

Resumen
La dermatomiositis positiva contra el gen . asociado a la diferenciación de melanoma (DM anti-MDA5) es una enfermedad rara que representa menos del 2%. La prevalencia de DM anti-MDA5 varía de 7 a 60%, con mayor prevalencia en asiáticos (11-60%) y mujeres. El cuadro clínico es muy variado y se acompaña por las características típicas de dermatomiositis, como la eritema en heliotropo, con edema, exantema eritematoso en la cara o la parte anterior del tórax (signo de V) y en la espalda y los hombros (signo del chal), las pápulas de Gottron en la parte dorsal de las articulaciones metacarpofalángicas o interfalángicas, que son patognomónicas por definición, pero uno de los signos más llamativos es la ulceración cutánea dolorosa que se encuentra hasta en un 82% de los casos, es profunda y en sacabocados muestran costras hiperqueratósicas. Para el diagnóstico son necesarias las erupciones típicas de la DM (pápulas de Gottron o signo de Gottron y erupción de heliotropo), junto con una patología cutánea de "dermatitis de interfase" o evidencia de miositis o un MSA (autoanticuerpos específicos de miositis). La inmunoprecipitación es el método de referencia para detectar MSA. Para su tratamiento se usan combinaciones de glucocorticoides e inmunosupresores; ademas, es necesaria la detección de enfermedad intersticial rápidamente progresiva (RP-ILD) con tomografía computarizada de alta resolución por su alta asociación con un pronóstico fatal.
Palabras clave: Dermatomiositis, Anticuerpos, Úlcera
Abstract
Dermatomyositis positive anti-melanoma differentiation-associated gene . (anti-MDA5 DM) is a rare disease that represents less than 2%. The prevalence of anti-MDA5 DM ranges from 7 to 60%, with higher prevalence in Asian (11-60%) and women. The clinical picture may be variable and is accompanied by the typical features of dermatomyositis, such as periorbital heliotrope (blue–purple) rash with edema, erythematous rash on the face, or the anterior chest (in a V-sign), and back and shoulders (in a shawl sign), violaceous papules or plaques located on the dorsal part of the metacarpophalangeal or interphalangeal joints, which are pathognomonic by definition; yet, one of the most striking signs is the painful ulceration skin that is found in 82% of cases, which is deep and in punching holes or showing hyperkeratotic crusts. For diagnosis is necessary the typical DM rashes (Gottron’s papules or Gottron’s sign and heliotrope rash), along with either an “interface dermatitis” skin pathology or evidence of myositis or a MSA (myositis-specific autoantibodies). Immunoprecipitation is the gold standard method to detect MSA. Combinations of glucocorticoids and immunosuppressants are used for treatment; besides, it is necessary the detection of rapidly progressive interstitial disease (RP-ILD) with a high-resolution CT because of its high association with fatal prognosis.
Keywords: Dermatomyositis, Antibodies, Ulcer
Introducción
Las miopatías inflamatorias idiopáticas (MII) son un grupo heterogéneo de enfermedades raras del tejido conjuntivo, caracterizadas por la inflamación de varios órganos y tejidos. Tienen cuatro subgrupos muy heterogéneos en sus características clínicas, pronósticas y patológicas, lo que dificulta el diagnóstico y el tratamiento. Se incluyen la miositis necrosante inmunomediada, la miositis por cuerpos de inclusión, el síndrome antisintetasa y la dermatomiositis (DM). Sin embargo, el descubrimiento y la inclusión de autoanticuerpos específicos de miositis (MSA) en el diagnóstico de la miositis permitió una mejor definición de subgrupos de pacientes en términos de fenotipos clínicos, pronóstico y respuesta al tratamiento. 1 Se han descrito cinco para DM: anti-Mi-2, anti-factor intermediario de transcripción 1 γ (TIF1γ), anti-proteína de matriz nuclear 2 (NXP2), anti-antígeno de diferenciación de melanoma 5 (MDA5) y anticuerpos anti-enzima activadora modificadora similar a la ubiquitina (SAE). 2
La dermatomiositis positiva contra el gen 5 asociado a la diferenciación de melanoma (DM anti-MDA5) es predominante en la región de Asia oriental. En cuanto al cuadro clínico, este suele ser muy variado, con rasgos mucocutáneos distintivos como ulceración oral, pápulas palmares dolorosas, alopecia difusa no cicatrizal, paniculitis, úlceras cutáneas, artritis y menor incidencia de miositis (DM hipo o amiopática). Por tal razón, se dificulta el diagnóstico. El pronóstico puede ser extremadamente malo, debido a la alta incidencia de enfermedad intersticial rápidamente progresiva (EPI-RP) si no se diagnostica de manera temprana, aunado a la mala calidad de vida de los pacientes debido a las ulceraciones. 3,4
Metodología
Este artículo de revisión se ha realizado mediante una búsqueda bibliográfica de los últimos cinco años hasta la actualidad y se basa en la selección de estudios y revisiones de la enfermedad y de estudios publicados de diferentes bases de datos. Como fuentes de información se han utilizado:
· Bases de datos bibliográficas: Pubmed, ScienceDirect, ClinicalKey.
· Guias o protocolos de sociedades científicas: EULAR/ACR.
· Documentos de organismos oficiales: el 239th ENMC International Workshop.
Resultados
La DM anti-MDA5 es una enfermedad con mal pronóstico, debido a su relación con la enfermedad intersticial rápidamente progresiva, la cual se presenta en pacientes en los que no se tuvo una sospecha diagnóstica al principio, por lo que en la siguiente revisión se abordarán los aspectos generales de esta entidad, que en primera instancia son manifestaciones cutáneas.
Epidemiología
La DM anti-MDA5 es una enfermedad rara que representa menos del 2% de las MII en Europa. Entre el subgrupo de DM, la prevalencia varía de 7 a 60%, con mayor prevalencia en asiáticos (11-60%) que en caucásicos (7-16%). Al igual que otras enfermedades autoinmunes, ocurre principalmente en mujeres, con una relación mujer/hombre que varía de 0.6 a 7.3 (F/M > 1 en 14 de 16 estudios). 1
Etiología
Al igual que con otros tipos de DM, la etiología y la patogenia de la DM anti-MDA5 siguen siendo desconocidas. Generalmente, la enfermedad puede ocurrir como resultado de la exposición a factores ambientales en individuos genéticamente susceptibles. El MDA5 es codificado por el gen IFIH1, que es uno de los receptores similares al gen I (RIG-I) inducible por ácido retinoico (RLR), que funciona como un sensor de proteína clave del RNAds viral. Los picornavirus, como el virus de la hepatitis A, el virus coxsackie B, el enterovirus y el rinovirus, podrían activar MDA5 con la producción de IFN tipo I (IFNα e IFNβ) y otras citocinas inflamatorias involucradas en la respuesta antiviral. La sobreactivación de la vía del IFN tipo I podría conducir a enfermedades autoinflamatorias. Por lo tanto, se propuso que las infecciones virales podrían ser un evento desencadenante en el desarrollo de DM anti-MDA5. También el IFN-κ, secretado por los queratinocitos, estaba elevado en la piel de pacientes con DM anti-MDA5 y podría contribuir a la fisiopatología de sus lesiones cutáneas. 3
Con respecto a los factores de riesgo genéticos, aunque no se observa asociación de anti-MDA5 y la región del alelo HLA en caucásicos, se ha descrito la susceptibilidad del HLA-DRB*01:01/*04:05 en población japonesa. Un estudio reciente de GWAS (Genome-Wide Association Studies) encontró una variante de empalme del gen WDYF4 en pacientes con DM anti-MDA5, la cual podría potenciar la apoptosis inducida por MDA5 a partir de la activación de NK-κB. El Mx1, un gen característico de IFN de tipo I, se distribuía en los vasos sanguíneos y se extendía a la dermis profunda, lo que implicaba una vasculopatía relacionada con IFN en pacientes con esta patología. 3
La hiperinflamación, evidenciada por hiperferritinemia pronunciada y aumento de citocinas, es otra característica importante pero no existe un patrón universal y se plantea que tenga un patrón dinámico en el trascurso de la enfermedad. Las interleucinas involucradas son IL-8, IL-15, IL-6 e IL-18; en cuanto a EPI-RP se elevan IFNα, IFN-γ, IL-1β e IL-12. 3
Cuadro clínico
La DM anti-MDA5 tiene predilección por población asiática. Se acompaña por las características típicas de dermatomiositis, como el eritema en heliotropo, edema, exantema eritematoso en la cara o la parte anterior del tórax (signo de la V) (figura 1) y en la espalda y hombros (signo del chal), las pápulas de Gottron en la parte dorsal de las articulaciones metacarpofalángicas o interfalángicas (figura 2), que son patognomónicas por definición; sin embargo, uno de los signos más llamativos es la ulceración cutánea dolorosa, que se encuentra hasta en un 82% de los casos y generalmente es profunda en sacabocados o muestra costras hiperqueratósicas. Las úlceras cutáneas representan la convergencia de varios procesos patogénicos independientes, que incluyen vasculopatía, dermatitis de interfase severa y vasculitis. Estas muestran una predilección por ciertos sitios, como el de las pápulas de Gottron, los pulpejos de los dedos y el área periungueal, o sobre el signo de Gottron en las rodillas, los codos o ambos (figura 3). También pueden afectar zonas fotoexpuestas al sol. Estos pacientes tienen un riesgo significativamente mayor de desarrollar EPI (enfermedad pulmonar intersticial). Se pueden presentar las pápulas de Gottron inversas, ubicadas en la superficie palmar de las articulaciones interfalángicas (a diferencia de las clásicas pápulas de Gottron), que se ven como hiperqueratosis triangular blanca. 5 La gangrena o la osteomielitis pueden sobrevenir y provocar complicaciones más graves, como la amputación digital. 6
Figura 1. Signo de la V en la región anterior del tórax y eritema en heliotropo que afecta área periorbitaria en una paciente del INCMNSZ.
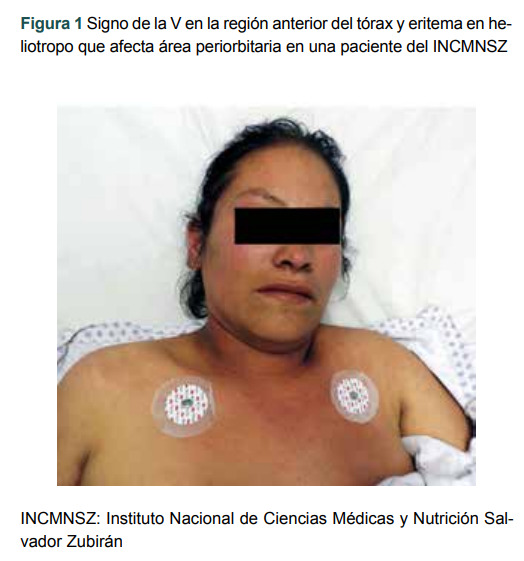
INCMNSZ: Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán
Figura 2. Pápulas de Gottron en la parte dorsal de las articulaciones metacarpofalángicas o interfalángicas en una paciente del INCMNSZ.

INCMNSZ: Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán
Nótese cómo las articulaciones metacarpofalángicas o interfalángicas están acompañadas de una costra hiperqueratósica en la quinta articulación metacarpofalángica
Figura 3. Signo de Gottron: afectación de codos en una paciente del INCMNSZ.

INCMNSZ: Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán
Las pápulas palmares representan otra característica única de la dermatomiositis anti-MDA5 en el 60% de los casos. Son secundarias a vasculopatía oclusiva y pueden presentarse como máculas y parches en las palmas y sobre los pliegues de las articulaciones metacarpofalángicas o interfalángicas. Típicamente son dolorosas, inflamatorias, pueden ser pálidas, atróficas y desarrollarse sobre un fondo púrpura con una coloración marfileña central o livedoide e incluso ulcerarse. 7 También se han descrito lesiones cutáneas auriculares menos específicas, caracterizadas por máculas violáceas en antihélix/hélix y pápulas eritematosas auriculares. Si se presentan, son de mal pronóstico. 1
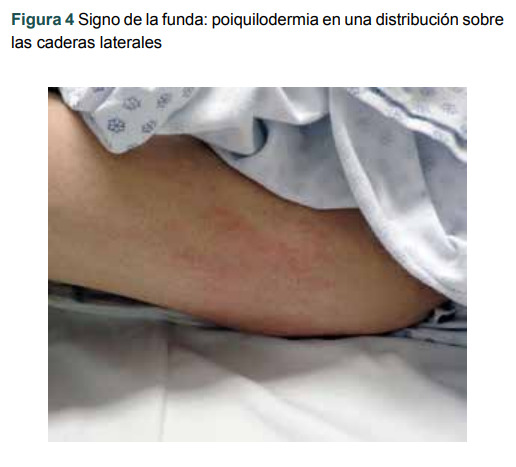
La poiquilodermia también es un rasgo característico. Consiste en atrofia, edema, eritema, hiperpigmentación, hipopigmentación y telangiectasias, y comúnmente se presenta en partes del cuerpo expuestas al sol. 7 En una distribución sobre las caderas laterales, la poiquilodermia se conoce como el "signo de la funda" (figura 4). 8 Asimismo, se pueden observar rayas lineales, también conocidas como eritema flagelado o rayas de cebra. 9
Figura 4. Signo de la funda: poiquilodermia en una distribución sobre las caderas laterales.
La alopecia no cicatrizal difusa se encuentra como parte de una exacerbación de esta enfermedad y se caracteriza por placas atróficas, eritematosas y escamosas. 10 Otras son úlceras orales dolorosas (50%) y paniculitis (nódulos indurados, aislados o confluentes en glúteos, brazos, muslos y abdomen) en brazos y muslos (20%).7
Los síntomas constitucionales y musculoesqueléticos son comunes y se presentan como fiebre en el 74% de los casos, artralgias y mialgias. La artritis es simétrica, erosiva y afecta las pequeñas articulaciones de las manos, muñecas y tobillos; se asemeja a la artritis reumatoide y puede dejar deformidad permanente. Se puede presentar ronquera o dolor de garganta y disfagia. Los pacientes con dermatomiositis con asociación a MDA5 tienen enfermedad muscular mínima o ausente en comparación con los pacientes con dermatomiositis típica, la cual afecta principalmente a los músculos proximales. 1,3,7
La enfermedad pulmonar intersticial (EPI) es la complicación sistémica que aumenta la mortalidad en estos pacientes. Su prevalencia es del 42 al 100% y con desenlace fatal tiene una prevalencia estimada de 39-92%. Se debe a la lesión endotelial causada por anticuerpos MDA5 y el aumento de la expresión de citocinas profibróticas del parénquima pulmonar. Se define por fibrosis u opacificación en vidrio esmerilado, la cual se observa en la tomografía computarizada (TC); también se pueden utilizar pruebas de función pulmonar (PFT) con capacidad de difusión del pulmón para monóxido de carbono (DLCO): la presencia de ambas restricciones (capacidad vital forzada < 80% de los niveles predichos en ausencia de obstrucción) y la disminución de la DLCO (< 80% del valor teórico) son sugerentes. 7
La malignidad es poco común; en el 25% de los casos nuevos se ha encontrado carcinoma metastásico de células pequeñas y cáncer de tiroides.1El cáncer se detecta un año antes o hasta tres años después del diagnóstico y tiene una correlación inversa con la presencia de EPI. 7
También se enumeran otras complicaciones raras, como la afectación cardiaca (por ejemplo, la fibrilación auricular de nueva aparición o la elevación de la troponina cardiaca) y la hemorragia intramuscular espontánea. 3
Algunos pacientes tienen un fenotipo cercano al síndrome antisintetasa (es decir, artritis, síndrome de Raynaud, manos de mecánico, EPI), lo que complica aún más el diagnóstico. 1
Diagnóstico
Los criterios de Sontheimer y los criterios de la European League Against Rheumatism/American College of Rheumatology (EULAR/ACR) se basan en la presencia de autoanticuerpos específicos de la miositis (MSAs), por lo que no son muy prácticos. 11 Por otro lado, los criterios propuestos por el 239th ENMC International Workshop (European Neuromuscular Centre) son más aplicables para la DM anti-MDA5. 12 Los antes mencionados toman para el diagnóstico las erupciones típicas de la DM (pápulas de Gottron o signo de Gottron y erupción de heliotropo), junto con una patología cutánea de "dermatitis de interfase" o evidencia de miositis o un MSA. En otras palabras, con erupciones típicas de DM y un anti-MDA5 positivo es suficiente para clasificar al paciente como dermatomiositis anti-MDA. 3
El diagnóstico de dermatomiositis anti-MDA5 depende de la detección de anticuerpos MDA5 por inmunoprecipitación o ELISA. La inmunoprecipitación es el método de referencia para detectar MSA, pero esta técnica no es factible en la práctica actual. 2 Los niveles de ferritina sérica suelen estar elevados en personas con dermatomiositis anti-MDA5, con valores de corte informados que varían de 450 a 2000 ng/mL. Estos niveles parecen correlacionarse con la actividad de la enfermedad, en particular con la EPI, por lo que son útiles en la evaluación de la respuesta al tratamiento. 3,7
En la dermatomiositis juvenil, los anticuerpos anti-MDA5 son los terceros anticuerpos identificados en frecuencia en miopatías inflamatorias en niños (6-7%), antecedidos por anti-TIF1γ (18%) y NXP2 (15%). 13,14 La enzima lactato deshidrogenasa (LDH) está relacionada con la actividad de la enfermedad de los pacientes con DM, especialmente para aquellos con CK normal. 6 Hay elevación de ALT y GGT. 1
Krebs von den Lungen-6 (KL-6), una glicoproteína de alto peso molecular, similar a la mucina, se libera del epitelio bronquial dañado y de las células alveolares regenerativas de tipo II. La concentración sérica de KL-6 en pacientes con DM-EPI fue significativamente mayor que en pacientes sin EPI. El valor del suero KL-6 cambia junto con la progresión o mejora de la EPI. 7
El nivel sérico de IL-15 aumentó significativamente en anti-MDA5 con EPI-RP y se relacionó con una supervivencia deficiente. 3
Otros hallazgos son la linfopenia, la velocidad de sedimentación globular elevada y las concentraciones sanguíneas periféricas elevadas de interleucina 18. Un bajo porcentaje puede dar positivo a anticuerpos antinucleares. 7 La positividad para anti-TRIM21 (Ro52) es de mal pronóstico. 1
Clasificación
Existen tres fenotipos: el primero está compuesto principalmente por mujeres con RP-EPI asociada a manos de mecánico, con la mayor tasa de mortalidad (80%). El segundo (55% de los casos) también lo forman mayoritariamente mujeres con artralgias o artritis (83%), menos frecuentes RP-EPI (17%) y de mejor pronóstico. El tercer fenotipo clínico engloba principalmente a hombres con síntomas dominados por vasculopatías cutáneas, incluido el fenómeno de Raynaud (82 %), úlceras cutáneas (77%), necrosis digital (32%) y calcinosis (23%). Se asocia a debilidad muscular proximal (68%). 1
Tratamiento
La terapia de primera línea para dermatomiositis en general debe incluir fotoprotección agresiva, agentes antipruriginosos y medicamentos antiinflamatorios tópicos (corticosteroides e inhibidores de la calcineurina). 15
Es necesario hacer la detección de enfermedad pulmonar para los casos sospechosos o confirmados. Si bien la tomografía computarizada de alta resolución del tórax es el estándar de oro para diagnosticar la EPI, su uso puede verse limitado por la alta exposición a la radiación acumulada. Por lo tanto, PFT con DLCO podría ser una prueba de detección inicial y repetir cada 3-6 meses durante el primer año. 7
Se ha demostrado que el diagnóstico temprano de la dermatomiositis anti-MDA5 y su tratamiento intensivo mejoran los resultados de los pacientes con una tasa de supervivencia que puede llegar a ser del 100%. 16 El régimen de tratamiento específico dependerá de los sistemas de órganos involucrados y la gravedad de la afectación. Se prefiere utilizar micofenolato de mofetilo como agente inicial en pacientes con dermatomiositis anti-MDA5, debido a que reduce la expresión de citoquinas profibróticas y parece atenuar la progresión de la enfermedad pulmonar. Se usa la asociación de glucocorticoides con un inhibidor de la calcineurina (ciclosporina A o tacrolimus) o una terapia triple con la adición de ciclofosfamida o micofenolato de mofetilo intravenosos con una tasa de mortalidad de 40%. Como terapia de mantenimiento se utiliza micofenolato de mofetilo con o sin tacrolimus. 17 Debido a que la enfermedad cutánea en la dermatomiositis anti-MDA5 puede ser difícil de manejar, a menudo también se usa inmunoglobulina intravenosa junto con micofenolato de mofetilo porque parece ser eficaz para tratar la dermatomiositis cutánea refractaria. En pacientes con enfermedad pulmonar grave o recalcitrante, se puede usar rituximab, ciclofosfamida u otros agentes. Existen terapias auxiliares destinadas a mejorar la vasculopatía subyacente (por ejemplo, vasodilatadores como la nifedipino y el sildenafil), así como los agentes que mejoran la circulación periférica (como la aspirina y la pentoxifilina), los cuales también pueden ser útiles para tratar la DM anti-MDA5. 7
Se reporta un caso donde las ulceraciones digitales fueron refractarias al tratamiento con hidroxicloroquina, metotrexato, prednisona, ciclosporina, inmunoglobulina intravenosa, triamcinolona intralesional y clobetasol tópico durante varios meses, por lo que se inició tratamiento con ciclofosfamida intravenosa en pulsos (15 mg/kg), y se continuó con metotrexato oral, ciclosporina y prednisona. Después de la primera infusión de ciclofosfamida, hubo una rápida mejoría de las úlceras digitales y el dolor; el paciente tuvo una resolución completa de las úlceras digitales sin efectos adversos del tratamiento a las tres semanas. 18
En caso de refractariedad al tratamiento y deterioro a pesar del soporte con ventilación mecánica en pacientes con EPI-RP, como última instancia se puede optar por una terapia puente con la ECMO (oxigenación por membrana extracorpórea) que se emplea para el trasplante de pulmón; se han descrito cuatro casos con resultados favorables. La terapia de mantenimiento posterior al trasplante sería una combinación de micofenolato de mofetilo y tacrolimus. 17,19,20,21
Los pacientes con niveles de anticuerpos anti-MDA5 superiores a 500 U/mL (umbral de positividad a 8 U/mL) son más resistentes al tratamiento con glucocorticoides/ciclofosfamida o inmunoglobulinas intravenosas y tienen altas tasas de mortalidad. Por el contrario, los pacientes con niveles de anti-MDA5 inferiores a 500 U/mL tuvieron lesiones pulmonares menos graves y los síntomas cutáneos mejoraron después del tratamiento. 1
Se ha planteado como terapia adyuvante el recambio plasmático con buenos resultados en pacientes que no responden a terapia combinada. 22
La vía de señalización JAK/STAT es activada por IFN y conduce a la transcripción de genes estimulados por interferón, incluido MDA5. El tofacitinib y el ruxolitinib inhiben esta vía y dismuyen la expresión y activación de MDA5. Como efecto adverso hay reactivación del citomegalovirus (100% de los pacientes), reactivación del virus varicela-zóster (60%) e infecciones respiratorias bacterianas (80%). 1
Finalmente, a menudo se necesita un enfoque multidisciplinario para abordar adecuadamente las diversas complicaciones de la dermatomiositis anti-MDA5. 7
Pronóstico
Las úlceras digitales en la dermatomiositis anti-MDA5 se asocian con una mala calidad de vida debido a los síntomas incapacitantes. 18
La supervivencia de DM anti-MDA5 con EPI es generalmente pobre. La mayoría de las muertes ocurren dentro de la primera mitad del año de la enfermedad debido a la EPI-RP y a pesar del tratamiento inmunosupresor intensivo, lo que lleva a una alta mortalidad a los seis meses, que varía entre el 33 y el 66 %. 7 A pesar de los avances en el diagnóstico oportuno y un tratamiento más eficaz, la mortalidad a los seis meses sigue siendo de alrededor del 40%. 3
Algunos investigadores han encontrado que la presencia de anti-MDA5 por sí sola es un predictor independiente de supervivencia deficiente. 7 Aunque también se ha visto que MDA5 positivo con valores altos (a diferencia de positivo medio o débil) se asocia a enfermedad grave, pero su disminución no predice la respuesta al tratamiento, debido a que se reduce de igual manera en pacientes que tienen buen o mal desenlace de la enfermedad. 23 La presencia de anti-Ro52 y anti-MDA5 positivos tiende a presentar un fenotipo clínico más agresivo con EPI grave y ulceración cutánea, lo que se traduce en un peor pronóstico. 17,24
Se puede utilizar el puntaje de FLAIR para predecir mortalidad. Este incluye ferritina (< 636 ng/mL, 0; ≥ 636 ng/mL, 2), lactato deshidrogenasa (< 355 U/L, 0; ≥ 355 U/L, 2), anticuerpo contra el gen 5 asociado a la diferenciación de antimelanoma (negativo, 0; +, 2; ++, 3; +++, 4), puntaje de imágenes de TC de alta resolución (< 133, 0; ≥ 133 , 3) y EPI-RP (no EPI-RP, 0; EPI-RP, 2). Esto da tres grupos de riesgo según la puntuación FLAIR: bajo, de 0 a 4; medio, de 5 a 9; y alto, de 10 a 13. 25
Discusión
La DM anti-MDA5 es una entidad rara en nuestro medio. Por tal motivo se llevó a cabo esta revision; sin embargo, por la alta mortalidad con la que se asocia, es muy importante tomar en cuenta esta patología como diagnóstico diferencial, principalmente en pacientes que manifiesten lesiones cutáneas y deterioro de la función respiratoria. Esto ayudaría mucho a establecer un diagnóstico temprano de acuerdo con el cuadro clínico, la serología y hacer tamizaje de enfermedad intersticial pulmonar con pruebas de función pulmonar o tomografía, en caso de contar con ella, en todos los pacientes con diagnóstico sospechoso o confirmado, y de esta manera iniciar un tratamiento temprano y agresivo.
Por otro lado, en cuanto a las manifestaciones cutáneas, estas tienen un alto componente emocional para los pacientes y esto se debe a la afección de la calidad de vida, principalmente por las úlceras que presentan, para las cuales hay muchas recomendaciones en la literatura, aunque ninguna se considera de elección, por lo que sería conveniente comparar más estudios.
Conclusiones
Si consideramos todo lo anterior, es importante tomar en cuenta esta enfermedad reumatológica en casos futuros y darle un enfoque multidisciplinario tanto médico como psicológico con seguimiento estrecho, principalmente por la afeccion pulmonar, que es una causa de alta mortalidad en estos pacientes. La deteccion temprana de anticuerpos nos ayudaría mucho para confirmar de manera temprana a estos pacientes.
Agradecimientos
Agradecemos al Departamento de Dermatología del Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán.
Notas
los autores han completado y enviado la forma traducida al español de la declaración de conflictos potenciales de interés del Comité Internacional de Editores de Revistas Médicas, y no fue reportado alguno relacionado con este artículo.
Referencias
- 1.Nombel A, Fabien N, Coutant F. Dermatomyositis With Anti-MDA5 Antibodies: Bioclinical Features, Pathogenesis and Emerging Therapies. Front Immunol. 2021;12:773352. doi: 10.3389/fimmu.2021.773352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bolko L, Gitiaux C, Allenbach Y. Dermatomyosites Nouveaux anticorps, nouvelle classification. Med Sci MS. 2019;35:18–23. doi: 10.1051/medsci/2019178. [DOI] [PubMed] [Google Scholar]
- 3.Wu W, Guo L, Fu Y, Wang K, Zhang D, Xu W, et al. Interstitial Lung Disease in Anti-MDA5 Positive Dermatomyositis. Clin Rev Allergy Immunol. 2021;60(2):293–304. doi: 10.1007/s12016-020-08822-5. [DOI] [PubMed] [Google Scholar]
- 4.Gupta R, Kumar S, Gow P, Hsien-Cheng Chang L, Yen L. Anti-MDA5-associated dermatomyositis. Intern Med J. 2020;50(4):484–487. doi: 10.1111/imj.14789. [DOI] [PubMed] [Google Scholar]
- 5.Sampaio AL, Bressan AL, Vasconcelos BN, Gripp AC. Manifestações cutâneas associadas a doenças sistêmicas – Parte I. An Bras Dermatol Port. 2021;96(6):655–671. doi: 10.1016/j.abd.2021.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DeWane ME, Waldman R, Lu J. Dermatomyositis: Clinical features and pathogenesis. J Am Acad Dermatol. 2020;82(2):267–281. doi: 10.1016/j.jaad.2019.06.1309. [DOI] [PubMed] [Google Scholar]
- 7.Kurtzman DJB, Vleugels RA. Anti-melanoma differentiation-associated gene 5 (MDA5) dermatomyositis: A concise review with an emphasis on distinctive clinical features. J Am Acad Dermatol. 2018;78(4):776–785. doi: 10.1016/j.jaad.2017.12.010. [DOI] [PubMed] [Google Scholar]
- 8.Gerami P, Walling HW, Lewis J, Doughty L, Sontheimer RD. A systematic review of juvenile-onset clinically amyopathic dermatomyositis. Br J Dermatol. 2007;157(4):637–644. doi: 10.1111/j.1365-2133.2007.08055.x. [DOI] [PubMed] [Google Scholar]
- 9.Udkoff J, Cohen PR. Amyopathic Dermatomyositis: A Concise Review of Clinical Manifestations and Associated Malignancies. Am J Clin Dermatol. 2016;17(5):509–518. doi: 10.1007/s40257-016-0199-z. [DOI] [PubMed] [Google Scholar]
- 10.Vastarella M, Gallo L, Cantelli M, Nappa P, Fabbrocini G. An Undetected Case of Tinea Capitis in an Elderly Woman Affected by Dermatomyositis: How Trichoscopy Can Guide to the Right Diagnosis. Skin Appendage Disord. 2019;5(3):186–188. doi: 10.1159/000495805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bottai M, Tjärnlund A, Santoni G, Werth VP, ilkington C, de Visser M, et al. EULAR/ACR classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups: a methodology report. RMD Open. 2017;3(2):e000507. doi: 10.1136/rmdopen-2017-000507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mammen AL, Allenbach Y, Stenzel W, Benveniste O ENMC 239th Workshop Study Group. 239th ENMC International Workshop: Classification of dermatomyositis, Amsterdam, the Netherlands, 14-16 December 2018. Neuromuscul Disord NMD. 2018;30(1):70–92. doi: 10.1016/j.nmd.2019.10.005. [DOI] [PubMed] [Google Scholar]
- 13.Tansley SL, Simou S, Shaddick G, Betteridge ZE, Almeida B, Gunawardena H, et al. Autoantibodies in juvenile-onset myositis: Their diagnostic value and associated clinical phenotype in a large UK cohort. J Autoimmun. 2017;84:55–64. doi: 10.1016/j.jaut.2017.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McHugh NJ, Tansley SL. Autoantibodies in myositis. Nat Rev Rheumatol. 2018;14(5):290–302. doi: 10.1038/nrrheum.2018.56. [DOI] [PubMed] [Google Scholar]
- 15.Cobos GA, Femia A, Vleugels RA. Dermatomyositis: An Update on Diagnosis and Treatment. Am J Clin Dermatol. 2020;21(3):339–353. doi: 10.1007/s40257-020-00502-6. [DOI] [PubMed] [Google Scholar]
- 16.Matsushita T, Mizumaki K, Kano M, Yagi N, Tennichi M, Takeuchi A, et al. Antimelanoma differentiation-associated protein 5 antibody level is a novel tool for monitoring disease activity in rapidly progressive interstitial lung disease with dermatomyositis. Br J Dermatol. 2017;176(2):395–402. doi: 10.1111/bjd.14882. [DOI] [PubMed] [Google Scholar]
- 17.Huang K, Vinik O, Shojania K, Yeung J, Shupak R, Nimmo M, et al. Clinical spectrum and therapeutics in Canadian patients with anti-melanoma differentiation-associated gene 5 (MDA5)-positive dermatomyositis: a case-based review. Rheumatol Int. 1981;39(11):1971–1981. doi: 10.1007/s00296-019-04398-2. [DOI] [PubMed] [Google Scholar]
- 18.Kim Y, Sykes AJ, Tugnet N, Cheng H. Digital ulcerations in anti-MDA5 dermatomyositis: Complete resolution following treatment with cyclophosphamide. Australas J Dermatol. 2020;61(2):e251–e252. doi: 10.1111/ajd.13220. [DOI] [PubMed] [Google Scholar]
- 19.Shoji T, Bando T, Fujinaga T, Chen F, Sasano H, Yukawa N, et al. Living-donor lobar lung transplantation for rapidly progressive interstitial pneumonia associated with clinically amyopathic dermatomyositis: report of a case. Gen Thorac Cardiovasc Surg. 2013;61(1):32–34. doi: 10.1007/s11748-012-0106-3. [DOI] [PubMed] [Google Scholar]
- 20.Kim J, Kim YW, Lee SM, Kim YS, Kim YT, Song YW. Successful lung transplantation in a patient with dermatomyositis and acute form of interstitial pneumonitis. Clin Exp Rheumatol. 2009;27(1):168–169. [PubMed] [Google Scholar]
- 21.Leclair V, Labirua-Iturburu A, Lundberg IE. Successful Lung Transplantation in a Case of Rapidly Progressive Interstitial Lung Disease Associated with Antimelanoma Differentiation-associated Gene 5 Antibodies. J Rheumatol. 2018;45(4):581–583. doi: 10.3899/jrheum.171047. [DOI] [PubMed] [Google Scholar]
- 22.Shirakashi M, Nakashima R, Tsuji H, Tanizawa K, Handa T, Hosono Y, et al. Efficacy of plasma exchange in anti-MDA5-positive dermatomyositis with interstitial lung disease under combined immunosuppressive treatment. Rheumatol Oxf Engl. 2020;59(11):3284–3292. doi: 10.1093/rheumatology/keaa123. [DOI] [PubMed] [Google Scholar]
- 23.Abe Y, Matsushita M, Tada K, Yamaji K, Takasaki Y, Tamura N. Clinical characteristics and change in the antibody titres of patients with anti-MDA5 antibody-positive inflammatory myositis. Rheumatol Oxf Engl. 2017;56(9):1492–1497. doi: 10.1093/rheumatology/kex188. [DOI] [PubMed] [Google Scholar]
- 24.Xu A, Ye Y, Fu Q, Lian X, Chen S, Guo Q, et al. Prognostic values of anti-Ro52 antibodies in anti-MDA5-positive clinically amyopathic dermatomyositis associated with interstitial lung disease. Rheumatol Oxf Engl. 2021;60(7):3343–3351. doi: 10.1093/rheumatology/keaa786. [DOI] [PubMed] [Google Scholar]
- 25.Lian X, Zou J, Guo Q, Chen S, Lu L, Wang R, et al. Mortality Risk Prediction in Amyopathic Dermatomyositis Associated with Interstitial Lung Disease: The FLAIR Model. Chest. Chest. 2020;158(4):1535–1545. doi: 10.1016/j.chest.2020.04.057. [DOI] [PubMed] [Google Scholar]