

Keywords: environmental metal, high-resolution metabolomics, lung inflammation and fibrosis, oxidative stress, vanadate
Abstract
Vanadium is available as a dietary supplement and also is known to be toxic if inhaled, yet little information is available concerning the effects of vanadium on mammalian metabolism when concentrations found in food and water. Vanadium pentoxide (V+5) is representative of the most common dietary and environmental exposures, and prior research shows that low-dose V+5 exposure causes oxidative stress measured by glutathione oxidation and protein S-glutathionylation. We examined the metabolic impact of V+5 at relevant dietary and environmental doses (0.01, 0.1, and 1 ppm for 24 h) in human lung fibroblasts (HLFs) and male C57BL/6J mice (0.02, 0.2, and 2 ppm in drinking water for 7 mo). Untargeted metabolomics using liquid chromatography-high-resolution mass spectrometry (LC-HRMS) showed that V+5 induced significant metabolic perturbations in both HLF cells and mouse lungs. We noted 30% of the significantly altered pathways in HLF cells, including pyrimidines and aminosugars, fatty acids, mitochondrial and redox pathways, showed similar dose-dependent patterns in mouse lung tissues. Alterations in lipid metabolism included leukotrienes and prostaglandins involved in inflammatory signaling, which have been associated with the pathogenesis of idiopathic pulmonary fibrosis (IPF) and other disease processes. Elevated hydroxyproline levels and excessive collagen deposition were also present in lungs from V+5-treated mice. Taken together, these results show that oxidative stress from environmental V+5, ingested at low levels, could alter metabolism to contribute to common human lung diseases.
NEW & NOTEWORTHY We used relevant dietary and environmental doses of Vanadium pentoxide (V+5) to examine its metabolic impact in vitro and in vivo. Using liquid chromatography-high-resolution mass spectrometry (LC-HRMS), we found significant metabolic perturbations, with similar dose-dependent patterns observed in human lung fibroblasts and male mouse lungs. Alterations in lipid metabolism included inflammatory signaling, elevated hydroxyproline levels, and excessive collagen deposition were present in V+5-treated lungs. Our findings suggest that low levels of V+5 could trigger pulmonary fibrotic signaling.
INTRODUCTION
Vanadium (V) exposures in environmental and occupational settings have drawn attention in the past few decades (1, 2), yet little information is available concerning V effects on lung metabolism. Because V exposures can be both ingested and inhaled and the nature of exposures through each route are varied, documented exposures can be difficult to compare. To facilitate comparisons, reported exposures are summarized in Fig. 1 in terms of total daily exposure (μg/kg body weight). In drinking water, V can naturally reach up to 200 ppb, depending on geological locations (3). There is no drinking water standard set by the US Environmental Protection Agency (EPA); it is currently an unregulated contaminant, and concentrations can become much higher in extreme situations. Notably, significant quantities of V, up to 899 ppm, have been observed in drinking water as a result of iron pipe corrosion, and this could pose severe health risks (4). Substantial amounts of V enter through water and food consumption, with an estimated daily dietary V intake of up to 20 μg/kg body weight for adults (5), and 11 μg/kg body weight for infants, children, and adolescents (6). In addition to normal dietary exposures, sodium metavanadate (125 mg/day) has been used as a supplement for patients with diabetes (7). This results in intake thousands of times higher than usual and nearly 70 times higher than the Tolerable Upper Intake Level for V (1.8 mg/day; 8). In these individuals, major side effects have been observed, including vomiting, diarrhea, hypoglycemia, and increased salivation; however, no information has been provided on side effects related to the lungs (7). Inhaled exposures will more directly affect the lungs. In cities in the Eastern United States, individuals are estimated to inhale 0.12 μg V/kg body weight daily (9). Exposure to V may occur during the production and processing of steel alloys and catalysts (10), mining, and oil refinery processing (11). Boilermakers have been shown to be exposed to inhaled V at 10–500 μg/m3 while on the job. In an 8 h shift, this approximates to as much as 33 μg V/kg body weight in a day, without considering any increased intake due to labored breathing (12). Inhaled exposures to V also occur through tobacco smoke (13) and vehicle exhaust (14). Among literature, V concentrations reported in human blood range from 0.032 to 0.095 ppb, but individuals with occupational exposures have been shown to have circulating V concentrations as high as 217 ppb (15–17). Epidemiological data concerning the number of individuals with high levels of V exposure is not available, but these numbers are expected to resemble those of individuals in industries that carry high risk of V inhalation and of individuals taking V in dietary supplements.
Figure 1.

Summary of levels and routes of V exposure documented in literature. Exposures are represented in a common unit (μg V/kg body weight), and in vitro and in vivo exposures utilized in this study are shown in relation to documented exposures. Estimates of daily exposure are based on 1) an adult human’s resting breath rate of 15–18 breaths/min and 0.5 L of air exchanged in each breath, 2) an adult human’s lung alveolar surface area of 1 m2/kg body weight, 3) an average adult human weight of 60 kg and water consumption of 3 L/day, and 4) an average adult mouse weight of 30 g and water consumption of 3 mL/day.
Vanadium pentoxide (V+5) is the main form of V to which humans are exposed. Studies have shown that pulmonary inflammation (18) and fibrosis (19) could be induced by inhaled V+5, meaning V+5 could be linked to idiopathic pulmonary fibrosis (IPF), a disease that impacts 5.6 individuals per 100,000 each year (20). However, less is known about the adverse health effects of V+5 incorporated into the lungs following ingestion; research on ingested V has largely focused on its use in the management of diabetes (7, 21–23). In those studies, V+5 was found to decrease water intake and blood glucose levels at 100–800 ppm level (21, 23). It was also found that the glutamic pyruvic transaminase, glutamic oxaloacetate transaminase, blood urea nitrogen, triglyceride, high-density lipoprotein, and total cholesterol levels in plasma were lower in groups receiving V+5 at 100 ppm (21). Nonetheless, V+5 intake through dietary and environmental exposure has greatly raised concerns over its toxic effects in humans recently (24), likely attributed to its strong oxidative power (25–28) or action as a phosphate analog (2, 29). Ingested V+5 will still impact the lungs through deposition from the blood. In our recent in vitro study, we found that V+5 caused lung fibroblast senescence through impairing glutathione (GSH) and protein thiols redox homeostasis (28), indicating that dietary V+5 exposure at an environmentally relevant level could contribute to lung fibrosis (30–34). It is therefore imperative to systematically investigate the potential toxicological effects in vivo and compare it to effects on lung fibroblasts following low-dose V+5 exposure.
In this study, we conducted a detailed metabolomic analysis with lungs from V+5-exposed male mice and human lung fibroblasts (HLFs). Earlier studies reported that higher estrogen levels in female mice may protect against oxidative stress induced by V+5 exposure (35, 36), showing noticeable pulmonary inflammation (37), reproductive toxicity (38), genotoxicity (38, 39), and carcinogenicity (37) in male mice receiving V+5 through inhalation. However, little information is available on the effect of V+5 on the lung metabolism and physiology through ingestion. Therefore, we focus on male mice in the current study to investigate metabolic perturbations induced by ingested V+5 without counteracting effects from high estrogen. The calculated V+5 intake in mouse through drinking water was less than 1 µg/day, which was below the estimated dietary intake of V (∼10 µg/day; 5, 6), and much lower than the tolerable upper intake level for V (1.8 mg/day; 8). Liquid chromatography-high-resolution mass spectrometry (LC-HRMS) was utilized in this study to profile the metabolic responses induced by V+5 exposure in both mouse lungs and HLF cells. Importantly, LC-HRMS provides a sensitive way to detect early effects on critical pulmonary responses against V+5 at low noncytotoxic exposure levels. Our study provides a foundation for linking lung fibroblast metabolic responses to that of whole lung tissue exposed to V+5, and to help understand relevant mechanisms in pulmonary inflammation, fibrosis, and associated respiratory disorders.
MATERIALS AND METHODS
Chemicals
Vanadium pentoxide (V+5, Cat. No. 22189), trichloroacetic acid (T6399), acetonitrile (34998), sodium iodoacetate (19148), KOH (417661), NaOH (221465), potassium tetraborate (P5754), chloroform (650498), and dansyl chloride (D2625) were purchased from Sigma-Aldrich (St. Louis, MO). Nitric acid (NX0409) was purchased from MilliporeSigma (Burlington, MA). Dithiothreitol (DTT, 161-0611) was purchased from Bio-Rad (Hercules, CA). γ-Glutamylglutamate (γ-Glu-Glu, AC228812500), perchloric acid (A2296), and boric acid (A73-1) were purchased from Fisher Scientific (Fair Lawn, NJ).
An internal standard mixture for LC-HRMS consisted of [13C6]-d-glucose (CLM-1396), [15N]-indole (NLM-792), [2-15N]-l-lysine dihydrochloride (NLM-143), [13C5]-l-glutamic acid (CLM-1800-H), [3,4-13C2]-cholesterol (CLM-792), [15N]-l-tyrosine (NLM-590), [trimethyl-13C3]-caffeine (CLM-514), [15N2]-uracil (NLM-637), [3,3-13C2]-cystine (CLM-520), [1,2-13C2]-palmitic acid (CLM-214), and [15N, 13C5]-l-methionine (CNLM-759-H) were purchased from Cambridge Isotope Laboratories, Inc (Andover, PA). [13C7]-benzoic acid (586234) and [15N]-choline chloride (609269) were purchased from Sigma-Aldrich. All reagents were analytical grade or above unless otherwise stated. All reagents were analytical grade or above unless otherwise stated.
Cell Culture
HLF was purchased from the American Type Culture Collection [HFL1 (CCL-153), passages 6–9, ATCC, Rockville, MD]. HLFs were cultured using F-12K medium (Kaighn’s Modification of Ham’s F-12 medium) with 10% fetal bovine serum (FBS) and 100 U/mL penicillin-streptomycin and maintained in a humidified incubator at 5% CO2 and 37°C. Once cells reached 80% confluency, cell medium was replaced with low FBS (0.5% FBS) media containing V+5 (0, 0.01, 0.1, or 1 ppm). Low FBS medium was used during treatments to prevent potential interaction of V+5 with serum albumin. Exposures were performed for 24 h, based on previous studies (28). Cells were collected immediately after 24 h of exposure for the assays described in High-Resolution Metabolomics; Transmission Electron Microscopy; GSH, GSSG, and Redox State Measurement; and Protein S-Glutathionylation. This time point assured that metabolomics and other studies were performed on viable cells but do not exclude longer-term effects on cell viability.
Animal Care and V+5 Exposure
Experimental protocols for animal studies were approved by Emory University Institutional Animal Care and Use Committees, and experiments were performed in accordance with the guidelines and regulations. Male C57BL/6J mice were purchased from Jackson Laboratory (Bar Harbor, ME) at 6 wk old and housed in clean facilities. Mice were fed a standardized mouse diet (Laboratory Rodent Diet 5001, LabDiet, St Louis, MO) to ensure control for any effects from dietary components on V toxicity (40). Beginning at 7 wk of age, mice were exposed to V+5 in sterile-filtered drinking water at 0, 0.02, 0.2, and 2 ppm for 7 mo, with three mice treated per exposure. Water intake and mouse weights were monitored on a weekly basis.
Quantification of V Deposit in Mouse Lung, Liver, and Kidney
51V deposition in mouse lung, liver, and kidney tissue was quantified using inductively coupled plasma mass spectrometry (ICP-MS, iCap Q, Thermo Fisher Scientific, Waltham, MA). Accuracy (100 ± 10%) and precision standards (RSD < 12%) were maintained. Briefly, ∼60 mg tissues were collected and digested in 45% nitric acid and 10% hydrogen peroxide using a programmable 1,200 W microwave (MARS 5, CEM Corp., Matthews, NC) with a rotor for 40 Teflon-lined vessels rated at 210°C and 350 psi (HP-500 Plus, CEM Corp., Matthews, NC). A standard curve was generated by serial dilution of V standard (1,000 ppm, 2% HNO3, Cat. No. CGV1, Inorganic Ventures, Christiansburg, VA).
High-Resolution Metabolomics
Mouse tissue (∼20 mg) and HLF cells from six-well plates were collected in 300 µL acetonitrile: water (2:1) containing internal standards following the procedures as described previously (41, 42). Samples were randomized and analyzed using a Q Exactive HF Hybrid Quadrupole Orbitrap mass spectrometer (120,000 resolution, 85–1,275 m/z, Thermo Fisher, Waltham, MA), coupled with hydrophilic interaction liquid chromatography [HILIC (+)] and C18 reversed phase chromatography paired with negative electrospray ionization [C18 (−)]. Each sample was run in triplicate in batches of 40 samples. Metabolic features comprised of mass-to-charge ratio (m/z) and retention time (rt) were extracted using apLCMS (43) and xMSanalyzer (44). Data were median-summarized among replicates. Data were filtered to retain features with nonzero values in greater than 70% of all samples and greater than 80% in each group, log2 transformed and quantile normalized. NIST SRM1950 and Q-Standard 3 (Qstd3) were run alongside samples as reference standards for quality control and quality assurance, as described previously (45).
Metabolomics Data Analysis
Statistical significance of metabolic features between groups was computed using one-way ANOVA (LIMMA) and partial least squares (PLS)-based feature selection using xmsPANDA (https://github.com/kuppal2/xmsPANDA, last accessed July 15, 2022). Statistical P values and PLS variable importance on projection (VIP) scores were used to select the most important features. Untargeted analysis of the significant features differentiating treatment groups (P < 0.05 and VIP > 2) was performed by hierarchical clustering analysis (HCA), partial least-squares discriminant analysis (PLS-DA), and correlation analysis using MetaboAnalyst (46).
Metabolic Pathway Enrichment Analysis
Significant metabolic features (P < 0.05 and VIP > 2) were further studied by pathway enrichment analysis using mummichog (47). Permutation testing was performed using 1,000 permutations to protect against type 1 statistical error (48). Analysis was performed using 5 ppm mass error tolerance and enforcing the presence of primary ions (M+H) and (M−H) for positive and negative ionization modes, respectively.
Metabolite Annotation and Identification
Metabolite annotation was carried out using xMSannotator (48). xMSannotator uses a multilevel scoring algorithm to determine annotation of metabolic features. xMSannotator generates annotations with a confidence score from 0 (no confidence) to 3 (high confidence), based on adduct/isotope patterns, adduct coelution, and elemental or abundance ratio checks. Annotations were made against Kyoto Encyclopedia of Genes and Genomes (https://www.kegg.jp, last accessed July 22, 2022) and the Human Metabolome Database (http://www.hmdb.ca/ last accessed July 22, 2022). Only annotated metabolites with a confidence score of two or greater were selected for further analyses, and considered as “putatively annotated” compounds (49).
Mitochondrial Function Assessment
HLF bioenergetics and mitochondrial function were determined using Seahorse extracellular flux analyzer (Seahorse Bioscience, MA), as previously described (50). HLF cells were diluted to approximately 107 cells/mL in 75 μL XF DMEM assay buffer (DMEM with 1 mM pyruvate, 10 mM d-glucose, 2 mM l-glutamine, and HEPES, pH 7.4) and were seeded onto XF96 microplates. The plate was then transferred to the XFe96 extracellular flux analyzer, and after an oxygen consumption rate (OCR) baseline measurement, three sequential injections of compounds that affect bioenergetics were performed, as follows: 25 μL of oligomycin (1 μM), 25 μL of carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (0.6 μM), and 25 μL of antimycin A/rotenone (5 μM) at injection in port C. Each treatment was carried out in 12 replicates, normalized to protein via the bicinchoninic acid (BCA) protein assay, and the results were expressed as pmol O2 consumed/min/µg protein.
Transmission Electron Microscopy
Mouse lung tissues and HLF cells were washed with PBS and fixed in Karnovsky’s fixative overnight. Postfixation was carried out using 1% osmium tetroxide in 0.1 M phosphate buffer (pH 7.4) for 1 h. Mouse lung tissues and HLF cells (a cell monolayer grown in six-well plates) were dehydrated via graded ethanol, infiltrated, embedded, and then polymerized in Eponate 12 resin (Ted Pella Inc., Redding, CA). Ultrathin sections (80 nm) were processed using a Leica-Reichert Ultracut S ultramicrotome (Leica, Deerfield, IL). Ultrathin sections were stained with 5% uranyl acetate and 2% lead citrate and imaged on a JEOL JEM-1400 TEM (JEOL Ltd., Japan) equipped with a Gatan US1000 2k × 2k high-resolution charge-coupled device (CCD) camera (Gatan, Pleasanton, CA) at 80 kV. Three mouse lung tissues per group were randomly selected for transmission electron microscopy (TEM) imaging.
GSH, GSSG, and Redox State Measurement
Fresh mouse lung tissues and HLF cells were collected, extracted, and quantified for GSH and glutathione disulfide (GSSG) using high-performance liquid chromatography (HPLC) with a gradient HPLC module 2695 (Waters, Milford, MA). A Waters 2475 fluorescence detector (Waters, Milford, MA) was used at a 335 nm excitation wavelength and 518 nm emission wavelength of 518 nm. Chromatography was performed using a SUPELCOSIL LC-NH2 HPLC Column (25 cm × 4.6 mm, 5 μm), and data were captured using Empower software, as previously described (51). Briefly, mouse lung tissues were placed in 350 μL ice-cold 5% perchloric acid with 0.2 M boric acid and 10 μM γ-Glu-Glu internal standard, and homogenized using an Active Motif Q120AM probe sonicator (Carlsbad, CA). After rinsing three times in ice-cold PBS, HLF cells were collected in 375 μL ice-cold 5% perchloric acid containing 0.2 M boric acid and 10 μM γ-Glu-Glu internal standard. Lung homogenate and cell lysate were centrifuged at 16,000 g, 4°C for 5 min. The supernatant (300 μL) was then transferred to new tubes, and 60 μL of 50 mM sodium iodoacetate was added at room temperature. pH was adjusted to nine using KOH-tetraborate (1 M KOH with 0.38 M potassium tetraborate), and samples were incubated at room temperature for 20 min. Three-hundred microliter of 75 mM dansyl chloride (in acetone) were added, and all tubes were left in the dark at room temperature overnight. Chloroform (500 μL) was then added into the solution. Samples were vortexed and centrifuged at 14,000 g, 4°C for 2 min. Aqueous fraction (50 μL) was injected, and concentrations of GSH and GSSG were calculated relative to the internal standard γ-Glu-Glu and normalized to the protein concentration. The individual concentrations were used with the Nernst equation to calculate redox potentials for GSH/GSSG (EhGSSG/GSH) with E0 = −240 mV, pH 7.0.
Protein S-Glutathionylation
Protein S-glutathionylation was quantified by measuring protein-bound GSH relative to total protein concentration, as previously described (52). For mouse lungs, tissue pellets were saved after removing lysate for GSH and GSSG measurements. Protein was precipitated from pellets using 10% trichloroacetic acid and centrifugation at 16,100 g for 5 min, at 4°C. After rinsing with ice-cold PBS, HLF cell lysates were collected in 200 μL of 10% trichloroacetic acid and centrifuged at 16,800 g for 5 min, at 4°C. Mouse lung tissue pellets and cell pellets were then washed in 25% trichloroacetic acid. After removing the supernatant by centrifuging at 16,100 g for 5 min, at 4°C, mouse lung and cell proteins were resolubilized in 200 μL of 1 M NaOH. Sample (125 μL) was mixed 1:1 with 5 mM DTT in 0.1 M sodium phosphate buffer (pH 6) and incubated for 30 min at room temperature. Reduced protein was reprecipitated by mixing samples 1:1 with 10% perchloric acid containing 0.2 M boric acid and 10 μM γ-Glu-Glu internal standard. GSH released from protein was quantified by HPLC with fluorescence detection (Waters 2695, Milford, MA), as described earlier. Five microliter solubilized protein was analyzed for protein concentration using the Pierce BCA protein assay.
BCA Protein Assay
Protein concentrations were determined using 5 μL of samples using Pierce BCA Protein Kit (Thermo Scientific) in a 96-well plate reader. After addition of 200 μL of working reagent to each well, mixing and incubation for 30 min, absorbance was measured at 562 nm (SpectraMax M2, Molecular Devices, San Jose, CA). A protein standard curve was generated using a serial dilution of bovine serum albumin standard.
Statistics
Data analyses were carried out in R and Maltab R2021a (MathWorks, Inc., Natick, MA). Graphs were generated using OriginPro 2021 b (OriginLab Corp., Northampton, MA). Results are presented as means ± standard deviation. One-way ANOVA with post hoc testing using Fisher’s least significant difference was performed to obtain statistical significance. Statistical P values < 0.05 were considered statistically significant, and P < 0.001 were extremely significant.
RESULTS
Metabolic Perturbations of V+5 Exposure in Human Lung Fibroblasts
Prior studies showed that exposures of HLF to concentrations of V+5 up to 1 ppm caused minimal to no measurable cell death (28). Because of potential effects on cell number, we used a WST-1 (also known as 4-[3-(4-iodophenyl)-2-(4-nitrophenyl)-2H-5-tetrazolio]-1,3-benzene disulfonate) assay to determine whether this range of V+5 concentration impacted cell proliferation. Results showed an apparent dose-response in cell survival and proliferation with V+5 exposure at low levels (0, 0.01, 0.1, and 1 ppm, Supplemental Fig. S1), with a 7% increase in cell proliferation in HLF exposed to 0.1 ppm V+5, and a decrease (7%) at the highest V+5 dose (1 ppm). Because these effects on cell number over the time course of incubation were small relative to other sources of experimental variation and did not appear to impact directionality of metabolic changes (see Fig. 2), we did not adjust for these differences in subsequent analyses.
Figure 2.

Alterations in metabolic functions of HLF induced by V+5 exposure. A: features with significance (P < 0.05) or PLS importance [variable importance on projection (VIP) > 2] from HILIC (+) and C18 (−) chromatography after data filtering in HLF cells exposed to V+5. B: PLS-DA of selected metabolites from HILIC (+) chromatography altered by V+5 exposure at P < 0.05 and VIP > 2 in HLF cells. C: HCA of metabolites from HILIC (+) chromatography altered by V+5 exposure at P < 0.05 and VIP > 2, showing two distinct clusters I and II. Warm color and positive z-score indicate higher abundance (C). D: pathway analysis revealed a total of 32 pathways, which were enriched in HLF cells exposed to V+5. Red color in D indicates shared pathways by mouse lung tissue and HLF cells exposed to V+5. Differences in metabolomes between groups were detected using an empirical Bayes extension of a one-way ANOVA design with limma. No post hoc testing was performed. Bars represent −log-transformed P values, resulting from pathway enrichment analysis with n = 5–12/group. HCA, hierarchical clustering analysis; HILIC (+), hydrophilic interaction liquid chromatography; HLF, human lung fibroblasts; PLS, partial least squares; PLS-DA, partial least-squares discriminant analysis; V+5, vanadium pentoxide.
HRM was used to investigate the metabolic responses of HLF cells to low-level V+5 exposure (0.01, 0.1, and 1 ppm). A total of 11516 HILIC (+) features and 9209 C18 (–) features were obtained for HLF cells exposed to V+5 (Fig. 2A). ANOVA (see Supplemental Fig. S2 for Manhattan plots) and VIP scores from the PLS model were used to select mass spectral features with P < 0.05 and VIP > 2, resulting in 391 [HILIC (+)] and 281 [C18 (–)] metabolites changed by V+5 in HLF cells by these criteria (Fig. 2A). PLS-DA showed that selected metabolites separated groups of samples with different V+5 exposures (Fig. 2B), and HCA-heatmap also showed separation into two clusters of metabolites according to directionality of change, with a greater number of metabolites decreased by V+5 than the number that increased (Fig. 2C).
Pathway enrichment analyses showed widespread effects on metabolism, with 32 pathways altered (P < 0.05) in association with V+5 treatment (Fig. 2D). The top three pathways included aminosugars (P = 9.2 × 10−5), tryptophan (P = 1.8 × 10−4), and urea cycle/amino group metabolism (P = 4.7 × 10−4). Besides tryptophan, a large number of other amino acid pathways were affected, including histidine (P = 5.2 × 10−4), alanine and aspartate (P = 6.1 × 10−4), glycine, serine, alanine, and threonine (P = 7.1 × 10−4), β-alanine (P = 0.002), glutamate (P = 0.003), arginine and proline (P = 0.003), tyrosine (P = 0.004), methionine and cysteine (P = 0.008), aspartate and asparagine (P = 0.01), and lysine (P = 0.04). Other pathways included both nucleotide metabolism pathways for pyrimidine (P = 0.0018) and purine (P = 0.016), and folate metabolism (P = 0.018). These pathways support cell repair and regeneration as well as metabolism of fatty acids, carbohydrates, and amino acids, and have well-established cellular functions relevant to human health (53, 54). Changes were also present in nucleotide sugar metabolism (P = 0.019), indicating metabolic disruption in glycan metabolism, and potential effects on cell signaling (55) and energy metabolism (56). Disruption in energy metabolism is also indicated by alterations in vitamin B3 (niacin; P = 0.008), hexose phosphorylation (P = 0.03), glycolysis (P = 0.028), and TCA cycle (P = 0.004), pathways commonly disrupted in fibrotic lungs (57). A series of carbohydrate pathways was also changed by V+5 exposures, including sialic acid (P = 0.005), galactose (P = 0.03), glyoxylate and dicarboxylate (P = 0.03), dibasic acid (P = 0.037), starch and sucrose (P = 0.045), further indicating a systematic disruption in cellular energy metabolism and cell functioning (58). Furthermore, glycerophospholipid (P = 0.008) metabolism was also affected by V+5, indicating metabolic disturbance in cell membrane functioning (59). Alterations in redox pathways were seen in neuroprostanes (P = 0.045) and glutathione (P = 0.02) metabolism, which are often involved in regulating cellular redox homeostasis and redox signal transduction, and used as biomarkers for oxidative stress (60, 61). Finally, drug metabolism (P = 0.02), nitrogen metabolism (P = 6 × 10−4), and carbon fixation pathways (P = 0.03) were also affected in HLF cells exposed to V+5.
Mouse Model to Study V+5 Effects on Lung Metabolism In Vivo
To determine whether similar effects of V+5 occur in lungs in vivo, a small study was designed to treat mice with 0, 0.02, 0.2, or 2 ppm V+5 in drinking water. Mouse weight gain (Fig. 3A) was less relative to controls in all three treatment groups, showing 2.6-, 2.2-, and 2.6-fold increase in mice treated with 0.02, 0.2, and 2 ppm V+5, respectively, compared with threefold increase in control mice (Fig. 3B). The result of suppressed body weight gain by V+5 was consistent with earlier studies (9, 21, 62, 63). Because V+5 was delivered via drinking water, we also monitored the water consumption weekly to track the actual V intake weekly from the given V doses. There was no measured difference in water consumption between V-treated mice and controls (Fig. 3C). V+5 intake via drinking water was higher in the first few weeks, due to the higher water consumption per mouse body weight (Fig. 3D). The V intake reached a relatively stable target after 10 wk, showing ∼0.03 mg V/d/kg body weight for those given 2 ppm V+5. This is below the no-observed-adverse-effect level (NOAEL) (10.5 mg V/d/kg body weight) and lowest-observed-adverse-effect (LOAEL) (16.4 mg V/d/kg body weight) values identified following oral exposure to V2O5 estimated by US EPA (64), or NOAEL (1–17 mg V/d/kg body weight) and LOAEL (2.1–25 mg V/d/kg body weight), varying on different endpoints, for rats and mice through intermediate-duration exposures.
Figure 3.

Body weight changes and tissue vanadium contents in mice receiving V+5 in drinking water. Weekly mouse body weight by treatment groups (A) and the fold of body weight changes at the end of exposure (B). C: weekly water consumption by treatment group. D: calculated daily V intake throughout the experiment. V content in lung (E), liver (F), and kidney (G) measured by ICP-MS. Data are presented as means ± standard deviation. Statistical analyses were performed using the one-way ANOVA with Fisher’s least significant difference post hoc testing. Statistical significance for each condition vs. control is indicated: *P < 0.05, **P < 0.01, and ***P < 0.001; n = 3/group. ICP-MS, inductively coupled plasma mass spectrometry; V, vanadium; V+5, vanadium pentoxide.
We determined the impact of oral V+5 exposure on mouse lung V+5 content using ICP-MS analysis. Lung V contents were substantially higher in mice given V in drinking water compared with no V (Fig. 3E), showing 1.3-, 2.5-, and 4.4-fold increase in lung V content treated with 0.02, 0.2, and 2 ppm V+5, respectively. Considering the measured V content in mouse food (0.5 ppm), the administered V in drinking water increased total V intake (food + water) by 30%, 150%, and 340% at the administered drinking water contents. Thus, one can suggest that the bioavailability of V+5 from drinking water was greater than that from food; none-the-less, the overall lung accumulation of V did not reflect the difference between the lowest and highest V+5 doses administered (i.e., approximately sixfold compared with the 100-fold difference between 0.02 and 2 ppm). In comparison, liver (Fig. 3F) and kidney (Fig. 3G) showed a greater increase in tissue V content with increased content in drinking water, suggesting important functions of liver and kidney in V distribution and homeostasis.
Metabolic Perturbations to V+5 Exposure in Mouse Lung
HRM of lungs from mice treated with 0, 0.02, 0.2, and 2 ppm V+5 in drinking water for 7 mo yielded 20,018 and 19,592 metabolic features for HILIC (+) and C18 (−) chromatography after data filtering (Fig. 4A). Selection of top discriminatory features by ANOVA P < 0.05 and VIP > 2 for PLS provided 305 [HILIC (+)] and 252 [C18 (−)] metabolites altered by V+5 in mouse lung tissues (Fig. 4A). Associated Manhattan plots are available in Supplemental Fig. S2. PLS-DA showed that the separation between V+5 exposures and vehicle controls was mostly significant in mouse lung tissues (Fig. 4B), and HCA-heatmap showed separation of the selected HILIC (+) metabolites from mouse lung tissues into subclusters with different directionalities in intensity with increasing V+5 exposure (Fig. 4C).
Figure 4.

Alterations in metabolic functions of mouse lungs induced by V+5 exposure. A: features with significance (P < 0.05) or PLS importance [variable importance on projection (VIP) > 2] from HILIC (+) and C18 (–) chromatography after data filtering in mouse lung tissue exposed to V+5. B: PLS-DA of selected metabolites from HILIC (+) chromatography altered by V+5 exposure at P < 0.05 and VIP > 2 in mouse lung tissue. C: HCA of metabolites from HILIC (+) chromatography altered by V+5 exposure at P < 0.05 and VIP > 2 in mouse lung tissue. Warm color and positive z-score indicates higher abundance (C). D: pathway analysis revealed a total of 21 pathways, which were enriched in HLF cells exposed to V+5. Red color in D indicates shared pathways by mouse lung tissue and HLF cells exposed to V+5. Differences in metabolomes between groups were detected using an empirical Bayes extension of a one-way ANOVA design with limma. No post hoc testing was performed. Bars represent −log-transformed P values, resulting from pathway enrichment analysis with n = 3/group. HCA, hierarchical clustering analysis; HILIC (+), hydrophilic interaction liquid chromatography; HLF, human lung fibroblasts; PLS, partial least squares; PLS-DA, partial least-squares discriminant analysis; V+5, vanadium pentoxide.
Pathway enrichment analyses showed 21 metabolic pathways were altered (P < 0.05) in lungs in association with in vivo V+5 exposure (Fig. 4D). Pyrimidine (P = 7 × 10−4), C21-steroid biosynthesis (P = 0.007), and fatty acid β-oxidation (P = 0.008) metabolism were the top three altered metabolic pathways. Other fatty acid pathways included fatty acid activation (P = 0.033), linoleate (P = 0.035), phytanic acid oxidation (P = 0.039), de novo fatty acid biosynthesis (P = 0.042), arachidonic acid (P = 0.04), and leukotriene (P = 0.014). In addition, aminosugars (P = 0.028) were also affected by V+5 exposure, indicating a strong metabolic disturbance in cell membrane functioning (59). Other pathways were changed in mouse lung that were also associated with V+5 exposure in HLF cells, including pyrimidine and purine (P = 0.02) pathways associated with metabolic signaling and cell growth (65, 66). Changes in major amino acid [histidine (P = 0.015), methionine and cysteine (P = 0.019), aspartate and asparagine (P = 0.04)], and pyruvate (P = 0.02), thiamine (P = 0.025), ubiquinone (P = 0.048), and ascorbate and aldarate (P = 0.014) pathways also suggest potential impact on systems with critical roles in growth, development and cell function (67–70), and antioxidant defenses (71). Finally, xenobiotic metabolism (P = 0.027), commonly associated with environmental exposure (72), was also affected by V+5 exposure.
Lung fibroblast activation is an important factor associated with IPF pathogenesis (73, 74), but the large number of shared pathways for V+5 effects in HLF and mouse lung was not anticipated because there are more than 40 cell types in lung. We selected 1,902 metabolites annotated by xMSannotator with confidence score ≥ 2 to test for V+5-associated metabolites common to mouse lung tissues and HLF cells (Supplemental Fig. S3A). ANOVA simultaneous component analysis (ASCA) explained 44% of the variation corresponding to the factor “tissue type × dose” with a clear dose-response pattern in both mouse lung tissue and HLF cells exposed to V+5 (Supplemental Fig. S3B). This evidence that mouse lung and HLF cells share metabolites altered by V+5 exposure prompted us to look at specific metabolites in more detail. We selected pathways previously linked to human lung diseases, i.e., pyrimidine, fatty acid, and energy-related pathways (75) to more specifically examine metabolic changes common to HLF and mouse lungs.
Disruption of Pyrimidine Metabolism
As noted in the pathway enrichment analyses, pyrimidine metabolism was significantly altered in both HLF cells (Fig. 2D) and mouse lung (Fig. 4D). Major metabolites involved in the pyrimidine pathway include orotic acid, cytidine monophosphate (CMP), cytidine triphosphate (CTP), uridine monophosphate (UMP), uridine diphosphate (UDP) glucuronate, uridine, and 5,6-dihydrouracil (Fig. 5A). Feeding into the pathway, orotic acid was decreased in both HLF and mouse lung (not shown), perhaps reflecting the sensitivity of preceding reactions to mitochondrial oxidative stress (28). Other metabolites showed increasing trends in response to increasing V+5 both in HLF cells (Fig. 5, B–F) and mouse lung (Fig. 5, G–K), except for CTP (Fig. 5, E and J), which was increased in HLF (Fig. 5E) and decreased in mouse lung (Fig. 5J). Significant increases in metabolite levels were observed in UDP glucuronate (Fig. 5B), uracil (Fig. 5C), 5,6-dihydrouracil (Fig. 5, D and I), CTP (Fig. 5E), and CMP (Fig. 5K). Pyrimidine metabolites play important roles in nucleic acid homeostasis and energy metabolism (65, 66) as well as providing CDP-choline as a precursor for phospholipid biosynthesis, UDP-sugars for glucuronidation and oligosaccharide biosynthesis, and dihydrouracil for β-alanine biosynthesis, as shown in the trending UDP glucuronate levels (Fig. 5, B and G). Notably, the trends of CMP (Fig. 5, F and K), uracil (Fig. 5, C and H), and 5,6-dihydrouracil (Fig. 5, D and I) with V+5 exposure in both mouse lung tissues and HLF cells, could reflect decreased activities of related processes functioning in repair and tissue homeostasis.
Figure 5.

Metabolic disruption in pyrimidine metabolism altered by V+5 exposure. A: major metabolites in pyrimidine metabolism affected by V+5 exposure. Directional changes are shown for metabolites from mouse lungs. Selected metabolites are individually plotted in B–K: UDP glucuronate (M − H) (B), UDP glucuronate [M − H(−1)] (G); uracil (M + H) (C), uracil (M − H) (H); 5,6-dihydrouracil (M − H) (D and I); CTP (M − H) (E and J); CMP (M + H) (F), CMP (M − H) (K). B–E are from human lung fibroblast, G–K are from mouse lung tissue. Data are presented as means ± standard deviation. Statistical analyses were performed using the one-way ANOVA with Fisher’s least significant difference post hoc testing. Statistical significance for each condition vs. control is indicated: *P < 0.05, **P < 0.01, and ***P < 0.001; n = 5–12 for human lung fibroblast, n = 3 for mouse lung tissue. M + H, positive mode; M − H, negative mode. CMP, cytidine monophosphate; CTP, cytidine triphosphate; UDP, uridine diphosphate; V+5, vanadium pentoxide.
Disruption of Lipid and Fatty Acid Metabolism
Fatty acid and lipid pathways were changed in both HLF (Fig. 2D) and mouse lung (Fig. 4D) following V+5 exposure (Fig. 6A). Significant decreases in metabolite levels were observed in linoleic acid (Fig. 6G), prostaglandins (Fig. 6I), sphingosine (Fig. 6J), and ceramides (Fig. 6K), whereas significant increases were observed in leukotrienes (Fig. 6, C and H). V+5-dose-dependent decrease in linoleic acid (Fig. 6G) is representative of free fatty acids in mouse lung tissue, with a similar trend observed in HLF cells (Fig. 6B). Changes in leukotrienes and prostaglandins are well-known to be associated with IPF (76–79). Leukotriene A4 in HLF (Fig. 6C) and N-acetyl-leukotriene E4 in mouse lung tissue (Fig. 6H) were increased with V+5. An increase in leukotriene C4 was also observed in both mouse lung tissue (m/z 624.2935, rt 182 s, M − H) and HLF (m/z 626.3093, rt 157 s, M + H; not shown). Decreases in prostaglandin D1/E1/F2b/H1 in HLF (Fig. 6D) and prostaglandin D2/E2/F3a/H2/I2 in mouse lung (Fig. 6I) occurred in response to V+5, along with other metabolic features matching prostaglandins in mouse lung, including prostaglandins J2/A2/B2/C2 (m/z 335.2214, rt 22 s, M + H; m/z 317.2109, rt 24 s, M + H-H2O; m/z 333.2078, rt 23 s, M − H), E3/D3 (m/z 351.2165, rt 24 s, M + H; m/z 349.2029, rt 23 s, M − H), E1 (m/z 353.234, rt 23 s, M − H), A1/C1 (m/z 335.2234, rt 23 s, M + H), and G1 (m/z 369.2292, rt 22 s, M − H).
Figure 6.

Metabolic disruption in lipid and fatty acid metabolism altered by V+5 exposure. A: major metabolites in pyrimidine metabolism affected by V+5 exposure. Directional changes are shown for metabolites from mouse lungs. Selected metabolites are individually plotted in B–K: linolenic acid (M − H) (B), linolenic acid [M − H(+3)] (G); leukotrienes A4 (M + H) (C), N-acetyl-leukotriene E4 (M − H) (H); prostaglandins D1/E1/F2b/H1 (M − H) (D), prostaglandins D2/E2/F3a/H2/I2 (M − H) (I); sphingosine (M + H) (E and J); ceramide (d18:1,16:0) (M + H) (F), ceramide (d18:1,20:0) (M + H) (K). B–E are from human lung fibroblast, G–K are from mouse lung tissue. Data are presented as means ± standard deviation. Statistical analyses were performed using the one-way ANOVA with Fisher’s least significant difference post hoc testing. Statistical significance for each condition vs. control is indicated: *P < 0.05, and **P < 0.01; n = 5–12 for human lung fibroblast, n = 3 for mouse lung tissue. M + H, positive mode; M − H, negative mode. V+5, vanadium pentoxide.
Other lipids also decreased in HLF and mouse lung with V+5, including sphingosine, ceramide, carnitine, and acyl-carnitines, illustrated by changes in sphingosine (Fig. 6J) and mass spectral signals matching ceramide (d18:1,20:0) in mouse lung (Fig. 6K). Similar nonsignificant trends were observed in HLF (Fig. 6, E and F). Carnitine (m/z 162.1125, rt 153 s, M + H) and acetylcarnitine (m/z 204.1230, rt 128 s, M + H) decreased in HLF exposed to V+5, whereas changes in mouse lung included different responses for short chain and long chain acylcarnitines (not shown).
Disruption of Glycolysis, TCA Cycle, Redox Pathway, and Energy Metabolism
Following exposure to V+5, metabolic perturbations in glycolysis and TCA cycle were observed in HLF, and redox pathway and energy pathway changes were observed in both HLF and mouse lung (Fig. 2D and Fig. 4D). Selected metabolites from glycolysis, TCA cycle, redox pathway, and energy metabolism are illustrated in Fig. 7A. A significant decrease occurred in HLF phosphocreatine (Fig. 7D) and mouse pyruvate (Fig. 7J), whereas a significant increase in metabolite level was observed in 4-hydroxyproline (Fig. 7, C and H), HLF pyruvate (Fig. 7E), and HLF citrate (Fig. 7F). Albeit nonsignificant, the decreasing trend observed in GSH in both HLF and mouse lungs (Fig. 7, B and G) was consistent with prior evidence that V+5 reacts directly with GSH and causes decreased values due to oxidation (28). Increased levels of 4-hydroxyproline were seen in both mouse lung tissue and HLF treated with V+5 (Fig. 7, C and H), suggesting changes that could be related to collagen deposition and fibrosis signaling (80–82). Phosphocreatine significantly decreased with V+5 exposure in HLF cells, and a similar, nonsignificant trend occurred in mouse lung (Fig. 7, D and I). Other metabolites differed in direction of change with V+5, and some differed in direction for HLF and lung. For instance, pyruvate (Fig. 7, E and J) and citrate (Fig. 7, F and K) increased significantly in HLF but had a nonsignificant decreasing trend in mouse lung. Other decreased metabolites (not shown) in HLF included hexose phosphate (m/z 261.0369, rt 218 s, M + H), aspartate (m/z 132.0303, rt 49 s, M − H), proline (m/z 114.0561, rt 58 s, M − H), citrulline (m/z 176.1029, rt 189 s, M + H), and fumarate (m/z 115.0038, rt 287 s, M + H), and in mouse lung included hexose 6-phosphate (m/z 261.0370, rt 115 s, M + H), hexose biphosphate (m/z 341.0032, rt 294 s, M + H), glutamine (m/z 147.0764, rt 72 s, M + H), citrulline (m/z 176.1029, rt 80 s, M + H). Increased metabolites in HLF included hexose biphosphate (m/z 341.0033, rt 247 s, M + H), lactate (m/z 89.0245, rt 62 s, M − H), succinate (m/z 117.0194, rt 284 s, M − H), and malate (m/z 133.0143, rt 45 s, M − H), and in mouse lung, increased ornithine (m/z 133.0971, rt 139 s, M + H) and lactate (m/z 89.0243, rt 21 s, M − H).
Figure 7.

Metabolic disruption in glycolysis, TCA cycle, redox pathway, and energy metabolism altered by V+5 exposure. A: major metabolites affected by V+5 exposure. Directional changes are shown for metabolites from mouse lungs. Selected metabolites are individually plotted in B–K: GSH (M + H) (B and G); 4-hydroxyproline (M − H) (C), 4-hydroxyproline (M + H) (H); phosphocreatine (M + H) (D and I); pyruvate (M − H) (E and J); citrate (M − H) (F and K). B–E are from human lung fibroblast, G–K are from mouse lung tissue. Data are presented as means ± standard deviation. Statistical analyses were performed using the one-way ANOVA with Fisher’s least significant difference post hoc testing. Statistical significance for each condition vs. control is indicated: *P < 0.05, **P < 0.01, and ***P < 0.001; n = 5–12 for human lung fibroblast, n = 3 for mouse lung tissue. M + H, positive mode; M − H, negative mode. GSH, glutathione; V+5, vanadium pentoxide.
Dysregulation of Mitochondrial Function following Low-Dose V+5 Exposure
The changes in energy metabolism and TCA cycle intermediates following V+5 exposure suggested disruption in mitochondrial function. We thus investigated the effects of 24-h V+5 exposure on mitochondrial function in HLF cells using a Seahorse flux analyzer. Results indicated increased mitochondrial activity following exposure to V+5, with increase in basal respiration (Fig. 8A), ATP-dependent respiration (Fig. 8B), maximal respiration (Fig. 8C), spare respiration capacity (Fig. 8D), and nonmitochondrial respiration (Fig. 8E). This is in agreement with increased cell proliferation in HLF exposed to V+5 (Supplemental Fig. S1), as well as the increased levels in metabolites discussed earlier. Increase in proton leak was also observed in HLF cells exposed to V+5 (Fig. 8F), suggesting increased mitochondrial ion transport at this low-level exposure. Evidence for decreased cristae abundance and increased intercristal space was obtained from TEM analysis (see Fig. 9) of lung sections (Fig. 9C, orange arrows), with decreased electron density and unidentifiable structures (Fig. 9C, blue arrows) also apparent.
Figure 8.

Modulation of mitochondrial respiration by low-dose V+5 in HLF cells for 24 h. OCR was assessed using the Seahorse XF-24 extracellular flux analyzer. A: basal respiration. B: ATP-dependent respiration. C: maximal respiration. D: spare respiration capacity. E: nonmitochondrial respiration. F: proton leak. Data are presented as means ± standard deviation (n = 6–22). Statistical analyses were performed using the one-way ANOVA with Fisher’s least significant difference post hoc testing. Statistical significance for each condition vs. control is indicated: *P < 0.05, **P < 0.01, and ***P < 0.001. HLF, human lung fibroblasts; OCR, oxygen consumption rate; V+5, vanadium pentoxide.
Figure 9.

TEM analysis of mouse lung tissues. Control is shown in A with no apparent collagen deposition. B–D: massive collagen deposition was observed in mouse lung tissue following the exposure to V+5 at 2 ppm. B and C: large deposition of collagen is demonstrated in enclosed, dash red lines. B and D: red arrows indicate collagen deposition elsewhere. C shows disruption of mitochondrial cristae architecture (depicted by orange curves) with decreased cristae abundance and increased intercristal space (orange arrows), reduced electron density and unidentifiable structure (blue arrows). Normal mitochondria are indicated by yellow arrow with well-defined cristae (depicted by yellow curves) in A. D shows collagen production within lung fibroblast. Images displayed are representative of n = 3 lungs examined per group. Data are presented as means ± standard deviation. Statistical analyses were performed using the one-way ANOVA with Fisher’s least significant difference post hoc testing. AS, alveolar space; C, capillary; LF, lung fibroblast; N, nucleus; V+5, vanadium pentoxide.
Collagen Deposition in Mouse Lung Tissue Exposed to V+5
To examine whether the increased level in hydroxyproline is associated with stimulation of fibrosis signaling, we examined for collagen deposition in lungs as a profibrotic marker. We utilized TEM to examine the lung tissues from mice receiving 2 ppm V+5 (Fig. 9, B and D) compared with vehicle control lungs (Fig. 9A). Excessive collagen deposition is clinically associated with pulmonary fibrosis (80–82), and numerous studies have shown that collagen-producing lung fibroblasts play critical roles in the pathogenesis of pulmonary fibrosis (81–84). Thus, these pilot study results showing lung fibroblast collagen production (Fig. 9D) suggest that V+5 effects on lung fibroblasts could contribute to fibrosis processes through increased collagen deposition.
Oxidative Stress Induced by V+5 Exposure
Earlier studies showed that TGFβ, one of the most potent inducers of collagen deposition and plays a central role in the pathogenesis of pulmonary fibrosis (85), induces NADPH oxidase-4 in lung fibroblasts (86), implicating oxidative stress in the pathology of fibrosis. Indeed, oxidative stress in pulmonary fibrosis has been extensively studied and well-documented (87, 88). From our metabolomics analysis, we observed a nonsignificant trend of decreasing GSH levels in both HLF cells and mouse lung exposed to V+5, indicating that V+5 exposure could cause oxidative stress (Fig. 7, B and G). To investigate the disruption in redox homeostasis induced by V+5 exposure, we further examined the GSH/GSSG redox system in mouse lung tissue and HLF cells using high-performance liquid chromatography (28). The data showed that GSH was nonsignificantly decreased in HLF cells (Fig. 10A) and significantly decreased in mouse lung tissue (Fig. 10E). No change of glutathione disulfide (GSSG) was observed in HLF (Fig. 10B), but significantly decreased GSSG was observed at the highest V+5 exposure in mouse lungs (Fig. 10F). Changes in the redox potential for the GSSG/GSH couple showed a similar trend between HLF and mouse lung, with significant oxidation due to the highest V+5 dose in HLF cells (Fig. 10, C and G). Protein S-glutathionylation (PrSSG) was significantly elevated in HLF cells with all V+5 doses (Fig. 10D), but this was not observed in mouse lung tissue (Fig. 10H).
Figure 10.

GSH redox changes in human lung fibroblast (A–D) and mouse lung tissue (E–H) following the exposure to V+5. GSH (A and E). GSSG (B and F). EhGSSG/GSH (C and G). PrSSG (D and H). Data are presented as means ± standard deviation. Statistical analyses were performed using the one-way ANOVA with Fisher’s least significant difference post hoc testing. Statistical significance for each condition vs. control is indicated: *P < 0.05 and ***P < 0.001; n = 6 for human lung fibroblast, n = 3 for mouse lung tissue. V+5, vanadium pentoxide.
DISCUSSION
Increasing evidence shows that environmental and occupational exposure to V+5 poses risks to induce fibrotic lung injury (1, 18, 89). Experimental studies of inhaled V+5 show that V+5 deposits primarily in lungs (90) and causes oxidative stress (28, 37, 91), but the biology of common exposures at low levels through food and water has not been studied in detail. Based upon studies demonstrating that lung fibroblasts play a central role in lung fibrosis (74, 92), we previously examined the effects of V+5 exposures to HLF. Results showed that low-level V+5 causes GSH oxidation and induces growth arrest and cellular senescence (28). The results are consistent with other findings that HLF provides a useful in vitro model system to study the pathogenesis of lung fibrosis (93). The cumulative data show that V+5 can interfere with lung fibroblast growth and functions (91, 94, 95), potentially increasing susceptibility to lung fibrosis. In this study, we exposed HLF to V+5 at 0.01, 0.1, and 1 ppm for 24 h to examine metabolic perturbation of lung fibroblasts, the lowest of which approximates an acute occupational inhalation of V or accumulation of V in the lung from the diet (Fig. 1). A small, in vivo study was also designed to investigate the effects of V+5 in lungs, in which we exposed male mice to 0, 0.02, 0.2, and 2 ppm V+5 in drinking water for 7 mo, mimicking levels found in human diets and drinking water (Fig. 1). The doses used in vitro are higher than what would have reached the mouse lungs after ingestion, absorption, and distribution; however, metabolomics results demonstrate that similar effects were achieved with a chronic exposure in mice compared with an in vitro exposure performed within the time afforded by cell culture for a metabolomics study.
The present study provides a direct comparison between metabolic effects of V+5 in HLF with metabolic alterations in lungs of mice exposed to V+5 at relevant low exposure levels, thereby helping to bridge the knowledge gap between in vitro and in vivo effects following low-dose V+5 exposure. The quantified V intake by mice through drinking water and food was below the NOAEL and LOAEL values following oral exposure to V2O5 estimated by the US EPA (64). The NOAEL and LOAEL values estimated by the US EPA were based on decreased red blood cell counts in Wistar rats. Considering room for differences between rats and mice as well as the strength of untargeted metabolomics for detection of prepathologic outcomes, observation of effects at exposures below the estimated NOAEL is unsurprising. With only an estimated 2% increase in total V (food + water) due to addition of V+5 to drinking water, the lung V content was increased in mice. Thus, bioavailability of V+5 in drinking water is greater than forms of V in mouse food. In terms of total V burden after 7 mo treatment with 2 ppm V+5 in drinking water, levels in mouse lung were on the lower end of measured lung V in nondiseased individuals and patients with pulmonary diseases (MR Smith, DP Jones, YM Go, unpublished). Thus, mice exposed to V+5 at 2 ppm in drinking water for 7 mo are appropriate for studies of low-level exposure, but higher exposures or durations would also appear suitable to achieve tissue burdens comparable with human levels.
Untargeted metabolomics analyses showed that V+5 exposure induced widespread metabolic changes in both HLF cells and mouse lungs at these low exposure levels. Thirty percent of the pathways altered in mouse lung tissues overlapped with the ones from HLF cells, showing similar response characteristics in many pathways. Decreased abundance of glycolytic metabolites in mouse lungs is consistent with previous findings on glycolysis inhibition by vanadate (96). Observation of altered HLF cell proliferation (Supplemental Fig. S1) could associate with dysregulation of pyrimidine, glycolytic, and TCA cycle pathway and altered mitochondrial activities, e.g., increased proton leak and decreased respiratory efficiency (97). Altered mitochondrial structures in mouse lungs receiving V+5 further support a potentially compromised mitochondrial respiratory efficiency (98). Similarly, altered mitochondrial metabolism and activity were observed in IPF lung, e.g., disrupted glycolysis, TCA cycle, mitochondrial β-oxidation, phospholipid, sphingolipid, arginine, glutamate, and aspartate metabolism (99, 100). This suggests that V+5-caused metabolic disruption may contribute to subsequent pathological lung development such as IPF.
The findings from HLF align well with prior evidence for altered lung fibroblast proliferation associated with fibrotic processes leading to IPF (74, 84, 101, 102) and commonly seen in IPF lungs (74, 103). In particular, leukotrienes and prostaglandins as inflammatory markers play important roles in the pathogenesis of IPF (76–79), and previous research shows that human lung fibroblasts from patients with IPF showed diminished capacity to synthesize prostaglandins (76, 77, 104). Prostaglandin D2/E2/F3a/H2/I2, decreased in mouse lung and HLF, is one of the most abundant prostaglandins produced in the body via cyclooxygenase (COX)-1 and COX-2 from arachidonic acid, and plays important roles in regulating immune responses (105) and mediating anti-inflammatory actions in lungs (106–112). Leukotriene A4, increased in mouse lungs by V+5, is formed from arachidonic acid degradation following infection or inflammation (113), which can be converted to leukotriene B4 through leukotriene A4 hydrolase, or by addition of glutathione to leukotriene C4 by leukotriene C4 synthase (114); leukotriene C4 was also increased in mouse lungs. Leukotrienes A4, B4, and C4 are biologically active profibrotic lipids, and all exhibit potent proinflammatory effects in lungs (114). A recent study revealed that cysteinyl leukotriene levels are increased in senescent lung fibroblasts, bleomycin-induced fibrotic lung, and patients with IPF (77). Thus, the present evidence for increased leukotrienes in HLF and mouse lungs in response to low environmental levels of ingested V+5 complements our previous finding that the same V+5 exposures induce human lung fibroblast cellular senescence (28).
Stimulation of inflammation signaling is closely related to increased oxidative stress. Impaired redox regulation and oxidative stress have been well-documented in lung fibrosis (115, 116) and are thought to be an important mediator of pulmonary fibrosis and reduced lung function in humans (87, 117, 118). Metabolomic and proteomic analyses in human IPF lung samples found upregulated levels of proteins relevant to oxidative stress (99, 100). Our previous studies demonstrated that V+5 oxidizes GSH and protein thiols and causes lung fibroblast senescence (28). The present study extends these findings to show other major redox pathways impacted by low levels of V+5, include methionine and cysteine, ubiquinone biosynthesis, and GSH metabolism. Decreased methionine and cysteine levels are associated with IPF (119) and cystic fibrosis (120, 121). Ubiquinone (i.e., CoQ10) serves as a link between the β-oxidation of fatty acids and the mitochondrial respiratory chain. Disruption in CoQ10 biosynthesis is associated with increased oxidative stress and mitochondrial dysfunction (122), disruption in energy metabolism (69), and human diseases (70, 123, 124). Moreover, increased levels of hydroxyproline in HLF cells and increased collagen production in mouse lungs following V+5 exposure support the interpretation that V+5 stimulates fibrosis signaling (80–82). Due to limited quantities of mouse lung tissue, preferred methods for assessing collagen deposition were not available, and our characterization of collagen production was limited to metabolomics and TEM-based evidence. Future work will be needed to support this connection.
In response to observed changes in energy metabolism and TCA cycle intermediates following V+5 exposure, we investigated the effects of V+5 on mitochondrial function in HLF cells and on mitochondrial morphology using TEM of mouse lungs. Our observations of increased mitochondrial activity, proton leak, and disruption of mitochondrial cristae morphology are consistent with existing literature on V+5 exposures. An earlier study showed that V+5 causes mitochondrial membrane damage through p53 activation (125). Richelmi et al. (126) demonstrated that V+5 could cause Ca2+ overload in mitochondria, leading to a progressive decrease in mitochondrial membrane potential, and causing a decrease in intracellular ATP level, formation of surface blebs, and cytotoxicity (126). In addition, vanadate as a phosphate analog interferes with various ATPases, phosphatases, and phosphate-transfer enzymes. Henderson et al. (127) reported that vanadate could inhibit mitochondrial respiration through interacting with H+ ATPase activity in Saccharomyces cerevisiae (127). Alterations of mitochondrial function are commonly linked to cell metabolism and function. The observed disruptions on glycolysis, TCA cycle, fatty acid oxidation, ubiquinone, and major amino acid metabolisms are tightly linked to mitochondrial dysfunction caused by V+5 in this study.
Although we have emphasized the utility of HRM methods to provide a sensitive way to detect functional changes, resulting from ingestion of low-dose V+5 and test for common responses in cell and mouse models, ability for comparison of these models is limited. For instance, only 30% of the metabolic pathways observed in HLF were also found in mice. Part of this discrepancy is because fibroblasts are only one of more than 30 cell types in lungs. Thus, future study of other cell types will help explain differences in pathway responses between the model systems, such as those revealed by comparison of glycolysis and energy metabolism. There was also limited ability to simulate exposure to ingested V+5 to lung fibroblasts in vitro. Addition of V+5 to HLF media is both a model of inhaled V+5 exposure and of exposure from circulation after ingestion. As the focus of this study was to investigate effects due to ingested V+5, this makes the in vivo model used in this study the more appropriate model of the two. The in vitro experiment was informative and mostly aligned with observations from the mouse experiment, but differences in effects may be attributed to differences in toxic mechanism when V+5 is applied directly to fibroblasts instead of through systemic circulation. Also, whether the extent of effects of V+5 at these low levels is sufficient to contribute to human disease is not clear; however, the results warrant more detailed investigation of ingested V-associated changes in critical metabolites and their contributions to disease etiology. Finally, this study only investigated male mice, as a pilot in vivo study, and the cell line used was male derived. Sex differences in physiological responses to V+5 exposure are likely to exist (35, 36). Earlier studies have reported that higher estrogen levels may be protective against oxidative stress induced by V+5 exposure. Future work will be needed to determine to which degree the effects reported here are observed in females.
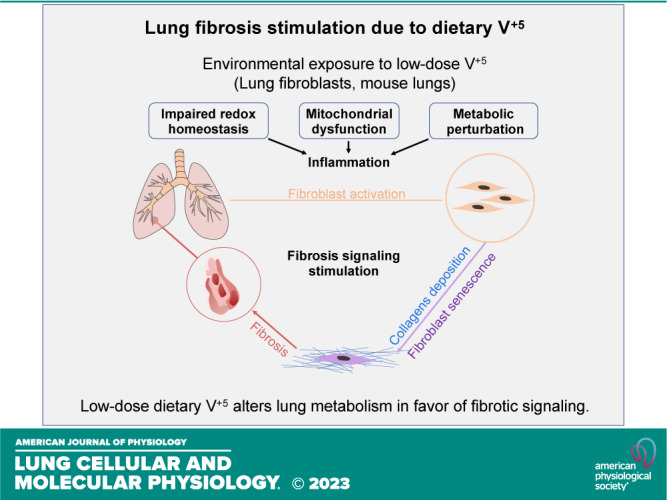
In summary, we used HRMS of HLF cells and male mouse lungs to examine metabolic alterations in response to ingested V+5. V+5 caused widespread dose-response effects of critical metabolites for lung physiology, e.g., cell proliferation, mitochondrial function, and redox homeostasis in both HLF cells and mouse lungs. Similar responses were observed in the cell and mouse models for increased oxidation of the glutathione redox couple and stimulation of inflammatory and fibrosis signaling, but divergent responses were seen for some mitochondrial and energy-related metabolites. As shown in a scheme (Fig. 11), the redox characterizations in mouse lungs and HLF cells as well as a more detailed deciphering of the metabolic pathways involved in the effects of V+5 could provide an asset for better understanding the roles of inflammatory responses and oxidative stress induced by V+5 in the pathogenesis of pulmonary fibrosis.
Figure 11.

Schematic diagram shows low-dose V+5 exposure causes impaired redox homeostasis, metabolic perturbation, mitochondrial dysfunction, and lung inflammation. Lung fibroblasts are activated, and collagen deposition is largely increased following V+5 exposure. Fibroblast senescence is likely to play a major role in the fibrosis signaling. V+5, vanadium pentoxide.
DATA AVAILABILITY
The data in this study will be provided upon reasonable request.
SUPPLEMENTAL DATA
Supplemental Figs. S1–S3: https://doi.org/10.6084/m9.figshare.21125392.v1.
GRANTS
This study was supported by National Institute of Environmental Health Sciences Grants R21 ES031824 (to D.P.J. and Y.-M.G.), R01 ES031980 (to Y.-M.G.), P30 ES019776 (to D.P.J.), R01 ES032189 (to D.P.J.), F32 ES033908 and T32 ES012870 (to Z.J.), and National Institute of Diabetes and Digestive and Kidney Diseases Grants RC2 DK118619 (to D.P.J.) and R01 DK125246 (to D.P.J. and Y.-M.G.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
X.H., Y.-M.G., and D.P.J conceived and designed research; X.H., Z.R.J., M.R.S., V.T.L., Y.L., and M.O. performed experiments; X.H., M.R.S., V.T.L., and V.S. analyzed data; X.H., M.R.S., X.H., V.S., Y.-M.G., and D.P.J. interpreted results of experiments; X.H., Y.-M.G., and D.P.J. prepared figures; X.H. drafted manuscript; X.H., Z.R.J., M.R.S., X.H., Y.-M.G., and D.P.J. edited and revised manuscript; X.H., Z.R.J., M.R.S., V.T.L., X.H., V.S., Y.L., M.O., Y.-M.G., and D.P.J. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Ricardo Guerrero and Jeannette Taylor [The Robert P. Apkarian Integrated Electron Microscopy Core (IEMC), Emory University] for helping TEM imaging.
Present address of X. He: Center for Toxicology and Human Health, Chemical Insights Research Institute, UL Research Institutes, Marietta, GA.
REFERENCES
- 1. Fortoul TI, Rojas-Lemus M, Rodriguez-Lara V, Gonzalez-Villalva A, Ustarroz-Cano M, Cano-Gutierrez G, Gonzalez-Rendon SE, Montano LF, Altamirano-Lozano M. Overview of environmental and occupational vanadium exposure and associated health outcomes: an article based on a presentation at the 8th International Symposium on Vanadium Chemistry, Biological Chemistry, and Toxicology, Washington DC, August 15–18, 2012. J Immunotoxicol 11: 13–18, 2014. doi: 10.3109/1547691X.2013.789940. [DOI] [PubMed] [Google Scholar]
- 2. Assem FL, Levy LS. A review of current toxicological concerns on vanadium pentoxide and other vanadium compounds: Gaps in knowledge and directions for future research. J Toxicol Environ Health B Crit Rev 12: 289–306, 2009. doi: 10.1080/10937400903094166. [DOI] [PubMed] [Google Scholar]
- 3. Lagerkvist BJ-S, Oskarsson A. Vanadium. In: Handbook on the Toxicology of Metals, edited by Nordberg GF, Fowler BA, Nordberg M, Friberg LT.. San Diego, CA: Academic Press, 2007, p. 905–923. [Google Scholar]
- 4. Gerke TL, Scheckel KG, Maynard JB. Speciation and distribution of vanadium in drinking water iron pipe corrosion by-products. Sci Total Environ 408: 5845–5853, 2010. doi: 10.1016/j.scitotenv.2010.08.036. [DOI] [PubMed] [Google Scholar]
- 5.European Food Safety Authority (EFSA). Opinion of the scientific panel on dietetic products, nutrition and allergies on a request from the Commission related to the tolerable upper intake level of vanadium. EFSA J 33: 1–22, 2004. doi: 10.2903/j.efsa.2004.33. [DOI] [Google Scholar]
- 6. Pennington JA, Jones JW. Molybdenum, nickel, cobalt, vanadium, and strontium in total diets. J Am Diet Assoc 87: 1644–1650, 1987. doi: 10.1016/S0002-8223(21)03381-2. [DOI] [PubMed] [Google Scholar]
- 7. Goldfine AB, Simonson DC, Folli F, Patti ME, Kahn CR. In vivo and in vitro studies of vanadate in human and rodent diabetes mellitus. Mol Cell Biochem 153: 217–231, 1995. doi: 10.1007/BF01075941. [DOI] [PubMed] [Google Scholar]
- 8.Institute of Medicine. Arsenic, boron, nickel, silicon, and vanadium. In: Dietary Reference Intakes for Vitamin A, Vitamin K, Arsenic, Boron, Chromium, Copper, Iodine, Iron, Manganese, Molybdenum, Nickel, Silicon, Vanadium, and Zinc. Washington, DC: National Academies Press, 2001, p. 502–553. doi: 10.17226/10026. [DOI] [PubMed] [Google Scholar]
- 9.Agency for Toxic Substances and Disease Registry. Toxicological Profile for Vanadium. Atlanta, GA: U.S. Department of Health and Human Services, 2012. [PubMed] [Google Scholar]
- 10. Schuler D, Chevalier HJ, Merker M, Morgenthal K, Ravanat JL, Sagelsdorff P, Walter M, Weber K, McGregor D. First steps towards an understanding of a mode of carcinogenic action for vanadium pentoxide. J Toxicol Pathol 24: 149–162, 2011. doi: 10.1293/tox.24.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nadal M, Schuhmacher M, Domingo JL. Metal pollution of soils and vegetation in an area with petrochemical industry. Sci Total Environ 321: 59–69, 2004. doi: 10.1016/j.scitotenv.2003.08.029. [DOI] [PubMed] [Google Scholar]
- 12. Liu Y, Woodin MA, Smith TJ, Herrick RF, Williams PL, Hauser R, Christiani DC. Exposure to fuel-oil ash and welding emissions during the overhaul of an oil-fired boiler. J Occup Environ Hyg 2: 435–443, 2005. doi: 10.1080/15459620591034529. [DOI] [PubMed] [Google Scholar]
- 13. Adachi A, Asai K, Koyama Y, Matsumoto Y, Kobayashi T. Vanadium content of cigarettes. Bull Environ Contam Toxicol 61: 276–280, 1998. doi: 10.1007/s001289900759. [DOI] [PubMed] [Google Scholar]
- 14. Riediker M, Williams R, Devlin R, Griggs T, Bromberg P. Exposure to particulate matter, volatile organic compounds, and other air pollutants inside patrol cars. Environ Sci Technol 37: 2084–2093, 2003. doi: 10.1021/es026264y. [DOI] [PubMed] [Google Scholar]
- 15. Kucera J, Byrne AR, Mravcová A, Lener J. Vanadium levels in hair and blood of normal and exposed persons. Sci Total Environ 115: 191–205, 1992. doi: 10.1016/0048-9697(92)90329-q. [DOI] [PubMed] [Google Scholar]
- 16. Lin TS, Chang CL, Shen FM. Whole blood vanadium in Taiwanese college students. Bull Environ Contam Toxicol 73: 781–786, 2004. doi: 10.1007/s00128-004-0495-9. [DOI] [PubMed] [Google Scholar]
- 17. Sabbioni E, Kuèera J, Pietra R, Vesterberg O. A critical review on normal concentrations of vanadium in human blood, serum, and urine. Sci Total Environ 188: 49–58, 1996. doi: 10.1016/0048-9697(96)05164-9. [DOI] [PubMed] [Google Scholar]
- 18. Ehrlich VA, Nersesyan AK, Atefie K, Hoelzl C, Ferk F, Bichler J, Valic E, Schaffer A, Schulte-Hermann R, Fenech M, Wagner K-H, Knasmüller S. Inhalative exposure to vanadium pentoxide causes DNA damage in workers: results of a multiple end point study. Environ Health Perspect 116: 1689–1693, 2008. [Erratum in Environ Health Perspect 117: A15, 2009]. doi: 10.1289/ehp.11438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Knecht EA, Moorman WJ, Clark JC, Lynch DW, Lewis TR. Pulmonary effects of acute vanadium pentoxide inhalation in monkeys. Am Rev Respir Dis 132: 1181–1185, 1985. doi: 10.1164/arrd.1985.132.6.1181. [DOI] [PubMed] [Google Scholar]
- 20. Glass DS, Grossfeld D, Renna HA, Agarwala P, Spiegler P, DeLeon J, Reiss AB. Idiopathic pulmonary fibrosis: current and future treatment. Clin Respir J 16: 84–96, 2022. doi: 10.1111/crj.13466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ding W, Hasegawa T, Hosaka H, Peng D, Takahashi K, Seko Y. Effect of long-term treatment with vanadate in drinking water on KK mice with genetic non-insulin-dependent diabetes mellitus. Biol Trace Elem Res 80: 159–174, 2001. doi: 10.1385/BTER:80:2:159. [DOI] [PubMed] [Google Scholar]
- 22. Shechter Y. Insulin-mimetic effects of vanadate: possible implications for future treatment of diabetes. Diabetes 39: 1–5, 1990. doi: 10.2337/diacare.39.1.1. [DOI] [PubMed] [Google Scholar]
- 23. Heyliger CE, Tahiliani AG, McNeill JH. Effect of vanadate on elevated blood glucose and depressed cardiac performance of diabetic rats. Science 227: 1474–1477, 1985. doi: 10.1126/science.3156405. [DOI] [PubMed] [Google Scholar]
- 24. Rehder D. The role of vanadium in biology. Metallomics 7: 730–742, 2015. doi: 10.1039/c4mt00304g. [DOI] [PubMed] [Google Scholar]
- 25. Capella LS, Gefé MR, Silva EF, Affonso-Mitidieri O, Lopes AG, Rumjanek VM, Capella MA. Mechanisms of vanadate-induced cellular toxicity: role of cellular glutathione and NADPH. Arch Biochem Biophys 406: 65–72, 2002. doi: 10.1016/s0003-9861(02)00408-3. [DOI] [PubMed] [Google Scholar]
- 26. Liu J, Cui H, Liu X, Peng X, Deng J, Zuo Z, Cui W, Deng Y, Wang K. Dietary high vanadium causes oxidative damage-induced renal and hepatic toxicity in broilers. Biol Trace Elem Res 145: 189–200, 2012. doi: 10.1007/s12011-011-9185-8. [DOI] [PubMed] [Google Scholar]
- 27. Nnama AU, Ekeh FN, Aguzie IO, Udegbunam SO, Nwani CD. Vanadium pentoxide induces hematological, oxidative stress and histological changes in Oryctolagus cuniculus. J Hazard Mater Adv 5: 100048, 2022. doi: 10.1016/j.hazadv.2022.100048. [DOI] [Google Scholar]
- 28. He X, Jarrell ZR, Liang Y, Ryan Smith M, Orr ML, Marts L, Go Y-M, Jones DP. Vanadium pentoxide induced oxidative stress and cellular senescence in human lung fibroblasts. Redox Biol 55: 102409, 2022. doi: 10.1016/j.redox.2022.102409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Karginov AV, Fokina AV, Kang HA, Kalebina TS, Sabirzyanova TA, Ter-Avanesyan MD, Agaphonov MO. Dissection of differential vanadate sensitivity in two Ogataea species links protein glycosylation and phosphate transport regulation. Sci Rep 8: 16428, 2018. doi: 10.1038/s41598-018-34888-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Waters DW, Blokland KE, Pathinayake PS, Burgess JK, Mutsaers SE, Prele CM, Schuliga M, Grainge CL, Knight DA. Fibroblast senescence in the pathology of idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 315: L162–L172, 2018. doi: 10.1152/ajplung.00037.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lin Y, Xu Z. Fibroblast senescence in idiopathic pulmonary fibrosis. Front Cell Dev Biol 8: 593283, 2020. doi: 10.3389/fcell.2020.593283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hernandez-Gonzalez F, Faner R, Rojas M, Agustí A, Serrano M, Sellarés J. Cellular senescence in lung fibrosis. Int J Mol Sci 22: 7012, 2021. doi: 10.3390/ijms22137012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kellogg DL, Kellogg DL Jr, Musi N, Nambiar AM. Cellular senescence in idiopathic pulmonary fibrosis. Curr Mol Biol Rep 7: 31–40, 2021. doi: 10.1007/s40610-021-00145-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Parimon T, Hohmann MS, Yao C. Cellular senescence: pathogenic mechanisms in lung fibrosis. Int J Mol Sci 22: 6214, 2021. doi: 10.3390/ijms22126214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rodriguez-Lara V, Cambas AM-R, Villalva AG, Fortoul TI. Sex-based differences in lymphocyte proliferation in the spleen after vanadium inhalation. J Immunotoxicol 13: 498–508, 2016. doi: 10.3109/1547691X.2015.1134731. [DOI] [PubMed] [Google Scholar]
- 36. Rojas-Lemus M, Altamirano-Lozano M, Fortoul TI. Sex differences in blood genotoxic and cytotoxic effects as a consequence of vanadium inhalation: micronucleus assay evaluation. J Appl Toxicol 34: 258–264, 2014. doi: 10.1002/jat.2873. [DOI] [PubMed] [Google Scholar]
- 37. Rondini EA, Walters DM, Bauer AK. Vanadium pentoxide induces pulmonary inflammation and tumor promotion in a strain-dependent manner. Part Fibre Toxicol 7: 9, 2010. doi: 10.1186/1743-8977-7-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Altamirano‐Lozano M, Alvarez‐Barrera L, Basurto‐Alcántara F, Valverde M, Rojas E. Reprotoxic and genotoxic studies of vanadium pentoxide in male mice. Teratog Carcinog Mutagen 16: 7–17, 1996. doi:. [DOI] [PubMed] [Google Scholar]
- 39. Altamirano‐Lozano M, Valverde M, Alvarez‐Barrera L, Molina B, Rojas E. Genotoxic studies of vanadium pentoxide (V2O5) in male mice. II. Effects in several mouse tissues. Teratog Carcinog Mutagen 19: 243–255, 1999. doi:. [DOI] [PubMed] [Google Scholar]
- 40. Zwolak I. Protective effects of dietary antioxidants against vanadium-induced toxicity: a review. Oxid Med Cell Longev 2020: 1490316, 2020. doi: 10.1155/2020/1490316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Go YM, Kim CW, Walker DI, Kang DW, Kumar S, Orr M, Uppal K, Quyyumi AA, Jo H, Jones DP. Disturbed flow induces systemic changes in metabolites in mouse plasma: a metabolomics study using apoe(-)/(-) mice with partial carotid ligation. Am J Physiol Regul Integr Comp Physiol 308: R62–R72, 2015. doi: 10.1152/ajpregu.00278.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jarrell ZR, Smith MR, He X, Orr M, Jones DP, Go Y-M. Firsthand and secondhand exposure levels of maltol-flavored electronic nicotine delivery system vapors disrupt amino acid metabolism. Toxicol Sci 182: 70–81, 2021. doi: 10.1093/toxsci/kfab051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yu T, Park Y, Johnson JM, Jones DP. apLCMS—adaptive processing of high-resolution LC/MS data. Bioinformatics 25: 1930–1936, 2009. doi: 10.1093/bioinformatics/btp291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Uppal K, Soltow QA, Strobel FH, Pittard WS, Gernert KM, Yu T, Dp J. xMSanalyzer: automated pipeline for improved feature detection and downstream analysis of large-scale, non-targeted metabolomics data. BMC Bioinformatics 14: 15, 2013. doi: 10.1186/1471-2105-14-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Go Y-M, Walker DI, Liang Y, Uppal K, Soltow QA, Tran V, Strobel F, Quyyumi AA, Ziegler TR, Pennell KD, Miller GW, Jones DP. Reference standardization for mass spectrometry and high-resolution metabolomics applications to exposome research. Toxicol Sci 148: 531–543, 2015. doi: 10.1093/toxsci/kfv198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chong J, Soufan O, Li C, Caraus I, Li S, Bourque G, Wishart DS, Xia J. Metaboanalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res 46: W486–W494, 2018. doi: 10.1093/nar/gky310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Li S, Park Y, Duraisingham S, Strobel FH, Khan N, Soltow QA, Jones DP, Pulendran B. Predicting network activity from high throughput metabolomics. PLoS Comput Biol 9: e1003123, 2013. doi: 10.1371/journal.pcbi.1003123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Uppal K, Walker DI, Liu K, Li S, Go YM, Jones DP. Computational metabolomics: a framework for the million metabolome. Chem Res Toxicol 29: 1956–1975, 2016. doi: 10.1021/acs.chemrestox.6b00179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sumner LW, Amberg A, Barrett D, Beale MH, Beger R, Daykin CA, Fan TW-M, Fiehn O, Goodacre R, Griffin JL, Hankemeier T, Hardy N, Harnly J, Higashi R, Kopka J, Lane AN, Lindon JC, Marriott P, Nicholls AW, Reily MD, Thaden JJ, Viant MR. Proposed minimum reporting standards for chemical analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics 3: 211–221, 2007. doi: 10.1007/s11306-007-0082-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Smith MR, Chacko BK, Johnson MS, Benavides GA, Uppal K, Go Y-M, Jones DP, Darley-Usmar VM. A precision medicine approach to defining the impact of doxorubicin on the bioenergetic-metabolite interactome in human platelets. Redox Biol 28: 101311, 2020. doi: 10.1016/j.redox.2019.101311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jones DP. Redox potential of GSH/GSSG couple: assay and biological significance. Methods Enzymol 348: 93–112, 2002. doi: 10.1016/s0076-6879(02)48630-2. [DOI] [PubMed] [Google Scholar]
- 52. Go YM, Ziegler TR, Johnson JM, Gu L, Hansen JM, Jones DP. Selective protection of nuclear thioredoxin-1 and glutathione redox systems against oxidation during glucose and glutamine deficiency in human colonic epithelial cells. Free Radic Biol Med 42: 363–370, 2007. doi: 10.1016/j.freeradbiomed.2006.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sauve AA. NAD+ and vitamin B3: from metabolism to therapies. J Pharmacol Exp Ther 324: 883–893, 2008. doi: 10.1124/jpet.107.120758. [DOI] [PubMed] [Google Scholar]
- 54. Lyon P, Strippoli V, Fang B, Cimmino L. B vitamins and one-carbon metabolism: implications in human health and disease. Nutrients 12: 2867, 2020. doi: 10.3390/nu12092867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mikkola S. Nucleotide sugars in chemistry and biology. Molecules 25: 5755, 2020. doi: 10.3390/molecules25235755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Engström W, Zetterberg A. The relationship between purines, pyrimidines, nucleosides, and glutamine for fibroblast cell proliferation. J Cell Physiol 120: 233–241, 1984. doi: 10.1002/jcp.1041200218. [DOI] [PubMed] [Google Scholar]
- 57. Weckerle J, Picart-Armada S, Klee S, Bretschneider T, Luippold AH, Rist W, Haslinger C, Schlüter H, Thomas MJ, Krawczyk B, Fernandez-Albert F, Kästle M, Veyel D. Mapping the metabolomic and lipidomic changes in the bleomycin model of pulmonary fibrosis in young and aged mice. Dis Model Mech 15: dmm049105, 2022. doi: 10.1242/dmm.049105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Dashty M. A quick look at biochemistry: carbohydrate metabolism. Clin Biochem 46: 1339–1352, 2013. doi: 10.1016/j.clinbiochem.2013.04.027. [DOI] [PubMed] [Google Scholar]
- 59. Wakil SJ, Abu-Elheiga LA. Fatty acid metabolism: target for metabolic syndrome. J Lipid Res 50: S138–S143, 2009. doi: 10.1194/jlr.R800079-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Miller E, Morel A, Saso L, Saluk J. Isoprostanes and neuroprostanes as biomarkers of oxidative stress in neurodegenerative diseases. Oxid Med Cell Longev 2014: 572491, 2014. doi: 10.1155/2014/572491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Aquilano K, Baldelli S, Ciriolo MR. Glutathione: new roles in redox signaling for an old antioxidant. Front Pharmacol 5: 196, 2014. doi: 10.3389/fphar.2014.00196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Imura H, Shimada A, Naota M, Morita T, Togawa M, Hasegawa T, Seko Y. Vanadium toxicity in mice: possible impairment of lipid metabolism and mucosal epithelial cell necrosis in the small intestine. Toxicol Pathol 41: 842–856, 2013. doi: 10.1177/0192623312467101. [DOI] [PubMed] [Google Scholar]
- 63. Wang J-P, Cui R-Y, Ding X-M, Bai S-P, Zeng Q-F, Peng H-W, Zhang K-Y. Vanadium in high-fat diets sourced from egg yolk decreases growth and antioxidative status of Wistar rats. Anim Nutr 5: 307–313, 2019. doi: 10.1016/j.aninu.2019.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.U.S. Environmental Protection Agency. Toxicological Review of Vanadium Pentoxide (V2O5) (CAS No. 1314-62-1). In Support of Summary Information on the Integrated Risk Information System (IRIS) (Online). Washington, DC.: U.S. Environmental Protection Agency, 2011. [2022 Jul 15]. https://ofmpub.epa.gov/eims/eimscomm.getfile?p_download_id=504127. [Google Scholar]
- 65. Handford M, Rodriguez-Furlán C, Orellana A. Nucleotide-sugar transporters: structure, function and roles in vivo. Braz J Med Biol Res 39: 1149–1158, 2006. doi: 10.1590/s0100-879x2006000900002. [DOI] [PubMed] [Google Scholar]
- 66. Lecca D, Ceruti S. Uracil nucleotides: from metabolic intermediates to neuroprotection and neuroinflammation. Biochem Pharmacol 75: 1869–1881, 2008. doi: 10.1016/j.bcp.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 67. Lonsdale D. A review of the biochemistry, metabolism and clinical benefits of thiamin(e) and its derivatives. Evid Based Complement Alternat Med 3: 49–59, 2006. doi: 10.1093/ecam/nek009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Carr AC, Maggini S. Vitamin C and immune function. Nutrients 9: 1211, 2017. doi: 10.3390/nu9111211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Crane FL. Biochemical functions of coenzyme Q10. J Am Coll Nutr 20: 591–598, 2001. doi: 10.1080/07315724.2001.10719063. [DOI] [PubMed] [Google Scholar]
- 70. Acosta MJ, Fonseca LV, Desbats MA, Cerqua C, Zordan R, Trevisson E, Salviati L. Coenzyme Q biosynthesis in health and disease. Biochim Biophys Acta 1857: 1079–1085, 2016. doi: 10.1016/j.bbabio.2016.03.036. [DOI] [PubMed] [Google Scholar]
- 71. Padayatty SJ, Katz A, Wang Y, Eck P, Kwon O, Lee J-H, Chen S, Corpe C, Dutta A, Dutta SK, Levine M. Vitamin C as an antioxidant: evaluation of its role in disease prevention. J Am Coll Nutr 22: 18–35, 2003. doi: 10.1080/07315724.2003.10719272. [DOI] [PubMed] [Google Scholar]
- 72. Esteves F, Rueff J, Kranendonk M. The central role of cytochrome P450 in xenobiotic metabolism—A brief review on a fascinating enzyme family. J Xenobiot 11: 94–114, 2021. doi: 10.3390/jox11030007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. King TE Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet 378: 1949–1961, 2011. doi: 10.1016/S0140-6736(11)60052-4. [DOI] [PubMed] [Google Scholar]
- 74. Pardo A, Selman M. Lung fibroblasts, aging, and idiopathic pulmonary fibrosis. Annals ATS 13: S417–S421, 2016. doi: 10.1513/AnnalsATS.201605-341AW. [DOI] [PubMed] [Google Scholar]
- 75. Smith MR, Jarrell ZR, Orr M, Liu KH, Go Y-M, Jones DP. Metabolome-wide association study of flavorant vanillin exposure in bronchial epithelial cells reveals disease-related perturbations in metabolism. Environ Int 147: 106323, 2021. doi: 10.1016/j.envint.2020.106323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bauman KA, Wettlaufer SH, Okunishi K, Vannella KM, Stoolman JS, Huang SK, Courey AJ, White ES, Hogaboam CM, Simon RH, Toews GB, Sisson TH, Moore BB, Peters-Golden M. The antifibrotic effects of plasminogen activation occur via prostaglandin E2 synthesis in humans and mice. J Clin Invest 120: 1950–1960, 2010. doi: 10.1172/JCI38369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wiley CD, Brumwell AN, Davis SS, Jackson JR, Valdovinos A, Calhoun C, Alimirah F, Castellanos CA, Ruan R, Wei Y, Chapman HA, Ramanathan A, Campisi J, Saux CJL. Secretion of leukotrienes by senescent lung fibroblasts promotes pulmonary fibrosis. JCI Insight 4: e130056, 2019. doi: 10.1172/jci.insight.130056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Liu X, Sun SQ, Ostrom RS. Fibrotic lung fibroblasts show blunted inhibition by cAMP due to deficient cAMP response element-binding protein phosphorylation. J Pharmacol Exp Ther 315: 678–687, 2005. doi: 10.1124/jpet.105.090324. [DOI] [PubMed] [Google Scholar]
- 79. Castelino FV. Lipids and eicosanoids in fibrosis: emerging targets for therapy. Curr Opin Rheumatol 24: 649–655, 2012. doi: 10.1097/BOR.0b013e328356d9f6. [DOI] [PubMed] [Google Scholar]
- 80. Organ LA, Duggan A-MR, Oballa E, Taggart SC, Simpson JK, Kang’ombe AR, Braybrooke R, Molyneaux PL, North B, Karkera Y, Leeming DJ, Karsdal MA, Nanthakumar CB, Fahy WA, Marshall RP, Jenkins RG, Maher TM. Biomarkers of collagen synthesis predict progression in the PROFILE idiopathic pulmonary fibrosis cohort. Respir Res 20: 148, 2019. doi: 10.1186/s12931-019-1118-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Wells RG. How collagen becomes ‘stiff’. eLife 11: e77041, 2022. doi: 10.7554/eLife.77041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Staab-Weijnitz CA. Fighting the fiber: Targeting collagen in lung fibrosis. Am J Respir Cell Mol Biol 66: 363–381, 2022. doi: 10.1165/rcmb.2021-0342TR. [DOI] [PubMed] [Google Scholar]
- 83. Tsukui T, Sun K-H, Wetter JB, Wilson-Kanamori JR, Hazelwood LA, Henderson NC, Adams TS, Schupp JC, Poli SD, Rosas IO, Kaminski N, Matthay MA, Wolters PJ, Sheppard D. Collagen-producing lung cell atlas identifies multiple subsets with distinct localization and relevance to fibrosis. Nat Commun 11: 1920, 2020. doi: 10.1038/s41467-020-15647-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Kendall RT, Feghali-Bostwick CA. Fibroblasts in fibrosis: novel roles and mediators. Front Pharmacol 5: 123, 2014. doi: 10.3389/fphar.2014.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Yue X, Shan B, Lasky JA. TGF-β: Titan of lung fibrogenesis. Curr Enzym Inhib 6: 67–77, 2010. doi: 10.2174/157340810791233033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Amara N, Goven D, Prost F, Muloway R, Crestani B, Boczkowski J. NOX4/NADPH oxidase expression is increased in pulmonary fibroblasts from patients with idiopathic pulmonary fibrosis and mediates TGFβ1-induced fibroblast differentiation into myofibroblasts. Thorax 65: 733–738, 2010. doi: 10.1136/thx.2009.113456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Kliment CR, Oury TD. Oxidative stress, extracellular matrix targets, and idiopathic pulmonary fibrosis. Free Radic Biol Med 49: 707–717, 2010. doi: 10.1016/j.freeradbiomed.2010.04.036. [DOI] [PubMed] [Google Scholar]
- 88. Cheresh P, Kim SJ, Tulasiram S, Kamp DW. Oxidative stress and pulmonary fibrosis. Biochim Biophys Acta 1832: 1028–1040, 2013. doi: 10.1016/j.bbadis.2012.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Irsigler GB, Visser PJ, Spangenberg PA. Asthma and chemical bronchitis in vanadium plant workers. Am J Ind Med 35: 366–374, 1999. doi:. [DOI] [PubMed] [Google Scholar]
- 90. Stokinger HE. Vanadium. In: Patty's Industrial Hygiene and Toxicology, edited by Clayton GD, Clayton FE.. New York: John Wiley and Sons, 1981, p. 2013–2033. [Google Scholar]
- 91. Antao-Menezes A, Turpin EA, Bost PC, Ryman-Rasmussen JP, Bonner JC. STAT-1 signaling in human lung fibroblasts is induced by vanadium pentoxide through an IFN-β autocrine loop. J Immunol 180: 4200–4207, 2008. doi: 10.4049/jimmunol.180.6.4200. [DOI] [PubMed] [Google Scholar]
- 92. Razzaque MS, Taguchi T. Pulmonary fibrosis: cellular and molecular events. Pathol Int 53: 133–145, 2003. doi: 10.1046/j.1440-1827.2003.01446.x. [DOI] [PubMed] [Google Scholar]
- 93. Rodriguez LR, Emblom-Callahan M, Chhina M, Bui S, Aljeburry B, Tran LH, Novak R, Lemma M, NathaN SD, Grant GM. Global gene expression analysis in an in vitro fibroblast model of idiopathic pulmonary fibrosis reveals potential role for CXCL14/CXCR4. Sci Rep 8: 3983, 2018. doi: 10.1038/s41598-018-21889-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Ingram JL, Rice AB, Santos J, Houten BV, Bonner JC. Vanadium-induced HB-EGF expression in human lung fibroblasts is oxidant dependent and requires MAP kinases. Am J Physiol Lung Cell Mol Physiol 284: L774–L782, 2003. doi: 10.1152/ajplung.00189.2002. [DOI] [PubMed] [Google Scholar]
- 95. Ingram JL, Antao-Menezes A, Turpin EA, Wallace DG, Mangum JB, Pluta LJ, Thomas RS, Bonner JC. Genomic analysis of human lung fibroblasts exposed to vanadium pentoxide to identify candidate genes for occupational bronchitis. Respir Res 8: 34, 2007. doi: 10.1186/1465-9921-8-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Benabe JE, Echegoyen LA, Pastrana B, Martínez-Maldonado M. Mechanism of inhibition of glycolysis by vanadate. J Biol Chem 262: 9555–9560, 1987. [PubMed] [Google Scholar]
- 97. Zhang H, Alder NN, Wang W, Szeto H, Marcinek DJ, Rabinovitch PS. Reduction of elevated proton leak rejuvenates mitochondria in the aged cardiomyocyte. eLife 9: e60827, 2020. doi: 10.7554/eLife.60827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Cogliati S, Frezza C, Soriano ME, Varanita T, Quintana-Cabrera R, Corrado M, Cipolat S, Costa V, Casarin A, Gomes LC, Perales-Clemente E, Salviati L, Fernandez-Silva P, Enriquez JA, Scorrano L. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 155: 160–171, 2013. doi: 10.1016/j.cell.2013.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Zhao YD, Yin L, Archer S, Lu C, Zhao G, Yao Y, Wu L, Hsin M, Waddell TK, Keshavjee S, Granton J, de Perrot M. Metabolic heterogeneity of idiopathic pulmonary fibrosis: a metabolomic study. BMJ Open Respir Res 4: e000183, 2017. doi: 10.1136/bmjresp-2017-000183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Korfei M, Schmitt S, Ruppert C, Henneke I, Markart P, Loeh B, Mahavadi P, Wygrecka M, Klepetko W, Fink L, Bonniaud P, Preissner KT, Lochnit G, Schaefer L, Seeger W, Guenther A. Comparative proteomic analysis of lung tissue from patients with idiopathic pulmonary fibrosis (IPF) and lung transplant donor lungs. J Proteome Res 10: 2185–2205, 2011. doi: 10.1021/pr1009355. [DOI] [PubMed] [Google Scholar]
- 101. Vancheri C. Idiopathic pulmonary fibrosis: an altered fibroblast proliferation linked to cancer biology. Proc Am Thorac Soc 9: 153–157, 2012. doi: 10.1513/pats.201203-025AW. [DOI] [PubMed] [Google Scholar]
- 102. Pierce EM, Carpenter K, Jakubzick C, Kunkel SL, Evanoff H, Flaherty KR, Martinez FJ, Toews GB, Hogaboam CM. Idiopathic pulmonary fibrosis fibroblasts migrate and proliferate to CC chemokine ligand 21. Eur Respir J 29: 1082–1093, 2007. doi: 10.1183/09031936.00122806. [DOI] [PubMed] [Google Scholar]
- 103. Li X, Zhang W, Cao Q, Wang Z, Zhao M, Xu L, Zhuang Q. Mitochondrial dysfunction in fibrotic diseases. Cell Death Discov 6: 80, 2020. doi: 10.1038/s41420-020-00316-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Wilborn J, Crofford LJ, Burdick MD, Kunkel SL, Strieter RM, Peters-Golden M. Cultured lung fibroblasts isolated from patients with idiopathic pulmonary fibrosis have a diminished capacity to synthesize prostaglandin E2 and to express cyclooxygenase-2. J Clin Invest 95: 1861–1868, 1995. doi: 10.1172/JCI117866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Martin JG, Suzuki M, Maghni K, Pantano R, Ramos-Barbón D, Ihaku D, Nantel F, Denis D, Hamid Q, Powell WS. The immunomodulatory actions of prostaglandin E2 on allergic airway responses in the rat. J Immunol 169: 3963–3969, 2002. doi: 10.4049/jimmunol.169.7.3963. [DOI] [PubMed] [Google Scholar]
- 106. Liu T, Laidlaw TM, Feng C, Xing W, Shen S, Milne GL, Boyce JA. Prostaglandin E2 deficiency uncovers a dominant role for thromboxane A2 in house dust mite-induced allergic pulmonary inflammation. Proc Natl Acad Sci USA 109: 12692–12697, 2012. doi: 10.1073/pnas.1207816109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Lundequist A, Nallamshetty SN, Xing W, Feng C, Laidlaw TM, Uematsu S, Akira S, Boyce JA. Prostaglandin E2 exerts homeostatic regulation of pulmonary vascular remodeling in allergic airway inflammation. J Immunol 184: 433–441, 2010. doi: 10.4049/jimmunol.0902835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Church RJ, Jania LA, Koller BH. Prostaglandin E2 produced by the lung augments the effector phase of allergic inflammation. J Immunol 188: 4093–4102, 2012. doi: 10.4049/jimmunol.1101873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Birrell MA, Maher SA, Dekkak B, Jones V, Wong S, Brook P, Belvisi MG. Anti-inflammatory effects of PGE2 in the lung: role of the EP4 receptor subtype. Thorax 70: 740–747, 2015. doi: 10.1136/thoraxjnl-2014-206592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Yamanaka K, Saluja AK, Brown GE, Yamaguchi Y, Hofbauer B, Steer ML. Protective effects of prostaglandin E1 on acute lung injury of caerulein-induced acute pancreatitis in rats. Am J Physiol 272: G23–G30, 1997. doi: 10.1152/ajpgi.1997.272.1.G23. [DOI] [PubMed] [Google Scholar]
- 111. Fantone JC, Marasco WA, Elgas LJ, Ward PA. Anti-inflammatory effects of prostaglandin E1: in vivo modulation of the formyl peptide chemotactic receptor on the rat neutrophil. J Immunol 130: 1495–1497, 1983. doi: 10.4049/jimmunol.130.4.1495. [DOI] [PubMed] [Google Scholar]
- 112. Lai Y-J, Hsu H-H, Chang G-J, Lin S-H, Chen W-J, Huang C-C, Pang JS. Prostaglandin E1 attenuates pulmonary artery remodeling by activating phosphorylation of CREB and the PTEN signaling pathway. Sci Rep 7: 9974, 2017. [Erratum in Sci Rep 8: 11383, 2018]. doi: 10.1038/s41598-017-09707-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Marx JL. The leukotrienes in allergy and inflammation. Science 215: 1380–1383, 1982. doi: 10.1126/science.6278589. [DOI] [PubMed] [Google Scholar]
- 114. Samuelsson B. Leukotrienes: mediators of immediate hypersensitivity reactions and inflammation. Science 220: 568–575, 1983. doi: 10.1126/science.6301011. [DOI] [PubMed] [Google Scholar]
- 115. Kinnula VL, Fattman CL, Tan RJ, Oury TD. Oxidative stress in pulmonary fibrosis: a possible role for redox modulatory therapy. Am J Respir Crit Care Med 172: 417–422, 2005. doi: 10.1164/rccm.200501-017PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Day BJ. Antioxidants as potential therapeutics for lung fibrosis. Antioxid Redox Signal 10: 355–370, 2008. doi: 10.1089/ars.2007.1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Chia SB, Elko EA, Aboushousha R, Manuel AM, van de Wetering C, Druso JE, van der Velden J, Seward DJ, Anathy V, Irvin CG, Lam Y-W, van der Vliet A, Janssen-Heininger YMW. Dysregulation of the glutaredoxin/S-glutathionylation redox axis in lung diseases. Am J Physiol Cell Physiol 318: C304–C327, 2020. doi: 10.1152/ajpcell.00410.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Teramoto S, Fukuchi Y, Uejima Y, Shu CY, Orimo H. Superoxide anion formation and glutathione metabolism of blood in patients with idiopathic pulmonary fibrosis. Biochem Mol Med 55: 66–70, 1995. doi: 10.1006/bmme.1995.1033. [DOI] [PubMed] [Google Scholar]
- 119. Maier K, Leuschel L, Costabel U. Increased levels of oxidized methionine residues in bronchoalveolar lavage fluid proteins from patients with idiopathic pulmonary fibrosis. Am Rev Respir Dis 143: 271–274, 1991. doi: 10.1164/ajrccm/143.2.271. [DOI] [PubMed] [Google Scholar]
- 120. Chandler JD, Margaroli C, Horati H, Kilgore MB, Veltman M, Liu HK, Taurone AJ, Peng L, Guglani L, Uppal K, Go Y-M, Tiddens HAWM, Scholte BJ, Tirouvanziam R, Jones DP, Janssens HM. Myeloperoxidase oxidation of methionine associates with early cystic fibrosis lung disease. Eur Respir J 52: 1801118, 2018. doi: 10.1183/13993003.01118-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Innis SM, Davidson AGF, Chen A, Dyer R, Melnyk S, James SJ. Increased plasma homocysteine and S-adenosylhomocysteine and decreased methionine is associated with altered phosphatidylcholine and phosphatidylethanolamine in cystic fibrosis. J Pediatr 143: 351–356, 2003. doi: 10.1067/S0022-3476(03)00326-3. [DOI] [PubMed] [Google Scholar]
- 122. Cotán D, Cordero MD, Garrido-Maraver J, Oropesa-Ávila M, Rodríguez-Hernández A, Gómez Izquierdo L, De la Mata M, De Miguel M, Lorite JB, Infante ER, Jackson S, Navas P, Sánchez-Alcázar JA. Secondary coenzyme Q10 deficiency triggers mitochondria degradation by mitophagy in MELAS fibroblasts. FASEB J 25: 2669–2687, 2011. doi: 10.1096/fj.10-165340. [DOI] [PubMed] [Google Scholar]
- 123. Gempel K, Topaloglu H, Talim B, Schneiderat P, Schoser BGH, Hans VH, Pálmafy B, Kale G, Tokatli A, Quinzii C, Hirano M, Naini A, DiMauro S, Prokisch H, Lochmüller H, Horvath R. The myopathic form of coenzyme Q10 deficiency is caused by mutations in the electron-transferring-flavoprotein dehydrogenase (ETFDH) gene. Brain 130: 2037–2044, 2007. doi: 10.1093/brain/awm054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Cornelius N, Byron C, Hargreaves I, Guerra PF, Furdek AK, Land J, Radford WW, Frerman F, Corydon TJ, Gregersen N, Olsen RKJ. Secondary coenzyme Q10 deficiency and oxidative stress in cultured fibroblasts from patients with riboflavin responsive multiple acyl-CoA dehydrogenation deficiency. Hum Mol Genet 22: 3819–3827, 2013. doi: 10.1093/hmg/ddt232. [DOI] [PubMed] [Google Scholar]
- 125. Huang C, Zhang Z, Ding M, Li J, Ye J, Leonard SS, Shen HM, Butterworth L, Lu Y, Costa M, Rojanasakul Y, Castranova V, Vallyathan V, Shi X. Vanadate induces p53 transactivation through hydrogen peroxide and causes apoptosis. J Biol Chem 275: 32516–32522, 2000. doi: 10.1074/jbc.M005366200. [DOI] [PubMed] [Google Scholar]
- 126. Richelmi P, Mirabelli F, Salis A, Finard G, Berte F, Bellomo G. On the role of mitochondria in cell injury caused by vanadate-induced Ca2+ overload. Toxicology 57: 29–44, 1989. doi: 10.1016/0300-483x(89)90032-2. [DOI] [PubMed] [Google Scholar]
- 127. Henderson GE, Evans IH, Bruce IJ. Vanadate inhibition of mitochondrial respiration and H+ ATPase activity in Saccharomyces cerevisiae. Yeast 5: 73–77, 1989. doi: 10.1002/yea.320050109. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figs. S1–S3: https://doi.org/10.6084/m9.figshare.21125392.v1.
Data Availability Statement
The data in this study will be provided upon reasonable request.