

Abstract
Current clinical products delivering the osteogenic growth factor bone morphogenetic protein 2 (BMP-2) for bone regeneration have been plagued by safety concerns due to a high incidence of off-target effects resulting from bolus release and supraphysiological doses. Layer-by-layer (LbL) film deposition offers the opportunity to coat bone defect-relevant substrates with thin films containing proteins and other therapeutics; however, control of release kinetics is often hampered by interlayer diffusion of drugs throughout the film during assembly, which causes burst drug release. In this work, we present the design of different laponite clay diffusional barrier layer architectures in self-assembled LbL films to modulate the release kinetics of BMP-2 from the surface of a biodegradable implant. Release kinetics were tuned by incorporating laponite in different film arrangements and with varying deposition techniques to achieve release of BMP-2 over 2 days, 4 days, 14 days, and 30 days. Delivery of a low dose (0.5 micrograms) of BMP-2 over 2 days and 30 days using these LbL film architectures was then compared in an in vivo rat critical size calvarial defect model to determine the effect of BMP-2 release kinetics on bone regeneration. After 6 weeks, sustained release of BMP-2 over 30 days induced 3.7 times higher bone volume and 7.4 times higher bone mineral density as compared with 2-day release of BMP-2, which did not induce more bone growth than the uncoated scaffold control. These findings represent a crucial step in the understanding of how BMP-2 release kinetics influence treatment efficacy and underscore the necessity to optimize protein delivery methods in clinical formulations for bone regeneration. This work could be applied to the delivery of other therapeutic proteins for which careful tuning of the release rate is a key optimization parameter.
Keywords: bone regeneration, controlled release, drug delivery, BMP-2, layer-by-layer
Graphical Abstract
Introduction
Tunability of release kinetics in drug delivery is a key parameter in the overarching design and efficacy of drug-eluting materials. Depending on the drug and application, optimal release kinetics might range from a burst release over a few hours or days to a sustained release spanning weeks or months. Bone morphogenetic protein 2 (BMP-2) has been used widely in the bone regeneration space for treatment of fractures and bone defects, and it has been incorporated into numerous clinical products. While BMP-2 has the potential to be an effective agent in promoting bone growth within defects, its current clinical use has relied upon sub-optimal delivery systems, such as collagen sponges soaked in a BMP-2 solution. These delivery systems release much of their payload within a few days of implantation and deliver milligram-sized quantities of BMP-2, which are orders of magnitude higher than physiological concentrations (1–4). Using these delivery systems, BMP-2 has been linked to adverse events and concerns over patient safety (5–8), many of which can be tied to this bolus release and high-dose systemic exposure to BMP-2 (6, 7, 9–11). This has motivated the development of treatments that can deliver the protein in a more controlled manner that retains activity at the site of interest. Therefore, establishing a modular system that can deliver BMP-2 with a sustained release profile from a defect-relevant scaffold is a key design challenge towards increasing the safety and efficacy of BMP-2 as a bone regeneration treatment.
Recently, others have employed numerous techniques to extend the release kinetics of BMP-2 including core-shell nanoparticles nanofiber scaffolds (12–14), cryoelectrospun mats (15), microsphere-embedded in hydrogels (16), gelatin-based hydrogels (17), and others. While some of these techniques exhibit sustained delivery, the release kinetics are not particularly tunable and are likely a fixed function of the materials themselves. Additionally, some of these methods are rather complex or do not achieve high loading efficiency. An attractive technique to meet this delivery challenge is highly tunable layer-by-layer (LbL)-mediated film deposition. LbL involves the alternating adsorption of charged polyelectrolytes and therapeutics to build a film on the surface of a substrate by employing electrostatic interactions between film components. It is an ideal technique to meet the current challenges of BMP-2 delivery because: 1) films are formed conformally and can be built on a variety of substrates allowing for selection of application-relevant implants; 2) mild aqueous conditions are used in the dipping process, maintaining the bioactivity of the growth factor protein; 3) engineering of film design and architecture can achieve differential release kinetics of the drug (18–20). In previous work, we demonstrated the potential for LbL BMP-2 delivery alone and in combination with a mitogenic factor to enhance bone regeneration and vascularization in bone defects (21). In that study, we introduced BMP-2 in a singular formulation, and did not study the impact of film construction and architecture on release properties. Here, we examine how release kinetics can be manipulated within these films and optimized to achieve desired osteogenic properties.
One aspect to address with LbL film-mediated delivery is interlayer diffusion during film assembly, a well-known phenomenon whereby weakly charged and/or low molecular weight film components, such as proteins or other drugs, can diffuse through previously assembled layers and cause rearrangement during LbL film formation. This leads to disorganized films with uncontrolled release kinetics (22–24). This is a particularly critical consideration for BMP-2 since it is a basic protein with an isoelectric point of approximately 8–9 and molecular weight around 32 kDa (5); thus, due to the relatively small size and low charge density of BMP-2 along with the specific film deposition conditions, interlayer diffusion is likely.
Numerous strategies to overcome interlayer diffusion to better modulate release kinetics in LbL films have been explored in our lab primarily for the controlled release of small molecules, including covalently crosslinked polymer barrier layers using heat (24) or chemical moieties (25), functionalized graphene oxide sheets (26), and laponite clay interlayer barriers (27). Laponite clay is an attractive choice for achieving control of protein release in this application because it does not require extreme processing conditions during film formation that could denature the protein, can be easily purchased as-is from the manufacturer, and does not require additional chemical modification. Additionally, laponite is widely accepted as a non-toxic material with inherent osteoinductive properties (28) and excellent potential for use in biomedical applications (29, 30). These disc or platelet-like nanomaterials create barriers that block interlayer diffusion and control the release of components upon breakdown or delivery. To fully realize the potential presented with such nanomaterials, it is important to understand how laponite may influence interlayer diffusion and drug release kinetics when incorporated into films with different architectures through various arrangements and degrees of incorporation of the nanomaterial into the film.
In this work, we modulate the arrangement of laponite within the layers to introduce four distinct LbL film architectures that each create unique release kinetic profiles. LbL is conducive to testing laponite as a barrier layer since it is highly modular and since it enables the nano-sized platelets to deposit naturally on the implant surface; platelets of laponite’s size might not be useful for nanoparticle-based delivery approaches given that the size of the nanoparticle is too close to the size of the spherical laponite for appropriate incorporation. We demonstrate how the systematic inclusion of laponite clay enables BMP-2 delivery over time periods ranging from 2 days to 30 days, all with the same biocompatible polyelectrolyte components and underlying application-relevant polymeric substrate. The different diffusional barrier architectures introduced here provide an optimization platform for researchers creating drug-eluting implant coatings in applications where drug release kinetics are crucial safety and efficacy considerations, such as in the case of BMP-2 delivery for bone regeneration.
We then applied our fastest and slowest LbL architecture to an in vivo rat critical size calvarial defect to evaluate the effect of BMP-2 release kinetics on bone regeneration using a relatively low BMP-2 dose (0.5 μg). Bone growth was assessed over 6 weeks post-implantation using intravital microcomputed tomography (microCT), followed by histological examination of the bone at the 6-week study endpoint. We found that 0.5 μg of BMP-2 was effective in inducing bone regeneration and full or near-full coverage of the defects, but only when delivered in a sustained manner over 30 days. In contrast, delivery of this same BMP-2 dose in a burst fashion did not enhance bone regeneration as compared to an uncoated scaffold control scaffold. These results outline the importance of controlled release kinetics of BMP-2 in the treatment of critical size bone defects.
Materials
Materials were obtained from the following vendors, with catalog numbers included when available: Laponite XLG (Southern Clay Products, Rockwood Additives). Dextran Sulfate (Fluka BioChemika, Catalog #66786, Mr~100,000). Chitosan, low molecular weight (50,000–190,000 Da, Sigma-Aldrich #448869, CAS #9012-76-4). N,N-Dimethylformamide (DMF) (VWR Chemicals BDH, CAS #68-12-2). Sodium acetate buffer solution, 3M pH 5.2±0.1 (Sigma-Aldrich, CAS #126-96-5). Hexanes (Sigma-Aldrich, CAS #110-54-3). 4,4’-Trimethylenedipiperidine (Sigma-Aldrich, CAS #16898-52-5). Bovine serum albumin (BSA) (Fisher scientific, CAS #9048-46-8). Tetrahydrofuran (THF) (Fisher scientific, CAS #109-99-9). 1,6-Hexanediol diacrylate (Alfa Aesar, CAS #13048-33-4). Spectra/Por (R) 3 Dialysis Membranes, MWCO 3500 (Spectrum Laboratories, Supplier #132724). Resomer® RG 504, Poly(D,L-lactide-co-glycolide), lactide:glycolide 50:50, ester terminated, Mw 38,000–54,000 (PLGA) (Sigma-Aldrich, CAS #26780-50-7). Phosphate buffered saline (Lonza, BioWhittaker, Catalog #17–516F). Deionized water was obtained through a MilliQ Ultrapure Water System at 18.2 Ωcm. Linear Polyethyleneimine, MW 25000 (Polysciences, Inc., CAS #9002-98-6). Poly(4-styrenesulfonic acid) solution, Mw ~75,000, 18 wt. % in H2O (Sigma-Aldrich, CAS #28210-41-5). Alpha-MEM (Gibco, Catalog #A1049001). MC3T3-E1 subclone 4 (ATCC, CRL-2593). 100 mm single side polished silicon wafers (WaferPro, Catalog #C04004).
Recombinant human bone morphogenetic protein 2 (BMP-2) was generously donated by Dr. Howard Seeherman at Bioventus.
Methods
Poly2 synthesis
Poly2 synthesis followed the protocol from Lynn & Langer (31). 6.51 g of 4,4’-trimethylene dipiperidine (0.0309 moles) was added to a 150 mL round bottom flask. 50 mL of anhydrous THF was added to the flask and the contents were heated at 50°C with magnetic stirring until fully dissolved. 6.85 g of 1,6-hexanediol diacrylate (0.0303 moles) was then added dropwise to the stirring solution, and the flask headspace was purged with dry nitrogen gas for 5 minutes and sealed. The reaction was allowed to proceed with stirring at 50°C for 48 hours. The polymer solution was added to excess ice-cold hexanes to precipitate the polymer, and the resultant solid polymer was then air dried. To remove oligomers and any unreacted monomer, the polymer was then re-dissolved into THF and dialyzed in a 3.5 kDa molecular weight cutoff dialysis membrane against inhibitor-free THF to remove remnant monomer before being precipitated again into ice-cold hexanes and air-dried. The polymer was stored in a vacuum desiccator until further use.
Tetrahydrofuran-phase gel permeation chromatography (GPC, Agilent, Styragel column) was used to determine the molecular weight of the polymer. When compared against polystyrene standards, the molecular weight (Mn) was approximately 6.0 kDa with a PDI of 1.47. 1H-NMR of Poly2 in deuterated chloroform was used to confirm synthesis based on peak shifts previously reported (32).
PLGA scaffold fabrication
PLGA membranes were made using phase-inversion synthesis, as described previously (21). Briefly, PLGA was dissolved in dimethylformamide at 25% w/w. The PLGA solution was then spread onto a clean 75×50 mm glass slide (Corning Cat #2947–75×50) using a doctor blade, set to 1 mm thickness relative to the glass slide surface. The slide and solution were gently lowered into a large beaker of deionized water. The water was changed every few minutes in the first hour, then occasionally over the next 48 hours, then removed from the water and allowed to air dry overnight. Membranes were stored in a vacuum desiccator until use. The average thickness of the membranes used was 0.54 mm (standard deviation 0.11 mm) as measured by taking six caliper measurements along the perimeter of each PLGA sheet.
Polyelectrolyte solution preparation
Poly2 and Dextran Sulfate (DxS) dipping solutions were made at 1 mg/mL in pH 5.1 100 mM sodium acetate solution. The dipping solution of BMP-2 was prepared at 10 μg/mL for the No Barrier Film, Capping Barrier, and Interrupting Barriers formulations used in the in vitro release kinetics profiles or pilot in vivo study. For the Integrated Barriers formulation, BMP-2 was dissolved at 100 μg/mL in 100 mM sodium acetate that was pre-titrated to pH 4 using 1 M hydrochloric acid. To adjust the BMP-2 dose in the No Barrier Film for use in the full-scale in vivo study to match that of the Integrated Barriers formulation, the BMP-2 dip solution was prepared at 1 μg/mL in the pre-titrated pH 4 100 mM sodium acetate buffer. The laponite dipping solution was prepared by dispersing 1 mg/mL laponite into deionized water, then titrating to pH 9 with 1 N sodium hydroxide. For the sprayed barrier layers, laponite was prepared similarly. Chitosan was dissolved at 0.2 mg/mL in pH 5.1 10mM Sodium Acetate and titrated to pH 4 with 1 M hydrochloric acid.
BMP-2 LbL film formation
For profilometry and in vitro release kinetics studies, films were deposited on silicon chips. Silicon chips (WaferPro/Item #C04004) were cut to approximately 0.25” by 1.25” (~0.64 cm × 3.18 cm) rectangles. The silicon chips were sonicated for 10 minutes in a solution of 70% ethanol, rinsed vigorously with deionized water, and dried with nitrogen gas. Silicon chips were plasma treated using a Harrick plasma cleaner/sterilizer (PDC-32G) with oxygen plasma at 400 millitorr pressure for 60 seconds, then soaked in Poly2 solution for 15 minutes before starting the dip cycle. The films were formed using a Zeiss DS-50 Slide Stainer, which automated alternately dipping the silicon chips as outlined below.
For in vivo rat studies, films were formed on PLGA membrane scaffolds, cut to approximately 2.5 cm × 2 cm rectangles. The PLGA was plasma treated for 10 seconds with oxygen plasma at 400 mm Hg pressure, then soaked in the Poly2 solution for at least 15 minutes prior to commencing the dipping sequences below.
No Barrier Film, Capping Barrier, Interrupting Barriers BMP-2 films:
The film deposition sequence was as follows: Poly2 solution for 5 minutes, rinse for 30 s in deionized water twice, dextran sulfate for 5 minutes, rinse for 30 s in deionized water twice, BMP-2 for 5 minutes, rinse for 30 s in deionized water twice, dextran sulfate for 5 minutes, and rinse for 30 s in deionized water twice. This film cycle was notated as (Poly2/DxS/BMP-2/DxS)n where n is the number of cycle iterations. For profilometry, in vitro release kinetics studies, and the pilot-scale in vivo study with the No Barrier Film, this cycle was repeated 30 times (n=30). For the full-scale in vivo study with the No Barrier Film, this cycle was repeated 10 times (n=10). For the Capping Film, this cycle was repeated 30 times (n=30), followed by the laponite sprayed barrier layer, outlined below. For the Interrupting Barriers Film, this cycle was repeated 10 times (n=10), followed by one round of the laponite sprayed barrier (below) to make one iteration of the film pattern. This iteration was then repeated three times for a total of 30 BMP-2 film layers.
Integrated Barriers film:
The film deposition sequence was as follows: Poly2 solution for 5 minutes, rinse for 30 s in deionized water twice, laponite solution for 5 minutes, rinse for 30 s in deionized water twice, BMP-2 solution for 5 minutes, rinse for 30 s in deionized water twice, laponite for 5 minutes, and rinse for 30 s in deionized water twice. This film cycle was notated as (Poly2/laponite/BMP-2/laponite)m where m is the number of cycle iterations. This dipping cycle was repeated 30 times (m=30).
A summary of each formulation’s LbL architecture can be found in Table 1. A summary of the LbL formulations used for the full-scale in vivo study with matched dosing can be found in Table S1.
Table 1.
Summary of film architectures for profilometry, in vitro release kinetics studies, and pilot-scale in vivo study.
| Film name | LbL architecture |
|---|---|
| No Barrier Film | (Poly2/DxS/BMP-2/DxS)30D |
| Capping Barrier | (Poly2/DxS/BMP-2/DxS)30D + (Chitosan/laponite)10S |
| Interrupting Barrier | (Poly2/DxS/BMP-2/DxS)10D + (Chitosan/laponite)10S + (Poly2/DxS/BMP-2/DxS)10D + (Chitosan/laponite)10S + (Poly2/DxS/BMP-2/DxS)10D + (Chitosan/laponite)10S |
| Integrated Barrier | (Poly2/laponite/BMP-2/laponite)30D |
Film sections are listed in order of deposition sequence on substrate. Superscripts denote deposition method of each film segment as follows: D: dipped, S: sprayed.
Laponite sprayed barrier layer formulation
The laponite barrier layers were formed using a programmable LbL spray system (Svaya Nanotechnologies), as previously reported (35). The BMP-2 film-coated scaffold was rotated on a harness throughout the process to prevent accumulation of solution on one section of the film. The chitosan solution was sprayed at a rate of 0.25 mL/s for 2 seconds, followed by a 5-minute passive air dry step. Next, the laponite solution was sprayed at a rate of 0.22 mL/s for 2 seconds, followed by a 5-minute passive air dry step, completing one bilayer sequence. This sequence was repeated 10 times for a total of 10 bilayers.
Profilometry
A Veeco Dektak 150 profilometer was used to measure thickness of the LbL films on silicon chips. The dried films were scratched twice length-wise with evenly spaced scratches using a razor blade and thickness was measured at three evenly spaced locations on each film.
In vitro release kinetics
Layer-by-layer films were placed in 1 mL of 1% bovine serum albumin in 1X PBS solution at 37°C in 2 mL low adhesion microcentrifuge tubes. At predetermined time points, the films were gently removed from the solution and placed in fresh solution. The release samples from each time point were analyzed using a Peprotech Human/Murine/Rat BMP-2 Standard ABTS ELISA Development Kit (Cat# 900-K255) per the supplier’s provided protocol. The fractional cumulative release profiles were obtained by summing the mass eluted up to each time point and normalizing to the total cumulative mass released at the end point of the release kinetics study, which we decided based on plateauing of the release curve.
The final cumulative mass of BMP-2 at the final timepoint of the kinetic release study for each formulation was as follows (mean ± standard deviation): No Barrier Film, 1.83 ± 0.75 μg/cm2; Capping Barrier, 0.74 ± 0.10 μg/cm2; Interrupting Barrier, 1.15 ± 0.10 μg/cm2; Integrated Barrier, 0.32 ± 0.04 μg/cm2. These films were then used in vitro and for the pilot-scale study.
Total loading for full-scale in vivo study
In contrast to the final cumulative mass released used to quantify loading for the in vitro and pilot-scale in vivo study, loading on the films used for the full-scale in vivo study were quantified by completely disrupting the film en masse. No Barrier Film and Integrated Barrier films were placed in 1% BSA in 10x PBS solution to destabilize the films using high salt concentration. Films were placed in a 37°C incubator with rotational shaking at 150 RPM for two weeks prior to ELISA quantification. Total mass eluted was normalized to surface area of the respective film sample (1.01 cm2 total for both sides of the scaffold). The measured dose of BMP-2 used in the full-scale in vivo studies for each formulation was as follows (mean ± standard deviation): No Barrier Film, 0.49 ± 0.06 μg/cm2; Integrated Barriers Film, 0.57 ± 0.07 μg/cm2. The total release method can capture BMP-2 that may be retained in the film even after release has plateaued.
SEM Sample Preparation and Imaging
PLGA implants, coated and uncoated, were used as-is for SEM. The PLGA implant sample coated with the Integrated Barriers film or the No Barrier film was dipped into liquid nitrogen and immediately snapped in half using forceps to produce a cross-section of the coated implant. Samples were sputter-coated with gold and then observed with a scanning electron microscope (Zeiss Crossbeam 540) under an operating voltage of 5 kV.
In vivo rat critically-sized calvarial defect
The procedures performed in this study were approved by the Committee on Animal Care at the Massachusetts Institute of Technology (protocol 0718-057-21, PI: Dr. Paula Hammond).
Implant prep:
Implants for each treatment group were prepared as described above with some additional considerations. All polymer and buffer solutions were sterile filtered through 0.2 μm cellulose acetate syringe filters (VWR Cat #28145–477). The deionized water rinse bath solution was autoclaved prior to use or filtered using a 0.2 μm polyethersulfone vacuum filtration unit (VWR Cat#10040–436). The BMP-2, which was provided in sterile aliquots from Bioventus, was directly added to the filtered pH 4 100 mM sodium acetate bath to avoid protein loss in a filter or intermediate transfer container. Any other equipment was either purchased pre-sterilized, or wiped 3x with 70% ethanol. After dipping, an 8 mm-diameter biopsy punch was used to produce 8 mm-diameter implants to use in vivo.
Rat cranial defects were conducted using the protocol outlined by Spicer et al. (36), which is briefly described below.
Pre-op:
Male Sprague Dawley rats, 300–350 g, were ordered from Taconic Biosciences and allowed to acclimate for at least 72 hours prior to surgery. To prepare for surgery, rats were anesthetized using 2–3% isoflurane with flowing oxygen using an anesthesia machine. Once anesthetized, they were dosed with 1 mg/kg sustained release buprenorphine and 2 mg/kg meloxicam subcutaneously. Eye ointment was applied to the eyes. The surgical site was shaved and cleaned aseptically 3 times alternating between betadine solution and 70% ethanol solution using clean cotton balls. The rats were then transferred to a heating pad covered with a sterile drape to sustain body temperature. A toe pinch was used to confirm depth of anesthesia before beginning surgery.
Surgery:
All gloves and materials were ordered pre-sterile or sterilized in sterilization pouches using the dry setting on an autoclave. Sterile surgical gloves were donned, and a drape was cut to fit the surgical site. An approximately 1.5 cm scalp incision was made using a scalpel to expose the calvarium. A scalpel and cotton swab were used to gently free the periosteum from the calvarium. An 8 mm-diameter hollow trephine was used to carefully create an 8 mm-diameter circular defect in the exposed bone with the midline at the level of the suture. The 8 mm-diameter excised bone was removed and an 8 mm-diameter PLGA scaffold of the appropriate treatment group was press-fitted to the defect site. The periosteum and skin were each closed using resorbable 5–0 poliglecaprone 25 (Patterson Veterinary, Catalog #Q303) sutures.
Post-op:
5 mL of warm, sterile saline was injected subcutaneously to replace lost fluids and the surgical site was cleaned. The rat was then placed in a clean cage on a warm heating pad. Once awake, the rat was allowed unrestricted activity and unlimited food and water. DietGel was also provided at the bottom of the cage. Meloxicam at 2 mg/kg was subcutaneously injected 24-, 48-, and 72-hours post-op for pain.
microCT analysis of bone growth:
Rats were scanned using a Bruker Skyscan 1276 microCT system, which allows for in vivo bone imaging of live rats. Rats were scanned at 2-, 4-, and 6-weeks post-op. Rats were anesthetized using 2–3% isoflurane. Eye ointment was applied, and toe pinch was used to confirm anesthetic depth prior to scanning. Animals were scanned at 0.5° rotation angles over 180°, 200 μA current, 565 ms exposure time, 2×2 binning, 100 kV source voltage and 20.1-micron image pixel size. The filter was an aluminum and copper filter. The scan images were reconstructed with NRecon software (Bruker).
Post-scan animals were returned to their cages and allowed unrestricted activity and unlimited food and water. Bone mineral density phantoms were scanned to produce a standard curve used to calculate bone mineral density of samples with the average grayscale value. Qualitative isosurface images were produced using MicroView open-source software (http://microview.parallax-innovations.com/), with the grayscale threshold set to 20,000. Quantitative bone analysis was performed with MicroView’s bone analysis function, using a cylindrical ROI of 8 mm in diameter and 3 mm in depth to ensure the rounded calvarial bone would be within the ROI (Figure S1). Using the bone analysis function, bone volume (BV) and mean grayscale value within the ROI were determined. The mean grayscale value was converted to bone mineral density (BMD) by comparison to bone phantoms scanned using the same microCT protocol. The bone phantoms of known BMD were used to produce a calibration curve in mg hydroxyapatite (HA)/cm3. To obtain the bone volume fraction, bone volume for each defect was normalized to that of an uninjured rat calvarium ROI of 8 mm diameter size.
An important note is that the calculated BMD for some of the measurements was less than zero, which is a nonphysical result. We attribute this to limitations in our calibration methods. The calibration curve for BMD was made using bone phantoms of known BMD value in similar size to the samples of interest. However, these phantoms are not perfect representations of the rat calvarium; thus, the calibration curve based on these measurements is imperfect as well, which could account for the negative BMD calculated values. However, since all the BMD values presented in this work were calculated with the same calibration curve, the relative values may still be compared.
Calvarium excision and storage:
At the 6-week timepoint, rats were euthanized using CO2 asphyxiation, followed by bilateral diaphragm puncture. Calvaria were excised and stored in 10% neutral-buffered formalin for 24 hours for tissue fixation, followed by storage in 70% ethanol until further use. For histology, samples were placed in 10% (v/v) formic acid solution to decalcify the bone. The formic acid solution was changed daily for at least 7 days, until no calcium precipitate was observed when mixed with ammonium oxalate solution (37).
Histological analysis of bone growth:
Decalcified calvaria were bisected using a sharp razor blade, submerged in 70% ethanol, and submitted to the Koch Institute for Integrative Cancer Research, Hope Babette Tang (1983) Histology Facility for paraffin embedding, sectioning, and staining. Sections were 5 μm in thickness and stained with hematoxylin and eosin (H&E) stain or Masson’s trichrome stain. QuPath open-source software (https://qupath.github.io/) was used for examination and quantitative analysis of scanned histology slides. Qualitative examination of stained slides was performed by a pathologist, Dr. Robert Padera, who was blinded to the identity of the samples. For quantitative analysis, new bone tissue within the defect area was encircled using the magic wand and brush tools on QuPath, and the area of bone was quantified. Lacunae filled with osteocytes were not subtracted. However, pockets of space associated with empty areas, Haversian canals, or soft tissue were subtracted from the overall area measurement. To account for imperfect bisection of the defects or folding of tissue in preparation of histology slides, line segments were drawn across the span of the defect area and their lengths added up for a measurement of length of defect in that specific sectioning of the bone. Finally, the area of newly grown bone was divided by the length of the defect to provide a measurement of average bone thickness.
Statistical methods:
5–6 animals were used per surgical group in the full-scale in vivo study. The number of samples for other studies is noted in the text. All statistical data analysis was performed using GraphPad Prism 9. Bone growth data was analyzed using an ordinary two-way ANOVA with Tukey’s multiple comparisons test. Bone thickness measurements on histological slides were analyzed using an ordinary one-way ANOVA with Tukey’s multiple comparisons test. Biocompatibility of the materials was analyzed using an ordinary one-way ANOVA with Dunnett’s post-hoc comparison to the 5 μg/mL group. p values less than 0.05 were considered significant and were reported as follows: *p≤0.05, **p≤0.01, ***p≤0.001, ****p≤0.0001.
Supplemental Methods:
See the supplemental methods for information on the cell viability assay, dynamic light scattering and zetasizer measurements of laponite, NBT/BCIP alkaline phosphatase staining, RT-qPCR gene expression, and the alkaline phosphatase activity assay.
Results & Discussion
Design of LbL Films with Differential Release Kinetics of BMP-2
The layer-by-layer film formation process provides numerous opportunities to tune film properties by selection of deposition parameters and film materials. In this study, we focused on engineering BMP-2 release kinetics by carefully selecting film carrier materials, deposition technique, and film patterning or “architecture.” Structures or schematics of each material described below are shown in Figure 1.
Figure 1.
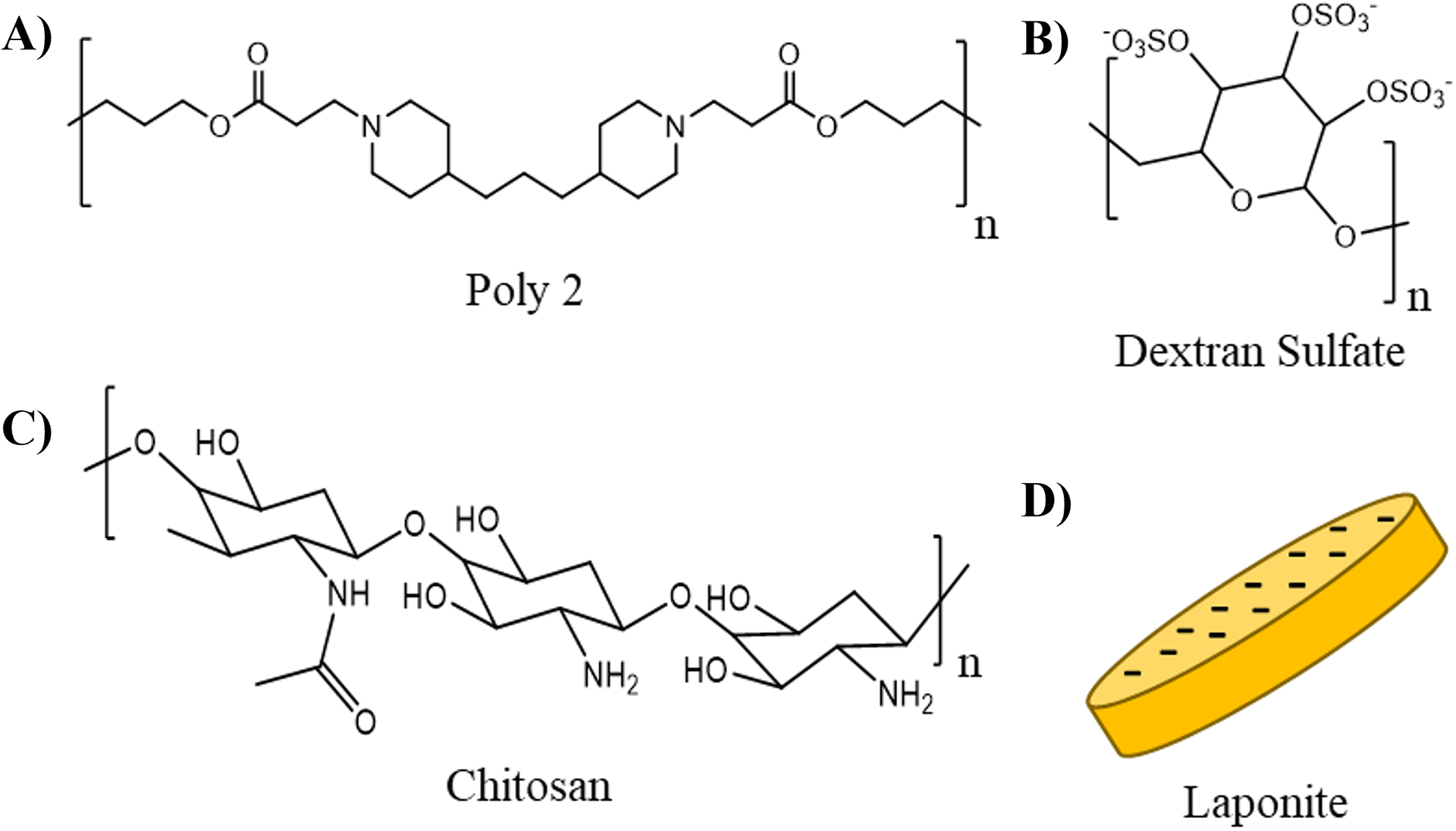
Chemical structures or schematics of layer-by-layer film components. A) Poly2; B) dextran sulfate; C) chitosan; D) laponite.
Cationic poly(β-amino ester) polymer “Poly2”, used in all of the film architectures presented here, was selected for its excellent biocompatibility and extensive use in biologic drug delivery applications (21, 27, 32, 35, 38–46). A key characteristic is that Poly2 is hydrolytically degradable, allowing an LbL film containing Poly2 to be broken down under physiological conditions to release its contents. Dextran sulfate, an anionic polymer, is non-toxic, has previous use in LbL drug delivery systems (35, 46) and has been shown to interact with and enhance the activity of BMPs (47). Chitosan is a naturally-derived, biodegradable polysaccharide and exhibits excellent biocompatibility (48). It has previously been used as a partner polycation to laponite in discrete barrier films (27). Laponite was used as the diffusional barrier molecule due to demonstrated diffusional barrier capabilities (27) and inherent osteoinductive properties (28). Laponite’s diffusional barrier properties are due to the high aspect ratio and surface area of its platelet morphology (49). The flat sides of the clay are negatively charged (49), allowing it to adsorb parallel relative to the surface of underlying positively-charged LbL film layers (50). Since the platelet itself is impermeable to macromolecules, the laponite deposited in recurring layers creates a tortuous path in the film through which film components, such as BMP-2, need to travel to diffuse through the film (27, 49). This slows the diffusion of loaded cargo from both exiting the film and rearranging within the film via interlayer diffusion. In this way, laponite’s intrinsic structure gives rise to its barrier-like function.
Dynamic light scattering measurements showed a number-average size of 47 nm with a polydispersity index of 0.274 (Figure S2A), suggesting a small amount of aggregation since the well-characterized dimensions of a laponite platelet are about 20–25 nm by 1 nm (51, 52). Deposition of aggregates may have also contributed to furthering the tortuosity of BMP-2’s diffusional path through the film. At pH 9, the laponite has a zeta potential of −33 mV (Figure S2B).
Films in this study were deposited using a combination of dip adsorption and spray deposition techniques. Dip adsorption relies on the attraction of molecules in solution to the surface of the substrate or existing LbL film components through electrostatic adsorption (53). When conditions during assembly favor interdiffusion, dip adsorption provides the timeframes that allow such processes to take place, permitting the diffusion and absorption of film components during the adsorption steps (24, 35, 53). In contrast, spray deposition involves alternately spraying the substrate or pre-existing film for very short time periods with solutions containing the film components. This deposition technique is significantly faster than dip LbL with a timescale on the order of seconds (54). Spray LbL kinetically “traps” molecules during assembly, greatly limiting interlayer diffusion processes (35). Multiple review articles can be referenced for further details on the different LbL film assembly techniques and their applications to drug delivery systems (18, 53, 54).
In this work, LbL film segments containing BMP-2 were assembled using the dip LbL technique to conserve expensive growth factor solution. In contrast, spray LbL was used to deposit the discrete laponite barrier layer segments to maximize kinetic trapping of molecules beneath the barrier and to speed the film deposition process. An additional consideration for utilizing spray deposition technique for the discrete laponite barrier layers is that the laponite-containing solution was kept at pH 9 to maintain a strong negative charge for film formation, whereas Poly2, BMP-2, and Dextran Sulfate were all deposited at pH 4–5. Spraying of the discrete laponite barriers avoided significant “stripping” of the underlying layers by prolonged exposure to pH 9 laponite solution. In the Integrated Barriers formulation, the film was assembled using a pH 9 laponite dip bath throughout the entire film formation process while maintaining the BMP-2 and Poly2 baths at pH 4–5. While the film was not “stripped” away with each laponite dip, we did observe that a higher BMP-2 dip bath concentration was required to achieve sufficient BMP-2 loading, likely due to BMP-2 losing some charge when exposed to the pH 9 dip bath and desorbing from the film.
Profilometry (Figure 2) was used to investigate the growth patterns of the films and better understand their mechanism of build-up. Specifically, multiple studies have shown that films undergoing interlayer diffusion during the deposition process exhibit exponential growth of LbL film thickness (22–24). Since film rearrangement during interlayer diffusion can alter the location of different LbL film components along the thickness of the film, and influence the drug release kinetics from the coating, in vitro release kinetics studies were performed to better understand the diffusional barrier capabilities of laponite clay in each film architecture. LbL films can release their drug payload through a variety of different mechanisms including diffusion, swelling, surface erosion, and dissolution (18). Interlayer diffusion of the drug molecule towards the surface of the film is expected to result in a more rapid release rate regardless of the dominating release mechanism because higher concentrations of protein exist at or near the top surface of the film in such cases. Modulating the extent to which interlayer diffusion can occur is a key factor in controlling the release kinetics of the drug from the LbL film, motivating our investigation into incorporating laponite into different LbL film architectures as a potential tool to achieve differential drug release kinetics.
Figure 2.
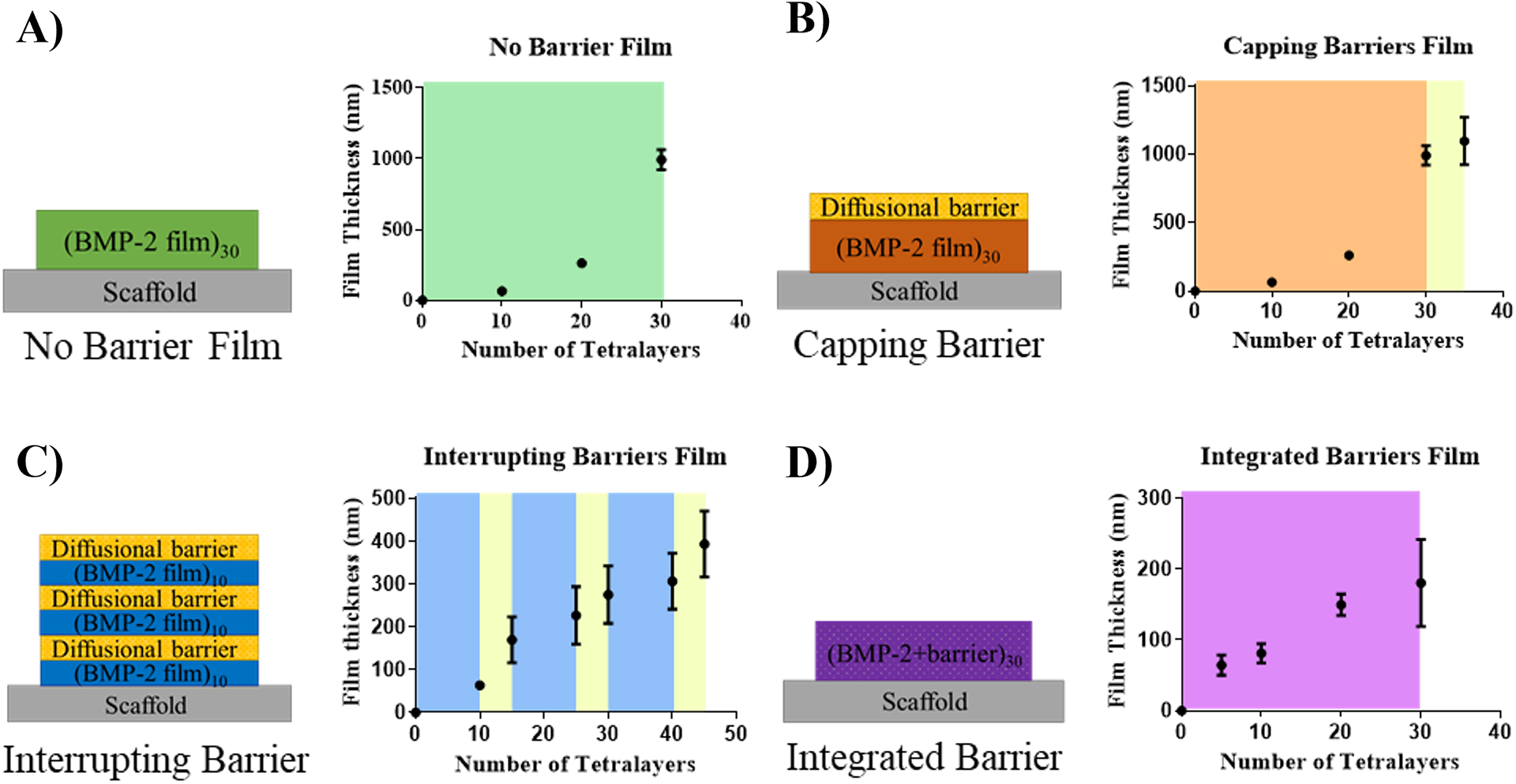
Film architecture schematics and film thickness curves measured with profilometry of formulations: A) No Barrier Film; B) Capping Barrier; C) Interrupting Barrier; D) Integrated Barrier.
The No Barrier film formulation, which did not contain laponite clay material, exhibited exponential growth in thickness with an increasing numbers of layers (Figure 2A) and released its payload in 2 days (Figure 3), suggesting that interlayer diffusion was occurring during the film fabrication process and was a likely contributor to the burst release kinetics. When this film was capped with a sprayed laponite diffusional barrier (Capping Barrier film), film thickness increased slightly with the barrier layer addition as compared to the underlying No Barrier film (Figure 2B) and the release was extended to four days (Figure 3). The release kinetics slowed only slightly since the clay barrier layer “cap” in this formulation merely delayed the release of BMP-2 from the highly inter-diffused film underneath it.
Figure 3.
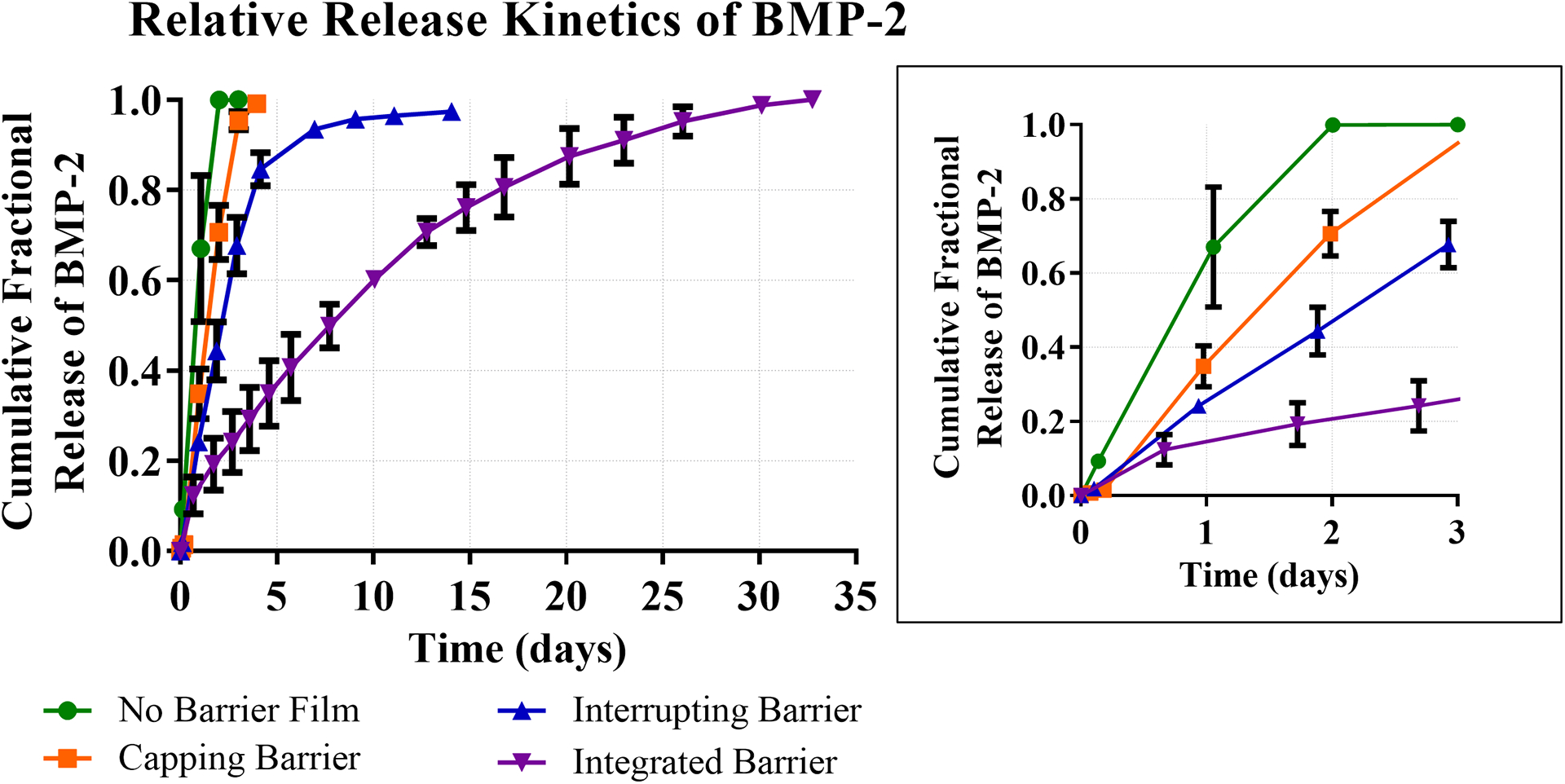
Relative release kinetics of BMP-2 from four different LBL film architectures. Zoomed panel shows expanded release kinetics over first three days.
To further slow the release kinetics, we interrupted the No Barrier film formulation with discrete sprayed barriers, splitting the total number of layers into thirds and incorporating two diffusional barriers between the layers and one on top (Interrupting Barriers). In this architecture, we expected to observe further slowing of the release kinetics due to the interrupting barriers reducing interlayer diffusion between film segments. Indeed, the release kinetics were lengthened to approximately 2 weeks for total payload release, supporting this hypothesis (Figure 3). The profilometry measurements for this formulation suggested non-exponential film thickness growth, which indicated interlayer diffusion occurring to a lesser extent than with the No Barrier film and Capping Barrier architectures (Figure 2C).
To further explore the barrier capabilities of laponite, we replaced anionic dextran sulfate with laponite in the Integrated Barrier films. In this way, we were able to maximize the ratio of diffusional barrier material, laponite, to the other components of the film and extended the total release window to 30 days (Figure 3). Additionally, film thickness build-up was relatively linear, indicating that interlayer diffusion was likely limited during film assembly (Figure 2D).
The sustained release results measured in this study are consistent with the diffusional barrier properties exhibited by laponite in other drug delivery work. Ordikhani et al. showed that electrodeposited films containing chitosan and laponite had a slower release of the antibiotic vancomycin than films with chitosan, alone (55), while Shi et al. demonstrated sustained release of methylene blue using polyacrylic acid-laponite nanocomposites in LbL films (56). These literature precedents both demonstrate that laponite can be used to successfully slow the release of small molecules. In our previous work, we used laponite in discrete barrier layers within LbL film conformations to temporally separate the release of BMP-2 and small molecule antibiotic gentamicin for combination therapy bone regeneration films (27), showing the promise of laponite in the field of LbL films for drug delivery in the bone regeneration space and beyond.
In vitro cell viability assay with film components
Cell viability experiments were conducted by treating MC3T3-E1 murine pre-osteoblast cells with the different film components to confirm the materials were not cytotoxic. Linear polyethyleneimine (LPEI) and sulfonated polystyrene (PSS) were included as reference polymers since they are commonly used for drug delivery applications. LPEI is known to induce cytotoxicity (57).
The cells showed little to no loss in viability when treated with Chitosan, Dextran Sulfate, and PSS. Poly2 and laponite were generally not cytotoxic and only resulted in lower cell viability at the highest concentration tested in this study, 50 μg/mL. In contrast, LPEI was cytotoxic even at the lowest concentration in this study, 5 μg/mL, and was more cytotoxic at increasing concentrations. These results indicate that the polymers and film components used in this study to create LbL films were not significantly cytotoxic to pre-osteoblast cells, especially as compared with widely used LPEI (Figure S3).
Imaging of PLGA implant coated with LbL film
The morphology of the implants themselves may play a role in promoting bone growth, as surface porosity and topography have been shown to play a role in osteogenesis (58, 59). A macrograph of the coated PLGA implants compared to an uncoated implant shows the micro-scale features, but it is hard to discern morphology of the deposited film (Figure S4). As such, SEM imaging was used to visualize the micro- and nano-scale features of the PLGA implants coated with LbL films. Uncoated PLGA has noticeable pores (Figure 4A) that were fully or partially covered after coating with either the Integrated Barrier or No Barrier film (Figure 4B, D). The substrate pores that have been covered by the conformal thin film are still visible as “craters” on the surface. Some pores were not completely covered by the LbL coating. These findings are consistent with previous results in Shah et al. (21) for coating a porous PLGA substrate with a LbL film. Looking at the cross-sectional views of the LbL-coated PLGA implant demonstrated the presence of an intact LbL film on both sides of the substrate, without altering the inner morphology of the PLGA implant (Figure 4C, E). Overall, SEM imaging confirms conformal coating of both sides of the porous PLGA substrate using layer-by-layer film deposition in both the Integrated Barrier and No Barrier film formulations.
Figure 4.

SEM images of coated and uncoated PLGA scaffolds. Images show conformal coating of substrate with layer-by-layer (LbL) film deposition on both sides of implant. A) uncoated PLGA; B) PLGA coated with Integrated Barrier LbL film; C) Cross-section of coated PLGA showing Integrated Barrier LbL film (indicated with arrows); D) PLGA coated with No Barrier LbL film; E) Cross-section of coated PLGA showing No Barrier LbL film (indicated with arrows).
In vivo pilot bioactivity study with four different BMP-2 release kinetic formulations
To assess the bioactivity of BMP-2 release from LbL films, we tested four film formulations coated on a biodegradable PLGA implant in a pilot-scale rat calvarial critical size defect model (n=2–3 per group). The dextran sulfate-based films (No Barrier, Capping Barrier, and Interrupting Barrier films) contained approximately 1.3 μg of BMP-2 and the Integrated Barriers film formulation contained approximately 0.6 μg of BMP-2. We included an uncoated PLGA scaffold negative control group (n=3) and a healthy 4-week rat control (n=1) as a positive control.
In analyzing the microCT data qualitatively, we observed that the uncoated PLGA control group did not exhibit a large amount of bone growth, nor total bridging of the defect gap, at 2 or 4 weeks. In contrast, the BMP-2-containing LbL film groups appeared to grow more bone than the PLGA control at 2 and 4 weeks, with some animals exhibiting near full-closure of the defect area (Figure S5). Additionally, the uniformity of bone coverage and defect closure appeared improved at 2- and 4-weeks for the slower releasing Integrated and Interrupted Barrier formulations, particularly in comparison to the No Barrier and Capping Barrier formulations.
Quantification of the microCT data confirmed these observations, with the barrier layer BMP-2-eluting formulations outperforming the uncoated PLGA control in bone volume at 4 weeks and trending towards increased bone volume in the No Barrier formulations compared to the uncoated PLGA control. This suggests that the BMP-2 retained its osteogenic properties after the LbL film deposition process and its subsequent release (Figure S6). Additionally, though this pilot-scale study is underpowered, we observed a trend that treatment with the slow-releasing Interrupting Barriers and Integrated Barriers formulations resulted in higher bone volume and bone mineral density after 4 weeks of healing as compared to the fast-releasing No Barrier Film formulations at the same timepoint.
Overall, these results suggest that released BMP-2 retained bioactivity and that sustained BMP-2 release may enable better bone growth than more rapid release. The results from this pilot study motivated further examination of the effects of BMP-2 release kinetics on bone regeneration utilizing modularly assembled LbL films.
In vivo low dose full-scale study of bone growth with sustained versus rapid release of BMP-2
We decided to focus a separate full-scale study with appropriate statistical power on comparing the efficacy of rapid release of a pre-determined low BMP-2 dose over 2 days (No Barrier Film) with sustained release of the same dose over 30 days (Integrated Barriers Film), given that these were the two extremes of release kinetics in our study. No Barrier and Integrated Barrier formulations containing 0.5 μg of BMP-2 were compared to uncoated PLGA scaffolds. As in the pilot-scale study, a rat calvarial critical-sized defect model was used to evaluate the efficacy of the BMP-2 scaffold formulations. To ensure a properly powered study, additional data from rats in the pilot study were included for the uncoated PLGA and Integrated Barrier layer groups. Rats were also treated with the No Barrier scaffolds containing a matching dose of BMP-2 to the Integrated Barrier formulation.
MicroCT imaging analysis was used to examine bone growth qualitatively and quantitatively over 6 weeks (Figures 5 and 6) via measurement of bone volume (BV) and calculation of bone mineral density (BMD). Qualitative examination of the generated 3D bone isosurface in MicroView showed that the uncoated PLGA control did exhibit some bone growth across the defect over 2, 4, and 6 weeks, but the bone did not fully bridge the defect. Biocompatible scaffolds, even without the supplemental delivery of osteogenic factors, provide a mechanical structure over which progenitor cells can travel and deposit bone, which could explain this observation (60). The fast releasing No Barrier architecture also exhibited some bone growth into the defect over the 6 weeks, but qualitatively did not appear to be different than the uncoated PLGA control. In contrast, sustained BMP-2 release from the Integrated Barrier formulation exhibited quite substantial bone growth even at 2 weeks. Over 4–6 weeks, many of these samples showed nearly full coverage of the 8 mm defect and qualitatively more bone than both the uncoated control and the fast release No Barrier formulation (Figure 5).
Figure 5.
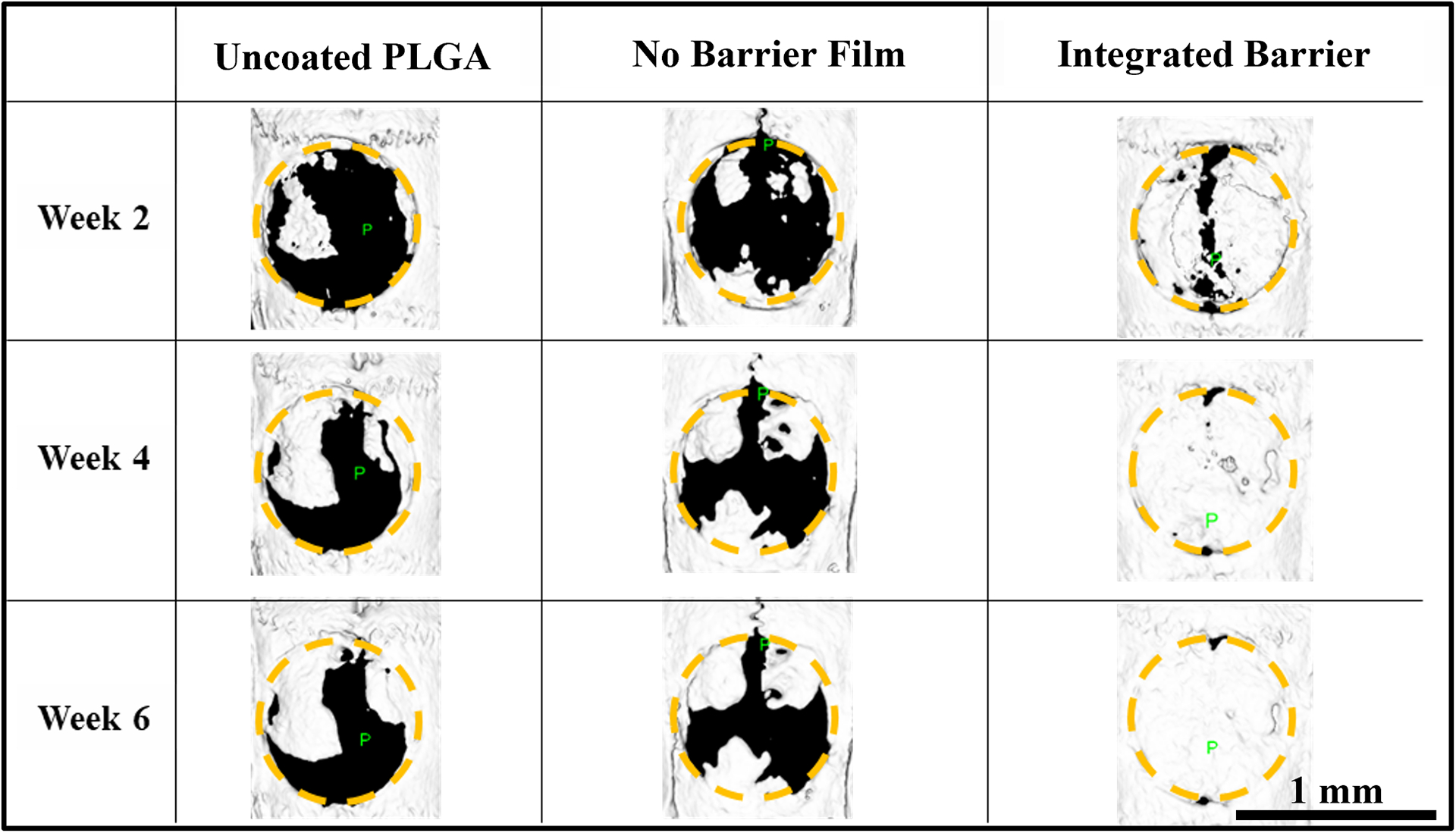
Representative images of rat calvarial defects. microCT isosurfaces of bone growth in each treatment group, 8 mm diameter ROI indicated with yellow circle.
Figure 6.
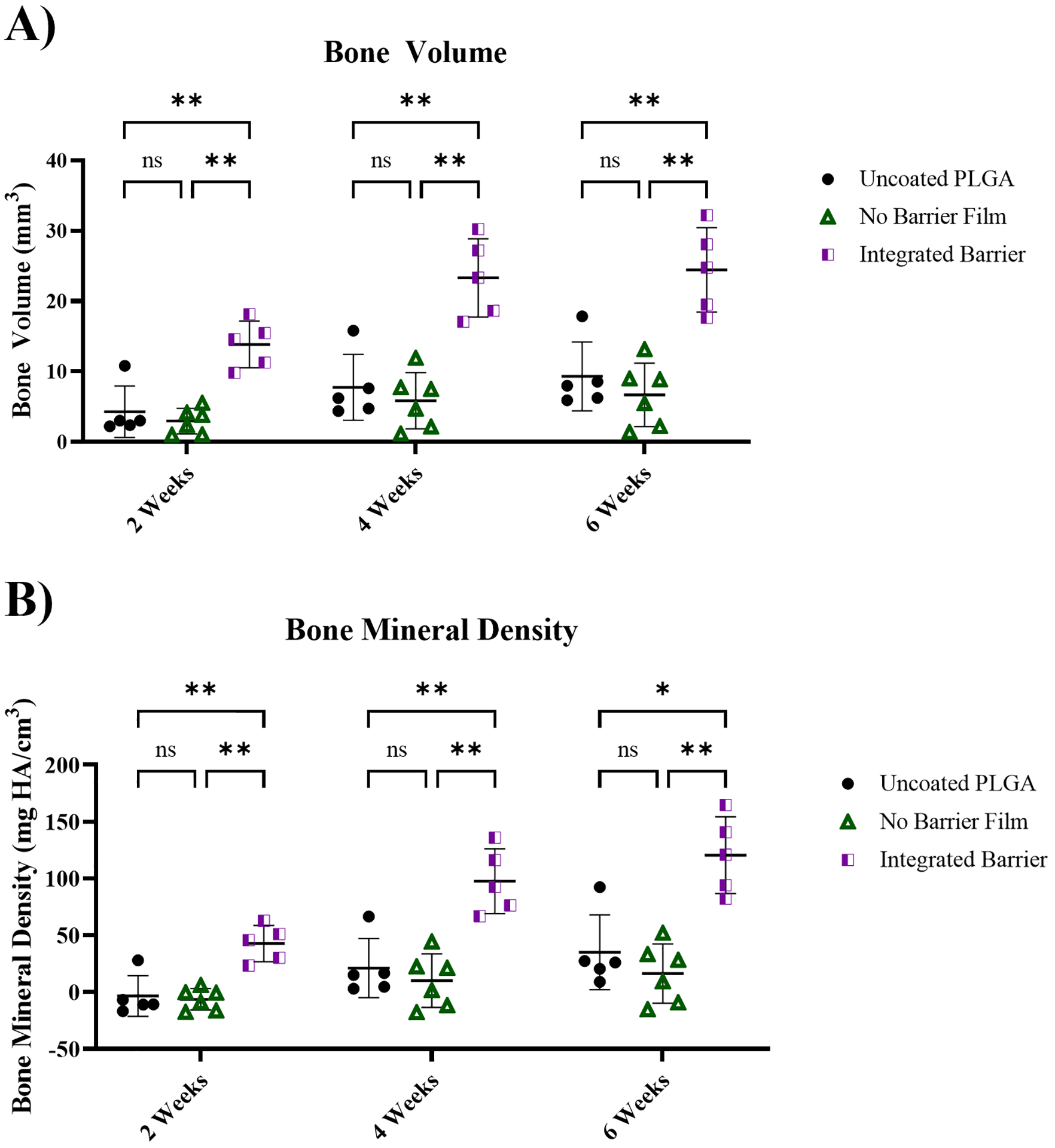
Quantitative bone growth measured with microCT imaging. A) Quantitative measurement of bone volume of each treatment group at 2, 4, and 6 weeks; B) Quantitative measurement of bone mineral density of each treatment group at 2, 4, and 6 weeks. Statistical analysis conducted in GraphPad Prism 9 using 2-way ordinary ANOVA with Tukey’s multiple comparison’s test. *p≤0.05, **p≤0.01. n=5–6.
Quantitatively, at 2-, 4-, and 6-weeks, treatment with the Integrated Barrier formulation resulted in significantly higher BV (Figure 6A) and BMD (Figure 6B) while the No Barrier Film and uncoated PLGA control did not differ significantly in BV and BMD. In comparing the mean bone volume at each timepoint, the Integrated Barriers formulation was 3.7x and 2.6x higher at 6 weeks compared to the uncoated PLGA control and No Barrier Film formulation, respectively. For bone mineral density, the Integrated Barrier formulation was 7.4x and 3.4x higher at 6 weeks compared to the uncoated formulation and No Barrier formulation, respectively, indicating a clear difference in bone mass and density. Normalizing the BV to the tissue volume to obtain a bone volume fraction, similar trends in bone growth are observed. The bone volume fraction values within the calvarial defects following treatment with the Integrated Barrier formulation were 0.45, 0.75, and 0.8 at 2, 4, and 6 weeks, respectively (Figure S7).
The microCT analysis provides evidence that sustained release of BMP-2 from Integrated Barrier films resulted in the greatest bone growth over 6 weeks, with higher bone regeneration even at early time points. These results are also a clear indication that a 0.5 μg dose of BMP-2 can be sufficient in a defect of this size to promote significant bone growth, but only when the release kinetics are sustained. In contrast, the fast release formulation delivering a similar dose of BMP-2 did not elicit significant bone growth as compared with an uncoated implant control. The sustained release of BMP-2 is hypothesized to lower the risk of toxicity and off-target side effects by keeping BMP-2 concentrations below unsafe levels, as well as to improve the efficacy of the treatment by maintaining BMP-2 presence in the defect area for longer periods of time, allowing for sustained osteogenic activity. Establishing flexibility and control over local BMP-2 delivery kinetics using LbL coating design is an attractive method for minimizing dose of BMP-2 required while maximizing efficacy for bone regeneration applications.
Histological examination of bone growth
As shown in the representative images of histological slides at 6 weeks in Figure 7, no samples in any of the treatment groups showed signs of substantial inflammation or foreign-body response, indicating that the PLGA implant and the coatings were non-toxic and non-immunogenic. Examination of the uncoated PLGA samples showed evidence of incomplete bone coverage with fibrous tissue spanning the defect and bone islands of varying length and thickness. Bone islands were generally mature lamellar bone with few osteoblasts present at the surface, indicating that further substantial bone healing would be unlikely (Figure 7A, C).
Figure 7.
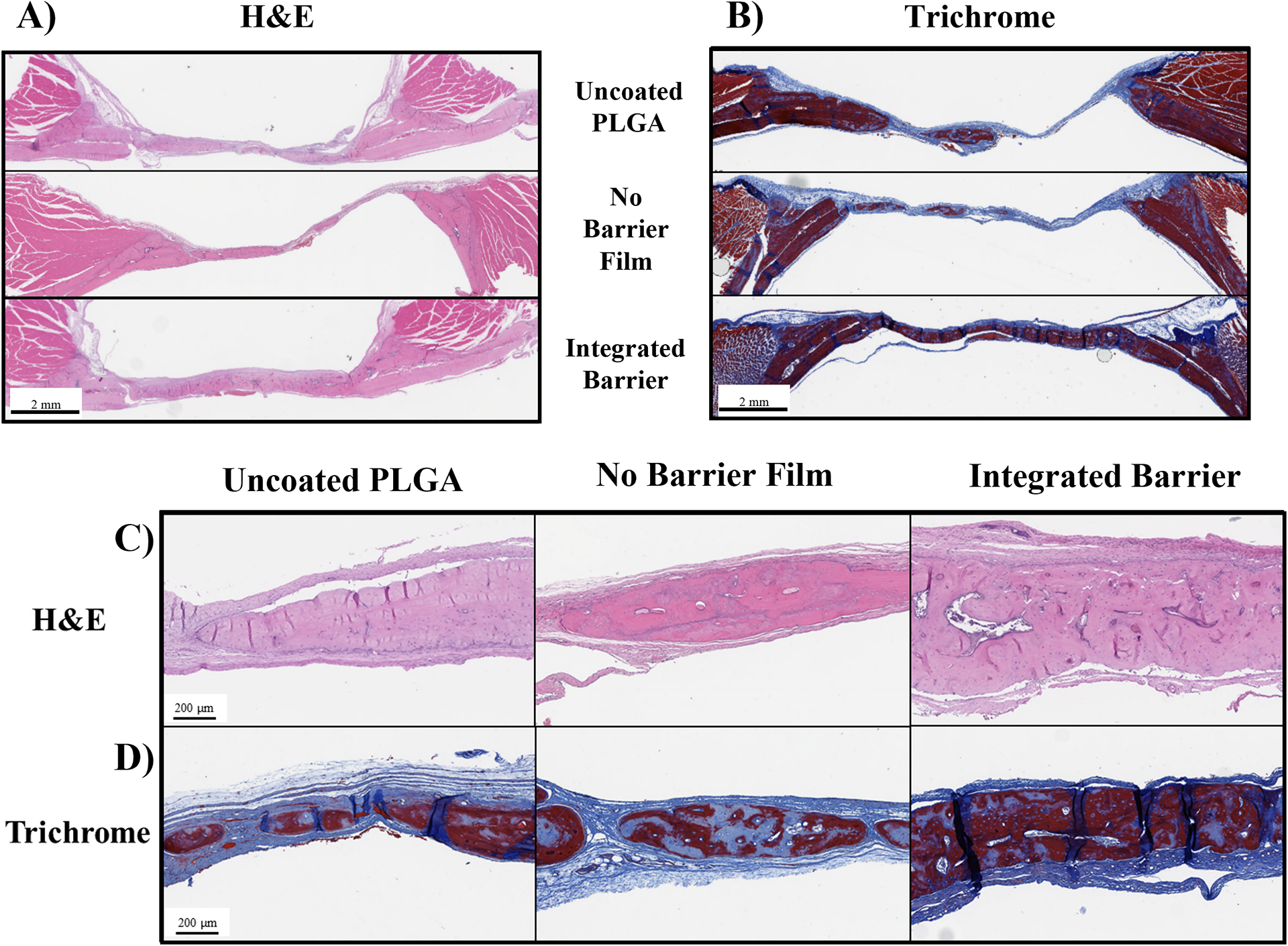
Histological examination of calvarial explants. A&C) Representative images of bone defects stained with H&E, scalebar at 2 mm in A), scalebar at 200 μm in C); B&D) Representative images of slides stained with trichrome stain, scalebar at 2 mm in B, scalebar at 200 μm in D.
The fast-releasing No Barrier film group similarly did not have any defects fully bridged by bone tissue and were comprised of fibrous tissue spanning the defect with interspersed bone islands. The bone islands were a mix of mature and immature bone and had substantial osteoblasts present on the edges of the bone tissue (Figure 7A, C).
The slow-releasing Integrated Barrier group had full bridging of the defect with bone in two of the five samples. The bone had fibrous tissue over the entire top surface, indicative of normal healing and soft tissue presence. In two of the samples, the bone was mostly mature lamellar bone, and in three of the samples the tissue was a mix of mature and woven bone with osteoblast presence on the outer edges of the bone, indicating that further bone deposition and remodeling could occur with longer healing times. The bone in this group had substantial vascularization and Haversian systems present, and multiple samples showed evidence of marrow and fat, hallmarks of high quality, viable bone (Figure 7A, C).
Masson’s Trichrome staining allowed for visual differentiation of lamellar versus woven bone, where mature bone appeared dark red while immature, woven bone or fibrous soft tissue appeared light blue. Examination of the Masson’s Trichrome-stained samples confirmed the observations made with H&E staining regarding bone tissue maturity. All treatment groups showed a combination of mature lamellar bone and immature woven bone (Figure 7B, D).
Using QuPath software, average bone thickness at 6 weeks was also quantified. While the microCT analysis provided a measurement of the bulk bone volume and mineral density, this average cross-sectional thickness provides an additional perspective on not only how much of the defect surface area was regenerated, but also how much of that bone spanned the thickness of the defect. In agreement with results from the microCT analysis, we found that the average thickness of bone in rats treated with the Integrated Barrier formulation was significantly higher than the thickness of bone in rats treated with the No Barrier Film formulation and the uncoated PLGA formulation. Additionally, the No Barrier Film and uncoated PLGA groups were not significantly different from each other, in agreement with the microCT results (Figure S8).
In vitro assessment of No Barrier and Integrated Barrier films
BMP-2 is a well-known mediator of cellular osteogenesis both in vitro and in vivo (1, 2, 61). We were interested if the differences in healing with the No Barrier and Integrated Barrier films were due in part to altered levels of osteoprogenitor cell differentiation. To assess this, along with the in vitro temporal bioactivity of BMP-2 on progenitor cells, we treated MC3T3-E1 pre-osteoblast cells with film releasate collected from discrete time points and measured alkaline phosphatase activity (Figure S9). We also incubated MC3T3-E1 cells with the two coated formulations or an uncoated implant for seven days and then stained the alkaline phosphatase to get a qualitative assessment of osteogenesis (Figure S10). Both cellular staining and quantitative measurements of alkaline phosphatase activity indicate that Integrated Barrier films promote cellular osteogenesis, while the No Barrier formulations promote relatively limited bioactivity over time. Finally, we confirmed these results by looking at the gene expression of 4 genes known to be upregulated in a pro-osteogenic environment: Runx2, Sp7 (osterix), Alp (alkaline phosphatase), and Bglap (bone gamma-carboxyglutamate protein, osteocalcin) after BMP-2 loaded implant release into MC3T3-E1 cells (Figure S11). While all four markers of osteogenesis are upregulated at day 7 when BMP-2 is released from the Integrated Barrier implants, they are not upregulated within error when released from No Barrier implants.
These results align with the known phenomenon whereby bolus BMP-2 acts as a chemoattractant to osteoprogenitor cells, whereas sustained release promotes their differentiation. The performed assays primarily measure differentiation over chemoattraction. Thus, these results corroborate our in vivo findings that low dose BMP-2 delivered from implants requires sustained release for bioactivity and suggest that this is in part due to differences in the differentiation of osteoblast progenitor cells.
Discussion of BMP-2 dose, release kinetics, and efficacy
In comparing the 0.5 μg dose used here to other work with the same rat 8 mm diameter calvarial defect delivering BMP-2 from cell-free scaffolds, the amount of BMP-2 used in this study is similar to or lower than other therapies that induce significant bone regeneration. Studies have investigated BMP-2 delivery in this animal model ranging from 0.2 μg (21) up to 40 μg (62), with many showing significant bone growth and union of the defects. Of particular relevance to this study, Young et al. studied dose dependence of BMP-2 delivery from poly(propylene fumarate) scaffolds and found that 0.5 μg, 1 μg, and 2 μg of BMP-2 induced full union of 0/8, 1/8, and 3/8 defects at 12 weeks, respectively, indicating a clear dose dependence and that in this case, 0.5 μg was not sufficient to induce full union of the defect (63). In our work, we found that 0.5 μg of BMP-2 could induce full coverage in 2/5 calvarial defects, as described in the histological analysis section. The reason for this difference in healing response could be attributed to varying BMP-2 release kinetics between the two studies or other experimental differences, such as scaffold material. Previous work from our lab has shown that delivery of 0.2 and 2 μg of BMP-2 from similar LbL coated scaffolds over 30 days from a PLGA scaffold in the same rat injury model resulted in substantial bone growth after 4 weeks, as compared with the implant control (21). This agrees with our findings that a 0.5 μg dose (within the 0.2–2 μg range) can induce significant bone growth when delivered with a sustained release profile.
In comparison with studies using absorbable collagen sponges, which are the clinically used material for BMP-2 delivery, Lee et al. studied dose dependence by varying BMP-2 loading in an absorbable collagen sponge from 2.5 to 20 μg in an 8 mm diameter rat calvarial defect with 2 and 8-week study endpoints. They found that at these doses, all of the BMP-2 delivery treatment groups induced significantly higher bone growth than the implant control based on histological analysis with 80–100% closure of the defect for all of the BMP-2-treated groups as compared to 10–20% in the collagen sponge control group (64). Of note, even the smallest dose used in this collagen sponge was approximately 5 times higher than the dose presented here. Similarly, Pelaez et al. studied BMP-2 loading in an absorbable collagen sponge carrier with doses ranging from 1.25 to 20 μg in the 8 mm diameter rat calvarial defect and found that BMP-2 delivery increased rate of bone growth into the defect based on radiographic analysis, with significantly more bone observed at 2 and 4 weeks with BMP-2 delivery compared to carrier control. Interestingly, at the 8-week timepoint, the percentage of bone fill in the collagen sponge carrier control was not significantly different compared to the BMP-2 delivery groups (65). While BMP-2 release kinetics are not reported in this paper, it is important to recall that collagen as a BMP-2 carrier is well-known for its burst release properties, and the collagen sponge system used in their work likely also exhibited rapid release kinetics of BMP-2. Thus, their finding that implants exhibiting burst release of BMP-2 did not outperform the carrier control, even with doses ranging from 2–33 times higher than the dose used in our study, is consistent with our observations.
While it is challenging to directly compare the efficacy from our study with that of other studies investigating sustained release systems due to differences in the total loading of BMP-2 and intrinsic material properties, we note that our Integrated Barrier release formulation provide nearly complete bone healing faster than is observed in other studies employing bone regeneration biomaterials solutions in a rat calvarial defect model. For example, He et al. delivered BMP-2 and mesenchymal stem cells to bone defects using freeze-dried chitosan/alginate/hydroxyapatite scaffolds. While we achieved 80% healing of the defects with the Integrated Barriers at 6 weeks, they required 12 weeks to achieve 80% healing (66). Similarly, Li et al. delivered BMP-2 and the steroid dexamethasone using nanoparticles embedded in an electrospun mat. While they achieved sustained release over similar time scales as our system, defects were only about 30% closed at 4 weeks in comparison to closure of about 75% with the Integrated Barrier formulation (67). Finally, Lee et al. explored the sequential delivery of BMP-2 and alendronate, another pro-osteogenic therapy, from PLGA microspheres loaded onto a collagen-hydroxyapatite scaffold. They demonstrated only about 50% defect coverage, even with the addition of alendronate, at 4 weeks (68). We suspect that highly tuned release along with a biocompatible scaffold makes our approach so effective, as the results we report here support previous results in our lab with sustained release of BMP-2 from scaffolds (21).
Addressing the safety aspect of BMP-2 delivery, current clinical products utilizing a soaked absorbable collagen sponge exhibit a burst release of supraphysiological amounts of BMP-2 which have been linked to adverse events (1). Considering these serious safety concerns, our findings more generally suggest that release kinetics are a key parameter in BMP-2-eluting implant design that must be considered in future product design. Furthermore, this release kinetic control can be achieved in a localized fashion from the surfaces of degradable implants using LbL approaches for delivery with our approach.
In interpreting the differences in treatment efficacy between the fast and slow releasing formulations presented here, the sustained release architecture developed in this study provided a continuous supply of BMP-2 to the local defect area for weeks, allowing ample time for progenitor cells to migrate to the injury site and differentiate to produce new bone. In contrast, the fast-release formulation dosed the area with the same total amount of BMP-2 over only a few days, which may not provide progenitor cells the opportunity to reach the area before the growth factor is cleared from the body. Though the effect of release kinetics is already apparent in this work using the small rodent model, larger animals including humans often exhibit slower growth and metabolism, potentially making sustained release even more crucial (2).
While these findings indicate that growth factor release kinetics are very important for bone healing applications in a healthy animal model, they could be even more critical to consider in the treatment of patients with impaired healing, such as those with diabetes (69) or those with polytrauma (70). While this work did not explore application of these LbL-coated biodegradable PLGA scaffolds to an animal model with impaired healing, this could be an interesting avenue for future exploration and would certainly be a more challenging injury model in which to investigate our findings. The modularity afforded by the LbL technique would allow for the tuning of the dose and release kinetics to fit an impaired healing defect, as well as selection of the underlying scaffold to optimize bone regeneration in an impaired healing scenario to inform improvements on the current standard of care for these patients.
Conclusion
In this work, we demonstrated the facility of LbL self-assembly to produce nanoscale thickness films incorporating osteogenic growth factor BMP-2, a biologic drug relevant for bone regeneration applications. LbL films formed on a defect-relevant PLGA scaffold could be engineered to deliver BMP-2 over four distinct time windows ranging from 2 days to 30 days by incorporating a laponite clay barrier material in different architectural conformations. In a rat calvarial critical size defect model, we found that 0.5 μg of BMP-2 could induce effective bone regeneration, but only when using a sustained release formulation. In contrast, rapid release of a matched dose of BMP-2 did not provide significant differences in defect healing compared to an uncoated PLGA implant. The sustained release BMP-2 formulations also demonstrated growth of well-formed mature bone with Haversian canals and significantly higher average thickness spanning the defect based on histological examination.
This work has implications for the design of clinical therapeutic products aiming to deliver BMP-2 for bone regeneration applications. LbL formulations with barrier layers can be used to tune release kinetics, and these principles can be applied to the delivery of other therapeutics. Specifically, this work indicates that release kinetics are a crucial factor in the successful delivery of BMP-2, and that the total administered dose of BMP-2 can be reduced by slowing the release rate of the growth factor. Reducing the dose of BMP-2 is attractive in that manufacturers can significantly cut the cost associated with these treatments by reducing the amount of expensive growth factors used while also reducing the potential of harmful side effects. Additionally, the sustained release of BMP-2 may be an even more significant factor to consider for patients with underlying conditions causing impaired bone healing and could provide possibilities for specialty products in this treatment space.
Supplementary Material
Highlights.
Clinical osteogenic protein formulations exhibit burst release of excessive doses
Assembly of protein-containing films via layer-by-layer deposition enables controlled release
Incorporation of diffusional barrier layers enables release over 2, 4, 14, or 30 days
An extended-release formulation enhances bone healing over a rapid release formulation
Acknowledgements
We thank the Koch Institute Swanson Biotechnology Center for technical support, specifically the Animal Imaging and Preclinical Testing Center, especially Dr. Virginia Spanoudaki and Milton Cornwall-Brady, and the Hope Babette Tang (1983) Histology Facility, especially Kathleen Cormier, for their help in sample processing, data acquisition, and data analysis. This work was supported in part by the Koch Institute Support (core) Grant P30-CA14051 from the National Cancer Institute. The authors would like to thank the Swager Lab and the MIT Department of Chemistry Instrumentation Facility for use of their equipment. The authors would also like to thank Dr. Jennifer Haupt for surgical training in animal studies. We are very grateful to Dr. Apoorv Shanker at the Koch Institute for Integrative Cancer Research at MIT for the helpful scientific discussions and comments on this manuscript. Special thanks go to Dr. Howard Seeherman and Bioventus LLC for the generous donation of rhBMP-2 to make this work possible. The graphical abstract was generated with Biorender.
Funding Sources
This work was funded by the National Institutes of Health (NIH) grants R01DE024747 and the Army Research Office grant W911NF-18-2-0048. J.R.M. acknowledges support from NIH grant F32DE027877. A.G.B. acknowledges support from NIH grant F30DK130564. R.F.P. acknowledges work as a consultant to Medtronic, PLC on topics unrelated to the work described herein. P.T.H. acknowledges support from the David H. Koch (1962) Chair Professorship in Engineering. P.T.H. discloses membership on the scientific advisory boards of Moderna Therapeutics and Performance Indicator, a board member of Alector, and as a cofounder and Board member of LayerBio, Inc.
Abbreviations
- BMP-2
Bone morphogenetic protein-2
- LbL
Layer-by-layer
- PLGA
Poly(lactic-co-glycolic acid)
- DMF
Dimethylformamide
- THF
Tetrahydrofuran
- BSA
Bovine serum albumin
- GPC
Gel Permeation Chromatography
- microCT
Micro-computed tomography
- BMD
Bone mineral density
- HA
Hydroxyapatite
- PDI
Polydispersity Index
- SEM
Scanning Electron Microscopy
- BV
Bone volume
- LPEI
Linear polyethyleneimine
- PSS
Sulfonated polystyrene
- RT-qPCR
Reverse transcription quantitative real-time polymerase chain reaction
- ALP
Alkalione phosphatase
Contributor Information
MayLin T. Howard, Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA 02139; Department of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139.
Sheryl Wang, Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA 02139; Department of Biological Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139.
Adam G. Berger, Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA 02139 Division of Health Sciences and Technology, Massachusetts Institute of Technology, Cambridge, MA 02139.
John R. Martin, Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA 02139; Department of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139.
Sasan Jalili-Firoozinezhad, Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA 02139; Department of Biological Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139.
Robert F. Padera, The Harvard-MIT Division of Health Sciences and Technology, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, United States Department of Pathology, Brigham and Women’s Hospital and Harvard Medical School, Boston, Massachusetts 02215, United States.
Paula T. Hammond, Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA 02139 Department of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139.
Data Availability
Raw/processed data used to generate the figures in this manuscript are available upon request from the authors.
References
- 1.Lo KWH, Ulery BD, Ashe KM, Laurencin CT, Studies of bone morphogenetic protein-based surgical repair. Advanced Drug Delivery Reviews 64, 1277–1291 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li RH, Wozney JM, Delivering on the promise of bone morphogenetic proteins. Trends Biotechnol 19, 255–265 (2001). [DOI] [PubMed] [Google Scholar]
- 3.Mckay WF, Peckham SM, Badura JM, A comprehensive clinical review of recombinant human bone morphogenetic protein-2 (INFUSE (R) Bone Graft). International Orthopaedics 31, 729–734 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sleiman Razzouk D, Sarkis R, BMP-2: biological challenges to its clinical use. New York State Dental Journal 78, 37 (2012). [PubMed] [Google Scholar]
- 5.El Bialy I, Jiskoot W, Reza Nejadnik M, Formulation, Delivery and Stability of Bone Morphogenetic Proteins for Effective Bone Regeneration. Pharmaceutical research 34, 1152–1170 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shields LB et al. , Adverse effects associated with high-dose recombinant human bone morphogenetic protein-2 use in anterior cervical spine fusion. Spine 31, 542–547 (2006). [DOI] [PubMed] [Google Scholar]
- 7.James AW et al. , A Review of the Clinical Side Effects of Bone Morphogenetic Protein-2. Tissue Eng Part B Rev 22, 284–297 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Epstein NE, Complications due to the use of BMP/INFUSE in spine surgery: The evidence continues to mount. Surg Neurol Int 4, S343–S352 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tollemar V et al. , Review article: Stem cells, growth factors and scaffolds in craniofacial regenerative medicine. Genes & Diseases 3, 56–71 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kolambkar YM et al. , Spatiotemporal delivery of bone morphogenetic protein enhances functional repair of segmental bone defects. Bone 49, 485–492 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Epstein NE, Pros, cons, and costs of INFUSE in spinal surgery. Surg Neurol Int 2, 10–10 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kareem MM, Hodgkinson T, Sanchez MS, Dalby MJ, Tanner KE, Hybrid core-shell scaffolds for bone tissue engineering. Biomed Mater 14, 025008 (2019). [DOI] [PubMed] [Google Scholar]
- 13.Cheng G et al. , Controlled Co-delivery of Growth Factors through Layer-by-Layer Assembly of Core-Shell Nanofibers for Improving Bone Regeneration. ACS Nano 13, 6372–6382 (2019). [DOI] [PubMed] [Google Scholar]
- 14.Su Y et al. , Controlled release of bone morphogenetic protein 2 and dexamethasone loaded in core-shell PLLACL-collagen fibers for use in bone tissue engineering. Acta Biomater 8, 763–771 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Lee SS, Santschi M, Ferguson SJ, A Biomimetic Macroporous Hybrid Scaffold with Sustained Drug Delivery for Enhanced Bone Regeneration. Biomacromolecules 22, 2460–2471 (2021). [DOI] [PubMed] [Google Scholar]
- 16.Datta S et al. , Microsphere embedded hydrogel construct - binary delivery of alendronate and BMP-2 for superior bone regeneration. J Mater Chem B 9, 6856–6869 (2021). [DOI] [PubMed] [Google Scholar]
- 17.Chen X et al. , Enhanced bone regeneration via spatiotemporal and controlled delivery of a genetically engineered BMP-2 in a composite Hydrogel. Biomaterials 277, 121117 (2021). [DOI] [PubMed] [Google Scholar]
- 18.Alkekhia D, Hammond PT, Shukla A, Layer-by-Layer Biomaterials for Drug Delivery. Annual Review of Biomedical Engineering 22, null (2020). [DOI] [PubMed] [Google Scholar]
- 19.Hammond PT, Building biomedical materials layer-by-layer. Materials Today 15, 196–206 (2012). [Google Scholar]
- 20.Martin JR, Howard MT, Wang S, Berger AG, Hammond PT, Oxidation-Responsive, Tunable Growth Factor Delivery from Polyelectrolyte-Coated Implants. Adv Healthc Mater 10, e2001941 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shah NJ et al. , Adaptive growth factor delivery from a polyelectrolyte coating promotes synergistic bone tissue repair and reconstruction. Proceedings of the National Academy of Sciences of the United States of America 111, 12847–12852 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lavalle P et al. , Comparison of the structure of polyelectrolyte multilayer films exhibiting a linear and an exponential growth regime: An in situ atomic force microscopy study. Macromolecules 35, 4458–4465 (2002). [Google Scholar]
- 23.Picart C et al. , Molecular basis for the explanation of the exponential growth of polyelectrolyte multilayers. Proceedings of the National Academy of Sciences 99, 12531 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wood KC, Chuang HF, Batten RD, Lynn DM, Hammond PT, Controlling interlayer diffusion to achieve sustained, multiagent delivery from layer-by-layer thin films. Proceedings of the National Academy of Sciences 103, 10207–10212 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hsu BB et al. , Ordered and kinetically discrete sequential protein release from biodegradable thin films. Angewandte Chemie 53, 8093–8098 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hong J et al. , Graphene multilayers as gates for multi-week sequential release of proteins from surfaces. ACS Nano 6, 81–88 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Min J, Braatz RD, Hammond PT, Tunable staged release of therapeutics from layer-by-layer coatings with clay interlayer barrier. Biomaterials 35, 2507–2517 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gaharwar AK et al. , Bioactive Silicate Nanoplatelets for Osteogenic Differentiation of Human Mesenchymal Stem Cells. Advanced Materials 25, 3329–3336 (2013). [DOI] [PubMed] [Google Scholar]
- 29.Das SS et al. , Laponite-based nanomaterials for biomedical applications: a review. Current pharmaceutical design 25, 424–443 (2019). [DOI] [PubMed] [Google Scholar]
- 30.Tomás H, Alves CS, Rodrigues J, Laponite®: A key nanoplatform for biomedical applications? Nanomedicine: Nanotechnology, Biology and Medicine 14, 2407–2420 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Lynn DM, Langer R, Degradable Poly(β-amino esters): Synthesis, Characterization, and Self-Assembly with Plasmid DNA. Journal of the American Chemical Society 122, 10761–10768 (2000). [Google Scholar]
- 32.Shah NJ et al. , Surface-Mediated Bone Tissue Morphogenesis from Tunable Nanolayered Implant Coatings. Science Translational Medicine 5, 191ra183–191ra183 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abdurrahman R. C. A. Pierozan; Gregorio Luis Liva Araujo; Jorge Gabriel Zornberg, Optical and Physical Properties of Laponite for Use as Clay Surrogate in Geotechnical Models. Geotechnical Testing Journal 45, 79–100 (2022). [Google Scholar]
- 34.Ravi Kumar, K. NVNM; Joshi Yogesh M., On the refractive index of ageing dispersions of Laponite. Applied Clay Science 42, 326–330 (2008). [Google Scholar]
- 35.Shukla A, Avadhany SN, Fang JC, Hammond PT, Tunable vancomycin releasing surfaces for biomedical applications. Small 6, 2392–2404 (2010). [DOI] [PubMed] [Google Scholar]
- 36.Spicer PP et al. , Evaluation of bone regeneration using the rat critical size calvarial defect. Nat. Protocols 7, 1918–1929 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.An YH, Martin KL, Handbook of histology methods for bone and cartilage. (Springer, 2003). [Google Scholar]
- 38.Han U, Hong J, Structure of a Multilayer Nanofilm To Increase the Encapsulation Efficiency of Basic Fibroblast Growth Factor. Molecular Pharmaceutics 15, 1277–1283 (2018). [DOI] [PubMed] [Google Scholar]
- 39.Wong SY et al. , Dual Functional Polyelectrolyte Multilayer Coatings for Implants: Permanent Microbicidal Base with Controlled Release of Therapeutic Agents. Journal of the American Chemical Society 132, 17840–17848 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chuang HF, Smith RC, Hammond PT, Polyelectrolyte multilayers for tunable release of antibiotics. Biomacromolecules 9, 1660–1668 (2008). [DOI] [PubMed] [Google Scholar]
- 41.Macdonald ML, Rodriguez NM, Shah NJ, Hammond PT, Characterization of Tunable FGF-2 Releasing Polyelectrolyte Multilayers. Biomacromolecules 11, 2053–2059 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Macdonald ML et al. , Tissue integration of growth factor-eluting layer-by-layer polyelectrolyte multilayer coated implants. Biomaterials 32, 1446–1453 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Min J et al. , Designer Dual Therapy Nanolayered Implant Coatings Eradicate Biofilms and Accelerate Bone Tissue Repair. ACS Nano 10, 4441–4450 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shah NJ, Hong J, Hyder MN, Hammond PT, Osteophilic Multilayer Coatings for Accelerated Bone Tissue Growth. Advanced Materials 24, 1445–1450 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shah NJ et al. , Tunable dual growth factor delivery from polyelectrolyte multilayer films. Biomaterials 32, 6183–6193 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shukla A, Fuller RC, Hammond PT, Design of multi-drug release coatings targeting infection and inflammation. Journal of controlled release 155, 159–166 (2011). [DOI] [PubMed] [Google Scholar]
- 47.Takada T et al. , Sulfated polysaccharides enhance the biological activities of bone morphogenetic proteins. Journal of Biological Chemistry 278, 43229–43235 (2003). [DOI] [PubMed] [Google Scholar]
- 48.VandeVord PJ et al. , Evaluation of the biocompatibility of a chitosan scaffold in mice. Journal of Biomedical Materials Research 59, 585–590 (2002). [DOI] [PubMed] [Google Scholar]
- 49., B. A. Instruments, Ed. (BYK.com). [Google Scholar]
- 50.Lutkenhaus JL et al. , Anisotropic Structure and Transport in Self-Assembled Layered Polymer−Clay Nanocomposites. Langmuir 23, 8515–8521 (2007). [DOI] [PubMed] [Google Scholar]
- 51.Tomas H, Alves CS, Rodrigues J, Laponite(R): A key nanoplatform for biomedical applications? Nanomedicine 14, 2407–2420 (2018). [DOI] [PubMed] [Google Scholar]
- 52.Huang X-B et al. , Laponite: a promising nanomaterial to formulate high-performance water-based drilling fluids. Petroleum Science 18, 579–590 (2021). [Google Scholar]
- 53.Hammond PT, Engineering materials layer‐by‐layer: Challenges and opportunities in multilayer assembly. AIChE Journal 57, 2928–2940 (2011). [Google Scholar]
- 54.Richardson JJ, Björnmalm M, Caruso F, Technology-driven layer-by-layer assembly of nanofilms. Science 348, (2015). [DOI] [PubMed] [Google Scholar]
- 55.Ordikhani F, Dehghani M, Simchi A, Antibiotic-loaded chitosan–Laponite films for local drug delivery by titanium implants: cell proliferation and drug release studies. Journal of Materials Science: Materials in Medicine 26, 269 (2015). [DOI] [PubMed] [Google Scholar]
- 56.Shi H, Zhang R, Feng S, Wang J, Influence of laponite on the drug loading and release performance of LbL polyurethane/poly(acrylic acid) multilayers. Journal of Applied Polymer Science 136, 47348 (2019). [Google Scholar]
- 57.Brunot C et al. , Cytotoxicity of polyethyleneimine (PEI), precursor base layer of polyelectrolyte multilayer films. Biomaterials 28, 632–640 (2007). [DOI] [PubMed] [Google Scholar]
- 58.Kelly CN et al. , High-strength, porous additively manufactured implants with optimized mechanical osseointegration. Biomaterials 279, 121206 (2021). [DOI] [PubMed] [Google Scholar]
- 59.Dai Y et al. , Promoting osteoblasts responses in vitro and improving osteointegration in vivo through bioactive coating of nanosilicon nitride on polyetheretherketone. J Orthop Translat 24, 198–208 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Berner A et al. , Effects of scaffold architecture on cranial bone healing. International journal of oral and maxillofacial surgery 43, 506–513 (2014). [DOI] [PubMed] [Google Scholar]
- 61.Reddi AH, Role of morphogenetic proteins in skeletal tissue engineering and regeneration. Nat Biotechnol 16, 247–252 (1998). [DOI] [PubMed] [Google Scholar]
- 62.Cho TH et al. , Early and Marked Enhancement of New Bone Quality by Alendronate-Loaded Collagen Sponge Combined with Bone Morphogenetic Protein-2 at High Dose: A Long-Term Study in Calvarial Defects in a Rat Model. Tissue Engineering Part A, (2017). [DOI] [PubMed] [Google Scholar]
- 63.Young S et al. , Dose Effect of Dual Delivery of Vascular Endothelial Growth Factor and Bone Morphogenetic Protein-2 on Bone Regeneration in a Rat Critical-Size Defect Model. Tissue Engineering Part A 15, 2347–2362 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee J-H et al. , The induction of bone formation in rat calvarial defects and subcutaneous tissues by recombinant human BMP-2, produced in Escherichia coli. Biomaterials 31, 3512–3519 (2010). [DOI] [PubMed] [Google Scholar]
- 65.Pelaez M et al. , Effect of rh BMP‐2 dose on bone formation/maturation in a rat critical‐size calvarial defect model. Journal of clinical periodontology 41, 827–836 (2014). [DOI] [PubMed] [Google Scholar]
- 66.He X, Liu Y, Yuan X, Lu L, Enhanced healing of rat calvarial defects with MSCs loaded on BMP-2 releasing chitosan/alginate/hydroxyapatite scaffolds. PLoS One 9, e104061 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li L et al. , Controlled dual delivery of BMP-2 and dexamethasone by nanoparticle-embedded electrospun nanofibers for the efficient repair of critical-sized rat calvarial defect. Biomaterials 37, 218–229 (2015). [DOI] [PubMed] [Google Scholar]
- 68.Lee D et al. , Sequential dual-drug delivery of BMP-2 and alendronate from hydroxyapatite-collagen scaffolds for enhanced bone regeneration. Sci Rep 11, 746 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jiao H, Xiao E, Graves DT, Diabetes and Its Effect on Bone and Fracture Healing. Current Osteoporosis Reports 13, 327–335 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bastian OW et al. , Impaired bone healing in multitrauma patients is associated with altered leukocyte kinetics after major trauma. J Inflamm Res 9, 69–78 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hwang PW, Horton JA, Variable osteogenic performance of MC3T3-E1 subclones impacts their utility as models of osteoblast biology. Sci Rep 9, 8299 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rahman SU, Park CH, Baek JH, Ryoo HM, Woo KM, Fibrin-Enhanced Canonical Wnt Signaling Directs Plasminogen Expression in Cementoblasts. Int J Mol Sci 18, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw/processed data used to generate the figures in this manuscript are available upon request from the authors.