

Abstract
Kir6.2/SUR1 is an ATP-regulated potassium channel that acts as an intracellular metabolic sensor, controlling insulin and appetite-stimulatory neuropeptides secretion. In this Letter, we present the SAR around a novel Kir6.2/SUR1 channel opener scaffold derived from an HTS screening campaign. New series of compounds with tractable SAR trends and favorable potencies are reported.
To create your abstract, type over the instructions in the template box below. Fonts or abstract dimensions should not be changed or altered.9.0_/0
Keywords: KATP Channels, Channel opener, Kir6.2/SUR1, Insulin homeostasis, Structure-Activity, Relationship (SAR)
Graphical Abstract
ATP-regulated potassium channels (KATP channels) are important intracellular metabolic sensors consisting of a heterooctameric assembly of four Kir6 and four SUR subunits.1,2 Figure 1A summarizes the tissue-specific expression of various KATP isoforms. In particular, the Kir6.2/SUR1 subfamily channel has been considered an important drug target due to its role in glucose homeostasis through the control of insulin secretion in pancreatic β-cells.3–7 Recent studies have also shown that Kir6.2/SUR1 channels may be involved in the secretion of appetite-stimulatory neuropeptides in hypothalamic neurons, further exemplifying the importance of the channel as a valuable therapeutic target.8
Figure 1.

KATP channel isoforms and selected Kir6.2/SUR1 channel openers. (A) Diversity of KATP isoforms and their tissue-specific expression. (B) Structures of representative Kir6.2/SUR1 channel openers 1–4.6,11,12,13,16 aReported EC50 value; EC50 was 0.75 ± 0.08 μM (Mean ± SEM, n = 6 experiments) in thallium flux assays with T-REx-HEK293 cells
Although Diazoxide (1), an FDA-approved KATP channel opener, is still widely used in the clinic, its off-target effects on vascular KATP channels limit its utility to efficiently treat abnormalities (such as hyperinsulinism, and hypoglycemia).6,9,10 As such, there remains an unmet need in the field for alternative therapeutics. In 2003, NN414 (2) was discovered by Novo Nordisk as a next-generation KATP channel opener. NN414 (EC50 = 0.45 μM; ref 5) was over sixty-fold more potent than Diazoxide (EC50 = 30 μM), while showing good Kir6.2/SUR1 channel selectivity and giving hope to the field.4,11 However, the clinical development of NN414 was suspended during phase 2 clinical trials due to an adverse effect on the liver.3 Besides NN414 (and its close analogs) and (±)-Cromakalim derivative BMS-191095 (3), only a limited number of Kir6.2/SUR1 channel opener chemotypes have been reported in the public domain,6,11–16 many of which only show moderate potencies (e.g. 4, EC50 = 7 μM)6 or contained pharmaceutically disadvantageous moieties. In this Letter, we describe the synthesis and structure-activity relationship (SAR) of a structurally distinct Kir6.2/SUR1 channel opener scaffold derived from an HTS screening campaign.
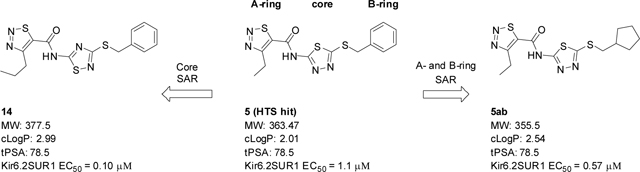
Our ongoing HTS efforts to find potent and selective Kir6.2/SUR1 channel openers have identified several hits of interest including 5-8 (Figure 2A–C); 20,480 compounds were screened at a single dose of 10 μM via high-throughput thallium flux assay and 513 hits were confirmed with 2.5% hit rate. This 20,480 compound library is a chemically diverse subset of the Vanderbilt Discovery Collection. The Discovery Collection (~100,000 total) is made up of lead-like motifs, minimum panassay interference, and maximum chemically diverse small molecules (mean MW = 378) from Life Chemicals. Compounds 5, 7, and 8 showed complete subtype selectivity against Kir6.1/SUR2B channel (a vascular KATP channel), while 6 showed weak activity (EC50 > 10 μM). Especially, compound 5 (EC50 = 1.2 μM) was about five-fold more potent than our first-generation tool compound (VU0071063 (4), EC50 = 7 μM) and was selected as our new starting point. To understand the basic SAR texture around 5, we divided the molecule into three parts, the A-ring, the core, and the B-ring, and explored each part separately (Figure 2D).
Figure 2.

Summary of HTS screening results and hit to lead optimization strategy (A) Summary of Primary Screening results revealed small molecules that increase the measurement of thallium flux in cells containing Kir6.2SUR1 channels. Hits were identified as Activators when the increase was greater than 3 Standard deviations from the mean plate response (N = 320). Hits selected for retesting in duplicate included the 327 Activators (>3 SD in blue) and 249 weaker activators (B-score > 5). Retest positives remaining after the duplicated testing included 513 of the 576 tested. Comparison to Counter screening reduced these to 494 promoting to candidates for Concentration Response Curve studies (Omit 63 retest negatives or 19 actives in comparison to Kir6.1SUR2B or HEK). Values are displayed normalized to the plate control VU0071063 (positive control in purple n = 32 per plate). Total tested small molecules were 20480. Not shown (B-score Activators (1254), B-score Inhibitors (272). (B) Concentration Response Curve of selected Compounds 5-8. Triplicate values of 9 concentrations ranging 5 nM-30 μM*, plots using GraphPad Prism 9.2 *30 μM omitted on 5. (C) Structures of selected HTS hits 5 – 8. (D) Compound Optimization Strategy.
The synthesis of 5 and its related analogs is outlined in Scheme 1. An EDCI coupling reaction between commercially available aminothiadiazoles 10 and readily available carboxylic acids 9 provided HTS hit 5 as well as A-ring and B-ring variants in low to moderate yields. Several B-ring variants were synthesized in two steps starting from 11 through either an amidation-alkylation sequence (Scheme 1b) or an alkylation-amidation sequence (Scheme 1c).
Scheme 1.

Synthesis of Kir6.2/SUR1 channel openersa
aReagents and conditions: (a) EDCI·HCl, HOBt, DMAP, DMF, 50 °C, o/n; (b) alkyl bromides, KOH, EtOH/H2O (3:1), rt, 1 h.
Our initial efforts were focused on the A-ring pendant group. As shown in Table 1, the replacement of the ethyl moiety on the 4-position of the 1,2,3-thiadiazole ring with a methyl-group was well tolerated (5a, EC50 = 1.8 μM), while slightly larger substituents marginally improved potency (5b-d). Interestingly, a phenyl ring on the 4-position was also well tolerated (5e, EC50 = 0.78 μM). This result indicates there may be adequate room in the binding pocket to accommodate larger substitutions. As our initial screen of analogs demonstrated that the 4-position of the 1,2,3thiadiazole tolerated simple substituents, we shifted our focus to evaluate the SAR of other areas of the scaffold. The A-ring SAR could then be revisited in the context of an optimized B-ring and core.
Table 1.
Structures and activities for A-ring variants 5a-5e
| |||
|---|---|---|---|
| Cmpd | R= | Synthetic Route | Kir6.2/SUR1 EC50 (μM) [Emax (%)] |
| 5HTS hit |
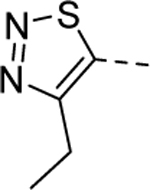
|
1 | 1.1a [93] |
| 5a |
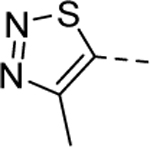
|
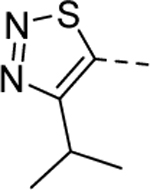
1 | 1.8a,c [91] |
| 5b |
|
1 | 0.46b [82] |
| 5c |
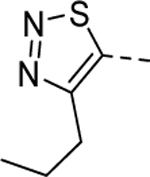
|
1 | 0.25b [87] |
| 5d |
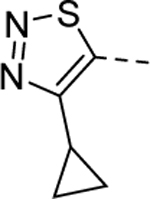
|
1 | 0.66b [90] |
| 5e |
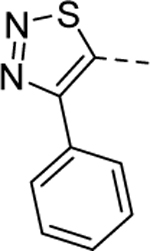
|
1 | 0.78b [87] |
Thallium flux assays with T-REx-HEK293 cells; values represent means from one experiment performed in triplicate. 4 was used as a positive control. 0.48 mM Tl2SO4 was used.
Thallium flux assays with T-REx-HEK293 cells; values represent means from one experiment performed in triplicate. 2 (EC50 = 0.75 ± 0.08 μM) was used as a positive control.
Values represent means from two experiments performed in triplicate.
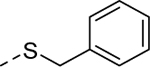
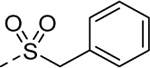
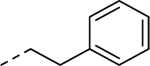
Holding the A-ring and the core constant (4-ethyl-1,2,3-thiadiazole and 2-amino-1,3,4-thiadiazole, respectively), our SAR focus then moved to the B-ring (Table 2). The first library (5f-5j) was aimed at replacing the thioether linker while maintaining the terminal phenyl ring. Although replacement of the sulfur atom with -SO2- (5f, EC50 = 35 μM) or -NH- (5g, inactive) were not tolerated, the regioisomeric thioether (5h, EC50 = 2.0 μM), ether (5i, EC50 = 3.3 μM), and simple ethylene linker (5j, EC50 = 4.1 μM) maintained good potencies. We were encouraged by this result as it shows that the thioether, a well-known metabolic soft spot, can be replaced with a pharmaceutically more desirable moiety.
Table 2.
Structures and activities for B-ring variants 5f-5ab
| |||||||
|---|---|---|---|---|---|---|---|
| Cmpd | R= | Synthetic Route | Kir6.2/SUR1 EC50 (μM) [Emax (%)] | Cmpd | R= | Synthetic Route | Kir6.2/SUR1 EC50 (μM) [Emax (%)] |
|
5 HTS hit |
|
1 | 1.1a [93] |
5q |
|
3 | 1.14b [105] |
| 5f |
|
1c | 35a [150] |
5r |
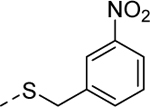
|
3 | 3.1a,e [82] |
| 5g |
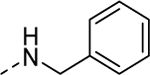
|
1d | Inactiveb | 5s |
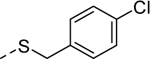
|
1 | 0.88b [83] |
| 5h |
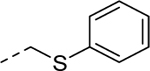
|
1 | 2.0a [89] |
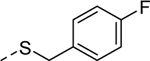
5t |
|
1 | 0.55b [85] |
| 5i |
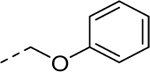
|
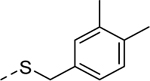
1 | 3.3a[100] | 5u |
|
2 | 0.97b [106] |
| 5j |
|
1 | 4.1a [96] |
5v |
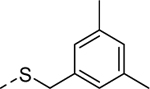
|
2 | 0.94b [98] |
| 5k |
|
1 | 6.5a [101] | 5w |
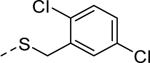
|
1 | 0.56b [105] |
| 5l |
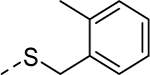
|
2 | 1.0a [69] |
5x |
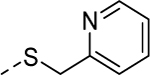
|
1 | Inactivea |
| 5m |
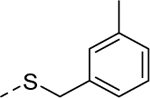
|
1 | 0.53b [83] |
5y |
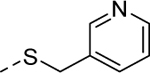
|
1 | Inactivea |
| 5n |
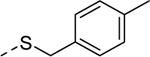
|
2 | 0.71b [105] |
5z |
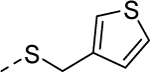
|
2 | 1.1a [77] |
| 5o |
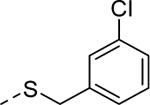
|
1 | 0.71b [100] |
5aa |
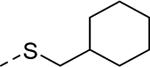
|
3 | 0.31a,e [54] |
| 5p |
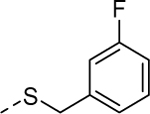
|
3 | 1.00b [109] |
5ab |
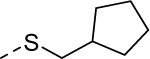
|
3 | 0.57b [111] |
Thallium flux assays with T-REx-HEK293 cells; values represent means from one experiment performed in triplicate. 4 was used as a positive control. 0.48 mM Tl2SO4 was used.
Thallium flux assays with T-REx-HEK293 cells; values represent means from one experiment performed in triplicate. 2 (0.75 ± 0.08 μM) was used as a positive control.
Intermediate, 5-(benzylsulfonyl)-1,3,4-thiadiazol-2-amine, was prepared using 2.2 equiv. of m-CPBA.
Intermediate, N2-benzyl-1,3,4-thiadiazole-2,5-diamine, was prepared by heating a mixture of 5-bromo-1,3,4-thiadiazol-2-amine (1 equiv.), benzylamine (1.5 equiv.), and triethylamine (2.5 equiv.) at 66 °C for 6 h.
Values represent means from two experiment performed in triplicate.
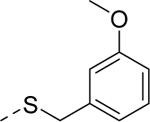
Because the des-phenyl analog (5k, EC50 = 6.5 μM) gave about 4-fold loss in potency, our attention turned to phenyl ring decoration instead (5l-5w). Although m- and p-mono methylated analogs (5m, EC50 = 0.53 μM and 5n, EC50 = 0.71 μM) were favored compared to an o-methylated compound (5l, EC50 = 1.0 μM), dimethylated analogs (5u, EC50 = 0.97 μM and 5v, EC50 = 0.94 μM) were not as potent as the mono methylated analogs. Among synthesized analogs, the only unexpected outlier to this trend came out from the 2,5-dichlorinated analog 5w (EC50 = 0.56 μM). As expected, we were able to use halogens as methyl surrogates (5o, 5p, 5s, and 5t). However, halogenated compounds were slightly less potent than their methylated comparators (5m and 5n). Interestingly, polar substituent-containing analogs also showed comparable potency to HTS hit 5 (5q, EC50 = 1.14 μM and 5r, EC50 = 3.1 μM).
Our efforts were then directed towards the replacement of the phenyl ring, exploring both aromatic heterocycles (such as pyridine and thiophene) and aliphatic carbocycles (cyclohexane and cyclopentane). 2- and 3-pyridyl compounds (5x and 5y, respectively) proved inactive. However, the more hydrophobic thiophene analog (5z, EC50 = 1.1 μM) showed comparable potency to HTS hit 5. Interestingly, both carbocycles gave a slight boost in potency (5aa, EC50 = 0.31 μM and 5ab, EC50 = 0.57 μM). This is noteworthy in that this substitution provides an opportunity to increase the sp3 character of the current chemotype while maintaining its potency. To that end, we designed our next-generation library based on the cyclopentyl moiety from 5ab.
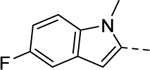
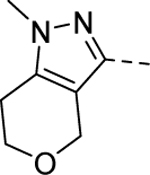
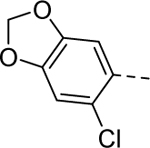
Maintaining the B-ring and the core as constants (cyclopentane and 2-amino-1,3,4-thiadiazol, respectively), we revisited the A-ring SAR (Table 3). However, synthesized analogs containing heterocycles other than a 4-substituted 1,2,3-thiadiazole were all inactive (5ac-5ae). The only exception to this trend was 5af (EC50 > 10 μM). Interestingly, only 5af contained a nitrogen atom (bolded) that could potentially mimic the 3-position nitrogen atom of 1,2,3-thiadiazole. Based on this result, we suspect that the 3-position nitrogen atom of the 1,2,3-thiadiazole ring may play an important role in binding. It certainly warrants a follow-up SAR study.
Table 3.
Structures and activities for alternative A-rings 5ac-5afa
| |||
|---|---|---|---|
| Cmpd | R = | SyntheticRoute | Kir6.2/SUR1 EC50 (μM) [Emax (%)] |
| 5ac |
|
1 | Inactive |
| 5ad |
|
1 | Inactive |
| 5ae |
|
1 | Inactive |
| 5af |
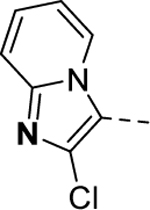
|
1 | >10 [97] |
Thallium flux assays with T-REx-HEK293 cells; values represent means from one experiment performed in triplicate. 2 (0.75 ± 0.08 μM) was used as a positive control.
Lastly, we altered the heterocyclic core (Figure 3). Although our attempt to replace the 1,2,3-thiadiazole core with a pyridazine core was unsuccessful (15, inactive), the 1,2,4-thiadiazole 14 was well tolerated and showed notable improvement (~10-fold) in potency compared to 5c and HTS hit 5 (14, EC50 = 0.10 μM).
Figure 3.

Structures and activities for core variations. aPrepared via Scheme 1a. bPrepared through an SNAr between 2-amino-6-chloropyridazine and benzylamine followed by amidation.
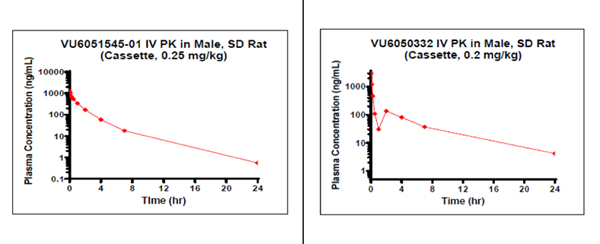
Table 4 presents the detailed ADME profiles as well as the subtype selectivity of 14 and 5ab. Although both compounds showed favorable on-target potencies (0.10 μM and 0.57 μM respectively), their free fraction (both plasma and brain) were quite low (rat, human fu < 0.01) indicating obvious room for improvement. Rat IV PK cassette study suggests both compounds were peripherally restricted (Kp = 0.03 and 0.04 respectively). Interestingly, both compounds showed in vitro-in vivo (IVIV) disconnect; the observed in vivo clearance (CLp, 3.02 and 2.43 mL min−1 kg−1, respectively) was significantly lower than the predicted clearance in the in vitro assay (Predicted Clhep, 66 and 55.3 mL min−1 kg−1, respectively). Although the low free fraction (fu) may contribute to this IVIV disconnect, follow-up experiments are warranted to elucidate the origin of the disconnection.
Table 4.
Tier 1 DMPK profile data for the selected compounds
| Property | 14, VU6051545 | 5ab, VU6050332 |
|---|---|---|
| Kir6.2/SUR1 EC50 (μM) | 0.10 ± 0.01 (n=3) | 0.57 ± 0.02 (n=3) |
| Kir6.1/SUR2B EC50 (μM) | inactive | inactive |
| MW | 377.5 | 355.5 |
| cLogP | 2.99 | 2.54 |
| TPSA | 81 | 81 |
| Fu, plasma (rat) | <0.01 | <0.01 |
| Fu, plasma (human) | <0.01 | <0.01 |
| Fu, brain (rat) | <0.01 | 0.01 |
| Kp | 0.03 | 0.04 |
| Clint (mL/min/kg) | 120 (h), 1168 (r) | 363 (h), 264 (r) |
| Predicted Clhep (mL/min/kg) | 17.9 (h), 66 (r) | 19.9 (h), 55.3 (r) |
| Rat IV PK cassette | ||
| CLp (mL/min/kg) | 3.02 | 2.43 |
| t1/2 (h) | 3.11 | 4.89 |
| MRT (h) | 2.12 | 4.11 |
| Vss (L/kg) | 0.38 | 0.60 |
| ||
In summary, we discovered a novel Kir6.2/SUR1 channel opener series from an HTS screening campaign. Our initial SAR exercise demonstrated tractable SAR textures that provide helpful insight for next-generation analog designs. Several compounds within this series showed improved potencies compare to NN414 (and Diazoxide). Potent and pharmaceutically favorable compounds within this series will be the subject of comprehensive subsequent pharmacological studies related to insulin secretion from the pancreatic β-cells, which will be published in due course.
Acknowledgments
We thank the following funding agent for their generous support of this work: Soleno therapeutics (to C.W.L.). We thank the William K. Warren Family and Foundation for funding the William K. Warren, Jr. Chair in Medicine and supporting our program. We also thank Dr. Christopher C. Presley for assistance with HRMS. The Vanderbilt HTS Core (VHTSC) receives support from the Vanderbilt Institute of Chemical Biology and the Vanderbilt Ingram Cancer Center (P30CA68485). The WaveFront Biosciences Panoptic kinetic imaging plate reader within the VHTSC was funded by NIH Shared Instrumentation Grant (S10OD021734). J.A.B. is supported by an NCI award (R50CA211206).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- (1).Shyng S; Nichols CG Octameric stoichiometry of the KATP channel complex. J Gen Physiol 1997, 110, 655–664. DOI: 10.1085/jgp.110.6.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Wang M; Wu J-X; Ding D; Chen L. Structural insights into the mechanism of pancreatic KATP channel regulation by nucleotides. Nat Commun 2022, 13, 2770. DOI: 10.1038/s41467-022-30430-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).B. HJ. Towards selective Kir6.2/SUR1 potassium channel openers, medicinal chemistry and therapeutic perspectives. Curr Med Chem 2006, 13, 361–376. DOI: 10.2174/092986706775527947. [DOI] [PubMed] [Google Scholar]
- (4).Zdravkovic M; Kruse M; Rost KL; Møss J; Kecskes A; Dyrberg T. The effects of NN414, a SUR1/Kir6.2 selective potassium channel opener, in healthy male subjects. J Clin Pharmacol 2005, 45, 763–772. DOI: 10.1177/0091270005276947. [DOI] [PubMed] [Google Scholar]
- (5).Dabrowski M; Larsen T; Ashcroft FM; Hansen JB; Wahl P. Potent and selective activation of the pancreatic beta-cell type K(ATP) channel by two novel diazoxide analogues. Diabetologia 2003, 46, 1375–1382. DOI: 10.1007/s00125-003-1198-1. [DOI] [PubMed] [Google Scholar]
- (6).Kharade SV; Sanchez-Andres JV; Fluton MG; Shelton EL; Blobaum AL; Engers DW; Hofmann CS; Dadi PK; Lantier L; Jacobson DA; et al. Structure-activity relationships, pharmacokinetics, and pharmacodynamics of the Kir6.2/SUR1-specific channel opener VU0071063. J Pharmacol Exp Ther 2019, 370, 350–359. DOI: 10.1124/jpet.119.257204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Raphmot R; Swale DR; Dadi PK; Jacobson DA; Cooper P; Wojtovich AP; Banerjee S; Nichols CG; Denton JS Direct activation of β-cell KATP channels with a novel xanthine derivative. Mol Pharmacol 2014, 85, 858–865. DOI: 10.1124/mol.114.091884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Cowen N; Bhatnagar A. The potential role of activating the ATP-sensitive potassium channel in the treatment of hyperphagic obesity. Genes 2020, 11, 450. DOI: 10.3390/genes11040450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Coetzee WA Multiplicity of effectors of the cardioprotective agent, diazoxide. Pharmacology & therapeutics 2013, 140, 167–175. DOI: 10.1016/j.pharmthera.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Standen NB; Quayle JM; Davies NW; Brayden JE; Huang Y; Nelson MT Hyperpolarizing vasodilators activate ATPsensitive K+ channels in arterial smooth muscle. Science 1989, 245, 177–180. DOI: 10.1126/science.2501869. [DOI] [PubMed] [Google Scholar]
- (11).Dabrowski M; Larsen T; Ashcroft FM; J. BH; Wahl P. Potent and selective activation of the pancreatic beta-cell type K(ATP) channel by two novel diazoxide analogues. Diabetologia 2003, 46, 1375–1382. DOI: 10.1007/s00125-003-1198-1. [DOI] [PubMed] [Google Scholar]
- (12).Raphemot R; Swale DR; Dadi PK; Jacobson DA; Cooper P; Wojtovich AP; Banerjee S; Nichols CG; Denton JS Direct activation of β-cell KATP channels with a novel xanthine derivative. Mol Pharmacol 2014, 85, 858–865. DOI: 10.1124/mol.114.091884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Schwanstecher M; Sieverding C; Dörschner H; Gross I; Aguilar-Bryan L; Schwanstecher C; Bryan J. Potassium channel openers require ATP to bind to and act through sulfonylurea receptors. EMBO 1998, 17, 5529–2235. DOI: 10.1093/emboj/17.19.5529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Please refer to ref 14, 15 and their references for more information: Mannhold, R. KATP channel openers: structure-activity relationships and therapeutic potential. Med. Res. Rev. 2004, 24, 213266. [DOI] [PubMed] [Google Scholar]
- (15).Dyhring T; Jansen-Olesen I; Christophersen P; Olesen J. Pharmacological profiling of KATP channel modulators: an outlook for new treatment opportunities for migraine. Pharmaceuticals 2023, 26, 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Cho M-R; Park J-W; Jung I-S; Yi K-Y; Yoo S-E; Chung H-J; Yun Y-P; Kwon S-H; Shin H-S BMS-191095, a cardioselective mitochondrial KATP opener, inhibits human platelet aggregation by opening mitochondrial KATP channels. Arch. Pharm. Res. 2005, 28, 61–67. [DOI] [PubMed] [Google Scholar]