

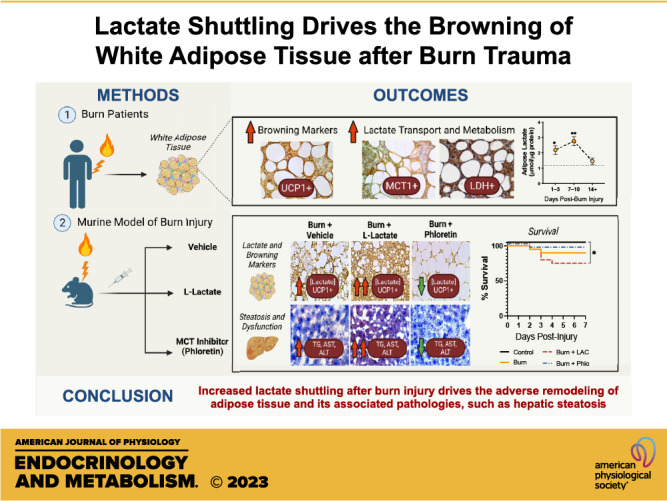
Keywords: adipose, browning, burns, hypermetabolism, lactate
Abstract
High levels of plasma lactate are associated with increased mortality in critically injured patients, including those with severe burns. Although lactate has long been considered a waste product of glycolysis, it was recently revealed that it acts as a potent inducer of white adipose tissue (WAT) browning, a response implicated in mediating postburn cachexia, hepatic steatosis, and sustained hypermetabolism. Despite the clinical presentation of hyperlactatemia and browning in burns, whether these two pathological responses are linked is currently unknown. Here, we report that elevated lactate plays a causal signaling role in mediating adverse outcomes after burn trauma by directly promoting WAT browning. Using WAT obtained from human burn patients and mouse models of thermal injury, we show that the induction of postburn browning is positively correlated with a shift toward lactate import and metabolism. Furthermore, daily administration of l-lactate is sufficient to augment burn-induced mortality and weight loss in vivo. At the organ level, increased lactate transport amplified the thermogenic activation of WAT and its associated wasting, thereby driving postburn hepatic lipotoxicity and dysfunction. Mechanistically, the thermogenic effects of lactate appeared to result from increased import through MCT transporters, which in turn increased intracellular redox pressure, [NADH/NAD+], and expression of the batokine, FGF21. In fact, pharmacological inhibition of MCT-mediated lactate uptake attenuated browning and improved hepatic function in mice after injury. Collectively, our findings identify a signaling role for lactate that impacts multiple aspects of postburn hypermetabolism, necessitating further investigation of this multifaceted metabolite in trauma and critical illness.
NEW & NOTEWORTHY To our knowledge, this study was the first to investigate the role of lactate signaling in mediating white adipose tissue browning after burn trauma. We show that the induction of browning in both human burn patients and mice is positively correlated with a shift toward lactate import and metabolism. Daily l-lactate administration augments burn-induced mortality, browning, and hepatic lipotoxicity in vivo, whereas pharmacologically targeting lactate transport alleviates burn-induced browning and improves liver dysfunction after injury.
INTRODUCTION
The continuous increase in the worldwide incidence of metabolic-associated diseases including diabetes, cardiovascular disease, and cancer has spurred renewed interest in the cell biology of the adipose organ (1–3). Although previously regarded as a passive energy reservoir, white adipose tissue (WAT) is now considered to be a remarkably complex endocrine organ capable of regulating a variety of biological functions (3, 4). In addition to secreting a plethora of adipokines, WAT plays a major part in inflammation, mechanical organ protection, and thermoregulation (4). Indeed, it was discovered that certain depots of WAT could undergo substantial remodeling when subjected to certain physiological stimuli in a process termed “browning” (5, 6). This results in the recruitment of “brown-in-white,” or beige adipocytes that are characterized by a multilocular appearance, increased mitochondrial biogenesis, and the expression of uncoupling protein 1 (UCP1)—the master regulator of nonshivering thermogenesis (7).
Owing to its high degree of plasticity, WAT beiging has garnered significant attention over the past decade for its potential to be exploited for the treatment of obesity and other metabolic disorders (8–10). Since its discovery, numerous studies have demonstrated that increasing the presence and activity of UCP1-positive cells confers a number of metabolic benefits in obese mice, namely, increases in energy expenditure, improved insulin sensitivity, and weight loss (11–13). However, these potentially beneficial aspects of browning are detrimental to hypermetabolic patients, including those with cancer and trauma, who also undergo a browning response (14–16). In fact, in these hypermetabolic patients, WAT browning has been shown to drive the development of cancer-associated cachexia, a state characterized by severe weight loss and muscle catabolism (15). Likewise, a previous study from our group has implicated WAT browning-induced lipolysis in mediating hepatic steatosis and dysfunction in mice postburn, thereby increasing morbidity and mortality (17). Given the health implications of WAT browning in various settings, there is thus considerable interest in understanding the cellular pathways that underpin the thermogenic phenotype (18).
Recently, a study by Carriere et al. (19) revealed a new role for lactate, a major product of anaerobic respiration in mediating the browning process. In this setting, lactate import by the monocarboxylate transporters (MCTs) was found to be critical for initiating the cold-induced thermogenic program via a redox-dependent process involving mitochondrial lactate oxidation (19, 20). Specifically, the increase in [NADH/NAD+] within adipocytes that occurs following lactate import triggers the expression of UCP1, perhaps as an adaptive response to prevent oxidative damage (19, 20). This points to a novel protective function for beige adipocytes and extends the role of lactate as a signaling molecule with important regulatory functions in energy metabolism (21). Interestingly, this signaling pathway could also be involved in the emergence of UCP1-expressing adipocytes in WAT in hypermetabolic conditions, such as burns and cancer, where increased levels of lactate have been shown to correlate with morbidity and mortality (22–24). Despite the clinical presentation of hyperlactatemia and browning in these patients, whether or how these two pathological responses are linked is currently unknown.
The goal of the current study was to investigate whether lactate could directly regulate the browning of WAT during the postburn hypermetabolic period. Based on recent evidence suggesting that expression of lactate transporters is controlled by physiological stimuli of browning, we hypothesized that elevated lactate levels after a burn injury can contribute to increased morbidity and mortality by promoting the adverse remodeling of WAT and its associated pathologies, such as hepatic steatosis. Thus, pharmacological inhibition of lactate flux activity by targeting MCT transporters may provide an effective means to abrogate postburn WAT dysfunction and potentially improve outcomes. To test this, we first characterized changes in lactate transport and metabolism associated with the induction of browning using adipose tissue samples from a clinical population of human burn patients and a well-established murine burn model. Subsequently, we investigated the metabolic effects of increasing and decreasing lactate flux activity on the postburn browning response in vivo. Our findings unveil an unexpected role of lactate as a signaling molecule after major trauma and open new perspectives for the treatment of hypermetabolism and its related disorders.
MATERIALS AND METHODS
Patient Samples
Burn patients admitted to the Ross Tilley Burn Center at Sunnybrook Hospital (Toronto, Canada) consented preoperatively for tissue collection before undergoing surgery. Approval for our study was obtained from the Research Ethics Board at Sunnybrook Hospital (Approval No. 194–2010). We enrolled a total of 12 burn patients (44 ± 7 yr) with burns encompassing an average of 50% ± 16% of their total body surface area (TBSA). Subcutaneous white adipose tissue (sWAT) samples were obtained at various time points after burn (1 specimen per patient) and were immediately frozen at −80°C or transferred into fixative until the time of analysis. For controls, we obtained fat from seven nonburn patients (47 ± 4 yr) undergoing elective surgery.
Animals and Model
Animal experiments were conducted in accordance with and approved by the Sunnybrook Research Institute Animal Care Committee (Toronto, ON, Canada; AUP No. 22–467). Male wild-type C57BL/6J (WT) mice (8–10 wk old, n = 6 or 7 per group) were purchased from Jackson Laboratories (Bar Harbor, Maine) and housed at ambient temperature, and cared for in accordance with the Guide for the Care and Use of Laboratory Animals. All mice were anesthetized with 2.5% isoflurane and shaved along the dorsal spine region. Saline (2–3 mL) was injected in the dorsal region subcutaneously in all mice to protect the spine and buprenorphine (0.05–0.1 mg/kg body wt) was administered for pain management. A full-thickness, third-degree scald burn encompassing 30% of total body surface area (TBSA) was achieved by immersing the dorsum of the mice in 95°C water for 10 s and the ventral region for 2 s. Burned mice were subsequently housed individually in sterile cages and food and water were given ad libitum. Sham mice (control) underwent identical experimental procedures, with the exception of burn injury. In vivo injections of sodium l-lactate (1 g/kg; Alfa Aesar; Cat. No. L14500) or Phloretin (50 mg/kg; Cayman Chemical; Cat. No. 14452) were administered intraperitoneally once every 24 h and continued for 7 days. Mice not receiving treatment were given vehicle (saline) injections. All injured rodents were health-scored by both certified veterinarians and laboratory staff daily to minimize animal pain and distress. Health scoring was based on a scale of 15, with up to 3 points given for eyes and nose, activity, food intake, grooming, and hydration. All tissues were stored in −80°C until further analysis.
Histology and Immunohistochemistry
Inguinal WAT (iWAT) was collected and immediately fixed in 10% formalin and then maintained in 70% ethanol before paraffin embedding. Subsequently, tissues were sectioned and incubated with hematoxylin and eosin (H&E), anti-UCP1 (Abcam; Cat. No. 155117), anti-MCT1 (Abcam; Cat. No. 90582), or anti-lactate dehydrogenase (LDH) (Abcam; Cat. No. 52488) antibody followed by DAB staining. Imaging was performed on laser scanning microscopy (LSM) confocal microscope (Zeiss, Germany). For quantification of adipocyte size, sections were imaged at a ×20 magnification. The smallest and largest diameters of each adipocyte were manually measured using Leica Application Suite Version 4.3.0 (Leica Microsystems, Switzerland), and the average of the two values was used for subsequent analyses.
Gene Expression Using RT-PCR
RNA was isolated from tissue using TRIzol-chloroform (Life Technologies) with subsequent purification using the RNeasy Kit (Qiagen) according to the manufacturer’s instructions. RNA (2 µg) was transcribed to cDNA using the high-capacity cDNA reverse transcription kit (Applied Biosystems). RT-PCR was performed using the Applied Biosystems Step One Plus Real-Time PCR System. Gene expression was expressed relative to β-actin, and levels were determined using the following formula: 2^(−ΔΔCt). Housekeeping gene expression was stable between groups, and samples with threshold cycle (Ct) values > 35 were not included in our analyses. Primer sequences used are available upon request.
Mitochondrial LDH Activity
Freshly excised tissue was minced in 10 volumes of mitochondrial isolation buffer [MHSE + BSA; 210 mM mannitol, 70 mM sucrose, 5 mM HEPES, 1 mM EGTA, 0.5% (w/v) fatty acid-free BSA, pH 7.2] as described previously (25). The tissue was then homogenized using a Teflon glass homogenizer with six strokes and filtered through three layers of cheesecloth. Mitochondria were isolated via differential centrifugation. Briefly, the homogenate was centrifuged at 600 g for 10 min and the supernatant was decanted into a new tube. This fraction was centrifuged at 9,000 g for 10 min to afford a mitochondrial pellet. The resultant supernatant, which contains soluble proteins, was subjected to a bicinchoninic acid (BCA) protein assay (Thermo Fisher Scientific) and set aside for other assays. A clear lipid layer surrounding the mitochondria was carefully aspirated and the pellet was resuspended in 200 µL of MHSE + BSA. A second centrifugation was performed to purify mitochondria as per the protocol by Rogers et al (26). Mitochondrial lactate dehydrogenase (mLDH) activity was then measured using an LDH activity assay kit according to the manufacturer’s instructions (Sigma Aldrich; Cat. No. MAK066). All samples were normalized to total mitochondrial protein levels as determined by the BCA protein assay kit (Pierce).
Lipids and Organ Damage Markers
Rodent plasma was collected and circulating levels of free fatty acids (FFA; FFA Colorimetric Assay Kit, Abcam; Cat. No. 65341), alanine aminotransferase (ALT; ALT Colorimetric Assay Kit, Abcam; Cat. No. 105134), and aspartate transaminase (AST; AST ELISA Kit, Abcam; Cat. No. 263882) were measured using assay kits according to the manufacturer’s instructions. Tissue triglycerides (TGs, TG Colorimetric Assay Kit, Abcam; Cat. No. 65336) were quantified using a colorimetric assay kit according to the manufacturer’s instructions.
Lactate and NADH: NAD+ Measurements
Plasma and adipose tissue lactate levels were quantified using an l-Lactate colorimetric assay kit according to the manufacturer’s instructions (Abcam; Cat. No. 65331). Total NAD and NADH were extracted from tissue and quantified using a NAD/NADH colorimetric assay kit (Abcam; Cat. No. 65348). Intracellular levels of NAD+ were then calculated by subtracting NADH from total NAD, and the NADH-to-NAD+ ratio was subsequently assessed. All samples were normalized to total protein concentration.
Statistical Analysis
All data are presented as means ± SE. Statistical analysis was performed using two-tailed unpaired t test with Welch’s correction and Mann–Whitney U test, where appropriate, for single variables. Statistical differences between three or more groups were evaluated using a one or two-way ANOVA followed by Tukey’s post hoc tests for multiple comparisons. Survival curves were also estimated for each group, considered separately, using the Kaplan–Meier method and compared statistically using the two-sided log-rank test. All graphs were created using GraphPad Prism 6.0 (San Diego, CA) and analyzed statistically using SPSS 20 (IBM Corp., NY, NY), with significance accepted at P < 0.05 (*), P < 0.01 (**), P < 0.001(***), P < 0.0001(****) for comparison between burn groups and corresponding shams, and P < 0.05 (#), P < 0.01 (##), P < 0.001 (###), and P < 0.0001 (####) for comparison between time points and within burn treatment groups.
RESULTS
Postburn WAT Browning Is Associated with Increased Lactate Transport and Metabolism
Burn patients.
We first examined the changes in lactate transport and metabolism associated with the induction of browning in a cohort of adults with burn injuries. Specifically, we enrolled 12 severely burn patients with an average burn encompassing 50% ± 16% TBSA (Table 1). Seven metabolically healthy adults who did not have a burn injury were compared as controls. Immunohistochemical staining for the key browning marker, UCP1, demonstrated increased expression in sWAT isolated from burn patients compared with nonburn (control) patients (Fig. 1A, top). This is in line with previous work and was further confirmed with our gene expression studies, demonstrating increased expression of several thermogenic markers (Ucp1, Pgc1a, Ppary, and Cidea) compared with normal WAT (P < 0.05; Fig. 1B). Having established burn-induced browning in these patients, we next compared the expression of MCT1 and MCT4, the main transporters enabling lactate uptake and export, respectively. Surprisingly, burn patients displayed significantly increased expression of the former lactate-importing isoform with no change in the latter (P < 0.001; Fig. 1C). These experiments also revealed a tight and significant positive correlation between Mct1 and Ucp1 mRNA levels, suggesting postburn browning is associated with an increased lactate import system (R2 = 0.719, P < 0.0001; Fig. 1D). Consistent with these findings, we observed enhanced MCT-1 staining in burn WAT, which corresponded with the presence of UCP-1 positive cells (Fig. 1A, middle). Furthermore, immunohistochemical staining for lactate dehydrogenase (LDH), the major enzyme involved in catalyzing the reversible conversion of lactate to pyruvate with the reduction of NAD+ to NADH and vice versa, was also significantly increased in burn compared with normal WAT (Fig. 1A, bottom). Although no change in LDHa was observed, gene expression of LDHb was significantly upregulated in human burn WAT compared with controls (P < 0.05; Fig. 1C), suggesting imported lactate is being imported and readily converted into pyruvate.
Table 1.
Demographics and outcomes of burn patients
| No. of Burn Patients | 12 |
|---|---|
| Age, means ± SD | 40 ± 19 |
| Males, no. (%) | 7 (58%) |
| TBSA, means ± SD | 50 ± 16 |
| Days postinjury, means ± SD | 14 ± 8 |
| Inhalation injury, no. (%) | 10 (83%) |
| Etiology, no. (%) | |
| Flame | 11 (92%) |
| Scald | 1 (8%) |
| Sepsis, no. (%) | 7 (58%) |
| Mortality, no. (%) | 2 (17%) |
TBSA, total body surface area.
Figure 1.

Upregulated UCP1, MCT1, and LDH in human adipose tissue after burn. A: immunohistochemical staining for UCP1, MCT1, and LDH in normal and burn human subcutaneous WAT (sWAT). Gene expression of browning markers UCP1, PGC1α, PPARγ, and CIDEA (B) and lactate transporters, MCT1 and MCT4, and dehydrogenases, LDHa and LDHb, in normal and burn human sWAT (C). D: correlation graph between MCT1 and mRNA expression. E: plasma lactate levels in burn patients over the course of admission relative to threshold of hyperlactatemia (dotted line) defined as levels >2 µmol/L. Changes in tissue lactate (F) and [NADH/NAD+] (G) levels burn sWAT at 1–3, 7–10, and 14+ days after injury relative to nonburn (dotted line). Values are presented as means ± standard error. Student’s t test and one-way ANOVA, Burn vs. nonburn *P < 0.05; **P < 0.01; ***P < 0.001, ****P < 0.0001 burn at different time points ###P < 0.001. CIDEA, cell death-inducing DNA fragmentation factor-alpha-like effector A; LDH, lactate dehydrogenase; MCT1, monocarboxylate transporter 1; PGC1α, peroxisome proliferator-activated receptor-γ coactivator 1-α; PPARγ, peroxisome proliferator-activated receptor γ; UCP1, uncoupling protein 1; WAT, white adipose tissue.
To confirm the metabolic significance of increased MCT1 and LDH after injury, we next assessed for alterations in plasma and tissue lactate distribution. As illustrated in Fig. 1E, burn patients showed evidence of hyperlactatemia, defined clinically as blood lactate >2 mmol/L (dotted line) (23), particularly at earlier time points after injury (P < 0.001). Although circulating lactate decreased at 2–4 days, intracellular levels increased in burn sWAT at 1–3 days (P < 0.05) and predominantly at 7–10 days (P < 0.01) when compared with nonburn sWAT (dotted line; Fig. 1F). In fact, the aforementioned changes in adipose lactate levels were also accompanied by a profound increase in [NADH/NAD+] at 1–3 days (P < 0.001) and 7–10 days (P < 0.01; Fig. 1G). Together, these findings indicate that levels of lactate and its oxidation by-product are increased in adipose tissue after burn, implicating a potential role for lactate-specific redox alterations in the induction of browning in after injury in humans.
Mouse model of burn injury.
To further uncover the significance of lactate in burn-induced browning, we utilized a burn injury mouse model and assessed the browning process and changes in lactate distribution and redox homeostasis in iWAT. Immunohistochemical staining for UCP1 demonstrated increased positivity in the iWAT at 3 days after burn with the greatest expression at 7 days postburn compared with sham mice (Fig. 2A, top). Our gene expression data corroborated these findings, indicating increased Ucp1 mRNA gene expression in iWAT from burn mice at 7 days after injury (P < 0.01; Fig. 2B). In addition, burn mice demonstrated increased MCT1 and LDH positive cells relative to sham as early as 3 days after injury (Fig. 2A, middle and bottom). In fact, gene expression of both Mct1 (P < 0.0001) and Ldhb (P < 0.01) was greatest at 7 days postburn, mirroring the peak expression of Ucp1 (Fig. 2, C and D). Consistent with our clinical data, burn mice also showed evidence of hyperlactatemia during the acute phase (P < 0.0001). In fact, lactate levels were still significantly elevated relative to sham at 7 days (P < 0.05) after injury, and even more so at 14 days (P < 0.001; Fig. 2E). At the tissue level, lactate levels significantly increased in iWAT of burn mice at 7 days (P < 0.001), which was also accompanied by increased intracellular [NADH/NAD+] (P < 0.01; Fig. 2, F and G). Taken together, our findings suggest that postburn browning is associated with a shift toward lactate import and metabolism, demonstrated by the differential profiles for MCT and LDH expression coupled with dynamic changes in WAT lactate and redox homeostasis in both patients and mice after injury.
Figure 2.

Upregulated UCP1, MCT1, and LDH in murine adipose tissue after burn. A: immunohistochemical staining for UCP1, MCT1, and LDH in inguinal WAT (iWAT) isolated from sham and burn mice at <1, 3, 7, and 14 days after injury. Gene expression of UCP1 (B), MCT1 (C), and Ldhb (D) in iWAT from sham and burn mice at <1, 3, 7, and 14 days after injury. Changes in plasma lactate (E), iWAT lactate (F), and iWAT [NADH/NAD+] (G) in sham (dotted line) and burn mice at <1, 3, 7, and 14 days after injury. Values are presented as means ± standard error. One-way ANOVA, Burn vs. sham *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001, n = 7/group. LDH, lactate dehydrogenase; MCT1, monocarboxylate transporter 1; PGC1α, peroxisome proliferator-activated receptor-γ coactivator 1-α; PPARγ, peroxisome proliferator-activated receptor γ; UCP1, uncoupling protein 1; WAT, white adipose tissue.
Daily l-Lactate Administration Augments Burn-Induced Mortality and Indices of Hypermetabolism In Vivo
Thus far having shown that burn-induced browning is associated with increased expression of MCT1 and LDHB expression that parallels UCP1 upregulation, we next sought to directly assess the effects of lactate shuttling on postburn browning in vivo. Based on previous evidence implicating lactate as a strong browning inducer, we hypothesized that increased lactate shuttling via MCTs is sufficient to instigate the postburn WAT browning response. To test this, burn mice were then treated with l-lactate daily for 7 consecutive days. In addition, to confirm that lactate is a key metabolite involved in postburn browning, we also treated burn mice with a pharmacological MCT inhibitor, Phloretin, that has previously been shown to be effective in reducing lactate influx into cells in several studies (19, 27, 28).
As illustrated in Fig. 3A, vehicle-treated burn mice showed a 90% survival over the course of 7 days. Although no significant difference in mortality was observed in Phloretin-treated burn mice, the combination of lactate and burn further decreased survival rate to 75% (P < 0.05). Interestingly, the adverse effects of daily l-lactate administration on postburn mortality were accompanied by augmented changes in systemic hypermetabolism, namely, a severe loss in body weight and fat wasting. Specifically, total body weight in vehicle-treated mice decreased by 2.36%, whereas lactate-treated burn mice lost 5.90% (P < 0.05; Fig. 3B). Similarly, daily l-lactate administration significantly exacerbated the effect of burn on iWAT weight after injury (P < 0.05), suggesting it may have increased the browning response postinjury (Fig. 3C). Surprisingly and consistent with this hypothesis, burn mice treated with Phloretin showed no change in body weight and iWAT weight relative to control mice at 7 days postinjury (Fig. 3, B and C). To confirm Phloretin’s beneficial effects were attributed to a reduction in lactate flux activity, we compared plasma and iWAT concentrations of lactate between groups (Fig. 3, D and E). Indeed, both circulating (P < 0.01) and intracellular levels (P < 0.05) of lactate were drastically increased in lactate-treated relative to vehicle-treated burn mice at 7 days postinjury. Although no change in plasma lactate was observed in Phloretin-treated mice, intracellular levels were substantially reduced in iWAT at this time point (P < 0.05). Together, these findings suggest that elevated lactate is not only a by-product of anaerobic respiration but also plays a causal signaling role in mediating poor survival and indices of systemic hypermetabolism (weight loss and fat wasting) following burn injury.
Figure 3.

Effect of daily l-lactate and Phloretin treatment on postburn mortality and body composition in vivo. A: Kaplan–Meier survival curves of control, vehicle-treated, l-lactate-treated, and Phloretin-treated burn mice over the course of 7 days. Changes in body weight (B), inguinal WAT (iWAT) expressed as a percentage of body weight (C), plasma lactate (D), and tissue lactate (E) in control, vehicle-treated, l-lactate-treated, and Phloretin-treated burn mice at 7 days after injury. Values are presented as means ± standard error. Log-rank test and one-way ANOVA, Burn vs. sham *P < 0.05; **P < 0.01; ***P < 0.001; ****P <0.0001, vehicle vs. treated burn #P < 0.05, ##P < 0.01, n = 7/group. WAT, white adipose tissue.
Increased Lactate Shuttling via MCTs Promotes Postburn Beige Adipocyte Formation via Redox-Dependent and -Independent Mechanisms
To assess how changes in lactate shuttling affect the burn-induced WAT browning response after injury, we performed immunohistochemical staining for the key browning gene Ucp1 in the subcutaneous iWAT depot from control, burn, and burn mice injected with l-lactate and Phloretin for 7 days. As illustrated in Fig. 4A, histological examination of the iWAT of vehicle-treated burned mice at 7 days showed the presence of multilocular UCP-1 + adipocytes—key indicators of the phenotypic switch from white to beige adipose. Consistent with our hypothesis, postburn l-lactate administration effectively heightened this pathological browning response, indicated by enhanced UCP-1 protein staining and increased presence of multilocular cells in the iWAT of lactate-treated relative to vehicle-treated burn mice at the same time point. Remarkably, postburn mice treated with Phloretin showed no evidence of WAT remodeling, as demonstrated by the appearance of reduced UCP-1 protein staining and larger lipid droplet sizes relative to vehicle-treated burn mice at 7 days after injury (P < 0.0001; Fig. 4B). In contrast, lactate-treated mice also showed substantially smaller lipid droplet sizes, a phenotype consistent with increased WAT browning and remodeling (P < 0.0001). This was further corroborated by our gene expression findings, which showed significantly increased expression of several key browning genes including Ucp1 (P < 0.01) after postburn l-lactate treatment (Fig. 4C). Conversely, no change in gene expression of browning markers was observed between control and Phloretin-treated burn mice at 7 days, suggesting decreasing lactate shuttling by targeting MCT transporters effectively ameliorates the pathological browning response to injury.
Figure 4.

Increased MCT-mediated lactate shuttling augments postburn beige adipocyte formation. A: immunohistochemical staining for UCP1 in iWAT from control, vehicle-treated, l-lactate-treated, and Phloretin-treated burn mice at 7 days after injury. B: postburn changes in adipocyte diameter control (n = 116), vehicle-treated (n = 241), l-lactate-treated (n = 225), and Phloretin-treated (n = 160) burn mice at 7 days after injury. C: quantitative RT-PCR analysis of browning genes in inguinal WAT at 7 days postinjury. Changes in mitochondrial LDH activity (D), relative [NADH/NAD+] (E), and FGF21 gene expression (F) at 7 days postinjury. G: schematic depicting redox-dependent and -independent mechanisms of lactate-induced browning. Values are presented as means ± standard error. One-way ANOVA, Sham vs. burn *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001, vehicle vs. treated burn #P < 0.05, ##P < 0.01, ####P < 0.0001, n = 7/group. WAT, white adipose tissue.
Given that lactate-mediated WAT browning is associated with an increased redox state, we next assessed whether administering the l-lactate or Phloretin had any effect on lactate metabolism by comparing activity levels of mitochondrial LDH (mLDH) and intracellular [NADH/NAD+] (Fig. 4, D and E). Compared with vehicle-treated mice, lactate-treated burn mice showed substantially higher levels of mLDH activity (P < 0.01) and [NADH/NAD+] (P < 0.05), likely as a result of increased substrate delivery. Indeed, both mLDH (P < 0.05) and [NADH/NAD+] (P < 0.05) were reduced in Phloretin-treated burn mice at the same time point, suggesting that lactate exerts its thermogenic effects, at least in part, by altering the intracellular redox state. Interestingly, FGF-21, a key regulator of the differentiation of WAT to brown adipocytes previously shown to be stimulated in response to lactate (29), was also increased in burn mice after l-lactate treatment (P < 0.05), consistent with a synergistic, nonredox effect of lactate on the postburn browning process (Fig. 4, F and G).
Lactate-Mediated WAT Browning Aggravates the Development of Postburn Hepatic Steatosis and Dysfunction
Another key consequence of postburn hypermetabolism includes browning-induced lipotoxicity and its impact on essential organs, namely, the liver (17). We previously implicated WAT browning and the associated efflux of lipids during this remodeling period in facilitating hepatic steatosis, a leading cause of morbidity and mortality in burn patients (30). Given that daily l-lactate administration amplified the pathological browning response in postburn mice, we next sought to determine if lactate could also drive its adverse lipotoxic effects on the liver after injury. To assess this, we examined liver sections from control, burn, and burn mice treated with l-lactate and Phloretin using Oil Red O staining for lipid droplets. As shown in Fig. 5A, the livers of untreated burn mice showed both increased lipid infiltration and fat deposition at 7 days postburn, as demonstrated by increased Oil Red O staining relative to control mice. Consistent with an increased postburn browning response, treatment with l-lactate augmented the extent of ectopic fat accumulation in postburn mice, which was further confirmed by the increase in liver weight at this time point (P < 0.01; Fig. 5B). Moreover, hepatic triglyceride (TG) content (P < 0.05) and plasma free fatty acid (FFA; P < 0.001) levels were both significantly increased in lactate-treated burn mice relative to the vehicle-treated burn group after injury (Fig. 5, C and D).
Figure 5.

Lactate-induced browning aggravates lipid-induced hepatic dysfunction postburn injury. A: Oil Red O staining for lipid droplets in liver sections from control, vehicle-treated, l-lactate-treated, and Phloretin-treated burn mice at 7 days postinjury. B: liver weights normalized to body weight at 7 days postinjury. C: liver triglyceride (TG) content at 7 days postinjury. Plasma free fatty acid (FFA; D), aspartate transaminase (AST; E), and alanine aminotransferase (ALT; F) concentrations at 7 days postinjury. Values are presented as means ± standard error. One-way ANOVA, Sham vs. burn *P < 0.05; **P < 0.01; ****P < 0.0001, vehicle vs. treated burn #P < 0.05; ##P < 0.01; ###P < 0.001, n = 7/group.
To assess how this altered liver functions after injury, we then measured circulating levels of hepatic damage markers, alanine transaminase (ALT), and aspartate aminotransferase (AST). As expected, both ALT (P < 0.01) and AST (P < 0.01) levels were drastically elevated in lactate-treated burn mice relative to vehicle, indicating that lactate-induced WAT browning augments organ steatosis and dysfunction after severe injury (Fig. 5, E and F). Remarkably, we observed a robust decrease in hepatic fat infiltration in the livers of Phloretin-treated burn mice at 7 days postburn, as assessed by the decreased presence of Oil Red O-stained lipid droplets and reduced liver weight (P < 0.05) relative to vehicle-treated burn mice. This was accompanied by decreased plasma FFA (P < 0.05) and AST (P < 0.001) levels at 7 days postinjury, suggesting that blocking lactate uptake not only mitigated burn-induced WAT browning but also mitigated the effects of lipid-induced hepatic dysfunction. These results are consistent with our previous findings that inhibiting WAT browning has the beneficial effect of attenuating hepatic steatosis to minimize postburn hypermetabolism.
DISCUSSION
Metabolic rewiring occurs after a severe trauma to better optimize nutrient usage (31, 32). Of great importance is the unique hypermetabolic response of a severely burned patient, which far surpasses that induced by any other injury or disease in terms of its magnitude and persistence (33). Although initially ubiquitous and essential for maintaining organ function under demanding trauma conditions, prolonged hypermetabolism becomes detrimental, leading to a profound catabolic state in almost every system of the body, multiorgan failure, and eventually death (33, 34). This autodestructive response is thought to be driven by the uncontrolled futile cycling of energy substrates after injury, which, in turn, causes pronounced metabolic derangements in vital organs such as the adipose and liver (35). Among intermediate substrates, circulating levels of lactate are elevated by two- to fourfold in severely burned patients due to a devastating and unique derangement called hypovolemic shock, which causes inadequate perfusion and global tissue hypoxia (oxygen insufficiency) (36, 37).
Although lactate has long been considered a waste product of anaerobic respiration, our findings suggest that elevated lactate can contribute to increased morbidity and mortality after injury by directly promoting the adverse browning response, via both redox-dependent and independent mechanisms. Using molecular and pharmacological approaches, we show that 1) circulating lactate levels are elevated in burn patients and mice even 2 wk after injury; 2) burn-induced browning is associated with increased lactate-importing MCT1 expression that parallels UCP1 upregulation; 3) increasing lactate uptake is sufficient to augment postburn mortality, WAT browning, and its associated downstream lipotoxic effects in the liver in vivo; and 4) pharmacological inhibition of MCT-mediated lactate shuttling diminishes WAT remodeling and improves hepatic dysfunction in postburn mice after injury.
Although no redox metabolite has previously been shown to directly regulate the postburn browning process, these findings are consistent with previous studies demonstrating that lactate acts as a potent inducer of UCP1 activation in white adipocytes in response to cold exposure and β-adrenergic stimulation (19, 20). Similarly, administration of lactate protects mice against high-fat-diet-induced obesity and improves dyslipidemia by effectively inducing adipose browning and enhancing thermogenesis (38–40). Mechanistically, this might be part of a redox regulatory and adaptive loop to alleviate a high redox (NADH/NAD+) pressure generated subsequent to its transport, just as mitochondrial reactive oxygen species positively control UCP1 protein activity (41, 42). Indeed, we show that lactate-based adipocyte remodeling occurs via increased import through MCT transporters, which in turn increases mitochondrial LDH activity leading to [NADH/NAD+] accumulation.
One can speculate that protein sensors whose activities are dependent on the NADH/NAD+ ratio synchronize cell signaling and transcriptional events with the cellular metabolic state (43). Thus, redox-regulated proteins in cells with high UCP1 and low NAD+ levels will become less responsive, impairing mitochondrial adaptation, and ultimately leading to metabolic dysfunction. Indeed, numerous studies have highlighted a crucial role for NAD+-dependent enzymes in the regulation of WAT and brown adipose tissue (BAT)–selective gene transcription, including the transcriptional coregulator NAD+-dependent Sirtuin-1 (SIRT-1) (44–46). As such, other metabolites that similarly impact the redox state (e.g., ketone body β-HB, involved in newborn BAT) may also constitute strong browning inducers following injury (19, 47). In addition to short-term effects via redox modulation, our findings herein suggest lactate also has the potential to produce long-term metabolic changes by upregulating expression of the brown adipokine, FGF21, highlighting the diversity of signaling mechanisms responsive to lactate after injury.
Our data indicate that blocking lactate transport with Phloretin not only blunts the pathological browning response in postburn mice but also alleviates hepatic steatosis and dysfunction. We have previously implicated WAT browning and the associated efflux of lipids during this remodeling period in driving ectopic lipid deposition and dysfunction of essential organs, including the liver (17). In fact, hepatic steatosis remains a leading cause of morbidity and mortality in burn patients (30). Therefore, it is not unreasonable to assume that Phloretin indirectly exerts its beneficial effects by counteracting browning-induced lipotoxicity and its impact on the liver. Consistent with this, we also show that augmenting the postburn WAT browning response via daily l-lactate treatments increased hepatic fatty infiltration, impaired liver function, and reduced survival in mice. Although precise mechanisms underlying this adipose liver cross talk after burn and the role lactate plays are still unclear and await further clarification, accumulating evidence suggests that Phloretin ameliorates hepatic steatosis through regulation of lipogenesis via the redox-dependent SIRT-1 pathway (48). Thus, in conjunction with WAT, the identification of redox-sensitive signaling pathways in the liver after burn requires further investigation.
An important caveat of our study is that only male mice were used. Although the potential for sex differences to affect outcomes following burn injury has not been well explored, future studies with female mice will establish if the effect of lactate shuttling on postburn adipose dysfunction, hepatic steatosis, and survival is sexually dimorphic. Future studies should also consider controlling for injection osmolarity and the coinjected sodium ions when studying the effects of exogenous sodium l-lactate administration on postburn browning in vivo, as counterions can have confounding effects on thermogenesis beyond lactate pharmacology, which were not accounted for in this study (49). Similarly, it is worth noting that we only used a nonspecific pharmacological MCT inhibitor without tissue or cell type-specific genetic inhibitory approaches to fully illustrate that the inhibitory thermogenic effects of Phloretin were specific to reducing WAT lactate uptake, rather than exerting other unknown effects. To circumvent this issue, subsequent studies should be conducted with adipose-specific MCT1-deficient mice to validate our results. Additional studies are also still needed to examine the effects of blocking MCT-mediated lactate shuttling on wound healing and the plethora of other organs known to preferentially utilize lactate in conditions of stress, including the brain, cardiac, and muscle tissue, targets not covered in this study, as this will help to characterize the full safety profile of using MCT inhibitors in burn trauma. Notwithstanding this limitation, the positive effects of Phloretin on the adipose and liver metabolism remain important as these two organs have been implicated as major drivers of postburn outcomes relative to others. Finally, at higher temperatures, it is possible that the effects of both burn and lactate on the browning of WAT are less drastic, reducing the lipotoxic burden of fat placed on the liver, although this remains to be explored. Studies are currently underway to address the potential metabolic impact of temperature relative to whole body energetics postburn with the use of metabolic caging under thermoneutral conditions.
In summary, our results suggest that WAT remodeling in pathophysiological situations where intermediate metabolites such as lactate are released could be due to direct signaling effects on adipocytes. Our study brings novel insights to the field of browning in the context of hypermetabolism and critical illness. To our knowledge, this is the first study demonstrating that lactate signaling has a direct impact on what is considered pathological WAT phenomena. Interestingly, since redox metabolites such as lactate or β-HB promote a metabolic switch toward an oxidative phenotype, mitochondrial uncoupling, and browning in burns may represent key elements of an adaptive process to alleviate redox pressure. Although additional studies are needed to understand if WAT browning is truly detrimental or beneficial, we surmise that limited browning after trauma may have an unexpected protective role. The ability to derail the metabolic spiral of hypermetabolism could have a profound impact in a therapeutic setting, necessitating further investigation of these redox metabolites in trauma and critical illness.
DATA AVAILABILITY
Data will be made available upon reasonable request.
GRANTS
This work was supported by a grant from the National Institutes of Health (NIH 2R01GM087285-05A1). D.B. is a recipient of the Frederick Banting and Charles Best Canada Graduate Scholarship (Canadian Institutes of Health Research (CIHR)—Institute of Aging; No. 181563).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
D.B. and M.G.J. conceived and designed research; D.B., C.M.K., F.K., and S.R. performed experiments; D.B., A.A., C.M.K., F.K., S.R., and M.G.J. analyzed data; D.B., A.A., R.A.S., and M.G.J. interpreted results of experiments; D.B. prepared figures; D.B., A.A., and M.G.J. drafted manuscript; D.B., A.A., C.M.K., F.K., S.R., R.A.S., and M.G.J. edited and revised manuscript; D.B., A.A., C.M.K., F.K., S.R., R.A.S., and M.G.J. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank all the burn patients who participated in this study and the Ross Tilley Burn Centre staff for support.
REFERENCES
- 1. Bray GA, Bellanger T. Epidemiology, trends, and morbidities of obesity and the metabolic syndrome. Endocrine 29: 109–117, 2006. doi: 10.1385/ENDO:29:1:109. [DOI] [PubMed] [Google Scholar]
- 2. Bartelt A, Heeren J. Adipose tissue browning and metabolic health. Nat Rev Endocrinol 10: 24–36, 2014. doi: 10.1038/nrendo.2013.204. [DOI] [PubMed] [Google Scholar]
- 3. Guilherme A, Virbasius JV, Puri V, Czech MP. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat Rev Mol Cell Biol 9: 367–377, 2008. doi: 10.1038/nrm2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Galic S, Oakhill JS, Steinberg GR. Adipose tissue as an endocrine organ. Mol Cell Endocrinol 316: 129–139, 2010. doi: 10.1016/j.mce.2009.08.018. [DOI] [PubMed] [Google Scholar]
- 5. Cristancho AG, Lazar MA. Forming functional fat: a growing understanding of adipocyte differentiation. Nat Rev Mol Cell Biol 12: 722–734, 2011. doi: 10.1038/nrm3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Peirce V, Carobbio S, Vidal-Puig A. The different shades of fat. Nature 510: 76–83, 2014. doi: 10.1038/nature13477. [DOI] [PubMed] [Google Scholar]
- 7. Rosen ED, Spiegelman BM. What we talk about when we talk about fat. Cell 156: 20–44, 2014. doi: 10.1016/j.cell.2013.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Otero-Díaz B, Rodríguez-Flores M, Sánchez-Muñoz V, Monraz-Preciado F, Ordoñez-Ortega S, Becerril-Elias V, Baay-Guzmán G, Obando-Monge R, García-García E, Palacios-González B, Villarreal-Molina MT, Sierra-Salazar M, Antuna-Puente B. Exercise induces white adipose tissue browning across the weight spectrum in humans. Front Physiol 9: 1781, 2018. doi: 10.3389/fphys.2018.01781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lim S, Honek J, Xue Y, Seki T, Cao Z, Andersson P, Yang X, Hosaka K, Cao Y. Cold-induced activation of brown adipose tissue and adipose angiogenesis in mice. Nat Protoc 7: 606–615, 2012. doi: 10.1038/nprot.2012.013. [DOI] [PubMed] [Google Scholar]
- 10. Abdullahi A, Jeschke MG. White adipose tissue browning: a double edge sword. Trends Endocrinol Metab 27: 542–552, 2016. doi: 10.1016/j.tem.2016.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xue Y, Xu X, Zhang X-Q, Farokhzad OC, Langer R. Preventing diet-induced obesity in mice by adipose tissue transformation and angiogenesis using targeted nanoparticles. Proc Natl Acad Sci USA 113: 5552–5557, 2016. doi: 10.1073/pnas.1603840113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang S, Liang X, Yang Q, Fu X, Rogers CJ, Zhu M, Rodgers BD, Jiang Q, Dodson MV, Du M. Resveratrol induces brown-like adipocyte formation in white fat through activation of AMP-activated protein kinase (AMPK) α1. Int J Obes (Lond) 39: 967–976, 2015. doi: 10.1038/ijo.2015.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu X, Wang S, You Y, Meng M, Zheng Z, Dong M, Lin J, Zhao Q, Zhang C, Yuan X, Hu T, Liu L, Huang Y, Zhang L, Wang D, Zhan J, Jong Lee H, Speakman JR, Jin W. Brown adipose tissue transplantation reverses obesity in ob/ob mice. Endocrinology 156: 2461–2469, 2015. doi: 10.1210/en.2014-1598. [DOI] [PubMed] [Google Scholar]
- 14. Patsouris D, Qi P, Abdullahi A, Stanojcic M, Chen P, Parousis A, Amini-Nik S, Jeschke MG. Burn induces browning of the subcutaneous white adipose tissue in mice and humans. Cell Rep 13: 1538–1544, 2015. doi: 10.1016/j.celrep.2015.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Petruzzelli M, Schweiger M, Schreiber R, Campos-Olivas R, Tsoli M, Allen J, Swarbrick M, Rose-John S, Rincon M, Robertson G, Zechner R, Wagner EF. A switch from white to brown fat increases energy expenditure in cancer-associated cachexia. Cell Metab 20: 433–447, 2014. doi: 10.1016/j.cmet.2014.06.011. [DOI] [PubMed] [Google Scholar]
- 16. Sidossis LS, Porter C, Saraf MK, Børsheim E, Radhakrishnan RS, Chao T, Ali A, Chondronikola M, Mlcak R, Finnerty CC, Hawkins HK, Toliver-Kinsky T, Herndon DN. Browning of subcutaneous white adipose tissue in humans after severe adrenergic stress. Cell Metab 22: 219–227, 2015. doi: 10.1016/j.cmet.2015.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Abdullahi A, Samadi O, Auger C, Kanagalingam T, Boehning D, Bi S, Jeschke MG. Browning of white adipose tissue after a burn injury promotes hepatic steatosis and dysfunction. Cell Death Dis 10: 870, 2019. doi: 10.1038/s41419-019-2103-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wu J, Cohen P, Spiegelman BM. Adaptive thermogenesis in adipocytes: is beige the new brown? Genes Dev 27: 234–250, 2013. doi: 10.1101/gad.211649.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Carrière A, Jeanson Y, Berger-Müller S, André M, Chenouard V, Arnaud E, Barreau C, Walther R, Galinier A, Wdziekonski B, Villageois P, Louche K, Collas P, Moro C, Dani C, Villarroya F, Casteilla L. Browning of white adipose cells by intermediate metabolites: an adaptive mechanism to alleviate redox pressure. Diabetes 63: 3253–3265, 2014. doi: 10.2337/db13-1885. [DOI] [PubMed] [Google Scholar]
- 20. Lagarde D, Jeanson Y, Barreau C, Moro C, Peyriga L, Cahoreau E, Guissard C, Arnaud E, Galinier A, Bouzier-Sore A-K, Pellerin L, Chouchani ET, Pénicaud L, Ader I, Portais J-C, Casteilla L, Carrière A. Lactate fluxes mediated by the monocarboxylate transporter-1 are key determinants of the metabolic activity of beige adipocytes. J Biol Chem 296: 100137, 2021. doi: 10.1074/jbc.RA120.016303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Carrière A, Lagarde D, Jeanson Y, Portais J-C, Galinier A, Ader I, Casteilla L. The emerging roles of lactate as a redox substrate and signaling molecule in adipose tissues. J Physiol Biochem 76: 241–250, 2020. doi: 10.1007/s13105-019-00723-2. [DOI] [PubMed] [Google Scholar]
- 22. Holroyde CP, Gabuzda TG, Putnam RC, Paul P, Reichard GA. Altered glucose metabolism in metastatic carcinoma. Cancer Res 35: 3710–3714, 1975. [PubMed] [Google Scholar]
- 23. Herrero E, Sánchez M, Cachafeiro L, Agrifoglio A, Galván B, Asensio M, de Lorenzo AG. Lactate in the burn patient. Crit Care 19: P145, 2015. doi: 10.1186/cc14225. [DOI] [Google Scholar]
- 24. Mokline A, Abdenneji A, Rahmani I, Gharsallah L, Tlaili S, Harzallah I, Gasri B, Hamouda R, Messadi AA. Lactate: prognostic biomarker in severely burned patients. Ann Burns Fire Disasters 30: 35–38, 2017. [PMC free article] [PubMed] [Google Scholar]
- 25. Auger C, Appanna VD. A novel ATP-generating machinery to counter nitrosative stress is mediated by substrate-level phosphorylation. Biochim Biophys Acta 1850: 43–50, 2015. doi: 10.1016/j.bbagen.2014.09.028. [DOI] [PubMed] [Google Scholar]
- 26. Rogers GW, Brand MD, Petrosyan S, Ashok D, Elorza AA, Ferrick DA, Murphy AN. High throughput microplate respiratory measurements using minimal quantities of isolated mitochondria. PLoS One 6: e21746, 2011. doi: 10.1371/journal.pone.0021746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sonveaux P, Copetti T, De Saedeleer CJ, Végran F, Verrax J, Kennedy KM, Moon EJ, Dhup S, Danhier P, Frérart F, Gallez B, Ribeiro A, Michiels C, Dewhirst MW, Feron O. Targeting the lactate transporter MCT1 in endothelial cells inhibits lactate-induced HIF-1 activation and tumor angiogenesis. PLoS One 7: e33418, 2012. doi: 10.1371/journal.pone.0033418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Carstensen MH, Leichtweiss HP, Schröder H. Lactate carriers in the artificially perfused human term placenta. Placenta 4: 165–174, 1983. doi: 10.1016/s0143-4004(83)80029-0. [DOI] [PubMed] [Google Scholar]
- 29. Jeanson Y, Ribas F, Galinier A, Arnaud E, Ducos M, André M, Chenouard V, Villarroya F, Casteilla L, Carrière A. Lactate induces FGF21 expression in adipocytes through a p38-MAPK pathway. Biochem J 473: 685–692, 2016. doi: 10.1042/BJ20150808. [DOI] [PubMed] [Google Scholar]
- 30. Jeschke MG. The hepatic response to thermal injury: is the liver important for postburn outcomes? Mol Med 15: 337–351, 2009. doi: 10.2119/molmed.2009.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Şimşek T, Şimşek HU, Cantürk NZ. Response to trauma and metabolic changes: posttraumatic metabolism. Ulus Cerrahi Derg 30: 153–159, 2014. doi: 10.5152/UCD.2014.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Williams FN, Jeschke MG, Chinkes DL, Suman OE, Branski LK, Herndon DN. Modulation of the hypermetabolic response to trauma: temperature, nutrition, and drugs. J Am Coll Surg 208: 489–502, 2009. doi: 10.1016/j.jamcollsurg.2009.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jeschke MG, Gauglitz GG, Kulp GA, Finnerty CC, Williams FN, Kraft R, Suman OE, Mlcak RP, Herndon DN. Long-term persistance of the pathophysiologic response to severe burn injury. PLoS One 6: e21245, 2011. doi: 10.1371/journal.pone.0021245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jeschke MG. Postburn hypermetabolism: past, present, and future. J Burn Care Res 37: 86–96, 2016. doi: 10.1097/BCR.0000000000000265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wolfe RR, Herndon DN, Jahoor F, Miyoshi H, Wolfe M. Effect of severe burn injury on substrate cycling by glucose and fatty acids. N Engl J Med 317: 403–408, 1987. doi: 10.1056/NEJM198708133170702. [DOI] [PubMed] [Google Scholar]
- 36. Ishihara H, Otomo N, Suzuki A, Takamura K, Tsubo T, Matsuki A. Detection of capillary protein leakage by glucose and indocyanine green dilutions during the early post-burn period. Burns 24: 525–531, 1998. doi: 10.1016/s0305-4179(98)80004-1. [DOI] [PubMed] [Google Scholar]
- 37. Schaefer TJ, Nunez Lopez O. Burn resuscitation and management. In: StatPearls. Treasure Island, FL. StatPearls Publishing, 2023. [PubMed] [Google Scholar]
- 38. Yao Z, Yan Y, Zheng X, Wang M, Zhang H, Li H, Chen W. Dietary lactate supplementation protects against obesity by promoting adipose browning in mice. J Agric Food Chem 68: 14841–14849, 2020. doi: 10.1021/acs.jafc.0c05899. [DOI] [PubMed] [Google Scholar]
- 39. Cai H, Wang X, Zhang Z, Chen J, Wang F, Wang L, Liu J. Moderate l-lactate administration suppresses adipose tissue macrophage M1 polarization to alleviate obesity-associated insulin resistance. J Biol Chem 298: 101768, 2022. doi: 10.1016/j.jbc.2022.101768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li G, Xie C, Lu S, Nichols RG, Tian Y, Li L, Patel D, Ma Y, Brocker CN, Yan T, Krausz KW, Xiang R, Gavrilova O, Patterson AD, Gonzalez FJ. Intermittent fasting promotes white adipose browning and decreases obesity by shaping the gut microbiota. Cell Metab 26: 672–685.e4, 2017. doi: 10.1016/j.cmet.2017.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang Q, Zou M-H. Measurement of reactive oxygen species (ROS) and mitochondrial ROS in AMPK knockout mice blood vessels. Methods Mol Biol 1732: 507–517, 2018. doi: 10.1007/978-1-4939-7598-3_32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jastroch M. Uncoupling protein 1 controls reactive oxygen species in brown adipose tissue. Proc Natl Acad Sci USA 114: 7744–7746, 2017. doi: 10.1073/pnas.1709064114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chiarugi A, Dölle C, Felici R, Ziegler M. The NAD metabolome–a key determinant of cancer cell biology. Nat Rev Cancer 12: 741–752, 2012. doi: 10.1038/nrc3340. [DOI] [PubMed] [Google Scholar]
- 44. Kajimura S, Seale P, Tomaru T, Erdjument-Bromage H, Cooper MP, Ruas JL, Chin S, Tempst P, Lazar MA, Spiegelman BM. Regulation of the brown and white fat gene programs through a PRDM16/CtBP transcriptional complex. Genes Dev 22: 1397–1409, 2008. doi: 10.1101/gad.1666108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Qiang L, Wang L, Kon N, Zhao W, Lee S, Zhang Y, Rosenbaum M, Zhao Y, Gu W, Farmer SR, Accili D. Brown remodeling of white adipose tissue by SirT1-dependent deacetylation of Pparγ. Cell 150: 620–632, 2012. doi: 10.1016/j.cell.2012.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang Q, Wang S-Y, Nottke AC, Rocheleau JV, Piston DW, Goodman RH. Redox sensor CtBP mediates hypoxia-induced tumor cell migration. Proc Natl Acad Sci USA 103: 9029–9033, 2006. doi: 10.1073/pnas.0603269103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Srivastava S, Kashiwaya Y, King MT, Baxa U, Tam J, Niu G, Chen X, Clarke K, Veech RL. Mitochondrial biogenesis and increased uncoupling protein 1 in brown adipose tissue of mice fed a ketone ester diet. FASEB J 26: 2351–2362, 2012. doi: 10.1096/fj.11-200410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Liou C-J, Wu S-J, Shen S-C, Chen L-C, Chen Y-L, Huang W-C. Phloretin ameliorates hepatic steatosis through regulation of lipogenesis and Sirt1/AMPK signaling in obese mice. Cell Biosci 10: 114, 2020. doi: 10.1186/s13578-020-00477-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lund J, Breum AW, Gil C, Falk S, Sass F, Isidor MS, Dmytriyeva O, Ranea-Robles P, Mathiesen CV, Basse AL, Johansen OS, Fadahunsi N, Lund C, Nicolaisen TS, Klein AB, Ma T, Emanuelli B, Kleinert M, Sørensen CM, Gerhart-Hines Z, Clemmensen C. The anorectic and thermogenic effects of pharmacological lactate in male mice are confounded by treatment osmolarity and co-administered counterions. Nat Metab 5: 677–698, 2023. doi: 10.1038/s42255-023-00780-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data will be made available upon reasonable request.