

Abstract
Large‐scale genomic sequencing of colorectal cancers has been reported mainly for Western populations. Differences by stage and ethnicity in the genomic landscape and their prognostic impact remain poorly understood. We investigated 534 Japanese stage III colorectal cancer samples from the Phase III trial, JCOG0910. Targeted‐capture sequencing of 171 potentially colorectal cancer‐associated genes was performed, and somatic single‐nucleotide variants and insertion–deletions were determined. Hypermutated tumors were defined as tumors with MSIsensor score >7 and ultra‐mutated tumors with POLE mutations. Genes with alterations associated with relapse‐free survival were analyzed using multivariable Cox regression models. In all patients (184 right‐sided, 350 left‐sided), mutation frequencies were TP53, 75.3%; APC, 75.1%; KRAS, 43.6%; PIK3CA, 19.7%; FBXW7, 18.5%; SOX9, 11.8%; COL6A3, 8.2%; NOTCH3, 4.5%; NRAS, 4.1%; and RNF43, 3.7%. Thirty‐one tumors were hypermutated (5.8%; 14.1% right‐sided, 1.4% left‐sided). Modest associations were observed: poorer relapse‐free survival was seen with mutant KRAS (hazard ratio 1.66; p = 0.011) and mutant RNF43 (2.17; p = 0.055), whereas better relapse‐free survival was seen with mutant COL6A3 (0.35; p = 0.040) and mutant NOTCH3 (0.18; p = 0.093). Relapse‐free survival tended to be better for hypermutated tumors (0.53; p = 0.229). In conclusion, the overall spectrum of mutations in our Japanese stage III colorectal cancer cohort was similar to that in Western populations, but the frequencies of mutation for TP53, SOX9, and FBXW7 were higher, and the proportion of hypermutated tumors was lower. Multiple gene mutations appeared to impact relapse‐free survival, suggesting that tumor genomic profiling can support precision medicine for colorectal cancer.
Keywords: clinical trial, colorectal cancer, ethnicity, genomics, precision medicine
We demonstrated that the overall spectrum of mutations in our Japanese stage III colorectal cancer cohort (534 patients) was largely similar to that in Western populations, but the frequencies of mutation for TP53 (75.3%), SOX9 (11.8%), and FBXW7 (18.5%) were higher, and the proportion of hypermutated tumors (5.8%) was lower. We also revealed that poorer relapse‐free survival was seen with mutant KRAS (hazard ratio 1.66; p = 0.011) and mutant RNF43 (2.17; p = 0.055) as well as hypermutated tumors (0.53; p = 0.229), whereas better relapse‐free survival was seen with mutant COL6A3 (0.35; p = 0.040) and mutant NOTCH3 (0.18; p = 0.093), suggesting that tumor genomic profiling has the potential to support precision medicine for patients with colorectal cancer.
Abbreviations
- CI
confidence interval
- CNV
copy number variation
- CRC
colorectal cancer
- DFS
disease‐free survival
- dMMR
defective DNA mismatch repair
- Indel
insertion–deletion
- MSI
microsatellite instability
- OS
overall survival
- RFS
relapse‐free survival
- SNV
single‐nucleotide variants
- TCGA
The Cancer Genome Atlas
1. INTRODUCTION
Identifying somatic mutations is vital to understanding the molecular mechanisms of cancers and developing novel therapeutics for cancer, and comprehensive genome sequencing shows tremendous promise in advancing precision medicine for patients with cancer worldwide. The first comprehensive next‐generation sequencing studies in colorectal cancer (CRC) were performed in the early 2010s by several groups, including The Cancer Genome Atlas (TCGA) Research Network and a Dutch group. 1 , 2 Large‐scale genomic sequencing studies have thus far been reported mainly in Western countries, that is, in Caucasian populations. For example, the TCGA Research Network analyzed 276 CRC samples and reported common mutations in the expected genes, including APC, TP53, KRAS, PIK3CA, FBXW7, and SMAD4, as well as in more recently detected genes, such as ARID1A, SOX9, and FAM123B. 1
Although limited, there have been some reports on genomic sequencing of CRC in Asia. One paper from China showed that, in addition to five CRC‐related genes that are well‐known in Western populations (APC, TP53, KRAS, FBXW7, and SMAD4), three novel recurrently mutated genes, namely, CDH10, FAT4, and DOCK2, exhibited a high prevalence of mutation in an Asian CRC cohort. 3 A large‐scale observational nationwide study in Japan showed that the frequency of KRAS mutation was 37.6% 4 in the Asian population, which seemed to be similar to that in the RASCAL study (37.7%) conducted in several Western countries. 5 Interestingly, in that study, there was a significant association between the frequency of KRAS mutation and age, which did not appear to be the case in the Western population. Furthermore, a study based on real‐world Japanese data detected high microsatellite instability (MSI‐H) in 3.78% of unresectable or metastatic CRCs, 6 which seemed to be lower than the rate in Western countries. Therefore, so far, the contribution of ethnicity to mutations in CRC‐related genes is not well understood. Moreover, the prognostic impact of the genomic landscape in CRC remains unclear.
Stage III CRC is CRC with lymph node involvement but without distant metastases. 7 Adjuvant therapy is recommended for patients with stage III CRC in the treatment guidelines in many countries, including Japan. 8 JCOG0910 was a nationwide multicenter randomized Phase III trial that assessed the non‐inferiority of S‐1 to capecitabine as adjuvant chemotherapy. 9 Using samples from that trial, we aimed to define the genomic landscape of stage III CRC in an Asian population and to identify prognostic biomarkers.
2. MATERIALS AND METHODS
2.1. Study population
This study involved patients with stage III CRC from the JOG trial, which was conducted by the Colorectal Cancer Study Group of the Japan Clinical Oncology Group (JCOG). 9 Permission to carry out this research was obtained from the medical director of each participating institution with approval from the institutional review board or ethics committee at the participating institutions. Signed informed consent for the collection of biological samples and testing for CRC‐related genes was obtained from surviving patients who participated in JCOG0910. When informed consent could not be secured because of death or loss to follow‐up, samples were collected with the permission of the director of each participating institution. Finally, samples from 534 (34%) of 1564 patients with stage III CRC from the JCOG0910 study were collected for this ancillary study, known as JCOG1506A1.
The study was conducted in collaboration with BioBank Japan.
2.2. JCOG0910 study
JCOG0910 was a multicenter, open‐label, randomized Phase III trial conducted at 57 Japanese institutions to demonstrate the non‐inferiority of S‐1 to capecitabine as adjuvant chemotherapy in terms of disease‐free survival (UMIN Clinical Trial Registry number UMIN000003272). 9 As reported previously for JCOG0910, 9 the eligibility criteria were as follows: age 20–80 years; a diagnosis of stage III colorectal adenocarcinoma, defined by the presence of a distal margin of the primary tumor above the peritoneal reflection; R0 resection; and colectomy with D3 or D2 lymph node dissection. Between March 1, 2010 and August 23, 2013, 1564 patients were randomly assigned to receive capecitabine (n = 782) or S‐1 (n = 782); all patients were included in the intention‐to‐treat analysis of efficacy. At the prespecified second interim analysis after final enrollment, 258 (48.2%) of the 535 required events were reported. The Data and Safety Monitoring Committee recommended the early publication of the study because S‐1 was considered to be unlikely to show non‐inferiority to capecitabine in terms of DFS. With a median follow‐up of 23.7 months (interquartile range 14.1, 35.2), 3‐year DFS was 82.0% (95% confidence interval [CI] 78.5–85.0) in the capecitabine arm and 77.9% (95% CI 74.1–81.1) in the S‐1 arm (hazard ratio [HR] 1.23, 99.05% CI 0.89–1.70; one‐sided p‐value for non‐inferiority = 0.46). Therefore, it was concluded that adjuvant S‐1 is not non‐inferior to adjuvant capecitabine in terms of DFS. Adjuvant capecitabine remains one of the standard treatments for stage III CRC in Japan; S‐1 is not recommended. 9
2.3. Target‐capture sequencing
Formalin‐fixed paraffin‐embedded (FFPE) sections from primary CRC tissues and matched normal tissues were obtained to extract genomic DNA. All samples analyzed were resection specimens and obtained before any chemotherapy was administered. Tissue DNA was extracted from the sliced FFPE surgical tissue samples using the FFPE Tissue LEV DNA Purification Kit (Promega) and quantified using a NanoDrop spectrophotometer.
The SureSelect custom capture library (Agilent Technologies) was used to capture all exons of the 171 potentially CRC‐associated genes shown in Table S1. Sequence libraries were prepared using a combination of the Hyper Prep Kit (Kapa Biosystems) and SureSelect Enrichment Plus Adapter TPFD‐KB, ILM (Agilent Technologies). We followed the KAPA Hyper Prep Kit protocol until the first library amplification and the standard SureSelect kit protocol after hybridization. In brief, 200–2000 ng of DNA (DIN ≥ 2.0) was fragmented using an enzymatic reaction followed by end repair and A‐tailing. Adapter ligation was performed using SureSelect Adapter Oligo Mix (Agilent Technologies). The adapter‐ligated libraries were purified using Agencourt AMPure XP beads (Beckman Coulter). Pre‐capture amplification of the adapter‐ligated library was performed by polymerase chain reaction using the SureSelect kit primer and purified using Agencourt AMPure XP beads. The quantity and size distribution of the library were confirmed using TapeStation 2200 (Agilent Technologies). Next, 750 ng of the amplified libraries were hybridized with the SureSelect custom capture library (Agilent Technologies) for 16 h and then purified using Dynabeads MyOne Streptavidin T1 (Thermo Fisher Scientific). Post‐capture amplification was performed for 13 cycles, and amplified libraries were purified using Agencourt AMPure XP beads. The quantity and size distribution of the amplified libraries were determined using TapeStation 2200 (Agilent Technologies) and the KAPA Library Quantification Kit (KAPA Biosystems). The prepared libraries were sequenced on Illumina HiSeq 2500 platforms (Illumina Inc.) in paired‐end mode. Targeted‐capture sequencing was performed on both tumor samples and normal tissue samples. Median coverage of targeted NGS for tumor tissue and normal tissue was 530.5 and 272.2, respectively.
Before alignment, the sequenced reads were trimmed to remove adapter contamination using the Cutadapt software package. The trimmed reads for both tumor samples and normal tissue samples were aligned to the human reference genome (GRCh37) using the Burrows–Wheeler Aligner. 10 Probable polymerase chain reaction duplications were removed, in which paired‐end reads were aligned to the same genomic positions, and pile‐up files were generated using SAMtools 11 and a program developed in‐house. Given that sequence errors occur in a sequence‐specific manner, the read information from all non‐tumor samples was pooled into a so‐called normal panel for accurate discrimination between true positives and false positives. The details of our filtering conditions have been reported previously. 12 Somatic single‐nucleotide variants (SNVs) and insertion–deletion mutations (Indels) were determined in tumor samples using the method described above. In this study, gene mutation was defined as one or more somatic SNVs or Indels detected in the gene. Microsatellite instability was evaluated from aligned BAM files of tumor–normal tissue pairs using the MSIsensor tool 13 with default parameter settings. Briefly, MSIsensor assesses the aligned sequencing data for available microsatellite regions with sufficient coverage in a tumor–normal tissue pair where variation in deletion length is identified. The c2 test is used to determine loci with significant variation, and after multiple testing corrections of the p‐values, the percentage of unstable loci is reported as the MSIsensor score. In this study, tumors with an MSIsensor score >7 were defined as MSI‐H.
2.4. One hundred and seventy‐one potentially colorectal cancer‐associated genes
The 171 potentially CRC‐associated genes are shown in Table S1. The 171 genes were selected based on previous studies as genes that have been described to be, or could be, potential colorectal cancer‐associated genes. First, 43 genes were selected from the colorectal cancer‐related TCGA paper. 1 Second, 24 genes were added from the study on breast cancer and colorectal cancer conducted at the Johns Hopkins Kimmel Cancer Center. 14 Third, six genes were added from a study of colorectal cancer in Asian populations, 3 and four genes were added from a study on adenomatous polyposis and colorectal cancer. 15 Fourth, since genes associated with other cancers may also be potential colorectal cancer‐associated genes, we added 22 genes related to biliary tract cancer, 16 22 genes related to liver cancer, 17 17 genes related to esophageal cancer, 18 and eight genes related to gastric cancer. 19 Finally, another 25 genes considered by our study group to be potential colorectal cancer‐associated genes were added to the list. Targeted‐capture sequencing of these genes was performed on both normal tissue and tumor samples, and SNVs and Indels were determined.
2.5. Pathway abnormality
After the exclusion of one gene (CADPS), 170 candidate genes were classified in 17 pathways. When there were one or more mutations (any SNV or Indel) in at least one gene in the pathway, it was defined as having a pathway abnormality.
2.6. Gene mutations stratified by sidedness of the primary tumor
A primary tumor in the cecum, ascending colon, or transverse colon was classified as right‐sided, and a primary tumor in the splenic flexure, descending colon, sigmoid colon, rectosigmoid, or rectum was classified as left‐sided. Frequencies of gene mutations stratified by sidedness of the primary tumor were examined.
2.7. Hypermutated and non‐hypermutated tumors
In this study, tumors with an MSIsensor score >7 and ultra‐mutated tumors with known POLE exonuclease domain mutations (P286R, S459F, or V411L) 20 were grouped as hypermutated tumors. The remaining samples were then grouped as non‐hypermutated tumors. POLE is a catalytic subunit of DNA polymerase epsilon and is involved in replication and repair of nuclear DNA. In this study, ultra‐mutated tumors were defined as tumors with a hotspot mutation in POLE at P286R, S459F, or V411L. 21
2.8. Data collection
Data were collected on sex, age, body mass index, sidedness of the primary tumor, histological differentiation, and type of adjuvant chemotherapy regimen (capecitabine in arm A and S‐1 in arm B).
2.9. Statistical analysis
The frequencies of gene mutations are shown with the exact 95% confidence intervals (CIs). Genes with a mutation frequency of ≥3% were targeted for analysis in this study. The mutation frequency for each gene was compared between right‐sided and left‐sided primary tumors using Fisher's exact test.
Relapse‐free survival (RFS) was defined as days from randomization to relapse or death from any cause and was censored at the last day when the patient was alive without any evidence of relapse. Relapse was diagnosed radiologically or pathologically based on a biopsy sample. Elevated tumor marker levels were not regarded as relapse, and additional imaging was required.
The probability of RFS was estimated using the Kaplan–Meier method. Greenwood's formula was used to calculate the 95% CIs. Genes with alterations that were associated with RFS were evaluated using multivariable Cox regression models. These models, which included age (younger or older than 65 years), sex, tumor depth (T0, T1, T2/T3/T4), nodal status (N1/N2), and tumor location (right‐sided/left‐sided, colon/rectum) and were stratified by treatment arm (capecitabine or S‐1), were used to estimate HRs with 95% CIs and two‐sided p‐values for each gene with a mutation frequency of ≥3%. The family‐wise error for gene‐level analysis for RFS was controlled at 0.05 using the Bonferroni method.
3. RESULTS
3.1. Characteristics of the study cohort
Patient characteristics are summarized in Table 1. Median age was 67 years (range, 29–80); 283 patients (53.0%) were male, and 184 (34.5%) had right‐sided tumors. Of the 534 patients, 109 relapsed or died during the study period. The 3‐year RFS rate was 81.0% (95% CI 77.4–84.1).
TABLE 1.
Patient characteristics
| Category | Number | Percentage |
|---|---|---|
| JOG treatment arm | ||
| Capecitabine | 270 | 50.6 |
| S‐1 | 264 | 49.4 |
| Age | ||
| Median (range) | 67 years | 29–80 |
| Sex | ||
| Male | 283 | 53.0 |
| Female | 251 | 47.0 |
| Body mass index | ||
| Median (range) | 21.9 | 14.8–33.9 |
| Tumor location | ||
| Right‐sided (C/A/T) | 184 (41/107/36) | 34.5 |
| Left‐sided (D/S/rectosigmoid/rectum) | 350 (22/141/116/71) | 65.5 |
| Histology | ||
| Well‐differentiated adenocarcinoma | 125 | 23.4 |
| Moderately differentiated adenocarcinoma | 369 | 69.1 |
| Poorly differentiated/mucinous/papillary | 15/13/12 | 7.5 |
| Stage | ||
| IIIA | 94 | 17.6 |
| IIIB | 397 | 74.3 |
| IIIC | 43 | 8.1 |
| Pathological tumor depth a | ||
| T1 | 34 | 6.4 |
| T2 | 61 | 11.4 |
| T3 | 321 | 60.1 |
| T4a | 103 | 19.3 |
| T4b | 15 | 2.8 |
| Pathological nodal status a | ||
| N1a | 244 | 45.7 |
| N1b | 209 | 39.1 |
| N2a | 64 | 12.0 |
| N2b | 17 | 3.2 |
Abbreviations: A, ascending colon; C, cecum; D, descending colon; S, sigmoid colon; T, transverse colon.
UICC‐TNM 7th edition.
3.2. Frequency of colorectal cancer‐associated gene mutations
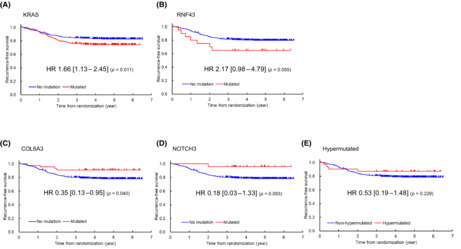
In this study, as described in Section 2, gene mutation was defined as one or more SNVs or Indels in the gene. Among 171 potentially CRC‐associated genes, 49 were found to have a mutation frequency of ≥3%. The 10 most frequently mutated genes were TP53 (75.3%), APC (75.1%), KRAS (43.6%), PIK3CA (19.7%), FBXW7 (18.5%), FAT4 (13.3%), LRP1B (11.8%), SOX9, COL6A3 (8.2%), and FAM123B (8.1%). ATM (7.5%), ARID1A (6.2%), NOTCH3 (4.5%), NRAS (4.1%), and RNF43 (3.7%) were also frequently mutated (Figure 1).
FIGURE 1.
Mutation frequencies in 534 patients with stage III colorectal cancer from the JOG study. Forty‐nine genes with a mutation frequency of ≥3% are shown.
3.3. Distribution of genetic alterations in right‐sided and left‐sided colorectal cancer
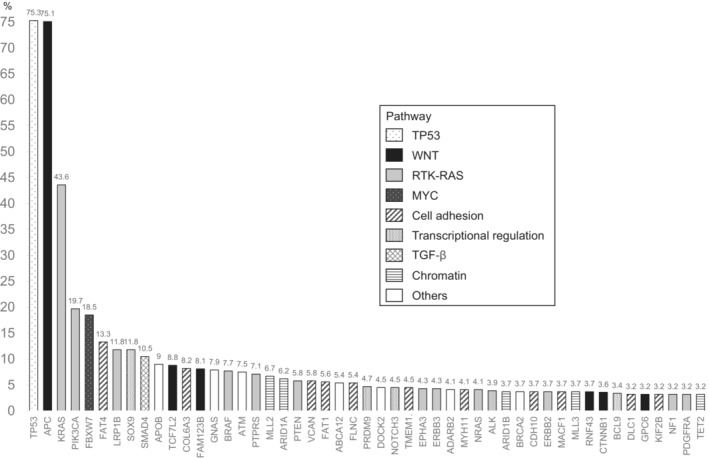
Figure 2 shows the mutation frequency and exact 95% CI for each CRC‐related gene according to the sidedness of the primary tumor. Thirty‐seven genes differed by sidedness (p < 0.05). In 34 of these genes, including PIK3CA and KRAS, the frequency of mutation was higher for right‐sided CRC than for left‐sided CRC. In three genes (TP53, APC, and FBXW7), the mutation frequency was lower for right‐sided CRC than for left‐sided CRC.
FIGURE 2.
Distribution of genetic alterations in right‐sided and left‐sided colorectal cancer. Thirty‐seven genes showed a significant difference in the frequency of genetic alterations according to sidedness (p < 0.05, Fisher's exact test). The error bars represent exact 95% CIs.
3.4. Hypermutated tumors
Twenty‐nine (5.4%) of the 534 tumors had an MSIsensor score >7. Two (0.4%) of the tumors were ultra‐mutated with a hotspot mutation in POLE at P286R and had an MSIsensor score <7. In total, 31 tumors (5.8%) were found to be hypermutated and 503 (94.2%) to be non‐hypermutated. Twenty‐six of the 31 hypermutated tumors were right‐sided (14.1%; 26/184), and five were left‐sided (1.4%; 5/350).
3.5. Mutated genes in hypermutated and non‐hypermutated tumors
Several genes (BRAF, IGF1R, ERBB2, RNF43, ERBB3, SOX9, ACVR1B, and TGFBR2) showed recurrent mutations in hypermutated cancers but not in non‐hypermutated samples: BRAF (52% vs. 5%), IGF1R (32% vs. 0%), ERBB2 (32% vs. 2%), RNF43 (29% vs. 2%), ERBB3 (23% vs. 3%), SOX9 (29% vs. 11%), ACVR1B (19% vs. 1%), and TGFBR2 (16% vs. 2%). In contrast, three genes that were frequently mutated in non‐hypermutated cancers were significantly less frequently mutated in hypermutated tumors: TP53 (35% vs. 78%), APC (45% vs. 77%), and KRAS (23% vs. 43%).
3.6. Pathway abnormalities in colorectal cancer
The diversity and frequency of genetic changes leading to deregulation of six major signaling pathways, namely, Wnt signaling, P53 signaling, RTK/RAS signaling, cell adhesion pathway, transcriptional regulation pathway, and transforming growth factor (TGF)‐β signaling, in the 534 patients with stage III CRC are shown in Figure 1; these pathways exhibited genetic alterations in 83.3%, 75.3%, 73.2%, 43.4%, 27.0%, and 22.1% of CRC samples, respectively. Overall, 16 of 17 pathways were mutated in ≥3% of cases.
3.7. Gene mutations associated with relapse‐free survival
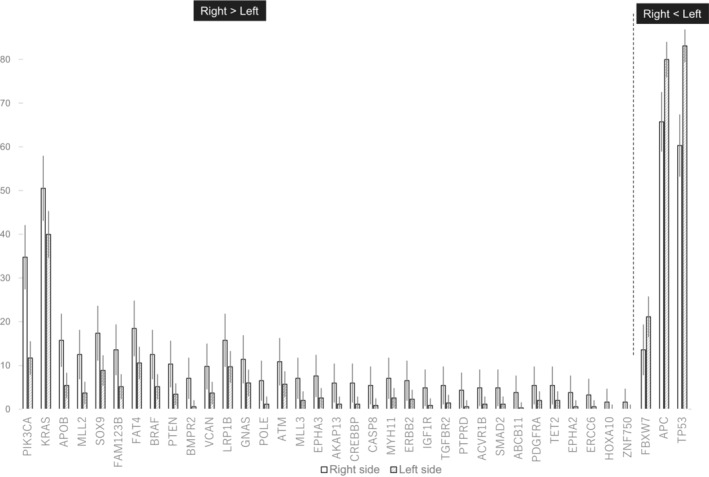
Of the 49 genes with a mutation frequency of ≥3%, none was significantly associated with RFS after Bonferroni's correction for multiple comparisons (in this situation, gene‐level significance level = 0.05/49 = 0.001) (Table 2). Modest associations were observed for the following: mutant KRAS (HR 1.66; 95% CI 1.13 to 2.45; p = 0.011) and mutant RNF43 (HR 2.17; 95% CI 0.98 to 4.79; p = 0.055) had poorer RFS, whereas mutant COL6A3 (HR 0.35; 95% CI 0.13 to 0.95; p = 0.040) and mutant NOTCH3 (HR 0.18; 95% CI 0.03–1.33; p = 0.093) had better RFS (Table 2, Figure 3). RFS tended to be better in patients with hypermutated tumors than in those with non‐hypermutated tumors (HR 0.53; 95% CI 0.19–1.48; p = 0.229; Table 2, Figure 3).
TABLE 2.
Hazard ratios with 95% confidence intervals and p‐values for associations between genes with a mutation frequency ≥3% and relapse‐free survival
| Gene | Hazard ratio a | 95% LCL | 95% UCL | p‐Value | Gene | Hazard ratio a | 95% LCL | 95% UCL | p‐Value |
|---|---|---|---|---|---|---|---|---|---|
| KRAS | 1.66 | 1.13 | 2.45 | 0.011 | ARID1B | 1.39 | 0.56 | 3.47 | 0.477 |
| COL6A3 | 0.35 | 0.13 | 0.95 | 0.04 | KIF2B | 1.41 | 0.51 | 3.85 | 0.508 |
| RNF43 | 2.17 | 0.98 | 4.79 | 0.055 | ERBB2 | 0.7 | 0.22 | 2.24 | 0.543 |
| NOTCH3 | 0.18 | 0.03 | 1.33 | 0.093 | ALK | 0.7 | 0.22 | 2.22 | 0.545 |
| SOX9 | 0.55 | 0.27 | 1.16 | 0.116 | FAT1 | 0.76 | 0.31 | 1.88 | 0.553 |
| ERBB3 | 1.68 | 0.77 | 3.69 | 0.193 | VCAN | 0.74 | 0.27 | 2.03 | 0.554 |
| MLL2 | 0.55 | 0.22 | 1.36 | 0.195 | BRCA2 | 1.3 | 0.53 | 3.23 | 0.566 |
| CTNNB1 | 1.64 | 0.74 | 3.6 | 0.223 | NRAS | 0.76 | 0.28 | 2.07 | 0.587 |
| DLC1 | 0.42 | 0.1 | 1.73 | 0.233 | EPHA3 | 1.23 | 0.56 | 2.72 | 0.605 |
| FAT4 | 0.7 | 0.38 | 1.28 | 0.242 | ABCA12 | 0.82 | 0.36 | 1.88 | 0.64 |
| GPC6 | 0.44 | 0.11 | 1.8 | 0.254 | MYH11 | 1.24 | 0.5 | 3.08 | 0.644 |
| MLL3 | 1.7 | 0.67 | 4.27 | 0.262 | GNAS | 0.87 | 0.45 | 1.69 | 0.673 |
| PDGFRA | 1.7 | 0.61 | 4.71 | 0.308 | APOB | 0.86 | 0.41 | 1.81 | 0.694 |
| CDH10 | 1.52 | 0.65 | 3.52 | 0.33 | SMAD4 | 0.88 | 0.47 | 1.66 | 0.698 |
| TCF7L2 | 0.68 | 0.3 | 1.54 | 0.351 | APC | 0.93 | 0.6 | 1.43 | 0.739 |
| FAM123B | 1.35 | 0.69 | 2.64 | 0.377 | ADARB2 | 1.14 | 0.41 | 3.15 | 0.796 |
| MACF1 | 1.5 | 0.6 | 3.73 | 0.387 | ATM | 0.91 | 0.44 | 1.88 | 0.801 |
| BCL9 | 0.54 | 0.13 | 2.21 | 0.394 | BRAF | 0.93 | 0.44 | 1.95 | 0.844 |
| TP53 | 1.22 | 0.77 | 1.95 | 0.395 | FBXW7 | 0.95 | 0.58 | 1.57 | 0.846 |
| PRDM9 | 0.66 | 0.24 | 1.8 | 0.416 | LRP1B | 1.03 | 0.57 | 1.84 | 0.932 |
| TET2 | 0.62 | 0.19 | 2 | 0.423 | PTPRS | 1.02 | 0.47 | 2.23 | 0.953 |
| TMEM132D | 1.39 | 0.6 | 3.2 | 0.439 | FLNC | 0.98 | 0.43 | 2.24 | 0.955 |
| DOCK2 | 1.39 | 0.6 | 3.21 | 0.442 | ARID1A | 0.98 | 0.45 | 2.15 | 0.968 |
| NF1 | 1.43 | 0.57 | 3.56 | 0.445 | PIK3CA | 1.01 | 0.62 | 1.64 | 0.97 |
| PTEN | 1.31 | 0.63 | 2.74 | 0.473 |
Abbreviations: LCL, lower confidence limit; UCL, upper confidence limit.
Adjusted for treatment arm (capecitabine/S‐1), age (older or younger than 65 years), sex, tumor depth (T0, T1, T2/T3/T4), nodal status (N1/N2), and tumor location (right‐sided/left‐sided). Genes with a gene mutation frequency of ≥3% were targeted for this analysis.
FIGURE 3.
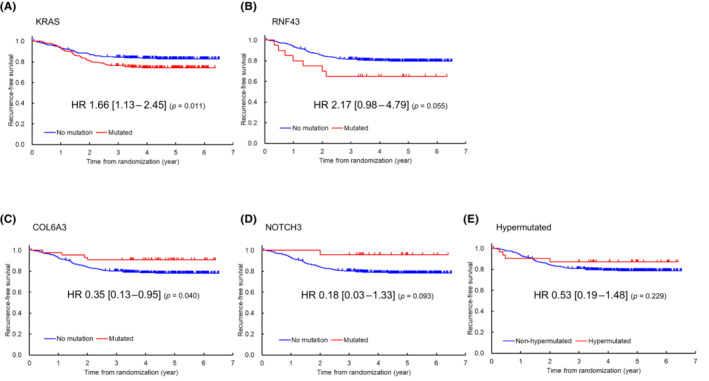
Kaplan–Meier curves for relapse‐free survival. (A) KRAS, (B) RNF, (C) COL6A3, and (D) NOTCH3. Blue: wild‐type. Red: mutated. (E) Hypermutated tumors (red) vs. non‐hypermutated tumors (blue).
3.8. KRAS mutations associated with RFS
KRAS mutation is a well‐known gene alteration in colorectal cancer, including several hot spot variants. Of the 534 patients, 233 had KRAS exon 2 mutations, including 62 with the KRAS G12D mutation, 17 with the KRAS G12C mutation, 52 with the KRAS G12V mutation, and 53 with the KRAS G13D mutation. We examined the association of these KRAS mutations with RFS. Two patients with double mutations were excluded from this analysis (i.e., one patient with both G12D and G13D mutations and another patient with both G12V and G13D mutations), resulting in a final analysis population of 532 patients (61 with the KRAS G12D mutation, 17 with the KRAS G12C mutation, 51 with the KRAS G12V mutation, 51 with the KRAS G13D mutation, 51 with other KRAS mutations, and 301 without KRAS mutations). The analysis found that patients with the KRAS G12D mutation (HR 1.99; 95% CI 1.14–3.45; p = 0.015) and those with the KRAS G12V mutation (HR 2.12; 95% CI 1.13–4.00; p = 0.020) had poorer RFS than patients without KRAS mutations. Similarly, patients with the KRAS G12C mutation (HR 1.97; 95% CI 0.78–5.00; p = 0.152), those with other KRAS mutations (HR 1.45; 95% CI 0.76 to 2.76; p = 0.255) and those with the KRAS G13D mutation (HR 1.23; 95% CI 0.63–2.38; p = 0.546) tended to have poor RFS compared to patients without KRAS mutations (Figure 4).
FIGURE 4.
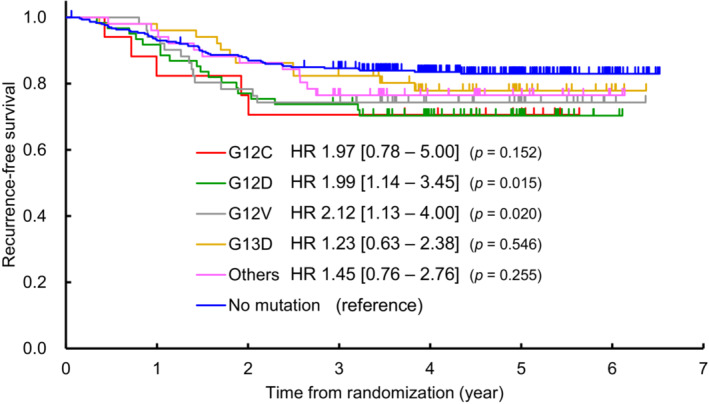
Kaplan–Meier curves for relapse‐free survival. Relapse‐free survival of 532 patients (61 with the KRAS G12D mutation, 17 with the KRAS G12C mutation, 51 with the KRAS G12V mutation, 51 with the KRAS G13D mutation, 51 with other KRAS mutations, and 301 without KRAS mutations) were analyzed. Blue: wild‐type (no KRAS mutation). Two patients were excluded from the analysis because they had double mutations.
4. DISCUSSION
Although sequencing studies of CRC have been reported, 1 , 22 , 23 to date, tumors from Asian populations have not been comprehensively evaluated except in a few papers. In this report, we present the results of targeted‐capture sequencing of 171 potentially CRC‐associated genes from an analysis of 534 Japanese patients with stage III CRC. First, we demonstrated that TP53, APC, KRAS, PIK3CA, and FBXW7 are the five most frequently mutated genes. The overall spectrum of gene mutations in our stage III CRC cohort is generally similar to that of the TCGA data obtained for Western populations. 1 However, in our Asian population, the mutation frequencies of TP53, SOX9, and FBXW7 were higher, and the proportion of hypermutated tumors was lower. Second, we found significant differences in mutation frequencies in 37 genes according to whether the CRC was right‐sided or left‐sided. For 34 of these 37 genes, the mutation frequency was higher in right‐sided CRC than left‐sided CRC. In contrast, the mutation frequency in three genes, namely, TP53, APC, and FBXW7, which were, respectively, the first, second, and fifth most frequently mutated genes, was lower for right‐sided CRC than for left‐sided CRC. Third, we found that the frequency of hypermutated tumors in Japanese patients with stage III CRC was 5.8%, which seems low compared with several reports from Western countries. However, this result is consistent with several previous reports on MSI‐high status in Asia. 24 Therefore, it seems true that the frequency of hypermutated tumors is lower in Asia than in Western countries. This finding suggests that the frequency of hypermutated tumors in patients with CRC varies according to ethnicity. Fourth, we found that most genes, including BRAF, IGF1R, ERBB2, RNF43, ERBB3, SOX9, ACVR1B, and TGFBR2, were more frequently recurrently mutated in hypermutated tumors than in non‐hypermutated tumors, whereas three genes, TP53, APC and KRAS, which were, respectively, the first, second, and third most frequently mutated genes, were frequently mutated in non‐hypermutated cancers and significantly less frequently mutated in hypermutated tumors. Fifth, after Bonferroni's correction for multiple comparisons, none of the 49 genes with mutation frequencies ≥3% was significantly associated with RFS, whereas mutant KRAS and RNF43 tended to be associated with poorer RFS and mutant COL6A3 and NOTCH3 tended to be associated with better RFS.
A large‐scale study in Japanese patients with CRC found that the overall frequency of MSI‐H was 5.9%. 24 However, the frequency varied according to pathological stage (5.9% in stage 0–I, 8.9% in stage II, 4.0% in stage III, and 3.7% in stage IV). 24 In our study of Japanese patients with stage III CRC, 29 (5.4%) of the 534 tumors had an MSIsensor score >7, indicating that this score is the appropriate threshold for the detection of MSI‐H tumors. In contrast, in Western countries, the frequency of MSI‐H was reported to be 20% in stage I–II, 12% in stage III, and 4% in stage IV, respectively. 25 Thus, taken together, the frequency of MSI‐H varies by race and by stage.
In this study, tumors with an MSIsensor score >7 and ultra‐mutated tumors with POLE mutations were grouped as hypermutated tumors. We found that RFS tended to be better in patients with hypermutated tumors than in those with non‐hypermutated tumors (HR 0.53; p = 0.229). The failure of this finding to reach statistical significance was a surprise considering that stage II–III colon cancer with defective DNA mismatch repair (dMMR) is known to have a favorable prognosis. 26 However, it is also known that defective dMMR is a marker for lack of efficacy of fluorouracil‐based adjuvant therapy in colon cancer; that is, patients with dMMR tumors who receive 5‐fluorouracil show no improvement in DFS, unlike those who are assigned to surgery alone. 26 In this study, all patients received fluorouracil‐based adjuvant therapy. Therefore, it is possible that adjuvant therapy, which is known to improve prognosis compared with the surgery without adjuvant therapy, was beneficial in patients with non‐hypermutated tumors but not in those with hypermutated tumors and that this benefit resulted in a narrowing of the difference in prognosis.
Our finding that mutant KRAS was associated with poorer RFS (HR1.66; p = 0.011) in patients with stage III CRC receiving adjuvant treatment is similar to that in a previous report on European patients with stage III CRC who received adjuvant treatment with FOLFOX (folinic acid [leucovorin calcium], 5‐fluorouracil, and oxaliplatin) with or without cetuximab. 27 In that study, the authors investigated 1869 patients with stage III colon cancer who participated in the Pan‐European Trials in Alimentary Tract Cancer (PETACC)‐8 randomized Phase III trial. Their multivariable analysis showed that patients with mutant RAS had significantly poorer DFS (HR 1.56; 95% CI 1.27–1.92; p < 0.0001). 27 We also found that patients with mutant RNF43 had poorer RFS (HR 2.17; p = 0.055). The ubiquitin ligase RNF43 downregulates Wnt signaling by removing frizzled receptor from the cell membrane, and its mutations lead to a loss of function of the ubiquitin E3 ligase. Therefore, activation of Wnt signaling by RNF43 mutations is thought to enhance tumor growth and promote relapse in patients with CRC. 28 Our study showed that the likelihood of relapse is greater in patients with CRC harboring mutations in RNF43 than in those without such mutations.
In this study, we also analyzed associations between OS and gene alterations. Of the 534 patients, 50 died during the study period. Of the 49 genes with a mutation frequency of ≥3%, none were significantly associated with OS after Bonferroni's correction for multiple comparisons (data not shown; in this situation, gene‐level significance level = 0.05/49 = 0.001). Modest associations with poorer OS were found with the following genes: mutant CTNNB1 (HR 2.62; 95% CI 0.98–7.03; p = 0.055), mutant BRCA2 (HR 2.74; 95% CI 0.96–7.82; p = 0.060), and mutant CDH10 (HR 2.47; 95% CI 0.86–7.15; p = 0.095). No association with OS was observed for the following four genes, which were modestly associated with RFS: KRAS (HR 1.42; 95% CI 0.79–2.52; p = 0.238), RNF43 (HR 1.53; 95% CI 0.45–5.15; p = 0.496), mutant COL6A3 (HR 0.67; 95% CI 0.21–2.18; p = 0.509), and mutant NOTCH3 (HR 0.00; p = 0.987). Discrepancies between the results for RFS and OS may be interesting to investigate in a future study but are beyond the scope of this study.
Recently, ERBB2 (HER2) amplification in colorectal cancer was reported as an attractive therapeutic target. 29 Thus, copy number variation (CNV) may be associated with the prognosis of colorectal cancer patients. In this study, CNV was examined for 171 potentially colorectal cancer‐associated genes. There were nine genes with an amplification frequency of ≥3%; HNF4A (15.9%), GNAS (6.8%), HOXA10 (5.7%), FLT3 (5.3%), KLF5 (4.0%), SOX9 (3.8%), RBM10 (3.6%), MYC (3.4%), and HOXA9 (3.0%). The amplification frequency of ERBB2 was 2.3% (12 cases). We also examined the association of the nine genes noted above and ERBB2 with RFS. While none of the nine genes was significantly associated with RFS after Bonferroni's correction for multiple comparisons (data not shown), a modest association was observed for ERRB2, with ERBB2 amplification being associated with poorer RFS (HR 2.54; 95% CI 0.92 to 6.97; p = 0.071). Our results also supported ERBB2 (HER2) amplification as an attractive therapeutic target in colorectal cancer. 29
The prognostic value of the sidedness of the primary tumor has been a topic of considerable interest in CRC. For unresectable stage IV CRC, pooled analyses of several randomized trials have shown that overall survival is significantly worse in patients with right‐sided tumors than in those with left‐sided tumors. 30 Furthermore, large‐scale nationwide multicenter retrospective studies in nonmetastatic colon cancer have consistently shown that tumor location has a prognostic impact after relapse, with right‐sided colon cancer being associated with significantly shorter cancer‐specific survival after relapse than left‐sided colon cancer in patients with stage II–III 31 and stage III 32 , 33 colon cancer.
We also found that the mutation frequency differed by sidedness in 34 of 37 genes, with a higher mutation frequency in right‐sided CRC than in left‐sided CRC. This would be one of the reasons why the prognosis is poorer for right‐sided CRC than for left‐sided CRC.
Several genes, including BRAF, IGF1R, ERBB2, RNF43, ERBB3, SOX9, ACVR1B, and TGFBR2, were found to show recurrent mutations in hypermutated cancers but not in the non‐hypermutated cancers, whereas three genes, namely, TP53, APC and KRAS, were frequently mutated in non‐hypermutated cancers but mutated significantly less often in hypermutated tumors. These findings indicate that hypermutated and non‐hypermutated tumors progress through different sequences of genetic events. Therefore, non‐hypermutated and hypermutated cancers require different treatment strategies. Indeed, nowadays, immune checkpoint inhibitors, including pembrolizumab (an anti‐PD‐1 inhibitor) and nivolumab (an anti‐PD‐1 inhibitor) with or without ipilimumab (an anti‐CTLA‐4 inhibitor), have been integrated into the standard of care for MSI‐high/dMMR metastatic CRC. 34
This study has several limitations. First, it included only 534 (34%) of the 1564 patients in the JCOG0910 study. Although the characteristics of the patients in this study are almost the same as those in the entire JCOG0910 study population, we cannot exclude the possibility our study population might not be fully representative of that in JCOG0910. Second, the study population comprised patients from JCOG0910, which was an adjuvant chemotherapy study. Thus, all the patients received adjuvant chemotherapy, which affects prognosis. This study could, therefore, only reveal the prognostic impact of the genomic landscape in Japanese patients with stage III CRC who have received adjuvant chemotherapy and not that in those who have not. Third, patients in this study received 5‐fluorouracil, capecitabine, or S‐1 as adjuvant chemotherapy. Nowadays, adjuvant chemotherapy strategies have changed, and oxaliplatin is used in addition to 5‐fluorouracil. Therefore, our findings might not be fully reflective of current medical practice.
In conclusion, the spectrum of gene mutations in our stage III CRC cohort in Japan was generally similar to that in the TCGA. However, the mutation frequencies of TP53, SOX9, and FBXW7 were higher, and the proportion of hypermutated tumors was lower. Multiple gene mutations seemed to impact RFS, suggesting that tumor genomic profiling has the potential to support precision medicine for patients with CRC.
AUTHOR CONTRIBUTIONS
Dai Shida, Aya Kuchiba, Tatsuhiro Shibata, Satoshi Yamasaki, Keisuke Kanato, and Yoshinori Murakami designed the trial, wrote the protocol, and coordinated the study. Dai Shida, Tetsuya Hamaguchi, Masaaki Ito, Takaya Kobatake, Tadahiko Masaki, Manabu Shiozawa, Yasumasa Takii, Hiroyuki Uetake, Shu Okamura, Hitoshi Ojima, Hiroshi Takeyama, Yasuhiro Shimada, and Yukihide Kanemitsu enrolled patients. Aya Kuchiba, Tatsuhiro Shibata, Satoshi Yamasaki, Keisuke Kanato, and Yukihide Kanemitsu managed, analyzed, and interpreted the data. Dai Shida drafted the manuscript. All authors reviewed and revised the paper, and approved the submitted version.
FUNDING INFORMATION
Supported by National Cancer Research and Development Funds (26‐A‐3, 29‐A‐3) and AMED under Grant No. 17km0405106h0005.
CONFLICT OF INTEREST STATEMENT
Tatsuhiro Shibata and Yoshinori Murakami are current editorial Board Members of Cancer Science. HT received honoraria from Ono Pharma and Taiho. The other authors have no conflicts of interest.
ETHICS STATEMENT
Approval of the research protocol: This study was approved by the institutional review board or ethical committees of all participating institutions.
Informed consent: All study participants gave informed consent.
Registration: UMIN Clinical Trial Registry, number UMIN000003272.
Animal studies: None.
Supporting information
Table S1.
ACKNOWLEDGMENTS
We sincerely appreciate all the patients who participated in this research and their families. We thank all the members of the JCOG Colorectal Cancer Study Group and the pathologists of each institution participating in this research. The authors also express sincere gratitude to the members of the JCOG Data Center, JCOG Operations Office, and BioBank Japan for their support in this study.
Shida D, Kuchiba A, Shibata T, et al. Genomic landscape and its prognostic significance in stage III colorectal cancer: JCOG1506A1, an ancillary of JCOG0910 . Cancer Sci. 2023;114:3352‐3363. doi: 10.1111/cas.15834
REFERENCES
- 1. Cancer Genome Atlas N . Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330‐337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Haan JC, Labots M, Rausch C, et al. Genomic landscape of metastatic colorectal cancer. Nat Commun. 2014;5:5457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yu J, Wu WK, Li X, et al. Novel recurrently mutated genes and a prognostic mutation signature in colorectal cancer. Gut. 2015;64:636‐645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Watanabe T, Yoshino T, Uetake H, et al. KRAS mutational status in Japanese patients with colorectal cancer: results from a nationwide, multicenter, cross‐sectional study. Jpn J Clin Oncol. 2013;43:706‐712. [DOI] [PubMed] [Google Scholar]
- 5. Andreyev HJ, Norman AR, Cunningham D, Oates JR, Clarke PA. Kirsten ras mutations in patients with colorectal cancer: the multicenter “RASCAL” study. J Natl Cancer Inst. 1998;90:675‐684. [DOI] [PubMed] [Google Scholar]
- 6. Akagi K, Oki E, Taniguchi H, et al. Real‐world data on microsatellite instability status in various unresectable or metastatic solid tumors. Cancer Sci. 2021;112:1105‐1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. UICC . TNM Classification of Malignant Tumours. 8th ed. John Wiley & Sons, Ltd; 2017. [Google Scholar]
- 8. Hashiguchi Y, Muro K, Saito Y, et al. Japanese Society for Cancer of the Colon and Rectum (JSCCR) guidelines 2019 for the treatment of colorectal cancer. Int J Clin Oncol. 2020;25:1‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hamaguchi T, Shimada Y, Mizusawa J, et al. Capecitabine versus S‐1 as adjuvant chemotherapy for patients with stage III colorectal cancer (JCOG0910): an open‐label, non‐inferiority, randomised, phase 3, multicentre trial. Lancet Gastroenterol Hepatol. 2018;3:47‐56. [DOI] [PubMed] [Google Scholar]
- 10. Li H, Durbin R. Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics. 2009;25:1754‐1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078‐2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Totoki Y, Tatsuno K, Covington KR, et al. Trans‐ancestry mutational landscape of hepatocellular carcinoma genomes. Nat Genet. 2014;46:1267‐1273. [DOI] [PubMed] [Google Scholar]
- 13. Niu B, Ye K, Zhang Q, et al. MSIsensor: microsatellite instability detection using paired tumor‐normal sequence data. Bioinformatics. 2014;30:1015‐1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wood LD, Parsons DW, Jones S, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108‐1113. [DOI] [PubMed] [Google Scholar]
- 15. Weren RD, Ligtenberg MJ, Kets CM, et al. A germline homozygous mutation in the base‐excision repair gene NTHL1 causes adenomatous polyposis and colorectal cancer. Nat Genet. 2015;47:668‐671. [DOI] [PubMed] [Google Scholar]
- 16. Nakamura H, Arai Y, Totoki Y, et al. Genomic spectra of biliary tract cancer. Nat Genet. 2015;47:1003‐1010. [DOI] [PubMed] [Google Scholar]
- 17. Fujimoto A, Furuta M, Totoki Y, et al. Whole‐genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat Genet. 2016;48:500‐509. [DOI] [PubMed] [Google Scholar]
- 18. Sawada G, Niida A, Uchi R, et al. Genomic landscape of esophageal squamous cell carcinoma in a Japanese population. Gastroenterology. 2016;150:1171‐1182. [DOI] [PubMed] [Google Scholar]
- 19. Kakiuchi M, Nishizawa T, Ueda H, et al. Recurrent gain‐of‐function mutations of RHOA in diffuse‐type gastric carcinoma. Nat Genet. 2014;46:583‐587. [DOI] [PubMed] [Google Scholar]
- 20. Church DN, Briggs SE, Palles C, et al. DNA polymerase epsilon and delta exonuclease domain mutations in endometrial cancer. Hum Mol Genet. 2013;22:2820‐2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cancer Genome Atlas Research N , Kandoth C, Schultz N, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schell MJ, Yang M, Teer JK, et al. A multigene mutation classification of 468 colorectal cancers reveals a prognostic role for APC. Nat Commun. 2016;7:11743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Brannon AR, Vakiani E, Sylvester BE, et al. Comparative sequencing analysis reveals high genomic concordance between matched primary and metastatic colorectal cancer lesions. Genome Biol. 2014;15:454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fujiyoshi K, Yamamoto G, Takenoya T, et al. Metastatic pattern of stage IV colorectal cancer with high‐frequency microsatellite instability as a prognostic factor. Anticancer Res. 2017;37:239‐247. [DOI] [PubMed] [Google Scholar]
- 25. Stadler ZK. Diagnosis and management of DNA mismatch repair‐deficient colorectal cancer. Hematol Oncol Clin North Am. 2015;29:29‐41. [DOI] [PubMed] [Google Scholar]
- 26. Sargent DJ, Marsoni S, Monges G, et al. Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil‐based adjuvant therapy in colon cancer. J Clin Oncol. 2010;28:3219‐3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Taieb J, Kourie HR, Emile JF, et al. Association of prognostic value of primary tumor location in stage III colon cancer with RAS and BRAF mutational status. JAMA Oncol. 2018;4:e173695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Eto T, Miyake K, Nosho K, et al. Impact of loss‐of‐function mutations at the RNF43 locus on colorectal cancer development and progression. J Pathol. 2018;245:445‐455. [DOI] [PubMed] [Google Scholar]
- 29. Nakamura Y, Okamoto W, Kato T, et al. Circulating tumor DNA‐guided treatment with pertuzumab plus trastuzumab for HER2‐amplified metastatic colorectal cancer: a phase 2 trial. Nat Med. 2021;27:1899‐1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Arnold D, Lueza B, Douillard JY, et al. Prognostic and predictive value of primary tumour side in patients with RAS wild‐type metastatic colorectal cancer treated with chemotherapy and EGFR directed antibodies in six randomized trials. Ann Oncol. 2017;28:1713‐1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ishihara S, Murono K, Sasaki K, et al. Impact of primary tumor location on postoperative recurrence and subsequent prognosis in nonmetastatic colon cancers: a multicenter retrospective study using a propensity score analysis. Ann Surg. 2018;267:917‐921. [DOI] [PubMed] [Google Scholar]
- 32. Kishiki T, Kuchta K, Matsuoka H, et al. The impact of tumor location on the biological and oncological differences of colon cancer: multi‐institutional propensity score‐matched study. Am J Surg. 2019;217:46‐52. [DOI] [PubMed] [Google Scholar]
- 33. Shida D, Inoue M, Tanabe T, et al. Prognostic impact of primary tumor location in stage III colorectal cancer‐right‐sided colon versus left‐sided colon versus rectum: a nationwide multicenter retrospective study. J Gastroenterol. 2020;55:958‐968. [DOI] [PubMed] [Google Scholar]
- 34. Hirano H, Takashima A, Hamaguchi T, Shida D, Kanemitsu Y, Colorectal Cancer Study Group of the Japan Clinical Oncology G . Current status and perspectives of immune checkpoint inhibitors for colorectal cancer. Jpn J Clin Oncol. 2021;51:10‐19. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1.