

Abstract
Heart failure with preserved ejection fraction (HFpEF) is now the most common form of heart failure and a significant public health concern for which limited effective therapies exist. Inflammation triggered by comorbidity burden is a critical element of HFpEF pathophysiology. Here, we discuss evidence for comorbidity-driven systemic and myocardial inflammation and the mechanistic role of inflammation in pathological myocardial remodeling in HFpEF.
Keywords: heart failure, inflammation, meta-inflammation, myocardial remodeling
Introduction
Cardiovascular diseases remain a leading cause of death globally marked by an ever-evolving landscape of challenges (1). As we have become increasingly successful in treating acute cardiovascular events, attention has turned to heart failure (HF) as the next major hurdle in cardiovascular medicine; more and more people are surviving the acutely lethal manifestations of cardiovascular diseases, returning to family and society with an injured heart, often culminating in HF.
The syndrome of HF can be broadly categorized into two clinically distinct subtypes: heart failure with reduced ejection fraction (HFrEF) and heart failure with preserved ejection fraction (HFpEF). A growing therapeutic arsenal has improved outcomes in HFrEF, yet our ability to treat HFpEF remains modestly effective. In light of the significant morbidity/mortality associated with HFpEF, coupled with the fact that it is now the most prevalent form of HF, deciphering the molecular underpinnings of this syndrome is paramount (2–4). Leveraging these findings to promote the development of new therapies is critical for improving patient outcomes.
HFpEF is characterized by multiple phenotypic disorders, including diastolic dysfunction mediated by passive myocardial stiffness, microvascular dysfunction, metabolic dyscrasias, and more (5). With respect to myocardial stiffness, underlying cellular and molecular changes remain poorly understood; however, a critical role for inflammation is emerging. Initial observations correlating elevated serum proinflammatory cytokine levels and adverse clinical outcomes in both HFpEF and HFrEF sparked interest in inflammation as a driver of HF (6–9). It is now believed that this inflammation is largely generated by comorbidity burden and plays a critical role in HFpEF myocardial remodeling (3, 4). Indeed, the majority of HFpEF patients are obese and hypertensive, with advanced age, chronic kidney disease, and chronic obstructive pulmonary disease also being highly prevalent (5, 10).
In this review, we discuss mechanisms of comorbidity-driven inflammation and its importance in HFpEF pathogenesis. We delve into how inflammation promotes the characteristic cellular and molecular changes seen in HFpEF.
HFpEF in Perspective: from Disease to Heterogenous Syndrome
Early simplistic views of HFpEF as a disease resulting from abnormalities in diastolic function have morphed into a more complex understanding that HFpEF is a comorbidity-driven clinical syndrome. Each comorbidity contributes uniquely to overall syndrome pathophysiology. In fact, patients diagnosed with HFpEF are a heterogenous group presenting with varying clinical manifestations, serum markers, and comorbidity burden. Furthermore, HFpEF not only involves the heart and cardiomyocytes but is a systemic syndrome affecting multiple organs and cell types via diverse pathophysiological processes.
In an attempt to subclassify this heterogenous population, it has become increasingly popular to classify HFpEF patients based on comorbidity phenogroups, which include autoimmune/inflammatory disease-associated HFpEF, cardiorenal HFpEF, cardiometabolic HFpEF, likely to be the most prevalent form, and several more (4). Evidence for the existence of these subgroups derives largely from pheno-mapping studies based on machine learning-derived clustering analysis (11–15). The importance of characterizing these phenogroups lies in their differing prognoses and responses to therapy (15, 16). Using a precision medicine-based approach to treat individual subgroups may provide better outcomes in future clinical trials.
Inflammation at the Heart of HFpEF Pathophysiology
Levine et al. (17) first linked inflammation to HF, reporting an elevation in circulating tumor necrosis factor (TNF) in patients with HFrEF. This work was corroborated over the following decades by multiple studies showing a strong correlation between various serum proinflammatory cytokines and adverse clinical outcomes in HF (7, 18–20). These findings ushered in strong interest in exploring inflammation as a key pathophysiological contributor to HF, culminating in a multitude of both successful and unsuccessful phase III clinical trials.
Inflammation in HFpEF is characterized as chronic and low grade, present at both the systemic and myocardial levels. Studies based on animal models and patient endomyocardial biopsies have led us to propose the following: initially, there is an increase in both circulating and myocardium-infiltrating immune cells, including macrophages (21). This increase in leukocyte migration may be mediated by increased adhesion receptor expression and activation (e.g., E-selectin and ICAM) (22). Next, evidence exists for a potential imbalance in inflammatory to anti-inflammatory immune cell populations such as circulating T cells (Th17/Treg ratio); however, more work is needed to confirm this imbalance (23). Increased inflammatory activity of immune cells results in an increase in systemic and myocardial proinflammatory cytokine abundance in HFpEF (23–25).
In its initial formulation, the comorbidity-inflammation paradigm stipulated that inflammation in HFpEF is largely comorbidity driven, resulting in endothelial inflammation and coronary microvascular dysfunction limiting nitric oxide (NO) bioavailability and PKG activity in cardiomyocytes. The resulting cardiomyocyte hypertrophy and reduced distensibility combined with ventricular fibrosis due to increased myofibroblast activity have been held to contribute to HFpEF pathogenesis (26). Whereas reduced endothelial NO production has yet to be definitively linked to reduced cardiomyocyte PKG activity in HFpEF, evidence continues to accumulate implicating comorbidity-driven inflammation in a wide range of the pathophysiological events observed in this syndrome. Recent proteomics mediation analysis from the PROMIS-HFpEF study further bolsters the comorbidity-inflammation paradigm by revealing that inflammation mediates the association between comorbidity burden and echocardiographic parameters in HFpEF (27). Furthermore, tumor necrosis factor receptor 1 (TNFR1), urokinase plasminogen activator receptor (UPAR), insulin-like growth factor binding protein 7 (IGFBP7), and growth differentiation factor-15 (GDF-15) have all been identified as strong mediators of this association (27) (FIGURE 1).
FIGURE 1.
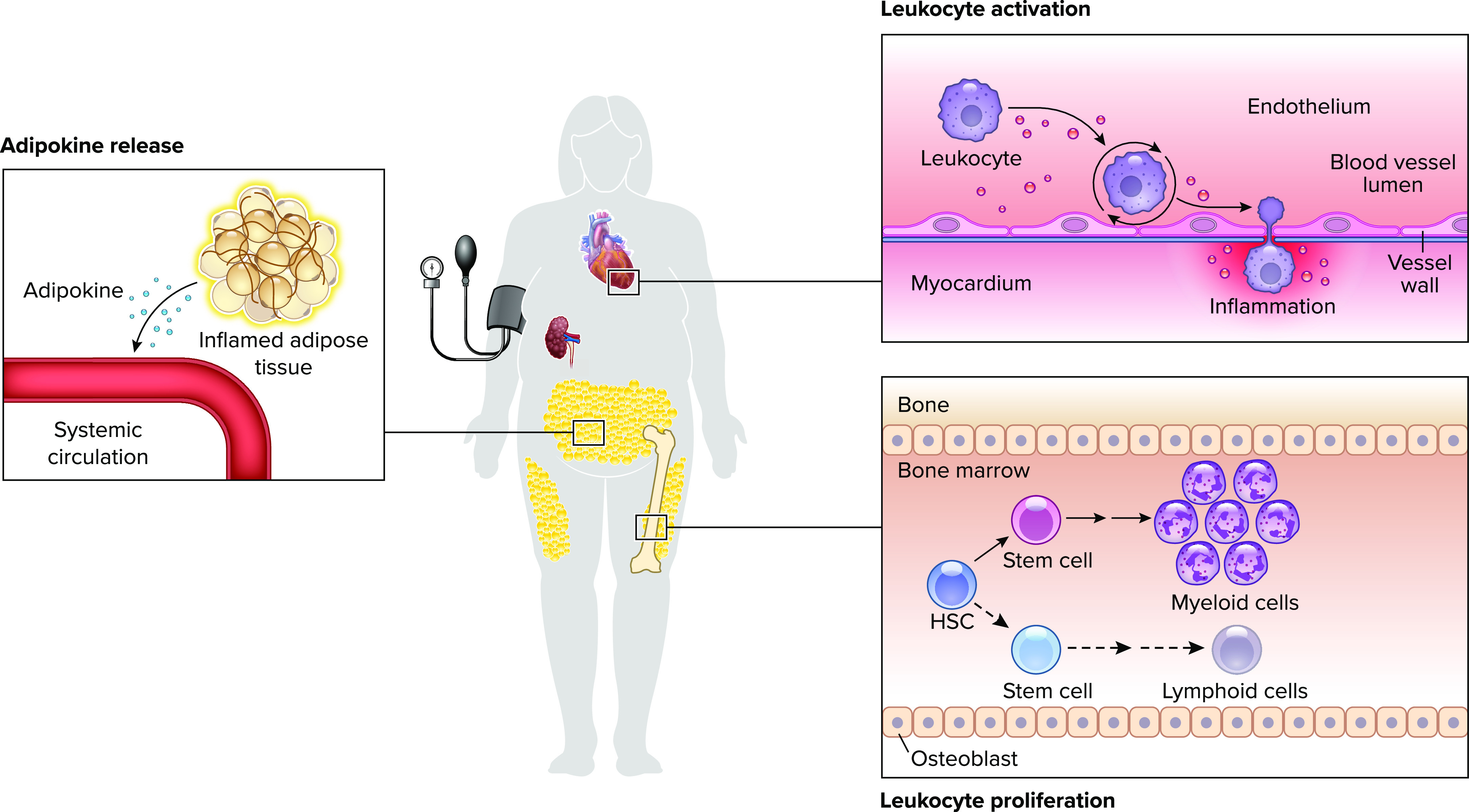
Heart failure with preserved ejection fraction-associated comorbidities increase circulating and activated, myocardium-infiltrating leukocytes Adipose tissue, in turn, contributes to circulating inflammatory signals. The resulting systemic and myocardial inflammation is characteristic of heart failure with preserved ejection fraction (HFpEF). HSC, hematopoietic stem cell.
Other bioinformatics studies have shed light on the importance of inflammation in overall syndrome severity and clinical outcomes. Clustering analysis performed with inflammatory biomarkers as input variables identified three inflammatory clusters in HFpEF: pan-inflammatory phenotype, noninflammatory phenotype, and obese-high CRP phenotype (14). Interestingly, patients with the pan-inflammatory phenotype exhibited the worst outcomes (14). Findings such as these highlight the likely importance of inflammation in HFpEF pathophysiology and identify patient subgroups that may benefit from anti-inflammatory therapies.
Cellular and molecular differences between the pathophysiologies of HFpEF and HFrEF continue to emerge with recent studies indicating that inflammation may play a more crucial role in HFpEF. Using data from the BIOSTAT-CHF program, network analysis enriched with data regarding protein-protein interactions revealed that pathways relating to inflammation and extracellular matrix (ECM) remodeling are overrepresented in HFpEF compared to HFrEF (28). Evidence remains inconclusive, however, as another study using transcriptomics data revealed similar importance of fibrosis, inflammation, hypertrophy, and oxidative stress in HFpEF and HFrEF, whereas protein homeostasis, endoplasmic reticulum stress, and angiogenesis were identified as unique to HFpEF (29). Most striking, however, was a clear distinction in gene expression patterns between HFpEF and HFrEF and the identification of a HFpEF phenogroup with inflammatory and matrix signatures (29). It is important to note that some of these differences may be attributable to the use of serum biomarkers in the former study and endomyocardial biopsies in the latter.
Mechanisms of Comorbidity-Driven, Low-Grade, Chronic Inflammation
Adipose Tissue Expansion
The majority of HFpEF patients are obese with an average body mass index above 35 kg/m2 (30). The roles of different adipose tissue (AT) depots in the pathogenesis of HFpEF are poorly understood, yet AT expansion and associated inflammation are recognized as important drivers of metabolic dysregulation and systemic inflammation. Visceral AT (VAT) expansion and inflammation occur simultaneously. Deciphering how these factors trigger this inflammatory transformation is incomplete but includes AT hypoxia-inducible factor 1α (HIF-1α) activation, accumulation of proinflammatory long chain fatty acids in adipocytes triggering various inflammatory pathways, and possibly metabolic endotoxemia (31–36). The consequence is a well-characterized accumulation and activation of diverse inflammatory cell types in VAT. The ensuing release of a multitude of inflammatory molecules translates into increased systemic and myocardial inflammation (31).
Whereas extensive evidence implicates visceral adipose depots in both systemic and myocardial inflammation in HFpEF, epicardial AT (EAT) has been less well studied. Notably, due to its anatomical proximity and intricate association with the myocardium, EAT expansion and associated inflammation have been proposed as important elements in HFpEF pathobiology (37). Clinically, it is observed that in patients with cardiometabolic HFpEF, EAT expansion is associated with impaired hemodynamic parameters and worse exercise capacity (38). In fact, EAT accumulation has been independently associated with all-cause mortality and HF hospitalizations in HFpEF patients (39).
The epicardial space is connected to the myocardium by a direct microcirculatory network allowing the transfer of adipokines and immune cells (40–42). EAT expansion occurs concurrently with adipocyte transformation to a predominantly white phenotype and the adoption of a proinflammatory state (40, 43). This transformation predisposes epicardial adipocytes to lipolysis and secretion of proinflammatory cytokines (including leptin, TNF-α, IL-1β, and IL-6) while decreasing the secretion of adiponectin (44–46). A direct relationship has been observed between epicardial adipose thickness and both myocardial and systemic inflammation in obese animals (47). EAT can also act as a depot of both innate and adaptive immune cell populations, including macrophages, mast cells, and B and T cells located in fat-associated lymphoid clusters (FALCs) and in the stromal vascular fraction of EAT (42, 48, 49). EAT-resident immune cells such as macrophages in HFpEF begin to display a proinflammatory phenotype with EAT expansion and inflammation (47).
Accumulating evidence points to obesity-induced metabolic stress as a driver of inflammation, a process termed “meta-inflammation.” Meta-inflammation involves bidirectional cross talk between inflammation and metabolic processes, whereby metabolic intermediates affect inflammatory pathways and vice versa (3, 4). Metabolic alterations observed in obesity can fuel immunologic activation via a wide range of metabolic intermediates that modulate multiple cellular pathways (50). This is especially evident in immune cell populations in which metabolism can robustly affect inflammatory activity (51). TCA cycle derangements, linked with obesity, can lead to the accumulation of various TCA cycle intermediates triggering macrophage proinflammatory polarization and activation of inflammatory pathways (52, 53). Succinate accumulation, for example, promotes macrophage polarization toward an M1-like phenotype and activates HIF-1α, upregulating IL-1β expression (54). Similarly, fumarate accumulation increases TNF-α and IL-6 expression by inducing epigenetic modifications through KDM5 histone demethylase inhibition (55). This bidirectional immune-metabolic cross talk also results in inflammation-induced metabolic alterations. Of note, whereas these have not been extensively explored in a preclinical model of HFpEF utilizing high-fat diet (HFD) and IL-6 infusion, AMP-activated protein kinase (AMPK) is suppressed, and glucose metabolism is impaired (56) (FIGURE 2).
FIGURE 2.

Depicted are common comorbidities associated with HFpEF Each comorbidity is associated with, and drives, low-grade, systemic inflammation.
Hemodynamic Overload
Hypertension is the most prevalent comorbidity in HFpEF, affecting the majority of patients with the syndrome (57–59). There is increasing evidence that hypertension-induced immune activation is a central element mediating myocardial damage in hypertension (60). As in HFpEF, hypertensive patients harbor increased circulating proinflammatory cytokines, including C-reactive protein (CRP), TNF-α, IL-1β, and IL-6 (61, 62).
Multiple animal models of hypertension manifest increased capillary permeability and immune cell recruitment to the myocardium with evidence of immune-mediated myocardial damage (63–65). Both innate and adaptive cellular immunity play crucial roles in mediating these effects. Like HFpEF, an imbalance in T-cell populations (Th17/Treg) skews the overall T-cell phenotype to proinflammatory. Dendritic cells play an important role in this imbalance by inducing polarization of T cells into proinflammatory Th17 cells, as was shown in the deoxycortisone acetate-salt (DOCA-salt) murine model (66). In fact, the presence of Th17 cells is necessary for the development of hypertension in angiotensin II (ANG II) models (67).
HTN can induce Nod-like receptor-containing pyrin domain 3 (NLRP-3) activation in antigen-presenting cells resulting in immune activation (68). Predominance of M1-like over M2-like macrophages in spontaneously hypertensive rats points to proinflammatory polarization of macrophages in hypertension (69, 70). Tissue infiltration of these inflammatory macrophages can then induce end-organ damage, increased CD11b+ macrophage adhesion, and migration in hypertension. Interestingly, the presence of these macrophages is also essential for development of hypertension (71).
Hypertension-induced myocyte damage promotes the release of damage-associated molecular patterns (DAMPs) capable of activating inflammatory cascades in both immune cells and in cardiomyocytes (72). DAMPs include high mobility group box 1 (HMGB1), heat shock proteins, DNA fragments, oxidized LDL, and mitochondrial content. DAMPs induce an inflammatory response via Toll-like receptors (TLRs), important mediators of inflammation in cardiovascular diseases (CVD) (73). In fact, hypertension results in increased cardiac TLR-4 expression and activity, contributing to chronic, myocardial, low-grade inflammation (74).
Aging
A strong association exists between aging and HFpEF, with the incidence of HFpEF doubling with every decade of life after 65 (75). Aging-associated systemic, chronic, low-grade inflammation, also known as “inflammaging,” has been recognized as an important pathophysiologic process increasing CVD risk in aging (76). Inflammaging is evidenced by the presence of elevated circulating proinflammatory cytokines, including IL-1, IL-6, IL-8, transforming growth factor-β (TGF-β), and TNF-α without the presence of active disease or comorbidities (77–80).
Secretion of senescence-associated secretory phenotype (SASP) signals by aging cells has emerged as a key driver of both local and systemic inflammation and a paracrine effector mediating senescent cardiomyocyte interaction with other myocardial cell populations. SASP signals include a wide range of bioactive molecules, such as proinflammatory cytokines, proteases, and extracellular matrix components that are secreted by senescent cells (81, 82). A specific proinflammatory subset of SASP signals that include chemotactic factors [monocyte chemoattractant protein 1–4 (MCP 1–4], proinflammatory cytokines (IL-1β, TNF-α, IL-6, etc.), DAMPs (e.g., HMGB1, DNA fragments, oxidized cardiolipin, etc.), and immune cell differentiation and maturation factors [macrophage colony-stimulating factor (M-CSF), granulocyte macrophage-colony-stimulating factor (GM-CSF), etc.] can contribute to inflammation (76). Activin, another proinflammatory SASP signal, for example, exacerbates age-related cardiac dysfunction (83). Interestingly, SASP-induced proliferation of CD38+ macrophages contributes to reductions in NAD+ levels seen in aging (84). As we have shown previously, NAD+ deficiency is an important feature of HFpEF (85).
Aging results in the infiltration of inflammatory cells into the myocardium, an indicator of myocardial inflammaging (21). Clonal hematopoiesis of indeterminate potential (CHIP) is another mechanism promoting immune dysregulation and subsequent cardiac dysfunction in aging (86, 87). Clinical studies have revealed a link between CHIP and increased 5-yr mortality in HF (88, 89). Numerous experimental studies have also linked CHIP driven by Dnmt3a, Tet2, or Jak2 to HF (87, 90–92), although a recent study has called into question CHIP-induced inflammation as a driver of CVD (93). Remarkably, Tet2 CHIP alone results in spontaneous cardiac dysfunction in aged mice (87, 92). This mutation has been directly linked to increased NLRP3/IL-1β signaling in the heart (91).
Chronic Kidney Disease
Like obesity and aging, chronic kidney disease (CKD) is associated with a persistent, systemic, proinflammatory state. As CKD progresses and the glomerular filtration rate (eGFR) declines, there is a proportional increase in inflammatory cytokines and acute phase reactant levels (94). Remarkably, in the background of CKD leading to increased levels of proinflammatory cytokines, acute phase reactants, or increased IL-1α expression in circulating monocytes, is associated with a higher risk of CVD (95, 96). Interestingly, the preclinical murine model of salty drinking water-unilateral nephrectomy-aldosterone (SAUNA) provides a clear indication that kidney-related disease can result in diastolic dysfunction, myocardial inflammation, and pathologic myocardial remodeling (21, 97–99).
Various factors have been implicated in driving systemic inflammation in CKD (100, 101). Increased production and decreased renal clearance of inflammatory cytokines contribute to elevated systemic levels (102). Uremia drives oxidative stress which in turn drives inflammation (103). Metabolic acidosis, a hallmark of CKD, and sodium overload can also induce inflammation (104, 105). Sympathetic nervous system stimulation (SNS) and renin-angiotensin-aldosterone (RAAS) activation, hallmarks of both HFpEF and CKD, have also been implicated in inflammation via multiple pathways (65, 106). Whereas both SNS and RAAS blockade strategies have failed in HFpEF, these two pathways remain contributors to overall inflammation.
Increased levels of circulating carbamylated LDL, a posttranslational modification of LDL that increases its inflammatory properties, have been reported in CKD (107, 108). Carbamylated LDL has been implicated in endothelial dysfunction and a subsequent decrease in endothelial nitric oxide (NO) release, a primary pathophysiologic mechanism in HFpEF (109). Similarly, symmetric dimethylarginine (SDMA) accumulates in HDL and has been implicated in TLR2-mediated endothelial inflammation and endothelial nitric oxide synthase (eNOS) inhibition (110–112).
Accumulation of proinflammatory microbiota-derived trimethylamine N-oxide (TMAO), an independent risk factor of HFpEF and predictor of renal dysfunction in HFpEF, occurs in CKD (113). A similar increase in other proinflammatory gut-derived toxins is also seen (114). SASP-driven inflammation occurs in CKD due to premature cellular senescence evidenced by the accumulation of cells expressing p16ink4a or p21cip1 (115).
Inflammation-Driven HFpEF Pathophysiology
Inflammation-Induced Extracellular Matrix Remodeling
Myocardial “reactive” fibrosis driven by ECM remodeling plays a key role in altering the mechanics of myocardial diastolic function (116). ECM remodeling is governed fundamentally by cardiac fibroblasts via the synthesis of fibrillar ECM proteins and secretion of degradative enzymes [matrix metalloproteases (MMPs) and ADAMTS] and regulators of collagen crosslinking (e.g., SPARC, osteopontin, lysyl oxidase) eliciting a net increase in ECM deposition (117).
Immune cell-fibroblast cross talk is a major driver of the differentiation of quiescent fibroblasts into activated myofibroblasts and thus a key element of pathological myocardial ECM remodeling in HFpEF. Various immune cell types, including macrophages, dendritic cells, mast cells, and B and T cells, have been implicated in fibroblast activation and myocardial fibrotic remodeling. An association between collagen deposition, immune cell infiltration, and diastolic dysfunction has been observed in HFpEF patients (118). In both HFpEF endomyocardial biopsies and different HFpEF preclinical models, it has been shown that both myocardial macrophage and neutrophil populations expand in HFpEF. Furthermore, macrophage-specific knockout of the profibrotic molecule IL-10 improved diastolic dysfunction and reduced fibrosis in these models (21). Inhibition of immune cell myocardial infiltration may provide a promising strategy to impede fibroblast activation and myocardial fibrosis. In fact, inhibition of macrophage migration to the myocardium with an anti-ICAM-1 monoclonal antibody (Nab) reduced myocardial fibrosis by inhibiting TGF-β expression and fibroblast proliferation (119). Similarly, treatment of ANG II-infused mice with MCP-1 monoclonal antibodies reduced myocardial macrophage accumulation and ameliorated diastolic dysfunction (120). As immune cells infiltrate the myocardium, the presence of a proinflammatory milieu triggers further inflammatory activation.
A variety of proinflammatory cytokines including IL-6, the IL-1 family, and IL-18 can induce fibroblast activation, several acting by converging on the TGF-β signaling pathway, a central mechanism governing fibroblast activation (121–123). TGF-β acting via both canonical and noncanonical pathways is central in downregulating MMPs, upregulating tissue inhibitors of matrix metalloproteases (TIMPs), and upregulating ECM protein expression, tipping the balance toward ECM expansion (124). Interestingly, elevations in the fibroblast activator short suppression of tumorigenicity 2 (sST2) and TIMP-1 have been associated with worse outcomes in HFpEF patients (125). Proinflammatory cytokines including TGF-β can also induce endothelial-to-mesenchymal transition (EndMT), which may be an important contributor to myocardial fibrosis in HFpEF (126).
Activation of cardiac fibroblasts can, in turn, contribute to myocardial inflammation provoking a positive feed-forward loop. NF-κB activation in myofibroblasts has been shown to increase Ly6Chi monocyte recruitment to the myocardium (127). In fact, cardiac fibroblasts can function as antigen presenting to CD4+ T cells via IFN-γ induced major histocompatibility complex type II (MHC II) (128). This points to a bidirectional cross talk between fibroblasts and immune cells that amplifies inflammation and fibrosis.
Whereas the PIROUETTE trial testing the anti-TGF-β drug pirfenidone in HFpEF revealed reduced ECM volume, there was no change in diastolic function (129). Limited reduction in ECM volume may underlie a lack of improvement in diastolic dysfunction. Interestingly, pirfenidone has anti-inflammatory properties and may still emerge as a useful tool (130, 131). Whereas traditional methods of detecting fibrosis have revealed more modest degrees of fibrosis in HFpEF than previously thought, a potentially less recognized role of “hidden fibrosis” resulting from an increase in ECM collagen deposition detectable by highly sensitive methods, such as quantitative mass spectrometry, may play an important role in diastolic dysfunction (24, 132, 133). Highly sensitive methods of detection of myocardial fibrosis may help to further decipher ECM remodeling in HFpEF. Furthermore, alternative strategies targeting fibrosis may prove fruitful. Chimeric antigen receptor T cell (CAR-T) therapy has been used to transiently target fibroblasts, reduce fibrosis, and improve cardiac function in different HF models (134, 135). Similarly, cardiosphere-derived cells (CDCs) reduced inflammatory infiltrates, myocardial fibrosis, and ventricular arrhythmias in Dahl salt-sensitive rats (136, 137).
Groundbreaking results from the EMPEROR-Preserved trial indicate empagliflozin can reduce HF hospitalizations in HFpEF (58). Among the diverse effects of SGLT2 inhibition is the ability to attenuate both systemic and myocardial inflammation. Recently, increased expression of HMGB1, a DAMP, was reported to be critical to neutrophil extracellular trap (NET) formation in HFpEF. This HMGB-NET axis can mediate diastolic dysfunction and myocardial fibrosis and is inhibited with SGLT2 inhibitor therapy (138). Similarly, SGLT2 inhibition has been shown to reduce systemic inflammation in both animal models and patient populations with diabetes (139–142). Whereas anti-inflammatory mechanisms remain under investigation, SGLT2 inhibitors have been shown to reduce NLRP3 activation in various tissues including the heart (143–146). Empagliflozin-mediated suppression of increases in β-hydroxybutyrate can block NLRP3 activation in diabetic individuals at high risk of cardiovascular disease (147). A similar effect on NLRP3 has been reported in rodent models of HF (146). Empagliflozin has also been shown to promote adipose tissue browning and reduce inflammatory M1 macrophage accumulation in adipose tissue, possibly limiting metabolic inflammation (147). Myocardial antifibrotic effects have also been reported in several models of cardiovascular disease (148–150) (FIGURE 3).
FIGURE 3.
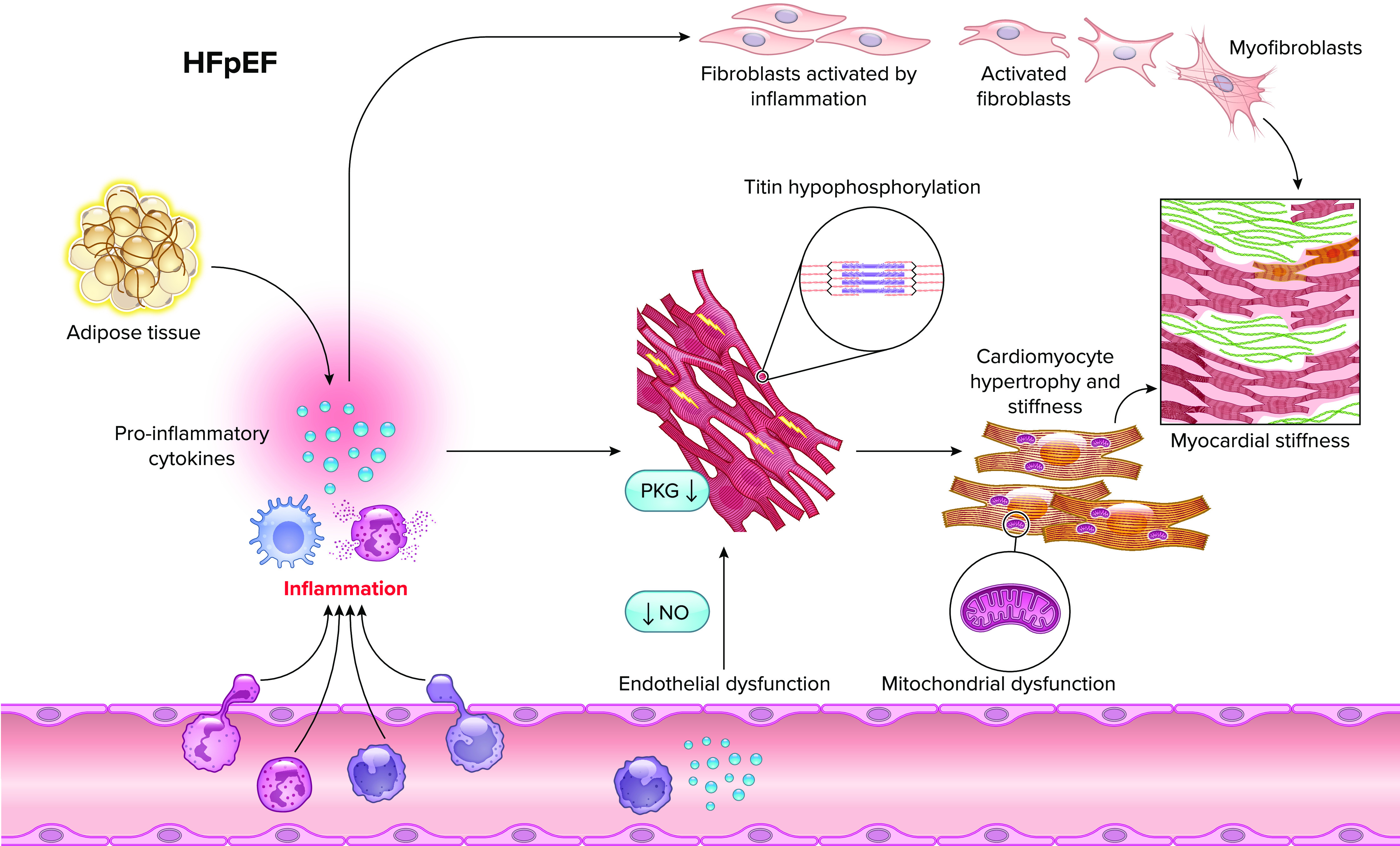
Inflammation underpins pathological cardiomyocyte and fibroblast alterations that contribute to myocardial stiffness in HFpEF NO, nitric oxide.
Inflammation-Induced Cardiomyocyte Dysfunction
Myocardial stiffness in HFpEF is multifactorial, and inflammation-driven alterations in cardiomyocyte morphology, homeostasis, distensibility, and electrophysiology are important contributors. Inflammation and nitro-oxidative stress are closely linked and important contributors to HFpEF pathobiology. NO signaling via iNOS is induced by inflammation and plays a fundamental role in mediating cardiomyocyte dysfunction in HFpEF. As we have reported in both patient samples and in a HFD plus Nω-nitro-l-arginine methyl ester (l-NAME ) mouse model of HFpEF (151), upregulation of cardiomyocyte inducible nitric oxide synthase (iNOS) leads to S-nitrosylation of the endonuclease inositol-requiring protein 1α (IRE1α) resulting in defective XBP1 splicing, accumulation of unfolded proteins, and cardiomyocyte dysfunction (151, 152).
The mitochondrial respiratory chain is the primary source of oxidative stress in CVD, with other mitochondrial proteins, such as NADPH oxidases (NOX2/4), xanthine oxidase, and cytochrome P450 enzymes contributing to the reactive oxygen species (ROS) pool (153). Inflammation can drive ROS production and vice versa, triggering a vicious cycle of inflammation and oxidative stress (154). Proinflammatory cytokine signaling is a culprit in macrophage-mediated inflammation and has been implicated in HFpEF in driving mitochondrial ROS via IL-1β (155). TGF-β can increase NOX4 activity and reduce antioxidant factors such as glutathione, driving ROS accumulation (156). IL-6 and TNF-α have been similarly implicated in increasing oxidative stress (154, 157).
Titin compliance is important in cardiomyocyte distensibility. Coronary endothelial inflammation resulting in dysregulation of the eNOS-cGMP-PKG axis with consequent hypophosphorylation of cardiomyocyte titin is a proposed mechanism of reduced passive cardiomyocyte elasticity in HFpEF; however, it is important to note that cardiomyocytes have an innate ability to stimulate PKG via NO production. Activation of PKG or PKA in vivo improves left ventricular (LV) chamber compliance via titin N2B segment phosphorylation (158). Interestingly, activation of PKG has a wide range of effects on the myocardium, including reduced inflammation, hypertrophy, and fibrosis (159–161). In addition, PKG is known to stimulate autophagy, improve mitochondrial function, and increase the clearance of misfolded proteins via proteasomal degradation, making it an attractive therapeutic target (162–164). Several phosphodiesterase (PDE)-5 and -9 inhibitors that augment cardiomyocyte cGMP, and thereby PKG activity, have been examined in multiple preclinical models of HFpEF (165–168). Whereas clinical trials using PDE5 inhibitors have revealed no improvement in patient clinical status, we are still awaiting results from PDE9 inhibitor trials (169). Important differential actions of PDE5 and PDE9 exist due to effects on NO-stimulated cGMP in the former and natriuretic peptide-stimulated cGMP in the latter (167, 170). Alternatively, targeting inflammation may prove to be successful in attenuating endothelial dysfunction and dysregulation of the eNOS-cGMP-PKG axis. The IL-1 antagonist anakinra reduced endothelial NOX and NF-κB activation and subsequently endothelial dysfunction in streptozotocin-induced diabetic rats (171). Whereas cytoskeletal α-tubulin abundance/posttranslational modifications and titin isoform ratios have been linked to increased cardiomyocyte stiffness in HFpEF, no link has been made with inflammation (172–174).
Proinflammatory cytokine signaling can alter cardiac electrophysiology contributing to abnormal diastolic function. In vitro studies examining myocyte relaxation time and contractility have identified IL-1β, IFN-γ, and poly(I:C) as inducers of abnormal cardiomyocyte relaxation time without affecting contractility (175). In vivo, inhibition of IL-1β or macrophage depletion ameliorated diastolic dysfunction in a HFD model of HFpEF, whereas infusion of IL-6 alone can induce cardiac hypertrophy, fibrosis, and diastolic dysfunction (155, 176). Although targeting individual cytokines in those who have already developed overt HFpEF has proven unsuccessful in clinical trials, a preventive strategy as seen in CANTOS may prove fruitful. The challenge, however, remains in identifying high-risk populations that may benefit from this strategy.
Cardiomyocyte hypertrophy is another important contributor to the HFpEF phenotype, and inflammation, via immune cell-cardiomyocyte interaction and secreted factors, is a key driver of LV hypertrophic remodeling. In mice exposed to increased afterload, macrophage-specific knockout of microRNA-155 reduced monocyte/macrophage myocardial infiltration, cardiomyocyte hypertrophy, and cardiac function (177). Innate and adaptive immune cell cross talk can also modulate LV hypertrophic remodeling. Suppression of CCR2+ macrophages, for example, attenuates lymph node CD3+ T cell expansion and cardiac hypertrophy (178). Interestingly, CXCL1-CXCR2 signaling has been implicated in myocardial monocyte infiltration and hypertrophic remodeling in ANG II-infused animals and may mediate the myocardial accumulation of inflammatory cells in HFpEF (179). Although the specific cell sources have not been identified, individual cytokines, such as IL-6, IL-1β, TNF-α, and TGF-β, have also been implicated in cardiomyocyte hypertrophy (180–182).
Adipoinflammatory Myocardial Remodeling
As discussed previously, AT remodeling is an important contributor to systemic and myocardial inflammation in cardiometabolic HFpEF. Adipocyte-immune cell cross talk is well characterized in obesity-induced AT inflammation and is beginning to emerge as an important player in myocardial remodeling.
Adipocyte-secreted free fatty acids trigger resident macrophage release of TNF-α, establishing a paracrine loop that worsens AT inflammation (183). Such cross talk can also act to promote myocardial remodeling as seen with a recently identified macrophage-adipocyte axis; macrophages recruited to the myocardium in HF secrete urokinase plasminogen activator (uPA), cleaving adipocyte-secreted full-length platelet-derived growth factor-D (PDGF-D ) and promoting pathologic myocardial remodeling (184, 185).
Adaptive immune cell-AT interactions have also been reported in CVD. FALCs, harboring B and T cells, are present in EAT and can expand in obesity and inflammation to mediate systemic and myocardial inflammation in pathological states (48). In mice, B-cell depletion from FALCs results in reduced bone marrow granulopoiesis and neutrophil infiltration to the myocardium after myocardial infarction, reducing fibrosis (42). Similarly, global B cell depletion in ANG II-infused mice using a CD22 antibody resulted in reduced myocardial remodeling and improved function (186). These clusters can also interact with myeloid cells potentially modulating their activity (48, 187). Such interactions are yet to be reported in HFpEF; furthermore, their existence is sporadic. These findings highlight the role of EAT as not only a source of inflammatory adipokines but an immune cell reservoir directly affecting the myocardium.
Exacerbation of EAT inflammation in obesity via ACE2 knockout results in worsened cardiac function and steatosis (47). Similarly, activation of hypoxia-inducible factor 2α (HIF2α ) in AT results in pathological cardiac hypertrophy, elevated levels of circulating proinflammatory cytokines, and myocardial inflammation via NF-κB and nuclear factor of activated T-cell (NFAT) activation (188). Myocardial homeostasis is directly affected via an array of secreted factors including inflammatory adipokines. Secreted retinol-binding protein 4 (RBP4) induces the expression of TLR4 and myeloid differentiation primary response gene 88 (MyD88) resulting in cardiomyocyte inflammation, hypertrophy, and oxidative stress. RBP4 also reduces the expression of glucose-transporter 4 (GLUT4) and mediates cardiomyocyte insulin resistance, a cardinal feature of cardiometabolic HFpEF (189).
AT also mediates protective homeostatic functions under normal conditions. Cross talk between brown AT (BAT) and the myocardium can prevent hypertension-induced cardiac hypertrophy via adenosine A2A receptor-derived secretion of FGF21 from BAT (190). Similarly, adiponectin is a cardioprotective adipokine, whose levels decrease in obesity (191). Reversal of AT inflammation and promoting BAT formation may prove productive in reducing pathologic changes seen in cardiometabolic HFpEF.
Inflammation-Mediated Metabolic and Energetic Dysfunction
Structural and bioenergetic abnormalities in cardiomyocyte mitochondria are characteristic of HFpEF, resulting in a mismatch between ATP demand/production and energy deficit (192). Mitochondrial dysfunction is accompanied by altered cardiomyocyte metabolism, including metabolic inflexibility, impaired fatty acid and glucose oxidation, and increased fatty acid uptake with myocardial steatosis (85, 152). ROS have been proposed as the major contributor to mitochondrial dysfunction and can promote inflammation, cardiac remodeling, and diastolic dysfunction (193, 194). However, evidence is emerging that implicates inflammation-induced mitochondrial dysfunction as an important driver of HFpEF.
Mitochondrial protein acetylation is a crucial posttranslational modification regulating mitochondrial energy metabolism. Mitochondrial hyperacetylation in HFpEF occurs via sirtuin 3 downregulation and a defective NAD+ salvage pathway and is linked to impairment of oxidative metabolism and metabolic inflexibility, both hallmarks of HFpEF (85, 195). Evidence is emerging that implicates hyperacetylation as a driver of inflammation. Mitochondrial hyperacetylation increases NLRP3 assembly triggering increased production of IL-1β/IL-18 and resulting in mitochondrial dysfunction in cardiometabolic HFpEF. Interestingly, β-hydroxybutyrate supplementation reverses diastolic dysfunction, NLRP3 activation, and mitochondrial dysfunction by reducing mitochondrial hyperacetylation (196).
The proinflammatory cytokines IL-6, IL-1β, TNF-α, and TGF-β have all been implicated in mitochondrial dysfunction via ROS generation (154–156). Similarly, iNOS-mediated nitrosative stress triggers mitochondrial dysfunction in HFpEF. iNOS inhibition ameliorated mitochondrial injury and dysfunction while also reducing Akt S-nitrosylation and thereby insulin resistance in HFpEF (151). Mitochondria can also contribute to inflammation via ROS generation and the release of DAMPs that trigger immune cell activation (197). This generates a cycle of inflammation and mitochondrial dysfunction. Thus, targeting inflammatory pathways may prove successful in ameliorating mitochondrial dysfunction in HFpEF.
Conclusions and Perspectives
In light of important experimental and clinical findings discussed here, targeting inflammation as a therapeutic strategy in HFpEF warrants further investigation. Whereas the DHART2 trial revealed reductions in NT-proBNP and high-sensitivity CRP with IL-1β blockade in HFpEF, it did not translate into improvement in clinical parameters (198). A large-scale, randomized clinical trial may be required to derive reliable conclusions regarding IL-1β blockade in HFpEF; in the meantime, reliance on alternate strategies that target inflammation differently or more broadly may prove fruitful. Examining anti-inflammatory mechanisms as a preventive strategy in a high-risk subset of patients may also prove successful and warrants further consideration. Also, worthy of further exploration is delineation of subsets of HFpEF patients manifesting strong inflammatory profiles who might benefit from anti-inflammatory therapies.
In summary, comorbidity-driven inflammation is emerging as a critical contributor to HFpEF pathogenesis and progression. Cardiomyocyte dysfunction and pathologic ECM remodeling occur as downstream effects of chronic low-grade inflammation, generating myocardial stiffness and reduced ventricular compliance. Discovery of novel inflammatory mechanisms driving key pathophysiological events in HFpEF is paramount to better understanding of syndrome development and progression and therapeutic intervention.
Acknowledgments
This work was supported by National Institutes of Health Grants HL128215 (to J.A.H.), HL147933 (to J.A.H.), HL155765 (to J.A.H., T.G.G.), HL164586 (to J.A.H., T.G.G.); American Heart Association: 23 POST1019228 (to D.D.); and Foundation for the National Institutes of Health Grant S10RR02372.
No conflicts of interest, financial or otherwise, are declared by the authors.
D.D. drafted manuscript; T.G.G. and J.A.H. edited and revised manuscript; T.G.G. and J.A.H. approved final version of manuscript.
References
- 1. Roth GA, Mensah GA, Johnson CO, Addolorato G, Ammirati E, Baddour LM, , et al. Global burden of cardiovascular diseases and risk factors, 1990-2019: update from the GBD 2019 study. J Am Coll Cardiol 76: 2982–3021, 2020. doi: 10.1016/j.jacc.2020.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dunlay SM, Roger VL, Redfield MM. Epidemiology of heart failure with preserved ejection fraction. Nat Rev Cardiol 14: 591–602, 2017. doi: 10.1038/nrcardio.2017.65. [DOI] [PubMed] [Google Scholar]
- 3. Schiattarella GG, Rodolico D, Hill JA. Metabolic inflammation in heart failure with preserved ejection fraction. Cardiovasc Res 117: 423–434, 2021. doi: 10.1093/cvr/cvaa217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schiattarella GG, Alcaide P, Condorelli G, Gillette TG, Heymans S, Jones EA, Kallikourdis M, Lichtman A, Marelli-Berg F, Shah S, Thorp EB, Hill JA. Immunometabolic mechanisms of heart failure with preserved ejection fraction. Nat Cardiovasc Res 1: 211–222, 2022. doi: 10.1038/s44161-022-00032-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Redfield MM, Borlaug BA. Heart failure with preserved ejection fraction: a review. JAMA 329: 827–838, 2023. doi: 10.1001/jama.2023.2020. [DOI] [PubMed] [Google Scholar]
- 6. Dick SA, Epelman S. Chronic heart failure and inflammation: what do we really know? Circ Res 119: 159–176, 2016. doi: 10.1161/CIRCRESAHA.116.308030. [DOI] [PubMed] [Google Scholar]
- 7. Vasan RS, Sullivan LM, Roubenoff R, Dinarello CA, Harris T, Benjamin EJ, Sawyer DB, Levy D, Wilson PW, D'Agostino RB, Framingham HS, Framingham Heart Study. Inflammatory markers and risk of heart failure in elderly subjects without prior myocardial infarction: the Framingham Heart Study. Circulation 107: 1486–1491, 2003. doi: 10.1161/01.cir.0000057810.48709.f6. [DOI] [PubMed] [Google Scholar]
- 8. Deswal A, Petersen NJ, Feldman AM, Young JB, White BG, Mann DL. Cytokines and cytokine receptors in advanced heart failure: an analysis of the cytokine database from the Vesnarinone trial (VEST). Circulation 103: 2055–2059, 2001. doi: 10.1161/01.cir.103.16.2055. [DOI] [PubMed] [Google Scholar]
- 9. Hage C, Michaelsson E, Linde C, Donal E, Daubert JC, Gan LM, Lund LH. Inflammatory biomarkers predict heart failure severity and prognosis in patients with heart failure with preserved ejection fraction: a holistic proteomic approach. Circ Cardiovasc Genet 10: e001633, 2017. doi: 10.1161/CIRCGENETICS.116.001633. [DOI] [PubMed] [Google Scholar]
- 10. Pandey A, Vaduganathan M, Arora S, Qamar A, Mentz RJ, Shah SJ, Chang PP, Russell SD, Rosamond WD, Caughey MC. Temporal trends in prevalence and prognostic implications of comorbidities among patients with acute decompensated heart failure: the ARIC study community surveillance. Circulation 142: 230–243, 2020. doi: 10.1161/CIRCULATIONAHA.120.047019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shah SJ, Katz DH, Selvaraj S, Burke MA, Yancy CW, Gheorghiade M, Bonow RO, Huang CC, Deo RC. Phenomapping for novel classification of heart failure with preserved ejection fraction. Circulation 131: 269–279, 2015. doi: 10.1161/CIRCULATIONAHA.114.010637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Segar MW, Patel KV, Ayers C, Basit M, Tang WH, Willett D, Berry J, Grodin JL, Pandey A. Phenomapping of patients with heart failure with preserved ejection fraction using machine learning-based unsupervised cluster analysis. Eur J Heart Fail 22: 148–158, 2020. doi: 10.1002/ejhf.1621. [DOI] [PubMed] [Google Scholar]
- 13. Hedman AK, Hage C, Sharma A, Brosnan MJ, Buckbinder L, Gan LM, Shah SJ, Linde CM, Donal E, Daubert JC, Malarstig A, Ziemek D, Lund L. Identification of novel pheno-groups in heart failure with preserved ejection fraction using machine learning. Heart 106: 342–349, 2020. doi: 10.1136/heartjnl-2019-315481. [DOI] [PubMed] [Google Scholar]
- 14. Sabbah MS, Fayyaz AU, de Denus S, Felker GM, Borlaug BA, Dasari S, Carter RE, Redfield MM. Obese-inflammatory phenotypes in heart failure with preserved ejection fraction. Circ Heart Fail 13: e006414, 2020. doi: 10.1161/CIRCHEARTFAILURE.119.006414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cohen JB, Schrauben SJ, Zhao L, Basso MD, Cvijic ME, Li Z, Yarde M, Wang Z, Bhattacharya PT, Chirinos DA, Prenner S, Zamani P, Seiffert DA, Car BD, Gordon DA, Margulies K, Cappola T, Chirinos JA. Clinical phenogroups in heart failure with preserved ejection fraction: detailed phenotypes, prognosis, and response to spironolactone. JACC Heart Fail 8: 172–184, 2020. doi: 10.1016/j.jchf.2019.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shah AM, Claggett B, Sweitzer NK, Shah SJ, Anand IS, O'Meara E, Desai AS, Heitner JF, Li G, Fang J, Rouleau J, Zile MR, Markov V, Ryabov V, Reis G, Assmann SF, McKinlay SM, Pitt B, Pfeffer MA, Solomon SD. Cardiac structure and function and prognosis in heart failure with preserved ejection fraction: findings from the echocardiographic study of the Treatment of Preserved Cardiac Function Heart Failure with an Aldosterone Antagonist (TOPCAT) Trial. Circ Heart Fail 7: 740–751, 2014. doi: 10.1161/CIRCHEARTFAILURE.114.001583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Levine B, Kalman J, Mayer L, Fillit HM, Packer M. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N Engl J Med 323: 236–241, 1990. doi: 10.1056/NEJM199007263230405. [DOI] [PubMed] [Google Scholar]
- 18. Torre-Amione G, Kapadia S, Lee J, Durand JB, Bies RD, Young JB, Mann DL. Tumor necrosis factor-alpha and tumor necrosis factor receptors in the failing human heart. Circulation 93: 704–711, 1996. doi: 10.1161/01.cir.93.4.704. [DOI] [PubMed] [Google Scholar]
- 19. Edelmann F, Holzendorf V, Wachter R, Nolte K, Schmidt AG, Kraigher-Krainer E, Duvinage A, Unkelbach I, Dungen HD, Tschope C, Herrmann-Lingen C, Halle M, Hasenfuss G, Gelbrich G, Stough WG, Pieske BM. Galectin-3 in patients with heart failure with preserved ejection fraction: results from the Aldo-DHF trial. Eur J Heart Fail 17: 214–223, 2015. doi: 10.1002/ejhf.203. [DOI] [PubMed] [Google Scholar]
- 20. Mann DL. Innate immunity and the failing heart: the cytokine hypothesis revisited. Circ Res 116: 1254–1268, 2015. doi: 10.1161/CIRCRESAHA.116.302317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hulsmans M, Sager HB, Roh JD, Valero-Munoz M, Houstis NE, Iwamoto Y, Sun Y, Wilson RM, Wojtkiewicz G, Tricot B, Osborne MT, Hung J, Vinegoni C, Naxerova K, Sosnovik DE, Zile MR, Bradshaw AD, Liao R, Tawakol A, Weissleder R, Rosenzweig A, Swirski FK, Sam F, Nahrendorf M. Cardiac macrophages promote diastolic dysfunction. J Exp Med 215: 423–440, 2018. doi: 10.1084/jem.20171274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Franssen C, Chen S, Unger A, Korkmaz HI, De Keulenaer GW, Tschope C, Leite-Moreira AF, Musters R, Niessen HW, Linke WA, Paulus WJ, Hamdani N. Myocardial microvascular inflammatory endothelial activation in heart failure with preserved ejection fraction. JACC Heart Fail 4: 312–324, 2016. doi: 10.1016/j.jchf.2015.10.007. [DOI] [PubMed] [Google Scholar]
- 23. Li N, Bian H, Zhang J, Li X, Ji X, Zhang Y. The Th17/Treg imbalance exists in patients with heart failure with normal ejection fraction and heart failure with reduced ejection fraction. Clin Chim Acta 411: 1963–1968, 2010. doi: 10.1016/j.cca.2010.08.013. [DOI] [PubMed] [Google Scholar]
- 24. Hahn VS, Yanek LR, Vaishnav J, Ying W, Vaidya D, Lee YZ, Riley SJ, Subramanya V, Brown EE, Hopkins CD, Ononogbu S, Perzel Mandell K, Halushka MK, Steenbergen C Jr, Rosenberg AZ, Tedford RJ, Judge DP, Shah SJ, Russell SD, Kass DA, Sharma K. Endomyocardial biopsy characterization of heart failure with preserved ejection fraction and prevalence of cardiac amyloidosis. JACC Heart Fail 8: 712–724, 2020. doi: 10.1016/j.jchf.2020.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kaye DM, Nanayakkara S, Wang B, Shihata W, Marques FZ, Esler M, Lambert G, Mariani J. Characterization of cardiac sympathetic nervous system and inflammatory activation in HFpEF patients. JACC Basic Transl Sci 7: 116–127, 2022. doi: 10.1016/j.jacbts.2021.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Paulus WJ, Tschope C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol 62: 263–271, 2013. doi: 10.1016/j.jacc.2013.02.092. [DOI] [PubMed] [Google Scholar]
- 27. Sanders-van Wijk S, Tromp J, Beussink-Nelson L, Hage C, Svedlund S, Saraste A, Swat SA, Sanchez C, Njoroge J, Tan RS, Fermer ML, Gan LM, Lund LH, Lam CS, Shah SJ. Proteomic evaluation of the comorbidity-inflammation paradigm in heart failure with preserved ejection fraction: results from the PROMIS-HFpEF study. Circulation 142: 2029–2044, 2020. doi: 10.1161/CIRCULATIONAHA.120.045810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tromp J, Westenbrink BD, Ouwerkerk W, van Veldhuisen DJ, Samani NJ, Ponikowski P, Metra M, Anker SD, Cleland JG, Dickstein K, Filippatos G, van der Harst P, Lang CC, Ng LL, Zannad F, Zwinderman AH, Hillege HL, van der Meer P, Voors AA. Identifying pathophysiological mechanisms in heart failure with reduced versus preserved ejection fraction. J Am Coll Cardiol 72: 1081–1090, 2018. doi: 10.1016/j.jacc.2018.06.050. [DOI] [PubMed] [Google Scholar]
- 29. Hahn VS, Knutsdottir H, Luo X, Bedi K, Margulies KB, Haldar SM, Stolina M, Yin J, Khakoo AY, Vaishnav J, Bader JS, Kass DA, Sharma K. Myocardial gene expression signatures in human heart failure with preserved ejection fraction. Circulation 143: 120–134, 2021. doi: 10.1161/CIRCULATIONAHA.120.050498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Obokata M, Reddy YN, Pislaru SV, Melenovsky V, Borlaug BA. Evidence supporting the existence of a distinct obese phenotype of heart failure with preserved ejection fraction. Circulation 136: 6–19, 2017. doi: 10.1161/CIRCULATIONAHA.116.026807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wu H, Ballantyne CM. Metabolic inflammation and insulin resistance in obesity. Circ Res 126: 1549–1564, 2020. doi: 10.1161/CIRCRESAHA.119.315896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lee YS, Kim JW, Osborne O, Oh DY, Sasik R, Schenk S, Chen A, Chung H, Murphy A, Watkins SM, Quehenberger O, Johnson RS, Olefsky JM. Increased adipocyte O2 consumption triggers HIF-1alpha, causing inflammation and insulin resistance in obesity. Cell 157: 1339–1352, 2014. doi: 10.1016/j.cell.2014.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT, Brickey WJ, Ting JP. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol 12: 408–415, 2011. doi: 10.1038/ni.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bleau C, Karelis AD, St-Pierre DH, Lamontagne L. Crosstalk between intestinal microbiota, adipose tissue and skeletal muscle as an early event in systemic low-grade inflammation and the development of obesity and diabetes. Diabetes Metab Res Rev 31: 545–561, 2015. doi: 10.1002/dmrr.2617. [DOI] [PubMed] [Google Scholar]
- 35. Tilg H, Zmora N, Adolph TE, Elinav E. The intestinal microbiota fuelling metabolic inflammation. Nat Rev Immunol 20: 40–54, 2020. doi: 10.1038/s41577-019-0198-4. [DOI] [PubMed] [Google Scholar]
- 36. Fujisaka S, Usui I, Ikutani M, Aminuddin A, Takikawa A, Tsuneyama K, Mahmood A, Goda N, Nagai Y, Takatsu K, Tobe K. Adipose tissue hypoxia induces inflammatory M1 polarity of macrophages in an HIF-1alpha-dependent and HIF-1alpha-independent manner in obese mice. Diabetologia 56: 1403–1412, 2013. doi: 10.1007/s00125-013-2885-1. [DOI] [PubMed] [Google Scholar]
- 37. Packer M. Epicardial adipose tissue may mediate deleterious effects of obesity and inflammation on the myocardium. J Am Coll Cardiol 71: 2360–2372, 2018. doi: 10.1016/j.jacc.2018.03.509. [DOI] [PubMed] [Google Scholar]
- 38. Koepp KE, Obokata M, Reddy YN, Olson TP, Borlaug BA. Hemodynamic and functional impact of epicardial adipose tissue in heart failure with preserved ejection fraction. JACC Heart Fail 8: 657–666, 2020. doi: 10.1016/j.jchf.2020.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. van Woerden G, van Veldhuisen DJ, Manintveld OC, van Empel VP, Willems TP, de Boer RA, Rienstra M, Westenbrink BD, Gorter TM. Epicardial adipose tissue and outcome in heart failure with mid-range and preserved ejection fraction. Circ Heart Fail 15: e009238, 2022. doi: 10.1161/CIRCHEARTFAILURE.121.009238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Iacobellis G, Barbaro G. The double role of epicardial adipose tissue as pro- and anti-inflammatory organ. Horm Metab Res 40: 442–445, 2008. doi: 10.1055/s-2008-1062724. [DOI] [PubMed] [Google Scholar]
- 41. Iacobellis G. Local and systemic effects of the multifaceted epicardial adipose tissue depot. Nat Rev Endocrinol 11: 363–371, 2015. doi: 10.1038/nrendo.2015.58. [DOI] [PubMed] [Google Scholar]
- 42. Horckmans M, Bianchini M, Santovito D, Megens RT, Springael JY, Negri I, Vacca M, Di Eusanio M, Moschetta A, Weber C, Duchene J, Steffens S. Pericardial adipose tissue regulates granulopoiesis, fibrosis, and cardiac function after myocardial infarction. Circulation 137: 948–960, 2018. doi: 10.1161/CIRCULATIONAHA.117.028833. [DOI] [PubMed] [Google Scholar]
- 43. van Dam AD, Boon MR, Berbee JF, Rensen PC, van Harmelen V. Targeting white, brown and perivascular adipose tissue in atherosclerosis development. Eur J Pharmacol 816: 82–92, 2017. doi: 10.1016/j.ejphar.2017.03.051. [DOI] [PubMed] [Google Scholar]
- 44. Cheng KH, Chu CS, Lee KT, Lin TH, Hsieh CC, Chiu CC, Voon WC, Sheu SH, Lai WT. Adipocytokines and proinflammatory mediators from abdominal and epicardial adipose tissue in patients with coronary artery disease. Int J Obes (Lond) 32: 268–274, 2008. doi: 10.1038/sj.ijo.0803726. [DOI] [PubMed] [Google Scholar]
- 45. Gruzdeva OV, Akbasheva OE, Dyleva YA, Antonova LV, Matveeva VG, Uchasova EG, Fanaskova EV, Karetnikova VN, Ivanov SV, Barbarash OL. Adipokine and cytokine profiles of epicardial and subcutaneous adipose tissue in patients with coronary heart disease. Bull Exp Biol Med 163: 608–611, 2017. doi: 10.1007/s10517-017-3860-5. [DOI] [PubMed] [Google Scholar]
- 46. Patel VB, Basu R, Oudit GY. ACE2/Ang 1-7 axis: a critical regulator of epicardial adipose tissue inflammation and cardiac dysfunction in obesity. Adipocyte 5: 306–311, 2016. doi: 10.1080/21623945.2015.1131881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Patel VB, Mori J, McLean BA, Basu R, Das SK, Ramprasath T, Parajuli N, Penninger JM, Grant MB, Lopaschuk GD, Oudit GY. ACE2 deficiency worsens epicardial adipose tissue inflammation and cardiac dysfunction in response to diet-induced obesity. Diabetes 65: 85–95, 2016. doi: 10.2337/db15-0399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Benezech C, Luu NT, Walker JA, Kruglov AA, Loo Y, Nakamura K, Zhang Y, Nayar S, Jones LH, Flores-Langarica A, McIntosh A, Marshall J, Barone F, Besra G, Miles K, Allen JE, Gray M, Kollias G, Cunningham AF, Withers DR, Toellner KM, Jones ND, Veldhoen M, Nedospasov SA, McKenzie AN, Caamano JH. Inflammation-induced formation of fat-associated lymphoid clusters. Nat Immunol 16: 819–828, 2015. doi: 10.1038/ni.3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Antonopoulos AS, Antoniades C. The role of epicardial adipose tissue in cardiac biology: classic concepts and emerging roles. J Physiol 595: 3907–3917, 2017. doi: 10.1113/JP273049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. McKnight SL. On getting there from here. Science 330: 1338–1339, 2010. doi: 10.1126/science.1199908. [DOI] [PubMed] [Google Scholar]
- 51. Norata GD, Caligiuri G, Chavakis T, Matarese G, Netea MG, Nicoletti A, O'Neill LA, Marelli-Berg FM. The cellular and molecular basis of translational immunometabolism. Immunity 43: 421–434, 2015. doi: 10.1016/j.immuni.2015.08.023. [DOI] [PubMed] [Google Scholar]
- 52. Zasłona Z, O'Neill LA. Cytokine-like roles for metabolites in immunity. Mol Cell 78: 814–823, 2020. doi: 10.1016/j.molcel.2020.04.002. [DOI] [PubMed] [Google Scholar]
- 53. Murphy MP, O'Neill LA. Krebs cycle reimagined: the emerging roles of succinate and itaconate as signal transducers. Cell 174: 780–784, 2018. doi: 10.1016/j.cell.2018.07.030. [DOI] [PubMed] [Google Scholar]
- 54. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, , et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 496: 238–242, 2013. doi: 10.1038/nature11986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Arts RJ, Novakovic B, Ter Horst R, Carvalho A, Bekkering S, Lachmandas E, Rodrigues F, Silvestre R, Cheng SC, Wang SY, Habibi E, Goncalves LG, Mesquita I, Cunha C, van Laarhoven A, van de Veerdonk FL, Williams DL, van der Meer JW, Logie C, O'Neill LA, Dinarello CA, Riksen NP, van Crevel R, Clish C, Notebaart RA, Joosten LA, Stunnenberg HG, Xavier RJ, Netea MG. Glutaminolysis and fumarate accumulation integrate immunometabolic and epigenetic programs in trained immunity. Cell Metab 24: 807–819, 2016. doi: 10.1016/j.cmet.2016.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ko HJ, Zhang Z, Jung DY, Jun JY, Ma Z, Jones KE, Chan SY, Kim JK. Nutrient stress activates inflammation and reduces glucose metabolism by suppressing AMP-activated protein kinase in the heart. Diabetes 58: 2536–2546, 2009. doi: 10.2337/db08-1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Solomon SD, Rizkala AR, Lefkowitz MP, Shi VC, Gong J, Anavekar N, , et al. Baseline Characteristics of patients with heart failure and preserved ejection fraction in the PARAGON-HF trial. Circ Heart Fail 11: e004962, 2018. doi: 10.1161/CIRCHEARTFAILURE.118.004962. [DOI] [PubMed] [Google Scholar]
- 58. Anker SD, Butler J, Packer M. Empagliflozin in heart failure with a preserved ejection fraction. Reply. N Engl J Med 386: e57, 2022. doi: 10.1056/NEJMc2118470. [DOI] [PubMed] [Google Scholar]
- 59. Solomon SD, McMurray JJ, Anand IS, Ge J, Lam CS, Maggioni AP, , et al. Angiotensin-neprilysin inhibition in heart failure with preserved ejection fraction. N Engl J Med 381: 1609–1620, 2019. doi: 10.1056/NEJMoa1908655. [DOI] [PubMed] [Google Scholar]
- 60. McMaster WG, Kirabo A, Madhur MS, Harrison DG. Inflammation, immunity, and hypertensive end-organ damage. Circ Res 116: 1022–1033, 2015. doi: 10.1161/CIRCRESAHA.116.303697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bautista LE, Vera LM, Arenas IA, Gamarra G. Independent association between inflammatory markers (C-reactive protein, interleukin-6, and TNF-alpha) and essential hypertension. J Hum Hypertens 19: 149–154, 2005. doi: 10.1038/sj.jhh.1001785. [DOI] [PubMed] [Google Scholar]
- 62. Dalekos GN, Elisaf M, Bairaktari E, Tsolas O, Siamopoulos KC. Increased serum levels of interleukin-1beta in the systemic circulation of patients with essential hypertension: additional risk factor for atherogenesis in hypertensive patients? J Lab Clin Med 129: 300–308, 1997. doi: 10.1016/s0022-2143(97)90178-5. [DOI] [PubMed] [Google Scholar]
- 63. Behr TM, Willette RN, Coatney RW, Berova M, Angermann CE, Anderson K, Sackner-Bernstein JD, Barone FC. Eprosartan improves cardiac performance, reduces cardiac hypertrophy and mortality and downregulates myocardial monocyte chemoattractant protein-1 and inflammation in hypertensive heart disease. J Hypertens 22: 583–592, 2004. doi: 10.1097/00004872-200403000-00022. [DOI] [PubMed] [Google Scholar]
- 64. Yoshida K, Kim-Mitsuyama S, Wake R, Izumiya Y, Izumi Y, Yukimura T, Ueda M, Yoshiyama M, Iwao H. Excess aldosterone under normal salt diet induces cardiac hypertrophy and infiltration via oxidative stress. Hypertens Res 28: 447–455, 2005. doi: 10.1291/hypres.28.447. [DOI] [PubMed] [Google Scholar]
- 65. Levick SP, Murray DB, Janicki JS, Brower GL. Sympathetic nervous system modulation of inflammation and remodeling in the hypertensive heart. Hypertension 55: 270–276, 2010. doi: 10.1161/HYPERTENSIONAHA.109.142042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Herrada AA, Contreras FJ, Marini NP, Amador CA, Gonzalez PA, Cortes CM, Riedel CA, Carvajal CA, Figueroa F, Michea LF, Fardella CE, Kalergis AM. Aldosterone promotes autoimmune damage by enhancing Th17-mediated immunity. J Immunol 184: 191–202, 2010. doi: 10.4049/jimmunol.0802886. [DOI] [PubMed] [Google Scholar]
- 67. Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med 204: 2449–2460, 2007. doi: 10.1084/jem.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Li X, Zhang Z, Luo M, Cheng Z, Wang R, Liu Q, Lv D, Yan J, Shang F, Luo S, Xia Y. NLRP3 inflammasome contributes to endothelial dysfunction in angiotensin II-induced hypertension in mice. Microvasc Res 143: 104384, 2022. doi: 10.1016/j.mvr.2022.104384. [DOI] [PubMed] [Google Scholar]
- 69. Wenzel P, Knorr M, Kossmann S, Stratmann J, Hausding M, Schuhmacher S, Karbach SH, Schwenk M, Yogev N, Schulz E, Oelze M, Grabbe S, Jonuleit H, Becker C, Daiber A, Waisman A, Munzel T. Lysozyme M-positive monocytes mediate angiotensin II-induced arterial hypertension and vascular dysfunction. Circulation 124: 1370–1381, 2011. doi: 10.1161/CIRCULATIONAHA.111.034470. [DOI] [PubMed] [Google Scholar]
- 70. Harwani SC. Macrophages under pressure: the role of macrophage polarization in hypertension. Transl Res 191: 45–63, 2018. doi: 10.1016/j.trsl.2017.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lin QY, Bai J, Zhang YL, Li HH. Integrin CD11b contributes to hypertension and vascular dysfunction through mediating macrophage adhesion and migration. Hypertension 80: 57–69, 2023. doi: 10.1161/HYPERTENSIONAHA.122.20328. [DOI] [PubMed] [Google Scholar]
- 72. Ghigo A, Franco I, Morello F, Hirsch E. Myocyte signalling in leucocyte recruitment to the heart. Cardiovasc Res 102: 270–280, 2014. doi: 10.1093/cvr/cvu030. [DOI] [PubMed] [Google Scholar]
- 73. Frantz S, Ertl G, Bauersachs J. Mechanisms of disease: Toll-like receptors in cardiovascular disease. Nat Clin Pract Cardiovasc Med 4: 444–454, 2007. doi: 10.1038/ncpcardio0938. [DOI] [PubMed] [Google Scholar]
- 74. Eissler R, Schmaderer C, Rusai K, Kuhne L, Sollinger D, Lahmer T, Witzke O, Lutz J, Heemann U, Baumann M. Hypertension augments cardiac Toll-like receptor 4 expression and activity. Hypertens Res 34: 551–558, 2011. doi: 10.1038/hr.2010.270. [DOI] [PubMed] [Google Scholar]
- 75. Roger VL, Go AS, Lloyd-Jones DM, Adams RJ, Berry JD, Brown TM, , et al. Heart disease and stroke statistics–2011 update: a report from the American Heart Association. Circulation 123: e18–e209, 2011. doi: 10.1161/CIR.0b013e3182009701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Ferrucci L, Fabbri E. Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat Rev Cardiol 15: 505–522, 2018. doi: 10.1038/s41569-018-0064-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ferrucci L, Corsi A, Lauretani F, Bandinelli S, Bartali B, Taub DD, Guralnik JM, Longo DL. The origins of age-related proinflammatory state. Blood 105: 2294–2299, 2005. doi: 10.1182/blood-2004-07-2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Ferrucci L, Semba RD, Guralnik JM, Ershler WB, Bandinelli S, Patel KV, Sun K, Woodman RC, Andrews NC, Cotter RJ, Ganz T, Nemeth E, Longo DL. Proinflammatory state, hepcidin, and anemia in older persons. Blood 115: 3810–3816, 2010. doi: 10.1182/blood-2009-02-201087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Cohen HJ, Pieper CF, Harris T, Rao KM, Currie MS. The association of plasma IL-6 levels with functional disability in community-dwelling elderly. J Gerontol A Biol Sci Med Sci 52: M201–208, 1997. doi: 10.1093/gerona/52a.4.m201. [DOI] [PubMed] [Google Scholar]
- 80. Newman AB, Sanders JL, Kizer JR, Boudreau RM, Odden MC, Zeki Al Hazzouri A, Arnold AM. Trajectories of function and biomarkers with age: the CHS All Stars Study. Int J Epidemiol 45: 1135–1145, 2016. doi: 10.1093/ije/dyw092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 6: 2853–2868, 2008. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Basisty N, Kale A, Jeon OH, Kuehnemann C, Payne T, Rao C, Holtz A, Shah S, Sharma V, Ferrucci L, Campisi J, Schilling B. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol 18: e3000599, 2020. doi: 10.1371/journal.pbio.3000599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Roh JD, Hobson R, Chaudhari V, Quintero P, Yeri A, Benson M, Xiao C, Zlotoff D, Bezzerides V, Houstis N, Platt C, Damilano F, Lindman BR, Elmariah S, Biersmith M, Lee SJ, Seidman CE, Seidman JG, Gerszten RE, Lach-Trifilieff E, Glass DJ, Rosenzweig A. Activin type II receptor signaling in cardiac aging and heart failure. Sci Transl Med 11: eaau8680, 2019. doi: 10.1126/scitranslmed.aau8680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Chini CC, Peclat TR, Warner GM, Kashyap S, Espindola-Netto JM, de Oliveira GC, , et al. CD38 ecto-enzyme in immune cells is induced during aging and regulates NAD(+) and NMN levels. Nat Metab 2: 1284–1304, 2020. doi: 10.1038/s42255-020-00298-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Tong D, Schiattarella GG, Jiang N, Altamirano F, Szweda PA, Elnwasany A, Lee DI, Yoo H, Kass DA, Szweda LI, Lavandero S, Verdin E, Gillette TG, Hill JA. NAD(+) repletion reverses heart failure with preserved ejection fraction. Circ Res 128: 1629–1641, 2021. doi: 10.1161/CIRCRESAHA.120.317046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Mooney L, Goodyear CS, Chandra T, Kirschner K, Copland M, Petrie MC, Lang NN. Clonal haematopoiesis of indeterminate potential: intersections between inflammation, vascular disease and heart failure. Clin Sci (Lond) 135: 991–1007, 2021. doi: 10.1042/CS20200306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Wang Y, Sano S, Yura Y, Ke Z, Sano M, Oshima K, Ogawa H, Horitani K, Min KD, Miura-Yura E, Kour A, Evans MA, Zuriaga MA, Hirschi KK, Fuster JJ, Pietras EM, Walsh K. Tet2-mediated clonal hematopoiesis in nonconditioned mice accelerates age-associated cardiac dysfunction. JCI Insight 5: e135204, 2020. doi: 10.1172/jci.insight.135204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Dorsheimer L, Assmus B, Rasper T, Ortmann CA, Ecke A, Abou-El-Ardat K, Schmid T, Brune B, Wagner S, Serve H, Hoffmann J, Seeger F, Dimmeler S, Zeiher AM, Rieger MA. Association of mutations contributing to clonal hematopoiesis with prognosis in chronic ischemic heart failure. JAMA Cardiol 4: 25–33, 2019. doi: 10.1001/jamacardio.2018.3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Pascual-Figal DA, Bayes-Genis A, Diez-Diez M, Hernandez-Vicente A, Vazquez-Andres D, de la Barrera J, Vazquez E, Quintas A, Zuriaga MA, Asensio-Lopez MC, Dopazo A, Sanchez-Cabo F, Fuster JJ. Clonal hematopoiesis and risk of progression of heart failure with reduced left ventricular ejection fraction. J Am Coll Cardiol 77: 1747–1759, 2021. doi: 10.1016/j.jacc.2021.02.028. [DOI] [PubMed] [Google Scholar]
- 90. Sano S, Wang Y, Yura Y, Sano M, Oshima K, Yang Y, Katanasaka Y, Min KD, Matsuura S, Ravid K, Mohi G, Walsh K. JAK2 (V617F)-mediated clonal hematopoiesis accelerates pathological remodeling in murine heart failure. JACC Basic Transl Sci 4: 684–697, 2019. doi: 10.1016/j.jacbts.2019.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Sano S, Oshima K, Wang Y, MacLauchlan S, Katanasaka Y, Sano M, Zuriaga MA, Yoshiyama M, Goukassian D, Cooper MA, Fuster JJ, Walsh K. Tet2-mediated clonal hematopoiesis accelerates heart failure through a mechanism involving the IL-1beta/NLRP3 Inflammasome. J Am Coll Cardiol 71: 875–886, 2018. doi: 10.1016/j.jacc.2017.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Sano S, Oshima K, Wang Y, Katanasaka Y, Sano M, Walsh K. CRISPR-mediated gene editing to assess the roles of Tet2 and Dnmt3a in clonal hematopoiesis and cardiovascular disease. Circ Res 123: 335–341, 2018. doi: 10.1161/CIRCRESAHA.118.313225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kessler MD, Damask A, O'Keeffe S, Banerjee N, Li D, Watanabe K, , et al. Common and rare variant associations with clonal haematopoiesis phenotypes. Nature 612: 301–309, 2022. doi: 10.1038/s41586-022-05448-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Oberg BP, McMenamin E, Lucas FL, McMonagle E, Morrow J, Ikizler TA, Himmelfarb J. Increased prevalence of oxidant stress and inflammation in patients with moderate to severe chronic kidney disease. Kidney Int 65: 1009–1016, 2004. doi: 10.1111/j.1523-1755.2004.00465.x. [DOI] [PubMed] [Google Scholar]
- 95. Schunk SJ, Triem S, Schmit D, Zewinger S, Sarakpi T, Becker E, Hutter G, Wrublewsky S, Kuting F, Hohl M, Alansary D, Prates Roma L, Lipp P, Mollmann J, Lehrke M, Laschke MW, Menger MD, Kramann R, Boor P, Jahnen-Dechent W, Marz W, Bohm M, Laufs U, Niemeyer BA, Fliser D, Ampofo E, Speer T. Interleukin-1alpha is a central regulator of leukocyte-endothelial adhesion in myocardial infarction and in chronic kidney disease. Circulation 144: 893–908, 2021. doi: 10.1161/CIRCULATIONAHA.121.053547. [DOI] [PubMed] [Google Scholar]
- 96. Batra G, Ghukasyan Lakic T, Lindback J, Held C, White HD, Stewart RA, Koenig W, Cannon CP, Budaj A, Hagstrom E, Siegbahn A, Wallentin L; STABILITY Investigators. Interleukin 6 and cardiovascular outcomes in patients with chronic kidney disease and chronic coronary syndrome. JAMA Cardiol 6: 1440–1445, 2021. doi: 10.1001/jamacardio.2021.3079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Valero-Munoz M, Li S, Wilson RM, Boldbaatar B, Iglarz M, Sam F. Dual endothelin-A/endothelin-B receptor blockade and cardiac remodeling in heart failure with preserved ejection fraction. Circ Heart Fail 9: e003381, 2016. doi: 10.1161/CIRCHEARTFAILURE.116.003381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Yoon S, Kim M, Lee H, Kang G, Bedi K, Margulies KB, Jain R, Nam KI, Kook H, Eom GH. S-Nitrosylation of histone deacetylase 2 by neuronal nitric oxide synthase as a mechanism of diastolic dysfunction. Circulation 143: 1912–1925, 2021. doi: 10.1161/CIRCULATIONAHA.119.043578. [DOI] [PubMed] [Google Scholar]
- 99. Roh J, Hill JA, Singh A, Valero-Munoz M, Sam F. Heart failure with preserved ejection fraction: heterogeneous syndrome, diverse preclinical models. Circ Res 130: 1906–1925, 2022. doi: 10.1161/CIRCRESAHA.122.320257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Zoccali C, Vanholder R, Massy ZA, Ortiz A, Sarafidis P, Dekker FW, Fliser D, Fouque D, Heine GH, Jager KJ, Kanbay M, Mallamaci F, Parati G, Rossignol P, Wiecek A, London G, European R; Cardiovascular Medicine Working Group of the European Renal Association-European Dialysis Transplantation Association. The systemic nature of CKD. Nat Rev Nephrol 13: 344–358, 2017. doi: 10.1038/nrneph.2017.52. [DOI] [PubMed] [Google Scholar]
- 101. Speer T, Dimmeler S, Schunk SJ, Fliser D, Ridker PM. Targeting innate immunity-driven inflammation in CKD and cardiovascular disease. Nat Rev Nephrol 18: 762–778, 2022. doi: 10.1038/s41581-022-00621-9. [DOI] [PubMed] [Google Scholar]
- 102. Rosengren BI, Sagstad SJ, Karlsen TV, Wiig H. Isolation of interstitial fluid and demonstration of local proinflammatory cytokine production and increased absorptive gradient in chronic peritoneal dialysis. Am J Physiol Renal Physiol 304: F198–F206, 2013. doi: 10.1152/ajprenal.00293.2012. [DOI] [PubMed] [Google Scholar]
- 103. Kim HJ, Vaziri ND. Contribution of impaired Nrf2-Keap1 pathway to oxidative stress and inflammation in chronic renal failure. Am J Physiol Renal Physiol 298: F662–F671, 2010. doi: 10.1152/ajprenal.00421.2009. [DOI] [PubMed] [Google Scholar]
- 104. Ori Y, Bergman M, Bessler H, Zingerman B, Levy-Drummer RS, Gafter U, Salman H. Cytokine secretion and markers of inflammation in relation to acidosis among chronic hemodialysis patients. Blood Purif 35: 181–186, 2013. doi: 10.1159/000346689. [DOI] [PubMed] [Google Scholar]
- 105. Rao AK, Djamali A, Korcarz CE, Aeschlimann SE, Wolff MR, Stein JH. Left atrial volume is associated with inflammation and atherosclerosis in patients with kidney disease. Echocardiography 25: 264–269, 2008. doi: 10.1111/j.1540-8175.2007.00589.x. [DOI] [PubMed] [Google Scholar]
- 106. Crowley SD, Rudemiller NP. Immunologic effects of the renin-angiotensin system. J Am Soc Nephrol 28: 1350–1361, 2017. doi: 10.1681/ASN.2016101066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Speer T, Ridker PM, von Eckardstein A, Schunk SJ, Fliser D. Lipoproteins in chronic kidney disease: from bench to bedside. Eur Heart J 42: 2170–2185, 2021. doi: 10.1093/eurheartj/ehaa1050. [DOI] [PubMed] [Google Scholar]
- 108. Wang Z, Nicholls SJ, Rodriguez ER, Kummu O, Horkko S, Barnard J, Reynolds WF, Topol EJ, DiDonato JA, Hazen SL. Protein carbamylation links inflammation, smoking, uremia and atherogenesis. Nat Med 13: 1176–1184, 2007. doi: 10.1038/nm1637. [DOI] [PubMed] [Google Scholar]
- 109. Speer T, Owala FO, Holy EW, Zewinger S, Frenzel FL, Stahli BE, Razavi M, Triem S, Cvija H, Rohrer L, Seiler S, Heine GH, Jankowski V, Jankowski J, Camici GG, Akhmedov A, Fliser D, Luscher TF, Tanner FC. Carbamylated low-density lipoprotein induces endothelial dysfunction. Eur Heart J 35: 3021–3032, 2014. doi: 10.1093/eurheartj/ehu111. [DOI] [PubMed] [Google Scholar]
- 110. Zewinger S, Drechsler C, Kleber ME, Dressel A, Riffel J, Triem S, Lehmann M, Kopecky C, Saemann MD, Lepper PM, Silbernagel G, Scharnagl H, Ritsch A, Thorand B, de las Heras Gala T, Wagenpfeil S, Koenig W, Peters A, Laufs U, Wanner C, Fliser D, Speer T, Marz W. Serum amyloid A: high-density lipoproteins interaction and cardiovascular risk. Eur Heart J 36: 3007–3016, 2015. doi: 10.1093/eurheartj/ehv352. [DOI] [PubMed] [Google Scholar]
- 111. Speer T, Rohrer L, Blyszczuk P, Shroff R, Kuschnerus K, Krankel N, Kania G, Zewinger S, Akhmedov A, Shi Y, Martin T, Perisa D, Winnik S, Muller MF, Sester U, Wernicke G, Jung A, Gutteck U, Eriksson U, Geisel J, Deanfield J, von Eckardstein A, Luscher TF, Fliser D, Bahlmann FH, Landmesser U. Abnormal high-density lipoprotein induces endothelial dysfunction via activation of Toll-like receptor-2. Immunity 38: 754–768, 2013. doi: 10.1016/j.immuni.2013.02.009. [DOI] [PubMed] [Google Scholar]
- 112. Zewinger S, Kleber ME, Rohrer L, Lehmann M, Triem S, Jennings RT, Petrakis I, Dressel A, Lepper PM, Scharnagl H, Ritsch A, Thorand B, Heier M, Meisinger C, de Las Heras Gala T, Koenig W, Wagenpfeil S, Schwedhelm E, Boger RH, Laufs U, von Eckardstein A, Landmesser U, Luscher TF, Fliser D, Marz W, Meinitzer A, Speer T. Symmetric dimethylarginine, high-density lipoproteins and cardiovascular disease. Eur Heart J 38: 1597–1607, 2017. doi: 10.1093/eurheartj/ehx118. [DOI] [PubMed] [Google Scholar]
- 113. Guo F, Qiu X, Tan Z, Li Z, Ouyang D. Plasma trimethylamine n-oxide is associated with renal function in patients with heart failure with preserved ejection fraction. BMC Cardiovasc Disord 20: 394, 2020. doi: 10.1186/s12872-020-01669-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Jovanovich A, Isakova T, Stubbs J. Microbiome and cardiovascular disease in CKD. Clin J Am Soc Nephrol 13: 1598–1604, 2018. doi: 10.2215/CJN.12691117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Docherty MH, O'Sullivan ED, Bonventre JV, Ferenbach DA. Cellular senescence in the kidney. J Am Soc Nephrol 30: 726–736, 2019. doi: 10.1681/ASN.2018121251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Paulus WJ, Zile MR. From systemic inflammation to myocardial fibrosis: the heart failure with preserved ejection fraction paradigm revisited. Circ Res 128: 1451–1467, 2021. doi: 10.1161/CIRCRESAHA.121.318159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Bowers SL, Meng Q, Molkentin JD. Fibroblasts orchestrate cellular crosstalk in the heart through the ECM. Nat Cardiovasc Res 1: 312–321, 2022. doi: 10.1038/s44161-022-00043-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Westermann D, Lindner D, Kasner M, Zietsch C, Savvatis K, Escher F, von Schlippenbach J, Skurk C, Steendijk P, Riad A, Poller W, Schultheiss HP, Tschope C. Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circ Heart Fail 4: 44–52, 2011. doi: 10.1161/CIRCHEARTFAILURE.109.931451. [DOI] [PubMed] [Google Scholar]
- 119. Kuwahara F, Kai H, Tokuda K, Niiyama H, Tahara N, Kusaba K, Takemiya K, Jalalidin A, Koga M, Nagata T, Shibata R, Imaizumi T. Roles of intercellular adhesion molecule-1 in hypertensive cardiac remodeling. Hypertension 41: 819–823, 2003. doi: 10.1161/01.HYP.0000056108.73219.0A. [DOI] [PubMed] [Google Scholar]
- 120. Kuwahara F, Kai H, Tokuda K, Takeya M, Takeshita A, Egashira K, Imaizumi T. Hypertensive myocardial fibrosis and diastolic dysfunction: another model of inflammation? Hypertension 43: 739–745, 2004. doi: 10.1161/01.HYP.0000118584.33350.7d. [DOI] [PubMed] [Google Scholar]
- 121. Zhang Y, Wang JH, Zhang YY, Wang YZ, Wang J, Zhao Y, Jin XX, Xue GL, Li PH, Sun YL, Huang QH, Song XT, Zhang ZR, Gao X, Yang BF, Du ZM, Pan ZW. Deletion of interleukin-6 alleviated interstitial fibrosis in streptozotocin-induced diabetic cardiomyopathy of mice through affecting TGFbeta1 and miR-29 pathways. Sci Rep 6: 23010, 2016. doi: 10.1038/srep23010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Narayan P, Trikantzopoulos E, Mezzaroma E, Mauro AG, Vohra H, Abbate A, Toldo S. The interleukin-1 receptor type I promotes the development of aging-associated cardiomyopathy in mice. Cytokine 151: 155811, 2022. doi: 10.1016/j.cyto.2022.155811. [DOI] [PubMed] [Google Scholar]
- 123. Carbone S, Lee PJ, Mauro AG, Mezzaroma E, Buzzetti R, Van Tassell B, Abbate A, Toldo S. Interleukin-18 mediates cardiac dysfunction induced by western diet independent of obesity and hyperglycemia in the mouse. Nutr Diabetes 7: e258, 2017. doi: 10.1038/nutd.2017.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Leask A. Getting to the heart of the matter: new insights into cardiac fibrosis. Circ Res 116: 1269–1276, 2015. doi: 10.1161/CIRCRESAHA.116.305381. [DOI] [PubMed] [Google Scholar]
- 125. Cunningham JW, Claggett BL, O'Meara E, Prescott MF, Pfeffer MA, Shah SJ, Redfield MM, Zannad F, Chiang LM, Rizkala AR, Shi VC, Lefkowitz MP, Rouleau J, McMurray JJ, Solomon SD, Zile MR. Effect of sacubitril/valsartan on biomarkers of extracellular matrix regulation in patients with HFpEF. J Am Coll Cardiol 76: 503–514, 2020. doi: 10.1016/j.jacc.2020.05.072. [DOI] [PubMed] [Google Scholar]
- 126. Alvandi Z, Bischoff J. Endothelial-mesenchymal transition in cardiovascular disease. Arterioscler Thromb Vasc Biol 41: 2357–2369, 2021. doi: 10.1161/ATVBAHA.121.313788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Abe H, Tanada Y, Omiya S, Podaru MN, Murakawa T, Ito J, Shah AM, Conway SJ, Ono M, Otsu K. NF-kappaB activation in cardiac fibroblasts results in the recruitment of inflammatory Ly6C(hi) monocytes in pressure-overloaded hearts. Sci Signal 14: eabe4932, 2021. doi: 10.1126/scisignal.abe4932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Ngwenyama N, Kaur K, Bugg D, Theall B, Aronovitz M, Berland R, Panagiotidou S, Genco C, Perrin MA, Davis J, Alcaide P. Antigen presentation by cardiac fibroblasts promotes cardiac dysfunction. Nat Cardiovasc Res 1: 761–774, 2022. doi: 10.1038/s44161-022-00116-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Lewis GA, Dodd S, Clayton D, Bedson E, Eccleson H, Schelbert EB, Naish JH, Jimenez BD, Williams SG, Cunnington C, Ahmed FZ, Cooper A, Rajavarma V, Russell S, McDonagh T, Williamson PR, Miller CA. Pirfenidone in heart failure with preserved ejection fraction: a randomized phase 2 trial. Nat Med 27: 1477–1482, 2021. doi: 10.1038/s41591-021-01452-0. [DOI] [PubMed] [Google Scholar]
- 130. Visner GA, Liu F, Bizargity P, Liu H, Liu K, Yang J, Wang L, Hancock WW. Pirfenidone inhibits T-cell activation, proliferation, cytokine and chemokine production, and host alloresponses. Transplantation 88: 330–338, 2009. doi: 10.1097/TP.0b013e3181ae3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Aimo A, Cerbai E, Bartolucci G, Adamo L, Barison A, Lo Surdo G, Biagini S, Passino C, Emdin M. Pirfenidone is a cardioprotective drug: mechanisms of action and preclinical evidence. Pharmacol Res 155: 104694, 2020. doi: 10.1016/j.phrs.2020.104694. [DOI] [PubMed] [Google Scholar]
- 132. Travers JG, Wennersten SA, Pena B, Bagchi RA, Smith HE, Hirsch RA, Vanderlinden LA, Lin YH, Dobrinskikh E, Demos-Davies KM, Cavasin MA, Mestroni L, Steinkuhler C, Lin CY, Houser SR, Woulfe KC, Lam MP, McKinsey TA. HDAC inhibition reverses preexisting diastolic dysfunction and blocks covert extracellular matrix remodeling. Circulation 143: 1874–1890, 2021. doi: 10.1161/CIRCULATIONAHA.120.046462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Gillette TG. HDAC inhibition in the heart: erasing hidden fibrosis. Circulation 143: 1891–1893, 2021. doi: 10.1161/CIRCULATIONAHA.121.054262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Rurik JG, Tombacz I, Yadegari A, Mendez Fernandez PO, Shewale SV, Li L, Kimura T, Soliman OY, Papp TE, Tam YK, Mui BL, Albelda SM, Pure E, June CH, Aghajanian H, Weissman D, Parhiz H, Epstein JA. CAR T cells produced in vivo to treat cardiac injury. Science 375: 91–96, 2022. doi: 10.1126/science.abm0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Aghajanian H, Kimura T, Rurik JG, Hancock AS, Leibowitz MS, Li L, Scholler J, Monslow J, Lo A, Han W, Wang T, Bedi K, Morley MP, Linares Saldana RA, Bolar NA, McDaid K, Assenmacher CA, Smith CL, Wirth D, June CH, Margulies KB, Jain R, Pure E, Albelda SM, Epstein JA. Targeting cardiac fibrosis with engineered T cells. Nature 573: 430–433, 2019. doi: 10.1038/s41586-019-1546-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Cho JH, Kilfoil PJ, Zhang R, Solymani RE, Bresee C, Kang EM, Luther K, Rogers RG, de Couto G, Goldhaber JI, Marban E, Cingolani E. Reverse electrical remodeling in rats with heart failure and preserved ejection fraction. JCI Insight 3: e121123, 2018. doi: 10.1172/jci.insight.121123. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 137. Gallet R, de Couto G, Simsolo E, Valle J, Sun B, Liu W, Tseliou E, Zile MR, Marban E. Cardiosphere-derived cells reverse heart failure with preserved ejection fraction (HFpEF) in rats by decreasing fibrosis and inflammation. JACC Basic Transl Sci 1: 14–28, 2016. doi: 10.1016/j.jacbts.2016.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Zhang XL, Wang TY, Chen Z, Wang HW, Yin Y, Wang L, Wang Y, Xu B, Xu W. HMGB1-promoted neutrophil extracellular traps contribute to cardiac diastolic dysfunction in mice. JAHA 11: e023800, 2022. doi: 10.1161/JAHA.121.023800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Kim SR, Lee SG, Kim SH, Kim JH, Choi E, Cho W, Rim JH, Hwang I, Lee CJ, Lee M, Oh CM, Jeon JY, Gee HY, Kim JH, Lee BW, Kang ES, Cha BS, Lee MS, Yu JW, Cho JW, Kim JS, Lee YH. SGLT2 inhibition modulates NLRP3 inflammasome activity via ketones and insulin in diabetes with cardiovascular disease. Nat Commun 11: 2127, 2020. doi: 10.1038/s41467-020-15983-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Aragon-Herrera A, Feijoo-Bandin S, Otero Santiago M, Barral L, Campos-Toimil M, Gil-Longo J, Costa Pereira TM, Garcia-Caballero T, Rodriguez-Segade S, Rodriguez J, Tarazon E, Rosello-Lleti E, Portoles M, Gualillo O, Gonzalez-Juanatey JR, Lago F. Empagliflozin reduces the levels of CD36 and cardiotoxic lipids while improving autophagy in the hearts of Zucker diabetic fatty rats. Biochem Pharmacol 170: 113677, 2019. doi: 10.1016/j.bcp.2019.113677. [DOI] [PubMed] [Google Scholar]
- 141. Garvey WT, Van Gaal L, Leiter LA, Vijapurkar U, List J, Cuddihy R, Ren J, Davies MJ. Effects of canagliflozin versus glimepiride on adipokines and inflammatory biomarkers in type 2 diabetes. Metabolism 85: 32–37, 2018. doi: 10.1016/j.metabol.2018.02.002. [DOI] [PubMed] [Google Scholar]
- 142. Steven S, Oelze M, Hanf A, Kroller-Schon S, Kashani F, Roohani S, Welschof P, Kopp M, Godtel-Armbrust U, Xia N, Li H, Schulz E, Lackner KJ, Wojnowski L, Bottari SP, Wenzel P, Mayoux E, Munzel T, Daiber A. The SGLT2 inhibitor empagliflozin improves the primary diabetic complications in ZDF rats. Redox Biol 13: 370–385, 2017. doi: 10.1016/j.redox.2017.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Benetti E, Mastrocola R, Vitarelli G, Cutrin JC, Nigro D, Chiazza F, Mayoux E, Collino M, Fantozzi R. Empagliflozin protects against diet-induced NLRP-3 inflammasome activation and lipid accumulation. J Pharmacol Exp Ther 359: 45–53, 2016. doi: 10.1124/jpet.116.235069. [DOI] [PubMed] [Google Scholar]
- 144. Tahara A, Kurosaki E, Yokono M, Yamajuku D, Kihara R, Hayashizaki Y, Takasu T, Imamura M, Li Q, Tomiyama H, Kobayashi Y, Noda A, Sasamata M, Shibasaki M. Effects of SGLT2 selective inhibitor ipragliflozin on hyperglycemia, hyperlipidemia, hepatic steatosis, oxidative stress, inflammation, and obesity in type 2 diabetic mice. Eur J Pharmacol 715: 246–255, 2013. doi: 10.1016/j.ejphar.2013.05.014. [DOI] [PubMed] [Google Scholar]
- 145. Lee YH, Kim SR, Han DH, Yu HT, Han YD, Kim JH, Kim SH, Lee CJ, Min BH, Kim DH, Kim KH, Cho JW, Lee WW, Shin EC, Park S. Senescent T cells predict the development of hyperglycemia in humans. Diabetes 68: 156–162, 2019. doi: 10.2337/db17-1218. [DOI] [PubMed] [Google Scholar]
- 146. Byrne NJ, Matsumura N, Maayah ZH, Ferdaoussi M, Takahara S, Darwesh AM, Levasseur JL, Jahng JW, Vos D, Parajuli N, El-Kadi AOS, Braam B, Young ME, Verma S, Light PE, Sweeney G, Seubert JM, Dyck JR. Empagliflozin blunts worsening cardiac dysfunction associated with reduced NLRP3 (nucleotide-binding domain-like receptor protein 3) inflammasome activation in heart failure. Circ Heart Fail 13: e006277, 2020. doi: 10.1161/CIRCHEARTFAILURE.119.006277. [DOI] [PubMed] [Google Scholar]
- 147. Xu L, Nagata N, Nagashimada M, Zhuge F, Ni Y, Chen G, Mayoux E, Kaneko S, Ota T. SGLT2 inhibition by empagliflozin promotes fat utilization and browning and attenuates inflammation and insulin resistance by polarizing M2 macrophages in diet-induced obese mice. EBioMedicine 20: 137–149, 2017. doi: 10.1016/j.ebiom.2017.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Li C, Zhang J, Xue M, Li X, Han F, Liu X, Xu L, Lu Y, Cheng Y, Li T, Yu X, Sun B, Chen L. SGLT2 inhibition with empagliflozin attenuates myocardial oxidative stress and fibrosis in diabetic mice heart. Cardiovasc Diabetol 18: 15, 2019. doi: 10.1186/s12933-019-0816-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Yang Z, Li T, Xian J, Chen J, Huang Y, Zhang Q, Lin X, Lu H, Lin Y. SGLT2 inhibitor dapagliflozin attenuates cardiac fibrosis and inflammation by reverting the HIF-2alpha signaling pathway in arrhythmogenic cardiomyopathy. FASEB J 36: e22410, 2022. doi: 10.1096/fj.202200243R. [DOI] [PubMed] [Google Scholar]
- 150. Lee TM, Chang NC, Lin SZ. Dapagliflozin, a selective SGLT2 Inhibitor, attenuated cardiac fibrosis by regulating the macrophage polarization via STAT3 signaling in infarcted rat hearts. Free Radic Biol Med 104: 298–310, 2017. doi: 10.1016/j.freeradbiomed.2017.01.035. [DOI] [PubMed] [Google Scholar]
- 151. Schiattarella GG, Altamirano F, Tong D, French KM, Villalobos E, Kim SY, Luo X, Jiang N, May HI, Wang ZV, Hill TM, Mammen PP, Huang J, Lee DI, Hahn VS, Sharma K, Kass DA, Lavandero S, Gillette TG, Hill JA. Nitrosative stress drives heart failure with preserved ejection fraction. Nature 568: 351–356, 2019. doi: 10.1038/s41586-019-1100-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Schiattarella GG, Altamirano F, Kim SY, Tong D, Ferdous A, Piristine H, Dasgupta S, Wang X, French KM, Villalobos E, Spurgin SB, Waldman M, Jiang N, May HI, Hill TM, Luo Y, Yoo H, Zaha VG, Lavandero S, Gillette TG, Hill JA. Xbp1s-FoxO1 axis governs lipid accumulation and contractile performance in heart failure with preserved ejection fraction. Nat Commun 12: 1684, 2021. doi: 10.1038/s41467-021-21931-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Peoples JN, Saraf A, Ghazal N, Pham TT, Kwong JQ. Mitochondrial dysfunction and oxidative stress in heart disease. Exp Mol Med 51: 1–13, 2019. doi: 10.1038/s12276-019-0355-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Aimo A, Castiglione V, Borrelli C, Saccaro LF, Franzini M, Masi S, Emdin M, Giannoni A. Oxidative stress and inflammation in the evolution of heart failure: from pathophysiology to therapeutic strategies. Eur J Prev Cardiol 27: 494–510, 2020. doi: 10.1177/2047487319870344. [DOI] [PubMed] [Google Scholar]
- 155. Liu H, Huang Y, Zhao Y, Kang G-J, Feng F, Wang X, Liu M, Shi G, Revelo X, Bernlohr D. Inflammatory macrophage interleukin-1β mediates high-fat diet-induced heart failure with preserved ejection fraction. JACC Basic Transl Sci 8: 174–185, 2022. doi: 10.1016/j.jacbts.2022.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Liu RM, Desai LP. Reciprocal regulation of TGF-beta and reactive oxygen species: a perverse cycle for fibrosis. Redox Biol 6: 565–577, 2015. doi: 10.1016/j.redox.2015.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Rosca MG, Hoppel CL. Mitochondrial dysfunction in heart failure. Heart Fail Rev 18: 607–622, 2013. doi: 10.1007/s10741-012-9340-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Kruger M, Kotter S, Grutzner A, Lang P, Andresen C, Redfield MM, Butt E, dos Remedios CG, Linke WA. Protein kinase G modulates human myocardial passive stiffness by phosphorylation of the titin springs. Circ Res 104: 87–94, 2009. doi: 10.1161/CIRCRESAHA.108.184408. [DOI] [PubMed] [Google Scholar]
- 159. Wang WZ, Jones AW, Wang M, Durante W, Korthuis RJ. Preconditioning with soluble guanylate cyclase activation prevents postischemic inflammation and reduces nitrate tolerance in heme oxygenase-1 knockout mice. Am J Physiol Heart Circ Physiol 305: H521–H532, 2013. doi: 10.1152/ajpheart.00810.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Holtwick R, van Eickels M, Skryabin BV, Baba HA, Bubikat A, Begrow F, Schneider MD, Garbers DL, Kuhn M. Pressure-independent cardiac hypertrophy in mice with cardiomyocyte-restricted inactivation of the atrial natriuretic peptide receptor guanylyl cyclase-A. J Clin Invest 111: 1399–1407, 2003. doi: 10.1172/JCI17061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Takimoto E, Champion HC, Li M, Belardi D, Ren S, Rodriguez ER, Bedja D, Gabrielson KL, Wang Y, Kass DA. Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nat Med 11: 214–222, 2005. doi: 10.1038/nm1175. [DOI] [PubMed] [Google Scholar]
- 162. Ranek MJ, Kokkonen-Simon KM, Chen A, Dunkerly-Eyring BL, Vera MP, Oeing CU, Patel CH, Nakamura T, Zhu G, Bedja D, Sasaki M, Holewinski RJ, Van Eyk JE, Powell JD, Lee DI, Kass DA. PKG1-modified TSC2 regulates mTORC1 activity to counter adverse cardiac stress. Nature 566: 264–269, 2019. doi: 10.1038/s41586-019-0895-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Ranek MJ, Terpstra EJ, Li J, Kass DA, Wang X. Protein kinase g positively regulates proteasome-mediated degradation of misfolded proteins. Circulation 128: 365–376, 2013. doi: 10.1161/CIRCULATIONAHA.113.001971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Inserte J, Garcia-Dorado D. The cGMP/PKG pathway as a common mediator of cardioprotection: translatability and mechanism. Br J Pharmacol 172: 1996–2009, 2015. doi: 10.1111/bph.12959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Methawasin M, Strom J, Borkowski T, Hourani Z, Runyan R, Smith JE 3rd, Granzier H. Phosphodiesterase 9a inhibition in mouse models of diastolic dysfunction. Circ Heart Fail 13: e006609, 2020. doi: 10.1161/CIRCHEARTFAILURE.119.006609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. McMurray JJ, Docherty KF. Phosphodiesterase-9 inhibition in heart failure: a further opportunity to augment the effects of natriuretic peptides? J Am Coll Cardiol 74: 902–904, 2019. doi: 10.1016/j.jacc.2019.07.008. [DOI] [PubMed] [Google Scholar]
- 167. Blanton RM. Phosphodiesterase 9 inhibition in models of heart failure with preserved left ventricular ejection fraction: should we focus on the positive or negative? Circ Heart Fail 13: e007107, 2020. doi: 10.1161/CIRCHEARTFAILURE.120.007107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Leite S, Moreira-Costa L, Cerqueira R, Sousa-Mendes C, Angelico-Goncalves A, Fontoura D, Vasques-Novoa F, Leite-Moreira AF, Lourenco AP. Chronic sildenafil therapy in the ZSF1 obese rat model of metabolic syndrome and heart failure with preserved ejection fraction. J Cardiovasc Pharmacol Ther 26: 690–701, 2021. doi: 10.1177/10742484211034253. [DOI] [PubMed] [Google Scholar]
- 169. Redfield MM, Chen HH, Borlaug BA, Semigran MJ, Lee KL, Lewis G, LeWinter MM, Rouleau JL, Bull DA, Mann DL, Deswal A, Stevenson LW, Givertz MM, Ofili EO, O'Connor CM, Felker GM, Goldsmith SR, Bart BA, McNulty SE, Ibarra JC, Lin G, Oh JK, Patel MR, Kim RJ, Tracy RP, Velazquez EJ, Anstrom KJ, Hernandez AF, Mascette AM, Braunwald E, RELAX trial. Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. JAMA 309: 1268–1277, 2013. doi: 10.1001/jama.2013.2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Lee DI, Zhu G, Sasaki T, Cho GS, Hamdani N, Holewinski R, Jo SH, Danner T, Zhang M, Rainer PP, Bedja D, Kirk JA, Ranek MJ, Dostmann WR, Kwon C, Margulies KB, Van Eyk JE, Paulus WJ, Takimoto E, Kass DA. Phosphodiesterase 9A controls nitric-oxide-independent cGMP and hypertrophic heart disease. Nature 519: 472–476, 2015. doi: 10.1038/nature14332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Vallejo S, Palacios E, Romacho T, Villalobos L, Peiro C, Sanchez-Ferrer CF. The interleukin-1 receptor antagonist anakinra improves endothelial dysfunction in streptozotocin-induced diabetic rats. Cardiovasc Diabetol 13: 158, 2014. doi: 10.1186/s12933-014-0158-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Schulz L, Werner S, Bottner J, Adams V, Lurz P, Besler C, Thiele H, Buttner P. Tubulin expression and modification in heart failure with preserved ejection fraction (HFpEF). Sci Rep 12: 15734, 2022. doi: 10.1038/s41598-022-19766-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. van Heerebeek L, Borbely A, Niessen HW, Bronzwaer JG, van der Velden J, Stienen GJ, Linke WA, Laarman GJ, Paulus WJ. Myocardial structure and function differ in systolic and diastolic heart failure. Circulation 113: 1966–1973, 2006. doi: 10.1161/CIRCULATIONAHA.105.587519. [DOI] [PubMed] [Google Scholar]
- 174. Borbely A, Falcao-Pires I, van Heerebeek L, Hamdani N, Edes I, Gavina C, Leite-Moreira AF, Bronzwaer JG, Papp Z, van der Velden J, Stienen GJ, Paulus WJ. Hypophosphorylation of the Stiff N2B titin isoform raises cardiomyocyte resting tension in failing human myocardium. Circ Res 104: 780–786, 2009. doi: 10.1161/CIRCRESAHA.108.193326. [DOI] [PubMed] [Google Scholar]
- 175. Mills RJ, Humphrey SJ, Fortuna PR, Lor M, Foster SR, Quaife-Ryan GA, , et al. BET inhibition blocks inflammation-induced cardiac dysfunction and SARS-CoV-2 infection. Cell 184: 2167–2182.e22, 2021.doi: 10.1016/j.cell.2021.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Melendez GC, McLarty JL, Levick SP, Du Y, Janicki JS, Brower GL. Interleukin 6 mediates myocardial fibrosis, concentric hypertrophy, and diastolic dysfunction in rats. Hypertension 56: 225–231, 2010. doi: 10.1161/HYPERTENSIONAHA.109.148635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Heymans S, Corsten MF, Verhesen W, Carai P, van Leeuwen RE, Custers K, Peters T, Hazebroek M, Stoger L, Wijnands E, Janssen BJ, Creemers EE, Pinto YM, Grimm D, Schurmann N, Vigorito E, Thum T, Stassen F, Yin X, Mayr M, de Windt LJ, Lutgens E, Wouters K, de Winther MP, Zacchigna S, Giacca M, van Bilsen M, Papageorgiou AP, Schroen B. Macrophage microRNA-155 promotes cardiac hypertrophy and failure. Circulation 128: 1420–1432, 2013. doi: 10.1161/CIRCULATIONAHA.112.001357. [DOI] [PubMed] [Google Scholar]
- 178. Patel B, Bansal SS, Ismahil MA, Hamid T, Rokosh G, Mack M, Prabhu SD. CCR2(+) monocyte-derived infiltrating macrophages are required for adverse cardiac remodeling during pressure overload. JACC Basic Transl Sci 3: 230–244, 2018. doi: 10.1016/j.jacbts.2017.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Wang L, Zhang YL, Lin QY, Liu Y, Guan XM, Ma XL, Cao HJ, Liu Y, Bai J, Xia YL, Du J, Li HH. CXCL1-CXCR2 axis mediates angiotensin II-induced cardiac hypertrophy and remodelling through regulation of monocyte infiltration. Eur Heart J 39: 1818–1831, 2018. doi: 10.1093/eurheartj/ehy085. [DOI] [PubMed] [Google Scholar]
- 180. Palmer JN, Hartogensis WE, Patten M, Fortuin FD, Long CS. Interleukin-1 beta induces cardiac myocyte growth but inhibits cardiac fibroblast proliferation in culture. J Clin Invest 95: 2555–2564, 1995. doi: 10.1172/JCI117956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Yokoyama T, Nakano M, Bednarczyk JL, McIntyre BW, Entman M, Mann DL. Tumor necrosis factor-alpha provokes a hypertrophic growth response in adult cardiac myocytes. Circulation 95: 1247–1252, 1997. doi: 10.1161/01.cir.95.5.1247. [DOI] [PubMed] [Google Scholar]
- 182. Schultz JE, Witt SA, Glascock BJ, Nieman ML, Reiser PJ, Nix SL, Kimball TR, Doetschman T. TGF-beta1 mediates the hypertrophic cardiomyocyte growth induced by angiotensin II. J Clin Invest 109: 787–796, 2002. doi: 10.1172/JCI14190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Suganami T, Nishida J, Ogawa Y. A paracrine loop between adipocytes and macrophages aggravates inflammatory changes: role of free fatty acids and tumor necrosis factor alpha. Arterioscler Thromb Vasc Biol 25: 2062–2068, 2005. doi: 10.1161/01.ATV.0000183883.72263.13. [DOI] [PubMed] [Google Scholar]
- 184. Yepuri G, Hasan SN, Schmidt AM, Ramasamy R. Macrophage-adipocyte communication and cardiac remodeling. J Exp Med 218: e20211098, 2021. doi: 10.1084/jem.20211098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Cheng YW, Zhang ZB, Lan BD, Lin JR, Chen XH, Kong LR, Xu L, Ruan CC, Gao PJ. PDGF-D activation by macrophage-derived uPA promotes AngII-induced cardiac remodeling in obese mice. J Exp Med 218: e20210252, 2021. doi: 10.1084/jem.20210252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Cordero-Reyes AM, Youker KA, Trevino AR, Celis R, Hamilton DJ, Flores-Arredondo JH, Orrego CM, Bhimaraj A, Estep JD, Torre-Amione G. Full expression of cardiomyopathy is partly dependent on B-cells: a pathway that involves cytokine activation, immunoglobulin deposition, and activation of apoptosis. J Am Heart Assoc 5: e002484, 2016. doi: 10.1161/JAHA.115.002484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Muir LA, Cho KW, Geletka LM, Baker NA, Flesher CG, Ehlers AP, Kaciroti N, Lindsly S, Ronquist S, Rajapakse I, O'Rourke RW, Lumeng CN. Human CD206+ macrophages associate with diabetes and adipose tissue lymphoid clusters. JCI Insight 7: e146563, 2022. doi: 10.1172/jci.insight.146563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Lin Q, Huang Y, Booth CJ, Haase VH, Johnson RS, Celeste Simon M, Giordano FJ, Yun Z. Activation of hypoxia-inducible factor-2 in adipocytes results in pathological cardiac hypertrophy. J Am Heart Assoc 2: e000548, 2013. doi: 10.1161/JAHA.113.000548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Gao W, Wang H, Zhang L, Cao Y, Bao JZ, Liu ZX, Wang LS, Yang Q, Lu X. Retinol-binding protein 4 induces cardiomyocyte hypertrophy by activating TLR4/MyD88 pathway. Endocrinology 157: 2282–2293, 2016. doi: 10.1210/en.2015-2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Ruan CC, Kong LR, Chen XH, Ma Y, Pan XX, Zhang ZB, Gao PJ. A(2A) Receptor activation attenuates hypertensive cardiac remodeling via promoting brown adipose tissue-derived FGF21. Cell Metab 32: 689, 2020. doi: 10.1016/j.cmet.2020.08.018. [DOI] [PubMed] [Google Scholar]
- 191. Arita Y, Kihara S, Ouchi N, Takahashi M, Maeda K, Miyagawa J, Hotta K, Shimomura I, Nakamura T, Miyaoka K, Kuriyama H, Nishida M, Yamashita S, Okubo K, Matsubara K, Muraguchi M, Ohmoto Y, Funahashi T, Matsuzawa Y. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun 257: 79–83, 1999. doi: 10.1006/bbrc.1999.0255. [DOI] [PubMed] [Google Scholar]
- 192. Phan TT, Abozguia K, Nallur Shivu G, Mahadevan G, Ahmed I, Williams L, Dwivedi G, Patel K, Steendijk P, Ashrafian H, Henning A, Frenneaux M. Heart failure with preserved ejection fraction is characterized by dynamic impairment of active relaxation and contraction of the left ventricle on exercise and associated with myocardial energy deficiency. J Am Coll Cardiol 54: 402–409, 2009. doi: 10.1016/j.jacc.2009.05.012. [DOI] [PubMed] [Google Scholar]
- 193. Dai DF, Chen T, Szeto H, Nieves-Cintron M, Kutyavin V, Santana LF, Rabinovitch PS. Mitochondrial targeted antioxidant peptide ameliorates hypertensive cardiomyopathy. J Am Coll Cardiol 58: 73–82, 2011. doi: 10.1016/j.jacc.2010.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Kumar AA, Kelly DP, Chirinos JA. Mitochondrial dysfunction in heart failure with preserved ejection fraction. Circulation 139: 1435–1450, 2019. doi: 10.1161/CIRCULATIONAHA.118.036259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Griffin TM, Humphries KM, Kinter M, Lim HY, Szweda LI. Nutrient sensing and utilization: Getting to the heart of metabolic flexibility. Biochimie 124: 74–83, 2016. doi: 10.1016/j.biochi.2015.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Deng Y, Xie M, Li Q, Xu X, Ou W, Zhang Y, Xiao H, Yu H, Zheng Y, Liang Y, Jiang C, Chen G, Du D, Zheng W, Wang S, Gong M, Chen Y, Tian R, Li T. Targeting mitochondria-inflammation circuit by beta-hydroxybutyrate mitigates HFpEF. Circ Res 128: 232–245, 2021. doi: 10.1161/CIRCRESAHA.120.317933. [DOI] [PubMed] [Google Scholar]
- 197. Marchi S, Guilbaud E, Tait SW, Yamazaki T, Galluzzi L. Mitochondrial control of inflammation. Nat Rev Immunol, 23: 159–173, 2023. doi: 10.1038/s41577-022-00760-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Van Tassell BW, Trankle CR, Canada JM, Carbone S, Buckley L, Kadariya D, Del Buono MG, Billingsley H, Wohlford G, Viscusi M, Oddi-Erdle C, Abouzaki NA, Dixon D, Biondi-Zoccai G, Arena R, Abbate A. IL-1 blockade in patients with heart failure with preserved ejection fraction. Circ Heart Fail 11: e005036, 2018. doi: 10.1161/CIRCHEARTFAILURE.118.005036. [DOI] [PMC free article] [PubMed] [Google Scholar]