

Abstract
A double mutation which converts nucleotide 1765 from A to T and nucleotide 1767 from G to A is frequently found in the hepatitis B virus (HBV) genome isolated from HBV patients with chronic hepatitis symptoms. This double mutation is located in the core promoter that controls the transcription of the precore RNA and the core RNA. In addition, this double mutation also resides in the X protein coding sequence, converting codon 130 from Lys to Met and codon 131 from Val to Ile. Previous studies indicate that this double mutation removes a nuclear receptor binding site in the core promoter, suppresses specifically precore RNA transcription, and enhances viral replication. In this study, we further investigated how this double mutation suppresses precore RNA transcription. We found that this double mutation not only removed the nuclear receptor binding site but also created an HNF1 transcription factor binding site. Further transfection studies using Huh7 hepatoma cells indicate that the removal of the nuclear receptor binding site has no effect on the transcription of HBV RNAs, the two-codon change in the X protein sequence suppresses the transcription of both precore and core RNAs, and the creation of the HNF1 binding site restores the core RNA level. Hence, the specific suppression of precore RNA transcription by this frequent double-nucleotide mutation is the combined result of multiple factors.
Hepatitis B virus (HBV) is a small DNA virus with a 3.2-kb, partially double stranded, circular genome. This genome, which is converted to a covalently closed circular DNA molecule after infection, contains four open reading frames that code for seven viral gene products (for reviews, see references 13 and 32). The expression of HBV genes is regulated by four different promoters. Of these, the core promoter controls transcription of the C gene, which produces core RNA and precore RNA (Fig. 1A) (32). The core RNA codes for the core protein, which is the major viral capsid protein, and for the DNA polymerase, which is a reverse transcriptase. In addition, the core RNA can also serve as the pregenomic RNA, which is packaged with the DNA polymerase to form the viral core particle. This packaged RNA is then converted by the DNA polymerase to the partially double stranded DNA genome. The precore RNA codes for the precore protein, which contains the entire coding sequence of the core protein plus an amino-terminal extension of 29 amino acids. This amino-terminal extension contains a signal peptide which targets the precore protein to the endoplasmic reticulum (ER) (for a review, see reference 23). After removal of the signal peptide by the signal peptidase in the ER lumen, the precore protein derivative p22e is translocated across the ER membrane, further cleaved at multiple sites at its carboxy terminus, and secreted (23). The secreted precore protein derivatives are called hepatitis B e antigen (HBeAg).
FIG. 1.

(A) Schematic illustration of the core promoter. The transcription initiation sites of precore and core RNAs are marked by (rightward arrows) and translation initiation codons (ATG) of precore and core proteins are marked. The nuclear receptor binding site is underlined, and the 1765-A→T 1767-G→A double mutation (M1) is denoted by the two downward arrows. CP, core promoter. (B) Sequence homologies between the WT HBV sequence and the DR1 nuclear receptor binding site (NRBS) (21) and between the M1 HBV sequence and the consensus HNF1 binding sequence. Arrows indicate locations of the mutations, and vertical lines indicate identical nucleotides.
Although the core protein and its RNA play very important roles in the replication of HBV, the precore protein gene is not an essential viral gene, as mutations which prevent it from being expressed do not abolish HBV replication in animal models (10, 11) or in patients (6, 9, 31). Studies conducted with cell cultures and transgenic mice indicate that the precore gene supplied in trans actually suppressed HBV replication (7, 16, 19). The precore gene appears to affect HBV replication at the RNA packaging step, as HBV mutants carrying the double mutation of A to T at nucleotide (nt) 1765 (1765-A→T) and G to A at nt 1767 (1767-G→A) were found to have a reduced level of precore gene expression and an increased efficiency of pregenomic RNA packaging (7). Since a small fraction of the precore derivative p22e fails to be translocated into the ER lumen (14, 24), it has been postulated that p22e, which contains the entire core protein sequence plus an amino-terminal extension of 10 amino acids, serves as a dominant negative factor of the core protein for packaging the viral pregenome (19). This hypothesis is supported by the observation that the overexpression of a p22e homologue in the cytosol can drastically reduce the replication rate of HBV (19, 28).
The transcription of precore RNA and core RNA can be differentially regulated, as the 1765-A→T 1767-G→A double mutation specifically affects precore RNA transcription but not core RNA transcription (7). It has been suggested that the transcription of precore and core RNAs is regulated by two different promoters which physically overlap (12, 34). The double mutation mentioned above is located in a nuclear receptor binding site (Fig. 1A), which is recognized by members of the steroid-thyroid hormone receptor superfamily including chicken ovalbumin upstream promoter transcription factors 1 and 2 (COUP-TFI and -II), hepatocyte nuclear factor 4 (HNF4), the heterodimer of retinoid X receptor alpha chain and peroxisome proliferator-activated receptor gamma chain (RXRα and PPARγ), and testicular receptor 2 (TR2) (8, 26, 35). Although they bind to the same site, these nuclear receptors have different effects on the transcription of precore and core RNAs: COUP-TFI suppresses the transcription of both RNAs, HNF4 or TR2 suppresses the transcription of precore RNA without affecting the transcription of core RNA, and RXRα and PPARγ together enhance the transcription of core RNA without affecting the transcription of precore RNA (35). Since the ratio of precore to core RNA can affect the replication efficiency of HBV (3, 7), different environmental factors that activate different nuclear receptors in the liver may have profound effects on the pathogenesis of HBV.
The HBV mutants carrying the 1765-A→T 1767-G→A double mutation are found in over 80% of HBeAg-positive patients with symptoms of chronic hepatitis (6, 23). The prevalence of this double mutation during chronic infection is likely due to its higher replication rate, which is caused by the reduced expression level of the precore gene. The nucleotide specificity of this double mutation is intriguing. Our previous studies indicate that this double mutation removes most, if not all, of the ability of the nuclear receptor binding site to bind nuclear receptors (8). However, this cannot be the sole reason for the selection of this double mutation during chronic infection, as mutations of nt 1765 and 1767 to other nucleotides or mutations in other locations of the nuclear receptor binding site can also abolish the binding of nuclear receptors (unpublished observation; also see below). The 1765-A→T 1767-G→A core promoter double mutation resides in the X protein coding sequence and converts codon 130 from Lys to Met (130-Lys→Met) and codon 131 from Val to Ile (131-Val→Ile) (23). The X protein is a transcriptional transactivator which can activate the expression of a number of viral and cellular genes (33). It is likely that this two-codon change affects X protein activity and plays a role in the selection of this double mutation. In this study, we investigated these possibilities. We found that the 1765-A→T 1767-G→A double mutation not only removed the nuclear receptor binding site but also created an HNF1 transcription factor binding site. Furthermore, our results indicate that while removal of the nuclear receptor binding site does not affect the transcription of precore and core RNAs, the change of the codons in the X protein sequence suppresses the transcription of both of these two RNAs, and the creation of the HNF1 site restores only core RNA transcription. The combination of these effects results in the specific suppression of the HBV precore RNA observed in cells transfected by the HBV DNA carrying this double-nucleotide mutation.
MATERIALS AND METHODS
DNA plasmids.
pWTD contains the wild-type HBV genome of the adw2 subtype (7). This plasmid was constructed by inserting a head-to-tail dimer of the HBV genome via its unique EcoRI site into the EcoRI cloning site of the pUC19 vector. pM1D is identical to pWTD except for containing the 1765-A→T 1767-G→A double mutation. pM4D contains the 1760-G→A single mutation, and pM5D contains the 1763-A→G single mutation in addition to the 1765-A→T 1767-G→A double mutation. The double mutation described herein corresponds to the 1762-A→T and 1764-G→A mutations reported previously (7, 8, 22, 30). pM1D was created by M13-based site-directed mutagenesis, and pM4D and pM5D were created by the PCR-based site-directed mutagenesis (15). The mutated sequences were verified by sequencing. pCMV-HNF1 was constructed by inserting the blunt-ended HincII-BstEII fragment of pON-HNF1 (a gift from B. Yen, University of California, San Francisco) (37), which contained the entire HNF1 coding sequence, into the XbaI site of pRc/CMV (Invitrogen). pCMX-COUP was a gift from R. M. Evans (Salk Institute). pXGH5 (Nichols Diagnostics) contains the human growth hormone coding sequence under the expression control of the mouse metallothionein promoter. This plasmid was used as an internal control for monitoring transfection efficiency, which was measured by determining the amount of human growth hormone secreted, using a commercial radioimmunoassay kit (Nichols Diagnostics).
Cell culture and DNA transfection.
Huh7 hepatoma cells were maintained in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum. Cells grown to approximately 80% confluence in a 10-cm-diameter dish were transfected with 20 μg of DNA by the calcium phosphate precipitation method (18). After 48 h, cells were lysed with RNAzolB (Tel-Test, Inc.), and the total cellular RNA was extracted as specified by the manufacturer. Occasionally, cells were lysed with Tris-saline (10 mM Tris [pH 7.0], 150 mM NaCl) containing 0.5% Nonidet P-40, and the cellular RNA was isolated by phenol extraction.
Primer extension analysis.
The sequence of the antisense primer used for the primer extension analysis was 5′GGTGAGCAATGCTCAGGAGACTCTAAGG 3′ (36), corresponding to nt 2051 to 2024 of the HBV genome. The primer was end labeled with [γ-32P]ATP and purified on an 8% sequencing gel. Approximately 106 cpm of the primer was mixed with 10 μg of total RNA in a 10-μl final volume containing 400 mM NaCl and 10 mM piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES) pH 6.4. The annealing reaction was carried out by heating the sample at 75°C for 3 min and then slowly cooling the sample to 42°C. After the addition of 80 μl of 1× avian myeloblastosis virus reverse transcriptase buffer (Promega) containing 0.5 mM deoxynucleoside triphosphates and 20 U of avian myeloblastosis virus reverse transcriptase (Promega), the mixture was further incubated at 42°C for 1 h for the primer extension reaction. The reaction was stopped by phenol-chloroform extraction and ethanol precipitation. The samples were then analyzed on an 8% sequencing gel.
EMSA.
Electrophoretic mobility shift analysis (EMSA) was conducted with Huh7 nuclear extracts or protein factors synthesized in vitro. The preparation of Huh7 nuclear extracts and synthesis of protein factors by using rabbit reticulocyte lysates have been described elsewhere (8). The sequences of the oligonucleotide probes used were as follows: (nucleotide changes are indicated in boldface):
WT 5′ GAGGAGATTAGGTTAAAGGTCTTTGTAT 3′3′ CTCTAATCCAATTTCCAGAAACATAATC 5′ M1 5′ GAGGAGATTAGGTTAATGATCTTTGTAT 3′ 3′ CTCTAATCCAATTACTAGAAACATAATC 5′ M4 5′ GAGGAGATTAGATTAAAGGTCTTTGTAT 3′ 3′ CTCTAATCTAATTTCCAGAAACATAATC 5′ M5 5′ GAGGAGATTAGGTTGATGATCTTTGTAT 3′ 3′ CTCTAATCCAACTACTAGAAACATAATC 5′
The DNA probes were end labeled with 32P and purified on a 4% nondenaturing polyacrylamide gel. The binding reaction mixture contained 5 μg of Huh7 nuclear extracts or 1 μl of reticulocyte lysates with or without the protein factor, 4 μl of 5× Stefan’s binding buffer (7), and water to 20 μl. The reaction mixture was preincubated on ice for 10 min and then, after addition of the probe, further incubated at room temperature for 20 min. For the supershift assay, 1 μl of antibody was added after the preincubation period. The sample was then further incubated on ice for 20 min before the probe was added. The HNF1 antibodies used were rabbit anti-HNF1 antibody A, which recognizes the N-terminal region of HNF1, and rabbit anti-HNF1 antibody B, which recognizes the C-terminal region (29a). The control antibody used was rabbit anti-hepatitis B core antigen antibody. The samples were then analyzed on a 4% nondenaturing, low-ionic-strength polyacrylamide gel as described previously (7).
RESULTS
Conversion of the nuclear receptor binding site to an HNF1 binding site by the double mutation.
We previously conducted EMSAs with nuclear extracts of Huh7 hepatoma cells and HBV DNA probes with or without the 1765-A→T 1767-G→A double mutation. Our results revealed that the double mutation removed the nuclear receptor bandshifts and created new bandshifts, suggesting that the double mutation might have created a new protein factor binding site (8). An examination of the mutated sequence indicates that it is highly homologous to the consensus binding sequence of HNF1, a liver-enriched transcription factor (Fig. 1B). To investigate whether this double mutation indeed had converted the nuclear receptor binding site to the HNF1 binding site, we performed EMSAs with both COUP-TF1 and HNF1, which had been synthesized in vitro rabbit reticulocyte lysates. As shown in Fig. 2A, COUP-TF1 could bind to the wild-type (WT) HBV DNA probe to generate a bandshift but could not bind to the HBV DNA probe (the M1 probe) containing the double mutation, consistent with our previous observation (8). In contrast, as shown in Fig. 2B, HNF1 synthesized in vitro was able to bind to the M1 probe but not to the WT probe; this result indicates that the double mutation had indeed converted the nuclear receptor binding site to an HNF1 binding site.
FIG. 2.

EMSA using the WT and M1 HBV DNA probes. Both COUP-TF (A) and HNF1 (B) were synthesized in vitro in rabbit reticulocyte lysates (for details, see Materials and Methods). Arrows denote bandshifts. Lanes 1 and 4, probe alone; lanes 2 and 5, control rabbit reticulocyte lysates added; lanes 3 and 6, rabbit reticulocyte lysates containing either COUP-TF (A) or HNF1 (B).
To ensure that the endogenous HNF1 in liver cells could also bind to the mutated sequence, we performed a supershift assay using two different anti-HNF1 antibodies. As shown in Fig. 3, the incubation of Huh7 nuclear extracts with the M1 DNA probe that contained the double mutation generated a cluster of bandshifts with similar electrophoretic mobilities. This cluster of bandshifts was not significantly affected by a control antibody, but it was removed by the anti-HNF1 antibody A and supershifted by the anti-HNF1 antibody B. Since these two anti-HNF1 antibodies are known to remove and supershift, respectively, HNF1 bandshifts (29a), the results shown in Fig. 3 indicate that the binding to the mutant probe was indeed due to HNF1. The cluster of bandshifts observed was likely due to the binding of the homodimers and the heterodimer of the two related HNF1α and HNF1β factors (2).
FIG. 3.
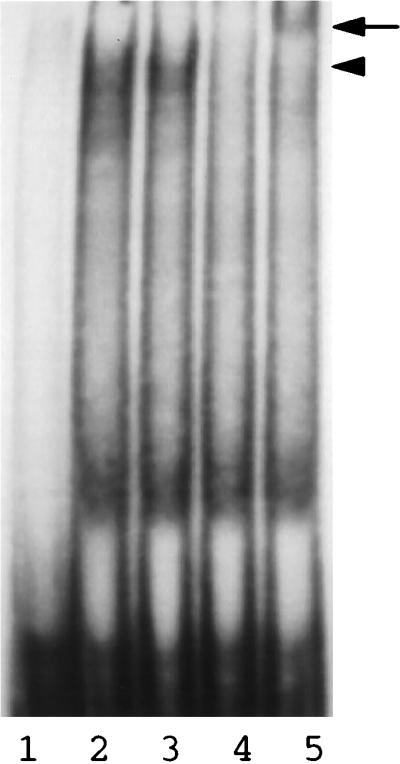
Supershift assay of the M1 double mutation probe, using Huh7 nuclear extracts. The M1 DNA probe was incubated with Huh7 nuclear extracts in the presence of a control antibody (lane 3), anti-HNF1 antibody A (lane 4), or anti-HNF1 antibody B (lane 5), followed by electrophoresis in a 4% low-ionic-strength, nondenaturing polyacrylamide gel. Lane 1, free DNA probe; lane 2, DNA probe incubated with nuclear extracts alone. Details of the procedures are given in Materials and Methods. Locations of the HNF1 bandshifts (arrowhead) and supershifted bands (arrow) are marked.
Regulation of the core promoter activity by HNF1 and the X protein mutant.
We previously reported that the double mutation specifically suppressed the transcription of the precore RNA without affecting the transcription of other HBV RNAs (7). This could be due to removal of the nuclear receptor binding site, creation of the HNF1 binding site, mutation of codons 130 and 131 in the X protein sequence, or different combinations of these factors. To investigate these possibilities, we created two additional mutants, M4 and M5. As shown in Fig. 4A, M4 is identical to WT except that it contains a G-to-A mutation at nt 1760. This mutation was created for the purpose of abolishing the nuclear receptor binding site without altering the X protein coding sequence. Comparison of the RNA phenotypes of WT and M4 allowed us to investigate the possible role of the nuclear receptor binding site in the transcription of HBV RNAs. Also shown in Fig. 4A is mutant M5, which is identical to M1 except that it contains an additional A-to-G mutation at nt 1763. This additional mutation, which is a silent mutation in the X protein sequence, was introduced for the purpose of abolishing the HNF1 binding site. Thus, comparison of the RNA phenotypes of M1 and M5 allowed us to investigate the role of the HNF1 binding site in the transcription of HBV RNAs. Since M5 differs from M4 by having the 130-Lys→Met and 131-Val→Ile changes, comparison of the RNA phenotypes of M4 and M5 also allowed us to examine the possible role of this two-codon change in the transcription of HBV RNAs. The X protein coding sequences as well as the predicted transcription factor binding properties of the WT and various mutants are also shown in Fig. 4A.
FIG. 4.

(A) Partial nucleotide sequences of WT, M1, M4, and M5 DNA probes. Locations of mutations are shown in boldface; the additional mutations introduced into M4 and M5 are underlined. The predicted binding properties of COUP-TF and HNF1 to the DNA probes are shown as + (binding) and − (no binding). The X protein coding sequences are shown in the single-letter amino acid code. (B) EMSA of various DNA probes without (−) and with (+) Huh7 nuclear extracts (NE).
To ensure that mutants M4 and M5 had lost their protein factor binding sites, we performed EMSAs using Huh7 nuclear extracts. As shown in Fig. 4B, a major bandshift was detected when the WT DNA probe was used. Our previous studies indicated that this bandshift was caused predominantly by binding of COUP-TF and, to a lesser degree, of HNF4 (8). This nuclear receptor bandshift was replaced by another bandshift with a different electrophoretic mobility when the M1 double-mutant probe was used for the experiment. As demonstrated in Fig. 3, this new bandshift was caused by binding of HNF1 to the M1 probe. In contrast, the M4 or M5 DNA probe did not generate any bandshift, confirming that the additional mutations created in these two probes had removed both the nuclear receptor and HNF1 binding sites without creating new protein factor binding sites.
To investigate the roles of nuclear receptors, HNF1, and the codon changes in the X protein on the transcription of HBV RNAs, head-to-tail HBV genomic dimers containing the WT sequence and the M1, M4, and M5 mutations were constructed and then transfected separately into Huh7 cells. Cells were lysed 2 days after transfection, and the HBV RNAs were analyzed by Northern blotting. As shown in Fig. 5A, mutants M1 and M4 expressed the WT levels of S and C gene RNAs, whereas M5 expressed the WT level of S RNA and approximately one-third of the WT level of the C gene RNA.
FIG. 5.

Effects of various mutations on transcription of HBV RNAs. (A) Northern blot analysis. Positions of HBV S gene (S), C gene (C), and X gene (X) transcripts are marked on the right. The RNA was isolated from Huh7 cells transfected with pWTD (lane 1), pM1D (lane 2), pM4D (lane 3), or pM5D (lane 4), subjected to electrophoresis in a 1% agarose gel. Northern blotted to a nitrocellulose membrane, and hybridized to the nick-translated, 3.2-kb HBV DNA probe. pXGH5 was used as an internal control to monitor transfection efficiency (see Materials and Methods). Note that although no significant difference in S gene transcript level was detected between various HBV samples, the level of C gene transcript of M5 was reduced to approximately one-third, as determined by phosphorimager analysis. This result was reproducible in at least six different experiments. (B) Primer extension analysis of precore (PC) and core (C) RNAs. Details of the procedures are given in Materials and Methods.
Since we could not determine by Northern blot analysis the expression levels of the precore RNA and core RNAs separately due to their similar sizes, we performed the more sensitive primer extension analysis to investigate the effects of various mutations on the transcription of these RNAs. As shown in Fig. 5B, the M1 double mutation reduced specifically the expression level of the precore RNA without significantly affecting the core RNA level, consistent with results reported before (7, 8). Mutant M4 expressed precore and core RNAs at levels similar to those of the WT, and in agreement with the Northern blot results, M5 expressed reduced levels of both precore and core RNAs.
Results of RNA expression for the WT and various HBV mutants (summarized in Fig. 6) show that the nuclear receptor binding site is not important for the transcription of precore and core RNAs in our cell culture system, as M4 differs from WT by lacking this particular site yet it expressed the WT levels of these two RNAs. Next, the two-codon change in the X protein sequence results in the suppression of transcription of both the precore and core RNAs, as M5 differs from M4 by having this two-codon change. Finally, creation of the HNF1 site restores the core RNA level but not the precore RNA level, as M1 differs from M5 by having the HNF1 site.
FIG. 6.

Schematic illustration of the effects of various factors on precore and core RNA transcription. Bold arrows, high transcriptional activities; thin arrows, low transcriptional activities; X, wild-type X protein; and XMl, X protein with the 130-Lys→Met and 131-Val→Ile double codon mutation; C, core RNA; PC, precore RNA; NRBS, nuclear receptor binding site; HNF1, HNF1 binding site.
DISCUSSION
HBV mutants carrying the 1765-A→T 1767-G→A double mutation are found in approximately 80% of HBeAg-positive patients with symptoms of chronic hepatitis (6, 23). The prevalence of this specific mutation indicates that there is a selective advantage for it. We have previously found that this double mutation removes a nuclear receptor binding site in the core promoter, reduces precore gene expression, and increases the viral replication rate (7). The increase of the viral replication rate is likely the reason why this double mutation is selected for during chronic infection. Removal of the nuclear receptor binding site is unlikely to be the sole reason for the selection of this double mutation, as mutations of these two nucleotides to other nucleotides or mutations in the vicinity of nt 1765 and 1767 can also abolish the nuclear receptor binding site (Fig. 4 and unpublished observation). As shown in Fig. 1B, the 1765-A→T 1767-G→A double mutation created a sequence that is highly homologous to the consensus HNF1 binding site. Indeed, in subsequent EMSAs, we found that this double mutation created a site which could be bound by HNF1 synthesized in vitro (Fig. 2) and by HNF1 present in Huh7 nuclear extracts (Fig. 3). Thus, our results demonstrate that the 1765-A→T 1767-G→A double mutation converted the nuclear receptor binding site to an HNF1 binding site. Gunther et al. (17) conducted an EMSA using Huh7 nuclear extracts and a DNA probe containing the HNF1 site derived from the pre-S1 promoter of HBV. They found that the oligonucleotide containing the M1 double-mutant sequence could not compete effectively with the pre-S1 DNA probe for binding to HNF1 and hence concluded that the M1 double mutation did not create an HNF1 site. We believe that Gunther et al. (17) have overlooked the HNF1 binding site created by the M1 double mutation because their competition assay was less sensitive and examined only the relative HNF1 binding affinity to the M1 site and to the pre-S1 site.
The need to convert the nuclear receptor binding site to an HNF1 site may be one of the reasons why the M1 double mutation is specifically selected. Since this double mutation also caused the 130-Lys→Met and 131-Val→Ile changes in the X protein sequence, this codon change may also be important for selection of the double mutation. We investigated these possibilities by creating two additional mutants, M4 and M5. The M4 mutant differed from the WT by having a single G-to-A mutation at nt 1760. This nucleotide mutation removed the nuclear receptor binding site and created a silent mutation in the X protein coding sequence (Fig. 4). Interestingly, this loss of the nuclear receptor binding site did not apparently affect the transcription of HBV RNAs (Fig. 5), which indicates that the nuclear receptor binding site in the core promoter is not important for HBV RNA transcription in Huh7 cells. Yu and Mertz (35) conducted a similar study using a subgenomic HBV DNA fragment containing the core promoter with and without mutations in the nuclear receptor binding site. They found that the nuclear receptor binding site did not affect the transcription of precore and core RNAs unless the amount of DNA plasmids used for transfection was low, in which case precore RNA transcription was suppressed. They therefore postulated that nuclear receptors could regulate precore and core RNA transcription in the early stage of infection when the copy number of the viral genome in the cell is low. In addition to this hypothesis, it is likely that this nuclear receptor binding site also allows HBV to respond to different environmental factors, which may activate different nuclear receptors, to regulate its gene expression and replication.
The 1765-A→T 1767-G→A double mutation resides in the X protein coding sequence and caused the 130-Lys→Met and 131-Val→Ile change. Thus, this two-codon change may also play a role during the selection of the M1 double mutant. The M5 mutant was created to investigate this possibility. M4 and M5 both lacked the binding sites for nuclear receptors, HNF1, and other transcription factors (Fig. 4). However, the X protein of the former has the WT sequence, and that of the latter has the two-codon change. Interestingly, although M4 expressed the WT level of precore and core RNAs, M5 expressed a reduced level of these two RNAs (Fig. 5), which indicates that the two-codon change in the X protein sequence can suppress the C gene transcription. This result is rather surprising, as previous studies indicate that mutations that prevent X gene expression do not affect HBV gene expression and replication in Huh7 cells (5, 38). It is unclear how the X protein affects C gene expression. It appears unlikely that the X protein mutant affects the transcription of precore and core RNAs by suppressing the two HBV enhancers, because the transcription of the S gene, which is also regulated by the two HBV enhancers (1, 29), was not affected (Fig. 5A). Since the X protein can interact with transcription factors (20), it is perhaps more likely that the X protein double mutant serves as a dominant negative factor to interact with transcription factors to suppress C gene transcription. Alternatively, the X protein mutant may affect the Ras–Raf–mitogen-activated protein kinase signaling pathway and indirectly suppress C gene transcription (4). Further research in this area is required to resolve this issue.
The M1 double mutant differed from the M5 mutant by having the HNF1 binding site. As shown in Fig. 5B and 6, creation of this HNF1 site restored the core RNA level but not the precore RNA level, which indicates that HNF1 can partially antagonize the suppressive effect of the X protein mutant. Recently, a number of HBV mutants with deletions, insertions, and base substitutions in the core promoter region have been isolated (17, 25). Interestingly, most of those deletions and insertions resulted in the creation of the HNF1 binding site (17, 25); the mutations were also found to correlate with a decrease in the precore RNA level and frequently an increase in the core RNA level and the viral replication rate. Since those mutations and insertions also affect the X protein coding sequence, it is likely that the alteration of precore and core RNA transcription of the mutants is also the result of the combined effects of HNF1 and the X protein mutations.
In summary, in this report we demonstrated that the 1765-A→T 1767-G→A double mutation in the HBV genome converted the nuclear receptor binding site in the core promoter to an HNF1 binding site. Removal of the nuclear receptor binding site did not affect the transcription of HBV RNAs in Huh7 cells, mutation of the X protein sequence due to this double mutation suppressed C gene expression, and creation of the HNF1 site restored the core RNA level but not the precore RNA level. The ability to create these combined effects, which lead to a higher viral replication rate, is likely the reason why this double mutation is specifically selected during chronic infection.
ACKNOWLEDGMENTS
We thank Riccardo Cortese, Gennaro Ciliberto, and Rosalba Tafi for the anti-HNF1 antibodies, R. M. Evans for the COUP-TF cDNA, Ben Yen for the HNF1 cDNA and for critical reading of the manuscript, and members of J.-H. Ou’s laboratory for helpful discussions.
This work was supported by research grants from the National Institutes of Health and the Council for Tobacco Research.
REFERENCES
- 1.Antonucci T K, Rutter W J. Hepatitis B virus (HBV) promoters are regulated by the HBV enhancer in a tissue-specific manner. J Virol. 1989;63:579–583. doi: 10.1128/jvi.63.2.579-583.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bach I, Mattei M G, Cereghini S, Yaniv M. Two members of an HNF1 homeoprotein family are expressed in human liver. Nucleic Acids Res. 1991;19:3553–3559. doi: 10.1093/nar/19.13.3553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baumert T F, Gogers S A, Hasegawa K, Liang T J. Two mutations identified in a hepatitis result in enhanced viral replication. J Clin Investig. 1996;98:2268–2276. doi: 10.1172/JCI119037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benn J, Schneider R J. Hepatitis B virus HBx protein deregulates cell cycle checkpoint controls. Proc Natl Acad Sci USA. 1995;92:11215–11219. doi: 10.1073/pnas.92.24.11215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blum H E, Zhang Z-S, Galun E, von Weizsacker F, Garner B, Liang T J, Wands J R. Hepatitis B virus X protein is not central to the viral life cycle in vitro. J Virol. 1992;66:1223–1227. doi: 10.1128/jvi.66.2.1223-1227.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buckwold V E. Regulation of the core promoter of hepatitis B virus. Ph.D. thesis. Los Angeles, Calif: University of Southern California; 1996. [Google Scholar]
- 7.Buckwold V E, Xu Z, Chen M, Yen T S B, Ou J-H. Effects of a naturally occurred mutation in the hepatitis B virus basal core promoter on precore gene expression and viral replication. J Virol. 1996;70:5845–5851. doi: 10.1128/jvi.70.9.5845-5851.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buckwold V E, Xu Z, Yen T S B, Ou J-H. Effects of a frequent double-nucleotide basal core promoter mutation and its putative single-nucleotide precursor mutation on hepatitis B virus gene expression and replication. J Gen Virol. 1997;78:2055–2065. doi: 10.1099/0022-1317-78-8-2055. [DOI] [PubMed] [Google Scholar]
- 9.Carman W F, Jacyna M R, Hadziyannis S, Karayiannis P, McGarvey M J, Makris A, Thomas H C. Mutations preventing formation of hepatitis B e antigen in patients with chronic hepatitis B infection. Lancet. 1989;ii:588–590. doi: 10.1016/s0140-6736(89)90713-7. [DOI] [PubMed] [Google Scholar]
- 10.Chang C, Enders G, Sprengel R, Peters N, Varmus H E, Ganem D. Expression of the precore region of an avian hepatitis B virus is not required for viral replication. J Virol. 1987;61:3322–3325. doi: 10.1128/jvi.61.10.3322-3325.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen H S, Kew M C, Hornbuckle W E, Tennant B C, Cote P J, Gerin J L, Purcell R H, Miller R H. The precore gene of the woodchuck hepatitis virus genome is not essential for viral replication in the natural host. J Virol. 1993;66:5682–5684. doi: 10.1128/jvi.66.9.5682-5684.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen I H, Huang C J, Ting L P. Overlapping initiator and TATA box functions in the basal core promoter of hepatitis B virus. J Virol. 1995;69:3647–3657. doi: 10.1128/jvi.69.6.3647-3657.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ganem D. Hepadnaviridae: the virus and their replication. In: Fields B N, Knipe D M, Howley P M, editors. Fields virology. 3rd ed. Philadelphia, Pa: Lippincott-Raven; 1995. pp. 2703–2736. [Google Scholar]
- 14.Garcia P D, Ou J-H, Rutter W J, Walter P. Targeting of the hepatitis B virus precore protein to the endoplasmic reticulum membrane: after signal peptide cleavage translocation can be aborted and the product released into the cytoplasm. J Cell Biol. 1988;106:1093–1104. doi: 10.1083/jcb.106.4.1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ge L, Rudolph P. Simultaneous introduction of multiple mutations using overlap extension PCR. BioTechniques. 1997;22:28–30. doi: 10.2144/97221bm03. [DOI] [PubMed] [Google Scholar]
- 16.Guidotti L G, Matzke B, Pasquinelli C, Shoenberger J M, Rogler C E, Chisari F V. The hepatitis B virus (HBV) precore protein inhibits HBV replication in transgenic mice. J Virol. 1996;70:7056–7061. doi: 10.1128/jvi.70.10.7056-7061.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gunther S, Piwon N, Iwanska A, Schilling R, Meisel H, Will H. Type, prevalence, and significance of core promoter/enhancer II mutation in hepatitis B viruses from immunosuppressed patients with severe liver disease. J Virol. 1996;70:8318–8331. doi: 10.1128/jvi.70.12.8318-8331.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo W, Chen M, Yen T S B, Ou J-H. Hepatocyte-specific expression of the hepatitis B virus core promoter depends on both positive and negative regulation. Mol Cell Biol. 1993;13:443–448. doi: 10.1128/mcb.13.1.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lamberts C, Nassal M, Velhagen I, Zentgraf H, Schroder C H. Precore-mediated inhibition of hepatitis B virus progeny DNA synthesis. J Virol. 1993;67:3756–3762. doi: 10.1128/jvi.67.7.3756-3762.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maguire H F, Hoeffler J P, Siddiqui A. HBV X protein alters the DNA binding specificity of CREB and ATF-2 by protein-protein interactions. Science. 1991;252:842–844. doi: 10.1126/science.1827531. [DOI] [PubMed] [Google Scholar]
- 21.Mangelsdorf D J, Kliewer S A, Kakizuka A, Umesono K, Evans R M. Retinoid receptors. Recent Prog Hormone Res. 1993;48:99–121. doi: 10.1016/b978-0-12-571148-7.50008-7. [DOI] [PubMed] [Google Scholar]
- 22.Okamoto H, Tsuda F, Akahane Y, Sugai Y, Yoshi M, Moriyama K, Tanaka T, Miyakawa Y, Mayumi M. Hepatitis B virus with mutations in the core promoter for an e antigen-negative phenotype in carriers with antibody to e antigen. J Virol. 1994;68:8102–8110. doi: 10.1128/jvi.68.12.8102-8110.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ou, J.-H. 1997. Molecular biology of hepatitis B virus e antigen. J. Gastroenterol. Hepatol. 12(Suppl.):S178–S187. [DOI] [PubMed]
- 24.Ou J-H, Yeh C-T, Yen T S B. Transport of hepatitis B virus precore protein into the nucleus after cleavage of its signal peptide. J Virol. 1989;63:5238–5243. doi: 10.1128/jvi.63.12.5238-5243.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pult I, Chouard T, Wieland S, Klemenz R, Yaniv M, Blum H E. A hepatitis B virus mutant with a new hepatocyte nuclear factor binding site emerging in transplant-transmitted fulminant hepatitis B. Hepatology. 1997;25:1507–1515. doi: 10.1002/hep.510250633. [DOI] [PubMed] [Google Scholar]
- 26.Raney A K, Johnson J L, Palmer C N, McLachlan A. Members of the nuclear receptor superfamily regulate transcription from the hepatitis B virus nucleocapsid promoter. J Virol. 1997;71:1058–1071. doi: 10.1128/jvi.71.2.1058-1071.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ringeisen F, Rey-Campos J, Yaniv M. The transactivation potential of variant hepatocyte nuclear factor 1 is modified by alternative splicing. J Biol Chem. 1993;268:25706–25711. [PubMed] [Google Scholar]
- 28.Scaglioni P P, Melegari M M, Wands J R. Biologic properties of hepatitis B viral genomes with mutations in the precore promoter and precore open reading frame. Virology. 1997;233:374–381. doi: 10.1006/viro.1997.8594. [DOI] [PubMed] [Google Scholar]
- 29.Su H, Yee J K. Regulation of hepatitis B virus gene expression by its two enhancers. Proc Natl Acad Sci USA. 1992;89:2708–2712. doi: 10.1073/pnas.89.7.2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29a.Tafi, R. Personal communication.
- 30.Takashi K, Aoyama K, Ohno N, Iwata K, Akahane Y, Baba K, Yoshizawa H, Mishiro S. The precore/core promoter mutant (T1762A1764) of hepatitis B virus: clinical significance and an easy method for detection. J Gen Virol. 1995;76:3159–3164. doi: 10.1099/0022-1317-76-12-3159. [DOI] [PubMed] [Google Scholar]
- 31.Tong S, Li J, Vitvitski L, Ktrepo C. Active hepatitis B virus replication in the presence of anti-HBe is associated with viral variants containing an inactive pre-C region. Virology. 1990;176:596–603. doi: 10.1016/0042-6822(90)90030-u. [DOI] [PubMed] [Google Scholar]
- 32.Yen T S B. Regulation of hepatitis B virus gene expression. Semin Virol. 1993;4:33–42. [Google Scholar]
- 33.Yen T S B. Hepadnaviral X protein: review of recent progress. J Biomed Sci. 1996;3:20–30. doi: 10.1007/BF02253575. [DOI] [PubMed] [Google Scholar]
- 34.Yu X, Mertz J E. Promoter for synthesis of the pre-C and pregenomic mRNAs of human hepatitis B virus are genetically distinct and differentially regulated. J Virol. 1996;70:8719–8726. doi: 10.1128/jvi.70.12.8719-8726.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu X, Mertz J E. Differential regulation of the pre-C and pregenomic promoters of human hepatitis B virus by members of the nuclear receptor superfamily. J Virol. 1997;71:9366–9374. doi: 10.1128/jvi.71.12.9366-9374.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zheng Y W, Yen T S B. Negative regulation of hepatitis B virus gene expression and replication by oxidative stress. J Biol Chem. 1994;269:8857–8862. [PubMed] [Google Scholar]
- 37.Zhou D X, Yen T S B. The ubiquitous transcription factor Oct-1 and the liver-specific factor HNF1 are both required to activate transcription of a hepatitis B virus promoter. Mol Cell Biol. 1991;11:1353–1359. doi: 10.1128/mcb.11.3.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zoulim F, Saputelli J, Seeger C. Woodchuck hepatitis virus X protein is required for viral infection in vivo. J Virol. 1993;68:2086–2030. doi: 10.1128/jvi.68.3.2026-2030.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]