

Abstract
Perhexiline is a prophylactic antianginal agent developed in the 1970s. Although, therapeutically, it remained a success, the concerns of its severe adverse effects including hepatotoxicity caused the restricted use of the drug, and eventually its withdrawal from the market in multiple countries. In the clinical setting, cytochrome P450 (CYP) 2D6 is considered as a possible risk factor for the adverse effects of perhexiline. However, the role of CYP-mediated metabolism in the toxicity of perhexiline, particularly in the intact cells, remains unclear. Using our previously established HepG2 cell lines that individually express 14 CYPs (1A1, 1A2, 1B1, 2A6, 2B6, 2C8, 2C9, 2C18, 2C19, 2D6, 2E1, 3A4, 3A5, and 3A7) and human liver microsomes, we identified that CYP2D6 plays a major role in the hydroxylation of perhexiline. We also determined that CYP1A2, 2C19, and 3A4 contribute to the metabolism of perhexiline. The toxic effect of perhexiline was reduced significantly in CYP2D6-overexpressing HepG2 cells, in comparison to the control cells. In contrast, overexpression of CYP1A2, 2C19, and 3A4 did not show a significant protective effect against the toxicity of perhexiline. Pre-incubation with quinidine, a well-recognized CYP2D6 inhibitor, significantly attenuated the protective effect in CYP2D6-overexpressing HepG2 cells. Furthermore, perhexiline-induced mitochondrial damage, apoptosis, and ER stress were also attenuated in CYP2D6-overexpressing HepG2 cells. These findings suggest that CYP2D6-mediated metabolism protects the cells from perhexiline-induced cytotoxicity and support the clinical observation that CYP2D6 poor metabolizers may have higher risk for perhexiline-induced hepatotoxicity.
Keywords: Drug metabolism, CYPs, Hepatotoxicity, Perhexiline
Introduction
Perhexiline is a drug developed in the 1970s for the treatment of angina (Ashrafian et al. 2007). As a coronary vasodilator, perhexiline was highly efficient in the treatment of angina and initially enjoyed widespread success. Life-threatening adverse effects of the drug, including neurotoxicity and hepatotoxicity, were subsequently reported, which eventually resulted in its withdrawal from many countries. Despite its adverse effects, perhexiline is still used in some countries, including Australia and New Zealand due to its high efficacy (Ashrafian et al. 2007; Dhakal et al. 2022; Inglis and Stewart 2006; Midei et al. 2021).
Using multiple hepatic cells, we reported that several mechanisms, including apoptosis, mitochondrial dysfunction, and endoplasmic reticulum (ER) stress, underlie the hepatotoxicity of perhexiline (Ren et al. 2021, 2020). We reported that activation of MAPK signaling pathway contributes to its cytotoxicity (Ren et al. 2021). Besides these molecular mechanisms been studied, it was of interest to investigate the metabolism of perhexiline because of the critical roles of metabolism in drug- and chemical-induced toxicity (He et al. 2021; Todorović Vukotić et al. 2021) and the importance of inter-individual variability in the expression and activity of drug metabolizing enzymes in the idiosyncratic susceptibility (Chen et al. 2015; Mueller-Schoell et al. 2021). Clinically, the severe side effects of perhexiline were mostly observed in a small group of patients that were poor metabolizers of the drug (Shah et al. 1982). These patients have polymorphisms in CYP2D6 genes that resulted in the absence or inactivation of CYP2D6 in their liver; and thus, excess accumulation of perhexiline or reduced amounts of its metabolites was detected in their plasma when taking the drug (Ashrafian et al. 2007; Cooper et al. 1984; Davies et al. 2006; Sallustio et al. 2002). Interestingly, using a liver microsomal system, several cytochrome P450 enzymes (CYPs) including CYP2D6 have been identified to catalyze the oxidative metabolism of perhexiline (Davies et al. 2007). Despite these above-mentioned reports, information is sparse regarding the precise roles of CYP-mediated metabolism in toxicity or detoxification of perhexiline, especially no data are available in the intact cells.
We previously established 14 HepG2 cell lines that stably overexpress individual CYP enzymes (1A1, 1A2, 1B1, 2A6, 2B6, 2C8, 2C9, 2C18, 2C19, 2E1, 3A4, 3A5, and 3A7) (Xuan et al. 2016). This system has been validated extensively and used by researchers for drug-related metabolism studies (Chen et al. 2018b, 2020, 2021; Hofman et al. 2019, 2021; Vagiannis et al. 2020; Wu et al. 2016). In the current study, we took the advantage of these cell lines to systematically explore the role of these 14 CYP enzymes in the metabolism and cytotoxicity of perhexiline.
Materials and methods
Chemicals and reagents
Williams’ Medium E, perhexiline maleate salt (racemic mixture), quinidine, β-nicotinamide adenine dinucleotide 2′-phosphate reduced tetrasodium salt hydrate (NADPH), formic acid, and dimethysulfoxide (DMSO) were purchased from Sigma-Aldrich (St. Louis, MO). Cis-OH-Phx and trans-OH-Phx were purchased from Toronto Research Chemicals (Toronto, Canada), and no information is available on the enantiomeric and diastereomeric composition of these standards. α-Naphthoflavone, nootkatone, and ketoconazole were obtained from Cayman Chemical (Ann Arbor, MI). Xtreme 200-donor Mixed Gender Human Liver Microsomes and EasyCYP bactosomes (recombinant human CYPs) were purchased from XenoTech (Lenexa, KS). Antibiotic–antimycotic was obtained from Life Technologies (Carlsbad, CA). PureCol® Type I Collagen Solution was from Advanced BioMatrix (San Diego, CA). Fetal Bovine Serum (FBS) was purchased from Atlanta Biologicals (Lawrenceville, GA). All solvents for liquid chromatography and mass spectrometry were of the highest grade commercially available.
Cell culture and drug treatment
The human cell line HepG2 was obtained from the American Type Culture Collection (ATCC, Manassas, VA). HepG2-derived CYP-overexpressing cell lines were previously established in our laboratory (Xuan et al. 2016). HepG2 and CYP-overexpressing HepG2 cells were cultured in Williams’ Medium E supplemented with 10% FBS and 1 × antibiotic–antimycotic (100 units/mL of penicillin, 100 μg/mL of streptomycin, and 250 ng/mL of amphotericin B).
For toxicity assays, HepG2 cells were seeded at a density of 2.5 × 105 cells/ml in volumes of 100 μL in each well of 96-well tissue culture plates. For biochemical assays, cells were seeded at a density of 3 × 105 cells/ml in volumes of 5 mL in 60 mm plates. Drug treatment with various concentrations started approximately 24 h after cells were seeded. DMSO-treated cells served as control with a final concentration of 0.1% v/v.
Primary human hepatocytes (pooled from 10 donors) were obtained from Discovery Life Sciences IVAL (Columbia, MD). Culture plates used for primary human hepatocytes were pre-coated overnight with 100 μg/mL of PureCol® Type I Collagen solution at room temperature. Primary human hepatocytes were thawed following the supplier’s instruction and plated at a density of 4 × 105 cells/mL in volumes of 100 μL in each well of 96-well tissue culture plates. Cells were maintained in Universal Primary Cell Plating Medium (UPCM™, Discovery Life Sciences IVAL).
The cell culture conditions for the glucose-galactose assay followed the description of Marroquin et al. (2007). DMEM (Life Technologies) deprived of glucose was used as base medium. Glucose-containing medium was DMEM supplemented with 25 mM glucose, 5 mM HEPES, 1 mM sodium pyruvate, 10% FBS, and 1× antibiotic–antimycotic. Galactose-containing medium was DMEM supplemented with 10 mM galactose, 5 mM HEPES, 1 mM sodium pyruvate, 10% FBS, and 1 × antibiotic–antimycotic. Cultures of HepG2 cells were switched to glucose-containing or galactose-containing medium upon drug treatment.
Mass spectrometry analysis of perhexiline and its metabolites from perhexiline-treated cells
CYP-overexpressing HepG2 cells were seeded in 6-well plates at a density of 1 × 106 cells/well in a volume of 2 mL and treated with 5 μM perhexiline for 24 h. Cell lysates were then obtained by adding 500 μL acetonitrile directly to each well in 6-well plates. The lysate mixtures were centrifuged at 15,000g for 15 min at 4 °C. The supernatants were then diluted using three volumes of methanol, vortexed, and then placed on ice for 15 min. The samples were centrifuged at 15,000g for 15 min at 4 °C and the supernatants were transferred into sample vials. Three μL of the sample was injected onto for UHPLC- Q Exactive MS System (ultrahigh-performance liquid chromatography–mass spectrometry) for analysis (Thermo Fisher Scientific, San Jose, CA). Perhexiline and its metabolites were separated on a Zorbax Eclipse XDB-C18 column (4.6 mm × 50 mm, 1.8 μm, Agilent, Santa Clara, CA). The column temperature was maintained at 40 °C. Water (A) and acetonitrile (B), both containing 0.1% formic acid, were used as mobile phase. The flow rate was 300 μL/min with an LC gradient from 30% B to 98% B from 0 to 2.5 min, maintaining at 98% B for 1 min, returning to 30% B, and maintained for 1.5 min to re-equilibrate the column. The Q Exactive MS was operated in the positive mode with an electrospray ionization. Ultra-pure nitrogen was applied as the sheath (45 arbitrary units), auxiliary (10 arbitrary units), and sweep (1.0 arbitrary units) gases. The capillary temperature was set at 350 °C and the capillary voltages was set at 3.7 kV. MS data were acquired from m/z 80 to 1200 in-profile mode and m/z 371.1012 was used as the reference ions for positive mode during acquisition.
Metabolism of perhexiline in human liver microsomes and recombinant CYPs
Incubations were conducted in 1 × PBS (pH 7.4) containing 30 μM perhexiline and 0.1 mg human liver microsomes or 1 pmol of each cDNA-expressed human CYP enzymes (control, CYP1A2, 2C19, 2D6, and 3A4) in a final volume of 95 μL. After a 5 min pre-incubation at 37 °C, 5 μL of 20 mM NADPH were added (final concentration 1.0 mM) and incubation was continued for 40 min with gentle shaking. Coincubations of perhexiline (30 μM) with α-naphthoflavone (CYP1A2 inhibitor, 3 μM), nootkatone (CYP2C19 inhibitor, 10 μM), quinidine (CYP2D6 inhibitor, 2 μM), or ketoconazole (CYP3A inhibitor, 4 μM) in human liver microsomes were performed to determine the role of CYPs in the formation of perhexiline metabolites. Incubations without inhibitors were used as controls. Incubations were performed in duplicate for cDNA-expressed CYP enzymes, and in triplicate for human liver microsomes experiments. Reactions were quenched by adding 100 μL of ice-cold methanol and vortexing for 30 s. The mixtures were then centrifuged at 15,000g for 15 min. Each supernatant was transferred to an auto-sampler vial, and 3 μL of the supernatants were injected onto a UHPLC-Q Exactive MS for analysis. Perhexiline and its metabolites were separated on an BEH-C18 column (2.1 mm × 100 mm, 1.7 μm, Waters, Milford, MA), and the column temperature was maintained at 40 °C. Water (A) and acetonitrile (B), both containing 0.1% formic acid, were used as mobile phase. The flow rate was 300 μL/min with a gradient from 2% B to 98% B from 0.25 to 12 min, maintaining at 98% B for 1 min, returning to 2% B, and maintained for 1.5 min to re-equilibrate the column. The Q Exactive MS was operated in the same conditions used in the analysis of cell samples described above.
Cellular ATP assay
The cellular ATP level was measured using a CellTiter-Glo® Luminescent Cell Viability Assay (Promega Corporation, Madison, WI). The intensity of luminescent signals was detected using a Cytation 5 Microplate Reader (BioTek Instruments, Winooski, VT) and the results in drug-treated cells were normalized to those in DMSO controls.
Lactate dehydrogenase assay
Cellular lactate dehydrogenase (LDH) release was measured and analyzed using a protocol described previously (Chen et al. 2018a). The 100% LDH release was defined as the LDH level in total cell lysate in each sample, and the LDH release after drug treatment was expressed as percentage of the total level.
Cell viability assay
Cell viability was measured using a CellTiter-Blue® Cell Viability Assay kit (Promega Corporation). Briefly, upon the completion of drug treatment, 20 μL CellTiter-Blue® reagent were added into the medium in each well of the 96-well cell culture dish and incubated for 1 h in 37 °C incubator. Fluorescence at 560/590 nm was read using a Cytation 5 Microplate Reader.
Measurement of caspase 3/7 activity
Caspase 3/7 activity was measured using a Caspase-Glo® 3/7 Assay System (Promega Corporation). The results in drug-treated cells were normalized to those in DMSO controls.
Measurement of mitochondrial membrane potential
Mitochondrial membrane potential (MMP) was measured using the cationic carbocyanine dye JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolyl-carbocyanine iodide) (Thermo Fisher Scientific). The results in drug-treated cells were normalized to those in DMSO controls.
RNA isolation and quantitative real-time PCR assay
Total RNA was isolated using RNeasy Mini Kits (Qiagen, Germantown, MD). The yield of the extracted RNA was measured using a NanoDrop™ 2000 spectrometer (Thermo Fisher Scientific) at 260 nm. Two micrograms of total RNA were used in the reverse transcription with a High-Capacity cDNA Reverse Transcription Kit (Applied Biosystem, Foster City, CA) to generate cDNAs. Quantitative real-time PCR assay for ATF6, ERN1, EIF2AK3, TNF, ATF4, and DDIT3 was performed as described previously (Chen et al. 2014). GAPDH was used as an internal control. All the probes were purchased from Thermo Fisher Scientific. The assay ID number of each probe is listed as below.
| Gene name | Assay ID | Gene name | Assay ID |
|---|---|---|---|
|
| |||
| ATF6 | Hs00232586_m1 | ATF4 | Hs00909569_g1 |
| ERN1 | Hs00176385_m1 | DDIT3 | Hs01090850_m1 |
| EIF2AK3 | Hs00178128_m1 | GAPDH | Hs02758991_g1 |
| TNF | Hs00174128_m1 | ||
Western blot analysis
Western blot analysis was conducted following the description in our previous reports (Ren et al. 2016, 2021). Briefly, cells were harvested directly using RIPA buffer supplemented with Halt Protease Inhibitor Cocktail (Thermo Fisher Scientific) and the concentration of the total proteins was measured using Bio-Rad Protein Assay (Bio-Rad Laboratories, Hercules, CA). Primary antibodies against CHOP (Cat# 2895, Cell Signaling Technology, Danvers, Massachusetts) and GAPDH (Cat# sc-47724, Santa Cruz Biotechnology, Santa Cruz, CA) were used. The bands were detected by FluorChem E Imager (ProteinSimple, San Jose, CA).
Statistics analyses
Data are presented as the mean ± standard deviation (SD) of at least three biological replicates. Analyses were conducted using GraphPad Prism 5 (GraphPad Software, San Diego, CA). Statistical significance was determined by one-way analysis of variance (ANOVA) followed by Dunnett’s test for pairwise-comparisons or two-way ANOVA followed by a Bonferroni post-test. The difference was considered statistically significant when p < 0.05.
Results
Perhexiline-induced cytotoxicity in hepatic cells
To obtain an overall cytotoxicity profile of perhexiline in hepatic cells (Fig. 1), we first exposed primary human hepatocytes to perhexiline at concentrations from 5 to 15 μM for 24 h (Fig. 2A). This exposure induced significant cellular damage, as evidenced by the reduction in cellular ATP content. Notably, a decrease in ATP was observed at concentration as low as 5 μM. Considerable ATP reduction upon perhexiline treatment was also found in HepG2 cells. When exposed to 2.5–10 μM perhexiline for 24 h, the cellular ATP levels in HepG2 cells showed a concentration-dependent decrease (Fig. 2B). At the higher concentration of 10 μM, cellular ATP levels were largely depleted, suggesting prominent cell death. Using the C ellTiter®-Blue cell viability assay, perhexiline caused significant decrease in cell viability in HepG2 cells (Fig. 2C). These observations were further confirmed by a concentration-dependent increase of LDH release, which indicated loss of cell membrane integrity (Fig. 2D). In our previous study, we demonstrated that apoptosis and mitochondrial dysfunction were the contributing factors in the cytotoxicity of perhexiline after a shorter-term exposure of 4 h (Ren et al. 2020). Consistent with the previous findings, treatment with 7.5 or 10 μM perhexiline for 24 h resulted in an increase of caspase 3/7 activity (Fig. 2E). Furthermore, the mitochondrial membrane potential level, measured via JC-1 staining, showed a significant concentration-dependent decrease upon perhexiline treatment (Fig. 2F). Taken together, these results demonstrated that perhexiline caused cytotoxicity in both primary human hepatocytes and HepG2 cells.
Fig. 1.
The chemical structure of perhexiline and the two major metabolites cis-OH-Phx and trans-OH-Phx
Fig. 2.
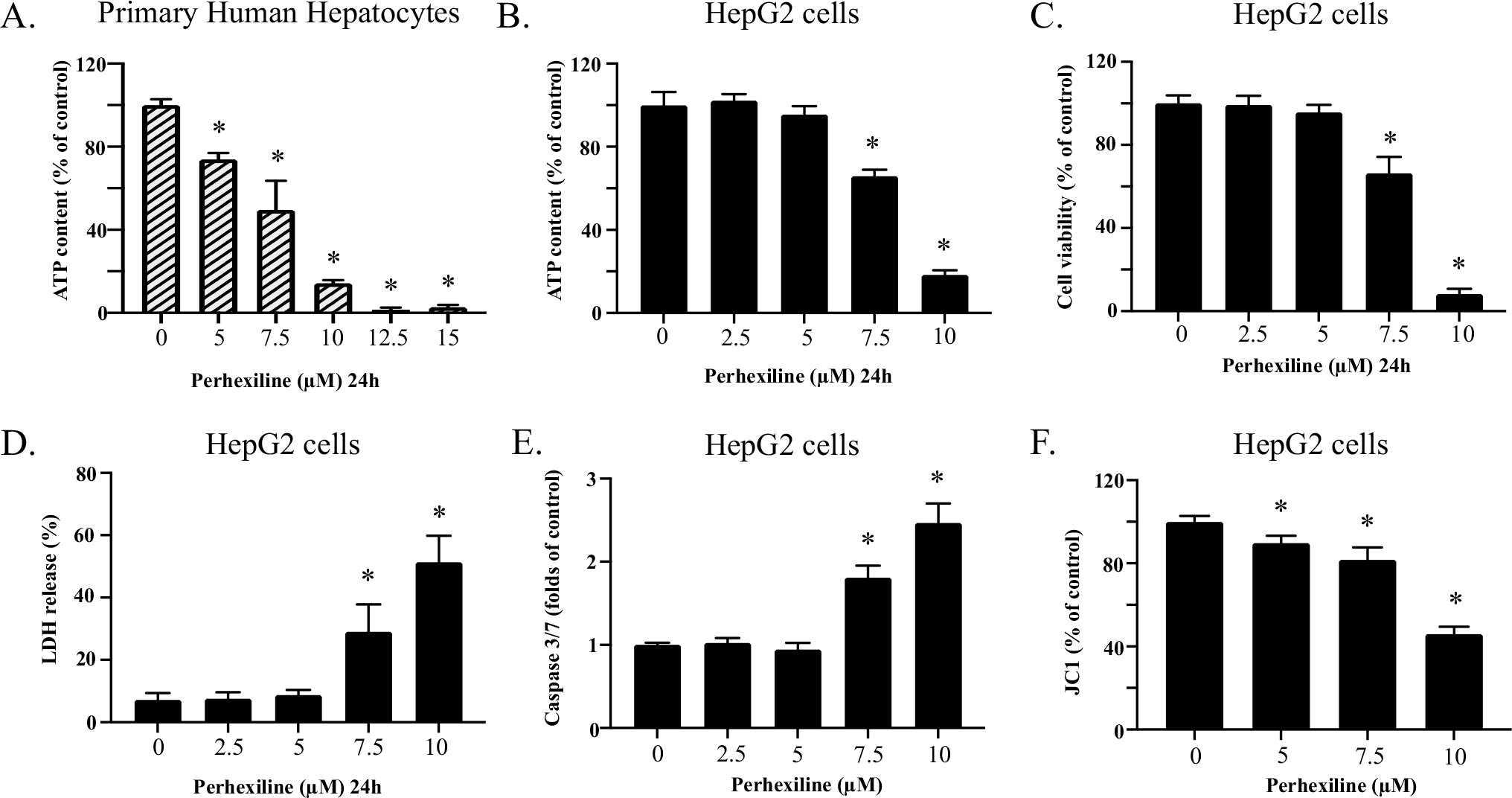
Perhexiline causes cellular damage in hepatic cells (A). Primary human hepatocytes were exposed to 0, 5, 7.5, 10, 12.5, or 15 μM perhexiline for 24 h. The cytotoxicity of perhexiline was measured using cellular ATP contents. B–F HepG2 cells were exposed to 0, 2.5, 5, 7.5, or 10 μM perhexiline for 24 h. Cellular ATP content (B), cell viability (C), LDH release (D), caspase 3/7 activity (E), and the change in mitochondrial membrane potential as visualized by JC-1 staining (F) were measured. The results shown are mean ± SD from 3 biological replicates. *p < 0.05 compared to DMSO control
Metabolism of perhexiline in CYP-overexpressing HepG2 cell lines
Metabolism may play a critical role in the toxicity of perhexiline. Therefore, we utilized our previously established 14 stable CYP-overexpressing HepG2 cell lines to explore the metabolism of perhexiline by CYPs. The stability and robustness of these CYP-overexpressing HepG2 cells were validated in our previous study (Chen et al. 2021). To obtain a metabolic profile by CYPs, we first used mass spectrometry analysis and measured the amount of the parent drug in each cell line after a 24 h exposure with 5 μM perhexiline, a concentration with minimal toxicity, as shown in Fig. 2B–F. A representative mass chromatogram for the parent drug perhexiline and its two major metabolites cis-OH-Phx and trans-OH-Phx in empty vector (EV) transduced control cells or CYP2D6- and 3A4-overepxressing HepG2 cells is shown in Fig. 3A. The results are presented in Fig. 3B–D. A significant reduction of the parent perhexiline level was observed in CYP3A4-, 1A2-, 2C19-, and 2D6-overexpressing HepG2 cells (Fig. 3B), implying the importance of these subtypes of CYPs in metabolizing perhexiline. Notably, the amount of parent perhexiline in CYP2D6-overexpressing cells was only 1% of that in EV control, suggesting an efficient metabolism of perhexiline by CYP2D6. We then measured the formation of cis-OH-Phx and trans-OH-Phx in each cell line (Fig. 3C, D). In CYP2D6-overexpressing cells, abundant cis-OH-Phx was formed, but very low formation of trans-OH-Phx was detected. In CYP2C19-overexpressing HepG2 cells, a significant amount of cis-OH-Phx was also detected; however, the amount of trans-OH-Phx showed a more prominent increase. Similar results were observed in CYP3A4-overexpressing cells, with a limited increase in cis-OH-Phx and a significant increase in trans-OH-Phx. Interestingly, no significant amount of cis-OH-Phx or trans-OH-Phx was detected in CYP1A2-overexpressing HepG2 cells, although there was a decrease in perhexiline, suggesting the existence of other metabolites.
Fig. 3.
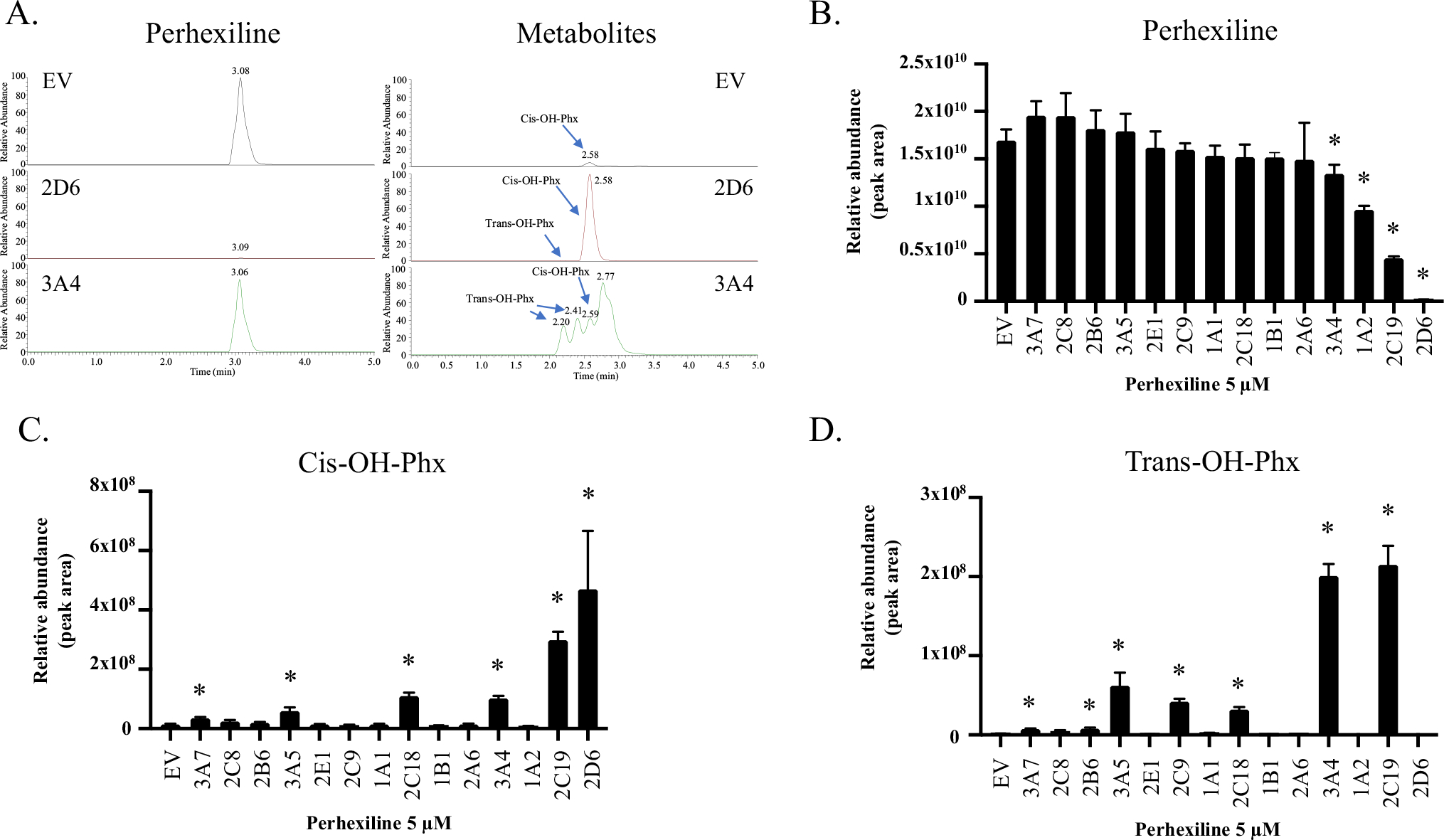
Metabolism of perhexiline in individual CYP-overexpressing HepG2 cells. Fourteen individual CYP-overexpressing HepG2 cell lines were treated with 5 μM perhexiline for 24 h. The cell extracts were then subjected to mass spectrometry to determine the abundance of perhexiline and its two major metabolites cis-OH-Phx and trans-OH-Phx. A Representative chromatograms for perhexiline and its two metabolites cis-OH-Phx and trans-OH-Phx. B The relative abundance (intensity of ions) of perhexiline, C cis-OH-Phx, and D trans-OH-Phx in the cell lysate. The results shown are mean ± SD from 4 separate incubations. *p < 0.05 compared to EV control
Role of CYP1A2, 2C19, 2D6, and 3A4 in the metabolism of perhexiline
Based on the mass spectrometry data, we explored further the role of CYP1A2, 2C19, 2D6, and 3A4 in the metabolism of perhexiline using recombinant human CYPs and chemical inhibition experiments in human liver microsomes. Perhexiline was incubated with bactosomes expressing CYP1A2, 2C19, 2D6, or 3A4, and the abundance of each metabolite from the incubation was analyzed. Six mono-hydroxylated metabolites were identified (Table 1). To demonstrate the relative contribution of individual CYP enzyme to each metabolite, the most abundance level observed in the CYP, based on the peak area of the metabolite, was set to 100% and the abundance generated by other CYP enzymes was expressed as percentage accordingly. Among them, CYP1A2 played a minor role in the formation of all the six metabolites. CYP2D6 was the predominant enzyme contributing to the formation of M1, M2, M4, and M5. Its role in the formation of M3 and M6, however, was marginal. The formation of M3 and M6 was mainly mediated by CYP3A4. CYP2C19 was involved in the formation of all 6 metabolites to a different extent (Table 1), while CYP2C19 showed a similar contribution to the formation of M3 as CYP3A4. According to the retention times and the exact masses of their corresponding standards, M1 and M3 were both identified as trans-OH-Phx (diastereomers), and M5 was identified as cis-OH-Phx (Fig. 4A). We did not identify the remaining three mono-hydroxylated metabolites.
Table 1.
Contribution of CYP isoforms in the formation of metabolites of perhexiline in cDNA-expressed CYP bactosomes
| M1 (trans-OH-Phx-1) | M2 | M3 (trans-OH-Phx-2) | M4 | M5 (cis-OH-Phx) | M6 | |
|---|---|---|---|---|---|---|
|
| ||||||
| EV | 0 | 0 | 0 | 0 | 0 | 0.2 |
| CYP1A2 | 0 | 0 | 0 | 2.4 | 0.2 | 7.8 |
| CYP2C19 | 19.5 | 27.5 | 92.6 | 62.5 | 3.1 | 2.9 |
| CYP2D6 | 100 | 100 | 6.5 | 100 | 100 | 0.6 |
| CYP3A4 | 91.3 | 0 | 100 | 0 | 22.9 | 100 |
To demonstrate the relative contribution of individual CYP enzymes, for each metabolite, the highest abundance level observed was set to 100% and the abundance generated by other CYP enzymes was expressed as percentages accordingly. All six metabolites detected were mono-hydroxylated derivatives of perhexiline based on their exact masses
Fig. 4.
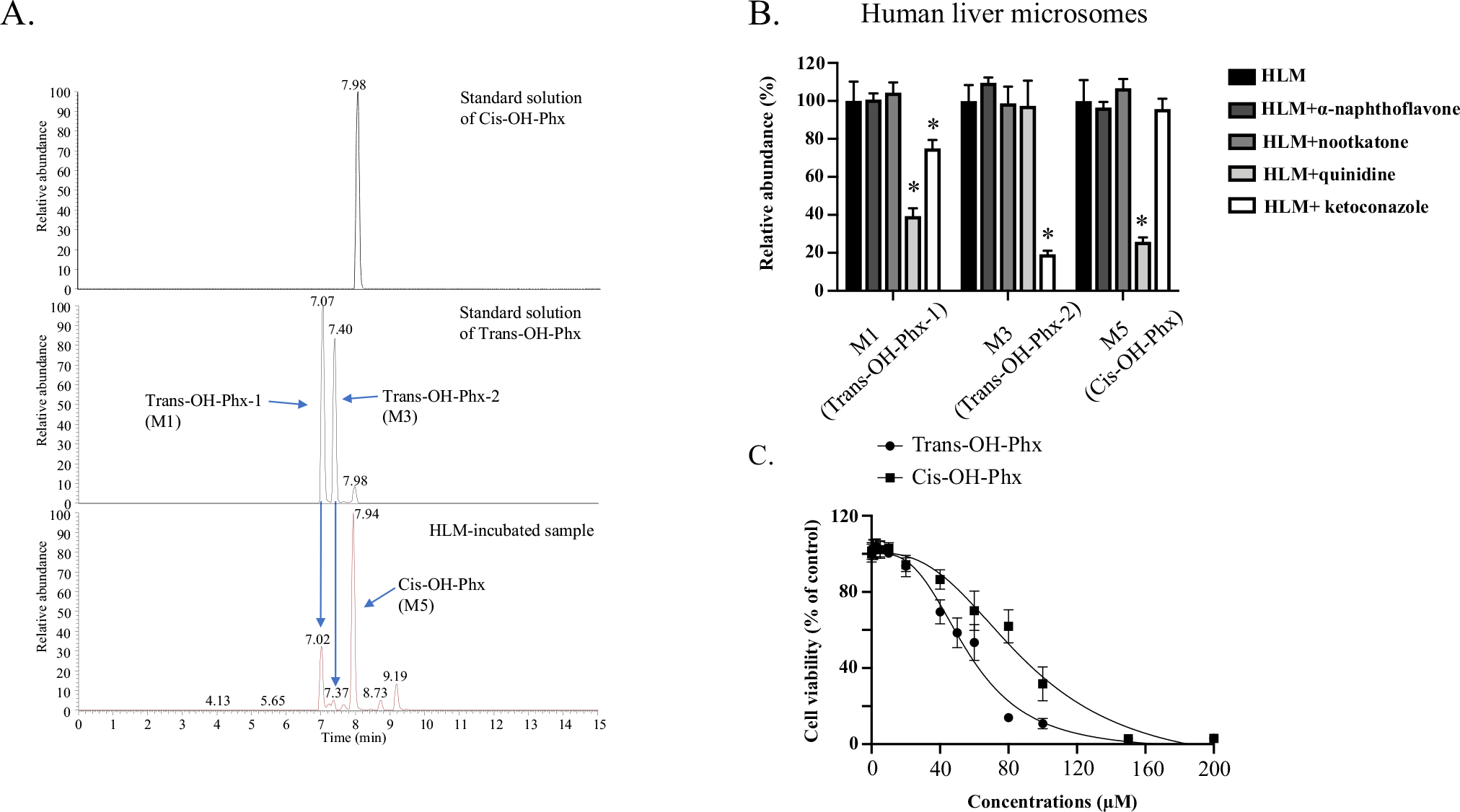
Cis-OH-Phx and trans-OH-Phx are generated by different CYPs and have different toxicity. A Representative chromatograms for perhexiline metabolites. Optimized LC–MS with a longer running time was used to better separate the multiple mono-hydroxylated metabolites, and the peaks of cis-OH-Phx (M5) and trans-OH-Phx (M1 and M3, diastereomers) were identified by the standard compound. B Perhexiline was co-incubated with the CYP1A2 inhibitor α-naphthoflavone (6 μM), the CYP2C19 inhibitor nootkatone (10 μM), the CYP2D6 inhibitor quinidine (2 μM), or the CYP3A4 inhibitor ketoconazole (2 μM) in human liver microsomes (HLM). The relative abundance of the two perhexiline metabolite M3 (trans-OH-Phx) and M5 (cis-OH-Phx) was quantified by LC–MS and normalized to the control group. *p < 0.05 compared to HLM without inhibitor. C HepG2 cells were exposed to cis-OH-Phx or trans-OH-Phx for 24 h and cell viability was measured. The results shown are mean ± SD from 3 independent experiments
The role of these four CYPs in the metabolism of perhexiline was further verified by co-incubation of perhexiline with specific chemical inhibitor for each CYP enzyme in human liver microsomes. The inhibitors used were the CYP1A2 inhibitor α-naphthoflavone, the CYP2C19 inhibitor nootkatone, the CYP2D6 inhibitor quinidine, and the CYP3A4 inhibitor ketoconazole (MacKenzie et al. 2020). As shown in Fig. 4B, ketoconazole (2 μM, CYP3A4 inhibitor) suppressed the formation of M3 (trans-OH-Phx-2) up to 80%; quinidine (2 μM, CYP2D6 inhibitor) suppressed 74% of M5 (cis-OH-Phx) formation. CYP3A4 and CYP2D6 inhibitors also reduced the formation of M1 (trans-OH-Phx-1), while CYP2D6 and CYP3A4 inhibitors did not have effect on M3 and M5, accordingly. The inhibitors of CYP1A2 and CYP2C19 did not show significant effect on the formation of M1, M3, and M5.
The cytotoxicity profile of cis-OH-Phx and trans-OH-Phx was then measured in HepG2 cells using CellTiter®-Blue cell viability assay at a 24 h exposure time. As shown in Fig. 4C, cis-OH-Phx (IC50=90 μM) was less toxic than trans-OH-Phx (IC50=56 μM). The IC50 value for perhexiline based on the results from the same assays was 8 μM, indicating that both metabolites were significantly less toxic than perhexiline.
CYP2D6-mediated metabolism suppressed perhexiline-induced cytotoxicity
The effect of CYP2D6, 2C19, 1A2, and 3A4-mediated metabolism on the cytotoxicity of perhexiline was then measured in the corresponding cells overexpressing the enzymes. The cellular ATP levels and LDH release upon treatment of 5–10 μM perhexiline for 24 h were measured in CYP2D6-, 2C19-, 1A2-, and 3A4-overexpressing HepG2 cells and then compared to the levels of EV control cells. As shown in Fig. 5, CYP2D6-mediated metabolism significantly attenuated the perhexiline-induced decrease in cellular ATP levels and increase in LDH release. In contrast, little protective effect was observed in CYP2C19-, CYP1A2-, and CYP3A4-overexpressing HepG2 cells.
Fig. 5.
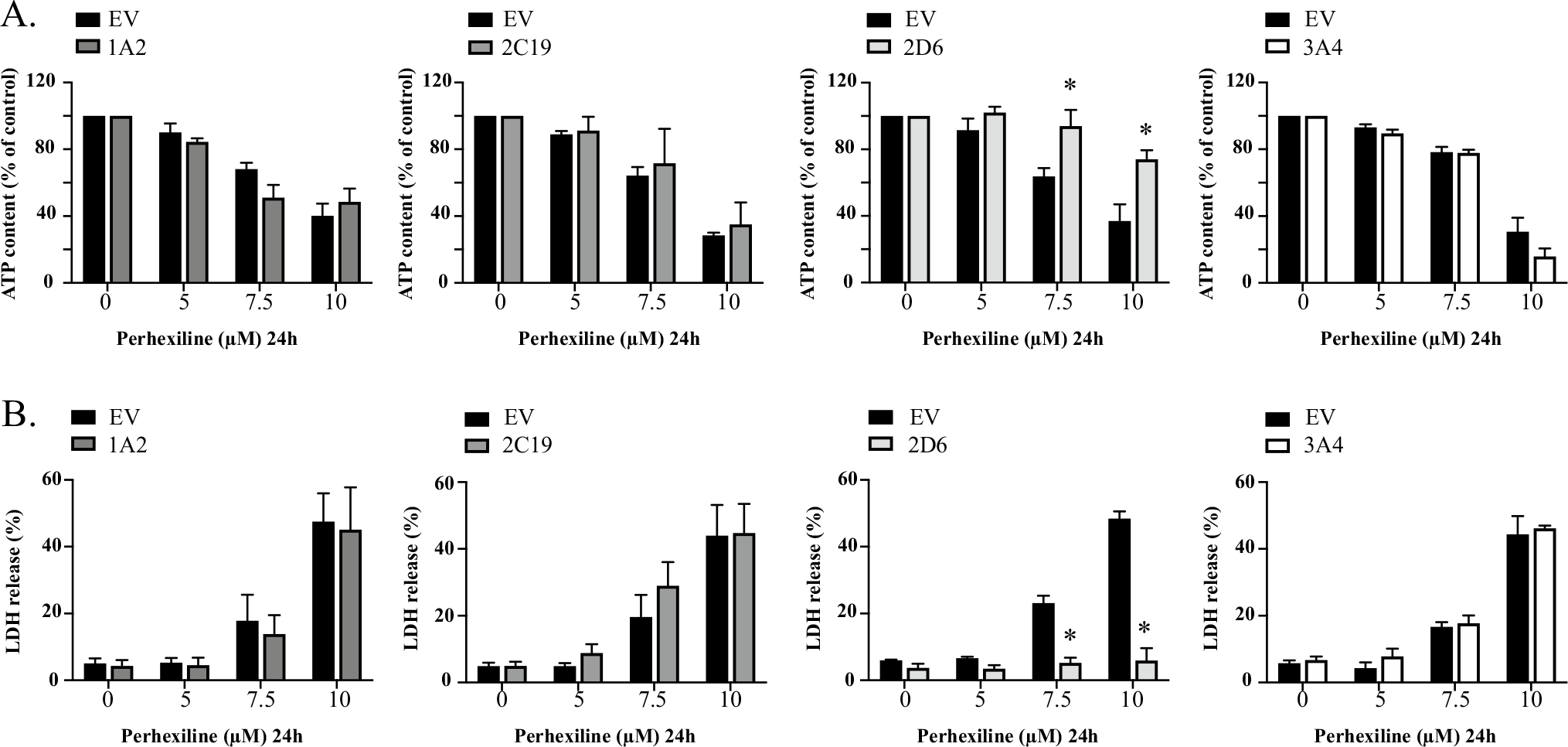
CYP2D6-overexpressing HepG2 cells attenuate the cytotoxicity of perhexiline. EV-, CYP1A2-, CYP2C19-, CYP2D6-, and CYP3A4-overexpressing HepG2 cells were exposed to 0, 5, 7.5, and 10 μM perhexiline for 24 h. Cellular ATP contents (A) and LDH release (B) were measured as toxicity endpoints. The results shown are mean ± SD from 3 biological replicates. *p < 0.05 compared to EV control at the same concentration of perhexiline
To confirm further the protective effect observed in CYP2D6-overexpressing HepG2 cells, we then applied a potent CYP2D6-specific inhibitor quinidine. Both CYP2D6-overexpressing and EV control cells were pre-incubated with 10 μM quinidine or DMSO for 2 h before exposure to various concentrations of perhexiline for 24 h. Upon the completion of the treatment, various endpoints including cellular ATP level, cell viability, and LDH release were measured. As shown in Fig. 6, EV control cells pre-incubated with quinidine showed a similar response towards perhexiline exposure, compared to those pretreated with DMSO control. In CYP2D6-overexpressing HepG2 cells, quinidine significantly diminished the protective effect by CYP2D6-mediated metabolism. The effect of the inhibitor was prominent for perhexiline exposure at 10 μM, the concentration that showed significant cytotoxicity. The CYP2D6 inhibitor did not completely reverse the reduction in cytotoxicity by CYP2D6, probably because of the high efficiency of CYP2D6-mediated metabolism of perhexiline and the incomplete inhibition during the process. Nevertheless, the results from the inhibitory study confirmed further that CYP2D6-mediated metabolism alleviates the perhexiline-induced toxicity.
Fig. 6.
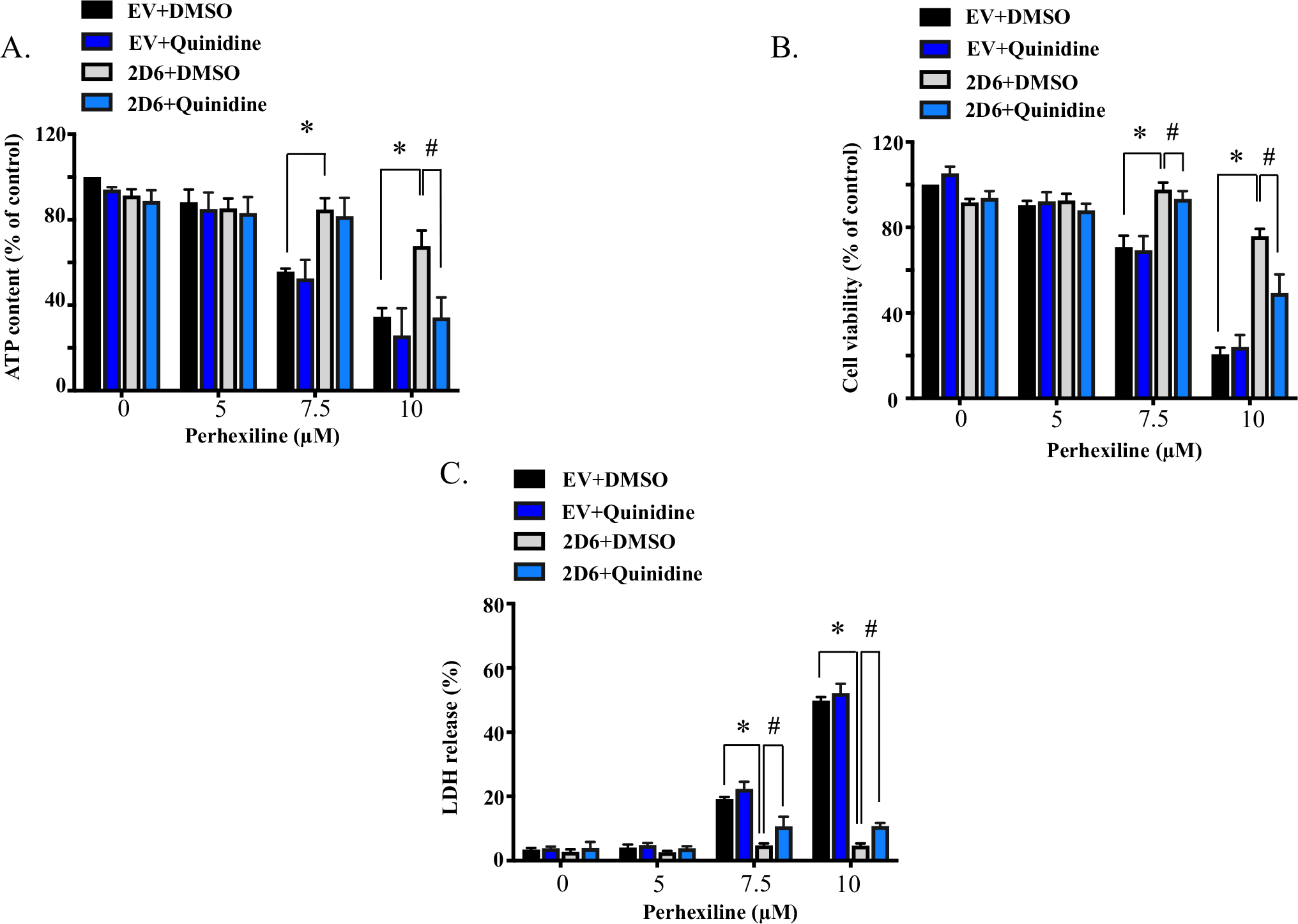
Quinidine diminishes the protective effect in CYP2D6-overexpressing HepG2 cells towards perhexiline. CYP2D6-overexpressing HepG2 cells were pretreated with CYP2D6-specific inhibitor quinidine (10 μM) or control DMSO for 2 h before exposed to various concentrations of perhexiline for 24 h. Cellular ATP contents (A), cell viability (B), and LDH release (C) were measured as toxicity endpoints. The results shown are mean ± SD from 3 biological replicates. *p < 0.05 for comparison between CYP2D6-overexpressing HepG2 cells and EV control cells with DMSO pretreatment. #p < 0.05 for comparison between DMSO and quinidine pretreated CYP2D6-overexpressing HepG2 cells
CYP2D6-mediated metabolism reduced perhexiline-induced mitochondrial dysfunction and ER stress
In our previous studies, we demonstrated that perhexiline exposure caused mitochondrial dysfunction (Ren et al. 2020). Since CYP2D6-overexpressing HepG2 cells demonstrated significant resistance towards the cytotoxicity of perhexiline and its specific inhibitor reversed such effect (Figs. 5, 6), we explored whether the mitochondrial dysfunction induced by perhexiline was also diminished in these cells. One classic assay for mitochondrial damage is the “glucose-galactose assay” (Marroquin et al. 2007). When the energy source in the medium is changed from glucose to galactose, HepG2 cells will be forced to rely on oxidative phosphorylation for the ATP production, and thus are more sensitive to mitochondrial toxicants. We conducted this assay in the CYP2D6-overexpressing HepG2 cells and EV control cells exposed to 5–10 μM perhexiline, and measured cell viability upon the completion of the treatment. As shown in Supplemental Fig. S1A, for EV control HepG2 cells, the cell viability levels in galactose-containing medium were decreased compared to that in glucose-containing medium. A significant difference between cells grown in the two media started to occur at 5 μM. When the same procedure was applied to CYP2D6-overexpressing cells, no obvious difference was observed on the cell viability levels between cells grown in the two types of media except 10 μM perhexiline, the highest concentration tested.
Next, we measured the membrane potential of mitochondria using JC-1 staining on cells exposed to various concentrations of perhexiline. As we reported in Fig. 2F, in HepG2 cells, treatment with perhexiline resulted in a significant decrease in mitochondrial membrane potential, suggesting the occurrence of mitochondrial damage. A similar reduction was seen in EV control HepG2 cells (Supplemental Fig. S1B); however, this reduction was significantly attenuated in CYP2D6-overexpressing HepG2 cells.
CYP2D6-mediated metabolism protected cells from perhexiline-induced apoptosis and ER stress
Perhexiline induced a significant increase in caspase 3/7 in HepG2 cells (Fig. 2E), which is considered a hallmark of apoptosis. A similar increase was observed in EV control HepG2 cells after perhexiline treatment for 24 h (Supplemental Fig. S1C). In CYP2D6-overexpressing HepG2 cells, however, this increase was abolished.
Our previous study also showed the involvement of ER stress in perhexiline-induced liver injury (Ren et al. 2021); thus, we investigated whether CYP2D6-mediated metabolism could protect the cells from the occurrence of ER stress. We examined the expression levels of several hallmark genes in ER stress pathways. In EV control cells, the mRNA levels of hallmark genes ATF6 (activating transcription factor 6), ERN1 (endoplasmic reticulum to nucleus signaling 1), EIF2AK3 (eukaryotic translation initiation factor 2 alpha kinase 3), ATF4 (activating transcription factor 4), and DDIT3 (DNA damage inducible transcript 3) were all significantly elevated when exposed to 10 μM perhexiline for 24 h, as measured by fold changes compared to the DMSO-treated control (Supplemental Fig. S1D). This increase was drastically attenuated in CYP2D6-overexpressing HepG2 cells. In addition, the ER stress marker CHOP (C/EBP homologous protein), the protein product of gene DDIT3, was markedly increased in EV control cells upon perhexiline treatment (Supplemental Fig. S1E). This increase was greatly reduced in CYP2D6-overexpressing HepG2 cells. Collectively, these observations suggest that CYP2D6-mediated metabolism reduced perhexiline-induced mitochondrial dysfunction, apoptosis, and ER stress, and thus protected hepatic cells from its toxicity.
CYP3A4-mediated metabolism showed marginal protective effect against perhexiline-induced mitochondrial damage and ER stress
Besides CYP2D6, in intact CYP3A4-overexpressing cells and microsomes, the mass spectrometry analysis indicated that CYP3A4 also played important role in the metabolism of perhexiline (Figs. 3, 4 and Table 1). Although CYP3A4-overexpressing HepG2 cells did not demonstrate significant resistance towards the overall cytotoxicity of perhexiline (Fig. 5), it was of interest to explore whether perhexiline-induced mitochondrial damage, apoptosis, and ER stress were changed in these cells. In the “glucose-galactose assay”, the viability of the CYP3A4-overexpressing HepG2 cells cultured in galactose medium started to decrease drastically at 7.5 μM perhexiline, whereas no difference between cells grown in the two media was observed at 5 μM perhexiline, in contrast to the results in EV controls (Supplemental Figs. S2A, S1A). Consistent with this observation, CYP3A4-overexpressing HepG2 cells also showed a marginal protective effect in JC-1 staining (Supplemental Fig. S2B). Measurement of the caspase 3/7 activities showed little difference between EV control cells and CYP3A4-overexpressing HepG2 cells (Supplemental Fig. S2C). These observations suggested that CYP3A4-mediated metabolism could also protect cells from perhexiline-induced mitochondrial damage, although to a much lesser extent in comparison to CYP2D6.
Discussion
Previous reports on the hepatic and neurological adverse effects of perhexiline occurred when the drug plasma concentrations reached between 720 and 2680 μg/L (2.59 and 9.65 μM) (Ashrafian et al. 2007). In the current study, the drug concentrations ranged from 2.5 μM to 15 μM, and cytotoxicity was observed starting at 5 μM perhexiline. Given the potential fluctuation of drug plasma concentrations before a steady status is reached, and a tendency for perhexiline to accumulate in liver (Ashrafian et al. 2007), the concentrations used here are clinically relevant and meaningful.
The reported hepatotoxicity of perhexiline occurred primarily in CYP2D6 poor metabolizers. In these patients, the plasma concentrations of the parent drug perhexiline were much higher in comparison to other patients, indicating a significant role of CYP2D6 in the metabolism and hepatotoxicity of perhexiline (Davies et al. 2006; Shah et al. 1982; Sørensen et al. 2003). In our study, when using mass spectrometry to analyze the amount of parent drug perhexiline in 14 CYPs-overexpressing HepG2 cells, we found that in CYP2D6-overexpressing HepG2 cells, the perhexiline level was reduced to near 1% of the level in EV control HepG2 cells (Fig. 3B). The results demonstrated a good metabolic efficiency of CYP2D6 in our cell system. This was consistent with the previous clinical report that in known extensive metabolizers, the plasma perhexiline was undetectable beyond 24 h after a single administration (Cooper et al. 1987). Further analysis using recombinant human CYPs also demonstrated that CYP2D6 played a pivotal role in the metabolism of perhexiline. In particular, among the four CYPs that potentially metabolize perhexiline, CYP2D6 is the predominant enzyme for the formation of cis-OH-Phx (M5) (Table 1). The high levels of cis-OH-Phx generated in intact HepG2 cells overexpressing CYP2D6 (Fig. 3C) cross-validated this result. Moreover, the CYP2D6 selective inhibitor quinidine suppressed the generation of cis-OH-Phx (M5) to 26% of control in human liver microsomes, which confirmed further the predominant role of CYP2D6 in the production of cis-OH-Phx. The contribution of CYP2D6 to the generation of cis-OH-Phx found in our current study is in line with several clinical reports (Amoah et al. 1986; Cooper et al. 1984, 1987).
Another enzyme identified to have a potential role in the metabolism of perhexiline is CYP3A4, as depicted in Table 1. CYP3A4 was the major enzyme responsible for the formation of M3, and contributed greatly to the formation of M1, both of which were identified as trans-OH-Phx. Inhibition of CYP3A4 in human liver microsomes reduced M1 to 75% and M3 to 19% of control (Fig. 4B), which confirmed further the contribution of CYP3A4 to the generation of trans-OH-Phx. The results in CYP3A4-overexpressing HepG2 cells also demonstrated a drastic 144-fold higher level of trans-OH-Phx, in comparison to EV-control cells. Notably, the metabolism by CYP3A4 was less efficient than that by CYP2D6, because abundant parent perhexiline was still detected in CYP3A4-overexpressing HepG2 cells after a 24 h exposure (Fig. 3B). These results were in good agreement with the findings by Davies et al. (2007), that CYP2D6 had a primary role in catalyzing the formation of cis-OH-Phx, and in the absence of CYP2D6, CYP3A4 could also metabolize perhexiline with low affinity (Davies et al. 2007).
It is worth noting that the role of CYP2D6 on the generation of trans-OH-Phx in the microsome study and in the cell study was less clear in our study. The results from recombinant human CYPs suggested that CYP2D6 also played a significant role in the formation of M1 (trans-OH-Phx) (Table 1), which was further supported by the inhibitory study with human liver microsomes (Fig. 4B). A previous study by Davies et al. (2007) using similar systems also demonstrated that CYP2D6 contributed considerably to the formation of trans-OH-Phx-1 with a high affinity, but not to trans-OH-Phx-2, whereas CYP3A4 contributed to the formation of both diastereomers of trans-OH-Phx (Davies et al. 2007). In CYP2D6-overexpressing HepG2 cells, however, after a 24 h perhexiline exposure, a very limited amount of trans-OH-Phx was detected. A closer look at the data suggested that CYP2D6 cells indeed generated more M1 compared to EV control cells, although it was only a 2.6-fold increase, much lower than that observed in CYP3A4 cells (Supplemental Fig. S3). Interestingly, multiple research groups have reported that the plasma trans-OH-Phx level was undetectable in most patients post-dosage, or very low in the few cases that it could be detected (Amoah et al. 1986; Cooper et al. 1984, 1987), whereas the generation of cisOH-Phx demonstrated a proportional relationship with the number of functional alleles in the patients (Davies et al. 2006). These reports suggested the low level of trans-OH-Phx in the in vivo metabolism of perhexiline supported the observation that cis-OH-Phx is the major metabolite in CYP2D6-mediated metabolism, similar to what we observed in CYP2D6-ovexpressing cells. One potential explanation for this discrepancy among the testing systems is that the drug presented in the CYP-overexpressing cells for a much longer period of time than in the human liver microsomes or the recombinant human CYP450s. As noted above, CYP3A4-mediated metabolism has much lower efficiency than that mediated by CYP2D6; therefore, the results in these two systems may not reflect the status of the metabolites and the contributions of the CYPs for a longer time frame. Also, longer incubation times could lead to secondary metabolite formation from trans-OH-Phx in CYP2D6-overexpressing cells, which resulted in low levels of trans-OH-Phx in the system. Another possibility is that the phase II metabolism or other cellular process could lead to the elimination of phase I metabolites. These enzymes and processes are absent in the bactosome system. Further studies using different cellular systems and more time points will be needed to answer this question.
Another point needs to be addressed is that although CYP3A4 demonstrated metabolizing capacity of perhexiline, in CYP3A4-overexpressing HepG2 cells, the cytotoxicity of perhexiline was not significantly reduced, compared to that in EV control cells (Fig. 5). The results were different from those observed in CYP2D6-overexpressing HepG2 cells. Mechanistic studies showed that the perhexiline-induced mitochondrial damage was marginally reduced in CYP3A4-overexpressing HepG2 cells (Supplemental Fig. S2). However, these modest effects did not result in an overall protective effect against the cytotoxicity of perhexiline. CYP2D6-mediated metabolism reduced the mitochondrial damage and diminished the increase in caspase 3/7 activity. It also attenuated the perhexiline-induced increase in ER stress markers (Supplemental Fig. S1). Overall CYP2D6-overexpressing cells showed significant resistance towards perhexiline-induced cytotoxicity. This might be explained by the difference between the capacity of CYP2D6 and CYP3A4 to metabolize perhexiline. During the 24 h treatment time in the current study, it was highly likely that in CYP3A4-overexpressing HepG2 cells, a large proportion of the parent perhexiline still remained, which contributed to the overall toxicity observed. In addition, CYP3A4 was more efficient in producing trans-OH-Phx than cis-OH-Phx (Table 1). Because trans-OH-Phx (IC50 value = 56 μM) was more cytotoxic than cis-OH-Phx (IC50 value = 90 μM), although both metabolites were less toxic than its parent drug (IC50 value = 8 μM) (Fig. 4C), it cannot be ruled out that the lower metabolic efficacy and the higher proportion of trans-OH-Phx were the reasons for the absence of an overall protective effect of CYP3A4-overexpressing HepG2 cells.
In addition to CYP3A4 and 2D6, we also detected a reduced level of perhexiline in CYP2C19- and 1A2-overexpressing cells, indicating a possible metabolic role of these two subtypes (Fig. 3B). Experiments in human liver microsomes indicated that CYP2C19 contributed significantly to the formation of M3 (trans-OH-Phx-2) (Table 1); this was consistent with the levels of trans-OH-Phx in CYPs-overexpressing cells (Fig. 3D). Application of the CYP2C19 inhibitor nootkatone, however, showed little effect on the relative abundance of trans-OH-Phx (Fig. 4B). In CYP2C19-overexpressing HepG2 cells, the cytotoxicity of perhexiline was not significantly attenuated (Fig. 5), suggesting a minor role of CYP2C19-mediated metabolism in the cytotoxicity of perhexiline. Similarly, although the parent drug level was decreased in CYP1A2-overexpressing HepG2 cells, the results in human liver microsomes and the cytotoxicity profile of perhexiline in CYP1A2-overexpressing cells did not suggest a pivotal role of CYP1A2 in the formation of M1–M6. No clinical assessment regarding the role of these two subtypes of CYPs in the metabolism of perhexiline has been documented. It is also worth mentioning that Davies et al (2007) identified CYP2B6 as one of the contributing enzymes for the metabolism of perhexiline (Davies et al. 2007). In the current study, we also observed a small increase in the formation of trans-OH-Phx (5.7-fold of EV-control, Fig. 3D) in CYP2B6-overexpressing HepG2 cells after perhexiline treatment; however, the decrease of perhexiline was not detected in CYP2B6 cells (Fig. 3B), and nor was a protective effect against the cytotoxicity of perhexiline observed (Supplemental Fig. S4). As Davies et al (2007) suggested in their study, the metabolism by CYP2B6 was with low affinity, which could be one explanation why we did not observe its effect in the time frame of our study (Davies et al. 2007). We also observed the production of cis-OH-Phx and trans-OH-Phx in other CYPs. For instance, we detected cis-OH-Phx in CYP3A7, 3A5, and 2C18 and trans-OH-Phx in CYP3A7, 3A5, 2C19, and 2C18. However, no significant decrease of parent perhexiline was detected, suggesting these CYPs metabolized perhexiline to a very minor extent. Therefore, the role of these CYPs in perhexiline’s cytotoxicity was presumably not critical, at least in the test systems used in our study.
Perhexiline is known to be a racemic mixture containing both (+) and (−) enantiomers. It has been shown that metabolizing enzymes including CYPs often demonstrate preferences among the enantiomers of a chiral drug, and thus result in enantioselectivity. Previous studies have suggested that differences might exist in the disposition of distinct enantiomers of perhexiline (Ashrafian et al. 2007; Davies et al. 2006, 2008). Stereoselective metabolism of drugs is often the major contributing factor to stereoselectivity in pharmacokinetics. Metabolizing enzymes display a preference for one enantiomer of a chiral drug over the other, resulting in enantioselectivity. In the case of perhexiline, the (−)-perhexiline has been shown to have a higher clearance rate, and this enantioselectivity could primarily be attributed to CYP2D6-mediated metabolism (Davies et al. 2008). Davies et al. (2007) suggested in their study that CYP2D6 catalyzed the formation of different cis-OH-Phx and trans-OH-Phx enantiomers from (+)- and (−)-perhexiline (Davies et al. 2007). Due to technical limit of our experiment setting, the detailed contribution of CYPs towards various diastereomers of perhexiline was not addressed in this current study and it warrants a further investigation.
In summary, to our best knowledge, this is the first study demonstrating the pivotal role of CYP2D6 in the metabolism and cytotoxicity of perhexiline in intact cells in culture. The underlying mechanisms of CYP2D6 protective effects against the cytotoxicity of perhexiline were unmasked. The identification of other CYPs could also improve our understanding of perhexiline-induced liver injury. Clinically, the hepatotoxicity of perhexiline was frequently observed in CYP2D6 poor metabolizers; however, not every CYP2D6 poor metabolizers developed the severe side effect. This suggests that other metabolizing enzymes and the balance of the multiple metabolites contribute to the metabolism and toxicity. Our study provided insights on the metabolic profile and toxicity of a drug like perhexiline, which may facilitate the mitigation of its undesired adverse effect.
Supplementary Material
Funding
This study was funded by the National Center for Toxicological Research/U.S. Food and Drug Administration. Drs. Feng Li and Xuan Qin were supported by the National Institute of Diabetes and Digestive and Kidney Diseases (R01-DK121970) and the Eunice Kennedy Shriver National Institute of Child Health and Human Development (R61HD099995) to Feng Li.
Footnotes
Declarations
Conflicts of interest The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Disclaimer
This article reflects the views of the authors and does not necessarily reflect those of the U.S. Food and Drug Administration. Any mention of commercial products is for clarification only and is not intended as approval, endorsement, or recommendation.
Supplementary Information The online version contains supplementary material available at https://doi.org/10.1007/s00204-022-03369-0.
References
- Amoah AGB, Gould BJ, Parke DV, Lockhart JDF (1986) Further studies on the pharmacokinetics of perhexiline maleate in humans. Xenobiotica 16(1):63–68 [DOI] [PubMed] [Google Scholar]
- Ashrafian H, Horowitz JD, Frenneaux MP (2007) Perhexiline. Cardiovasc Drug Rev 25(1):76–97 [DOI] [PubMed] [Google Scholar]
- Chen S, Xuan J, Wan L, Lin H, Couch L, Mei N, Dobrovolsky VN, Guo L (2014) Sertraline, an antidepressant, induces apoptosis in hepatic cells through the mitogen-activated protein kinase pathway. Toxicol Sci 137(2):404–415 [DOI] [PubMed] [Google Scholar]
- Chen M, Suzuki A, Borlak J, Andrade RJ, Lucena MI (2015) Drug-induced liver injury: interactions between drug properties and host factors. J Hepatol 63(2):503–514 [DOI] [PubMed] [Google Scholar]
- Chen S, Ren Z, Yu D, Ning B, Guo L (2018a) DNA damage-induced apoptosis and mitogen-activated protein kinase pathway contribute to the toxicity of dronedarone in hepatic cells. Environ Mol Mutagen 59(4):278–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Wu Q, Ning B, Bryant M, Guo L (2018b) The role of hepatic cytochrome P450s in the cytotoxicity of dronedarone. Arch Toxicol 92(6):1969–1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Wu Q, Li X, Li D, Fan M, Ren Z, Bryant M, Mei N, Ning B, Guo L (2020) The role of hepatic cytochrome P450s in the cytotoxicity of sertraline. Arch Toxicol 94(7):2401–2411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Wu Q, Li X, Li D, Mei N, Ning B, Puig M, Ren Z, Tolleson WH, Guo L (2021) Characterization of cytochrome P450s (CYP)-overexpressing HepG2 cells for assessing drug and chemical-induced liver toxicity. J Environ Sci Health C Toxicol Carcinog 39(1):68–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper RG, Evans DA, Whibley EJ (1984) Polymorphic hydroxylation of perhexiline maleate in man. J Med Genet 21(1):27–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper RG, Evans DA, Price AH (1987) Studies on the metabolism of perhexiline in man. Eur J Clin Pharmacol 32(6):569–576 [DOI] [PubMed] [Google Scholar]
- Davies BJ, Coller JK, James HM, Somogyi AA, Horowitz JD, Sallustio BC (2006) The influence of CYP2D6 genotype on trough plasma perhexiline and cis-OH-perhexiline concentrations following a standard loading regimen in patients with myocardial ischaemia. Br J Clin Pharmacol 61(3):321–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies BJ, Coller JK, Somogyi AA, Milne RW, Sallustio BC (2007) CYP2B6, CYP2D6, and CYP3A4 catalyze the primary oxidative metabolism of perhexiline enantiomers by human liver microsomes. Drug Metab Dispos 35(1):128–138 [DOI] [PubMed] [Google Scholar]
- Davies BJ, Herbert MK, Coller JK, Somogyi AA, Milne RW, Sallustio BC (2008) Steady-state pharmacokinetics of the enantiomers of perhexiline in CYP2D6 poor and extensive metabolizers administered Rac-perhexiline. Br J Clin Pharmacol 65(3):347–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhakal B, Li CMY, Li R, Yeo K, Wright JA, Gieniec KA, Vrbanac L, Sammour T, Lawrence M, Thomas M, Lewis M, Perry J, Worthley DL, Woods SL, Drew P, Sallustio BC, Smith E, Horowitz JD, Maddern GJ, Licari G, Fenix K (2022) The antianginal drug perhexiline displays cytotoxicity against colorectal cancer cells in vitro: a potential for drug repurposing. Cancers 14(4):1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Zhu L, Ma J, Lin G (2021) Metabolism-mediated cytotoxicity and genotoxicity of pyrrolizidine alkaloids. Arch Toxicol 95(6):1917–1942 [DOI] [PubMed] [Google Scholar]
- Hofman J, Sorf A, Vagiannis D, Sucha S, Kammerer S, Küpper JH, Chen S, Guo L, Ceckova M, Staud F (2019) Brivanib exhibits potential for pharmacokinetic drug-drug interactions and the modulation of multidrug resistance through the inhibition of human ABCG2 drug efflux transporter and CYP450 biotransformation enzymes. Mol Pharm 16(11):4436–4450 [DOI] [PubMed] [Google Scholar]
- Hofman J, Vagiannis D, Chen S, Guo L (2021) Roles of CYP3A4, CYP3A5 and CYP2C8 drug-metabolizing enzymes in cellular cytostatic resistance. Chem Biol Interac 340:109448. [DOI] [PubMed] [Google Scholar]
- Inglis S, Stewart S (2006) Metabolic therapeutics in angina pectoris: history revisited with perhexiline. Eur J Cardiovasc Nurs 5(2):175–184 [DOI] [PubMed] [Google Scholar]
- MacKenzie KR, Zhao M, Barzi M, Wang J, Bissig K-D, Maletic-Savatic M, Jung SY, Li F (2020) Metabolic profiling of norepinephrine reuptake inhibitor atomoxetine. Eur J Pharm Sci 153:105488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marroquin LD, Hynes J, Dykens JA, Jamieson JD, Will Y (2007) Circumventing the Crabtree effect: replacing media glucose with galactose increases susceptibility of HepG2 cells to mitochondrial toxicants. Toxicol Sci 97(2):539–547 [DOI] [PubMed] [Google Scholar]
- Midei MG, Darpo B, Ayers G, Brown R, Couderc J-P, Daly W, Ferber G, Sager PT, Camm AJ (2021) Electrophysiological and ECG effects of perhexiline, a mixed cardiac ion channel inhibitor, evaluated in nonclinical assays and in healthy subjects. J Clin Pharmacol 61(12):1606–1617 [DOI] [PubMed] [Google Scholar]
- Mueller-Schoell A, Michelet R, Weinelt F, Kloft C, Mikus G (2021) CYP2D6 phenotype explains reported yohimbine concentrations in four severe acute intoxications. Arch Toxicol 95(8):2867–2870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Z, Chen S, Zhang J, Doshi U, Li AP, Guo L (2016) Endoplasmic reticulum stress induction and ERK1/2 activation contribute to nefazodone-induced toxicity in hepatic cells. Toxicol Sci 154(2):368–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Z, Chen S, Seo JE, Guo X, Li D, Ning B, Guo L (2020) Mitochondrial dysfunction and apoptosis underlie the hepatotoxicity of perhexiline. Toxicol in Vitro 34:104987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Z, Chen S, Pak S, Guo L (2021) A mechanism of perhexiline’s cytotoxicity in hepatic cells involves endoplasmic reticulum stress and p38 signaling pathway. Chem Biol Interac 334:109353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallustio BC, Westley IS, Morris RG (2002) Pharmacokinetics of the antianginal agent perhexiline: relationship between metabolic ratio and steady-state dose. Br J Clin Pharmacol 54(2):107–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah RR, Oates NS, Idle JR, Smith RL, Lockhart JD (1982) Impaired oxidation of debrisoquine in patients with perhexiline neuropathy. Br Med J (clin Res Ed) 284(6312):295–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sørensen LB, Sørensen RN, Miners JO, Somogyi AA, Grgurinovich N, Birkett DJ (2003) Polymorphic hydroxylation of perhexiline in vitro. Br J Clin Pharmacol 55(6):635–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todorović Vukotić N, Đorđević J, Pejić S, Đorđević N, Pajović SB (2021) Antidepressants- and antipsychotics-induced hepatotoxicity. Arch Toxicol 95(3):767–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagiannis D, Novotna E, Skarka A, Kammerer S, Küpper J-H, Chen S, Guo L, Staud F, Hofman J (2020) Ensartinib (X-396) effectively modulates pharmacokinetic resistance mediated by ABCB1 and ABCG2 drug efflux transporters and CYP3A4 biotransformation enzyme. Cancers 12(4):813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q, Ning B, Xuan J, Ren Z, Guo L, Bryant MS (2016) The role of CYP 3A4 and 1A1 in amiodarone-induced hepatocellular toxicity. Toxicol Lett 253:55–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xuan J, Chen S, Ning B, Tolleson WH, Guo L (2016) Development of HepG2-derived cells expressing cytochrome P450s for assessing metabolism-associated drug-induced liver toxicity. Chem Biol Interact 255:63–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.