

Abstract
Escherichia coli is both a highly prevalent commensal and a major opportunistic pathogen causing bloodstream infections (BSI). A systematic analysis characterizing the genomic determinants of extra-intestinal pathogenic vs. commensal isolates in human populations, which could inform mechanisms of pathogenesis, diagnostic, prevention and treatment is still lacking. We used a collection of 912 BSI and 370 commensal E. coli isolates collected in France over a 17-year period (2000–2017). We compared their pangenomes, genetic backgrounds (phylogroups, STs, O groups), presence of virulence-associated genes (VAGs) and antimicrobial resistance genes, finding significant differences in all comparisons between commensal and BSI isolates. A machine learning linear model trained on all the genetic variants derived from the pangenome and controlling for population structure reveals similar differences in VAGs, discovers new variants associated with pathogenicity (capacity to cause BSI), and accurately classifies BSI vs. commensal strains. Pathogenicity is a highly heritable trait, with up to 69% of the variance explained by bacterial genetic variants. Lastly, complementing our commensal collection with an older collection from 1980, we predict that pathogenicity continuously increased through 1980, 2000, to 2010. Together our findings imply that E. coli exhibit substantial genetic variation contributing to the transition between commensalism and pathogenicity and that this species evolved towards higher pathogenicity.
Author summary
Escherichia coli is a usually harmless bacteria typically found in the human gut. However, certain strains can cause severe bloodstream infections (BSIs) with high mortality rates, especially in older and fragile patients. To develop more effective prevention and treatment measures, understanding the genetic factors that determine a strain’s capacity to cause infection is essential.
In this study, we analyzed 912 BSI and 370 commensal E. coli isolates collected in France over 17 years (2000–2017). Through genetic comparison, we identified notable differences in their genetic backgrounds, as well as the presence of virulence-associated genes (VAGs) and antimicrobial resistance (AMR) genes. Using a machine learning model, we distinguished BSI from commensal strains, thereby discovering genetic factors linked to pathogenicity.
Our findings indicate that E. coli exhibits significant genetic variation contributing to the switch between commensalism and pathogenesis. We also suggest that pathogenicity in France has steadily increased over time, presenting a potentially serious public health threat. Our work is an important step in guiding future research to enhance diagnostic, prevention, and treatment strategies.
Introduction
Escherichia coli bloodstream infections (BSI) are severe diseases with an incidence of around 5 × 10−4 to 1 × 10−3 per person-year in Europe and the United States and a mortality ranging from 10 to 30% [1–5]. They may account for a few percents of all deaths in these countries [4]. The increase in incidence of BSI [1,2], the global emergence of multidrug resistance clones such as ST131 [6–9], and the aging population all make BSI an important and growing public health problem. A better understanding of the bacterial genetic factors determining pathogenicity (the capacity to cause infection) and virulence (the severity of infection) [10] would improve our understanding of pathophysiology and potentially improve stewardship and control policies.
The primary niche of E. coli is the gut of vertebrates, especially humans, where it behaves as a commensal [11]. BSI are opportunistic infections resulting from two main routes of infection, digestive and urinary, corresponding to two distinct pathophysiologic entities. BSI with digestive portal of entry are more severe than urinary ones: for example, the respective death rates were 14.7 vs 7.6% in a study in France [12]. Host conditions and comorbidities affect the severity of infection [13–15]. A few bacterial genetic factors affecting virulence have been reported. In a genome-wide association study (GWAS) conducted on 912 patients, no bacterial genetic factor was associated with outcome (death, septic shock, admission to ICU), possibly because of insufficient power [16]. Alternatively, in a murine model of BSI, a GWAS conducted on 370 Escherichia strains have shown that the Yersinia pestis High Pathogenicity Island (HPI), and two additional groups of genes involved in iron uptake, were associated with a higher probability of mouse death [17].
There is a rich tradition of comparing E. coli strains sampled from commensal carriage vs. in infections to reveal the determinants of pathogenicity [18,19], classically defined as the propensity to cause infection [10]. The numerous previous studies investigating the bacterial genetic determinants of pathogenicity vary in their study design, and in the resolution of the bacterial genetic information. Many studies use a case-control design, where cases are the individuals with BSI, controls are the healthy individuals, and the exposure is E. coli sampled from the blood or stool, respectively [20–25]. The exposure is variable, because bacteria are genetically variable. In this design, it is important to adjust for any potential confounder. Indeed, host factors, such as age or co-morbidities, are important determinants of infection [12]. Without adjustment, it is possible that the bacterial genetic factors identified do not causally affect pathogenicity but rather are associated with colonization of at-risk host groups. A less frequent design consists in sampling E. coli from stools vs. from infections in the same individuals, similar to a case-crossover design [18,26–29]. This design is interesting because it removes the confounding effect of the host factors. The case-crossover design, however, has limited power to detect variants associated with infections because it only considers hosts with infections, limiting the possibility of comparison to the diversity of strains present in stools of these hosts. For example, if hosts are colonized by a single strain which is the source of infection, the case-crossover design has zero power to detect genetic variants affecting pathogenicity as the strains from stool and blood samples will be identical.
Previous studies also differ in the genetic characterization of E. coli. Many studies characterize known virulence or resistance genes and alleles, serotypes, phylogroups or sequence types. This prevents the discovery of new determinants of pathogenicity beyond the established lists of virulence and resistance genes. The limited genetic information also prevents controlling for bacterial population structure. Indeed, the increased availability of large whole genome sequence collections from BSI revealed that a small number of sequence types, mainly ST131, 73, 95, 69, 10, are involved in the majority of BSI [30]. These STs are rich in virulence associated genes (VAGs) encoding adhesins, iron acquisition systems, protectins and toxins [18,19]. Pinpointing potentially causal individual genetic determinants can only be done in a rigorous GWAS controlling for population structure. Such control would also estimate the heritability, which is the fraction of variance in pathogenicity explained by bacterial genetic factors.
Thus, no study has so far investigated the bacterial genetic determinants of pathogenicity by comparing large numbers of whole genome sequences of bacteria sampled from the gut (commensals) vs. sampled from infections. A large-scale, comprehensive and systematic picture of the bacterial genetic determinants of E. coli pathogenicity is missing. In the present work, we took advantage of two recently published collections of BSI [12,31] and commensal [32] strains gathered between 1980 and 2017 in France, with associated host metadata, and full genome sequences. We compared BSI and commensal strain genomes at three levels: phylogenomic composition, virulence and resistance gene content, and lastly unitig content in a GWAS. Our goal was to compare the diversity of commensal and BSI strains and to identify specific genomic features affecting the propensity to cause BSI, using both a targeted and a hypothesis-free approach.
Results
A dataset of 912 BSI and 370 commensal isolates
We compared the genomes of 912 strains from BSI in adults, originating from two prospective multicentric studies (Colibafi in 2005 and Septicoli in 2016–7 [12,31]) performed in the Paris area, to the genomes of 370 commensal strains gathered from stools of healthy adult subjects in 2000, 2001, 2002, 2010 and 2017 in Brittany and the Paris area (Fig 1A). In-hospital death (or at day 28) was 12.9 and 9.5% in the Colibafi and Septicoli studies, respectively. Most of the BSI were community acquired (79.6 and 54.3% in the two collections, respectively). To avoid biases, all strains were isolated with similar protocols adapted to the sample origin (BSI and commensal) and sequenced in our laboratory using a similar approach (Illumina technology). To reduce the influence of the origin of the different studies we introduced the date of the study as a covariate, encoding it as a binary variable with the studies collected before and in or after 2010. To account for host factors, we additionally included sex and age as binary variables. For age, the variable was recording if the individual was above 60 years old or not. Finally, we also focused on the reported portal of entry of the BSI strains, which has previously been associated with some genetic variants (Fig 1B) [16]. The two collections had similar distributions of these variables, with the important exception of the proportion of isolates corresponding to older individuals, which is higher (69.43%) in the BSI collection (Fig 1C).
Fig 1. Global representation of the strain data set and the associated variables.

(A) Core genome phylogenetic tree of the 1,282 E. coli isolates used in this study (see Materials and methods) with their phylogroup distribution (internal color ring) and commensal or BSI status (external color ring). (B) Proportion of commensal isolates, distribution of covariates (sex, age, collection date), and BSI isolates with the urinary tract and digestive tract as portal of entry within the full dataset. (C) Scatter plot of the distributions of all covariates in the two collections, colors matching that of panel B. PE: portal of entry.
We computed the power of our design to detect variants affecting pathogenicity with simulations. Our design achieves a 50 to 60% power to detect a variant increasing pathogenicity by +30%. In our design, unadjusted confounders would unwantedly increase power and inflate the effect size estimate (S1 Fig). To inform potential future studies, we also compared our case-control study design to the other commonly used case-crossover design. In the case-crossover design, the commensal strains would have been hypothetically isolated from the 912 stool samples of the same BSI patients (instead of 370 unrelated healthy individuals). The case-crossover design removes the undesirable effects of confounding (S1 Fig). However, it suffers from low power when the within-host diversity of E. coli takes on plausible values of 1–5 strains per host, and underestimates effect size.
Commensal strains are genetically more diverse than BSI strains and have a distinct phylogenetic composition
We first compared the global phylogenomic characteristics of the two collections. The pangenomes of the BSI (N = 912) and commensal (N = 370) collections were composed of 24,321 and 22,373 genes, respectively. For a comparable number of strains, commensal strains had a higher diversity in gene content than BSI strains (Fig 2A). Conversely, the core genomes of both collections were similar (3,133 and 2,985 genes, respectively), and close to the core genome of E. coli species as a whole. In terms of SNP diversity of the core genome, the commensal collection was more diverse (pairwise nucleotide diversity π = 2.10e-2) than the BSI collection (π = 2.05e-2, p-value << 10e-10).
Fig 2. Comparison of the global phylogenomic characteristics of the commensal and BSI collections.

(A) Pangenome sizes as a function of the number of genomes analyzed for the BSI (912 strains) and commensal (370 strains) collections, showing the greater pangenome size of the commensal collection. (B) Cumulative distribution of strain sequences within ST in commensal and BSI collections. To be able to compare the BSI collection with the smaller commensal collection (N = 370), we extracted 200 random sub-samples of 370 sequences from the BSI collection (grey curves). (C) Comparison of the distribution of the sequence types (STs) of the E. coli commensal and BSI collections isolates (see S2 Table). We show the odds ratio (OR with 95% CI) for the risk of infection associated with colonization by each ST (logistic model of infection status as a function of the ST). We selected the STs present in at least 5 strains in at least one of the two collections. STs are ordered by decreasing associated odds ratio for all strains.
Commensal strains belonged almost equally to A and B2 phylogroups (25.4% and 32.4%) whereas BSI strains belonged mainly to phylogroup B2 and D (51.2% and 15.8%) (Fig 1A and S1 Table). The commensal collection was more diverse in its ST composition, with a higher number of rare STs and a lower number of frequent STs compared to the BSI collection (Fig 2B). This greater phylogenetic diversity could explain both the larger diversity in gene content [33] and larger nucleotidic sequence diversity of the pangenomes of commensals.
As previously noted, the diversity of STs in commensal strains was very distinct to that in BSI strains (S2 Table). Notably, ST10 and ST59 were abundant in commensal strains (13.2% and 3.8%) but under-represented in BSI strains (3.7% and 0.6%); on the contrary, ST131, ST73, ST69, ST95 were less common in commensal strains than they are in BSI strains. This comparison can be translated in an odds ratio for the risk of infection associated with gut colonization by each ST, which can be seen as a quantitative measure of pathogenicity. The sequence type ST131 was the most pathogenic and ST59 the least pathogenic (Fig 2C and S2 Table). When the portal of entry was considered for the ST distribution, a similar pattern was observed for both portals of entry as for the whole collection, although the significance level of the risk of infection might change (Fig 2C and S2 Table).
The distribution of the O-group diversity also differed between the commensal and the BSI collections (S3 Table). The four O-groups targeted by the recently developed bioconjugate vaccine ExPEC4V [34,35], O1, O2, O6 and O25 are the most abundant O-groups in the BSI collection. However, unlike the O-groups O6 and O25, the O-groups O1 and O2 are not particularly associated with BSI strains (S3 Table). In other words, these two O-groups are frequent in BSI because they are the two most frequent O-groups in commensalism, but are not particularly pathogenic.
BSI strains are enriched in VAGs and antibiotic resistance genes (ARGs) as compared to commensal strains
Using a targeted approach, we next focused on the frequency of known VAGs and ARGs in both collections. A global comparison in the number of VAGs classified in functional categories showed a significantly higher presence of VAGs coding for adhesins, iron acquisition systems, protectins and toxins categories in BSI strains (Fig 3A and S4 Table). We found similar results when comparing against BSI strains with urinary portal of entry to commensals (Fig 3B). However, only the iron acquisition systems category remained significant when comparing against BSI strains with digestive portal of entry (Fig 3C). More precisely for the full dataset, the highest significance was observed for the pap genes with the papGII allele, followed by the sit, iuc and irp2/fyuA (HPI) genes, all with p-values << 10−10 (S4 Table). These analyses do not imply a causal role of these genes and alleles in BSI, as they are not adjusted for the distinct phylogenomic composition of commensal and BSI strains. However, it is possible to crudely adjust for this population structure by focusing on the B2 phylogroup strains which are known to exhibit the highest prevalence of VAGs within the E. coli species [19].
Fig 3. Comparison of known VAG and ARG characteristics studied in the targeted approach.

(A-C) Comparison of the distribution of VAGs per strain among the six main functional classes of virulence of the E. coli commensal and BSI collections isolates. We show the odds ratio (OR with 95% CI) for the risk of infection associated with the number of VAGs (logistic model of infection status as a function of the number of VAGs), for (A) all the strains (912 BSI strains), (B) BSI strains with urinary portal of entry (PE) to commensals (498 BSI strains) and (C) BSI strains with digestive portal of entry to commensals (310 BSI strains). Functional classes of virulence are ordered by increasing associated odds ratio for all strains. (D-F) Comparison of the distribution of resistant strains for eight antibiotics of clinical importance of the E. coli commensal and BSI collections isolates. We show the odds ratio (OR with 95% CI) for the risk of infection associated with the resistance of strains (logistic model of infection status as a function of the resistance of strains), for (D) all the strains (912 BSI strains), (E) BSI strains with urinary portal of entry (PE) to commensals (498 BSI strains) and (F) BSI strains with digestive portal of entry to commensals (310 BSI strains). Categories of antibiotics are ordered by increasing associated odds ratio for all strains. For AMK, FQ and FEP categories, we only show the lower bound of the CI because the estimated odds ratios are huge as none of the commensal isolates were resistant to these antibiotics. AMK, amikacin; AMP, ampicillin; CTX/CAZ, cefotaxime/ceftazidime; FEP, cefepime; FQ, fluoroquinolones; GEN, gentamicin; SXT, cotrimoxazole; TZP, piperacillin/tazobactam. (G) Repertoire of adhesins and (H) iron capture systems in BSI and commensal strains. We selected the four most significant systems in terms of complete genes and/or alleles when comparing commensal to BSI strains (S4 Table) and evaluated their combinations. A system was considered present when all its genes were detected. (I) Predictor coefficients of pathogenicity among adhesins and (J) iron capture systems determined with a lasso regression. We calculated confidence intervals using 1000 bootstrap resamples. Unselected genes and/or alleles (lasso coefficient close to zero) are shown in gray.
When only B2 phylogroup strains were compared, only VAGs coding for adhesins category remained significantly over-represented in BSI (S2 Fig). When comparing only B2 strains with urinary portal of entry to B2 commensals, again only adhesins were over-represented, and no differences were observed when comparing only against B2 strains with digestive portal of entry (S2 Fig). Regarding individual genes, interestingly, for two VAGs with experimentally validated role in urinary tract infection, pap genes [36] and fim genes [37], we found a higher level of significance in B2 strains with urinary portal of entry than in all B2 strains (pap) or in all strains (fimD-H) (S4 Table).
As virulence in E. coli is the result of additive gene effect [38], we further evaluated the repertoire of adhesins and iron capture systems, the two categories for which we found the higher significance (S4 Table). We restricted our analysis to the four most significant systems in terms of complete genes and/or alleles (< 10−7). For both categories, we found a different distribution of systems between BSI and commensal strains, with more co-occurences of systems in BSI strains (Fig 3 and S5 Table). For instance, 38% and 72% of BSI strains carry three or four systems of adhesins and iron capture systems respectively, compared to 13% and 42% respectively for commensal strains. We found two adhesins encoding genes, ecp and papGII and three iron capture systems, HPI and sit and iuc gene clusters to be the genes and/or alleles best explaining pathogenicity, using a lasso regression (with adhesins and iron capture systems evaluated separately or together) (Fig 3I and 3J). The ecp (or yag or mat) operon is highly prevalent within the E. coli species (more than 90%) and encodes a fimbrial adhesin (E. coli common pilus) used both by commensal and pathogenic strains [39,40]. In our work, the prevalence of ecp in commensal and BSI strains is 91 vs 98%, respectively, and the significance of its association with BSI strains disappeared when only the B2 were studied (S5 Table), suggesting a phylogenetic effect.
BSI strains were predicted to be more resistant to all classes of antibiotics than commensal strains (Fig 3D). This also holds true when specific portals of entry and/or phylogroup B2 were taken into account, with the exception of the resistance to amikacin when comparing B2 BSI strains with digestive portal of entry to B2 commensals (Figs 3E, 3F and S3). To verify that this over-representation of resistance in BSI was not explained by the fact that BSI isolates were slightly more recent on average than commensal isolates, we restricted our analysis to BSI Colibafi strains (sampled in 2005) and found the same results when considering all phylogroups and portals of entry, with the exception of amikacin and fluoroquinolones which had the lowest prevalence (S3 Fig, panel D).
No difference in VAG numbers (t-test, all Benjamini-Hochberg corrected p-value > 0.05), nor in resistance prevalences (Fisher’s exact test, all Benjamini-Hochberg corrected p value > 0.05), was found when comparing nosocomial and community BSI strains, considering both Septicoli (167 nosocomial and 296 community BSI strains) and Colibafi (75 nosocomial and 292 community BSI strains) collections together or individually.
Bacterial genetic factors explain a large fraction of the variation in the BSI phenotype
We then computed the heritability, as the proportion of the variance of a phenotype explained by variable genetic factors [41], to estimate whether we could expect to find bacterial genetic variants associated with commensalism vs. BSI in our dataset. We first measured the heritability using the ST information alone, to measure the influence of the genetic background on phenotypic variability. We then computed the heritability emerging from the individual genetic variants (Fig 4A). We found that STs could explain 24%, 28%, and 11% of the phenotypic variance in the full collection, the subset with BSI isolates with urinary tract as portal of entry and digestive tract as portal of entry, respectively. Genetic variants alone could explain a larger fraction of the phenotypic variability: 65%, 69%, and 39% in the full collection and the two subsets, respectively. This suggests that pathogenicity might not be solely determined by the sequence type but also by specific genetic variants within sequence types.
Fig 4. Main results of the hypothesis-free (GWAS) approach comparing commensal and BSI isolates using the portal of entry covariable.

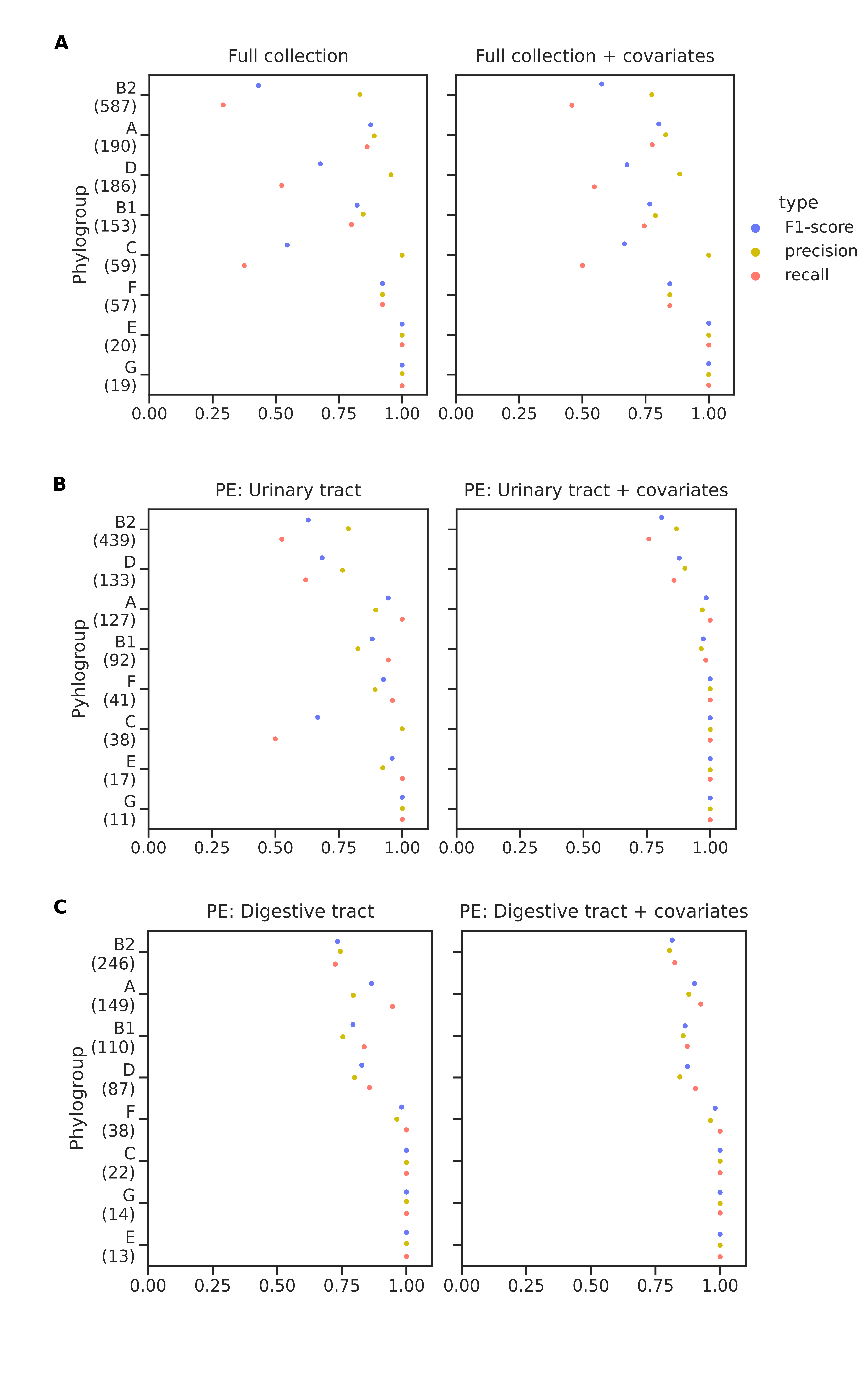
A) Heritability estimates for the commensal phenotype. B) wg-GWAS model performance within each phylogroup. F1-score representation for the naive analysis (blue dots), with covariates (yellow dots), and the one expected by chance (grey dots). Numbers within parentheses below each phylogroup indicate the sample size. C) Virulence associated genes enrichment analysis for the different functional categories. The significance threshold is represented over the dotted line (Fisher’s exact test, p<0.05). PE: portal of entry.
A whole-genome machine learning model differentiates commensals from BSI strains
We applied a machine learning model trained on both the core and accessory genome of the strains to differentiate between commensal and BSI strains and highlight the genetic variants that contribute the most to the discriminatory power of the model (whole genome wg-GWAS). We performed the analysis on three different datasets: the full strain collection, and two subsets of BSI isolates: one with urinary tract as portal of entry, and another one with digestive tract as portal of entry. We used all the genetic variants covering the pangenome compactly represented by unitigs. Unitigs are nodes of sequence in a compressed de Bruijn graph, usually longer than k-mers, reducing the computational burden and the redundancy present in k-mer counting. They are short sequence fragments that represent both gene content and nucleotidic variation across genomes, including coding and intergenic non-coding sequences. We associated unitigs with phenotype with the elastic net linear model implemented in pyseer [42]. This approach is similar to a logistic regression except that the number of predictors (all genetic variants in the form of unitigs presence/absence) vastly exceeds the number of observations (phenotype, commensal vs. BSI). To resolve this problem, the likelihood of observations (the function to be maximized in classical logistic regression) is complemented by a term penalizing large coefficients: the ‘elastic net’ regularization. In pyseer, the strength of the penalty is tuned such as to maximize the accuracy of the fit when performing ‘leave-one-strain-out’ cross-validation. Fitting the elastic net model results in a set of unitigs retained in the whole genome model, with associated coefficients. The procedure ensures implicit correction for population structure and has been found to outperform other methods to detect causal variants [43,44].
We used the following three binary variables as covariates to account for host factors and collection biases: the sex of the individual, their age (older than 60 years old), and the date of each collection (before or after 2010). To quantify model performance, we computed the precision (proportion of true BSI among the predicted BSI strains), recall (sensitivity) and F1-score (harmonic mean of precision and recall) on each phylogroup (Figs 4B and S4).
The model performance improved in all cases when the covariates were considered for the associations, potentially confirming that host factors also explained part of bacterial pathogenicity. Model performance also improved in the two subsets with BSI isolates with a specific portal of entry, compared to the full collection. Furthermore, the model did not perform better than expected by chance within the B2 phylogroups and without dividing BSI by portal of entry (Fig 4B). This suggests the presence of specific genetic variants associated with either portal of entry, and underlines the critical importance of considering the portal of entry when inferring the determinants of pathogenicity [16].
We found a number of unitigs associated with commensalism vs. BSI (i.e. with non-zero weight in the elastic net model). Overall, 107 and 59 unitigs passed the threshold for the model built naïvely and with covariates, respectively, which we then mapped back to 34 and 28 genes. Moreover, we checked for gene hits upstream and downstream of the unitigs found in intergenic regions, revealing an additional set of 11 genes, one being also identified on coding region (dhfr). We found that 9 out of the 39 genes (28 in genes + 11 intergenic) obtained through the analysis with covariates were clearly related to virulence and/or resistance, notably the iucB gene encoding an aerobactin siderophore biosynthesis protein (30) and papG encoding the adhesin at the tip of the P pilus (31). Both the iucB and papG genes have already been associated with invasive uropathogenic E. coli (UPEC) isolates (15, 32). Of note, we had identified these two genes in the targeted approach even after focusing on the B2 phylogroup strains (see above). In addition to these two genes, we also found the following genes: mltB, which is part of a network connecting resistance, membrane homeostasis, biogenesis of pili and fitness in Acinetobacter baumannii [45]; fliL, encoding for the flagellar protein FliL [46]; oprM, as part of the intergenic hits, described as a component of an efflux pump in Pseudomonas aeruginosa [47] potentially involved in resistance to puromycin, acriflavine, and tetraphenylarsonium chloride (S9 Table). Lastly, two unnamed orthologous groups (group_5900 and group_9261), described as the putative bacterial toxin ydaT [48], were identified.
A larger number of genes were associated to the phenotype when dividing the BSI strains according to their portal of entry. We found a total of 152 and 96 associated unitigs for the urinary and digestive tract subsets, respectively, which we then mapped back to 101 and 45 genes, some of which are known to be involved in pathogenicity and antimicrobial resistance (Table 1 and S5). Additionally, 17 and 4 gene hits upstream and downstream of the unitigs in intergenic regions were found for the urinary and digestive tract subsets, respectively (S9 Table). Of note, the orthologous group 16391, identified for the urinary tract subset, was annotated as a paralog of dhfR, which had hits in its coding region. Moreover, we find lysO and aqpZ in both subsets.
Table 1. Genes with functions related to pathogenicity and antimicrobial resistance with unitigs associated with the phenotype mapped to them for the two subsets (urinary and digestive portal of entries).
| Portal of entry: urinary tract | ||
|---|---|---|
| Gene | Relevance | Reference |
| papG | Tip adhesin. Belongs to the pap operon encoding for a type P pilus | [16,52] |
| papH | Adhesin anchor to the cell. Belongs to the pap operon encoding for a type P pilus | [53] |
| papF * | Adhesin adapter. Belongs to the pap operon encoding for a type P pilus | [16,52] |
| iucB/C | Iron acquisition. Aerobactin siderophore biosynthesis protein | [54] |
| mltC | Involved in release of peptidoglycan-derived pathogen-associated molecular patterns as a virulence mechanism | [55] |
| ompX | Might be involved in biofilm formation and curli production | [56,57] |
| dhfrI, Group 16391 * | Trimethoprim resistance gene | [58], S9 Table |
| fliD | Relevance in adhesion. Flagellar hook-associated protein. | [59] |
| dgcE | Involved in regulation of the switch from flagellar motility to sessile behavior and curli expression | [60] |
| Groups 10969 and 4151 | Type II/IV secretion system protein (T2SSE) | S5 Table |
| Group 9261 | Putative bacterial toxin | S5 Table |
| epsM | Involved in type II secretion systems (T2SS) | S5 Table |
| aceF | Involved in the virulence and oxidative response of P. aeruginosa | [61] |
| klcA | Antirestriction protein. Encoding gene present in the kilC operon found in IncP plasmids, which usually carry multiple AMR determinants | [62] |
| Portal of entry: digestive tract | ||
| Gene | Relevance | Ref |
| iucC | Iron acquisition. Aerobactin siderophore biosynthesis protein | [54] |
| Group 3130 | Tfp pilus assembly protein FimV | S5 Table |
| fliD | Relevance in adhesion. Flagellar hook-associated protein | [59] |
| epsE/F | Type II/IV secretion system protein | S5 Table |
| yehB | Relevance in adhesion. Encodes a type of putative fimbrial complex belonging to the chaperone-usher assembly pathway | [63] |
| oprM * | Possibly involved in resistance to puromycin, acriflavine, and tetraphenylarsonium chloride. Component of an efflux pump in Pseudomonas aeruginosa. | S9 Table [47] |
Intergenic (*) corresponds to gene hits upstream or downstream of associated unitigs within intergenic regions.
Taken as a whole, we found the associated genes to be enriched in the L COG category (replication, recombination and repair) for the three subsets, and in the K COG category (transcription) for the full dataset only. We also performed a Gene Ontology (GO) term enrichment analysis and found that for the subset with BSI isolates with urinary tract as portal of entry, the relevant (depth > 1) enriched GO terms include different categories related to metabolic processes, ion binding and intracellular anatomical structure (S6 Table). Similarly, to the targeted analysis described above, we found that the genes resulting from the three associations were enriched for VAGs and ARGs (Fig 4C); when considering all VAGs and ARGs together we found a significant (p-value < 0.05) enrichment for the full dataset and the urinary tract subset. We found VAGs related to iron acquisition to be enriched in all three datasets, while adhesins were enriched in the full dataset only. For the ARGs, only the resistance to cotrimoxazole (dfrA for SXT resistance) was enriched in the urinary tract subset.
The model can be used to predict the potential pathogenicity of other isolates based on the presence of the unitigs for which the model’s weight is different than zero. We predicted the pathogenicity of commensal strains collected at three time periods: 1980 [49], 2000–2002 and 2010. Interestingly, the model predicts a marked increase in pathogenicity of these commensal isolates, with the proportion doubling between the 1980s, the 2000s, and the 2010s (23% vs. 31% vs. 46%, S5 Fig). As the strains from the 1980 collection (VDG strains) have been stored in stabs between 1980–2000 before being frozen at -80°C, we verified that artifacts due to storage were not involved. We first confirmed the good quality of the strain sequences in all collections (S6 Fig, panel A). We then looked at the mutation patterns in the rpoS gene that are indicative of poor sample management [50] which may have affected genome content between sample isolation and sequencing (S6 Fig, panel B). As expected, we found a high rate of rpoS mutations in the VDG collection only. In addition, we compared the presence of two VAGs, hlyC (plasmid-borne or chromosomal) and papC (chromosomal), determined from this work by WGS and both phenotypically and via PCR in earlier studies [49,51]. We found a perfect match for PCR except one gene in one strain and slightly smaller percentages of presence of Hly and Pap assessed phenotypically (4.7% and 7%, respectively) versus 9% for both hly and pap assessed by WGS in this work. We speculated that different storage conditions could have caused the loss of virulence genes in the past collection, and biased downwards the predicted pathogenicity in 1980: although there was a weak (not significant) trend of increased gene content, this trend together with the inferred effects did not result in any change in predicted pathogenicity (Material and Methods). In sum, although the strains from 1980 were stored differently, there was no evidence that this could have affected our results on pathogenicity. Altogether, these data suggest that the commensal strains inhabiting the gut of healthy humans may have evolved towards higher pathogenicity in the past decades.
Discussion
It is known since the 1940s [64] that within the E. coli species, some strains with a specific genetic background have higher capacity to cause extra-intestinal diseases. Later on, pathogenicity has been associated with specific serotypes, STs, and the phylogroup B2, which are enriched in some VAGs [65,66]. However, disentangling the respective roles of causal genetic variants from the genetic background in a mostly clonal species is a difficult task [67], and a comprehensive and systematic view of the bacterial genetic determinants of pathogenicity is lacking. To do so, we systematically investigated the genomic differences between 912 E. coli strains from bloodstream infections and 370 strains sampled from the stools of healthy volunteers. The large size of the collection and case-control study design ensure a powerful examination of these determinants.
We revealed differences at three levels. First, at the phylogenetic level, strains from BSI are less diverse, dominated by a small number of highly pathogenic STs, and consequently have smaller pangenomes and lower genetic diversity than commensal strains. Second, strains from infections are enriched in VAGs, and are predicted to be more antibiotic resistant. Third, in a machine learning assisted GWAS designed to identify putative causal variants, we found 118 and 49 genes associated with BSI with urinary and digestive portal of entry, respectively, independently of the clonal background. Our analyses give several new insights on E. coli pathogenesis: pathogenicity is a highly heritable phenotype, with 69% heritability for urinary tract portal of entry BSI; tens of specific variants may causally impact pathogenicity; antimicrobial resistance genes are associated with, but do not play a causal role in infection; and pathogenicity may have increased in the past decades in France.
We discuss these four new results in detail in the following. However, note that an important limitation of our study is that we did not use available information on host co-morbidities in BSI patients for the comparison with commensal strains. In fact, the most frequent co-morbidity in the BSI collection is immunosuppression, which was an exclusion criterion for the commensal collection. Co-morbidities are associated with BSI [5,12,31]. It is possible that co-morbidities act as a confounder in our study, if they both increase the probability of BSI and influence the E. coli strains carried by individuals. If this is the case, the variants we identify may not be directly causal for infections. Rather, they may be bacterial variants that favor the colonization of individuals with co-morbidities. Age is also associated with BSI [5,15]. In this work, we do control for age, albeit in a crude way, with the covariate “above or below 60 years old”. If some of the variation associated with age is not captured by this covariate, some of the variants we identify could favor the colonization of older or younger individuals. For example, there is evidence of age-associated variants in Streptococcus pneumoniae [68]. To attenuate these concerns on confounding, we remind that several of the significant variants have an experimentally validated role in infection and virulence (Table 1).
The heritability of pathogenicity is estimated at 69% (urinary PE) and 39% (digestive PE), in agreement with the higher role of the host factors in BSI with digestive PE [12,31]. Thus, a large fraction of pathogenicity is explained by bacterial genetic factors. This is roughly double of the heritability when considering STs alone, suggesting that specific genetic variants at a finer phylogenetic scale than ST are determining pathogenicity. For comparison, age, a host factor strongly associated with BSI, explains 17.6% of the variance. Bacterial genetics has a significant role in determining pathogenicity, even after basic host factors (age and sex) have been accounted for. The present study compares E. coli whole genomes in colonization and in infection in a case-control study, as done before for Klebsellia pneumoniae [69], S. pneumoniae [70], Staphylococcus aureus [71,72], Enterococcus faecalis [73], Neisseria meningitidis [74]. These previous GWAS studies presented a range of results, from low heritability (2.6% for S. aureus carriage vs. BSI [71]), to intermediate (34% for E. faecalis intestinal colonization vs. extraintestinal infection, 36.5% for N. meningitidis carriage vs. invasive meningococcal disease), and an analogously large heritability of 70% for S. pneumoniae invasive disease vs. carriage, along with a handful of significant SNPs [70]. We find a large heritability for E. coli BSI vs. colonization, which suggests that a vaccine targeted at virulence determinants could reduce (at least temporarily) the burden of infection [34].
Some of the specific variants identified in the GWAS are involved in adhesion and in iron acquisition, as well as other functions. Generally, genes with a significant association are enriched in iron acquisition system, the L COG category (replication, recombination and repair) and GO terms including different categories related to metabolic processes, ion binding and intracellular anatomical structure.
Interestingly, 28% of identified genetic variants linked to pathogenicity between commensal and BSI isolates were located in intergenic regions. Non-translated intergenic regions compose 10–15% of bacterial genomes, and contain many regulatory elements with key functions. They have been shown to be under strong purifying selection in several bacterial species including E. coli [75]. This could indicate an important role of regulation of VAGs but also of core genome genes in pathogenicity [76]. Differences in gene (VAG, metabolic gene) regulation between anatomical sites have been reported in Campylobacter coli [77], Klebsiella, Staphylococcus aureus and Streptococcus pyogenes [78]. Also, differences between E. coli lineages have been described [40,79,80]. In the latter cases concerning differential expression of fimbriae (ECP, Ucl and P fimbriae, respectively), causal SNPs in the gene promoter region were identified. Further studies on the intergenic regions highlighted in the present analysis should be performed.
We found that strains from infections are more likely to be resistant to antimicrobials. What is the mechanism behind this association, also found in similar GWAS conducted on other pathogens [69,71]? Confounding is a first possibility: hosts with co-morbidities are more likely to develop a BSI and to use antibiotics frequently. Individuals may even be already treated by antibiotics at the time of infection, in which case only resistant strains would be able to cause this infection. If this mechanism operates, we could expect resistance to be more frequent in hospital-associated than in community-associated BSI, if hosts in hospitals are more likely to use antibiotics at the time of infection. However, we did not find any difference between resistance in hospital-associated and community-associated BSI. Second, antimicrobial resistance genes may have a causal role in infection. This seems unlikely given their very specific function. Third, there might be a genetic association (linkage disequilibrium) between resistance genes and genetic determinants of infection [69,81]. In the third case, we expect the association to disappear when controlling for population structure. With this control, we find that indeed, only one out of nine categories of resistance was significantly enriched in BSI compared to commensals. This suggests that antibiotic resistance genes are genetically linked with pathogenicity determinants, and opens the interesting possibility that antibiotic resistance coevolves with pathogenicity determinants associated with the clonal background of E. coli. The co-evolution of resistance and virulence elements may result in their co-localisation on the same genetic elements, such as plasmids, or nearby on the chromosome [82–84]. However, the properties of bacterial recombination enable associations to emerge even between physically distant genes [85]. We investigated the proximity between VAGs and AMR in our collections, and found that VAGs and AMR genes are never encoded in the same contig in the draft genomes used in this study, supporting the hypothesis of co-evolution of physically distant resistance and virulence elements through homologous recombination.
The large heritability of E. coli capacity to cause infection also implies that this trait can readily evolve. Evolution of E. coli pathogenicity would have important public health implications, given that E. coli BSI are a major cause of morbidity and death in Western countries. To investigate temporal trends in pathogenicity, we computed the pathogenicity score with the machine learning model (used to predict the commensal vs. BSI status of strains), in a dataset of commensals from 1980 to 2010 in France [32]. We found that the proportion of commensal E. coli isolates predicted to be pathogenic isolates with our trained model increased over time, from 23% in the collection from 1980 to 31% in 2000, and then to 46% in the collection from 2010 (S5 Fig). Even though sample storage issues may slightly alter the predictions for samples from 1980 (S6 Fig, panel B), we see an even higher increase between 2000 and 2010. The signal of increased pathogenicity would be worth replicating in independent datasets. In fact, applying this predictive model to the large collection of available E. coli genome sequences, which currently numbers to more than 200,000 genomes [86], could unravel the dynamics of pathogenicity across time and space. One would need to focus on collections with homogenous collection strategies, time and geographic information, and ideally more detailed metadata such as portal of entry—unfortunately such collections remain very rare. This effort would further need to be properly controlled for the biases in the isolates sampled and sequenced (most of them coming from infections), and the phylogroup-specific performance of the model.
What selective pressures might act on pathogenicity determinants? The capacity to cause extra-intestinal infection may not be selected per se, as infections are a relatively rare occurrence in the life cycle of E. coli and do not obviously confer a transmission advantage. Pathogenicity determinants have diverse functions and may therefore be selected for a variety of reasons. They may for example improve the ability to colonize the human gut, improve the ability to compete and replace existing strains, or allow longer persistence in the gut [87–90]. In addition, epistatic interactions between these determinants (Fig 3G–3J) and the genetic background of the strains may determine pathogenicity, as recently reported for iron capture systems for virulence in a mouse model [91]. Elucidating the selective pressures acting on these determinants is an important research question that would improve our understanding of E. coli pathogenicity.
This work opens perspectives to improve studies of the determinants of E. coli pathogenicity. First of all, genes identified as good pathogenicity candidates not previously reported in E. coli (mltC, ompX, etc.) should be validated experimentally in animal models by gene inactivation assays. Second, it remains difficult to pinpoint individual variants because of the clonal structure of E. coli, and confounding by host factors is a concern. One idea to alleviate clonal structure is to focus on specific STs. This would limit the dominant effect of STs belonging to phylogroup B2 and carrying many virulence genes. However, the genetic diversity within a single ST might also be limited. This makes it difficult to anticipate the results of such ST-focused studies. Another idea is to extend to whole genomes the line of work comparing strains from infections vs. colonization in the same individuals. The case-crossover design reduces concerns on confounding host factors. However, its power is contingent on the within-host diversity of strains present in colonization (S1 Fig). In addition, it is difficult in practice to perform a rectal swab in patients arriving at the emergency room for a suspicion of E. coli BSI and before any antibiotic is prescribed. Third, further help will also likely come from linking pathogen diversity to clinical and epidemiological phenotypes and including the genetic variation of the host into the association such as in a previous study of S. pneumoniae [70]. Lastly, similar studies should be conducted in low and middle-income countries, where a potentially very distinct diversity of E. coli circulates [11] and where the public health problem posed by BSI will escalate with the aging population in the years to come.
In conclusion, we elucidated in a systematic and quantitative manner the bacterial genetic determinants of pathogenicity of the major human pathogen E. coli. The capacity to cause BSI, particularly with urinary PE, is strongly determined by sequence types, additional genetic factors, and tens of specific variants. This implies that E. coli pathogenicity may evolve, informs future studies of E. coli mechanisms of pathogenicity, and opens the possibility to reduce the burden of E. coli with a vaccine targeted at these variants.
Material and methods
Ethics statement
All multicenter clinical trials were approved by the appropriate ethic committees. The Colibafi study was approved by the French Comité de Protection des Personnes of Hôpital Saint-Louis, Paris, France (approval #2004–06, June 2004). The Septicoli study was approved by the French Comité de Protection des Personnes Ile de France n°IV (IRB 00003835, March 2016). Because of their non-interventional nature, only an oral consent from patients was requested under French Law. The study on the commensal strains was approved by the ethics evaluation committee of Institut National de la Santé et de la Recherche Médicale (INSERM) (CCTIRS no. 09.243, CNIL no. 909277, and CQI no. 01–014).
Strain collections
We studied the whole genomes of 1282 E. coli strains divided in two datasets, 370 commensals strains and 912 BSI strains. Commensal strains were gathered from stools of 370 healthy adults living in the Paris area or Brittany (both locations in the Northern part of France) between 2000 to 2017. These strains come from five previously published collections: ROAR in 2000 (n = 50) [92] (Brittany–a region in the North-West of France), LBC in 2001 (n = 27) [93] (Brittany), PAR in 2002 (n = 27) [93] (Paris area), Coliville in 2010 (n = 246) [94] (Paris area) and CEREMI in 2017 (n = 20) [95] (Paris area) (S7 Table). In addition, a collection of 53 commensal strains sampled in 1980 from 53 healthy subjects in Paris (VDG collection) [49] was used to assess the temporal trend of pathogenicity. BSI isolates (Colibafi (n = 367) and Septicoli (n = 545) collections) were collected in 2005 and 2016–2017, respectively [96]. In all studies, one single E. coli colony randomly picked was retained per individual after plating the blood cultures on non-selective rich medium, or the stools on Drigalsky plates. After this first step, the protocol for all isolates was similar except for the collection from 1980. After one or two subcultures in rich medium, the strains were immediately stored with glycerol at -80°C. The 1980’s collection was stored in agar tubes left at room temperature until the beginning of the 2000s, when the strains were subcultured and stored with glycerol at -80°C.
For the collection of the commensal strains, all participants lived in the community and volunteered to self-collect a faecal swab sample. The inclusion criteria were: age of 18 years or more, no history of gastrointestinal disease, no symptoms of immunosuppression, no antibiotic therapy in the previous month and no hospitalisation in the 3 months preceding inclusion.
The Colibafi study was performed in eight hospitals representing a total of 3,900 adult acute care beds, whereas seven hospitals were included in the Septicoli study, accounting for 5,800 acute care beds. Four hospitals were common between the two studies (i.e. 2,900 acute care beds). All the hospitals belong to the same institution, the “Assistance Publique-Hôpitaux de Paris” network, which accounts for a total of 13,000 adult acute care beds with a homogenous management for most bacterial infections. Clinical data were prospectively collected by clinicians in each center on two separate visits: Visit 1 corresponded to the time of BSI (the day the blood culture was drawn; data were collected retrospectively 24-48h hours later, once the blood culture had grown) and Visit 2 corresponded to the day of discharge or in-hospital death (or day 28 if the patient was still hospitalized). For each episode, the first E. coli strain collected in the blood culture was identified. The primary endpoint was vital status at discharge or day 28 (i.e. Visit 2). The likely portal of entry was established according to clinical and/or radiological characteristics of the episodes and the isolation of E. coli from the presumed source of infection. When E. coli could be isolated from the source of infection, the portal of entry was assigned on the basis of firm clinical suspicion [97]. In each centre, an infectious diseases clinician and a microbiologist were in charge of including patients and completing the case report form (see Colibafi and Septicoli groups in S1 Text). A steering committee was in charge of implementation and a scientific committee responsible for scientific overview.
All the sequences were available (Bioproject PRJEB38489 (ROAR), PRJEB44819 (LBC), PRJEB44872 (PAR), PRJEB39252 (Coliville), PRJEB39260 (Colibafi), PRJEB35745 (Septicoli) and PRJEB44873 (VDG)) except the 20 samples of the CEREMI collection that were whole-genome sequenced in the present work, following the protocol detailed in [31] (Bioproject PRJEB55584).
Computing the power of case-control and case-crossover studies
We used simulations to compute the power of our case-control study, and compare it to the power achieved with a case-crossover study. We modeled a bacterial genetic variant increasing pathogenicity by +30% (S1 Fig). Several strains may independently colonize the gut of individuals. The BSI is caused by a single bacterial strain invading the blood, selected at random from the gut with weight proportional to strain pathogenicity. For simplicity, we assumed all individuals carry the same number of strains.
We first measured the effect of this variant in a simulated study of 912 cases and 370 distinct controls using logistic regression. In the case-control design, the bacterial genetic variant is measured in the strain from BSI and in one randomly chosen strain from the stool samples of controls. Next, we examined a case-crossover design of 912 cases and 912 controls, with controls consisting of one strain randomly chosen from the stool samples of the 912 cases.
We computed both the power to detect this variant across 1000 replicates, and the estimated effect size of the variant. We varied two factors: (i) the number of bacterial strains carried by each host, from 1 to 100. (ii) The presence of a confounding factor that we cannot measure, affecting both the genetic variant and the incidence of infection. The confounding factor is binary, and the variant is present in 30% of hosts with low incidence and 70% of hosts with high incidence. We assumed several strengths of confounding: no confounding, +10% increase in infection, +100% increase in infection.
Genomic diversity of the core genome
The 1282 assemblies were annotated with Prokka v1.14.6 [98]. We then performed pan-genome analysis from annotated assemblies with Panaroo v1.3.0 with strict clean mode and the removal of invalid genes [99]. We generated a core genome alignment spanning the whole set of core genes as determined by Panaroo, and a phylogenetic tree was computed using FastTree v2.1.11 [100] and visualized using Microreact [101].
Comparison of commensal and BSI E. coli collection
Multilocus sequence typing (MLST) was performed using an in-house script Petanc, that integrates several existing bacterial genomic tools [102]. We determined STs (Warwick MLST scheme) [103] and O types [104].
We evaluated the risk of infection associated to colonization by a specific ST and by a specific O-group. We compared the ST and O-group diversity from the collection of 912 BSI isolates with the 370 commensal isolates, for all STs with at least 5 strains in at least one of the two collections and for all O-groups with at least 5 strains in at least one of the two collections.
The odds ratios for the infection risk were computed by fitting a logistic model of infection status (commensal or BSI) as a function of the ST or the O-group (here and thereafter, “significant” refers to significance at the 0.05 level).
Next, we compared the phylogenetic distribution of the commensal collection with the BSI collection. For all strains, we calculated the cumulative frequency distribution of STs in the commensal collection, and we compared it to the same distribution in 200 random sub-samples of 370 sequences from the BSI collection.
We plotted the pangenome variation with the number of genomes analyzed (Panaroo output). We evaluated the pangenome variation between commensal and BSI isolates with Panstripe [105] using the output of FastTree (phylogeny of all strains) and Panaroo (gene presence absence matrix). We randomly subsampled 100 trees of 370 tips from the BSI phylogeny (n = 912) and compared the rate of gene gain and loss between those trees and the commensal tree (n = 370). To quantify the genetic diversity, we computed the pairwise nucleotide diversity (π) [106] in R (package ape) [107].
We also compared the number of virulence factors and the proportion of resistance strains between commensal and BSI isolates. We evaluated the number of VAGs for each of the six main functional classes (adhesin, invasin, iron acquisition, miscellaneous, protectin and toxin) and predicted phenotypic resistance as described in [96] for eight antibiotics of clinical importance (amikacin, ampicillin, cefotaxime/ceftazidime, cefepime, fluoroquinolones, gentamicin, cotrimoxazole and piperacillin/tazobactam). We excluded the resistance to carbapenems in this study because it was very rare (2 strains over 1282). The odds ratios for the infection risk were computed by fitting a logistic model of infection status (commensal or BSI) as a function of the number of VAGs or the status of resistance (resistant versus sensitive).
We evaluated the co-occurrences of the four major iron-capture systems and of the four major adhesins systems (complete genes or alleles), defined as the systems with a significance < 10−7 when comparing commensals against BSI strains (see S4 Table). The iron capture systems (HPI, operon iro, iuc and sit) and the adhesin systems (ecp and pap) were considered present when all their genes were detected, at the exception of papA which is very rare and papG for which we examined the allele papGII (see below). We detected the presence of genes with Abricate [108] with 75% identity and 50% coverage. The adhesin alleles, papGII and iha7, were detected with Abricate with 90% identity and 90% coverage. We performed a lasso regression to select the genes and/or alleles that best predict the pathogenicity among the four adhesins systems, the four iron capture systems and the eight systems simultaneously. We estimated the CIs with 1000 bootstrap replicates.
Heritability estimates
We estimated narrow-sense heritability for the target variable using 2 different covariance matrices: one built from the genetic variants using a kinship matrix, and another one with the sequence types membership. Limix v3.04 [109] was used, assuming normal errors for the point estimate.
Association analysis
We derived unitigs using unitig-counter v1.1.0 [43]. We tested locus effects using the wg (whole genome) model of pyseer v1.3.6 [42,110], which trains a linear model with elastic net regularization using the presence/absence patterns of all unitigs. We used the parameter alpha with value of 1 for the elastic net, which is equivalent to a lasso model. The model performance was assessed by computing three metrics using each phylogroup. The precision, as the measure of how many positive predictions made are correct; the recall, as the measure of how many positive cases the classifier predicted correctly over all the positive cases; and the F1-score, as the harmonic mean of the two metrics. The F1-score expected by chance was computed overall, for each phylogroup and for the different subsets, by randomly assigning the phenotype to the test samples and running 1000 randomizations. The unitigs with a non-zero model coefficient were mapped back to all input genomes, and gene families were annotated by taking a representative protein sequence from all genomes encoding each gene, which was then used as the input for eggnog-mapper v2.1.3 using the panaroo output to collapse gene hits to individual groups of orthologs. Genes downstream and upstream of unitig hits within intergenic regions were further mapped back. GO terms enrichment was determined using goatools v1.2.3 [111]. An in-house list of E. coli virulence genes and antibiotic resistance genes was used to annotate the virulence and antibiotic resistant genes within the collection, and a Fisher’s exact test was used to determine the enriched genes, with a multiple testing correction based on the Benjamini-Hochberg method, with a 5% family-wise error rate. For the COG and virulence genes enrichment analysis a random ST131 genome from the full dataset was picked up as background.
Prediction analysis
We used unitig-caller v1.3.0 [112] to make variant calls in the test population, and the elastic net regularization, previously trained, model using pyseer v1.3.6 [110] to predict the phenotype in new commensal samples from different time periods, divided in decades.
Genome sequence quality control and storage effect assessment
To quantify general genome assembly quality, we used the N50 metric, defined as the length of the shortest contig at half the total length of the assembly. A smaller N50 value indicates a genome assembly with shorter contigs.
We used the presence of genetic variants in the rpoS gene as a metric for appropriate sample storage between isolation and sequencing; it has been shown that repeated freeze/thaw cycles and long-term storage in agar stabs induce mutations in this gene, as well as a possible marker for deletions [50]. We used snippy v4.6.0 to call and annotate variants between each genome assembly and the E. coli str. K-12 substr. MG1655 reference (RefSeq NC_000913.3). We filtered out synonymous variants as well as one common non-synonymous variant (Gln33Glu) and counted the remaining ones for each sample.
For the oldest collection (VDG, 1980), we also compared the results of the typing for the presence of the hly and pap genes assessed phenotypically (production of alpha hemolysin using horse erythrocyte agar plates and presence of mannose-resistant hemagglutination using glass microscope slides [113]) and genetically (PCR method for hlyC and papC detection) performed in 1980 [51] and 2000 [49], respectively.
Lastly, we checked whether gene loss in older collections could have resulted in the loss of genes involved in virulence, and therefore bias pathogenicity downwards in the sequences from 1980. To first examine potential trends in gene content, we selected the genes present in 5 to 95% of the strains from 1980 to 2010 (total 4895 genes). For 100 randomly chosen genes, we used a linear model to assess the association between gene frequency and sampling date. The mean effect size was +0.0012 per year (+3.5% frequency over 30 years). This trend could be due to the loss of genes in older collections (possible if the bacteria replicate slowly, in particular in stab culture and for plasmid-borne genes), but also to changes in the phylogenetic composition of the population. To assess how this slight trend could have changed pathogenicity, we first identified the unitigs selected in the GWAS that reflect gene presence/absence. The criterion was a correlation > 0.5 between gene presence/absence and unitig presence/absence. 13 unitigs out of 59 met this criterion. We then simulated two synthetic datasets of size 10000, one reflecting our main dataset (2000–2010) and one reflecting a hypothetical dataset of 1980 affected by the -3.5% frequency of the 13 genes identified selected in the GWAS. This was done by randomly drawing unitig presence/absence for each sequence with the corresponding frequency, and neglecting linkage. We predicted that the synthetic dataset of 1980 counted 27.5% of commensals, and that of 2000–2010 27.1% of commensals. Thus, a bias in gene content in 1980 could not possibly have caused the observed trends in pathogenicity, both because the observed trend in gene content is weak, and because the GWAS model does not predict a strong net positive effect of more genes on pathogenicity.
Code availability
Apart from the software packages mentioned in the previous sections, the following were used to run the analysis and generate the visualizations presented in this work: pandas v1.3.4 [114], numpy v1.20.3 [114], scipy v1.7.1 [115], matplotlib v3.4.3 [116], seaborn v0.11.2 [117], biopython v1.80 [118] jupyterlab v3.2.1 [119]. Most of the analysis were incorporated in a reproducible pipeline using snakemake v7.18.1 [120] and conda v4.10.3 [121], which is available as a code repository on GitHub (https://github.com/jburgaya/2022_ecoli_commensal) under a permissive licence (MIT).
Supporting information
(XLSX)
The number of isolates and the percentage are presented in the table. We compared the ST diversity of E. coli isolates from BSI (all portals of entry, urinary portal of entry and digestive portal of entry) with a collection of commensal isolates, for all STs present in at least 5 strains in at least one of the two collections. We show the odds ratio (with 95% CI) for the risk of infection associated with colonization by each ST (logistic model of infection status as a function of the ST). STs with odds ratio significantly different from 1 are highlighted in bold.
(XLSX)
The number of isolates and the percentage are presented in the table. We compared the O-group diversity of E. coli isolates from BSI (all portals of entry, urinary portal of entry and digestive portal of entry) with the collection of commensal isolates, for all O-groups present in at least 5 strains in at least one of the two collections. We show the odds ratio (with 95% CI) for the risk of infection associated with colonization by each O-group (logistic model of infection status as a function of the O-group). O-groups with odds ratio significantly different from 1 are highlighted in bold.
(XLSX)
(XLSX)
VAG proportions of commensal and BSI strains are indicated between brackets. Significant differences are in bold (at the 0.05 level).
(XLSX)
Genes are ordered by their average LRT pvalue, annotation columns are derived from the eggnog-mapper.
(XLSX)
(XLSX)
Phylogroups, MLSTs (Warwick and Pasteur), serotypes and fimH alleles are indicated.
(XLSX)
Genes are ordered by their average LRT pvalue, annotation columns are derived from the eggnog-mapper.
(XLSX)
This is shown for three strengths of confounding: no confounding (left), weak confounding (middle; +10% increase in incidence of infection), strong confounding (right; +100% increase in incidence of infection). The case-control study consistently has better power but is subject to over-estimating the effect size of a variant because of confounding. The case-crossover study has very poor power when the number of strains is small, maintains the same power regardless of confounding, and can estimate properly the effect size with large number of colonizing strains. The case cross-over study can have higher power than the case-control study in the absence of confounding (top left graph) bcause it benefits from a larger sample size (912+912).
(PNG)
{kind=link}
(A-C) Comparison of the distribution of VAGs per strain among the six main functional classes of virulence of the E. coli commensal and BSI collections isolates. We show the odds ratio (OR with 95% CI) for the risk of infection associated with the number of VAGs (logistic model of infection status as a function of the number of VAGs), for (A) all the B2 strains (467 BSI strains and 120 commensal strains), (B) B2 BSI strains with urinary portal of entry to B2 commensals (304 BSI strains) and (C) B2 BSI strains with digestive portal of entry to B2 commensals (124 BSI strains). Functional classes of virulence are ordered by increasing associated odds ratio for all B2 strains.
(PNG)
{kind=link}
(A-D) Comparison of the distribution of resistant strains for eight antibiotics of clinical importance of the E. coli commensal and BSI collections isolates. We show the odds ratio (OR with 95% CI) for the risk of infection associated with the resistance of strains (logistic model of infection status as a function of the resistance of strains), for (A) all the B2 strains (467 BSI strains and 120 commensal strains), (B) B2 BSI strains with urinary portal of entry to B2 commensals (304 BSI strains), (C) B2 BSI strains with digestive portal of entry to B2 commensals (124 BSI strains) and (D) BSI Colibafi strains (sampled in 2005) to commensals (367 BSI strains). Categories of antibiotics are ordered by increasing associated odds ratio for all B2 strains. For AMK, FQ, GEN and FEP categories, we only show the lower bound of the CI because the estimated odds ratios are huge as none of the commensal isolates were resistant to these antibiotics. AMK, amikacin; AMP, ampicillin; CTX/ CAZ, cefotaxime/ceftazidime; FEP, cefepime; FQ, fluoroquinolones; GEN, gentamicin; SXT, cotrimoxazole; TZP, piperacillin/tazobactam.
(PNG)
{kind=link}
F1-score representation (blue dots), precision (yellow dots), and recall (red dots). A) For the full collection B) the subset of clinical isolates with urinary tract as portal of entry, and C) the subset of clinical isolates with digestive tract as portal of entry. The naive and the analysis with covariates are represented. PE: portal of entry.
(PNG)
{kind=link}
423 isolates from commensal collections were fitted to the trained ML model. The proportion of BSI isolates for the 3 different periods of time is colored in red and the percentage indicated above each bar. The total number of isolates per year is given in brackets.
(PNG)
{kind=link}
A) Genome assembly quality does not differ substantially across strain collections, as measured using the N50 metric. B) Putatively deleterious mutations in rpoS, a metric for sample storage quality is negligible for all collections used for the main analysis, and much higher for the excluded collection, for which we had a lower confidence on sample storage.
(PNG)
{kind=link}
(DOCX)
Data Availability
All the genome sequences described in this study are available under the following Bioproject IDs: PRJEB38489, PRJEB44819, PRJEB44872, PRJEB39252, PRJEB39260, PRJEB35745, PRJEB44873, and PRJEB55584.
Funding Statement
ED was partially supported by the “Fondation pour la Recherche Médicale” (Equipe FRM 2016, grant number DEQ20161136698). GR was supported by a “Poste d’accueil” funded by the “Assistance Publique-Hôpitaux de Paris” (AP-HP) and the “Commissariat à l'énergie atomique et aux énergies alternatives” (CEA) personal grant for his PhD. MG and JB were supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy - EXC 2155 - project number 390874280. JB was further supported by the Deutsche Forschungsgemeinschaft grant number GA 3191/1-1. F.B. was funded by the ERC StG 949208 EvoComBac. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Goto M, McDanel JS, Jones MM, Livorsi DJ, Ohl ME, Beck BF, et al. Antimicrobial Nonsusceptibility of Gram-Negative Bloodstream Isolates, Veterans Health Administration System, United States, 2003–2013. Emerging infectious diseases. 2017;23: 1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.de Kraker MEA, Davey PG, Grundmann H. Mortality and hospital stay associated with resistant Staphylococcus aureus and Escherichia coli bacteremia: Estimating the burden of antibiotic resistance in Europe. PLoS Medicine. 2011;8. doi: 10.1371/journal.pmed.1001104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abernethy JK, Johnson AP, Guy R, Hinton N, Sheridan EA, Hope RJ. Thirty day all-cause mortality in patients with Escherichia coli bacteraemia in England. Clinical Microbiology and Infection. 2015;21: 251.e1–251.e8. doi: 10.1016/j.cmi.2015.01.001 [DOI] [PubMed] [Google Scholar]
- 4.Feldman SF, Temkin E, Wullfhart L, Nutman A, Schechner V, Shitrit P, et al. A nationwide population-based study of Escherichia coli bloodstream infections: incidence, antimicrobial resistance and mortality. Clinical Microbiology and Infection. 2022;28: 879.e1–879.e7. doi: 10.1016/j.cmi.2021.12.009 [DOI] [PubMed] [Google Scholar]
- 5.Bonten M, Johnson JR, van den Biggelaar AHJ, Georgalis L, Geurtsen J, de Palacios PI, et al. Epidemiology of Escherichia coli Bacteremia: A Systematic Literature Review. Clinical Infectious Diseases. 2021;72: 1211–1219. doi: 10.1093/cid/ciaa210 [DOI] [PubMed] [Google Scholar]
- 6.Gladstone RA, McNally A, Pöntinen AK, Tonkin-Hill G, Lees JA, Skytén K, et al. Emergence and dissemination of antimicrobial resistance in Escherichia coli causing bloodstream infections in Norway in 2002–17: a nationwide, longitudinal, microbial population genomic study. The Lancet Microbe. 2021;2: e331–e341. doi: 10.1016/S2666-5247(21)00031-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kallonen T, Brodrick HJ, Harris SR, Corander J, Brown NM, Martin V, et al. Systematic longitudinal survey of invasive Escherichia coli in England demonstrates a stable population structure only transiently disturbed by the emergence of ST131. Genome research. 2017;27: 1437–1449. doi: 10.1101/gr.216606.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Petty NK, Zakour NLB, Stanton-Cook M, Skippington E, Totsika M, Forde BM, et al. Global dissemination of a multidrug resistant Escherichia coli clone. Proceedings of the National Academy of Sciences. 2014;111: 5694–5699. doi: 10.1073/pnas.1322678111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nicolas-Chanoine MH, Blanco J, Leflon-Guibout V, Demarty R, Alonso MP, Caniça MM, et al. Intercontinental emergence of Escherichia coli clone O25:H4-ST131 producing CTX-M-15. Journal of Antimicrobial Chemotherapy. 2008;61: 273–281. doi: 10.1093/jac/dkm464 [DOI] [PubMed] [Google Scholar]
- 10.Casadevall A, Pirofski LA. Host-pathogen interactions: redefining the basic concepts of virulence and pathogenicity. Infect Immun. 1999;67: 3703–3713. doi: 10.1128/IAI.67.8.3703-3713.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tenaillon O, Skurnik D, Picard B, Denamur E. The population genetics of commensal Escherichia coli. Nature Reviews Microbiology. 2010;8: 207. doi: 10.1038/nrmicro2298 [DOI] [PubMed] [Google Scholar]
- 12.Lefort A, Panhard X, Clermont O, Woerther P-L, Branger C, Mentré F, et al. Host factors and portal of entry outweigh bacterial determinants to predict the severity of Escherichia coli bacteremia. J Clin Microbiol. 2011;49: 777–783. doi: 10.1128/JCM.01902-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kang C-I, Song J-H, Chung DR, Peck KR, Ko KS, Yeom J-S, et al. Risk factors and treatment outcomes of community-onset bacteraemia caused by extended-spectrum β-lactamase-producing Escherichia coli. International Journal of Antimicrobial Agents. 2010;36: 284–287. doi: 10.1016/j.ijantimicag.2010.05.009 [DOI] [PubMed] [Google Scholar]
- 14.Blandy O, Honeyford K, Gharbi M, Thomas A, Ramzan F, Ellington MJ, et al. Factors that impact on the burden of Escherichia coli bacteraemia: multivariable regression analysis of 2011–2015 data from West London. Journal of Hospital Infection. 2019;101: 120–128. doi: 10.1016/j.jhin.2018.10.024 [DOI] [PubMed] [Google Scholar]
- 15.Laupland KB, Gregson DB, Church DL, Ross T, Pitout JDD. Incidence, risk factors and outcomes of Escherichia coli bloodstream infections in a large Canadian region. Clinical Microbiology and Infection. 2008;14: 1041–1047. doi: 10.1111/j.1469-0691.2008.02089.x [DOI] [PubMed] [Google Scholar]
- 16.Denamur E, Condamine B, Esposito-Farèse M, Royer G, Clermont O, Laouenan C, et al. Genome wide association study of Escherichia coli bloodstream infection isolates identifies genetic determinants for the portal of entry but not fatal outcome. PLOS Genetics. 2022;18: e1010112. doi: 10.1371/journal.pgen.1010112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Galardini M, Clermont O, Baron A, Busby B, Dion S, Schubert S, et al. Major role of iron uptake systems in the intrinsic extra-intestinal virulence of the genus Escherichia revealed by a genome-wide association study. PLOS Genetics. 2020;16: e1009065. doi: 10.1371/journal.pgen.1009065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johnson JR. Virulence factors in Escherichia coli urinary tract infection. Clin Microbiol Rev. 1991;4: 80–128. doi: 10.1128/cmr.4.1.80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clermont O, Couffignal C, Blanco J, Mentré F, Picard B, Denamur E. Two levels of specialization in bacteraemic Escherichia coli strains revealed by their comparison with commensal strains. Epidemiology & Infection. 2017;145: 872–882. doi: 10.1017/S0950268816003010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Opal SM, Cross AS, Gemski P, Lyhte LW. Aerobactin and α-Hemolysin as Virulence Determinants in Escherichia coli Isolated from Human Blood, Urine, and Stool. The Journal of Infectious Diseases. 1990;161: 794–796. doi: 10.1093/infdis/161.4.794 [DOI] [PubMed] [Google Scholar]
- 21.Hagberg L, Jodal U, Korhonen TK, Lidin-Janson G, Lindberg U, Svanborg Edén C. Adhesion, hemagglutination, and virulence of Escherichia coli causing urinary tract infections. Infection and Immunity. 1981;31: 564–570. doi: 10.1128/iai.31.2.564-570.1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Janson GL, Hanson LÅ, Kaijser B, Lincoln K, Lindberg U, Olling S, et al. Comparison of Escherichia coli from Bacteriuric Patients with Those from Feces of Healthy Schoolchildren. The Journal of Infectious Diseases. 1977;136: 346–353. doi: 10.1093/infdis/136.3.346 [DOI] [PubMed] [Google Scholar]
- 23.Källenius G, Möllby R, Svenson SB, Helin I, Hultberg H, Cedergren B, et al. Occurrence of P-fimbriated Escherichia coli in urinary tract infections. Lancet. 1981;2: 1369–1372. doi: 10.1016/s0140-6736(81)92797-5 [DOI] [PubMed] [Google Scholar]
- 24.Mao B-H, Chang Y-F, Scaria J, Chang C-C, Chou L-W, Tien N, et al. Identification of Escherichia coli Genes Associated with Urinary Tract Infections. Journal of Clinical Microbiology. 2020;50: 449–456. doi: 10.1128/jcm.00640-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kausar Y, Chunchanur SK, Nadagir SD, Halesh LH, Chandrasekhar MR. Virulence factors, serotypes and antimicrobial suspectibility pattern of Escherichia coli in urinary tract infections. Al Ameen Journal of Medical Sciences. 2009;2: 47–51. [Google Scholar]
- 26.Schlager TA, Hendley JO, Bell AL, Whittam TS. Clonal Diversity of Escherichia coli Colonizing Stools and Urinary Tracts of Young Girls. Infection and Immunity. 2002;70: 1225–1229. doi: 10.1128/IAI.70.3.1225-1229.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moreno E, Andreu A, Pigrau C, Kuskowski MA, Johnson JR, Prats G. Relationship between Escherichia coli Strains Causing Acute Cystitis in Women and the Fecal E. coli Population of the Host. Journal of Clinical Microbiology. 2008;46: 2529–2534. doi: 10.1128/JCM.00813-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ruppé E, Lixandru B, Cojocaru R, Büke Ç, Paramythiotou E, Angebault C, et al. Relative fecal abundance of extended-spectrum beta-lactamases-producing Escherichia coli and their occurrence in urinary-tract infections in women. Antimicrobial agents and chemotherapy. 2013; AAC-00238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Niki M, Hirai I, Yoshinaga A, Ulzii-Orshikh L, Nakata A, Yamamoto A, et al. Extended-spectrum β-lactamase-producing Escherichia coli strains in the feces of carriers contribute substantially to urinary tract infections in these patients. Infection. 2011;39: 467–471. doi: 10.1007/s15010-011-0128-2 [DOI] [PubMed] [Google Scholar]
- 30.Salipante SJ, Roach DJ, Kitzman JO, Snyder MW, Stackhouse B, Butler-Wu SM, et al. Large-scale genomic sequencing of extraintestinal pathogenic Escherichia coli strains. Genome Research. 2015;25: 119–128. doi: 10.1101/gr.180190.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Lastours V, Laouénan C, Royer G, Carbonnelle E, Lepeule R, Esposito-Farèse M, et al. Mortality in Escherichia coli bloodstream infections: antibiotic resistance still does not make it. J Antimicrob Chemother. 2020;75: 2334–2343. doi: 10.1093/jac/dkaa161 [DOI] [PubMed] [Google Scholar]
- 32.Marin J, Clermont O, Royer G, Mercier-Darty M, Decousser JW, Tenaillon O, et al. The population genomics of increased virulence and antibiotic resistance in human commensal Escherichia coli over 30 years in France. Applied and Environmental Microbiology. 2022;88: e00664–22. doi: 10.1128/aem.00664-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Horesh G, Taylor-Brown A, McGimpsey S, Lassalle F, Corander J, Heinz E, et al. Different evolutionary trends form the twilight zone of the bacterial pan-genome. Microbial Genomics. 7: 000670. doi: 10.1099/mgen.0.000670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Frenck RW, Ervin J, Chu L, Abbanat D, Spiessens B, Go O, et al. Safety and immunogenicity of a vaccine for extra-intestinal pathogenic Escherichia coli (ESTELLA): a phase 2 randomised controlled trial. The Lancet Infectious Diseases. 2019;19: 631–640. doi: 10.1016/S1473-3099(18)30803-X [DOI] [PubMed] [Google Scholar]
- 35.Huttner A, Hatz C, van den Dobbelsteen G, Abbanat D, Hornacek A, Frölich R, et al. Safety, immunogenicity, and preliminary clinical efficacy of a vaccine against extraintestinal pathogenic Escherichia coli in women with a history of recurrent urinary tract infection: a randomised, single-blind, placebo-controlled phase 1b trial. Lancet Infect Dis. 2017;17: 528–537. doi: 10.1016/S1473-3099(17)30108-1 [DOI] [PubMed] [Google Scholar]
- 36.Lane MC, Mobley HLT. Role of P-fimbrial-mediated adherence in pyelonephritis and persistence of uropathogenic Escherichia coli (UPEC) in the mammalian kidney. Kidney International. 2007;72: 19–25. doi: 10.1038/sj.ki.5002230 [DOI] [PubMed] [Google Scholar]
- 37.Snyder JA, Lloyd AL, Lockatell CV, Johnson DE, Mobley HLT. Role of Phase Variation of Type 1 Fimbriae in a Uropathogenic Escherichia coli Cystitis Isolate during Urinary Tract Infection. Infection and Immunity. 2006;74: 1387–1393. doi: 10.1128/IAI.74.2.1387-1393.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tourret J, Diard M, Garry L, Matic I, Denamur E. Effects of single and multiple pathogenicity island deletions on uropathogenic Escherichia coli strain 536 intrinsic extra-intestinal virulence. Int J Med Microbiol. 2010;300: 435–439. doi: 10.1016/j.ijmm.2010.04.013 [DOI] [PubMed] [Google Scholar]
- 39.Rendón MA, Saldaña Z, Erdem AL, Monteiro-Neto V, Vázquez A, Kaper JB, et al. Commensal and pathogenic Escherichia coli use a common pilus adherence factor for epithelial cell colonization. Proc Natl Acad Sci U S A. 2007;104: 10637–10642. doi: 10.1073/pnas.0704104104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lehti TA, Bauchart P, Kukkonen M, Dobrindt U, Korhonen TK, Westerlund-Wikström B. Phylogenetic group-associated differences in regulation of the common colonization factor Mat fimbria in Escherichia coli. Mol Microbiol. 2013;87: 1200–1222. doi: 10.1111/mmi.12161 [DOI] [PubMed] [Google Scholar]
- 41.Visscher PM, Hill WG, Wray NR. Heritability in the genomics era: concepts and misconceptions. Nature Reviews Genetics. 2008;9: 255–266. doi: 10.1038/nrg2322 [DOI] [PubMed] [Google Scholar]
- 42.Lees JA, Mai TT, Galardini M, Wheeler NE, Horsfield ST, Parkhill J, et al. Improved Prediction of Bacterial Genotype-Phenotype Associations Using Interpretable Pangenome-Spanning Regressions. mBio. 2020;11: e01344–20. doi: 10.1128/mBio.01344-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jaillard M, Lima L, Tournoud M, Mahé P, van Belkum A, Lacroix V, et al. A fast and agnostic method for bacterial genome-wide association studies: Bridging the gap between k-mers and genetic events. PLoS Genet. 2018;14: e1007758. doi: 10.1371/journal.pgen.1007758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saber MM, Shapiro BJ. Benchmarking bacterial genome-wide association study methods using simulated genomes and phenotypes. Microbial Genomics. 2020;6: e000337. doi: 10.1099/mgen.0.000337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Crépin S, Ottosen EN, Peters K, Smith SN, Himpsl SD, Vollmer W, et al. The lytic transglycosylase MltB connects membrane homeostasis and in vivo fitness of Acinetobacter baumannii. Mol Microbiol. 2018;109: 745–762. doi: 10.1111/mmi.14000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Raha M, Sockett H, Macnab RM. Characterization of the fliL gene in the flagellar regulon of Escherichia coli and Salmonella typhimurium. J Bacteriol. 1994;176: 2308–2311. doi: 10.1128/jb.176.8.2308-2311.1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Blanco P, Hernando-Amado S, Reales-Calderon JA, Corona F, Lira F, Alcalde-Rico M, et al. Bacterial Multidrug Efflux Pumps: Much More Than Antibiotic Resistance Determinants. Microorganisms. 2016;4: 14. doi: 10.3390/microorganisms4010014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bindal G, Krishnamurthi R, Seshasayee ASN, Rath D. CRISPR-Cas-Mediated Gene Silencing Reveals RacR To Be a Negative Regulator of YdaS and YdaT Toxins in Escherichia coli K-12. mSphere. 2017;2: e00483–17. doi: 10.1128/mSphere.00483-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Duriez P, Clermont O, Bonacorsi S, Bingen E, Chaventré A, Elion J, et al. Commensal Escherichia coli isolates are phylogenetically distributed among geographically distinct human populations. Microbiology. 2001;147: 1671–1676. doi: 10.1099/00221287-147-6-1671 [DOI] [PubMed] [Google Scholar]
- 50.Bleibtreu A, Clermont O, Darlu P, Glodt J, Branger C, Picard B, et al. The rpoS gene is predominantly inactivated during laboratory storage and undergoes source-sink evolution in Escherichia coli species. J Bacteriol. 2014;196: 4276–4284. doi: 10.1128/JB.01972-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goullet P, Picard B, Garcia JS. Electrophoretic Mobility of an Esterase from Escherichia coli Isolated from Extraintestinal Infections. The Journal of Infectious Diseases. 1986;154: 727–728. doi: 10.1093/infdis/154.4.727 [DOI] [PubMed] [Google Scholar]
- 52.Hultgren SJ, Lindberg F, Magnusson G, Kihlberg J, Tennent JM, Normark S. The PapG adhesin of uropathogenic Escherichia coli contains separate regions for receptor binding and for the incorporation into the pilus. Proceedings of the National Academy of Sciences. 1989;86: 4357–4361. doi: 10.1073/pnas.86.12.4357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Biggel M, Xavier BB, Johnson JR, Nielsen KL, Frimodt-Møller N, Matheeussen V, et al. Horizontally acquired papGII-containing pathogenicity islands underlie the emergence of invasive uropathogenic Escherichia coli lineages. Nat Commun. 2020;11: 5968. doi: 10.1038/s41467-020-19714-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.de Lorenzo V, Bindereif A, Paw BH, Neilands JB. Aerobactin biosynthesis and transport genes of plasmid ColV-K30 in Escherichia coli K-12. J Bacteriol. 1986;165: 570–578. doi: 10.1128/jb.165.2.570-578.1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Artola-Recolons C, Lee M, Bernardo-García N, Blázquez B, Hesek D, Bartual SG, et al. Structure and Cell Wall Cleavage by Modular Lytic Transglycosylase MltC of Escherichia coli. ACS Chem Biol. 2014;9: 2058–2066. doi: 10.1021/cb500439c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li B, Huang Q, Cui A, Liu X, Hou B, Zhang L, et al. Overexpression of Outer Membrane Protein X (OmpX) Compensates for the Effect of TolC Inactivation on Biofilm Formation and Curli Production in Extraintestinal Pathogenic Escherichia coli (ExPEC). Frontiers in Cellular and Infection Microbiology. 2018;8. Available from: https://www.frontiersin.org/articles/10.3389/fcimb.2018.00208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hirakawa H, Suzue K, Takita A, Kamitani W, Tomita H. Roles of OmpX, an Outer Membrane Protein, on Virulence and Flagellar Expression in Uropathogenic Escherichia coli. Infect Immun. 2021;89: e00721–20. doi: 10.1128/IAI.00721-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Maynard C, Bekal S, Sanschagrin F, Levesque RC, Brousseau R, Masson L, et al. Heterogeneity among Virulence and Antimicrobial Resistance Gene Profiles of Extraintestinal Escherichia coli Isolates of Animal and Human Origin. Journal of Clinical Microbiology. 2004;42: 5444–5452. doi: 10.1128/JCM.42.12.5444-5452.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sampaio SCF, Luiz WB, Vieira MAM, Ferreira RCC, Garcia BG, Sinigaglia-Coimbra R, et al. Flagellar Cap Protein FliD Mediates Adherence of Atypical Enteropathogenic Escherichia coli to Enterocyte Microvilli. Infect Immun. 2016;84: 1112–1122. doi: 10.1128/IAI.01001-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pfiffer V, Sarenko O, Possling A, Hengge R. Genetic dissection of Escherichia coli’s master diguanylate cyclase DgcE: Role of the N-terminal MASE1 domain and direct signal input from a GTPase partner system. PLOS Genetics. 2019;15: e1008059. doi: 10.1371/journal.pgen.1008059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li H, Xia Y, Tian Z, Jin Y, Bai F, Cheng Z, et al. Dihydrolipoamide Acetyltransferase AceF Influences the Type III Secretion System and Resistance to Oxidative Stresses through RsmY/Z in Pseudomonas aeruginosa. Microorganisms. 2022;10: 666. doi: 10.3390/microorganisms10030666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Serfiotis-Mitsa D, Herbert AP, Roberts GA, Soares DC, White JH, Blakely GW, et al. The structure of the KlcA and ArdB proteins reveals a novel fold and antirestriction activity against Type I DNA restriction systems in vivo but not in vitro. Nucleic Acids Res. 2010;38: 1723–1737. doi: 10.1093/nar/gkp1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ravan H, Amandadi M. Analysis of yeh Fimbrial Gene Cluster in Escherichia coli O157:H7 in Order to Find a Genetic Marker for this Serotype. Curr Microbiol. 2015;71: 274–282. doi: 10.1007/s00284-015-0842-6 [DOI] [PubMed] [Google Scholar]
- 64.Kauffman J. The Serology of the Coli Group. The Journal of Immunology. 1947;57: 71–100. [PubMed] [Google Scholar]
- 65.Johnson JR, Johnston BD, Porter S, Thuras P, Aziz M, Price LB. Accessory Traits and Phylogenetic Background Predict Escherichia coli Extraintestinal Virulence Better Than Does Ecological Source. J Infect Dis. 2019;219: 121–132. doi: 10.1093/infdis/jiy459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Picard B, Garcia JS, Gouriou S, Duriez P, Brahimi N, Bingen E, et al. The link between phylogeny and virulence in Escherichia coli extraintestinal infection? Infection and Immunity. 1999;67: 546–553. doi: 10.1128/IAI.67.2.546-553.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Johnson JR, Kuskowski M. Clonal Origin, Virulence Factors, and Virulence. Infect Immun. 2000;68: 424–425. doi: 10.1128/IAI.68.1.424-425.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kremer PHC, Ferwerda B, Bootsma HJ, Rots NY, Wijmenga-Monsuur AJ, Sanders EAM, et al. Pneumococcal genetic variability in age-dependent bacterial carriage. Elife. 2022;11: e69244. doi: 10.7554/eLife.69244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vornhagen J, Roberts EK, Unverdorben L, Mason S, Patel A, Crawford R, et al. Combined comparative genomics and clinical modeling reveals plasmid-encoded genes are independently associated with Klebsiella infection. Nat Commun. 2022;13: 4459. doi: 10.1038/s41467-022-31990-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lees JA, Ferwerda B, Kremer PHC, Wheeler NE, Serón MV, Croucher NJ, et al. Joint sequencing of human and pathogen genomes reveals the genetics of pneumococcal meningitis. Nat Commun. 2019;10: 2176. doi: 10.1038/s41467-019-09976-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Young BC, Wu C-H, Charlesworth J, Earle S, Price JR, Gordon NC, et al. Antimicrobial resistance determinants are associated with Staphylococcus aureus bacteraemia and adaptation to the healthcare environment: a bacterial genome-wide association study. Microb Genom. 2021;7. doi: 10.1099/mgen.0.000700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Young BC, Earle SG, Soeng S, Sar P, Kumar V, Hor S, et al. Panton-Valentine leucocidin is the key determinant of Staphylococcus aureus pyomyositis in a bacterial GWAS. Elife. 2019;8: e42486. doi: 10.7554/eLife.42486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chaguza C, Pöntinen AK, Top J, Arredondo-Alonso S, Freitas AR, Novais C, et al. The population-level impact of Enterococcus faecalis genetics on intestinal colonisation and extraintestinal infection. bioRxiv; 2022. p. 2022.09.26.509451. doi: 10.1101/2022.09.26.509451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Earle SG, Lobanovska M, Lavender H, Tang C, Exley RM, Ramos-Sevillano E, et al. Genome-wide association studies reveal the role of polymorphisms affecting factor H binding protein expression in host invasion by Neisseria meningitidis. PLoS Pathog. 2021;17: e1009992. doi: 10.1371/journal.ppat.1009992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Thorpe HA, Bayliss SC, Hurst LD, Feil EJ. Comparative Analyses of Selection Operating on Nontranslated Intergenic Regions of Diverse Bacterial Species. Genetics. 2017;206: 363–376. doi: 10.1534/genetics.116.195784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mellies JL, Barron AMS. Virulence Gene Regulation in Escherichia coli. EcoSal Plus. 2006;2. doi: 10.1128/ecosalplus.8.9.1 [DOI] [PubMed] [Google Scholar]
- 77.Johansson C, Nilsson A, Kaden R, Rautelin H. Differences in virulence gene expression between human blood and stool Campylobacter coli clade 1 ST828CC isolates. Gut Pathog. 2019;11: 42. doi: 10.1186/s13099-019-0322-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mu A, Klare WP, Baines SL, Ignatius Pang CN, Guérillot R, Harbison-Price N, et al. Integrative omics identifies conserved and pathogen-specific responses of sepsis-causing bacteria. Nat Commun. 2023;14: 1530. doi: 10.1038/s41467-023-37200-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hancock SJ, Lo AW, Ve T, Day CJ, Tan L, Mendez AA, et al. Ucl fimbriae regulation and glycan receptor specificity contribute to gut colonisation by extra-intestinal pathogenic Escherichia coli. PLoS Pathog. 2022;18: e1010582. doi: 10.1371/journal.ppat.1010582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Totsika M, Beatson SA, Holden N, Gally DL. Regulatory interplay between pap operons in uropathogenic Escherichia coli. Mol Microbiol. 2008;67: 996–1011. doi: 10.1111/j.1365-2958.2007.06098.x [DOI] [PubMed] [Google Scholar]
- 81.Biggel M, Moons P, Nguyen MN, Goossens H, Van Puyvelde S. Convergence of virulence and antimicrobial resistance in increasingly prevalent Escherichia coli ST131 papGII+ sublineages. Commun Biol. 2022;5: 752. doi: 10.1038/s42003-022-03660-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wyrsch ER, Bushell RN, Marenda MS, Browning GF, Djordjevic SP. Global Phylogeny and F Virulence Plasmid Carriage in Pandemic Escherichia coli ST1193. Microbiol Spectr. 2022;10: e0255422. doi: 10.1128/spectrum.02554-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.McKinnon J, Roy Chowdhury P, Djordjevic SP. Genomic analysis of multidrug-resistant Escherichia coli ST58 causing urosepsis. Int J Antimicrob Agents. 2018;52: 430–435. doi: 10.1016/j.ijantimicag.2018.06.017 [DOI] [PubMed] [Google Scholar]
- 84.Venturini C, Beatson SA, Djordjevic SP, Walker MJ. Multiple antibiotic resistance gene recruitment onto the enterohemorrhagic Escherichia coli virulence plasmid. FASEB J. 2010;24: 1160–1166. doi: 10.1096/fj.09-144972 [DOI] [PubMed] [Google Scholar]
- 85.Arnold BJ, Gutmann MU, Grad YH, Sheppard SK, Corander J, Lipsitch M, et al. Weak epistasis may drive adaptation in recombining bacteria. Genetics. 2018; genetics–300662. doi: 10.1534/genetics.117.300662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhou Z, Alikhan N-F, Mohamed K, Fan Y, Achtman M. The EnteroBase user’s guide, with case studies on Salmonella transmissions, Yersinia pestis phylogeny, and Escherichia core genomic diversity. Genome Res. 2020;30: 138–152. doi: 10.1101/gr.251678.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ostblom A, Adlerberth I, Wold AE, Nowrouzian FL. Escherichia coli pathogenicity island-markers, malX and usp and the capacity to persist in the infant’s commensal microbiota. Applied and environmental microbiology. 2011;77: 2303–2308. doi: 10.1128/AEM.02405-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nowrouzian F, Hesselmar B, Saalman R, Stranneg\a ard IL, AAberg N, Wold AE, et al. Escherichia coli in infants’ intestinal microflora: Colonization rate, strain turnover, and virulence gene carriage. Pediatric Research. 2003;54: 8–14. doi: 10.1203/01.PDR.0000069843.20655.EE [DOI] [PubMed] [Google Scholar]
- 89.Le Gall T, Clermont O, Gouriou S, Picard B, Nassif X, Denamur E, et al. Extraintestinal virulence is a coincidental by-product of commensalism in b2 phylogenetic group Escherichia coli strains. Molecular Biology and Evolution. 2007;24: 2373–2384. doi: 10.1093/molbev/msm172 [DOI] [PubMed] [Google Scholar]
- 90.Diard M, Garry L, Selva M, Mosser T, Denamur E, Matic I. Pathogenicity-associated islands in extraintestinal pathogenic Escherichia coli are fitness elements involved in intestinal colonization. Journal of Bacteriology. 2010;192: 4885–4893. doi: 10.1128/JB.00804-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Royer G, Clermont O, Marin J, Condamine B, Dion S, Blanquart F, et al. Epistatic interactions between the high pathogenicity island and other iron uptake systems shape Escherichia coli extra-intestinal virulence. Nat Commun. 2023;14: 3667. doi: 10.1038/s41467-023-39428-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Skurnik D, Clermont O, Guillard T, Launay A, Danilchanka O, Pons S, et al. Emergence of Antimicrobial-Resistant Escherichia coli of Animal Origin Spreading in Humans. Mol Biol Evol. 2016;33: 898–914. doi: 10.1093/molbev/msv280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Escobar-Páramo P, Grenet K, Le Menac’h A, Rode L, Salgado E, Amorin C, et al. Large-Scale Population Structure of Human Commensal Escherichia coli Isolates. Appl Environ Microbiol. 2004;70: 5698–5700. doi: 10.1128/AEM.70.9.5698-5700.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Massot M, Daubi?? AS, Clermont O, Jauréguy F, Couffignal C, Dahbi G, et al. Phylogenetic, virulence and antibiotic resistance characteristics of commensal strain populations of escherichia coli from community subjects in the paris area in 2010 and evolution over 30 years. Microbiology (United Kingdom). 2016;162: 642–650. doi: 10.1099/mic.0.000242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Burdet C, Grall N, Linard M, Bridier-Nahmias A, Benhayoun M, Bourabha K, et al. Ceftriaxone and Cefotaxime Have Similar Effects on the Intestinal Microbiota in Human Volunteers Treated by Standard-Dose Regimens. Antimicrob Agents Chemother. 2019;63: e02244–18. doi: 10.1128/AAC.02244-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Royer G, Darty MM, Clermont O, Condamine B, Laouenan C, Decousser J-W, et al. Phylogroup stability contrasts with high within sequence type complex dynamics of Escherichia coli bloodstream infection isolates over a 12-year period. Genome Med. 2021;13: 77. doi: 10.1186/s13073-021-00892-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Clermont O, Glodt J, Burdet C, Pognard D, Lefort A, Branger C, et al. Complexity of Escherichia coli bacteremia pathophysiology evidenced by comparison of isolates from blood and portal of entry within single patients. Int J Med Microbiol. 2013;303: 529–532. doi: 10.1016/j.ijmm.2013.07.002 [DOI] [PubMed] [Google Scholar]
- 98.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30: 2068–2069. doi: 10.1093/bioinformatics/btu153 [DOI] [PubMed] [Google Scholar]
- 99.Tonkin-Hill G, MacAlasdair N, Ruis C, Weimann A, Horesh G, Lees JA, et al. Producing polished prokaryotic pangenomes with the Panaroo pipeline. Genome Biology. 2020;21: 180. doi: 10.1186/s13059-020-02090-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Price MN, Dehal PS, Arkin AP. FastTree 2 –Approximately Maximum-Likelihood Trees for Large Alignments. PLOS ONE. 2010;5: e9490. doi: 10.1371/journal.pone.0009490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Argimón S, Abudahab K, Goater RJE, Fedosejev A, Bhai J, Glasner C, et al. Microreact: visualizing and sharing data for genomic epidemiology and phylogeography. Microbial Genomics. 2016;2: e000093. doi: 10.1099/mgen.0.000093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bourrel AS, Poirel L, Royer G, Darty M, Vuillemin X, Kieffer N, et al. Colistin resistance in Parisian inpatient faecal Escherichia coli as the result of two distinct evolutionary pathways. J Antimicrob Chemother. 2019;74: 1521–1530. doi: 10.1093/jac/dkz090 [DOI] [PubMed] [Google Scholar]
- 103.Wirth T, Falush D, Lan R, Colles F, Mensa P, Wieler LH, et al. Sex and virulence in Escherichia coli: an evolutionary perspective. Mol Microbiol. 2006;60: 1136–1151. doi: 10.1111/j.1365-2958.2006.05172.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ingle DJ, Valcanis M, Kuzevski A, Tauschek M, Inouye M, Stinear T, et al. In silico serotyping of E. coli from short read data identifies limited novel O-loci but extensive diversity of O:H serotype combinations within and between pathogenic lineages. Microb Genom. 2016;2: e000064. doi: 10.1099/mgen.0.000064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tonkin-Hill G, Gladstone RA, Pöntinen AK, Arredondo-Alonso S, Bentley SD, Corander J. Robust analysis of prokaryotic pangenome gene gain and loss rates with Panstripe. Genome Res. 2023;33: 129–140. doi: 10.1101/gr.277340.122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nei M. Molecular Evolutionary Genetics. Molecular Evolutionary Genetics. Columbia University Press; 1987. doi: 10.7312/nei-92038 [DOI] [Google Scholar]
- 107.Paradis E, Schliep K. ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics. 2019;35: 526–528. doi: 10.1093/bioinformatics/bty633 [DOI] [PubMed] [Google Scholar]
- 108.Seemann T. ABRicate. 2023. Available from: https://github.com/tseemann/abricate. [Google Scholar]
- 109.Lippert C, Casale FP, Rakitsch B, Stegle O. LIMIX: genetic analysis of multiple traits. bioRxiv; 2014. p. 003905. doi: 10.1101/003905 [DOI] [Google Scholar]
- 110.Lees JA, Galardini M, Bentley SD, Weiser JN, Corander J. pyseer: a comprehensive tool for microbial pangenome-wide association studies. Bioinformatics. 2018;34: 4310–4312. doi: 10.1093/bioinformatics/bty539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Klopfenstein DV, Zhang L, Pedersen BS, Ramírez F, Warwick Vesztrocy A, Naldi A, et al. GOATOOLS: A Python library for Gene Ontology analyses. Sci Rep. 2018;8: 10872. doi: 10.1038/s41598-018-28948-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Holley G, Melsted P. Bifrost: highly parallel construction and indexing of colored and compacted de Bruijn graphs. Genome Biology. 2020;21: 249. doi: 10.1186/s13059-020-02135-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ph Goullet, Picard B. Highly Pathogenic Strains of Escherichia coli Revealed by the Distinct Electrophoretic Patterns of Carboxylesterase B. Microbiology. 1986;132: 1853–1858. doi: 10.1099/00221287-132-7-1853 [DOI] [PubMed] [Google Scholar]
- 114.McKinney W. Data Structures for Statistical Computing in Python. Austin, Texas; 2010. pp. 56–61. doi: 10.25080/Majora-92bf1922-00a [DOI] [Google Scholar]
- 115.Virtanen P, Gommers R, Oliphant TE, Haberland M, Reddy T, Cournapeau D, et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat Methods. 2020;17: 261–272. doi: 10.1038/s41592-019-0686-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hunter JD. Matplotlib: A 2D Graphics Environment. Computing in Science and Engg. 2007;9: 90–95. doi: 10.1109/MCSE.2007.55 [DOI] [Google Scholar]
- 117.Waskom M. seaborn: statistical data visualization. JOSS. 2021;6: 3021. doi: 10.21105/joss.03021 [DOI] [Google Scholar]
- 118.Cock PJA, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, et al. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics. 2009;25: 1422–1423. doi: 10.1093/bioinformatics/btp163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kluyver T, Ragan-Kelley B, Pérez F, Bussonnier M, Frederic J, Hamrick J, et al. Jupyter Notebooks—a publishing format for reproducible computational workflows.
- 120.Mölder F, Jablonski KP, Letcher B, Hall MB, Tomkins-Tinch CH, Sochat V, et al. Sustainable data analysis with Snakemake. F1000Res. 2021;10: 33. doi: 10.12688/f1000research.29032.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Grüning B, Dale R, Sjödin A, Chapman BA, Rowe J, Tomkins-Tinch CH, et al. Bioconda: sustainable and comprehensive software distribution for the life sciences. Nat Methods. 2018;15: 475–476. doi: 10.1038/s41592-018-0046-7 [DOI] [PMC free article] [PubMed] [Google Scholar]