks). Scale bar, 10 mm (left) and 50 μm (center and right). (B) A macroscopic view of a necrotic GC case (top) and a periodic acid–Schiff‐stained section (bottom) of the collected tumor region (top, red box), showing accumulation of fungal hyphae on the surface. The blue box shows another resected region with successful spheroid culture (HG5T). Scale bar, 10 mm (top) and 50 μm (bottom). (C) Macroscopic views of representative GC cases indicating tumor regions for sample collection (blue boxes) before (conventional method, left) and after improving the method (improved method, right). Yellow dotted lines outline the tumor area. Note that wider regions across the tumor boundary were dissected for the improved method. Scale bar, 10 mm. (D) A cross‐sectional view of a representative GC case indicating the depth of tumor dissection for sample collection. Cutting along a dotted line can result in missing cancer cells in the tissue sample (conventional method). The cancer tissue should be cut deeply along a solid line to obtain enough cancer stem cells (improved method). Scale bar, 5 mm.
FIGURE 2.
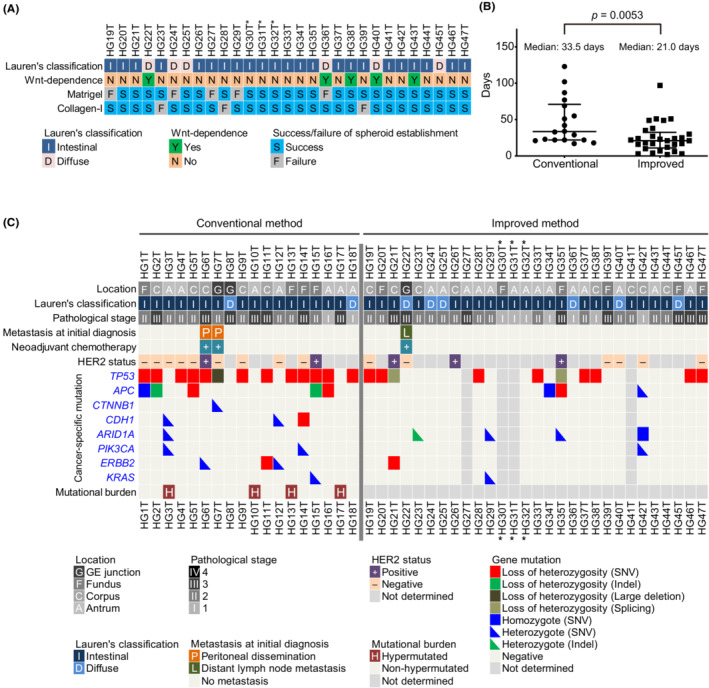
Establishment of patient‐derived gastric cancer stem cell (PD–GC‐SC) spheroids using an improved method. (A) Extracellular matrix (ECM) and Wnt ligand preference in primary culture. The spheroid lines were considered Wnt‐dependent when they perished in the cancer medium without L‐WRN CM during three serial passages. The three spheroid lines labeled with asterisks derived from a single patient. (B) Rapid establishment of GC‐SC spheroids in the improved culture condition. The duration time needed for expansion of each spheroid line from the patient sample to 12 wells of a 12‐well cell‐culture plate is plotted with the medians and interquartile ranges. p value, analyzed using Mann–Whitney U‐test. (C) Clinicopathological characteristics and mutational statuses of PD–GC‐SC spheroids. Shown are pathological features of 47 lines and representative genetic alterations of 43 lines. The pathological stage was determined by examination of surgically resected specimens. The HER2 status of the primary tumor was determined by immunohistochemistry or in situ hybridization. Cancer‐specific mutations were detected using a comprehensive cancer panel (HG1T–HG18T) or RNA sequencing (HG19T–HG47T). Indel, insertion/deletion variant; SNV, single nucleotide variant. The three spheroid lines labeled with asterisks derived from a single patient.
Typically, GC cells formed spherical aggregates in either Matrigel or collagen type‐I (Figure S2A), and they were highly proliferative in the cancer medium (Figure S2B). Their structures and expression of markers such as CDX2 and MUC2 recapitulated those in the epithelial components of their primary cancer tissues (Figure S2C). Consistent with a previous study, 33 culturing a Wnt‐dependent spheroid line (HG22T) in the Wnt‐free cancer medium accumulated signet‐ring cell‐like cells that were prominent in the primary tumor (Figure S2D). To assess the tumor‐initiating activity in vivo, we injected GC‐SC spheroids subcutaneously into immunodeficient mice, as we reported previously. 34 Three of the five GC‐SC spheroid lines formed subcutaneous tumors in nude or NSG mice, and their epithelial structures were similar to those of the primary tumors (Figure S3A,B), indicating that most of our GC‐SC spheroid lines contained abundant tumor‐initiating cells. Genetic alterations of TP53 and APC were detected frequently in the first patient cohort (13 and five lines, respectively, of 18), whereas they were less frequent in the second cohort (10 and three lines, respectively, of 25), suggesting that the improved culture condition helped propagate niche factor‐sensitive GC‐SCs that did not carry these key driver mutations (Figure 2C; Tables S4 and S5). Based on the estimated amounts of mutational burden, we identified four hypermutated GC‐SC spheroid lines in the first patient cohort (22%; four of 18 lines; Figure 2C; Figure S4A), which was confirmed for lack of mismatch repair proteins by immunohistochemistry (Figure S4B,C; Table S6).
Collectively, these results demonstrated that our revised method for GC‐SC spheroids was more efficient than our previous one.
3.2. Collagen type‐I stimulates the growth of some slow‐growing gastric cancer stem‐cell spheroids
A diffuse‐type GC‐SC spheroid line (HG18T) embedded in Matrigel grew very slowly in vitro compared with other lines in the first patient cohort. Diffuse‐type GC cells often invade the stromal layer of gastric mucosa, 4 suggesting that these cells have a higher affinity to collagen (e.g., collagen type‐I) than Matrigel extracellular scaffold rich in laminin‐1. 35 Therefore, we cultured HG18T and other spheroid lines separately in Matrigel and collagen type‐I. Notably, HG18T spheroids preferentially proliferated in collagen type‐I, whereas HG13T and HG15T in Matrigel. Other lines, HG6T, HG14T, and HG16T, showed little differences in growth between the two matrix materials without affecting the maintenance of spheroid lines because they more than quadrupled their cell volume in 6 days in either Matrigel or collagen type‐I (Figure 3A,B). Thus, we decided to try both Matrigel and collagen type‐I simultaneously but separately for primary culture of PD–GC‐SCs, and empirically determine the matrix best suited for each GC‐SC spheroid line.
FIGURE 3.
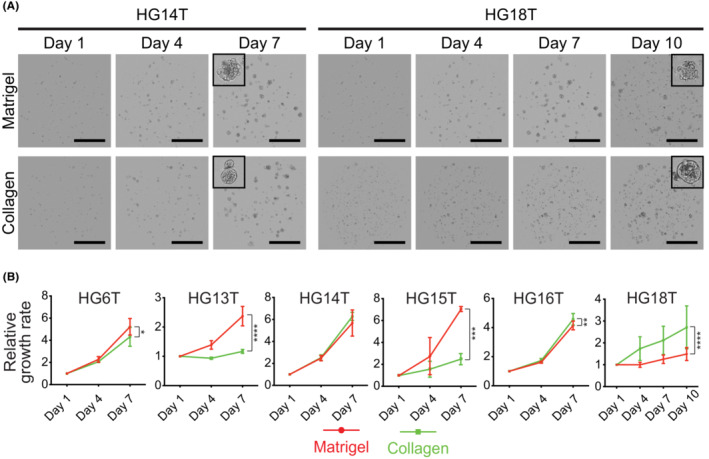
Effects of culture matrix materials on gastric cancer stem cell (GC‐SC) spheroid growth. (A) Representative cell scanning images of HG14T (left) and HG18T (right) spheroids cultured in Matrigel (top) and collagen type‐I (collagen, bottom). Scale bar, 1 mm. (B) Growth monitoring of spheroids with optical cell imaging. The total volumes of spheroids were estimated every 3 days during post‐passage days 1 to 7 or 10. Growth rates were calibrated to the initial cell volume on day 1. Shown are the mean growth rates ± standard deviation in three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001, statistical significance of the data difference (two‐way ANOVA followed by Tukey's post‐test).
3.3. Exogenous Wnt ligands stimulate the growth of some slow‐growing gastric cancer stem‐cell spheroids
Previous studies have shown that a subset of GC organoids is dependent on exogenous Wnt ligands such as Wnt and/or R‐spondin for growth. 13 , 14 However, Wnt ligands cause predominant growth of NGE‐SCs in primary culture, which necessitates another selection procedure to enrich GC‐SCs. 13 , 14 , 33 To resolve this problem, we hypothesized that a low concentration of L‐WRN CM that contained Wnt ligands could stimulate the growth of Wnt‐responsive GC‐SC spheroids without affecting NGE‐SCs. Before determining such a concentration of L‐WRN CM, we titrated its activity to ensure the reproducibility of culture conditions. We determined mRNA expression levels of MKI67 (proliferation marker) and LGR5 (stem cell marker) in normal colonic epithelial SC spheroids cultured with eL‐WRN media containing serially diluted L‐WRN CM according to the previous guidelines for quality control testing. 26 As a result, we found that low concentrations of L‐WRN CM (1%–10%) from two different sources (in‐house and commercial media) stimulated MKI67 mRNA expression in a dose‐dependent manner but failed to maintain LGR5 mRNA levels (Figure S5). Next, we conducted serial dilutions of in‐house L‐WRN CM with the cancer medium in the range of 0%–20% to titrate its effects on the growth of HG13T and HG18T, which showed the lowest growth rates among our GC‐SC lines that we have established so far (Figure 3B). In both spheroid lines, 5%–10% of L‐WRN CM supported the proliferation of GC‐SC spheroids, whereas 5% CM of NGE‐SC spheroids did not (Figure 4A,B; Figure S6A–C). Interestingly, 5% L‐WRN CM stimulated the expression of the stem cell marker LGR5 in both HG13T and HG18T but not in NGE‐SCs (Figure 4C). In contrast, L‐WRN CM had smaller effects on the expression of the proliferation marker MKI67 in GC‐SC lines than those in NGE‐SCs (Figure 4C). These results suggested that supplementation with a low concentration (e.g., at 5%) of L‐WRN CM should support self‐renewal of Wnt‐responsive GC‐SCs without allowing that of NGE‐SCs.
FIGURE 4.
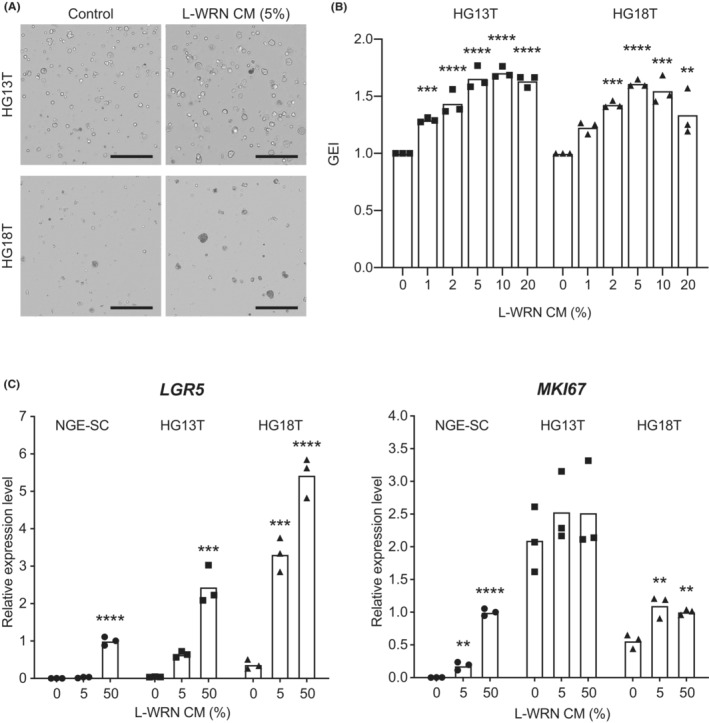
Effects of L‐WRN conditioned medium (CM) on gastric cancer stem cell (GC‐SC) spheroid growth. (A) Representative cell scanning images of HG13T (top) and HG18T (bottom) spheroids cultured with (right) and without (control, left) 5% L‐WRN CM for 6 days. Scale bar, 1 mm. (B) Growth monitoring of HG13T (left) and HG18T (right) spheroids with optical cell imaging. The GEI were calculated based on the growth rate of untreated spheroids (0%). The GEI in three independent experiments are plotted with the means. (C) Expression levels of LGR5 (left) and MKI67 (right) mRNAs determined by quantitative RT‐PCR analysis. Normal gastric epithelial stem cell (NGE‐SC) and GC‐SC (HG13T and HG18T) spheroids were cultured in the cancer media containing 0%, 5%, or 50% L‐WRN CM for 3 days. Relative expression levels in three independent experiments are plotted with the means. **p < 0.01; ***p < 0.001; ****p < 0.0001, statistical significance of the data difference between untreated (0%) and treated groups (one‐way ANOVA followed by Tukey's post‐test).
4. DISCUSSION
In this study, we propagated PD–GC‐SCs using our spheroid culture method modified from that originally developed for PD–CRC‐SCs. 21 Although non‐serum culture media are commonly used for organoid culture, 36 the present method takes advantage of the serum‐containing media that allow cost‐efficient propagation of pure populations of normal epithelial stem cells as undifferentiated spheroids. 22 , 24 We previously applied this strategy to culture PD–CRC‐SCs, and established more than 160 such spheroid lines at a high efficiency (up to approximately 90%). 21 Although the establishment of PD–GC‐SC lines was more challenging than CRC‐SC lines with the first patient cohort (25% success rate), we finally achieved a higher success rate (88%) by improving our previous culture protocol specifically for GC‐SCs (Table 1).
Importantly, we experienced difficulty in localizing the GC‐SCs by macroscopic observation of patient samples (Figure 1A) as well as more frequent contamination of fungi, likely Candida species (7%; in seven of 104 cases), 37 , 38 than in CRC (3%; in four of 148 cases). Therefore, we decided to sample tumor tissue pieces from a wider and deeper area, avoiding necrotic lesions as antifungal drugs appeared ineffective (Figure 1C,D). 38 We then re‐evaluated culture conditions and newly employed collagen type‐I matrix, which for the first time, shed light on the importance of ECM preference in the primary culture. Further studies are needed to determine the molecular features underlying the ECM preferences by GC‐SC lines.
We also overcame the previously addressed limitations of GC organoid culture, including the high cost of niche factors and concomitant propagation of NGE‐SCs, 18 , 39 , 40 , 41 by simply adding a low concentration of L‐WRN CM, a cost‐efficient source of stably active Wnt ligands (Figure S5). 26 These modifications should help propagate distinct populations of GC‐SCs that exhibit different dependencies on the niche factors without the need for negative selection to eliminate NGE‐SCs.
In conclusion, we developed a simple and efficient method to propagate PD–GC‐SC spheroids by improving our conventional sample collection protocol and culture conditions. Recent studies have shown that the drug sensitivity test on PD‐CRC organoids can predict patient outcomes with 100% sensitivity, 42 , 43 even if some intra‐tumor heterogeneity is lost in the spheroid/organoid line. 44 Our PD–GC‐SC spheroids can be utilized to investigate new molecular targeted therapies and their companion diagnostics for patient selection, 45 , 46 as we recently identified a subset of PD–CRC‐SC spheroid lines that responded to fibroblast growth factor receptor inhibitors. 47 , 48 Additionally, the genomic and expression profiles of GC‐SC spheroids will help determine novel molecular subtypes and diagnostic gene signatures. Thus, our improved method may open a new horizon for personalized GC diagnosis and treatment.
AUTHOR CONTRIBUTIONS
Conception and design, T. Morimoto (TMo), MMT, and H. Miyoshi (HMi); Development of methodology, TMo, YT, T. Miura (TMi), and HMi; Investigation, TMo, T. Yamamoto, FK, HA, H. Maekawa (HMa), T. Yamaura, and HMi; Analysis and interpretation of data, TMo, YT, TMi, and HMi; Administrative and material support, HMa, KK, YS, YY, HT, and KO; Manuscript writing, TMo, MMT, and HMi.
FUNDING INFORMATION
This work was supported by Grants‐in‐Aid for Scientific Research (JP18H02639 and JP22K07187 to H.Miyoshi and JP21K06948 to FK) from the Japan Society for the Promotion of Science; research funds from Kyo Diagnostics K.K. and SCREEN Holdings Co., Ltd. (to H.Miyoshi and KO); the Program for Creating Start‐ups from Advanced Research and Technology (ST261001TT) from the Japan Science and Technology Agency (to MMT); the Practical Research for Innovative Cancer Control (ck0106195h) from the Japan Agency for Medical Research and Development (to MMT); the Kyoto University Venture Incubation from the Kyoto University Office of Society‐Academia Collaboration for Innovation (to MMT); and the Dynamic Project for Colon Cancer Personalized Therapy from the Institute for Advancement of Clinical and Translational Science, KUHP (to MMT).
CONFLICT OF INTEREST STATEMENT
H. Miyoshi and KO received research funds from Kyo Diagnostics K.K. and SCREEN Holdings. MMT owns stock in Kyo Diagnostics K.K. YT and H.Maekawa belong to the Department of Personalized Cancer Medicine at the Graduate School of Medicine, Kyoto University, which is supported by Kyo Diagnostics K. K., AFI, and SCREEN Holdings. T.Miura is an employee of SCREEN Holdings. The other authors have no conflicts of interest to declare.
ETHICS STATEMENTS
Approval of the research protocol by an Institutional Reviewer Board: The study protocol was approved by Kyoto University Graduate School and Faculty of Medicine, Ethics Committee (No. R0915 and R0857) as well as that of Medical Research Institute Kitano Hospital (extension of the Kyoto University study as a collaboration).
Informed Consent: Written informed consent was obtained from all patients.
Registry and the Registration No. of the study/trial: N/A.
Animal Studies: All animal experiments were conducted according to the protocol approved by the Institutional Animal Care and Use Committee of Kyoto University Graduate School of Medicine (Nos 14546, 15091, 16047, 16654, 17086, 18080, and 19601).
Supporting information
Appendix S1.
ACKNOWLEDGMENTS
The authors thank members of the Department of Surgery at KUHP and Medical Research Institute Kitano Hospital for help collecting surgical specimens; Hiromi Kikuchi for technical assistance; and the Medical Research Support Center, Graduate School of Medicine, Kyoto University for the use of the facility. We also thank Dr. Thaddeus S. Stappenbeck for providing L‐WRN cells. We are grateful to Dr. Masanobu Oshima for comments on the manuscript.
Morimoto T, Takemura Y, Miura T, et al. Novel and efficient method for culturing patient‐derived gastric cancer stem cells. Cancer Sci. 2023;114:3259‐3269. doi: 10.1111/cas.15840
REFERENCES
- 1. Sung H, Ferlay J, Siegel RL, et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209‐249. [DOI] [PubMed] [Google Scholar]
- 2. Joshi SS, Badgwell BD. Current treatment and recent progress in gastric cancer. CA Cancer J Clin. 2021;71:264‐279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lauren P. The two histological main types of gastric carcinoma: Diffuse and so‐called intestinal‐type carcinoma. An attempt at a histo‐clinical classification. Acta Pathol Microbiol Scand. 1965;64:31‐49. [DOI] [PubMed] [Google Scholar]
- 4. Lokuhetty D, White AV, Watanabe R, Cree AI. Digestive system tumours: WHO classification of tumours. 5th ed. IARC; 2019. [Google Scholar]
- 5. Cancer Genome Atlas Research Network . Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202‐209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cristescu R, Lee J, Nebozhyn M, et al. Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat Med. 2015;21:449‐456. [DOI] [PubMed] [Google Scholar]
- 7. Bang YJ, Van Cutsem E, Feyereislova A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2‐positive advanced gastric or gastro‐oesophageal junction cancer (ToGA): a phase 3, open‐label, randomised controlled trial. Lancet. 2010;376:687‐697. [DOI] [PubMed] [Google Scholar]
- 8. Roviello G, Corona SP, D'Angelo A, Rosellini P, Nobili S, Mini E. Immune checkpoint inhibitors in pre‐treated gastric cancer patients: results from a literature‐based meta‐analysis. Int J Mol Sci. 2020;21:448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pietrantonio F, Randon G, Di Bartolomeo M, et al. Predictive role of microsatellite instability for PD‐1 blockade in patients with advanced gastric cancer: a meta‐analysis of randomized clinical trials. ESMO Open. 2021;6:100036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fujii M, Matano M, Nanki K, Sato T. Efficient genetic engineering of human intestinal organoids using electroporation. Nat Protoc. 2015;10:1474‐1485. [DOI] [PubMed] [Google Scholar]
- 11. Seino T, Kawasaki S, Shimokawa M, et al. Human pancreatic tumor organoids reveal loss of stem cell niche factor dependence during disease progression. Cell Stem Cell. 2018;22:454‐467.e456. [DOI] [PubMed] [Google Scholar]
- 12. Sato T, Stange DE, Ferrante M, et al. Long‐term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett's epithelium. Gastroenterology. 2011;141:1762‐1772. [DOI] [PubMed] [Google Scholar]
- 13. Nanki K, Toshimitsu K, Takano A, et al. Divergent routes toward Wnt and R‐spondin niche independency during human gastric carcinogenesis. Cell. 2018;174:856‐869.e817. [DOI] [PubMed] [Google Scholar]
- 14. Yan HHN, Siu HC, Law S, et al. A comprehensive human gastric cancer organoid biobank captures tumor subtype heterogeneity and enables therapeutic screening. Cell Stem Cell. 2018;23:882‐897.e811. [DOI] [PubMed] [Google Scholar]
- 15. Gao M, Lin M, Rao M, et al. Development of patient‐derived gastric cancer organoids from endoscopic biopsies and surgical tissues. Ann Surg Oncol. 2018;25:2767‐2775. [DOI] [PubMed] [Google Scholar]
- 16. Seidlitz T, Merker SR, Rothe A, et al. Human gastric cancer modelling using organoids. Gut. 2019;68:207‐217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Steele NG, Chakrabarti J, Wang J, et al. An organoid‐based preclinical model of human gastric cancer. Cell Mol Gastroenterol Hepatol. 2019;7:161‐184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wuputra K, Ku CC, Kato K, Wu DC, Saito S, Yokoyama KK. Translational models of 3‐D organoids and cancer stem cells in gastric cancer research. Stem Cell Res Ther. 2021;12:492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li G, Ma S, Wu Q, et al. Establishment of gastric signet ring cell carcinoma organoid for the therapeutic drug testing. Cell Death Discov. 2022;8:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Song H, Park JY, Kim JH, et al. Establishment of patient‐derived gastric cancer organoid model from tissue obtained by endoscopic biopsies. J Korean Med Sci. 2022;37:e220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Miyoshi H, Maekawa H, Kakizaki F, et al. An improved method for culturing patient‐derived colorectal cancer spheroids. Oncotarget. 2018;9:21950‐21964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Miyoshi H, Stappenbeck TS. In vitro expansion and genetic modification of gastrointestinal stem cells in spheroid culture. Nat Protoc. 2013;8:2471‐2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. VanDussen KL, Marinshaw JM, Shaikh N, et al. Development of an enhanced human gastrointestinal epithelial culture system to facilitate patient‐based assays. Gut. 2015;64:911‐920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wilson SS, Mayo M, Melim T, et al. Optimized culture conditions for improved growth and functional differentiation of mouse and human colon organoids. Front Immunol. 2020;11:547102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stelzner M, Helmrath M, Dunn JC, et al. A nomenclature for intestinal in vitro cultures. Am J Physiol Gastrointest Liver Physiol. 2012;302:G1359‐G1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. VanDussen KL, Sonnek NM, Stappenbeck TS. L‐WRN conditioned medium for gastrointestinal epithelial stem cell culture shows replicable batch‐to‐batch activity levels across multiple research teams. Stem Cell Res. 2019;37:101430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yamaura T, Miyoshi H, Maekawa H, et al. Accurate diagnosis of mismatch repair deficiency in colorectal cancer using high‐quality DNA samples from cultured stem cells. Oncotarget. 2018;9:37534‐37548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Reiter JG, Baretti M, Gerold JM, et al. An analysis of genetic heterogeneity in untreated cancers. Nat Rev Cancer. 2019;19:639‐650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Danecek P, Auton A, Abecasis G, et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156‐2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Res. 2010;38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Narahara M, Higasa K, Nakamura S, et al. Large‐scale East‐Asian eQTL mapping reveals novel candidate genes for LD mapping and the genomic landscape of transcriptional effects of sequence variants. PLoS ONE. 2014;9:e100924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Higasa K, Miyake N, Yoshimura J, et al. Human genetic variation database, a reference database of genetic variations in the Japanese population. J Hum Genet. 2016;61:547‐553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Togasaki K, Sugimoto S, Ohta Y, et al. Wnt signaling shapes the histologic variation in diffuse gastric cancer. Gastroenterology. 2021;160:823‐830. [DOI] [PubMed] [Google Scholar]
- 34. Maekawa H, Miyoshi H, Yamaura T, et al. A chemosensitivity study of colorectal cancer using xenografts of patient‐derived tumor‐initiating cells. Mol Cancer Ther. 2018;17:2187‐2196. [DOI] [PubMed] [Google Scholar]
- 35. Moreira AM, Pereira J, Melo S, et al. The extracellular matrix: an accomplice in gastric cancer development and progression. Cell. 2020;9:394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Drost J, Clevers H. Organoids in cancer research. Nat Rev Cancer. 2018;18:407‐418. [DOI] [PubMed] [Google Scholar]
- 37. Eras P, Goldstein MJ, Sherlock P. Candida infection of the gastrointestinal tract. Medicine (Baltimore). 1972;51:367‐379. [DOI] [PubMed] [Google Scholar]
- 38. Katzenstein AL, Maksem J. Candidal infection of gastric ulcers. Histology, incidence, and clinical significance. Am J Clin Pathol. 1979;71:137‐141. [DOI] [PubMed] [Google Scholar]
- 39. Dedhia PH, Bertaux‐Skeirik N, Zavros Y, Spence JR. Organoid models of human gastrointestinal development and disease. Gastroenterology. 2016;150:1098‐1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Seidlitz T, Stange DE. Gastrointestinal cancer organoids‐applications in basic and translational cancer research. Exp Mol Med. 2021;53:1459‐1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pang MJ, Burclaff JR, Jin R, et al. Gastric organoids: progress and remaining challenges. Cell Mol Gastroenterol Hepatol. 2022;13:19‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vlachogiannis G, Hedayat S, Vatsiou A, et al. Patient‐derived organoids model treatment response of metastatic gastrointestinal cancers. Science. 2018;359:920‐926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ooft SN, Weeber F, Dijkstra KK, et al. Patient‐derived organoids can predict response to chemotherapy in metastatic colorectal cancer patients. Sci Transl Med. 2019;11:eaay2574. [DOI] [PubMed] [Google Scholar]
- 44. Roerink SF, Sasaki N, Lee‐Six H, et al. Intra‐tumour diversification in colorectal cancer at the single‐cell level. Nature. 2018;556:457‐462. [DOI] [PubMed] [Google Scholar]
- 45. Choi S, Park S, Kim H, Kang SY, Ahn S, Kim KM. Gastric cancer: mechanisms, biomarkers, and therapeutic approaches. Biomedicine. 2022;10:543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li T, He Y, Zhong Q, Yu J, Chen X. Advances in treatment models of advanced gastric cancer. Technol Cancer Res Treat. 2022;21:153303382210903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yamamoto T, Miyoshi H, Kakizaki F, et al. Chemosensitivity of patient‐derived cancer stem cells identifies colorectal cancer patients with potential benefit from FGFR inhibitor therapy. Cancers (Basel). 2020;12:2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kitano S, Yamamoto T, Taketo MM. Novel parameter for cancer chemosensitivity to fibroblast growth factor receptor inhibitors. Cancer Sci. 2022;113:4005‐4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1.