

Abstract
The role of BRAF in tumor initiation has been established, however, the precise mechanism of autoinhibition has only been illustrated recently by several structural studies. These structures uncovered the basis by which the regulatory domains engage in regulating the activity of BRAF kinase domain, which lead to a more complete picture of the regulation cycle of RAF kinases. Small molecule BRAF inhibitors developed specifically to target BRAFV600E have proven effective at inhibiting the most dominant BRAF mutant in melanomas, but are less potent against other BRAF mutants in RAS-driven diseases due to paradoxical activation of the MAPK pathway. A variety of new generation inhibitors that do not show paradoxical activation have been developed. Alternatively, efforts have begun to develop inhibitors targeting the dimer interface of BRAF. A deeper understanding of BRAF regulation together with more diverse BRAF inhibitors will be beneficial for drug development in RAF or RASdriven cancers.
Keywords: RAS, RAF kinase, Autoinhibition, 14-3-3, RAS-RAF interaction, Paradoxical activation, Allosteric kinase inhibitor
Regulation of RAF kinases
RAF isoforms
RAF kinases (ARAF, BRAF, and CRAF/Raf-1), a family of Ser/Thr kinase, control the duration and amplitude of MAPK signaling, thus are subject to complex regulation and are considered as attractive targets for cancer therapy [1]. The RAF isoforms have three shared conserved regions (CR): CR1, CR2, and CR3 (Figure 1). In CR1, the RAS-binding domain (RBD) and cysteine-rich domain (CRD) are important for interactions with GTP-loaded active RAS proteins and the plasma membrane [2,3]. CR2, Ser/Thr-rich domain, contains a 14-3-3 binding site, which has been shown to negatively regulate RAF activity [4]. CR3 contains the kinase domain which is important for RAF dimerization, substrate binding, and phosphorylation of MEK1/2 (the only known protein substrates of RAF). The C-terminal tail possesses a second 14-3-3 binding site which is believed to promote RAF dimerization [5]. Unique to BRAF there is a region referred to as the BRAF-specific region (BSR). The activation of wild-type RAF kinase is contingent on RAS-initiated membrane recruitment and subsequent homo- or hetero-dimerization with other RAF isoforms. Both a “side-to-side” active dimer configuration [6] and an inactive monomer configuration of BRAF kinase domain [7] have been reported, underpinning an allosteric mechanism of activation.
Figure 1. Conserved regions of the RAF family.
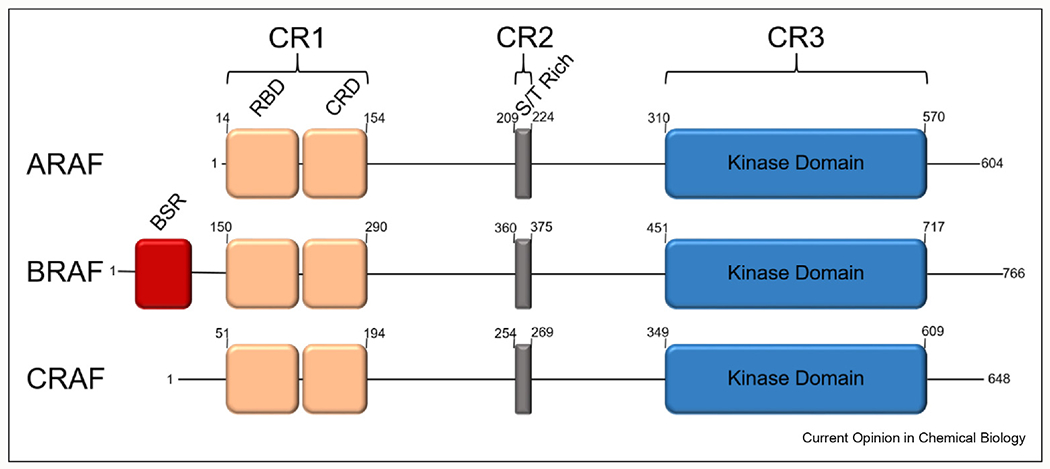
The RAF isoforms indicating the conserved regions (CR): CR1 (orange), CR2 (grey), and CR3 (blue). Labeled within the three regions are RAS-binding domain (RBD)/cysteine-rich domain (CRD), serine/threonine rich domain (S/T Rich), and kinase domain contained in CR1/CR2/CR3, respectively. On BRAF, there is a BRAF-specific region (red) located at the N-terminus. The amino acid numbers of the beginning and end of the RAF isoform and CR are shown.
RAS:RAF interactions
All three RAF isoforms share high sequence homology, thus regulation mechanisms identified from one isoform normally can be extended to other RAF kinases. However, they differ in catalytic activity and RAS-binding affinity [8,9]. BRAF has the highest basal activity [10] and is most frequently mutated in human cancers [11], which led to an extensive interest in investigating its regulation mechanism and development of BRAF-specific drugs. The role of RAS isoforms (HRAS, KRAS4A/B, and NRAS) in selectively regulating the biochemical activity of RAF dimers has been lately identified by live cell bioluminescence resonance energy transfer (BRET) [12] and structural analyses [13]. The N-terminal regulatory regions of RAF and the hypervariable region of RAS determine the selectivity toward each RAS:RAF pair [12]. The acidic characteristic of the BSR makes BRAF preferentially binds to KRAS whose C-terminal hypervariable region has a lysine-rich polybasic region, while CRAF, and to a lesser extent ARAF, have similar binding profiles toward all RAS isoforms. The HRAS:CRAF interaction is the strongest among all RAS:RAF interactions. Additionally, cells that induce BRAF/CRAF heterodimers, the most active RAF dimer, enhance the BRAF:HRAS interaction. It is well established that the RBD of RAF is directly involved in RAS binding and the CRD of RAF is interacting with the plasma membrane after RAS-initiated membrane recruitment. The significance of CRD in stabilizing the interaction between RAF and RAS was recently verified by the first crystal structure of KRAS in complex with RBD-CRD of CRAF, revealing the extensive interactions between CRD and KRAS [13].
The mechanism of autoinhibition and activation of RAF
In quiescent cells, RAF localizes to the cytoplasm and is believed to exist in an autoinhibited monomeric state with the N-terminal regulatory regions bound to the catalytic domain [14]. 14-3-3 proteins help maintain this autoinhibited state by binding to two sites: one site is located at the N-terminus, found in the CR2 (aa. ARAFS214/BRAFS365/CRAFS259), and the second 14-3-3 binding site is located at the C-terminus of RAF (aa. ARAFS582/BRAFS729/CRAFS621) [15,16]. Structural insights of the long-believed autoinhibitory mechanism have been provided by three independent studies recently published (Figure 2) [17–19]. The Cryo-EM structure of the active dimeric BRAF kinase domain:14-3-3 complex published by Kondo et al. reveals an asymmetrical interaction between BRAF and 14-3-3, suggesting that the C-terminal tail of one BRAF protomer that is ready for catalysis could play an auto-inhibitory role by occupying the active site of the other BRAF protomer (Figure 2a) [17]. Park et al. published the first Cryo-EM structure of autoinhibited full-length BRAF:MEK1:14-3-3 complex and revealed that the CRD of BRAF establishes several contacts with 14-3-3 and the catalytic domain which locks BRAF in an inactive monomeric conformation (Figure 2b) [18]. Notably, the dimer interface of the BRAF kinase domain is blocked by the position of CRD and is relieved in the structure of active dimeric BRAF:MEK1:14-3-3 complex, indicating that rearrangement of CRD upon association with RAS and plasma membrane is the key event involved in RAF activation. Therefore, the CRD displays dual roles in the regulation of RAF: it stabilizes the autoinhibitory interactions of RAF in the absence of active RAS through directly associating with the kinase domain and 14-3-3; it increases the interaction of RAF and RAS in the action process through directly associating with RAS and plasma membrane. Structures published by Martinez Fiesco et al. not only further verified the significance of CRD and 14-3-3 in maintaining the autoinhibited state but also identified the location and interactions of RBD (Figure 2c) [19]. In this autoinhibited monomeric full-length BRAF:MEK1:14-3-3 structure, the RBD interacts extensively with 14-3-3 while leaves the RAS-binding surface of RBD exposed so that it is feasible for association with GTP-loaded RAS. Collectively, these structures delineate the detailed molecular mechanism of RAF autoinhibition, dimerization, and activation, providing an integrated understanding of the transition between monomeric inactive state and dimeric active state, which is presented in Figure 3.
Figure 2. Cryo-EM structures of BRAF.
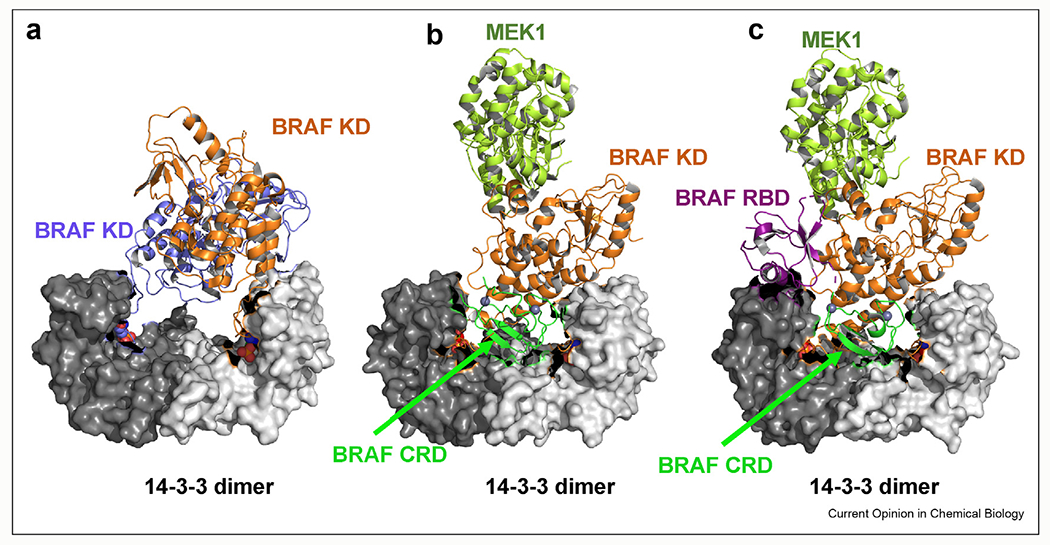
(a) The cryo-EM structure of a dimeric B-Raf:14-3-3 complex (PDB ID: 6UAN); (b) the cryo-EM structure of autoinhibited BRAF:14:3:3:MEK1 complex (PDB ID: 6NYB); (c) the cryo-EM structure of autoinhibited BRAF:14-3-3:MEK1 complex with RBD visible (PDB ID: 6MFD).
Figure 3. RAF activation mechanism.
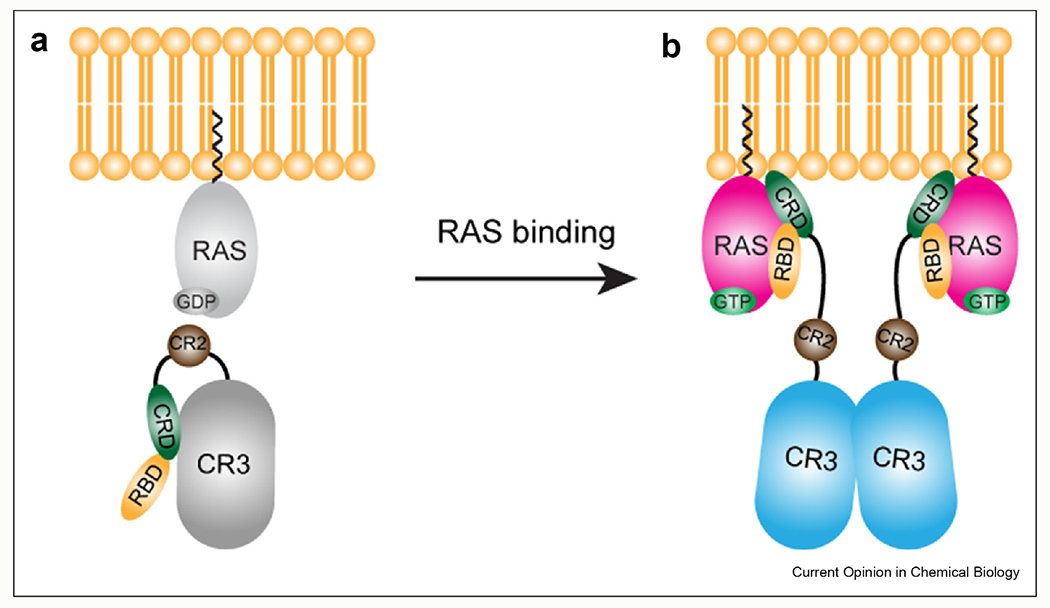
(a) Monomeric inactive state. When RAS exists in the GDP-bound inactive form (light gray), RAF is found in the cytosol as an autoinhibited inactive monomer with the CRD (green) binding to the CR3 (gray, kinase-domain). GDP-bound RAS (light gray) is inactive. (b) Dimeric active state. GTP-bound active RAS (magenta) binds to the RBD (orange) and CRD and recruits the RAF to plasma membrane, leading to the release of CRD from the dimer interface of CR3. RAS nano-clustering brings RAF molecules into close proximity for dimerization and activation of CR3 (blue). Both the autoinhibitory interaction and kinase domain dimerization are facilitated by 14-3-3 binding to the CR2 and the C-terminal tail of RAF, respectively. For clarity, 14-3-3 proteins and the C-terminal tail are not included to the schematic diagram.
Inhibition of BRAF
Mutations in BRAF
Among RAF family members, BRAF has the highest mutation propensity [11]. Many of the BRAF mutations gain activity by destabilizing the autoinhibited BRAF conformation resulting in a kinase primed for activation, dimerization, or both [20]. A missense mutation at V600 occurs in ~ 90% of BRAF mutations, and other notable non-V600 mutations occur at residues G464, G466, G469, D594, L597, and K601. Lung cancer has the highest rate of non-V600 mutations, with D594G and G469A having the highest mutation frequency [21]. All BRAF mutations activate the MAPK pathway, but have different biochemical characteristics, and the mutants have been classified into three classes [20]. Class 1 BRAF mutants are RAS independent, can signal as a monomer, and have a high kinase activity. Class 2 mutants are RAS independent but form a constitutive dimer with moderate/high kinase activity. Class 3 mutants are RAS and dimer dependent and have low/no kinase activity. In addition to the listed mutations, BRAF can also become activated by fusions with other proteins or truncations of the CR1/CR2 regions resulting in constitutive dimerization/activation [22].
RAF inhibitors
RAF kinases are central proteins in the MAPK pathway, and their high rates of mutation have led to decades of efforts for the development of inhibitors. Most of those inhibitors are ATP-competitive inhibitors and three of them have been FDA-approved for the treatment of BRAFV600E cancers [23,24]. RAF inhibitors are generally classified based on the conformational changes that they induce in the kinase domain upon binding [25]. Inhibitor binding leads to the change in position of the α-C helix and the catalytic DFG motif, which in turn affects the propensity of dimer formation and RAS association [26].
First-generation (α-C-in) RAF inhibitors
α-C-in inhibitors binding to one protomer of RAF dimer stabilize the α-C-helix in the IN conformation and sterically permit the binding of a second inhibitor molecule into the other protomer [7]. Therefore, α-C-in inhibitors bind both protomers of a dimer with equal affinity and have efficacy in inhibiting both monomeric and dimeric RAF homo- and heterodimers. Since wild-type BRAF also signals as a dimer, these inhibitors have a narrow therapeutic window. Examples of early α-C-in inhibitors include Sorafenib, ZM336372, and SB590885 [27] (Figure 4). Of these, Sorafenib is the only FDA-approved inhibitor for the treatment of advance renal cell carcinoma and hepatocellular carcinoma [28]. Although Sorafenib was originally developed as a RAF kinase inhibitor, it is approved for therapy against tyrosine kinases VEGFR and PDGFR.
Figure 4.
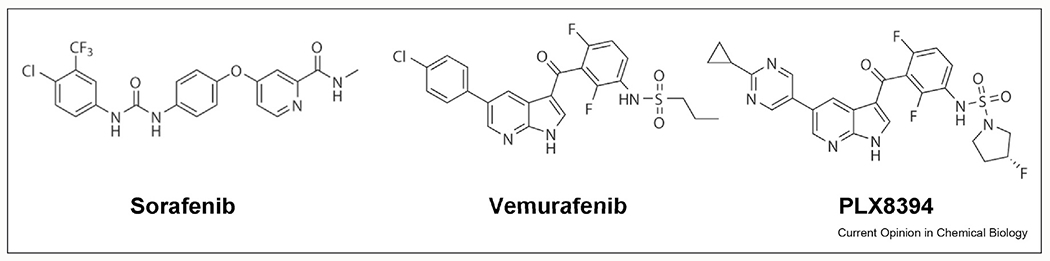
Chemical structures of representative RAF inhibitors.
Second-generation (α-C-out) RAF inhibitors
α-C-out inhibitors bind to a protomer and stabilize the α-C-helix in the OUT conformation which sterically hinders the movement of the α-C-helix of the second protomer, thereby locking it into the IN conformation [7]. α-C-out inhibitors have a reduced affinity for the second protomer leading to negative allostery. Therefore, α-C-out inhibitors have limited efficacy against dimeric RAF (BRAFWT, non-V600E BRAF mutants, such as BRAFG469A and BRAFL597V) [25]. They have a wide therapeutic window for the treatment of monomeric V600E, and three α-C-out inhibitors, vemurafenib, dabrafenib, and encorafenib have been FDA-approved for the treatment of BRAFV600E metastatic melanoma (Figure 4). Second-generation inhibitors are, however, characterized by the occurrence of paradoxical activation that binding of an inhibitor to one protomer, promotes dimerization and transactivation of the other drug-free protomer, leading to increased ERK signaling in wild-type BRAF cells [29]. This causes secondary cancers and squamous-cell carcinomas that are usually oncogenic RAS driven [30].
Third-generation (α-C-in and paradox breakers) RAF inhibitors
Third-generation RAF inhibitors in preclinical development have largely focused on reducing paradoxical activation. One group of molecules belonging to this class includes TAK632, MLN2480, and LY3009120, and crystal structures of these compounds with RAF show them binding in an α-C-in conformation much like the first-generation inhibitors [31,32]. Therefore, these inhibitors have a similar affinity for both monomeric and dimeric forms of RAF and are typically referred as pan RAF inhibitor. Interestingly, the second set of molecules belonging to this class include compounds that have been dubbed as “paradox breakers,” such as PLX7904 and PLX8394 (Figure 4) [33,34]. These α-C-out RAF inhibitors are more potent than vemurafenib against BRAFV600E while inducing dimer dissociation instead of dimer association, therefore, do not exhibit paradoxical activation.
Resistance to ATP-competitive inhibitors
Resistance to the current ATP-competitive inhibitors develops as a result of several mechanisms. One of the major mechanisms of resistance is through RAF dimerization [35]. There are other inhibitors still in the discovery stage that aim to target dimeric RAF for MAPK inhibition. Vem-BisAmide-2 is a compound containing two vemurafenib molecules joined with a bis-amide linker that promotes an inactive BRAFV600E dimer that is unable to signal downstream kinases [36]. PHI1 inhibitor was developed to target dimeric BRAF and displays positive cooperativity in which the occupation of the first promoter enhances the binding affinity toward the second protomer within the dimer [37]. Interestingly, PHI1 shows a different binding mode and recognizes a previously uncharacterized allosteric site next to the α-C helix. Another approach is the development of proteolysis-targeting chimera (PROTACs) that can selectively degrade and constrain oncogenic BRAF variants [38,39]. Since current kinase drugs are rendered ineffective due to dimerization, a significant effort is being made to develop inhibitors that can interfere with RAF dimerization. This can be achieved through improvements on existing α-C-out ATP-competitive inhibitors to prevent paradoxical activation or through direct inhibition of RAF dimerization.
Allosteric RAF inhibitors
In cancers with oncogenic RAS mutations, MAPK signaling is reliant on wild-type RAF dimerization. In cancers with a wild-type RAS and oncogenic BRAF, the necessity of an intact RAF dimer interface changes with specific BRAF variants. BRAFV600E/D/K mutants (class I) can function as monomers within the cells and are RAS-independent [40]. Non-V600E mutants, such as G469 A/V/R (class II) and G469E or D594G (class III), require an intact dimer interface and signal solely as constitutive dimers [41,42]. With current kinase inhibitors being resistant to these dimeric RAFs, targeting the dimer interface as an allosteric drug binding site can provide opportunities for inhibiting these oncogenic variants. The RAF dimer interface is composed of the α-C-helix-β4 loop that spans ~20 amino acids. This extensive surface is an unstructured loop that does not have well-defined binding pockets that can be exploited by small molecule inhibitors. Smaller peptides or peptidomimetics have several features that make them a good starting point for targeting such non-structured, protein–protein interaction surfaces. Freeman et al. used a ~30-residue peptide to show that blocking this interface could interrupt RAF kinase activity and MAPK signaling [43]. Gunderwala et al. reported a 10mer peptide inhibitor braftide designed to block RAF dimerization [42,44]. In vitro kinase assays with purified full-length wild-type BRAF and BRAFG469A demonstrate that braftide potently inhibits BRAFG469A and inhibits BRAFWT to a lesser extent. Beneker et al. designed and synthesized macrocyclic peptides that target the dimer interface of BRAF with high affinity [45]. Those lead compounds successfully block paradoxical activation induced by dabrafenib and vemurafenib in RAS mutated cells. The combination of ATP-competitive inhibitors and allosteric inhibitors eliminates paradoxical activation, suggesting an alternative strategy to improve the efficacy of current ATP-competitive inhibitors. Although these works are still in the very early stage, they establish the RAF dimer interface as a promising therapeutic target.
Concluding remarks
A comprehensive understanding of RAF regulation and inhibition is valuable for future drug development. The development of potent allosteric inhibitors has lagged behind; however, it posies great potential to overcome the limitations of current ATP-competitive inhibitors. The structure of the RAS:full-length RAF:14-3-3 complex is still missing and the orientation of each domain remains elusive, in particular the BSR of BRAF, probably due to its high flexibility in the absence of binding partners. Structural basis for how RAS binding promotes relief of the autoinhibitory RAF interactions, and the structural determinants of isoform-selectivity among RAF:RAS complexes remain to be defined.
Acknowledgements
This project was funded with federal funds from the NIGMS: NIGMS 1R01GM138671 (to Z. Wang) and NIGMS 1R15GM128099 (to Z. wang).
Declaration of competing interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: Zhihong Wang reports financial support was provided by National Institute of Health. Zhihong Wang reports financial support was provided by WW Smith Charitable Foundation.
Footnotes
This review comes from a themed issue on Mechanistic Biology
References
Papers of particular interest, published within the period of review, have been highlighted as:
* of special interest
** of outstanding interest
- 1.Dorard C, Vucak G, Baccarini M: Deciphering the RAS/ERK pathway in vivo. Biochem Soc Trans 2017, 45:27–36. [DOI] [PubMed] [Google Scholar]
- 2.Chuang E, Barnard D, Hettich L, Zhang XF, Avruch J, Marshall MS: Critical binding and regulatory interactions between Ras and Raf occur through a small, stable N-terminal domain of Raf and specific Ras effector residues. Mol Cell Biol 1994, 14:5318–5325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moodie SA, Willumsen BM, Weber MJ, Wolfman A: Complexes of Ras.GTP with Raf-1 and mitogen-activated protein kinase kinase. Science 1993, 260:1658–1661. [DOI] [PubMed] [Google Scholar]
- 4.Freed E, Symons M, Macdonald SG, McCormick F, Ruggieri R: Binding of 14-3-3 proteins to the protein kinase Raf and effects on its activation. Science 1994, 265:1713–1716. [DOI] [PubMed] [Google Scholar]
- 5.Dhillon AS, Yip YY, Grindlay GJ, Pakay JL, Dangers M, Hillmann M, Clark W, Pitt A, Mischak H, Kolch W: The C-terminus of Raf-1 acts as a 14-3-3-dependent activation switch. Cell Signal 2009, 21:1645–1651. [DOI] [PubMed] [Google Scholar]
- 6.Rajakulendran T, Sahmi M, Lefrancois M, Sicheri F, Therrien M: A dimerization-dependent mechanism drives RAF catalytic activation. Nature 2009, 461:542–545. [DOI] [PubMed] [Google Scholar]
- 7.Thevakumaran N, Lavoie H, Critton DA, Tebben A, Marinier A, Sicheri F, Therrien M: Crystal structure of a BRAF kinase domain monomer explains basis for allosteric regulation. Nat Struct Mol Biol 2015, 22:37–43. [DOI] [PubMed] [Google Scholar]
- 8.Tran NH, Wu X, Frost JA: B-Raf and Raf-1 are regulated by distinct autoregulatory mechanisms. J Biol Chem 2005, 280: 16244–16253. [DOI] [PubMed] [Google Scholar]
- 9.Garnett MJ, Rana S, Paterson H, Barford D, Marais R: Wild-type and mutant B-RAF activate C-RAF through distinct mechanisms involving heterodimerization. Mol Cell 2005, 20:963–969. [DOI] [PubMed] [Google Scholar]
- 10.Cope N, Candelora C, Wong K, Kumar S, Nan H, Grasso M, Novak B, Li Y, Marmorstein R, Wang Z: Mechanism of BRAF activation through biochemical characterization of the recombinant full-length protein. Chembiochem 2018, 19:1988–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417: 949–954. [DOI] [PubMed] [Google Scholar]
- 12.Terrell EM, Durrant DE, Ritt DA, Sealover NE, Sheffels E, Spencer-Smith R, Esposito D, Zhou Y, Hancock JF, Kortum RL, et al. : Distinct binding preferences between ras and raf family members and the impact on oncogenic ras signaling. Mol Cell 2019, 76:872–884. e875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.*.Tran TH, Chan AH, Young LC, Bindu L, Neale C, Messing S, Dharmaiah S, Taylor T, Denson JP, Esposito D, et al. : KRAS interaction with RAF1 RAS-binding domain and cysteine-rich domain provides insights into RAS-mediated RAF activation. Nat Commun 2021, 12:1176. [DOI] [PMC free article] [PubMed] [Google Scholar]; This work provides the first experimental structure of RAS-RBDCRD complex revealing the KRAS-CRD interaction interface. This structure provides key insights to undergstand RAS and RAF regulation.
- 14.Cutler RE Jr, Stephens RM, Saracino MR, Morrison DK: Autoregulation of the Raf-1 serine/threonine kinase. Proc Natl Acad Sci USA 1998, 95:9214–9219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roskoski R Jr: RAF protein-serine/threonine kinases: structure and regulation. Biochem Biophys Res Commun 2010, 399: 313–317. [DOI] [PubMed] [Google Scholar]
- 16.Fantl WJ, Muslin AJ, Kikuchi A, Martin JA, MacNicol AM, Gross RW, Williams LT: Activation of raf-1 by 14-3-3 proteins. Nature 1994, 371:612–614. [DOI] [PubMed] [Google Scholar]
- 17.Kondo Y, Ognjenović J, Banerjee S, Karandur D, Merk A, Kulhanek K, Wong K, Roose JP, Subramaniam S, Kuriyan J: Cryo-EM structure of a dimeric B-Raf:14-3-3 complex reveals asymmetry in the active sites of B-Raf kinases. Science 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.**.Park E, Rawson S, Li K, Kim BW, Ficarro SB, Pino GG, Sharif H, Marto JA, Jeon H, Eck MJ: Architecture of autoinhibited and active BRAF-MEK1-14-3-3 complexes. Nature 2019, 575: 545–550. [DOI] [PMC free article] [PubMed] [Google Scholar]; This work shows the first cryo-EM structures of full-length autoinhibited and active BRAF in complex with 14-3-3 and MEK, a major advance in understanding the autoinhibition and activation mechanism of RAF kinase.
- 19.*.Martinez Fiesco JA, Durrant DE, Morrison DK, Zhang P: Structural insights into the BRAF monomer-to-dimer transition mediated by RAS binding. Nat Commun 2022, 13:486. [DOI] [PMC free article] [PubMed] [Google Scholar]; The cryo-EM structure of monomeric full-length BRAF in complex with 14-3-3 and MEK shows for the first time the position and orientation of the RAS binding domain (RBD), providing valuable insights about how RAF autoinhibitory interactions are relieved upon RAS binding.
- 20.Yao Z, Torres NM, Tao A, Gao Y, Luo L, Li Q, de Stanchina E, Abdel-Wahab O, Solit DB, Poulikakos PI, et al. : BRAF mutants evade ERK-dependent feedback by different mechanisms that determine their sensitivity to pharmacologic inhibition. Cancer Cell 2015, 28:370–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Volckmar AL, Christopoulos P, Kirchner M, Allgauer M, Neumann O, Budczies J, Rempel E, Horak P, Glade J, Goldschmid H, et al. : Targeting rare and non-canonical driver variants in NSCLC - an uncharted clinical field. Lung Cancer 2021, 154:131–141. [DOI] [PubMed] [Google Scholar]
- 22.Ross JS, Wang K, Chmielecki J, Gay L, Johnson A, Chudnovsky J, Yelensky R, Lipson D, Ali SM, Elvin JA, et al. : The distribution of BRAF gene fusions in solid tumors and response to targeted therapy. Int J Cancer 2016,138:881–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lavoie H, Therrien M: Regulation of RAF protein kinases in ERK signalling. Nat Rev Mol Cell Biol 2015, 16:281–298. [DOI] [PubMed] [Google Scholar]
- 24.Karoulia Z, Gavathiotis E, Poulikakos PI: New perspectives for targeting RAF kinase in human cancer. Nat Rev Cancer 2017, 17:676–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karoulia Z, Wu Y, Ahmed TA, Xin Q, Bollard J, Krepler C, Wu X, Zhang C, Bollag G, Herlyn M, et al. : An integrated model of RAF inhibitor action predicts inhibitor activity against oncogenic BRAF signaling. Cancer Cell 2016, 30:501–503. [DOI] [PubMed] [Google Scholar]
- 26.Agianian B, Gavathiotis E: Current insights of BRAF inhibitors in cancer. J Med Chem 2018. [DOI] [PubMed] [Google Scholar]
- 27.King AJ, Patrick DR, Batorsky RS, Ho ML, Do HT, Zhang SY, Kumar R, Rusnak DW, Takle AK, Wilson DM, et al. : Demonstration of a genetic therapeutic index for tumors expressing oncogenic BRAF by the kinase inhibitor SB-590885. Cancer Res 2006, 66:11100–11105. [DOI] [PubMed] [Google Scholar]
- 28.Ben Mousa A: Sorafenib in the treatment of advanced hepatocellular carcinoma. Saudi J Gastroenterol 2008, 14:40–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, Ludlam MJ, Stokoe D, Gloor SL, Vigers G, et al. : RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 2010, 464:431–435. [DOI] [PubMed] [Google Scholar]
- 30.Su F, Viros A, Milagre C, Trunzer K, Bollag G, Spleiss O, Reis-Filho JS, Kong X, Koya RC, Flaherty KT, et al. : RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med 2012, 366:207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shao W, Mishina YM, Feng Y, Caponigro G, Cooke VG, Rivera S, Wang Y, Shen F, Korn JM, Mathews Griner LA, et al. : Antitumor properties of RAF709, a highly selective and potent inhibitor of RAF kinase dimers, in tumors driven by mutant RAS or BRAF. Cancer Res 2018, 78:1537–1548. [DOI] [PubMed] [Google Scholar]
- 32.Chen SH, Zhang Y, Van Horn RD, Yin T, Buchanan S, Yadav V, Mochalkin I, Wong SS, Yue YG, Huber L, et al. : Oncogenic BRaf deletions that function as homodimers and are sensitive to inhibition by raf dimer inhibitor LY3009120. Cancer Discov; 2016. [DOI] [PubMed] [Google Scholar]
- 33.Zhang C, Spevak W, Zhang Y, Burton EA, Ma Y, Habets G, Zhang J, Lin J, Ewing T, Matusow B, et al. : RAF inhibitors that evade paradoxical MAPK pathway activation. Nature 2015, 526:583–586. [DOI] [PubMed] [Google Scholar]
- 34.Yao Z, Gao Y, Su W, Yaeger R, Tao J, Na N, Zhang Y, Zhang C, Rymar A, Tao A, et al. : RAF inhibitor PLX8394 selectively disrupts BRAF dimers and RAS-independent BRAF-mutant-driven signaling. Nat Med 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, Shi H, Atefi M, Titz B, Gabay MT, et al. : RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 2011, 480:387–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grasso M, Estrada MA, Ventocilla C, Samanta M, Maksimoska J, Villanueva J, Winkler JD, Marmorstein R: Chemically linked vemurafenib inhibitors promote an inactive BRAFV600E conformation. ACS Chem Biol 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cotto-Rios XM, Agianian B, Gitego N, Zacharioudakis E, Giricz O, Wu Y, Zou Y, Verma A, Poulikakos PI, Gavathiotis E: Inhibitors of BRAF dimers using an allosteric site. Nat Commun 2020, 11:4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Posternak G, Tang X, Maisonneuve P, Jin T, Lavoie H, Daou S, Orlicky S, Goullet de Rugy T, Caldwell L, Chan K, et al. : Functional characterization of a PROTAC directed against BRAF mutant V600E. Nat Chem Biol 2020, 16:1170–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Han XR, Chen L, Wei Y, Yu W, Chen Y, Zhang C, Jiao B, Shi T, Sun L, Zhang C, et al. : Discovery of selective small molecule degraders of BRAF-V600E. J Med Chem 2020, 63:4069–4080. [DOI] [PubMed] [Google Scholar]
- 40.Roring M, Herr R, Fiala GJ, Heilmann K, Braun S, Eisenhardt AE, Halbach S, Capper D, von Deimling A, Schamel WW, et al. Distinct requirement for an intact dimer interface in wild-type, V600E and kinase-dead B-Raf signalling. EMBO J 2012, 31: 2629–2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yao Z, Yaeger R, Rodrik-Outmezguine VS, Tao A, Torres NM, Chang MT, Drosten M, Zhao H, Cecchi F, Hembrough T, et al. Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature 2017, 548:234–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cope NJ, Novak B, Liu Z, Cavallo M, Gunderwala AY, Connolly M, Wang Z: Analyses of the oncogenic BRAF(D594G) variant reveal a kinase-independent function of BRAF in activating MAPK signaling. J Biol Chem 2020, 295: 2407–2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Freeman AK, Ritt DA, Morrison DK: Effects of Raf dimerization and its inhibition on normal and disease-associated Raf signaling. Mol Cell 2013, 49:751–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.*.Gunderwala AY, Nimbvikar AA, Cope NJ, Li Z, Wang Z: Development of allosteric BRAF peptide inhibitors targeting the dimer interface of BRAF. ACS Chem Biol 2019, 14:1471–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.*.Beneker CM, Rovoli M, Kontopidis G, Roring M, Galda S, Braun S, Brummer T, McInnes C: Design and synthesis of type-IV inhibitors of BRAF kinase that block dimerization and overcome paradoxical MEK/ERK activation. J Med Chem 2019, 62:3886–3897. [DOI] [PMC free article] [PubMed] [Google Scholar]; The two labs concurrently developed and characterized small peptides targeting the dimer interface of BRAF, providing proof of concept that the dimer interface of BRAF is a novel drug target.