

Abstract
Estrogen Receptor is the driving transcription factor in about 75% of all breast cancers, which is the target of endocrine therapies, but drug resistance is a common clinical problem. ESR1 point mutations at the ligand binding domain are frequently identified in metastatic tumor and ctDNA (Circulating tumor DNA) derived from ER positive breast cancer patients with endocrine therapies. Although endocrine therapy and CDK4/6 inhibitor therapy have demonstrated preclinical and clinical benefits for breast cancer, the development of resistance remains a significant challenge and the detailed mechanisms, and potential therapeutic targets in advanced breast cancer yet to be revealed. Since a crosstalk between tumor and tumor microenvironment (TME) plays an important role to grow tumor and metastasis, this effect could serve as key regulators in the resistance of endocrine therapy and the transition of breast cancer cells to metastasis. In this article, we have reviewed recent progress in endocrine therapy and the contribution of TME to ER positive breast cancer.
Keywords: Breast cancer, ESR1, Endocrine resistance, CDK4/6, TME
Graphical Abstract

1. Introduction
Over 70% of breast cancers express estrogen receptor alpha (ER) (1). ER and ERβ both interact with their ligand estrogen to promote development and proliferation of breast cancer. Breast cancers can also be categorized via their histology, clinical presentation and molecular compositions. Five main subtypes have been identified including luminal B which is of interest as this subtype commonly is endocrine resistant (1–3).
Endocrine sensitive subtypes have been successfully treated for decades in ER positive breast cancer patients. Endocrine therapies use three mechanisms to treat breast cancer. One mechanism of action is inhibiting aromatase from converting androgens into estrogens via aromatization. Preventing estrogen with aromatase inhibitors (AIs) such as Anastrazole, Letrozole or Exemestane are effective because tumors need the estrogen to grow (1). Another mechanism is downregulating the estrogen receptors. Estrogens capacity to proliferate and grow the breast tumor will be reduced with down regulated receptors. Selective estrogen receptor down-regulators (SERD) such as fulvestrant act consistently as antagonist to estrogen and will degrade ER (1, 4).
In contrast, the third mechanism utilizes selective estrogen receptor modulators (SERM). SERMs could be antiestrogen or have greater estrogenicity in non-breast regions (5). By binding to the estrogen receptor and altering its binding domain estrogen can be prevented from binding. SERMs action is also tightly coupled with the presence or absence of co-regulators. SERM mechanism of action should be understood as the basis could be important in the development of the acquired resistance to SERMs like Tamoxifen. There are three phases of acquired SERM-resistance dependent on the length of SERM exposure (6). Tumors with the various phases of resistance are stimulated by or inhibited by different factors. Phase 1 resistance shows stimulation from both SERMs and estrogen. Phase 2 resistance shows stimulation from SERMs and inhibition from estrogen. Phase 3 resistance has autonomous growth but is estrogen inhibited.
2. ER structural/functional organization
2.1. LBD (Ligand Binding Domain)
On one end of ER there is the N terminus containing ligand independent activation function 1 (AF1) followed by the hinge region and DNA-binding domain (DBD) (1). The DBD functions as the facilitator of ER binding to the estrogen responsive elements (ERE) on the DNA. By binding to EREs the activity or function of ER can be regulated through kinase signaling. The kinase signaling is mediated through the nuclear localized signal within the hinge region (7). On the C end of the ER, the ligand binding domain (LBD) resides. In addition to LBD there is an activation function 2 domain (AF2). AF2 is pertinent in gene expression regulation and is dependent on ligand interaction.
The structural aspect of the LBD complex is what leads to either agonist or antagonist conformations. Conformational changes of the LBD are dependent on the vast spectrum of ligands that could bind to ER (1). The ligand binding domain on the estrogen receptor is known to interact with dozens of estrogen or antiestrogens resulting in the ability or inhibition of co-activators to bind. The LBD is of interest because the majority of mutations that lead to ligand-independent action of the ER occur in the LBD region, leading to significant effects on the endocrine therapy (8, 9).
2.2. Co-Regulatory Elements
Co-regulators could act as activators or repressors. Transcriptional activation or repression facilitated by SERMs or additional endocrine therapies is dependent on coregulatory factors. Ligands binding to the estrogen receptor will elicit the complex to assume a particular shape that will interact with a co-regulatory element in the target cell yielding an estrogenic or antiestrogenic response (6).
The p160 SRC family are some of the most studied transcriptional co-activators. Three family members of the p160 SRC transcription factors are: NCOA1, GRIP1, and AIB1. The relative concentration of NCOA1 in varying environments like uterine tissue versus breast cancer cells will mediate the tamoxifen response to be agonistic or antagonistic (6). SERMs such as tamoxifen and raloxifene will act as antagonist when bound to ER in the breast cancer cells by recruiting the co-repressors over co-activators, which is more common in the endometrium (10). With low NCOA1 in the endometrium, tamoxifen recruits coactivators to non-classical estrogen response elements (ERE). Several studies have identified coactivators such as p300, CBP, p160 and pCAF (11–14).
Using chromatin immunoprecipitation (ChIP) three main estrogen targets have been evaluated including, cathepsin D (CATD), TFF1, and c-Myc in MCF-7 cells (15). Shang et al. further analyzed the estrogen targets by starving the MCF-7 cells of estrogen for three days followed by treatment of saturated levels of 17β-estradiol. The promoter regions all showed significant increase in occupancy by ER and AIB1 (15). AIB1 is a known ER coactivator that facilitates cell growth (8). Additional promoters from the p160 family had increased occupancy.
In MCF-7 cells and Ishikawa cells, Tamoxifen and Raloxifene both elicit the recruitment of histone deacetylases (HDACs) to the CATD promoter to act as a corepressor (10). Interestingly, only in the endometrial adenocarcinoma Ishikawa cells and not in the MCF-7 cells, tamoxifen acts as a recruiter for coactivators. The coactivators NCOA1, AIB1 and CBP coalesce on the c-Myc promoter as mediated by the SERMs. The tamoxifen driven coactivation was also accompanied with histone acetylation (10).
Upregulation of pioneer factor, FOXA1 (16) and select transcriptional factors such as, activator protein 1 (AP1), specificity protein 1 (SP1), Homeobox7 (HOXB7), and nuclear factor-κβ (NFκβ) are often associated with endocrine resistance (17) (18). Musgrove et al. also found other co-activators that are over expressed or have increased phosphorylation with endocrine resistance are nuclear receptor co-activator 3, NCOA3, also known as AIB1 or SRC3.
Accordingly, antagonist-liganded ER recruit corepressors (10). When MCF-7 breast cancer cells bind with SERM Tamoxifen, corepressors (NCoR and SMRT) are recruited by the ER in addition to subsets of HDACs that will target promoters.
Patients with Tamoxifen resistance were observed to have overexpression of MYC or cyclin D1 (17, 19). The reason the cyclin D1 and cyclin E1 are overexpressed in the patients with tamoxifen resistance was hypothesized to be from an activation of upstream mitogenic signaling pathways (17, 20). In addition, there could also be a deregulation of transcriptional regulators, an example of a regulator would be E2F family (17).
The E2F transcriptional family is made inactive in normal cells through activated retinoblastoma susceptibility protein, Rb, binding to the E2F (8, 21). The inactivation of the E2F family stops the progression of cell cycle from G1-S phase (22). Cyclin-dependent kinases, CDKs, are also major regulators of the cell cycle. When CDK4/6 coordinates with cyclin D to form a complex, Rb can be phosphorylated which releases Rb from the E2F transcription factors allowing for cell progression. In cancer, the increased levels of CDK4 and cyclin D1 contribute to the enhanced cell cycle progression (8).
Interestingly, ER signaling upregulates cyclin D1 levels, stimulating signaling pathways leading to the upregulation of CDK4/6 in addition to acquired resistance pathways like PI3K/AKT (23, 24). Given the bidirectional nature of the crosstalk ER and cyclin D share, cyclin D1 can activate ER-mediated transcription independent of normal co-factors (24). Co-factors include, CDK interaction with ER, stimulating binding of ERE, or estrogen ligands binding to ER. Specifically, cyclin D1b, a splice variant identified in breast cancer, was shown to overcome cell cycle arrest from antiestrogen therapy via CDK4 (25). This is thought to contribute to therapy resistance (25). Additional abnormalities causing loss of cell cycle control or ER transcription is the loss of Rb. Loss of Rb would be lead to enhanced cell cycle progression because Rb acts as the inhibitor of E2F family (26).
The ER can also be influenced by methylation, phosphorylation and sumoylation (27). The methylation of the ER-mediated transcription has shown resistance in vitro and in xenograft models (28, 29). The methylation of the ER by protein arginine N-methyltransferase 1 (PRMT1) or the co-activator PELP1 lead to tamoxifen resistance (17). Methylation with PRMT1 leads to the formation of cytoplasm complexes containing ER, PI3K, SRC and focal adhesion kinase (FAK). FAK, PI3K and SRC are known to activate AKT/PI3K pathway. Instead of methylating the ER co-activator, PELP1 functions as a scaffolding that modifies the ER interaction with SRC. By manipulating the interaction between ER and SRC, the activation of SRC and the ERK family kinase allow for the promotion of estrogen activation of PI3K (30).
3. Endocrine Resistance Mechanisms
3.1. Cross talk with RTK (Receptor Tyrosine Kinase)
The extensive bi-directional communication between ER and additional kinase signaling pathways result in amplification or inhibition of signaling from the pathways (7). Interestingly, these pathways could exist prior to treatment or develop during treatment. One example of a frequently altered pathway, up to 70% of breast cancers, is the PI3K/AKT pathway especially in the luminal B subtype of ER positive breast cancer (8). Pathways such as PI3K and MAPK share cross-talk with ER via HER signaling to reduce ER levels/signaling or simultaneously activate the pathways via posttranslational modifications of ER and its transcriptional elements (7, 8, 17). Activation of ER by the pathway is significant as activation is occurring in the absence of estrogen, resulting in tumor resistance to the endocrine deprivation therapies. (7)
Within the PI3K/AKT/mTOR pathway, the AKT is significant. A serine/threonine kinase protein, AKT, acts as the predominant mediator of PI3k pathway (8, 31). Phosphorylated AKT will subsequently phosphorylate additional proteins such as mTOR. mTORC1 directly phosphorylates and activates ER. As MAPK/AKT are activated by EGFR or HER2, MAPK/AKT can then activate both ER and AIB1 (the ER coactivator) via phosphorylation. Activation initiates increased transcriptional activity of ER. From the increased transcriptional activity of ER, activity of the upstream pathway is also further increased (32).
The ER pathway can affect HER and additional GFR pathways by activating or inhibiting the pathways directly or indirectly (32). GFR ligands, (insulin-growth factor 1, its receptor (IGFR), transforming growth factor-α (TGF-α)) and receptors HER1 and HER2 can have their expression induced or inhibited by ER (33, 34). Consequently, when endocrine therapy is used to block this communication of ERα, the HER1 and HER2 can have increased expression leading to further endocrine resistance. Activated forms of the IGF1R is commonly expressed in the luminal A (84%) and luminal B (76%) ER positive breast cancers (35, 36). The bidirectional crosstalk between ER and the activity IGF1R allows for regulation of each pathway. IGF1R upregulates the transcription of ER by activation of the mTOR/S6K signaling (37).
Additional studies have shown when IGF1, IGF1R, EGFR and ERBB2 have increased expression, or their downstream signaling pathways (typically PI3K/AKT or RAS/MAPK/ERK) become activated, tamoxifen resistance can occur (38–44). With loss of deregulation of these signaling pathways, upstream or downstream modifications are often the answer as further explored below.
Further pathways associated with endocrine resistance in both preclinical and clinical trials, showed oxidative stress and stress-related signaling kinases could lead to alterations of ER activity. Signaling kinases including, JNK, p38, and JNK-dependent AP-1 activity and phosphorylation increase with stress related stimuli (45–48). The activation of these signaling kinases leads to increased activation of ER and/or AIB1, a coactivator, this in turn will change the ER activity and sensitivity to endocrine therapy or agents.
An interesting pathway includes those with fibroblast growth factor receptor 1 (FGFR1). Again, luminal B type breast cancers the aggressive ER positive type, frequently have a FGFR1 amplification of greater than 10% (7). With the increased expression of FGFR1, breast cancer cells show anchorage-independent proliferation and endocrine therapy resistance.
The common tyrosine kinase receptor pathway PI3K, is known for its crosstalk with ER leading to endocrine resistance. The PI3K pathway loses its regulation through the mutation and/or amplification of PIK3Ca, PIK3CB, PIK3RI, AKT1, AKT2 or because of diminished expression of negative regulators (PTEN or INPP4B) (49–51). Uncontrolled high expression of PI3K pathway is associated with reduced ER levels and poor response to endocrine therapy.
The deregulation of various aspects of estrogen signaling commonly leads to resistance or a proliferative and survival stimuli for the tumor (17).
3.2. ER gene discrepancies or mutations
The predominant mechanism of de novo resistance to tamoxifen is lack of expression of ER. The lack of ER expression affects only 15–20% of resistant breast cancer (47). Additional intrinsic mechanisms can also occur through in inactive allele of cytochrome P450 2D6, also called CYP2D6 (52). Resistance toward endocrine therapy is also associated with adaptations within the focal adhesion molecules, adapter proteins, and growth factors (6).
Through genetic reprogramming ERs actions are modified (7). Hyperactive growth factor receptor signaling can act as a reprogramming agent, initiating ER-DNA binding site shifts. Epidermal growth factor (EGF) triggers ER cistromes to not bind to ERE-motifs, but alternatively bind with sites associated with AP-1.
The ER gene ESR1, can be re-arranged with its adjacent gene CCDC170. ESR1 and CCDC170, encoding an unknown protein, can fuse to form ER-CCDC170 which encodes for shortened version of the CCDC170 protein that is accordingly transcribed from the ESR1 promoter constitutively (7). The ER-CCDC170 fusion gene is more commonly seen in the luminal B breast cancer subtype. In pre-clinical models the ER-CCDC170 expression was associated with enhancement of motility, tumor formation, and endocrine resistance (53).
Another significant fusion protein associated with endocrine resistance is ER-YAP1. As ESR1 fuses with the ER positive endocrine resistant associated gene YAP1 the resulting protein loses the LBD or ER. Consequently, the ER-YAP1 protein demonstrates ligand-independent activation of the receptor on classical ERE-dependent genes (54). Additional fusion proteins found with ESR1 also used the ER promoter and enhanced the ERE-independent activation and endocrine resistance.
Mutations of the ESR1 gene have been documented starting as early as over a decade ago with the controversial K303R ER mutation in the hinge region and more recent mutations looking at the “hotspot” of the LBD (55). In roughly 20% of metastatic breast cancer patients, mostly post endocrine treatment, ESR1 gain-of-function mutations were found in the LBD region (7). The missense mutations, predominately Y537 and D538 residues, lead to increased stabilization of the mutated protein in the antagonist conformation (56). By being in the antagonist conformation there is ligand-independent activation of ER and constitutive activity on the receptor producing a resistance to estrogen deprivation. The mutated receptor also showed a reduced sensitivity to tamoxifen and fulvestrant. Interestingly, ER mutations were found to increase with consequent lines of endocrine therapy signifying potential clonal selection of these mutations as a suggested facilitator for resistance (57).
In clinical trial done by Li and Levine group investigated the response of insulin like growth factor 1(IGF1) in Y537S and D538G ESR1 mutant breast cancer cells. Li et al, used genome edited MCF-7 and T47D cell lines with the Y537S and D538G ESR1 mutations and identified the altered IGF1 signaling. In both mutant models the insulin receptor substate-1 (IRS1) was enhanced and there were increased levels of IGF-regulated genes. Downstream effects were also apparent in mutated cells showing PI3K-AKT as the major signaling pathway affected. When the IGF1 receptors were blocked, there was reduced sensitivity to IGF 1. The authors suggest a combination treatment is reasonable to inhibit the IGF-1 receptor and the ER to synergistically inhibit the ESR1 mutant cells (58).
In an acquired resistance to tamoxifen model in athymic mice, a D351Y mutation was identified in the tamoxifen-stimulated tumors (59). In engineered cells, the amino acid D351Y can modify the estrogenic action of SERMs at estrogen response gene targets. Modification of the estrogenic action is significant because this was the first natural mutation to convert an antiestrogenic to an estrogen complex (6). An example of this behavior is D351Y converting the antiestrogenic raloxifene-ER complex to an estrogenic complex.
3.3. Epigenetics
Through epigenetic factors, ER has been showed to be transcriptionally activated from several cofactors. Coactivators with acetyltransferase activity, will acetylate histones leading to the destabilization of nucleosomes. Exposed nucleosomes will allow for the transcription of the nucleosomes. Conversely, ER expression can also be turned off with epigenetic factors, histone deacetylases (HDACs). Interestingly, by inhibiting the HDACs with HDAC inhibitors (HDACi) such as TSA, there is a growth arrest, differentiation and cell death in the breast cancer cells (8). TSA acts a regulator of ER causing decreased amounts of ER to accumulate in the absence of ER ligands.
3.4. GPER (G protein-coupled estrogen receptor)
The expression of GPER, β1-integrin, and mesenchymal biomarkers was higher in metastases compared to primary tumors in breast cancer patients. The upregulation of β1-integrin was induced by GPER agonists and was involved in the EGFR/ERK signaling pathway. Silencing of β1-integrin partially restored the sensitivity of MCF-7R cells to tamoxifen, and knockdown of β1-integrin reduced the migration and epithelial-mesenchymal transition induced by cancer-associated fibroblasts. The downstream kinases of β1-integrin, including focal adhesion kinase, Src, and AKT, were also activated in MCF-7R cells and may contribute to the interaction between cancer cells and cancer-associated fibroblasts. The study suggests that β1-integrin could be a potential target to improve anti-hormone therapy responses in breast cancer patients (60).
GPER is known to contribute to endocrine therapy resistance in breast cancer and has been found to have a complex role in various other cancers. Recent research suggests that targeting GPER in combination with immune checkpoint inhibition could be a promising approach, particularly in melanoma, as evidenced by the initiation of the first Phase I clinical trial for the GPER-selective agonist G-1. Furthermore, GPER’s involvement in metabolism and its link to obesity and diabetes highlight its therapeutic potential for targeting a range of diseases beyond cancer (61).
GPER1 has been found to play a critical role in breast cancer progression by interacting with estrogens or synthetic agonists like G-1 and influencing genes involved in various biological events such as cell proliferation, migration, apoptosis, and metastasis. Additionally, dysregulation of microRNAs (miRNAs), short sequences of non-coding RNA, has been implicated in several pathophysiological conditions, including breast cancer. Recent evidence suggests that estrogens may regulate miRNA expression and modulate their target genes not only through classical estrogen receptors (ERs) but also by activating GPER1 signaling, indicating an alternative molecular pathway involved in breast tumor progression (62).
Estrogen plays a vital role in physiological and pathophysiological processes through its interaction with three cellular receptors: ERα, ERβ, and GPER. Disruption of the control mechanisms that regulate estrogen bioavailability and receptor sensitivity can lead to various diseases, including cancer, cardiovascular and neurodegenerative diseases, obesity, insulin resistance, endometriosis, and systemic lupus erythematosus. Modulating estrogen bioavailability or receptor activity can effectively treat these pathological conditions. Blocking estrogen biosynthesis can prevent estrogen action at ERs and GPER, but resistance and ligand-independent receptor activation may still occur. Anti-estrogen drugs that antagonize ERs can function as GPER agonists. GPER is a promising therapeutic target for treating several health concerns, including metabolic dysregulation and advanced cancer. Selective ligands that specifically target GPER have been developed and may soon serve as pharmacological agents for treating human disease (63).
3.5. c-Src Kinase
Src kinase regulates cellular functions such as proliferation, migration, and invasion, and its activity is frequently elevated in human tumors. The study investigated the role of Src in MCF7 cells, a model of endocrine resistance. The tamoxifen-resistant (TamR) cells displayed elevated Src kinase activity, which was reduced by treatment with the novel Src inhibitor, AZD0530. The inhibitor also suppressed the motile and invasive nature of TamR cells in vitro, reduced activated FAK and paxillin levels, and promoted elongation of focal adhesions. The study suggests that Src plays a crucial role in mediating the motile and invasive phenotype observed in endocrine-resistant breast cancer cells and that Src inhibitors may prevent cancer progression and metastasis in combination with EGFR inhibitors such as gefitinib (64).
The Src kinase is increasingly implicated in cancer development due to its role in intracellular signaling pathways that control cell growth, adhesion, and migration. Src activity is often elevated in cancer cells, leading to enhanced tumour growth and/or migration. Src may also influence life or death decisions in cells, making it an attractive target for cancer therapy. Inhibitors of Src kinase activity or downstream effectors may be useful in altering cancer cell behavior and improving therapeutic outcomes (65).
Recent studies suggest that the ER-Src axis, which links ER with HER2 and PELP1, contributes to targeted therapy resistance. Inhibition of Src using shRNA or dasatinib, an inhibitor, reduced the growth of therapy-resistant cells in vitro and in vivo. The ER-Src axis plays a role in promoting hormonal resistance and blocking this axis prevents the development of hormonal independence in vivo. Combination therapies using endocrine agents and dasatinib may delay the development of hormonal resistance (66).
3.6. ER variants, 46 and 66 kDa
Breast cancer diagnosis studies have mostly focused on the full-length 66-kDa estrogen receptor (ER66) and ignored the shorter 46-kDa isoform (ER46), which lacks the transactivation function AF-1. This study investigated the expression levels of ER46 in breast tumors, its mechanism of generation, its specificities of coregulatory binding, and its functional activities. ER46 was expressed in over 70% of breast tumors, sometimes more abundant than ER66, particularly in lower-grade and smaller-sized tumors. The study found that ER46 can be generated via internal ribosome entry site-mediated translation in the context of endoplasmic reticulum stress. The binding affinities of both unliganded and fully-activated receptors towards co-regulator peptides revealed that ER46 and ER66 have different respective potencies, contributing to the differential transcriptional activity of target genes to 17β estradiol (E2).
Additionally, the study found that increasing amounts of ER46 decrease the proliferation rate of MCF7 tumor cells in response to E2. This overlooked ER46 isoform’s expression in a majority of breast tumors underlines the need to develop new technologies to discriminate between ER66 and ER46 expression in breast cancer diagnosis, which could have potential clinical relevance (67).
The reduced expression of the N-terminally truncated ERalpha46 protein in endocrine-resistant breast cancer cells compared to MCF-7 cells. Restoring ERalpha46 expression partially restored growth inhibition by tamoxifen in resistant cells. Overexpression of ER46 in MCF-7 cells reduced the transcription of various genes in response to estradiol (E2), including pS2, cyclin D1, nuclear respiratory factor-1 (NRF-1), and progesterone receptor. The expression of oncomiR miR-21 was lower in tamoxifen-resistant cells, but transfection with ERalpha46 altered the pharmacology of E2 regulation of miR-21 expression, indicating that ERalpha46 inhibits ER activity. The study also found that established miR-21 targets PTEN and PDCD4 were reduced in ERalpha46-transfected, E2-treated MCF-7 cells. Overall, the findings suggest that ERalpha46 may enhance endocrine responses by inhibiting specific ERalpha66 responses (68).
The study characterizes two estrogen receptor-alpha (ER) isoforms (ER66 and ER46) in ER-positive breast cancer cells, MCF7. ER66 mediates estrogen’s mitogenic effects, while ER46’s function remains undefined. The study shows that ER46 accumulates in the nucleus when cells reach confluence, and its overexpression in proliferating MCF7 cells leads to cell-cycle arrest. In PC12 cells, which lack ER, stable transfection of ER66 allows estrogen to act as a mitogen, while ER46 does not. The study shows that ER46 inhibits ER66-mediated estrogenic induction of AF-1-sensitive reporters such as c-fos and cyclin D1 and estrogen-responsive element-driven reporters in MCF7 cells, likely through functional competitions between both isoforms. Overall, the study suggests that ER46 antagonizes the proliferative action of ER66 in MCF7 cells by inhibiting ER66 AF-1 activity (69).
4. Current clinical impact in ER positive breast cancer
The drug therapy course for ERBC is dependent on many factors of each patient. D’Souza et al. discussed how to overcome endocrine resistant breast cancer while considering a patient’s prior adjuvant therapies, quality of life, side-effect profile and their disease-free interval (70). D’Souza and colleagues, discuss the current popularity of endocrine therapies like tamoxifen and the third generation aromatase inhibitors as the first and second line options for hormone receptor-positive metastatic breast cancer. However, with the endocrine resistance, SERDs such as fulvestrant are becoming increasingly popular options with clinical trial FIRST, FALCON and CONFIRM to support this use.
The CONFIRM trial done by Di Leo and colleagues confirms the appropriate use of Fulvestrant. The study’s population was post-menopausal women and compared the dosage strengths 250mg to 500mg in a double-blind, parallel-group, multicenter, phase III study (70). The previously approved dose, 250mg, was intended for post-menopausal women that had previously received endocrine therapy for advanced estrogen receptor breast cancer and experienced progression. The dosing frequency was once monthly, and the route is intramuscularly (IM).
The CONFIRM trial randomly assigned participants with the increased strength of 500 mg on day 0, 14, 28 and every 28 days after. The route was still IM. With the primary end-point of progression free survival (PFS), the fulvestrant 500mg (n= 362) showed significantly longer PFS over the fulvestrant 250mg (n=374) (70). Additional secondary markers such as clinical benefits rate (CBR) and overall survival (OS) also showed a increased benefit of the 500mg strength dose. The CBR for the 500mg dose was 45.6 % with the 250 mg strength at 39.6%. The OS was closer with the 500 mg to 250mg dose, 25.1 months and 22.8 months respectively (70). The 500mg dose was reportedly well tolerated with no dose-dependent adverse events.
In addition to selectively targeting the estrogen receptor such as SERDs and SERMs, groups such as Zhu et al. addressed benefits of tamoxifen with the addition of the adjuvant rapamycin to additionally target mTOR. Zhu et al. looked at the synergistic effects of rapamycin with tamoxifen (71). The rapamycin was already an FDA-approved agent to target mTOR in mammals and interestingly when administered in combination with tamoxifen, the required concentration for tamoxifen to inhibit cell growth was greatly reduced. The study also found the combination tamoxifen with rapamycin significantly inhibited tumor growth in vivo (71). Zhu et al. attributes this effect to the mechanism of which p73 is induced and therefore enhances the ER expression by direct binding onto the promoter region of the ER gene.
Recently, additional treatment groups are developing to address the endocrine resistance with CDK4/6 inhibitors. Drugs such Palbociclib, Ribociclib and Abemaciclib. In the trial MONARCH 2, Abemaciclib (LY2835219) was combined with fulvestrant in patients with hormone receptor positive HER2 negative breast cancer (72). This phase 3, randomized, double-blind, placebo-controlled study involved 669 participants. Experimental groups received, 150 mg Abemaciclib given orally every 12 hours in 28-day cycles with 500 mg fulvestrant administered as two 250 mg IM injections on days 1 and 15 of cycle 1 and then on day 1 of cycle 2. The experimental groups included the endocrine naïve cohort and a previously endocrine treated cohort. Main outcome report was progression free survival (PFS) and one of the secondary outcomes measuring overall survival (OS). The MONARCH 2 study concluded Abemaciclib 150 twice daily with fulvestrant was effective with PFS of 16.4 months compared to the median or 9.6 months (95% CI, 0.449 to 0.681; P < .001) (72, 73).
The MONALESSA-2 trial examined the use of Ribociclib with Letrozole (a non-steroidal aromatase inhibitor). The experimental group was treated with Ribociclib 600mg by mouth once daily (given as 3, 200 mg tablets) along with Letrozole 2.5 mg tablets by mouth once daily. The Ribociclib was given with rotation, 3 weeks on/ 1 week off. MONALEESA-2 looked at PFS as well as overall response rate (ORR). The PFS was increased with Ribociclib over placebo in the elderly population, demonstrating it was a valid treatment option.
POLARIS is a current trial that is looking at the use of the cyclin-dependent kinase 4/6 inhibitor Palbociclib (P). Investigators Tripathy et al. state how not all patients respond to P in the previous P trial PALMOA (74). Tripathy et al. hopes to identify biomarkers to indicate which patients would benefit from the addition of P to their endocrine therapy.
Moreover, In the PALOMA-2 and PALOMA-3 clinical trials, palbociclib plus ET (endocrine therapy) significantly improved progression-free survival (PFS) versus ET alone in patients with HR+ /HER2− ABC (75–77).
Another drug class of note is mTOR inhibitors. The PI3K/AKT/mTOR pathway is deeply implicated with the many cancers and almost certainly ERBC. Recent trials such as TAMRAD and MANTA along with the ongoing BOLERO-2 investigate mTOR inhibitors. The BALLET study looked to confirm the BOLERO-2 trial and examine the safety of everolimus plus exemestane. The patients included a broad range of women aged 18 and older with advanced breast cancer (ABC) that is recurring/progressing during/after prior non-steroidal aromatase inhibitors. The primary objective was addressing the safety as the BOLERO trial examines the efficacy (78). With over 2100 patients included in their analysis, the BALLET study concluded safety was consistent with the BOLERO-2 trial. Elderly patients ≥ 70 years of age demonstrated the highest adverse event (AE)-related treatment discontinuations versus non-elderly (23.8% to 13.0%). In all age groups the incidence of AEs of everolimus was reduced with first-line ABC over later lines.
Specific PI3K agents have also been developed to include Alpelisib, Buparlisib and Pictilisib from the SOLAR and BELLE trials. The BELLE-3 trial by investigators Di Leo et al., examined the use of a PI3K inhibitor buparlisib with fulvestrant in postmenopausal women. The participants were aromatase inhibitor treated with locally ABC or metastatic breast cancer that progressed on or after mTOR inhibitor-based treatment. Experimental groups received the buparlisib 100mg once daily (route not reported) with 500mg fulvestrant per standard of care. Placebo groups received equivalent placebo as well as equal fulvestrant. Results examined PFS and OS, putting both in favor of the experimental group. The 6-month PFS was 30.6% for experimental compared to 20.1% placebo. Investigators Di Leo et al. additionally concluded the greatest benefited group via PFS, was patients with PIK3CA-mut when compared to PIK3CA-wt tumors based on ctDNA and PCR (79).
5. Tumor microenvironment
The tumor microenvironment is becoming of increasing interest to researches as the roles of tumor progression, metastasis and drug resistance become illuminated. A dynamic conglomerate of heterogeneous population of stromal cells and tumor bulk comprise the tumor microenvironment (TME). An additional portion of the TME is the extracellular matrix (EMC). The EMC is critical not only to act as the scaffolding of the cell, but hold the abundance of key growth factors. As discussed below, like much of the TME, the EMC will become dysregulated as tumor progression occurs (80–82). One last portion of note of the TME is the vasculature. Tumor vasculature is notoriously inadequate to supply the growing needs of the mass. On top of being in short supply, the vessels often exhibit uneven vessel lumen and can even be leaky. Consequently, these short comings lead to rises in the interstitial fluid pressure causing uneven blood flow and dispersion of nutrients and drugs (80).
How the stroma is recruited by the tumor provides the first display of the crosstalk between the tumor and it’s microenvironment. Several groups suggest the maintenance and expansion of the tumor is dependent on the external signals from the tumor’s environment (80, 82–84). Within the TME, the stromal cells and fibroblasts secrete factors that promote growth, (fibroblast growth factor (FGF), CDCL12 chemokines) and act as chemoattractants stimulating migrations of additional cells into the TME (80).
A recent shift for therapeutic focus is now on the tumor associated stromal cells (TASC); immune cells, blood vessels, fibroblasts, and extracellular matrix contribute to the TASC. Dvorak et al. has described tumor recruitment or generation of stroma to be similar to normal wound healing involving neoangiogenesis, fibroblasts and immune cells (85). Bussard et al. proposes that with recent data TASC may be a necessary prerequisite for tumor (86). The origins of the stroma necessary for the tumor cell invasion and metastasis are from 6 diverse locations of fibroblasts, pericytes, bone marrow mesenchymal stromal cells, adipocytes, macrophages and immune cells (87–112).
5.1. Kynurenine pathway
Malignancies pose a significant public health challenge, and their treatment remains a major hurdle for modern medicine. Tumors have evolved various mechanisms to evade immune and therapeutic responses, leading to an urgent need for research to overcome these challenges. One such mechanism is the accumulation of tryptophan metabolites from the kynurenine pathway, which suppresses the immune system’s response to cancer cells and contributes to resistance to antitumor therapy. Kynurenine is one of the most potent immunosuppressive metabolites in this pathway and plays a significant role in the development of malignancies. Therefore, researchers are investigating whether targeting the enzymes responsible for its synthesis could be an effective therapeutic approach for various cancers. Several preclinical and clinical studies have been conducted, especially regarding the inhibition of indoleamine 2,3-dioxygenase activity, and the results are considered noteworthy (113).
The metabolic pathway of tryptophan known as the Kynurenine pathway is of significant importance, as its metabolites have been closely linked to several diseases. The first and rate-limiting step of this pathway is regulated by two enzymes, namely Indoleamine-2,3-dioxygenase (IDO) and Tryptophan-2,3-dioxygenase (TDO or TDO2). These enzymes are directly or indirectly involved in various diseases, such as inflammatory diseases, cancer, diabetes, and mental disorders. Recently, several potential mechanisms have been uncovered, highlighting the importance of the Kynurenine pathway in disease progression(114).
Tryptophan catabolite kynurenine induces CD8 T-cell death, and the tryptophan-catabolizing enzyme TDO is utilized by TNBC to inhibit CD8 T-cell viability. High TDO2 correlates with poor breast cancer clinical outcomes, while IDO1 does not. Metabolomic analysis reveals a strong correlation between substrate and catabolite of tryptophan and kynurenine. Interestingly, both tryptophan and kynurenine are lower in the plasma from patients with breast cancer, particularly in women with ER-negative and stage III and IV breast cancer. The study suggests that pharmacologic efforts should target both TDO and IDO1 to improve antitumor immunity in multiple cancers (115).
5.2. Secreted factors in ER positive breast cancer
CCL2 and CCL 5 are chemoattractants that attract tumor associated macrophage (TAM) into cancer tissues. ER positive breast cancer tissues show increased levels of CCL2 and CCL5 leading to increased TAM level compared to adjacent normal tissues. Those recruited TAMs have two phenotypes which may either increase or decrease cancer cell proliferation. The levels of CCL2 and CCL5 were decreased in cancer cell after 6 weeks of tamoxifen therapy followed by decreased TAM infiltration. This finding supports the hypothesis that CCL2 and CCL5 are important factors in estrogen receptor positive breast cancer and suggests novel therapeutic targets CCL2 and CCL5 (116).
Furthermore, in endocrine resistance breast cancer tissue, M2 polarization of TAM was induced leading to further endocrine resistance forming a positive feedback loop. Increased expression of CCL2 from M2 polarization leads to activation of PI3K, Akt, and mTOR signaling pathway resulting in endocrine resistance. Additionally, CCL2 induces aggregation of macrophages and monocytes. Since CCL2 from tumor associated macrophage plays an important role in endocrine resistance, these findings indicate novel therapeutic targets and corresponding therapeutic agents (117).
IL-33 as a cytokine that promotes endocrine resistance since level of IL-33 is elevated in patients with stage 4 breast cancer leading to poor prognosis. In normal tissue, IL-33 binds with ST2 receptor when cell is damaged or stressed. This leads to innate immunity causing inflammation. However, in cancer cell, IL-33/ST2 progress breast cancer causing tumorigenesis. The study was done to investigate role and effect of IL-33 in cancer cells and results proved that cancer cells had increased IL-33 level compared to normal tissue, promoting breast cancer stem cell properties. Since IL-33 is one of the factors that promotes resistance, they suggest a new therapeutic target to explore in the future to yield better results for breast cancer patients (118).
Tumor progression can result from inflammation and be induced by IL-6, IL-8 and TNF-a which are pro-inflammatory cytokines. Elevated levels of these cytokines lead to higher breast cancer stages and lymph node metastasis. Correlation of these pro-inflammatory factors with ER, PR, and HER2 are investigated to reveal effects of individual cytokines in breast cancers. The level of IL-6 was significantly increased in patients with breast cancers indicating lymph node metastasis. Also, high serum IL-6 level was observed in ER+ breast cancer compared to ER-breast cancer. IL-6 level was higher in HER2-breast cancer compared to HER2+, and IL-6 did not show any correlation with PR status. The level of IL-8 was higher in breast cancer with ER− and HER2+ patients, and it did not show correlation with PR. Lastly, TNF-a level was elevated in patients with stage 3 carcinoma, but it did not show correlation with all ER, PR, and HER2 status. TNF-a producing cells are thought to be not affected by ER, PR, and HER2. IL-6 secretion from immune cells is one major factor for elevated serum IL-6 level since, ER+ cell line secretes lower level of IL-6 than ER− cell line. ER+ breast cancer tissue downregulates the release of IL-8 resulting in reduced level. IL-8 and TNF-a are reported to have synergistic effects on cancer cell proliferation. These results suggests a few possible cancer biomarkers to better estimate prognosis of breast cancer with different stages (119).
IL-8 is a pro-inflammatory chemokine that binds to CXCR1 and CXCR2 leading to metastasis and worse prognosis. IL-8 level was significantly elevated in tamoxifen resistant breast cancer strengthening the hypothesis that IL-8 is can be a prognostic biomarker of resistance. The result indicated a worse relapse-free survival in patients with elevated IL-8 level in luminal A type breast cancer. However, in luminal B type breast cancer, expression of IL-8 did not show significant differences. Increase in IL-8 level in tamoxifen resistant cancer cell was observed with higher level of MEK and Akt activities. These two pathways led to increased IL-8 secretion. CXCR1/2 inhibitors such as SB225002 increased the number of apoptotic cells indicating that blockade of CXCR1/2 by its inhibitor will reduce growth of anchorage independent tamoxifen resistant cells (120).
IL-20 expression is regulated by elongation factor Ell3. ER(a), GATA3, and FOXA 1 forms transcriptional factors with Ell3 to induce or repress the expression of IL-20 in ER+ breast cancer. Induction of IL-20 leads to tumor progression and migration associated with inflammation reaction. ER induces IL-20 expression in ER+ breast cancer. IL-20 expression was decreased and the effect of estrogen inducing IL-20 was inhibited, suggesting IL-20 as a novel therapeutic target to develop a better treatment for ER+ breast cancer (121).
CXCR7 is highly expressed in luminal cells of ER+ breast cancer cell. CXCR7 was also shown to be closely related to EGFR in breast cancer cells. The interaction of CXCR7 and B-arrestins to phosphorylate EGFR and ERK1/2, indicating a relationship between EGFR and CXCR7 and suggesting a new therapeutic target for future breast cancer therapies (122).
CCL7, IL6, and IL8 are continuously secreted by tumor associated fibroblast, which induce PDGF-BB. PDGF-BB then ultimately induces IL-1B to make favorable tumor microenvironment. When paracrine signaling was blocked, tamoxifen was more effective in inhibiting cancer cell proliferation highlighting the importance of paracrine signaling of fibroblast in forming IL-1B rich tumor environment (123). Platelet-derived growth factor (PDGF) regulates cell proliferation which leads to growth and progression of cancer cells. Ponatinib and sunitinib, PDGFR inhibitors decreased the cell growth and proliferation by down regulating the phosphorylation of ERK and STAT3. Moreover, combination therapy with two PDGFR inhibitors with tamoxifen enhances the inhibitory effect in ER positive breast cancer and concurrent use of ponatinib ad sunitinib produced a better outcome compared to tamoxifen alone (124).
Our studies showed that CXCL1 plays a critical role in crosstalk between endocrine resistant breast cancer and fibroblast and adipsin, one of the cytokine in the complement system, controls ESR1 mutant cell growth (data not shown). These findings imply that crosstalk between ER positive breast cancer with stroma via. secreted factors enhance tumor growth and metastasis, which could be a potential target to treat ER positive breast cancer as well as endocrine resistant breast cancer.
5.3. microRNAs
As with cancer associated stromal cells, small, single stranded microRNAs (miRs) are also dysregulated in cancer. The dysregulation of the miR expression is connected with the pro-tumor behavior including poor patient outcome, tumor cell promotion, migration, and invasion (125, 126). In addition to the pro-tumor behavior, the dysregulated miRs have been shown by Miller et al. to disrupt common signaling cascades in TAFs such as the mitogen-activated protein kinase (MAPK)/ERK pathway (127). The MAPK/ERK pathway is a significant pathway in breast cancer as it is frequently hyperactive and subsequently associated with poor response to hormone therapy with both luminal A and B estrogen receptor breast cancer subtypes (86). Li et al. produced recent evidence that epigenetics is behind the dysregulation of miR in TAFs. Li et al. states epigenetic silencing due to the CpG island methylation may be responsible for miR dysregulation (105). The Zhang group identified a specific microRNA, miR-149 as a influential driver in the recruitment of typical fibroblast into the TME (128).
Of increasing interest is the exosomes containing miRNA. Melo et al. studied microRNA containing exosomes and their contributions to tumor progression in a breast cancer cell line MDA-MB-231. Results from the investigations suggests active biogenesis in exosomes and when exosomes originated from cancer lines oncogenic changes were induced compared to normal cell lines with no oncogenic changes (129).
6. Future Perspectives
In this review, we examine the tumor microenvironment of breast cancer, Diana Martinsa and Fernando Schmitt examine whether the environment, and the changes the cells that occur, are more hurtful or helpful to the body (130). Their findings along with concurring data show significant alterations of gene expression within the microenvironment throughout the development of the disease. Several stromal cells also become implicated in enhancing the trademarks of cancer.
Multiple groups have discussed the role of epigenetics being a key player in the changes through tumor progression in the microenvironment of breast cancer and how the stroma is altered through crosstalk. The stroma concurrently transforms with epithelial compartments throughout the development of the tumor. Creighton et al. shows the stimulus the stromal cells supply with crosstalk between cancerous cells and host is secreted through several ECM proteins, proteases, growth factors, cytokines, and protease inhibitors (131). Vargas et al. suggests what drives this crosstalk is less through genetic abnormalities and more epigenetic changes such as DNA methylation and chromatin modification (132).
Martinsa and Schmitt explain normal mammary epithelial tissue will experience changes in gene expression in non-carcinogenic scenarios. Lactogenic hormones will cause changes in the mammary epithelial tissue to produce milk proteins which work in collusion with other components of the microenvironment in mammary duct morphogenesis. We see a similar type of cellular response in the local environment in carcinogenic scenarios. The mammary microenvironment can degenerate neoplastic phenotypes of breast cancer through cellular differentiation and trigger accompany changes. Ronnov-Jessen states that with the tumor formation, the surrounding mammary microenvironment shows increased fibroblast proliferations and ECM remodeling along with the cellular differentiation of the mammary epithelial tissue (133). Bergamaschi et al. argue the stromal component, comprised of surplus of inflammatory cells and activated fibroblast, drive the expression of ECM components and growth factors in a paracrine fashion (134). This expression promotes survival and growth of tumor cells.
Because of the interactions between the transformed cells and the epithelial cells, Martinsa and Schmitt identify the microenvironment as less of a bystander and more of a facilitator of carcinogenesis. Ma et al. supports this classification and additionally recognizes the tumor epithelial cells are not the key players and rather the progression is driven by multiple key players (135). As for the main initiators of hormone therapy, Bolelens et al. looked at exosomes derived from stromal fibroblast along with miRNA, believing they may contribute to treatment resistance by the transfer of noncoding RNA to breast cancer cells (136).
Along the stromal cells and growth factors in the microenvironment, Polonia et al. shows in breast carcinomas the presence of tumor infiltrating lymphocytes. The tumor infiltrating lymphocytes are significantly correlated to high expression of Ki67, suggesting the involvement of the immune system (137). The immune system involvement and its accompanying cellular response is an important player in the tumor progression.
Ultimately, Martinsa and Schmitt appropriately concluded the complete role of the tumor microenvironment in breast cancer initiation and development is still under-recognized and incompletely understood. Our best therapeutic approach at this time would be to consider targeting the TME. By targeting the hypoxic niches, disrupting the crosstalk to the tumor cells, or inhibiting the abnormal pathways activated in the TME would be beneficial for ER positive breast cancer patient outcome.
Figure 1. Current endocrine therapy and potential therapeutic strategy in endocrine resistant breast cancer.

SERM (selective ER modulator), SERD (selective ER degrader), Als (aromatase inhibitors), CDK4/6 inhibitors are currently utilized in endocrine therapy regimen. However, after 3 to 5 year treatment, tumors recur in about 1/3 patients and have resistance to endocrine therapy. These endocrine resistant tumors displayed high RTK activation and its downstream signaling pathway, ESR1 fusion, ESR1 mutations, epigenetic alteration, and crosstalk with TME (tumor microenvironment). In order to block the advanced endocrine resistant breast cancer, SERD (fulvestrant), CDK4/6 inhibitors and RTK inhibitors are utilized. Recently, one of the immune therapy, ICI (immune checkpoint inhibitor) is emerging and could be an alternative therapeutic strategy. Furthermore, crosstalk between tumor and stroma via. secreted factors could be a potential target to attenuate endocrine resistance. Since combination therapy is the best way to enhance treatment efficacy, to inhibit the development of drug resistance, and to decrease the duration of treatment, we need to seek an optimal the combined inhibitors to treat patients with endocrine resistant breast cancer.
Figure 2. ER target gene expression is regulated by ER coactivators and corepressors.

When estrogen binds to estrogen receptors (ER), it recruits FOXA1, which binds to ER target gene enhancers and recruits ER and coactivators such as p300, CBP, p160, and pCAF. The interaction of the ER complex with transcription factors such as AP, SP1, NFkB, and HOXB7 promotes transcriptional activity. However, tamoxifen induces a conformational change in ER that leads to recruitment of corepressors such as NCOR and SMRT, repressing ER target gene expression. In some tamoxifen-resistant breast cancers, the balance between coactivators and corepressors shifts towards coactivators, leading to activation of ER target genes.
Figure 3. Schematic model of crosstalk between ER positive breast cancer and stroma.
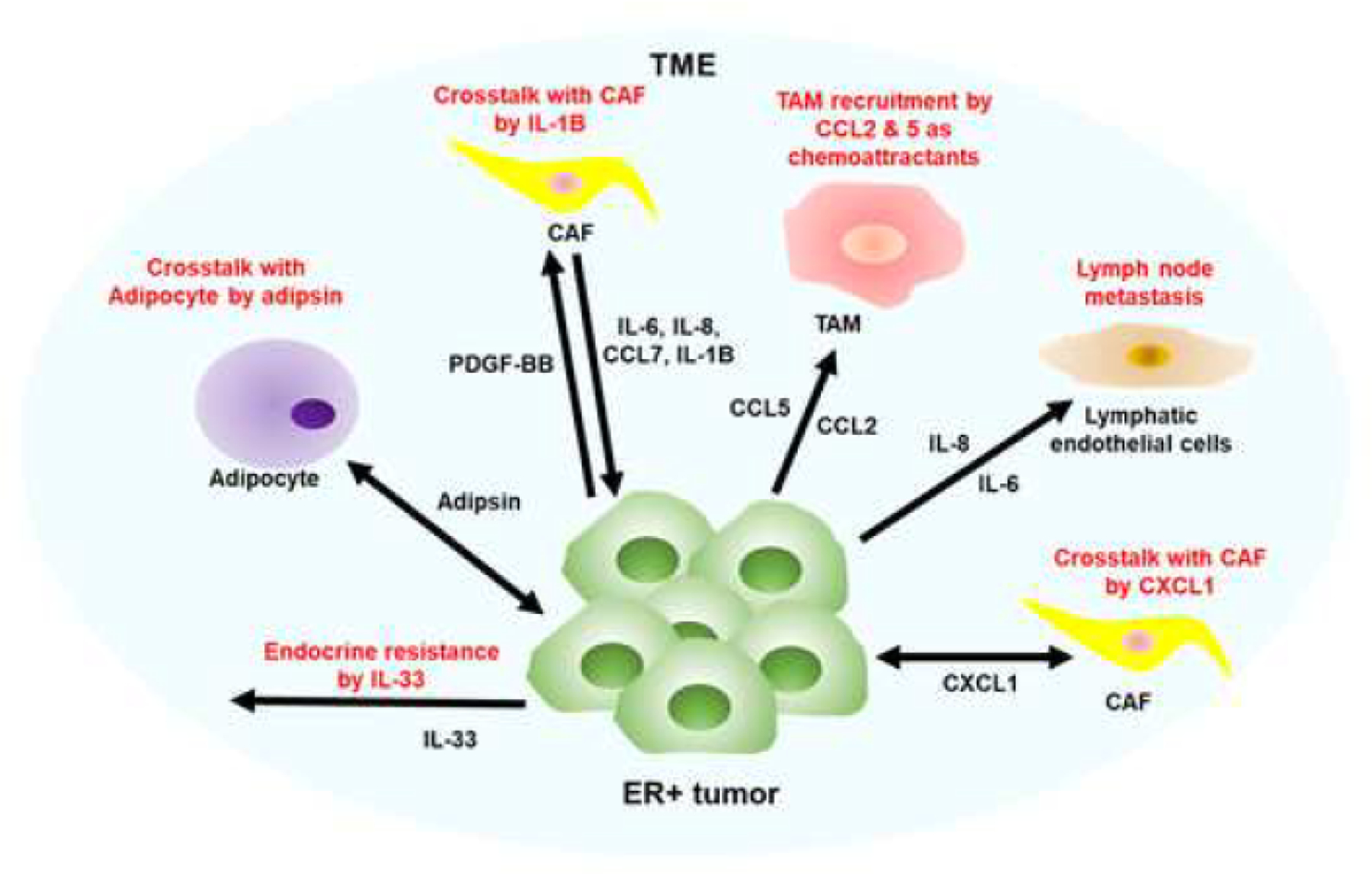
ER-positive breast cancer has been found to secrete a variety of cytokines and chemokines, including IL-6, CXCL1, IL-8, IL-20, CCL7, IL-1B, PDGF-BB, CCL2, CCL5, and IL-33. These secreted factors are responsible for recruiting various stromal components such as cancer-associated fibroblasts (CAFs), tumor-associated macrophages (TAMs), blood and lymphatic endothelial cells, and adipocytes. CCL2 and CCL5 act as chemoattractants for M2-type TAMs, which are known to promote tumor growth. Additionally, IL-8 and IL-6 secretion by ER-positive breast cancer enhances lymph node metastasis. PDGF-BB from tumors and IL-8, IL-6, and CCL7 from CAFs promote tumor growth by enhancing IL-1B secretion through crosstalk between tumors and CAFs. Endocrine resistance is also facilitated by the upregulation of IL-33. Furthermore, CXCL1 induces a positive feedback loop between endocrine-resistant tumors and fibroblasts, while adipsin is upregulated in the crosstalk of adipocytes to promote ER-positive tumor growth. However, the specific secreted factors from stromal and immune cells stimulated by various secreted factors from ER-positive breast cancer remain to be studied. Understanding these interactions between secreted factors and their receptors could provide potential therapeutic targets to attenuate ER-positive breast cancer growth and metastasis.
Table 1.
Active Pharmacological Intervention Trials in ER positive breast cancer.
| Clinical Trial | Drug | Dose | Phase |
|---|---|---|---|
| CONFIRM | fulvestrant | 250/500 mg | III |
| FALCON | fulvestrant+anastrozole | 500/1mg | III |
| FIRST | fulvestant+anstrozole | 500/1mg | II |
| MONARCH 2 | fulvestrant+abemaciclib | 500/150 mg | III |
| MONALESSA-2 | ribociclib+letrozole | 600/2.5 mg | III |
| PALOMA-2 | palbociclib+letrozole | 125/2.5 mg | III |
| PALOMA-3 | palbociclib+fulvestrant | 125/500 mg | III |
| TAMRAD | tamoxifen+everolimus | 20/10 mg | II |
| MANTA | fulvestrant+vistusertib fulvestrant+everolimus |
500/50 mg 500/10 mg |
II |
| BOLERO-2 | everolimus+exemestane | 5/25 mg | III |
| BALLET | everolimus+exemestane | 5–10/25 mg | III |
| SOLAR-1 | fulvestrant+alpelisib | 500/300 mg | III |
| BELLE-3 | fulvestrant+BMK120 | 500/100 mg | III |
| POLARIS | palbociclib |
Abbreviations:
- AIs
Aromatase Inhibitors
- CCL2
CC Motif Chemokine Ligand 2
- CCL5
CC Motif Chemokine Ligand 5
- CDK4/6
Cyclin dependent kinase-4/6
- CAF
Cancer associated fibroblast
- CXCL1
C-X-C Motif Chemokine Ligand
- CCL7
CC Motif Chemokine Ligand 7
- ER (ESR1)
Estrogen receptor
- IL-33
Interleukin-33
- IL-6
Interleukin-6
- 1; IL-1B
Interleukin-1beta
- IL-8
Interleukin-8
- IL-20
Interleukin-20
- ICI
N-n-butyl-N-methyl-11-(3,17β-dihydroxyestra-1,3,5(10)-trien-7α-yl)undecanamide
- PDGF-BB
Platelet-Derived Growth Factor-BB; SERM, Selective ER modulator
- SERD
Selective ER degrader
- TME
Tumor microenvironment
- TAM
Tumor associated macrophage
- RTK
Receptor tyrosine kinase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CRediT authorship contribution statement
Brianna Blakely, Seobum Shin, and Kideok Jin conceived the idea for writing this review and conducted literature review and contributed to the writing.
Declaration of Interest
Declarations of interest: none
References
- 1.Jeselsohn R, Buchwalter G, De Angelis C, Brown M, Schiff R. ESR1 mutations-a mechanism for acquired endocrine resistance in breast cancer. Nature reviews Clinical oncology 2015;12(10):573–83. Epub 2015/07/01. doi: 10.1038/nrclinonc.2015.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Creighton CJ. The molecular profile of luminal B breast cancer. Biologics : targets & therapy 2012;6:289–97. doi: 10.2147/BTT.S29923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS, Thorsen T, Quist H, Matese JC, Brown PO, Botstein D, Lonning PE, Borresen-Dale AL. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proceedings of the National Academy of Sciences of the United States of America 2001;98(19):10869–74. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Howell A Pure oestrogen antagonists for the treatment of advanced breast cancer. Endocrine-related cancer 2006;13(3):689–706. Epub 2006/09/07. doi: 10.1677/erc.1.00846. [DOI] [PubMed] [Google Scholar]
- 5.Peng J, Sengupta S, Jordan VC. Potential of selective estrogen receptor modulators as treatments and preventives of breast cancer. Anti-cancer agents in medicinal chemistry 2009;9(5):481–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fan P, Craig Jordan V. Acquired resistance to selective estrogen receptor modulators (SERMs) in clinical practice (tamoxifen & raloxifene) by selection pressure in breast cancer cell populations. Steroids 2014;90:44–52. Epub 2014/06/17. doi: 10.1016/j.steroids.2014.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nardone A, De Angelis C, Trivedi MV, Osborne CK, Schiff R. The changing role of ER in endocrine resistance. Breast (Edinburgh, Scotland) 2015;24 Suppl 2:S60–6. Epub 2015/08/15. doi: 10.1016/j.breast.2015.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Radhi S Molecular Changes During Breast Cancer and Mechanisms of Endocrine Therapy Resistance. Progress in molecular biology and translational science 2016;144:539–62. Epub 2016/11/21. doi: 10.1016/bs.pmbts.2016.09.009. [DOI] [PubMed] [Google Scholar]
- 9.Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, Davies S, Fauron C, He X, Hu Z, Quackenbush JF, Stijleman IJ, Palazzo J, Marron JS, Nobel AB, Mardis E, Nielsen TO, Ellis MJ, Perou CM, Bernard PS. Supervised risk predictor of breast cancer based on intrinsic subtypes. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2009;27(8):1160–7. doi: 10.1200/JCO.2008.18.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shang Y, Brown M. Molecular determinants for the tissue specificity of SERMs. Science 2002;295(5564):2465–8. doi: 10.1126/science.1068537. [DOI] [PubMed] [Google Scholar]
- 11.Halachmi S, Marden E, Martin G, MacKay H, Abbondanza C, Brown M. Estrogen receptor-associated proteins: possible mediators of hormone-induced transcription. Science 1994;264(5164):1455–8. [DOI] [PubMed] [Google Scholar]
- 12.Lemon BD, Freedman LP. Nuclear receptor cofactors as chromatin remodelers. Current opinion in genetics & development 1999;9(5):499–504. [DOI] [PubMed] [Google Scholar]
- 13.Glass CK, Rosenfeld MG. The coregulator exchange in transcriptional functions of nuclear receptors. Genes & development 2000;14(2):121–41. [PubMed] [Google Scholar]
- 14.Chen H, Tini M, Evans RM. HATs on and beyond chromatin. Current opinion in cell biology 2001;13(2):218–24. [DOI] [PubMed] [Google Scholar]
- 15.Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M. Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell 2000;103(6):843–52. [DOI] [PubMed] [Google Scholar]
- 16.Hurtado A, Holmes KA, Ross-Innes CS, Schmidt D, Carroll JS. FOXA1 is a key determinant of estrogen receptor function and endocrine response. Nat Genet 2011;43(1):27–33. doi: 10.1038/ng.730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Musgrove EA, Sutherland RL. Biological determinants of endocrine resistance in breast cancer. Nature reviews Cancer 2009;9(9):631–43. Epub 2009/08/25. doi: 10.1038/nrc2713. [DOI] [PubMed] [Google Scholar]
- 18.Jin K, Park S, Teo WW, Korangath P, Cho SS, Yoshida T, Gyorffy B, Goswami CP, Nakshatri H, Cruz LA, Zhou W, Ji H, Su Y, Ekram M, Wu Z, Zhu T, Polyak K, Sukumar S. HOXB7 Is an ERalpha Cofactor in the Activation of HER2 and Multiple ER Target Genes Leading to Endocrine Resistance. Cancer Discov 2015;5(9):944–59. doi: 10.1158/2159-8290.CD-15-0090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Caldon CE, Daly RJ, Sutherland RL, Musgrove EA. Cell cycle control in breast cancer cells. Journal of cellular biochemistry 2006;97(2):261–74. doi: 10.1002/jcb.20690. [DOI] [PubMed] [Google Scholar]
- 20.Butt AJ, McNeil CM, Musgrove EA, Sutherland RL. Downstream targets of growth factor and oestrogen signalling and endocrine resistance: the potential roles of c-Myc, cyclin D1 and cyclin E. Endocrine-related cancer 2005;12 Suppl 1:S47–59. doi: 10.1677/erc.1.00993. PubMed PMID: 1611309. [DOI] [PubMed] [Google Scholar]
- 21.Lundberg AS, Weinberg RA. Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Molecular and cellular biology 1998;18(2):753–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Verona R, Moberg K, Estes S, Starz M, Vernon JP, Lees JA. E2F activity is regulated by cell cycle-dependent changes in subcellular localization. Molecular and cellular biology 1997;17(12):7268–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Foster JS, Henley DC, Bukovsky A, Seth P, Wimalasena J. Multifaceted regulation of cell cycle progression by estrogen: regulation of Cdk inhibitors and Cdc25A independent of cyclin D1-Cdk4 function. Molecular and cellular biology 2001;21(3):794–810. doi: 10.1128/MCB.21.3.794-810.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Finn RS, Aleshin A, Slamon DJ. Targeting the cyclin-dependent kinases (CDK) 4/6 in estrogen receptor-positive breast cancers. Breast cancer research : BCR 2016;18(1):17. doi: 10.1186/s13058-015-0661-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Y, Dean JL, Millar EK, Tran TH, McNeil CM, Burd CJ, Henshall SM, Utama FE, Witkiewicz A, Rui H, Sutherland RL, Knudsen KE, Knudsen ES. Cyclin D1b is aberrantly regulated in response to therapeutic challenge and promotes resistance to estrogen antagonists. Cancer research 2008;68(14):5628–38. doi: 10.1158/0008-5472.CAN-07-3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dean JL, McClendon AK, Hickey TE, Butler LM, Tilley WD, Witkiewicz AK, Knudsen ES. Therapeutic response to CDK4/6 inhibition in breast cancer defined by ex vivo analyses of human tumors. Cell cycle (Georgetown, Tex) 2012;11(14):2756–61. Epub 2012/07/07. doi: 10.4161/cc.21195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jordan VC, O’Malley BW. Selective estrogen-receptor modulators and antihormonal resistance in breast cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2007;25(36):5815–24. doi: 10.1200/JCO.2007.11.3886. [DOI] [PubMed] [Google Scholar]
- 28.Ali S, Coombes RC. Endocrine-responsive breast cancer and strategies for combating resistance. Nature reviews Cancer 2002;2(2):101–12. doi: 10.1038/nrc721. [DOI] [PubMed] [Google Scholar]
- 29.Ring A, Dowsett M. Mechanisms of tamoxifen resistance. Endocrine-related cancer 2004;11(4):643–58. doi: 10.1677/erc.1.00776. [DOI] [PubMed] [Google Scholar]
- 30.Gururaj AE, Rayala SK, Vadlamudi RK, Kumar R. Novel mechanisms of resistance to endocrine therapy: genomic and nongenomic considerations. Clinical cancer research : an official journal of the American Association for Cancer Research 2006;12(3 Pt 2):1001s–7s. doi: 10.1158/1078-0432.CCR-05-2110. [DOI] [PubMed] [Google Scholar]
- 31.Cantley LC. The phosphoinositide 3-kinase pathway. Science 2002;296(5573):1655–7. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 32.Schiff R, Massarweh SA, Shou J, Bharwani L, Arpino G, Rimawi M, Osborne CK. Advanced concepts in estrogen receptor biology and breast cancer endocrine resistance: implicated role of growth factor signaling and estrogen receptor coregulators. Cancer chemotherapy and pharmacology 2005;56 Suppl 1:10–20. doi: 10.1007/s00280-005-0108-2. [DOI] [PubMed] [Google Scholar]
- 33.Bailey ST, Westerling T, Brown M. Loss of estrogen-regulated microRNA expression increases HER2 signaling and is prognostic of poor outcome in luminal breast cancer. Cancer research 2015;75(2):436–45. doi: 10.1158/0008-5472.CAN-14-1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Osborne CK, Schiff R. Mechanisms of endocrine resistance in breast cancer. Annual review of medicine 2011;62:233–47. Epub 2010/10/05. doi: 10.1146/annurev-med-070909-182917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Farabaugh SM, Boone DN, Lee AV. Role of IGF1R in Breast Cancer Subtypes, Stemness, and Lineage Differentiation. Frontiers in endocrinology 2015;6:59. doi: 10.3389/fendo.2015.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yerushalmi R, Gelmon KA, Leung S, Gao D, Cheang M, Pollak M, Turashvili G, Gilks BC, Kennecke H. Insulin-like growth factor receptor (IGF-1R) in breast cancer subtypes. Breast cancer research and treatment 2012;132(1):131–42. doi: 10.1007/s10549-011-1529-8. [DOI] [PubMed] [Google Scholar]
- 37.Rugo HS, Keck S. Reversing hormone resistance: have we found the golden key? Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2012;30(22):2707–9. doi: 10.1200/JCO.2012.42.1271. [DOI] [PubMed] [Google Scholar]
- 38.Faridi J, Wang L, Endemann G, Roth RA. Expression of constitutively active Akt-3 in MCF-7 breast cancer cells reverses the estrogen and tamoxifen responsivity of these cells in vivo. Clinical cancer research : an official journal of the American Association for Cancer Research 2003;9(8):2933–9. [PubMed] [Google Scholar]
- 39.DeGraffenried LA, Friedrichs WE, Fulcher L, Fernandes G, Silva JM, Peralba JM, Hidalgo M. Eicosapentaenoic acid restores tamoxifen sensitivity in breast cancer cells with high Akt activity. Annals of oncology : official journal of the European Society for Medical Oncology 2003;14(7):1051–6. [DOI] [PubMed] [Google Scholar]
- 40.deGraffenried LA, Friedrichs WE, Russell DH, Donzis EJ, Middleton AK, Silva JM, Roth RA, Hidalgo M. Inhibition of mTOR activity restores tamoxifen response in breast cancer cells with aberrant Akt Activity. Clinical cancer research : an official journal of the American Association for Cancer Research 2004;10(23):8059–67. doi: 10.1158/1078-0432.CCR-04-0035. [DOI] [PubMed] [Google Scholar]
- 41.Miller TW, Perez-Torres M, Narasanna A, Guix M, Stal O, Perez-Tenorio G, Gonzalez-Angulo AM, Hennessy BT, Mills GB, Kennedy JP, Lindsley CW, Arteaga CL. Loss of Phosphatase and Tensin homologue deleted on chromosome 10 engages ErbB3 and insulin-like growth factor-I receptor signaling to promote antiestrogen resistance in breast cancer. Cancer research 2009;69(10):4192–201. doi: 10.1158/0008-5472.CAN-09-0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McClelland RA, Barrow D, Madden TA, Dutkowski CM, Pamment J, Knowlden JM, Gee JM, Nicholson RI. Enhanced epidermal growth factor receptor signaling in MCF7 breast cancer cells after long-term culture in the presence of the pure antiestrogen ICI 182,780 (Faslodex). Endocrinology 2001;142(7):2776–88. doi: 10.1210/endo.142.7.8259. [DOI] [PubMed] [Google Scholar]
- 43.Knowlden JM, Hutcheson IR, Jones HE, Madden T, Gee JM, Harper ME, Barrow D, Wakeling AE, Nicholson RI. Elevated levels of epidermal growth factor receptor/cerbB2 heterodimers mediate an autocrine growth regulatory pathway in tamoxifen-resistant MCF-7 cells. Endocrinology 2003;144(3):1032–44. doi: 10.1210/en.2002-220620. [DOI] [PubMed] [Google Scholar]
- 44.Hutcheson IR, Knowlden JM, Madden TA, Barrow D, Gee JM, Wakeling AE, Nicholson RI. Oestrogen receptor-mediated modulation of the EGFR/MAPK pathway in tamoxifen-resistant MCF-7 cells. Breast cancer research and treatment 2003;81(1):81–93. doi: 10.1023/A:1025484908380. [DOI] [PubMed] [Google Scholar]
- 45.Johnston SR, Lu B, Scott GK, Kushner PJ, Smith IE, Dowsett M, Benz CC. Increased activator protein-1 DNA binding and c-Jun NH2-terminal kinase activity in human breast tumors with acquired tamoxifen resistance. Clinical cancer research : an official journal of the American Association for Cancer Research 1999;5(2):251–6. [PubMed] [Google Scholar]
- 46.Schiff R, Reddy P, Ahotupa M, Coronado-Heinsohn E, Grim M, Hilsenbeck SG, Lawrence R, Deneke S, Herrera R, Chamness GC, Fuqua SA, Brown PH, Osborne CK. Oxidative stress and AP-1 activity in tamoxifen-resistant breast tumors in vivo. Journal of the National Cancer Institute 2000;92(23):1926–34. [DOI] [PubMed] [Google Scholar]
- 47.Gutierrez MC, Detre S, Johnston S, Mohsin SK, Shou J, Allred DC, Schiff R, Osborne CK, Dowsett M. Molecular changes in tamoxifen-resistant breast cancer: relationship between estrogen receptor, HER-2, and p38 mitogen-activated protein kinase. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2005;23(11):2469–76. doi: 10.1200/JCO.2005.01.172. [DOI] [PubMed] [Google Scholar]
- 48.Zhou Y, Yau C, Gray JW, Chew K, Dairkee SH, Moore DH, Eppenberger U, Eppenberger-Castori S, Benz CC. Enhanced NF kappa B and AP-1 transcriptional activity associated with antiestrogen resistant breast cancer. BMC cancer 2007;7:59. doi: 10.1186/1471-2407-7-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ellis MJ, Lin L, Crowder R, Tao Y, Hoog J, Snider J, Davies S, DeSchryver K, Evans DB, Steinseifer J, Bandaru R, Liu W, Gardner H, Semiglazov V, Watson M, Hunt K, Olson J, Baselga J. Phosphatidyl-inositol-3-kinase alpha catalytic subunit mutation and response to neoadjuvant endocrine therapy for estrogen receptor positive breast cancer. Breast cancer research and treatment 2010;119(2):379–90. doi: 10.1007/s10549-009-0575-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, Neve RM, Kuo WL, Davies M, Carey M, Hu Z, Guan Y, Sahin A, Symmans WF, Pusztai L, Nolden LK, Horlings H, Berns K, Hung MC, van de Vijver MJ, Valero V, Gray JW, Bernards R, Mills GB, Hennessy BT. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer research 2008;68(15):6084–91. doi: 10.1158/0008-5472.CAN-07-6854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fu X, Creighton CJ, Biswal NC, Kumar V, Shea M, Herrera S, Contreras A, Gutierrez C, Wang T, Nanda S, Giuliano M, Morrison G, Nardone A, Karlin KL, Westbrook TF, Heiser LM, Anur P, Spellman P, Guichard SM, Smith PD, Davies BR, Klinowska T, Lee AV, Mills GB, Rimawi MF, Hilsenbeck SG, Gray JW, Joshi A, Osborne CK, Schiff R. Overcoming endocrine resistance due to reduced PTEN levels in estrogen receptor-positive breast cancer by co-targeting mammalian target of rapamycin, protein kinase B, or mitogen-activated protein kinase kinase. Breast cancer research : BCR 2014;16(5):430. doi: 10.1186/s13058-014-0430-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hoskins JM, Carey LA, McLeod HL. CYP2D6 and tamoxifen: DNA matters in breast cancer. Nature reviews Cancer 2009;9(8):576–86. doi: 10.1038/nrc2683. [DOI] [PubMed] [Google Scholar]
- 53.Veeraraghavan J, Tan Y, Cao XX, Kim JA, Wang X, Chamness GC, Maiti SN, Cooper LJ, Edwards DP, Contreras A, Hilsenbeck SG, Chang EC, Schiff R, Wang XS. Recurrent ESR1-CCDC170 rearrangements in an aggressive subset of oestrogen receptor-positive breast cancers. Nature communications 2014;5:4577. doi: 10.1038/ncomms5577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Balko JM, Giltnane JM, Wang K, Schwarz LJ, Young CD, Cook RS, Owens P, Sanders ME, Kuba MG, Sánchez V, Kurupi R, Moore PD, Pinto JA, Doimi FD, Gómez H, Horiuchi D, Goga A, Lehmann BD, Bauer JA, Pietenpol JA, Ross JS, Palmer GA, Yelensky R, Cronin M, Miller VA, Stephens PJ, Arteaga CL. Molecular profiling of the residual disease of triple-negative breast cancers after neoadjuvant chemotherapy identifies actionable therapeutic targets. Cancer Discov 2014;4(2):232–45. Epub 20131219. doi: 10.1158/2159-8290.CD-13-0286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Alluri PG, Speers C, Chinnaiyan AM. Estrogen receptor mutations and their role in breast cancer progression. Breast cancer research : BCR 2014;16(6):494. doi: 10.1186/s13058-014-0494-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Toy W, Shen Y, Won H, Green B, Sakr RA, Will M, Li Z, Gala K, Fanning S, King TA, Hudis C, Chen D, Taran T, Hortobagyi G, Greene G, Berger M, Baselga J, Chandarlapaty S. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nature genetics 2013;45(12):1439–45. doi: 10.1038/ng.2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jeselsohn R, Yelensky R, Buchwalter G, Frampton G, Meric-Bernstam F, Gonzalez-Angulo AM, Ferrer-Lozano J, Perez-Fidalgo JA, Cristofanilli M, Gomez H, Arteaga CL, Giltnane J, Balko JM, Cronin MT, Jarosz M, Sun J, Hawryluk M, Lipson D, Otto G, Ross JS, Dvir A, Soussan-Gutman L, Wolf I, Rubinek T, Gilmore L, Schnitt S, Come SE, Pusztai L, Stephens P, Brown M, Miller VA. Emergence of constitutively active estrogen receptor-alpha mutations in pretreated advanced estrogen receptor-positive breast cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 2014;20(7):1757–67. doi: 10.1158/1078-0432.CCR-13-2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li Z, Levine KM, Bahreini A, Wang P, Chu D, Park BH, Oesterreich S, Lee AV. Upregulation of IRS1 Enhances IGF1 Response in Y537S and D538G ESR1 Mutant Breast Cancer Cells. Endocrinology 2018;159(1):285–96. doi: 10.1210/en.2017-00693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wolf DM, Jordan VC. The estrogen receptor from a tamoxifen stimulated MCF-7 tumor variant contains a point mutation in the ligand binding domain. Breast cancer research and treatment 1994;31(1):129–38. [DOI] [PubMed] [Google Scholar]
- 60.Yuan J, Liu M, Yang L, Tu G, Zhu Q, Chen M, Cheng H, Luo H, Fu W, Li Z, Yang G. Acquisition of epithelial-mesenchymal transition phenotype in the tamoxifen-resistant breast cancer cell: a new role for G protein-coupled estrogen receptor in mediating tamoxifen resistance through cancer-associated fibroblast-derived fibronectin and beta1-integrin signaling pathway in tumor cells. Breast Cancer Res 2015;17(1):69. doi: 10.1186/s13058-015-0579-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pepermans RA, Sharma G, Prossnitz ER. G Protein-Coupled Estrogen Receptor in Cancer and Stromal Cells: Functions and Novel Therapeutic Perspectives. Cells 2021;10(3). doi: 10.3390/cells10030672. [DOI] [PMC free article] [PubMed]
- 62.Vivacqua A GPER1 and microRNA: Two Players in Breast Cancer Progression. Int J Mol Sci 2020;22(1). doi: 10.3390/ijms22010098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rouhimoghadam M, Lu AS, Salem AK, Filardo EJ. Therapeutic Perspectives on the Modulation of G-Protein Coupled Estrogen Receptor, GPER, Function. Front Endocrinol (Lausanne) 2020;11:591217. doi: 10.3389/fendo.2020.591217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hiscox S, Morgan L, Green TP, Barrow D, Gee J, Nicholson RI. Elevated Src activity promotes cellular invasion and motility in tamoxifen resistant breast cancer cells. Breast Cancer Res Treat 2006;97(3):263–74. doi: 10.1007/s10549-005-9120-9. [DOI] [PubMed] [Google Scholar]
- 65.Frame MC. Src in cancer: deregulation and consequences for cell behaviour. Biochim Biophys Acta 2002;1602(2):114–30. doi: 10.1016/s0304-419x(02)00040-9. [DOI] [PubMed] [Google Scholar]
- 66.Vallabhaneni S, Nair BC, Cortez V, Challa R, Chakravarty D, Tekmal RR, Vadlamudi RK. Significance of ER-Src axis in hormonal therapy resistance. Breast Cancer Res Treat 2011;130(2):377–85. doi: 10.1007/s10549-010-1312-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chantalat E, Boudou F, Laurell H, Palierne G, Houtman R, Melchers D, Rochaix P, Filleron T, Stella A, Burlet-Schiltz O, Brouchet A, Flouriot G, Metivier R, Arnal JF, Fontaine C, Lenfant F. The AF-1-deficient estrogen receptor ERalpha46 isoform is frequently expressed in human breast tumors. Breast Cancer Res 2016;18(1):123. doi: 10.1186/s13058-016-0780-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Klinge CM, Riggs KA, Wickramasinghe NS, Emberts CG, McConda DB, Barry PN, Magnusen JE. Estrogen receptor alpha 46 is reduced in tamoxifen resistant breast cancer cells and re-expression inhibits cell proliferation and estrogen receptor alpha 66-regulated target gene transcription. Mol Cell Endocrinol 2010;323(2):268–76. doi: 10.1016/j.mce.2010.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Penot G, Le Peron C, Merot Y, Grimaud-Fanouillere E, Ferriere F, Boujrad N, Kah O, Saligaut C, Ducouret B, Metivier R, Flouriot G. The human estrogen receptor-alpha isoform hERalpha46 antagonizes the proliferative influence of hERalpha66 in MCF7 breast cancer cells. Endocrinology 2005;146(12):5474–84. doi: 10.1210/en.2005-0866. [DOI] [PubMed] [Google Scholar]
- 70.D’Souza A, Spicer D, Lu J. Overcoming endocrine resistance in metastatic hormone receptor-positive breast cancer. Journal of Hematology & Oncology 2018;11(1):80. doi: 10.1186/s13045-018-0620-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhu L, Li XX, Shi L, Wu J, Qian JY, Xia TS, Zhou WB, Sun X, Zhou XJ, Wei JF, Ding Q. Rapamycin enhances the sensitivity of ERpositive breast cancer cells to tamoxifen by upregulating p73 expression. Oncology reports 2019;41(1):455–64. doi: 10.3892/or.2018.6842. [DOI] [PubMed] [Google Scholar]
- 72.A Study of Abemaciclib (LY2835219) Combined With Fulvestrant in Women With Hormone Receptor Positive HER2 Negative Breast Cancer Available from: https://ClinicalTrials.gov/show/NCT02107703.
- 73.Study of Efficacy and Safety of LEE011 in Postmenopausal Women With Advanced Breast Cancer.(MONALEESA-2)
- 74.Tripathy D, Bardia A, Blum J, Rocque G, Wilks S, Lakhanpal S, Migas J, Cappelleri J, Perkins J, Comstock G, Wang Y. Abstract OT3–05-03: POLARIS: Palbociclib (P) in hormone receptor-positive (HR+) advanced breast cancer: A prospective multicenter noninterventional study. Cancer research 2018;78(4 Supplement):OT3–05-3-OT3−-3. doi: 10.1158/1538-7445.sabcs17-ot3-05-03. [DOI] [Google Scholar]
- 75.Finn RS, Martin M, Rugo HS, Jones S, Im SA, Gelmon K, Harbeck N, Lipatov ON, Walshe JM, Moulder S, Gauthier E, Lu DR, Randolph S, Dieras V, Slamon DJ. Palbociclib and Letrozole in Advanced Breast Cancer. N Engl J Med 2016;375(20):1925–36. doi: 10.1056/NEJMoa1607303. [DOI] [PubMed] [Google Scholar]
- 76.Rugo HS, Finn RS, Dieras V, Ettl J, Lipatov O, Joy AA, Harbeck N, Castrellon A, Iyer S, Lu DR, Mori A, Gauthier ER, Bartlett CH, Gelmon KA, Slamon DJ. Palbociclib plus letrozole as first-line therapy in estrogen receptor-positive/human epidermal growth factor receptor 2-negative advanced breast cancer with extended follow-up. Breast cancer research and treatment 2019;174(3):719–29. doi: 10.1007/s10549-018-05125-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Turner NC, Slamon DJ, Ro J, Bondarenko I, Im SA, Masuda N, Colleoni M, DeMichele A, Loi S, Verma S, Iwata H, Harbeck N, Loibl S, Andre F, Puyana Theall K, Huang X, Giorgetti C, Huang Bartlett C, Cristofanilli M. Overall Survival with Palbociclib and Fulvestrant in Advanced Breast Cancer. N Engl J Med 2018;379(20):1926–36. doi: 10.1056/NEJMoa1810527. [DOI] [PubMed] [Google Scholar]
- 78.Canino F, Piacentini F, Omarini C, Toss A, Barbolini M, Vici P, Dominici M, Moscetti L. Role of Intrinsic Subtype Analysis with PAM50 in Hormone Receptors Positive HER2 Negative Metastatic Breast Cancer: A Systematic Review. Int J Mol Sci 2022;23(13). doi: 10.3390/ijms23137079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Angelo Di Leo GJ, Lubos Petruzelka, Roberto Torres, Igor N, Rustem Khasanov. Results of the CONFIRM Phase III trial Comparing Fulvestrant 250 mg with Fulvestrant 500 mg in Postmenopausal Women with Estrogen Receptor-Positive Advanced Breast Cancer Journal of Clinical Oncology 2010;28(30):4594–600. Epub September 20, 2010. doi: 10.1200/JCO.2010.28.8415. [DOI] [PubMed] [Google Scholar]
- 80.Hui L, Chen Y. Tumor microenvironment: Sanctuary of the devil. Cancer letters 2015;368(1):7–13. doi: 10.1016/j.canlet.2015.07.039. [DOI] [PubMed] [Google Scholar]
- 81.Lu P, Weaver VM, Werb Z. The extracellular matrix: a dynamic niche in cancer progression. The Journal of cell biology 2012;196(4):395–406. doi: 10.1083/jcb.201102147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Balkwill FR, Capasso M, Hagemann T. The tumor microenvironment at a glance. Journal of cell science 2012;125(Pt 23):5591–6. doi: 10.1242/jcs.116392. [DOI] [PubMed] [Google Scholar]
- 83.Konopleva MY, Jordan CT. Leukemia stem cells and microenvironment: biology and therapeutic targeting. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2011;29(5):591–9. doi: 10.1200/JCO.2010.31.0904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Klemm F, Joyce JA. Microenvironmental regulation of therapeutic response in cancer. Trends in cell biology 2015;25(4):198–213. doi: 10.1016/j.tcb.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dvorak HF, Harvey VS, Estrella P, Brown LF, McDonagh J, Dvorak AM. Fibrin containing gels induce angiogenesis. Implications for tumor stroma generation and wound healing. Laboratory investigation; a journal of technical methods and pathology 1987;57(6):673–86. [PubMed] [Google Scholar]
- 86.Bussard KM, Mutkus L, Stumpf K, Gomez-Manzano C, Marini FC. Tumor-associated stromal cells as key contributors to the tumor microenvironment. Breast cancer research : BCR 2016;18(1):84. doi: 10.1186/s13058-016-0740-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kojima Y, Acar A, Eaton EN, Mellody KT, Scheel C, Ben-Porath I, Onder TT, Wang ZC, Richardson AL, Weinberg RA, Orimo A. Autocrine TGF-beta and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proceedings of the National Academy of Sciences of the United States of America 2010;107(46):20009–14. doi: 10.1073/pnas.1013805107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Spaeth EL, Dembinski JL, Sasser AK, Watson K, Klopp A, Hall B, Andreeff M, Marini F. Mesenchymal stem cell transition to tumor-associated fibroblasts contributes to fibrovascular network expansion and tumor progression. PloS one 2009;4(4):e4992. doi: 10.1371/journal.pone.0004992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Medrek C, Ponten F, Jirstrom K, Leandersson K. The presence of tumor associated macrophages in tumor stroma as a prognostic marker for breast cancer patients. BMC cancer 2012;12:306. doi: 10.1186/1471-2407-12-306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Smith HA, Kang Y. The metastasis-promoting roles of tumor-associated immune cells. Journal of molecular medicine 2013;91(4):411–29. doi: 10.1007/s00109-013-1021-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Xiong Y, McDonald LT, Russell DL, Kelly RR, Wilson KR, Mehrotra M, Soloff AC, LaRue AC. Hematopoietic stem cell-derived adipocytes and fibroblasts in the tumor microenvironment. World journal of stem cells 2015;7(2):253–65. doi: 10.4252/wjsc.v7.i2.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ohlund D, Elyada E, Tuveson D. Fibroblast heterogeneity in the cancer wound. The Journal of experimental medicine 2014;211(8):1503–23. doi: 10.1084/jem.20140692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cirri P, Chiarugi P. Cancer associated fibroblasts: the dark side of the coin. American journal of cancer research 2011;1(4):482–97. [PMC free article] [PubMed] [Google Scholar]
- 94.Mao Y, Keller ET, Garfield DH, Shen K, Wang J. Stromal cells in tumor microenvironment and breast cancer. Cancer metastasis reviews 2013;32(1–2):303–15. doi: 10.1007/s10555-012-9415-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Acharyya S, Oskarsson T, Vanharanta S, Malladi S, Kim J, Morris PG, Manova-Todorova K, Leversha M, Hogg N, Seshan VE, Norton L, Brogi E, Massague J. A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell 2012;150(1):165–78. doi: 10.1016/j.cell.2012.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dirat B, Bochet L, Dabek M, Daviaud D, Dauvillier S, Majed B, Wang YY, Meulle A, Salles B, Le Gonidec S, Garrido I, Escourrou G, Valet P, Muller C. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer research 2011;71(7):2455–65. doi: 10.1158/0008-5472.CAN-10-3323. [DOI] [PubMed] [Google Scholar]
- 97.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell 2010;140(6):883–99. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wang YC, He F, Feng F, Liu XW, Dong GY, Qin HY, Hu XB, Zheng MH, Liang L, Feng L, Liang YM, Han H. Notch signaling determines the M1 versus M2 polarization of macrophages in antitumor immune responses. Cancer research 2010;70(12):4840–9. doi: 10.1158/0008-5472.CAN-10-0269. [DOI] [PubMed] [Google Scholar]
- 99.Ruffell B, Coussens LM. Macrophages and therapeutic resistance in cancer. Cancer cell 2015;27(4):462–72. doi: 10.1016/j.ccell.2015.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bruno A, Ferlazzo G, Albini A, Noonan DM. A think tank of TINK/TANKs: tumor-infiltrating/tumor-associated natural killer cells in tumor progression and angiogenesis. Journal of the National Cancer Institute 2014;106(8):dju200. doi: 10.1093/jnci/dju200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, Zhu Y, Wei S, Kryczek I, Daniel B, Gordon A, Myers L, Lackner A, Disis ML, Knutson KL, Chen L, Zou W. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nature medicine 2004;10(9):942–9. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 102.Nwabo Kamdje AH, Seke Etet PF, Vecchio L, Muller JM, Krampera M, Lukong KE. Signaling pathways in breast cancer: therapeutic targeting of the microenvironment. Cellular signalling 2014;26(12):2843–56. doi: 10.1016/j.cellsig.2014.07.034. [DOI] [PubMed] [Google Scholar]
- 103.Khin SS, Kitazawa R, Kondo T, Idei Y, Fujimoto M, Haraguchi R, Mori K, Kitazawa S. Epigenetic Alteration by DNA Promoter Hypermethylation of Genes Related to Transforming Growth Factor-beta (TGF-beta) Signaling in Cancer. Cancers 2011;3(1):982–93. doi: 10.3390/cancers3010982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Martinez-Outschoorn UE, Lin Z, Ko YH, Goldberg AF, Flomenberg N, Wang C, Pavlides S, Pestell RG, Howell A, Sotgia F, Lisanti MP. Understanding the metabolic basis of drug resistance: therapeutic induction of the Warburg effect kills cancer cells. Cell cycle (Georgetown, Tex) 2011;10(15):2521–8. Epub 2011/07/20. doi: 10.4161/cc.10.15.16584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ali HR, Glont SE, Blows FM, Provenzano E, Dawson SJ, Liu B, Hiller L, Dunn J, Poole CJ, Bowden S, Earl HM, Pharoah PD, Caldas C. PD-L1 protein expression in breast cancer is rare, enriched in basal-like tumours and associated with infiltrating lymphocytes. Ann Oncol 2015;26(7):1488–93. Epub 20150420. doi: 10.1093/annonc/mdv192. [DOI] [PubMed] [Google Scholar]
- 106.Osuala KO, Sameni M, Shah S, Aggarwal N, Simonait ML, Franco OE, Hong Y, Hayward SW, Behbod F, Mattingly RR, Sloane BF. Il-6 signaling between ductal carcinoma in situ cells and carcinoma-associated fibroblasts mediates tumor cell growth and migration. BMC cancer 2015;15:584. doi: 10.1186/s12885-015-1576-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Duluc C, Moatassim-Billah S, Chalabi-Dchar M, Perraud A, Samain R, Breibach F, Gayral M, Cordelier P, Delisle MB, Bousquet-Dubouch MP, Tomasini R, Schmid H, Mathonnet M, Pyronnet S, Martineau Y, Bousquet C. Pharmacological targeting of the protein synthesis mTOR/4E-BP1 pathway in cancer-associated fibroblasts abrogates pancreatic tumour chemoresistance. EMBO molecular medicine 2015;7(6):735–53. doi: 10.15252/emmm.201404346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lee KW, Yeo SY, Sung CO, Kim SH. Twist1 is a key regulator of cancer-associated fibroblasts. Cancer research 2015;75(1):73–85. doi: 10.1158/0008-5472.CAN-14-0350. [DOI] [PubMed] [Google Scholar]
- 109.Fang WB, Mafuvadze B, Yao M, Zou A, Portsche M, Cheng N. TGF-beta Negatively Regulates CXCL1 Chemokine Expression in Mammary Fibroblasts through Enhancement of Smad2/3 and Suppression of HGF/c-Met Signaling Mechanisms. PloS one 2015;10(8):e0135063. doi: 10.1371/journal.pone.0135063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Erez N, Truitt M, Olson P, Arron ST, Hanahan D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-kappaB-Dependent Manner. Cancer cell 2010;17(2):135–47. doi: 10.1016/j.ccr.2009.12.041. [DOI] [PubMed] [Google Scholar]
- 111.Xiang T, Long H, He L, Han X, Lin K, Liang Z, Zhuo W, Xie R, Zhu B. Interleukin-17 produced by tumor microenvironment promotes self-renewal of CD133+ cancer stem-like cells in ovarian cancer. Oncogene 2015;34(2):165–76. doi: 10.1038/onc.2013.537. [DOI] [PubMed] [Google Scholar]
- 112.Taguchi A, Kawana K, Tomio K, Yamashita A, Isobe Y, Nagasaka K, Koga K, Inoue T, Nishida H, Kojima S, Adachi K, Matsumoto Y, Arimoto T, Wada-Hiraike O, Oda K, Kang JX, Arai H, Arita M, Osuga Y, Fujii T. Matrix metalloproteinase (MMP)-9 in cancer-associated fibroblasts (CAFs) is suppressed by omega-3 polyunsaturated fatty acids in vitro and in vivo. PloS one 2014;9(2):e89605. doi: 10.1371/journal.pone.0089605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Mor A, Tankiewicz-Kwedlo A, Pawlak D. Kynurenines as a Novel Target for the Treatment of Malignancies. Pharmaceuticals (Basel) 2021;14(7). doi: 10.3390/ph14070606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ye Z, Yue L, Shi J, Shao M, Wu T. Role of IDO and TDO in Cancers and Related Diseases and the Therapeutic Implications. J Cancer 2019;10(12):2771–82. doi: 10.7150/jca.31727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Greene LI, Bruno TC, Christenson JL, D’Alessandro A, Culp-Hill R, Torkko K, Borges VF, Slansky JE, Richer JK. A Role for Tryptophan-2,3-dioxygenase in CD8 T-cell Suppression and Evidence of Tryptophan Catabolism in Breast Cancer Patient Plasma. Mol Cancer Res 2019;17(1):131–9. doi: 10.1158/1541-7786.MCR-18-0362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Svensson S, Abrahamsson A, Rodriguez GV, Olsson AK, Jensen L, Cao Y, Dabrosin C. CCL2 and CCL5 Are Novel Therapeutic Targets for Estrogen-Dependent Breast Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 2015;21(16):3794–805. doi: 10.1158/1078-0432.CCR-15-0204. [DOI] [PubMed] [Google Scholar]
- 117.Li D, Ji H, Niu X, Yin L, Wang Y, Gu Y, Wang J, Zhou X, Zhang H, Zhang Q. Tumor-associated macrophages secrete CC-chemokine ligand 2 and induce tamoxifen resistance by activating PI3K/Akt/mTOR in breast cancer. Cancer Sci 2020;111(1):47–58. doi: 10.1111/cas.14230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hu H, Sun J, Wang C, Bu X, Liu X, Mao Y, Wang H. IL-33 facilitates endocrine resistance of breast cancer by inducing cancer stem cell properties. Biochem Biophys Res Commun 2017;485(3):643–50. doi: 10.1016/j.bbrc.2017.02.080. [DOI] [PubMed] [Google Scholar]
- 119.Ma Y, Ren Y, Dai ZJ, Wu CJ, Ji YH, Xu J. IL-6, IL-8 and TNF-alpha levels correlate with disease stage in breast cancer patients. Adv Clin Exp Med 2017;26(3):421–6. doi: 10.17219/acem/62120. [DOI] [PubMed] [Google Scholar]
- 120.Kim S, Jeon M, Lee JE, Nam SJ. MEK activity controls IL-8 expression in tamoxifen-resistant MCF-7 breast cancer cells. Oncology reports 2016;35(4):2398–404. doi: 10.3892/or.2016.4557. [DOI] [PubMed] [Google Scholar]
- 121.Lee JY, Park YJ, Oh N, Kwack KB, Park KS. A transcriptional complex composed of ER(alpha), GATA3, FOXA1 and ELL3 regulates IL-20 expression in breast cancer cells. Oncotarget 2017;8(26):42752–60. doi: 10.18632/oncotarget.17459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Salazar N, Munoz D, Kallifatidis G, Singh RK, Jorda M, Lokeshwar BL. The chemokine receptor CXCR7 interacts with EGFR to promote breast cancer cell proliferation. Mol Cancer 2014;13:198. doi: 10.1186/1476-4598-13-198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Chatterjee S, Bhat V, Berdnikov A, Liu J, Zhang G, Buchel E, Safneck J, Marshall AJ, Murphy LC, Postovit LM, Raouf A. Paracrine Crosstalk between Fibroblasts and ER(+) Breast Cancer Cells Creates an IL1beta-Enriched Niche that Promotes Tumor Growth. iScience 2019;19:388–401. doi: 10.1016/j.isci.2019.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kim S, You D, Jeong Y, Yoon SY, Kim SA, Lee JE. Inhibition of platelet-derived growth factor receptor synergistically increases the pharmacological effect of tamoxifen in estrogen receptor alpha positive breast cancer. Oncol Lett 2021;21(4):294. doi: 10.3892/ol.2021.12555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Gangaraju VK, Lin H. MicroRNAs: key regulators of stem cells. Nature reviews Molecular cell biology 2009;10(2):116–25. doi: 10.1038/nrm2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kadera BE, Li L, Toste PA, Wu N, Adams C, Dawson DW, Donahue TR. MicroRNA-21 in pancreatic ductal adenocarcinoma tumor-associated fibroblasts promotes metastasis. PloS one 2013;8(8):e71978. doi: 10.1371/journal.pone.0071978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Miller PC, Clarke J, Koru-Sengul T, Brinkman J, El-Ashry D. A novel MAPK-microRNA signature is predictive of hormone-therapy resistance and poor outcome in ER-positive breast cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 2015;21(2):373–85. doi: 10.1158/1078-0432.CCR-14-2053. [DOI] [PubMed] [Google Scholar]
- 128.Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, Leiserson MDM, Miller CA, Welch JS, Walter MJ, Wendl MC, Ley TJ, Wilson RK, Raphael BJ, Ding L. Mutational landscape and significance across 12 major cancer types. Nature 2013;502(7471):333–9. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Melo SA, Sugimoto H, O’Connell JT, Kato N, Villanueva A, Vidal A, Qiu L, Vitkin E, Perelman LT, Melo CA, Lucci A, Ivan C, Calin GA, Kalluri R. Cancer exosomes perform cell-independent microRNA biogenesis and promote tumorigenesis. Cancer cell 2014;26(5):707–21. doi: 10.1016/j.ccell.2014.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Martins D, Schmitt F. Microenvironment in breast tumorigenesis: Friend or foe? Histology and histopathology 2018:18021. doi: 10.14670/HH-18-021. [DOI] [PubMed]
- 131.Creighton CJ, Bromberg-White JL, Misek DE, Monsma DJ, Brichory F, Kuick R, Giordano TJ, Gao W, Omenn GS, Webb CP, Hanash SM. Analysis of tumor-host interactions by gene expression profiling of lung adenocarcinoma xenografts identifies genes involved in tumor formation. Molecular cancer research : MCR 2005;3(3):119–29. doi: 10.1158/1541-7786.MCR-04-0189. [DOI] [PubMed] [Google Scholar]
- 132.Vargas AC, McCart Reed AE, Waddell N, Lane A, Reid LE, Smart CE, Cocciardi S, da Silva L, Song S, Chenevix-Trench G, Simpson PT, Lakhani SR. Gene expression profiling of tumour epithelial and stromal compartments during breast cancer progression. Breast cancer research and treatment 2012;135(1):153–65. Epub 2012/06/22. doi: 10.1007/s10549-012-2123-4. [DOI] [PubMed] [Google Scholar]
- 133.Ronnov-Jessen L, Petersen OW, Bissell MJ. Cellular changes involved in conversion of normal to malignant breast: importance of the stromal reaction. Physiological reviews 1996;76(1):69–125. doi: 10.1152/physrev.1996.76.1.69. [DOI] [PubMed] [Google Scholar]
- 134.Bergamaschi A, Tagliabue E, Sorlie T, Naume B, Triulzi T, Orlandi R, Russnes HG, Nesland JM, Tammi R, Auvinen P, Kosma VM, Menard S, Borresen-Dale AL. Extracellular matrix signature identifies breast cancer subgroups with different clinical outcome. The Journal of pathology 2008;214(3):357–67. doi: 10.1002/path.2278. [DOI] [PubMed] [Google Scholar]
- 135.Crane CA, Panner A, Murray JC, Wilson SP, Xu H, Chen L, Simko JP, Waldman FM, Pieper RO, Parsa AT. PI(3) kinase is associated with a mechanism of immunoresistance in breast and prostate cancer. Oncogene 2009;28(2):306–12. Epub 20081013. doi: 10.1038/onc.2008.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Boelens MC, Wu TJ, Nabet BY, Xu B, Qiu Y, Yoon T, Azzam DJ, Twyman-Saint Victor C, Wiemann BZ, Ishwaran H, Ter Brugge PJ, Jonkers J, Slingerland J, Minn AJ. Exosome transfer from stromal to breast cancer cells regulates therapy resistance pathways. Cell 2014;159(3):499–513. doi: 10.1016/j.cell.2014.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Polonia A, Pinto R, Cameselle-Teijeiro JF, Schmitt FC, Paredes J. Prognostic value of stromal tumour infiltrating lymphocytes and programmed cell death-ligand 1 expression in breast cancer. Journal of clinical pathology 2017;70(10):860–7. doi: 10.1136/jclinpath-2016-203990. [DOI] [PubMed] [Google Scholar]