

Abstract
Objectives:
Families that contain multiple siblings affected with childhood-onset of systemic lupus erythematosus (SLE) likely have strong genetic predispositions. We performed whole-exome sequencing (WES) to identify familial rare risk variants and to assess their effects in lupus.
Methods:
Sanger sequencing validated the two ultra-rare, predicted pathogenic risk variants discovered by WES and identified additional variants in 562 additional SLE patients. Effects of a splice site variant and a frameshift variant were assessed using a Minigene assay and CRISPR/Cas9-mediated knock-in (KI) mice, respectively.
Results:
The two familial ultra-rare, predicted loss-of-function (LOF) SAT1 variants exhibited X-linked recessive Mendelian inheritance in two unrelated African American families. Each LOF variant was transmitted from the heterozygous unaffected mother to her two sons with childhood-onset SLE. The p.Asp40Tyr variant affected a splice donor site causing deleterious transcripts. The young hemizygous male and homozygous female Sat1p.Glu92Leufs*6 KI mice spontaneously developed splenomegaly, enlarged glomeruli with leukocyte infiltration, proteinuria and elevated expression of type I interferon inducible genes. SAT1 is highly expressed in neutrophils and encodes spermidine/spermine-N1-acetyltransferase 1 (SSAT1), a rate-limiting enzyme in polyamine catabolism. Young male KI mice exhibited neutrophil defects and decreased proportions of Foxp3+CD4+ T-cell subsets. Circulating neutrophil counts and proportions of Foxp3+CD4+ T cells correlated with decreased plasma levels of spermine in treatment naïve, incipient SLE patients.
Conclusions:
We identified two novel SAT1 loss-of-function variants, showed the ability of the frameshift variant to confer murine lupus, highlighted the pathogenic role of dysregulated polyamine catabolism, and identified SAT1 LOF variants as new monogenic causes for SLE.
Introduction
Systemic lupus erythematosus (SLE or lupus) is a prototypic autoimmune disease with a multifactorial etiology contributed to by genetic, epigenetic, and environmental factors. SLE has a strong genetic component with more than 150 risk loci identified in genome-wide association studies (GWAS), and approximately 30 loci are associated with rare monogenic forms of lupus or lupus-like disease1,2. SLE is generally considered a polygenic trait contributed to by a large number of small-effect, non-coding, common GWAS-defined variants3,4. In a small proportion of childhood-onset SLE patients, Mendelian forms of disease can develop caused by rare, damaging variants, mainly in innate immunity, including deficiency of early complement components (C1Q, C1R/S, C2, C4A and C4B), type I interferon (IFN-I) signaling, and nucleic acid sensing and degradation5–7. It appears that GWAS-defined common variants and rare monogenic causes of illness often disrupt the same biological processes that lead to disease. The highly penetrant rare cases of monogenic lupus continue to provide new insights into lupus pathogenesis and potential treatment targets.
In this study, we identified two rare SAT1 loss-of-function variants on the X chromosome using WES that segregate with SLE phenotype in two unrelated families. SAT1 encodes the spermidine/spermine-N1-acetyltransferase (SSAT1), a rate-limiting enzyme that regulates the catabolism of polyamine and maintains cellular polyamine homeostasis. Dysregulated polyamine metabolism was previously described in SLE patients8. Here we present evidence for a causal role between LOF SAT1 variants in the pathogenesis of SLE.
Results
Patient history and genetic analysis.
We performed WES in two unrelated American African families that each had unaffected parents and two sons diagnosed with childhood-onset SLE (figure 1A, B, and Table 1). After filtering, we identified two rare predicted pathogenic X-linked SAT1 variants (c.118G>T, p.Asp40Tyr in family #1 and c.272_273dup, p.Glu92Leufs*6 in family #2. Reference Sequence transcript: NM_002970) (online supplemental figure 1A, B) were confirmed by Sanger sequencing. The unaffected heterozygous mothers passed the putative SAT1 LOF variant to the two hemizygous sons affected with SLE in each family and the wild type SAT1 allele to one unaffected son in family #2. These variants thus exhibit a X-linked recessive Mendelian inheritance pattern in which each familial SAT1 LOF variant co-segregated with the SLE-disease status of the affected sibling (figure 1A, B). The p.Asp40Tyr variant in exon 2, predicted to be deleterious by altering a splice donor site (figure 1C and online supplemental figure 2B), was confirmed using the minigene assay. Compared with a single normally spliced transcript generated from transiently transfected Asp40-containing minigene construct in 293T or HeLa cell lines, Tyr40-containing constructs yielded two aberrantly spliced transcripts (~30% exon 2 skipped, 30% intron 2 retention, and 40% normally spliced Tyr40-containing transcripts) (figure 1D, E and online supplemental figure 3A, B). The exon 2 skipped transcript resulted in premature termination, the intron 2 retention transcript was modeled to impair SSAT1 functions due to extra protein domains, and the normally spliced Tyr40-containing transcript was predicted to be deleterious (online supplemental figure 2B and figure 3C). The frameshift variant (p.Glu92Leufs*6) of SAT1 is predicted to trigger nonsense-mediated mRNA decay (figure 1C). Both SAT1 variants alter highly conserved residues (online supplemental figure 2A), are not previously known monogenic causes or GWAS-defined SLE-risk loci7,9, and are extremely rare in reported populations (absent in the gnomAD10, TOPMed11 and 1000 Genomes12 databases).
Figure 1. Identification of p.Asp40Tyr and p.Glu92Leufs*6 SAT1 variants segregating with disease status in each family.
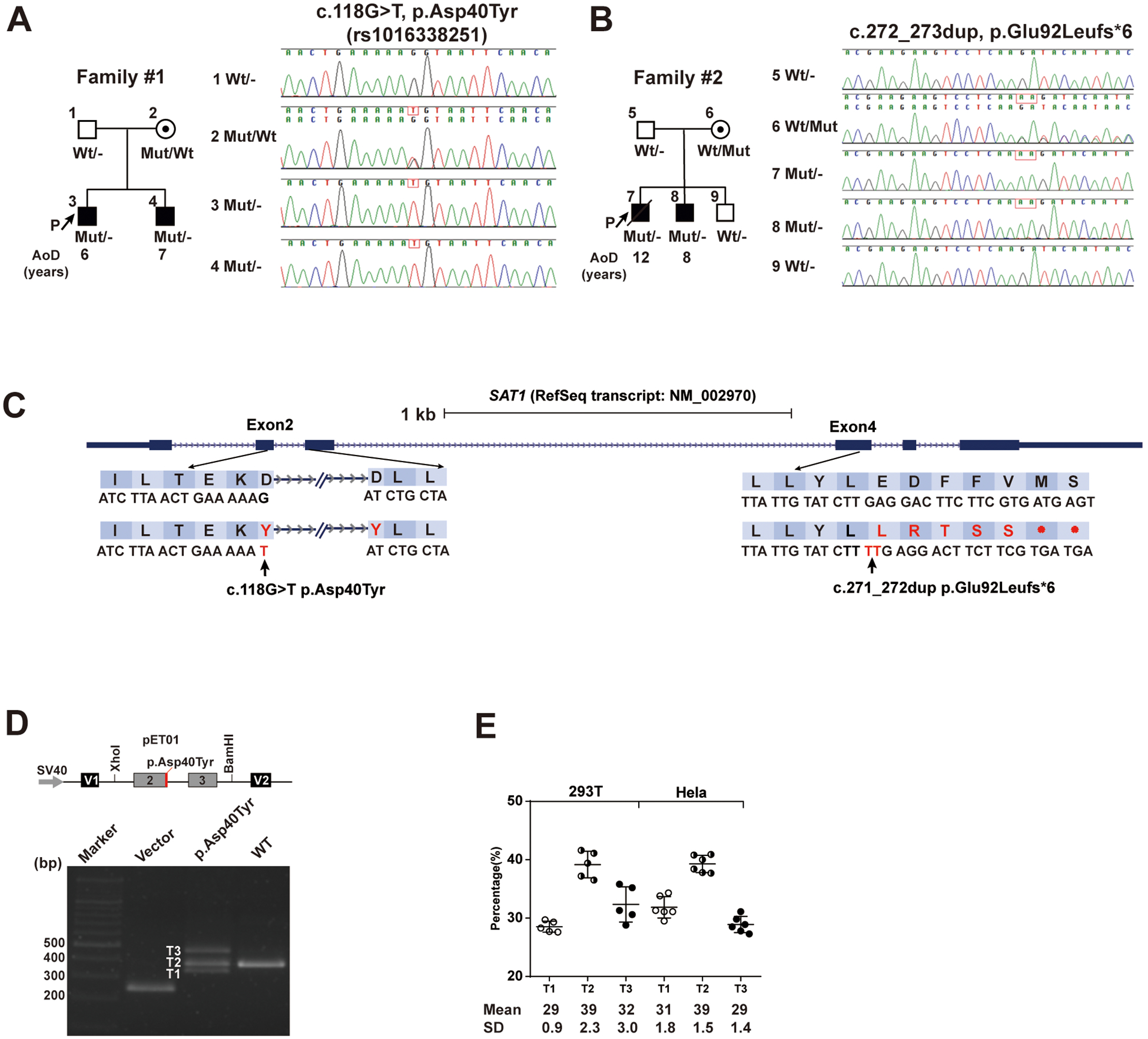
(A and B) Pedigree information of two unrelated African-American families containing SLE-affected sibpairs and Sanger sequencing data that confirmed Mendelian inheritance of either p.Glu92Leufs*6 or p.Asp40Tyr variant of X-linked SAT1 in each family. AoD, age of diagnosis. P, proband. p.Asp40Tyr is identified as rs1016338251 by Human Longevity company, but no annotation of any individual is available.
(C) Locations of the p.Asp40Tyr (NM_002970: c.118G>T, ChrX(GRCh38):g.23783709G>T) and p.Glu92Leufs*6 (NM_002970: c.272_273dup, ChrX(GRCh38):g.23785397–23785398dup) SAT1 variants and the corresponding sequences based on human reference genome build GRCh38/hg38.
(D and E) The genomic segment containing p.Asp40Tyr cloned into the Minigene assay vector resulted in aberrantly spliced transcripts in transfected 293T and HeLa cell lines. A schematic of the p.Asp40Tyr minigene plasmid and electrophoresed RT-PCR products from 293T cells transfected with a minigene plasmid with either Asp40- or Tyr40- containing genomic segment (D). The percentage of each spliced transcript from three independent transfection experiments is depicted. Data are mean ± SD (E).
Table 1.
Summary of clinical features in affected males from two families with SAT1 mutations.
| Patient (gender) | Family (ethnic origin) | Age of onset | Brief clinical symptoms | Renal disorder | Immunological disorder |
|---|---|---|---|---|---|
| 3 (M) | Family #1 (AA) | 6 y | Malar rash, Nonerosive arthritis, Serositis, Pleuritis and Pericarditis | Cellular casts | ANA, anti-dsDNA, anti-Smith, anti-CL, anti-Ro, anti-RNP; Low C3, C4 |
| 4 (M) | Family #1 (AA) | 7 y | Malar rash, Photosensitivity, Leukopenia, Lymphopenia | Proteinuria | ANA, anti-dsDNA, anti-Smith, Anti-CL, anti-Ro, anti-RNP; Low C3 |
| 7 (M) | Family #2 (AA) | 12 y | Nonerosive arthritis, Anemia, Fatigue | Proteinuria; Renal biopsy: membranous proliferative GN (Class V) | ANA, anti-dsDNA; Low C3, C4 |
| 14 y | Renal failure | ||||
| 8 (M) | Family #2 (AA) | 8 y | CNS lupus, Nonerosive arthritis | Proteinuria; Renal biopsy: focal proliferative GN (Class III) | Anti-dsDNA; ANA > 1:1280 |
| 14 y | Vasculitis affecting the eyes, 2/2 | Diffuse lupus glomerulonephritis with crescents | Anti-dsDNA, anti-Smith, anti-RNP, anti-β2GPI and anti-CL |
Abbreviation: M: male; AA: African American; y: Years old; GN: Glomerulonephritis; anti-dsDNA: anti-double stranded DNA; ANA: Anti-Nuclear Antibody; anti-CL: anti-cardiolipin; anti-RNP: anti- antinuclear ribonucleoprotein; anti-β2GPI: anti-β2 glycoprotein I; CNS: central nervous system.
Figure 3. Decreased levels and defective functions of bone marrow (BM)-isolated neutrophils from young male Sat1p.Glu92Leufs*6 mice.
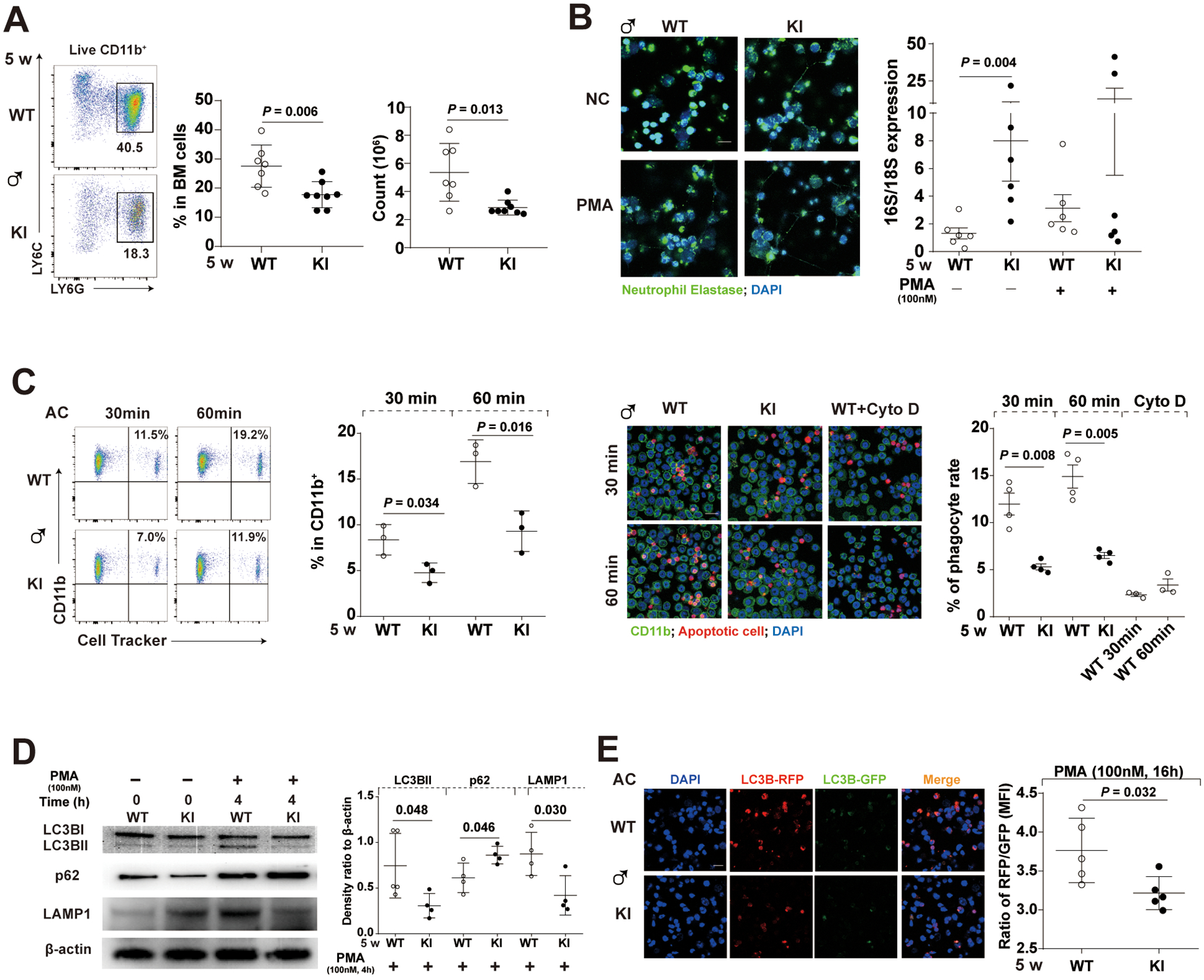
(A) Gating strategy, decreased percentage, and cell numbers of BM-isolated neutrophils (CD11b+LY6G+LY6Cint) in 5-week-old male KI mice compared to their WT littermates. Data are mean ± SD. Mann-Whiney U test.
(B) (Left) Representative images of the Neutrophil extracellular traps (NET) induced by PMA in BM-isolated neutrophils. (Right) Quantitation of mitochondrial (16S; officially known as MT-RNR2) and chromosomal (18S; officially known as RNA18S5) DNA in the immuno-precipitated total oxidized DNA from overnight culture supernatants of BM-isolated neutrophils of either WT or KI littermates incubated in the absence (spontaneous NETosis) or presence of PMA (induced NETosis). Green immunofluorescence represents neutrophil elastase and blue represents DNA (Hoechst_33342) of confocal images. Bar: 10μm; PMA, phorbol myristate acetate, 100nM; Incubation time, 24 hours. Data are mean ± SD. Mann-Whiney U test.
(C) Defective engulfment of Cell Tracker-labeled apoptotic cells (AC) by BM-isolated neutrophils from 5-week-old male KI mice after 30- or 60- min co-cultures assessed by either flow cytometry or confocal microscopy; BM-isolated neutrophils: AC=1:5. Cyto D, Cytochalasin D, an inhibitor of actin polymerization, at 10μM; Bar: 10μm. Data are mean ± SD. Unpaired t-test.
(D) Representative Western blot of LC3B, p62 (an autophagosome cargo protein), LAMP1 and β-actin, and quantification of relative levels of LC3B-II to LC3B-I, and relative levels of p62 or LAMP1 to β-actin in PMA-stimulated groups. PMA, 100nM. Bar: 10μm. Incubation time, 4 hours. Data are mean ± SD. Unpaired t-test.
(E) Decreased levels of autophagic flux in PMA-treated BM-isolated neutrophils from 5-week-old male KI mice. The left panel depicts representative fluorescence images of autophagic flux assays using an RFP-GFP-LC3B tandem construct that only the GFP signal could be quenched by the acidic lysosomal pH, and the right panel depicts relative ratios of RFP: GFP in each group. Bar: 10μm; PMA, 100nM; Incubation time, 16 hours. Data are mean ± SD. Unpaired t-test.
To explore if SAT1 coding variants were enriched in SLE patients, we sequenced the coding and splice regions of SAT1 in 562 SLE patients (422 male and 140 female, online supplemental table 2, 3), including 65 SLE probands from families containing multiple affected members, sporadic pediatric-onset lupus patients (disease onset ≤ 18 years old, 107 male and 104 female), and sporadic adult-onset lupus patients (disease onset >18 years old, 263 male and 23 female). The SAT1 sequencing data showed 3 additional rare variants and 12 common variants (online supplemental table 4), but none exhibited robust evidence for functional alterations based on HaploReg v413 and Regulome database14 (online supplemental table 5).
Mice carrying the frameshift variant in Sat1 have pathologies resembling SLE.
SAT1 encoded SSAT1 is a rate-limiting enzyme that regulates polyamine catabolism to maintain many functions of cellular polyamine homeostasis15. Polyamines, putrescine, spermidine and spermine are cationic aliphatic amines that regulate macromolecule interactions affecting critical cellular functions, including growth, differentiation, apoptosis, mobility, and resistance to oxidative and other stresses16,17. Considering that the SAT1 gene and SSAT1-regulated polyamine catabolism are not previously associated with SLE, we constructed Sat1p.Glu92Leufs*6 knock in (KI) mice in C57BL/6J background using CRISPR/Cas9 technology (online supplemental figure 4) to determine if this loss-of-function p.Glu92Leufs*6 variant (online supplemental figure 5) could spontaneously cause lupus-like features.
Compared to 5- to 10-week-old male WT littermates, hemizygous KI male mice spontaneously developed lupus-like features, including splenomegaly, increased ratio of spleen weight to body weight, increased levels of IgG anti-dsDNA, proteinuria, and blood urea nitrogen (BUN), and displayed glomerular enlargement with leukocyte infiltration and glomerular deposition of IgG and complement C3 (figure 2A, C, D, and online supplemental figure 6). The KI male mice also exhibited increased levels of antinuclear antibodies (ANA) (online supplemental figure 6C) and the inflammatory cytokine IL-17A (figure 2D and online supplemental figure 6D). No sex differences were observed between 5-week-old hemizygous male and homozygous female KI mice (figure 2). The KI spleen cells exhibited elevated expression of IFN-I stimulated genes (online supplemental figure 6E) presented as IFN scores (figure 2D), which positively correlated with quantities of IgG deposition in kidneys, proteinuria, serum anti-dsDNA and BUN (figure 2D). These spontaneously developed lupus-like features in young KI mice demonstrates causality of the Sat1 p.Glu92Leufs*6 variant, supporting Sat1p.Glu92Leufs*6 as a monogenic cause for lupus.
Figure 2. The young Sat1p.Glu92Leufs*6 KI male and female mice spontaneously develop lupus-like autoimmune disorder.
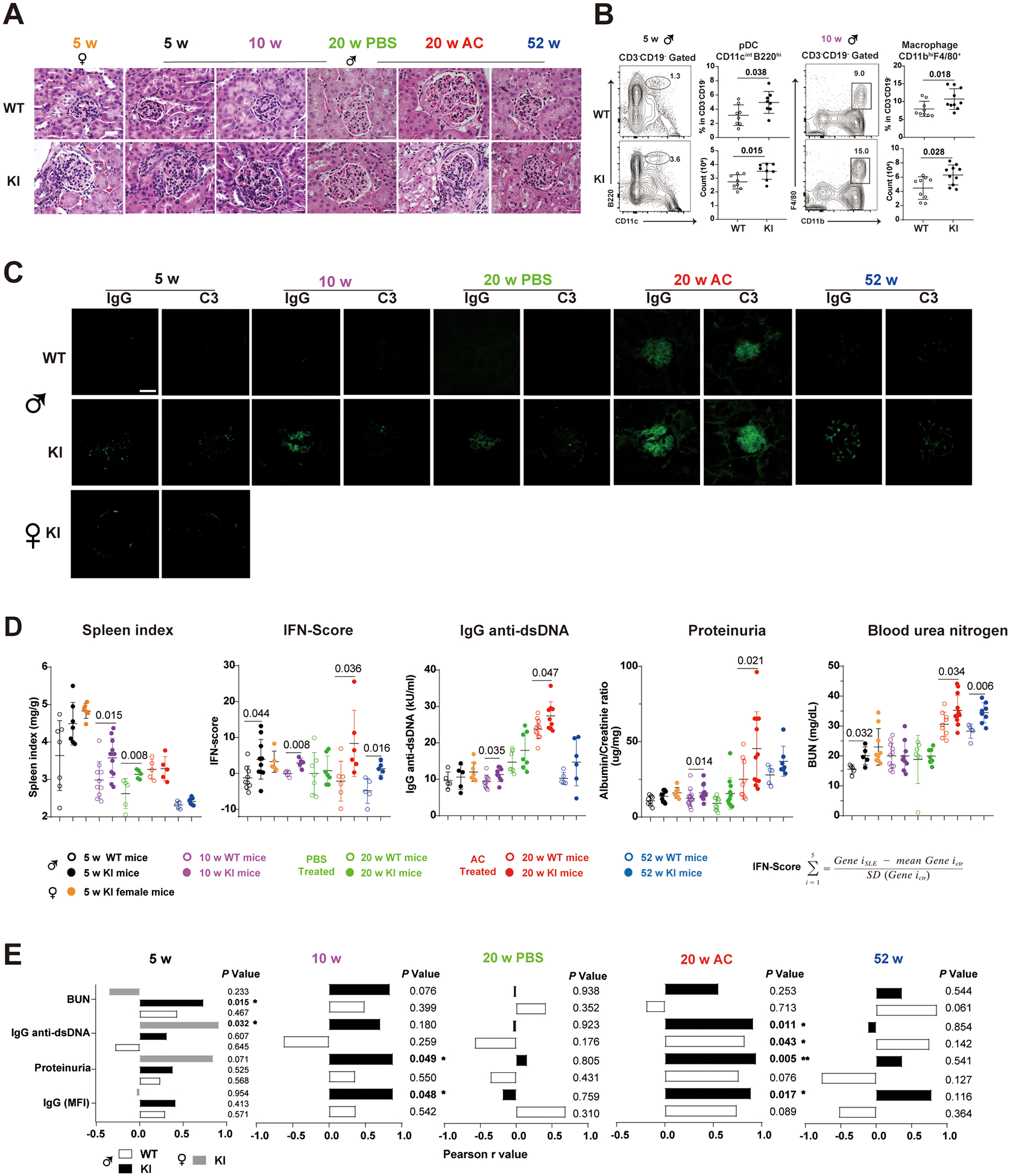
(A) Hematoxylin-eosin-stained kidney sections from female (5 wks-old) and male Sat1p.Glu92Leufs*6 KI mice and WT littermates that were naïve (5, 10 or 52 wks-old), or injected with either PBS (phosphate buffer saline) or apoptotic cells (AC) starting at 10 wks-old and sacrificed at 20 wks-old. Bar: 50μm.
(B) Gating strategy and percentage of plasmacytoid dendritic cells (pDC)(CD3−CD19−CD11cintB220hi) and macrophages (CD3−CD19−CD11bhiF4/80+) in spleen cells from 5- and 10-week-old naive male mice, respectively. Open circle, WT mice; closed circle, KI littermates. Data are mean ± SD. Mann-Whiney U test.
(C) Immunofluorescent staining of mouse Immunoglobulin G (IgG) and complement 3 (C3) depositions in the frozen kidney sections of Sat1p.Glu92Leufs*6 female KI mice, male KI and WT littermates. Bar: 50μm.
(D) Levels of Spleen index, type I IFN-Scores, serum IgG anti-dsDNA, proteinuria, and blood urea nitrogen (BUN) in 5 to 52-week-old female KI mice, male KI and WT littermates. Open circle, male WT mice; closed circle, male KI mice. Black, 5-week-old mice; purple, 10-week-old mice, green, 20-week-old mice injected with PBS; red, 20-week-old mice injected with apoptotic cells; blue, 52-week-old mice. Data are mean ± SD. Yellow closed circle, 5-week-old female KI mice. Mann-Whiney U test.
(E) Correlation analysis of levels of IgG deposition in the kidney, proteinuria, serum anti-dsDNA and BUN with type I IFN-Scores in splenocytes of 5 to 52-week-old KI mice. Closed box depicts statistically significant correlation. White box, male WT mice; Black box, male KI mice; Gray box, female KI mice.
Despite early development of spontaneous lupus-like kidney disorder, nephritis did not progress by one-year-of-age (figure 2A, C, and D), leading us to test whether an increased systemic exposure of syngeneic apoptotic cells, a condition that mimics the defective apoptotic cells (AC) clearance in SLE patients, as an induced lupus model18,19, could exacerbate glomerulonephritis. Compared with AC-treated WT littermates, 20-week-old AC-treated KI mice developed robust lupus-like manifestations, including elevated BUN, anti-dsDNA antibodies, ANA antibodies, proteinuria, increased serum creatinine levels, accelerated glomerulonephritis, and increased glomerular deposition of IgG and complement C3. These lupus manifestations were also positively correlated with increased IFN-I scores (figure 2E). Sat1p.Glu92Leufs*6 shifted immune phenotypes in spleen cells. Changes in the myeloid compartment included elevated macrophages (CD3−CD19−CD11bhiF4/80+) in 10-week-old male KI mice (figure 2B), and elevated proportions and cell numbers of plasmacytoid dendritic cells (pDCs, CD3−CD19−CD11cintB220low). These changes likely contribute to the upregulated IFN-I scores in 5- and 10-week-old male KI mice (figure 2B, D).
While the AC-treated Sat1p.Glu92Leufs*6 male mice had elevated Th17, Tph, and age-associated B cells (ABCs) indicative of extrafollicular activation, the female mice showed elevated T follicular helper cells (Tfh), Tfh/T follicular regulatory (Tfr) ratio, marginal zone B cells (MZ), follicular B cells (FO) and germinal center (GC) B cells responses characterized by activated adaptive immunity and follicular humoral responses (online supplemental figure 8).
Bone marrow (BM)-isolated Sat1p.Glu92Leufs*6 neutrophils show spontaneous NETosis and autophagy defect.
While SAT1 is fairly ubiquitously expressed, it is enriched in neutrophils of both human and mouse immune systems20,21 (online supplemental figure 10). We hypothesized that SAT1 expression is important in neutrophil functions. We observed bone marrow-isolated neutrophils from 5-week-old male KI mice had decreased cell numbers and percentages compared with the WT littermates (figure 3A). These KI neutrophils undergo spontaneous NETosis without phorbol myristate acetate (PMA) stimulation (figure 3B). They released oxidized mitochondrial DNA (mtDNA) into culture supernatants indicated by an increased ratio of mitochondrial (16s) to chromosomal (18s) DNA in anti-8-OHdG immunoprecipitated total oxidized DNA (figure 3B). These observed phenotypes resemble manifestations in SLE patients, including neutropenia, spontaneous NETosis and release of oxidized DNA from mitochondria that elicit anti-dsDNA responses22,23.
BM-isolated Sat1p.Glu92Leufs*6 neutrophils showed decreased AC ingestion at two time points using flowcytometry and confocal assays (figure 3C). Next, we performed immunoblot analysis to evaluate relative levels of LC3B, p62, and LAMP1 in BM-isolated neutrophils from 5-week-old WT and KI mice, with or without PMA administration. Compared with upregulated autophagy in PMA-stimulated WT neutrophils, p62 (an autophagosome cargo protein) was increased, and LC3-II (activated form of LC3) was reduced in KI neutrophils together with decreased LAMP1 levels (figure 3D). Using Autophagy RFP-GFP-LC3B Tandem Sensor, PMA-stimulated WT neutrophils exhibited elevated ratios of RFP (acid-insensitive) to GFP (acid-sensitive) indicative of autophagic flux with LC3B accumulation in acidic autophagolysomes, compared to KI (figure 3E).
Mice with the p.Glu92Leufs*6 variant exhibit perturbed Foxp3-related T cell subsets.
Recent publications report the critical role of polyamine metabolism in maintaining fidelity of T-cell lineage via epigenome regulation24–26. Given that the p.Glu92Leufs*6 LOF variant likely disturbs cellular polyamine homeostasis, we tested the differentiation of Foxp3-related T cell subsets from thymocytes and spleen cells. Compared with their WT counterpart, KI thymocytes had significantly decreased proportions of natural Treg cells at 5 weeks, a decreased trend at 10-weeks (online supplemental figure 9B), and a trend towards increased percentages of CD4+CD25+ T cells at 5 weeks (online supplemental figure 9A). In spleen cells, the percentage of CD4+Foxp3+ including T regulatory (Tregs) and Tfr subsets, was decreased in 10-week-old KI male mice (online supplemental figure 9C), resulting in an increased ratio of Tfh to Tfr compared with the WT littermates, a ratio that promotes humoral immune responses.
Plasma polyamine profiles of SLE patients and correlated with elevated disease activities in SLE patients.
To assess if our findings are relevant to SLE patients, we measured the levels of polyamine metabolites in treatment-naïve, newly diagnosed SLE patients. Figure 4A depicts polyamine metabolic pathways and intermediate metabolites. The percentage composition of nine polyamines was measured in plasma obtained from 26 patients with SLE and 20 HCs (online supplemental table 6, 7). In SLE patients, putrescine, N1-acetylspermidine and S-adenosyl-L-methionine were significantly increased, while levels of spermidine and spermine were significantly decreased compared with HCs (figure 4B). We explored potential links between proportions of polyamines and SLE manifestations. Plasma levels of putrescine acid were negatively correlated with cell-free DNA levels (r = −0.6, p = 0.01) and positively correlated with proportion of Tph cells (r = 0.7, p = 0.03). Spermidine levels correlated with levels of anti-dsDNA antibody (r = −0.42, p = 0.04), proportion of CD4+Foxp3+ cells (r = 0.48, p = 0.02) and Tfh cells (r = 0.53, p = 0.04). Spermine levels correlated with neutrophil count (r = −0.68, p < 0.01), titers of anti-dsDNA antibodies (r = −0.46, p = 0.02), and proportion of CD4+Foxp3+ cells (r = 0.67, p = 0.01) (figure 4C). Other correlations are shown in online supplemental Table 8.
Figure 4. Plasma concentration of nine metabolites related to polyamine homeostasis in treatment-naïve patients affected with SLE and in age- and sex- matched healthy controls (HCs).
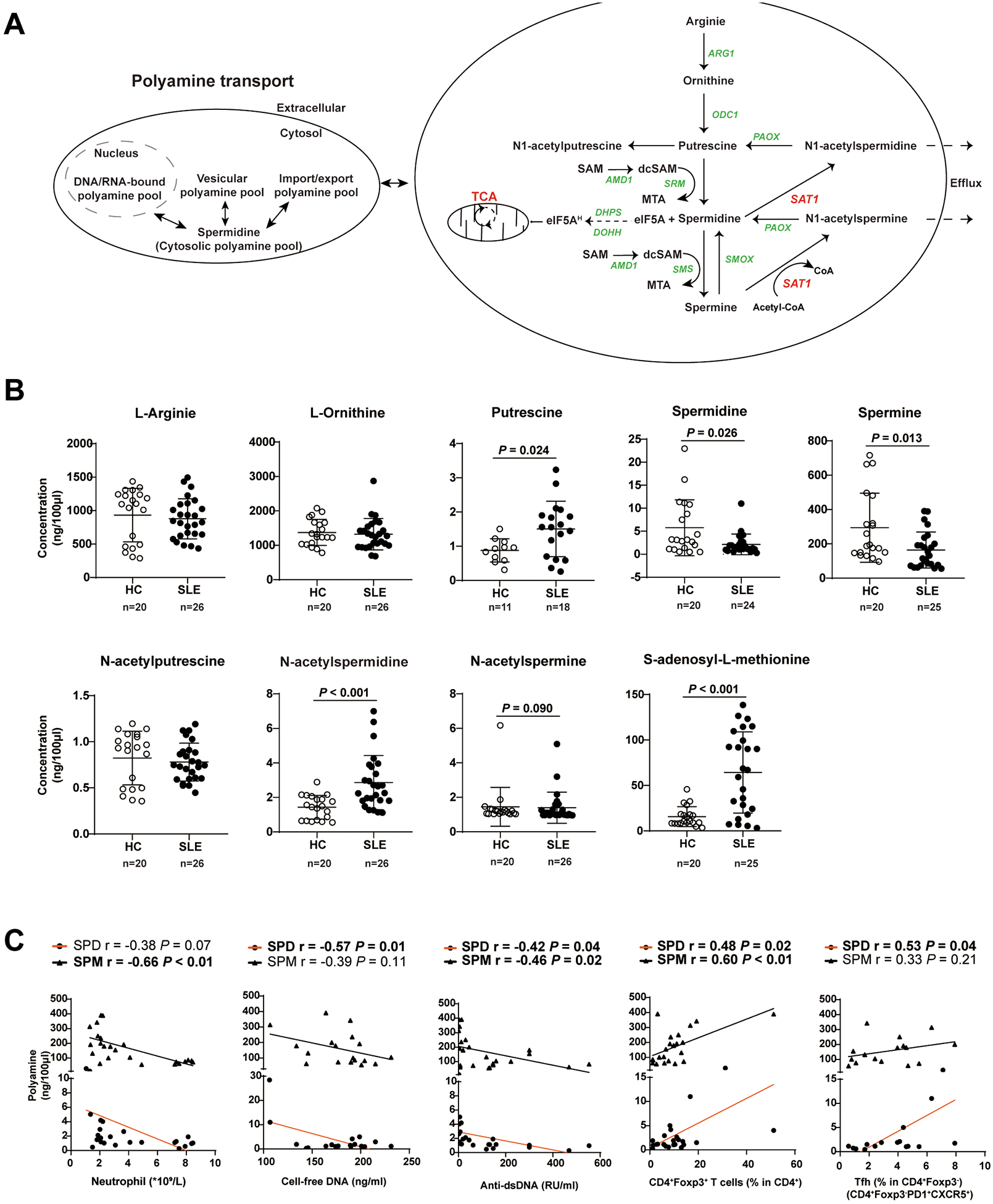
(A) The polyamine metabolism pathway. ARG1, arginase 1; ODC1, ornithine decarboxylase 1; PAOX, peroxisomal N(1)-acetyl-spermine/spermidine oxidase; SAT1, spermine/spermidine N1-acetyltransferase 1; SMS, spermine synthase; SMOX, Spermine oxidase; DHPS, deoxyhypusine synthase; DOHH, deoxyhypusine hydroxylase; eIF5A, eukaryotic initiation factor 5A; eIF5AH, eukaryotic initiation factor 5A hypusination; CoA, coenzyme A; dcSAM, decarboxylated SAM, S-adenosylmethionine; dcSAM, decarboxylated S-adenosylmethionine; SRM, spermidine synthase.
(B) Quantification of nine polyamine metabolites (ng/100μL) found in fasting plasma samples of treatment-naïve, newly diagnosed SLE patients (n = 26) and age-and sex-matched HCs (n = 20). Open circle, health control; closed circle, SLE patient. Data are mean ± SD. Mann-Whiney U test.
(C) Pearson correlations of polyamine concentrations (SPM and SPD, Spermine and Spermidine, respectively) with counts of blood neutrophils, percentages of Tfh and CD4+Foxp3+ T cells, serum levels of cell-free DNA and IgG anti-double-strand DNA antibodies in treatment-naïve SLE patients (n=26). Closed circle and orange line, spermidine; triangle and black line, spermine. Each symbol represents a sample from one individual subject.
Discussions
To the best of our knowledge, we show, for the first time, that loss-of-function SAT1 variants are likely monogenic causes of SLE by identifying two potential loss-of-function variants (p.Asp40Tyr and p.Glu92Leufs*6) that segregate with SLE in two unrelated families following the X-linked recessive inheritance model. Functional studies demonstrated that the p.Asp40Tyr variant caused aberrantly spliced, deleterious SAT1 transcripts. The hemizygous expression of the p.Glu92Leufs*6 variant in young C57BL/6J male mice induced glomerulonephritis, splenomegaly, the production of IgG anti-dsDNA antibodies, elevated type I IFN-scores, reduced numbers and proportions of bone marrow-isolated neutrophils, decreased phagocytosis of apoptotic cells and autophagic flux by neutrophils, increased spontaneous NETosis and release of oxidized mitochondrial DNA, and decreased proportions of Foxp3+CD4+ T cells. Given that these features are commonly found in SLE patients, recapitulating human SLE by a Mendelian inherited single LOF SAT1 variant on a non-autoimmune mouse background showed that it is a new monogenic cause for lupus. Similar to young hemizygous male KI mice, young homozygous female mice also exhibited lupus-like kidney features. By using both in vivo and in vitro studies, we showed defects in both innate (neutrophils) and adaptive immunity (T cells) induced by the p.Glu92Leufs*6 variant revealing a novel role of polyamine metabolism as a risk for SLE.
Monogenic forms of lupus or lupus-like syndromes are the most straightforward approach to unravel the molecular pathogenesis of childhood-onset SLE, especially in those with a more severe phenotype, with a family history of SLE, or from consanguineous marriages6,27,28. Recent advances in next-generation sequencing continues allow discovery of single gene rare variants that cause SLE/SLE-like syndromes exhibiting autosomal dominant/recessive model or de novo mutation2,7,29. While SAT1 is an IFN-I inducible gene30,31, the clinical characteristics of patients carrying the p.Asp40Tyr and p.Glu92Leufs*6 variants of the SAT1 fulfill more than 4 ACR criteria of SLE (Table 1), which differ from patients with some monogenic interferonopathies, who show clinical signs of lupus but do not fulfill classification criteria for SLE. The known causes for monogenic interferonopathies, including mutations in TREX1, SAMHD1, ADAR, IFIH1, and RNASEH2A/2B/2C, which disrupt proteasome degradation and cytoplasmic RNA and DNA sensing pathways32,33. Whole genome sequencing and WES applications in multi-case families with SLE identified a growing number of rare, likely pathogenic variants, but none clearly fulfilled a Mendelian inheritance pattern and/or demonstrated causality in vivo34–36. Higher genetic load for SLE, measured by individual polygenic risk scores (PRS; calculated from the effect size and number of common risk alleles), was reported in childhood-onset more than adult-onset SLE patients and in non-European ancestry more than European ancestry37,38. Our study design that focused on the combined high genetic load of childhood-onset, familial SLE, male lupus, and African-American ancestry38–41 contributed to our identification of the two rare LOF variants of SAT1 in these two unrelated families. These findings exhibit an X-linked recessive inheritance model in that the single copy of a knock-in frameshift variant is sufficient to induce murine lupus in young male mice on a non-autoimmune C57BL/6J background.
To assess if SAT1 variants were enriched in SLE patients in addition to these two LOF rare variants, we sequenced SAT1 coding regions in 562 SLE DNA samples enriched in male patients (422/562, 75.1%) and multiplex family cases (65/562, 11.6%). While we identified 3 additional rare variants and 12 common variants, none were predicted pathogenic, suggesting that the two identified LOF SAT1 variants were unique (online supplementary table 4 and 5). Most known loss-of-function SAT1 variants in the gnomAD database (with an average 50X sequencing depth) are located in coding regions of noncanonical transcripts that are expressed at low levels across all tissues12 (online supplementary figure 1B, C), implicating strong selection against LOF mutations in the highly expressed transcript. Given that both p.Asp40Tyr (in exon 2) and p.Glu92Leufs*6 (in exon 4) are located in highly conserved coding regions of SAT1 and are not present in > 200,000 individuals characterized in the gnomAD10, TOPMed11 and 1000 Genomes database42, these two variants were ultra-rare and highly penetrant in families with SLE patients, especially in male pediatric lupus.
How does this p.Glu92Leufs*6 variant of SAT1 affect the autoimmune responses? In the normal C57BL/6J background, the young male and female KI mice spontaneously developed lupus-like autoimmune disorders. While SAT1 has a broad spectrum of expression in many tissues and cell types, its expression in the immune system is mainly enriched in neutrophils in humans and C57BL/6 mice (online supplementary figure 10)20,21. We extended this study and found neutrophil defects, including decreased cell numbers and aberrant functions in young KI mice. These KI BM-isolated neutrophils spontaneously released NETs enriched in oxidized mtDNA (figure 3B), which were features previously described in neutrophils from SLE patients that could elicit IgG anti-dsDNA production and activate type I IFN responses (figure 2D)43,44. While mechanisms underlying decreased neutrophil counts were not characterized, it is plausible that autoantibodies and type I IFN in young KI male mice could cooperatively induce neutrophil ferroptosis and spontaneous NETosis as shown in murine and human lupus individuals23,45,46. A well-established phagocyte defect in SLE is the impaired clearance of apoptotic cells (termed efferocytosis), which results in accumulated apoptotic cell metabolites in various tissues as a source of self-antigens that could promote inflammation and the development of SLE47,48. We observed impaired KI neutrophil clearance of apoptotic cells via LC3-associated phagocytosis (LAP) (figure 3D, E) suggesting that a functional LAP is necessary to inhibit autoinflammatory, lupus-like responses to dying cells49. Additionally, it is plausible that dysfunctional SSAT1 encoded by Sat1p.Glu92Leufs*6 could perturb polyamine import and accumulation to diminish polyamine-mediated anti-inflammatory responses during efferocytosis50.
To our knowledge, none of monogenic lupus and SLE GWAS-defined risk loci have pointed to a role of polyamine metabolism in lupus pathogenesis1,51. Early connections between polyamines and lupus pathogenesis were shown using an inhibitor of polyamine synthesis that could reduce T-cell proliferation and prolong survival of lupus-prone MRL-lpr/lpr mice52,53 and could reduce pokeweed mitogen-induced cell proliferation and production of IgM and IgG in PBMC cultures54. Emerging evidence supports inverse correlations between polyamine levels and the extent of autoimmunity and inflammation55. Our findings of decreased plasma levels of spermidine and spermine in untreated, newly diagnosed SLE patients (figure 4) confirmed reduced levels previously reported in Korean SLE patients8. Recently, polyamine spermidine was shown to mediate metabolic and epigenetic regulation through translation factor eukaryotic translation initiation factor 5A-1 (eIF5A) hypusination governing the ability of CD4+ T cells to develop into specific functional subsets24. Additionally, the polyamine pathway is required for Th17 induction and Treg suppression26. These pivotal roles of polyamine metabolism in the differentiation of CD4+ T cells could help explain our observed decreased proportions of Foxp3+ T cells (including Treg and Tfr cells) in young naïve KI mice, and increased proportions of Tfh and Tfh/Tfr ratios in AC-induced exacerbated autoimmune disease of KI mice (online supplemental figure 9).
Interestingly, even though young, naïve, B6 KI mice of both sexes developed spontaneous lupus-like kidney disorder it did not progress into proliferative lupus-like kidney disease as they aged (figure 2). As bioactive polycations, polyamines bind nucleic acid and proteins and promote cell proliferation, it is plausible that the rapid growth phase early in life activates Sat1 expression. The maintenance phase of the adult mice kept on a normal chow diet confers relative low activation of SSAT1 enzyme activity, and B6 mice are a relatively lupus-resistant strain bred in a specific pathogen-free (SPF) environment30,56–58. Administration of syngeneic apoptotic thymocytes is established as an immune challenge that can induce mild lupus features in non-autoimmune mice and robust lupus-like autoimmunity in genetically predisposed mice18,19. When we perturbed the 10-week-old male and female KI mice with increased exposure to syngeneic apoptotic cells, robust production of IgG anti-dsDNA, diffuse proliferative glomerulonephritis, and proteinuria ensued, demonstrating the capability of the Sat1p.Glu92Leufs*6 variant to confer lupus-like disease upon immune activation. Of note, male KI mice appeared to have elevated extrafollicular immune activation and female KI mice more robust follicular humoral responses (figure 2 and online supplementary figure 8). The sex difference effect of Sat1 on activated B cells is consistent with the mechanisms that fine-tuned B-cell physiology imparts on sexual dimorphism in humoral responses and autoimmunity59.
Limitations of this study include the followings: (1) The highly penetrant LOF SAT1 variants are ultra-rare as we found only two familial variants in our study sample of 562 SLE patients enriched in male, childhood-onset, and family history of SLE. (2) The lack of cell sources from the two families and insufficient BM-isolated neutrophils from Sat1p.Glu92Leufs*6 mice prevented direct measurements of intracellular polyamine metabolites.
In summary, we showed, for the first time, SAT1 is a novel candidate causative gene of SLE by identifying two potentially loss-of-function variants (p.Asp40Tyr and p.Glu92Leufs*6), which segregated with the SLE disease status in two unrelated African-American families. Functional studies demonstrated that mice carrying the p.Glu92Leufs*6 variant exhibit lupus-like features, including immune cell dysplasia and dysfunction using both in vivo and in vitro studies. Our findings support LOF SAT1 variants as new monogenic causes for SLE and highlight the pathogenic role of disturbed polyamine metabolism in developing SLE.
Supplementary Material
Key messages.
What is already known about this subject?
Monogenic lupus is a subset of lupus associated with highly penetrant single gene variants, which offer new insights into lupus pathogenesis and help unravel potential treatment strategies. To our knowledge, none of monogenic lupus and SLE GWAS-defined risk loci have implicated polyamine metabolism in disease pathogenesis.
What does this study add?
We identified two previously undescribed, predicted loss-of-function (LOF) SAT1 variants (p.Asp40Tyr and p.Glu92Leufs*6) by whole-exome sequencing (WES) and in a X-linked recessive inheritance model in two unrelated African-American families. The p.Asp40Tyr variant caused aberrant splicing that resulted in deleterious transcripts assessed by in vitro assays. The p.Glu92Leufs*6 variant was introduced into the C57BL/6J mouse background (knock in, KI, by CRISPR/Cas9) to determine its role in lupus development.
Both young male and female Sat1p.Glu92Leufs*6 KI mice spontaneously developed lupus-like autoimmune disease, including splenomegaly, glomerular infiltration of leukocytes, proteinuria, and elevated type I interferon scores. Immune profiling showed functional neutrophil defects, and decreased proportions of Foxp3+CD4+ T-cell subsets in young KI mice. While nephritis did not progress up to one-year-of-age in a specific pathogen-free environment, apoptotic cell treatment resulted in exacerbated glomerulonephritis in both 20-week-old male and female Sat1p.Glu92Leufs*6 mice.
Compared to healthy controls, the treatment naïve, incipient SLE patients had decreased plasma levels of spermine that correlated with neutrophil counts negatively and with proportions of Foxp3+CD4+ T cells positively. These correlates implicate a link of polyamine metabolites with defects in both innate and adaptive immune responses in SLE patients.
How might this impact on clinical practice or future developments?
Our findings link dysregulated polyamine catabolism to the development of lupus manifestations and highlight potential monogenic contributions in multiplex families with childhood-onset of SLE. Our findings support SAT1 LOF variants as new monogenic causes for SLE.
Acknowledgments:
We thank Genome Engineering and Stem Cell Center (GESC) at Washington University in St. Louis with the design and validation of CRISPR reagents. We thank the participants of this study and appreciate the help in subject recruitment supported by NIAMS P60 AR062755 and P30 AR072582.
Funding:
This work was supported by the following grants: NIH R21074691 (grant to Betty P. Tsao)
Footnotes
Competing interests: None of the authors declare any conflicts of interest.
Ethics approval
Human DNA samples obtained from subjects enrolled in IRB-approved longitudinal cohorts from the University of California, Los Angeles (IRB number 10-001489), the Medical University of South Carolina (MUSC) (HR#10852), the MUSC Clinical and Community Core of the Core Center for Clinical Research (Pro00021985), and the Oklahoma Medical Research Foundation (95-12 and 06-12). Humans’ participants were recruited with informed consents approved by the Ethics Committee of the First Affiliated Hospital of Nanjing Medical University (2020-SR-044). Animal subjects were approved by the Medical University of South Carolina Institutional Animal Care (IACUC-2019-00711and IACUC-2018-00619).
Data and materials availability:
All data are available in the main text or the Online supplemental materials.
References
- 1.Ha E, Bae S-C, Kim K. Recent advances in understanding the genetic basis of systemic lupus erythematosus. Semin Immunopathol 2022;44:29–46. [DOI] [PubMed] [Google Scholar]
- 2.Harley ITW, Sawalha AH. Systemic lupus erythematosus as a genetic disease. Clin Immunol 2022;236:108953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deng Y, Tsao BP. Updates in lupus genetics. Curr Rheumatol Rep 2017;19:1–13. [DOI] [PubMed] [Google Scholar]
- 4.Cui Y, Sheng Y, Zhang X. Genetic susceptibility to SLE: recent progress from GWAS. J Autoimmun 2013;41:25–33. [DOI] [PubMed] [Google Scholar]
- 5.Truedsson L Classical pathway deficiencies - a short analytical review. Mol Immunol 2015;68:14–19. [DOI] [PubMed] [Google Scholar]
- 6.Omarjee O, Picard C, Frachette C, et al. Monogenic lupus: dissecting heterogeneity. Autoimmun Rev 2019;18:102361. [DOI] [PubMed] [Google Scholar]
- 7.Belot A, Rice GI, Omarjee SO, et al. Contribution of rare and predicted pathogenic gene variants to childhood-onset lupus: a large, genetic panel analysis of British and French cohorts. Lancet Rheumatol 2020;2:e99–109. [DOI] [PubMed] [Google Scholar]
- 8.Kim HA, Lee HS, Shin TH, et al. Polyamine patterns in plasma of patients with systemic lupus erythematosus and fever. Lupus 2018;27:930–8. [DOI] [PubMed] [Google Scholar]
- 9.Richard MLL, Tsao BP. Genes and genetics in human SLE. In: Systemic lupus erythematosus. Elsevier, 2021: 85–96. [Google Scholar]
- 10.Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020;581:434–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taliun D, Harris DN, Kessler MD, et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed program. Nature 2021;590:290–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.GTEx Consortium. The GTEx consortium atlas of genetic regulatory effects across human tissues. Science 2020;369:1318–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ward LD, Kellis M. HaploReg V4: systematic mining of putative causal variants, cell types, regulators and target genes for human complex traits and disease. Nucleic Acids Res 2016;44:D877–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boyle AP, Hong EL, Hariharan M, et al. Annotation of functional variation in personal genomes using regulomeDB. Genome Res 2012;22:1790–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Casero RA, Pegg AE. Polyamine catabolism and disease. Biochem J 2009;421:323–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pegg AE. Functions of polyamines in mammals. J Biol Chem 2016;291:14904–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Madeo F, Eisenberg T, Pietrocola F, et al. Spermidine in health and disease. Science 2018;359. doi: 10.1126/science.aan2788. [Epub ahead of print: 26 Jan 2018]. [DOI] [PubMed] [Google Scholar]
- 18.Mevorach D, Zhou JL, Song X, et al. Systemic exposure to irradiated apoptotic cells induces autoantibody production. J Exp Med 1998;188:387–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen X, Sun X, Yang W, et al. An autoimmune disease variant of IgG1 modulates B cell activation and differentiation. Science 2018;362:700–5. [DOI] [PubMed] [Google Scholar]
- 20.Karlsson M, Zhang C, Méar L, et al. A single-cell type transcriptomics map of human tissues. Sci Adv 2021;7:eabh2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alam O, Alam O. A single-cell-type transcriptomics map of human tissues. Nat Genet 2021;53:1275. [DOI] [PubMed] [Google Scholar]
- 22.Kaplan MJ. Neutrophils in the pathogenesis and manifestations of SLE. Nat Rev Rheumatol 2011;7:691–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lood C, Blanco LP, Purmalek MM, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med 2016;22:146–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Puleston DJ, Baixauli F, Sanin DE, et al. Polyamine metabolism is a central determinant of helper T cell lineage fidelity. Cell 2021;184:4186–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shi H, Chi H. Polyamine: a metabolic COMPASS for T helper cell fate direction. Cell 2021;184:4109–12. [DOI] [PubMed] [Google Scholar]
- 26.Wagner A, Wang C, Fessler J, et al. Metabolic modeling of single Th17 cells reveals regulators of autoimmunity. Cell 2021;184:4168–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alperin JM, Ortiz-Fernández L, Sawalha AH. Monogenic lupus: a developing paradigm of disease. Front Immunol 2018;9:2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Malattia C, Martini A. Paediatric-onset systemic lupus erythematosus. Best Pract Res Clin Rheumatol 2013;27:351–62. [DOI] [PubMed] [Google Scholar]
- 29.Brown GJ, Cañete PF, Wang H, et al. Tlr7 gain-of-function genetic variation causes human lupus. Nature 2022;605:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mounce BC, Poirier EZ, Passoni G, et al. Interferon-induced spermidine-spermine acetyltransferase and polyamine depletion restrict Zika and chikungunya viruses. Cell Host Microbe 2016;20:167–77. [DOI] [PubMed] [Google Scholar]
- 31.Li MMH, MacDonald MR. Polyamines: small molecules with a big role in promoting virus infection. Cell Host Microbe 2016;20:123–4. [DOI] [PubMed] [Google Scholar]
- 32.Crow YJ, Chase DS, Lowenstein Schmidt J, et al. Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am J Med Genet A 2015;167A:296–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rodero MP, Crow YJ. Type I interferon-mediated monogenic autoinflammation: the type I interferonopathies, a conceptual overview. J Exp Med 2016;213:2527–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Delgado-Vega AM, Martínez-Bueno M, Oparina NY, et al. Whole exome sequencing of patients from multicase families with systemic lupus erythematosus identifies multiple rare variants. Sci Rep 2018;8:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen J, Zhang P, Chen H, et al. Whole-genome sequencing identifies rare missense variants of WNT16 and ERVW-1 causing the systemic lupus erythematosus. Genomics 2022;114:110332. [DOI] [PubMed] [Google Scholar]
- 36.Wang Y, Chen S, Chen J, et al. Germline genetic patterns underlying familial rheumatoid arthritis, systemic lupus erythematosus and primary sjögren’s syndrome highlight T cell-initiated autoimmunity. Ann Rheum Dis 2020;79:268–75. [DOI] [PubMed] [Google Scholar]
- 37.Joo YB, Lim J, Tsao BP, et al. Genetic variants in systemic lupus erythematosus susceptibility loci, XKR6 and GLT1D1 are associated with childhood-onset SLE in a Korean cohort. Sci Rep 2018;8:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morris DL, Sheng Y, Zhang Y, et al. Genome-Wide association meta-analysis in Chinese and European individuals identifies ten new loci associated with systemic lupus erythematosus. Nat Genet 2016;48:940–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Webb R, Kelly JA, Somers EC, et al. Early disease onset is predicted by a higher genetic risk for lupus and is associated with a more severe phenotype in lupus patients. Ann Rheum Dis 2011;70:151–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kuo C-F, Grainge MJ, Valdes AM, et al. Familial aggregation of systemic lupus erythematosus and coaggregation of autoimmune diseases in affected families. JAMA Intern Med 2015;175:1518–26. [DOI] [PubMed] [Google Scholar]
- 41.Hughes T, Adler A, Merrill JT, et al. Analysis of autosomal genes reveals gene-sex interactions and higher total genetic risk in men with systemic lupus erythematosus. Ann Rheum Dis 2012;71:694–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42., Auton A, Brooks LD, et al. A global reference for human genetic variation. Nature 2015;526:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rongvaux A, Jackson R, Harman CCD, et al. Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell 2014;159:1563–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lei Y, Guerra Martinez C, Torres-Odio S, et al. Elevated type I interferon responses potentiate metabolic dysfunction, inflammation, and accelerated aging in mtDNAmutator mice. Sci Adv 2021;7:eabe7548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li P, Jiang M, Li K, et al. Glutathione peroxidase 4-regulated neutrophil ferroptosis induces systemic autoimmunity. Nat Immunol 2021;22:1107–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tay SH, Celhar T, Fairhurst A-M. Low-density neutrophils in systemic lupus erythematosus. Arthritis Rheumatol 2020;72:1587–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boada-Romero E, Martinez J, Heckmann BL, et al. The clearance of dead cells by efferocytosis. Nat Rev Mol Cell Biol 2020;21:398–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morioka S, Maueröder C, Ravichandran KS. Living on the edge: efferocytosis at the interface of homeostasis and pathology. Immunity 2019;50:1149–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Martinez J, Cunha LD, Park S, et al. Noncanonical autophagy inhibits the autoinflammatory, lupus-like response to dying cells. Nature 2016;533:115–9. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 50.Doran AC, Yurdagul A, Tabas I. Efferocytosis in health and disease. Nat Rev Immunol 2020;20:254–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Claverie N, Pasquali JL, Mamont PS, et al. Immunosuppressive effects of (2R,5R)-6-heptyne-2,5-diamine, an inhibitor of polyamine synthesis: II. beneficial effects on the development of a lupus-like disease in MRL-lpr/lpr mice. Clin Exp Immunol 1988;72:293. [PMC free article] [PubMed] [Google Scholar]
- 52.Thomas TJ, Messner RP. Beneficial effects of a polyamine biosynthesis inhibitor on lupus in MRL-lpr/lpr mice. Clin Exp Immunol 1989;78:239. [PMC free article] [PubMed] [Google Scholar]
- 53.Pasquali JL, Mamont PS, Weryha A, et al. Immunosuppressive effects of (2R,5R)- 6-heptyne-2,5-diamine an inhibitor of polyamine synthesis: I. effects on mitogeninduced immunoglobulin production in human cultured lymphocytes. Clin Exp Immunol 1988;72:141. [PMC free article] [PubMed] [Google Scholar]
- 54.Chia T-Y, Zolp A, Miska J. Polyamine Immunometabolism: central regulators of inflammation, cancer and autoimmunity. Cells 2022;11:896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Perry D, Sang A, Yin Y, et al. Murine models of systemic lupus erythematosus. J Biomed Biotechnol 2011;2011:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stohl W, Jacob N, Guo S, et al. Constitutive overexpression of BAFF in autoimmuneresistant mice drives only some aspects of systemic lupus erythematosus-like autoimmunity. Arthritis Rheum 2010;62:2432–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pegg AE. Spermidine/spermine-N(1)-acetyltransferase: a key metabolic regulator. Am J Physiol Endocrinol Metab 2008;294:E995–1010. [DOI] [PubMed] [Google Scholar]
- 58.Zhao R, Chen X, Ma W, et al. A GPR174-CCL21 module imparts sexual dimorphism to humoral immunity. Nature 2020;577:416–20. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.