

Abstract
Background
Lung adenocarcinoma (LUAD) is the leading cause of death among cancer diseases. The tumorigenic functions of AHNAK2 in LUAD have attracted more attention in recent years, while there are few studies which have reported its high molecular weight.
Methods
The mRNA‐seq data of AHNAK2 and corresponding clinical data from UCSC Xena and GEO was analyzed. LUAD cell lines were transfected with sh‐NC and sh‐AHNAK2, and cell proliferation, migration and invasion were then detected by in vitro experiments. We performed RNA sequencing and mass spectrometry analysis to explore the downstream mechanism and interacting proteins of AHNAK2. Finally, western blot, cell cycle analysis and CO‐IP were used to confirm our assumptions regarding previous experiments.
Results
Our study revealed that AHNAK2 expression was significantly higher in tumors than in normal lung tissues and higher AHNAK2 expression led to a poor prognosis, especially in patients with advanced tumors. AHNAK2 suppression via shRNA reduced the LUAD cell lines proliferation, migration and invasion and induced significant changes in DNA replication, NF‐kappa B signaling pathway and cell cycle. AHNAK2 knockdown also caused G1/S phase cell cycle arrest, which could be attributed to the interaction of AHNAK2 and RUVBL1. In addition, the results from gene set enrichment analysis (GSEA) and RNA sequencing suggested that AHNAK2 probably plays a part in the mitotic cell cycle.
Conclusion
AHNAK2 promotes proliferation, migration and invasion in LUAD and regulates the cell cycle via the interaction with RUVBL1. More studies of AHNAK2 are still needed to reveal its upstream mechanism.
Keywords: AHNAK2, cell cycle, LUAD, RUVBL1
Lung adenocarcinoma (LUAD) is the leading cause of death among cancer diseases. We found that in LUAD, AHNAK2 expression was significantly higher in tumor than in normal lung tissues and higher AHNAK2 expression led to poor prognosis. The cell phenotype and RNA sequencing results showed that AHNAK2 promoted the proliferation, migration and invasion of LUAD cells through the interaction with RUVBL1 and regulated the cell cycle pathway.
INTRODUCTION
Based on cancer statistics in 2021, lung cancer is the leading cause of death among cancer diseases. There were 235 760 new cases and 131 880 deaths of lung and bronchus cancer approximately. 1 Only 21.7% of all patients with non‐small cell lung cancer (NSCLC) are alive ≥5 years after diagnosis. 2 Lung adenocarcinoma (LUAD) accounts for around half of the total number of lung cancers, and its prevalence is increasing according to annual reports. 3 Therefore, it is necessary to discover unexplored mechanisms and new therapeutic targets.
As a giant nucleoprotein (molecular mass 616 kDa), AHNAK2 was first discovered in 2004 and belongs to the AHNAKs protein family (AHNAK and AHNAK2). Its sister nucleoprotein AHNAK has been thoroughly researched ever since it was discovered and appears to have a suppressive effect of tumorigenesis, such as melanoma, breast cancer and glioma. 4 , 5 , 6 AHNAK loss has previously been shown to be associated with LUAD development in a laboratory setting, 7 while we have no evidence that AHNAK has a clinical impact on the prognosis of patients with LUAD.
As an oncogene, AHNAK2 overexpression has been identified in various cancer cohorts in recent years. CCRC was the first cancer that has been systemically proven to be associated with upregulated AHNAK2, as well as concomitant epithelial‐mesenchymal transition (EMT) and stem cell‐like properties. 8 Furthermore, in several studies, AHNAK2 was found to be upregulated in pancreatic ductal adenocarcinoma, uveal melanoma, gastric cancer and thymic carcinoma and led to poor survival prognosis. 9 , 10 , 11 Nevertheless, the biological mechanism of AHNAK2 has not been elucidated until now. AHNAK2 was recently found to promote proliferation, migration and invasion in LUAD via the TGF‐β/Smad3 and MAPK pathways, resulting in a poor prognosis coincidently. 12 , 13 , 14 In addition, a bioinformatic analysis indicated that AHNAK2 expression was positively correlated with CD4+ T cells, macrophages and neutrophils, although there was no careful experimental data or any rigorous hypothesis testing to back it up. 15 The latest review of AHNAKs has been delivered by Zardab et al., and provides a detailed summary of protein structure and oncology functions. 16
In this investigation, we detected the AHNAK2 expression levels in tumor and paired normal tissues and explored cancer databases from The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) for gene expression and survival analysis. To verify our results from databases and explore the role of AHNAK2 in LUAD, we performed tumor cell phenotype assays, gene enrichment analysis and coimmunoprecipitation tests. Finally, we found that AHNAK2 interacted with RUVBL1 to regulate the proliferation, migration, invasion and cell cycle of LUAD cells, which might reveal upregulated AHNAK2 expression leads to poor prognosis in patients with LUAD. Furthermore, we provide evidence that AHNAK2 could cause G1/S phase cell cycle arrest by interaction with RUVBL1 and probably plays a part in mitotic progression through the α‐tubulin and β‐tubulin TGF–β/Smad3 pathway and MAPK pathway.
METHODS
Clinical samples and cell culture
We obtained 57 tumor tissues and the paired adjacent tissues from patients who received lung resections and were diagnosed with primary LUAD in the Beijing Chao‐Yang Hospital. All patients granted informed consent. Beijing‐ChaoYang Hospital ethics committee checked and approved this study.
LUAD cell‐lines NCI‐H1975 (1101HUM‐PUMC000252) and A549 (1101HUM‐PUMC000002) were obtained from the National Infrastructure of Cell Line Resource. NCI‐H1975 and A549 cells were cultured in 1640 supplemented with 10% fetal bovine serum (FBS: Gibco), 100 U/mL penicillin, and 100 μg /mL streptomycin (ThermoFisher) at 37°C in a humidified 5% CO2 atmosphere.
Data profile collection
mRNA‐seq data of TCGA LUAD patients and corresponding clinical data (overall survival time was more than 30 days) was downloaded from UCSC Xena (https://xena.ucsc.edu/). mRNA‐seq data and microarray data of Gene Expression Omnibus (GEO) LUAD patients and corresponding clinical data (overall survival time was more than 30 days) was downloaded from GEO DataSets (GSE81089 and GSE31210).
RNA isolation and quantitative real‐time polymerase chain reaction (qRT‐PCR)
We extracted total RNA from cultured cells with TRizol reagent (ThermoFisher) and synthesized cDNA was by reverse transcription with the PrimeScript RT reagent kit (Vazyme Biotech Co.) and subjected to qRT‐PCR with AHNAK2 via the system of CFX96 (Bio‐Rad) and ACTB primers in the presence of the qPCR SYBR Green Master Mix (Vazyme Biotech Co.). The primers of qRT‐PCR used are listed in Table 1.
TABLE 1.
The primer sequences of quantitative real‐time polymerase chain reaction (qRT‐PCR).
| Gene name | Primer name | Primer sequence (5′ to 3′) |
|---|---|---|
| AHNAK2 | AHNAK2‐homo‐F | GAGAAGGAGGACACGGATGTTGC |
| AHNAK2‐homo‐R | CCCCGCTTGCTCTTTATGGATTG | |
| GAPDH | GAPDH‐homo‐F | GGACTCATGACCACAGTCCA |
| GAPDH‐homo‐R | TCAGCTCAGGGATGACCTTG |
Plasmid constructs, lentivirus production and establishment of stable overexpression or knockdown cell lines
AHNAK2 shRNA were cloned into lentiviral expression vector Lenti‐Guide‐puro‐GFP. The shRNA‐AHNAK2 and negative control shRNA was ordered from Micro‐Helix Company. All of the constructs generated were confirmed by DNA sequencing. Lentiviral packaging plasmids psPAX2 and pMG2.G were cotransfected with the backbone plasmid into HEK293T cells for virus production. The H1975 and A549 cell line were cultured in six‐well tissue culture plates and infected with lentivirus at a multiplicity of infection of 10 for 24 h. Then the medium was replaced with fresh complete medium. Cells were selected in 2.5 μg/mL puromycin in the culture medium to generate stable transfections. The shRNA and siRNA are listed in Table 2.
TABLE 2.
The shRNA and siRNA sequences for AHNAK2 and RUVBL1.
| Gene name | shRNA and siRNA name | Sequence |
|---|---|---|
| AHNAK2 | sh‐AHNAK2‐A12 | CCGGAGTGTCCAGAGGCCAATATTGCTCGAGCAATATTGGCCTCTGGACACTTTTTTG |
| sh‐AHNAK2‐B1 | CCGGTCAGGCAGAGTGCGGTATATTCTCGAGAATATACCGCACTCTGCCTGATTTTTG | |
| RUVBL1 | si‐RUVBL1 | GCUGCGAAUAAAGGAGACCAA |
Western blotting
The LUAD cell lines were treated with lysis buffer (NP‐40, AMRESCO, E109; radioimmunoprecipitation assay [RIPA] buffer, ThermoFisher, R0278) and proteinase inhibitor (ThermoFisher, A32955). The protein concentration was examined by total cellular protein extraction using a BCA Protein Assay Kit (ThermoFisher, A53225). The equivalent protein quantities were sampled to SDS‐PAGE and transferred to PVDF membrane. Membranes were blocked with 5% BSA for 1 h at room temperature and then incubated with the indicated primary antibodies. The membranes were then treated with the appropriate anti‐rabbit secondary antibodies (Cell Signaling, 7074). The labeled protein bands were scanned and analyzed using the ECL chemiluminescence (GE, Las‐4000) detection system.
The primary antibodies are listed as follows: Anti‐β actin (Abcam, ab8227, 1:5000), Anti‐TIP49A (Abcam, ab226001, 1:2000), Anti‐p21 (Abcam ab109520, 1:2000), Anti‐PCNA (Cell Signaling, mAb#13110, 1:1000), Anti‐cyclin D1 (Cell Signaling, mAb#2978, 1:1000), Anti‐DDDDK (FLAG) tag (Abcam, ab205606,1:30).
Cell counting kit‐8 (CCK‐8) proliferation assay
We performed a CCK‐8 assay to detect the proliferation capacity of LUAD cell lines. The A549 and H1975 cells were cultured in 96‐well plates with 10% FBS medium for 24 h in the incubator (5 × 103 cells/well). We measured the cell viability every 24 h for three times, with CCK‐8 reagent (Promega Biotech) added into each well and cultured at 37°C for 2 h. OD value of cells was measured at 450 nm on an enzyme‐labeled instrument.
Migration and invasion assay
Transwell assay was performed to measure the invasion and migration capacity (with or without coated Matrigel [ThermoFisher]). First, the cells were trypsinized and resuspended in 200 μL serum‐free medium (RPMI‐1640). Then we measured the density of cells with a hemocytometer. A total of 1 × 104 LUAD cells with serum‐free medium were seeded into the top chambers, and 500 ul of serum‐free medium with 10% FBS was placed in the lower chamber. After 16 h, we fixed the cells on the lower surface of chambers with 4% paraformaldehyde for 10 min and stained with 0.1% crystal violet. Finally, we counted the quantity of migrated or invaded cells using a microscope.
Cell cycle analysis
The cultured A549 cells (5 × 102 cells/well) were synchronized in serum‐free medium for 24 h. Subsequently, the A549 cells were digested with trypsin and washed twice with phosphate buffered saline (PBS). Cells were collected by centrifugation at 300 × g at 4°C for 5 min, followed by fixation with 70% ethanol at 4°C for 24 h. After being washed with PBS, cells were stained with propidium iodide staining solution for 30 min at room temperature. The cell cycle was detected with the FACScan flow cytometer (BD Accuri, C6) and analyzed using CellQuest software (version 7.6.1; Flow Jo LLC.).
Coimmunoprecipitation (Co‐IP)
Co‐IP was performed using flag antibody as the molecular weight of AHNAK2 was too great to be tested with AHNAK2 antibody. The flag antibodies were added into cell lysates (2 μg flag antibody/1 mg protein) at 4°C for 8 h, and were then incubated with protein G beads at 4°C for 2 h. The beads were washed with washing buffer and the coimmunoprecipitated proteins were then eluted by western blotting with the antibody for RUVBL1 (Abcam, ab226001). The input was used as positive control, while IgG was used as negative control.
Statistical analysis
We performed statistical analysis using SPSS software (version 20.0, USA). Data are presented as the means ± SD from independent triplicate experiments. A Student's t‐test was used to compare the intergroup difference of measurement data. Western blot and q‐PCR results between intervention and control groups. p‐values < 0.05 were considered to be statistically significant.
RESULTS
AHNAK2 expression is upregulated in LUAD tissues and cell lines and leads to poor prognosis in patients with LUAD
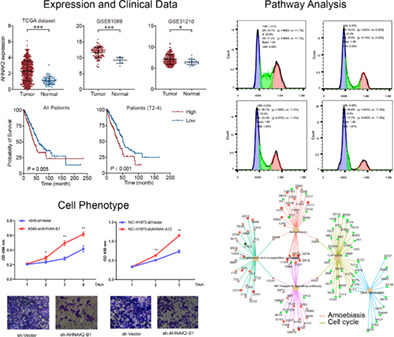
In the early stage, our interest in AHNAK2 can be traced back to a whole exon sequencing test in 2017 that showed AHNAK2 to be a high frequency mutated gene (p < 0.001 for tumor tissue compared to adjacent tissue). We then analyzed AHNAK2 expression at mRNA level in LUAD and normal tissues and its effect on survival time in TCGA and GEO datasets. We found that AHNAK2 expression was significantly higher in tumor than in normal lung tissues in both TCGA and GEO databases (Figure 1a). AHNAK2 was highly expressed in various tumor tissues (Figure S1). We then compared AHNAK2 expression levels in tumor samples with different T stages in TCGA LUAD samples and found that it was significantly higher in advanced tumors compared to early tumors (Figure 1b). We finally analyzed the impact of AHNAK2 expression on LUAD survival time and the results showed that higher AHNAK2 expression had significantly shorter LUAD survival time than lower AHNAK2 expression, especially in patients with advanced tumors (Figure 1c). Progression‐free survival time of patients with higher AHNAK2 expression was also significantly shorter than patients with lower expression (Figure 1d). To verify the results of the bioinformatic data, we performed qRT‐PCR of AHNAK2 in LUAD tumor tissues and adjacent normal tissues for the patients who received operations in our thoracic department. Consistent with TCGA and GEO data, our results also showed that AHNAK2 were highly expressed in tumor tissue and associated with poor survival (Figure 1e). However, our western blotting failed on account of the large molecular mass of AHNAK2 (616 kDa), and the bands could not be transferred and developed as usual. All these results demonstrated an oncogenic role of AHNAK2 in LUAD.
FIGURE 1.
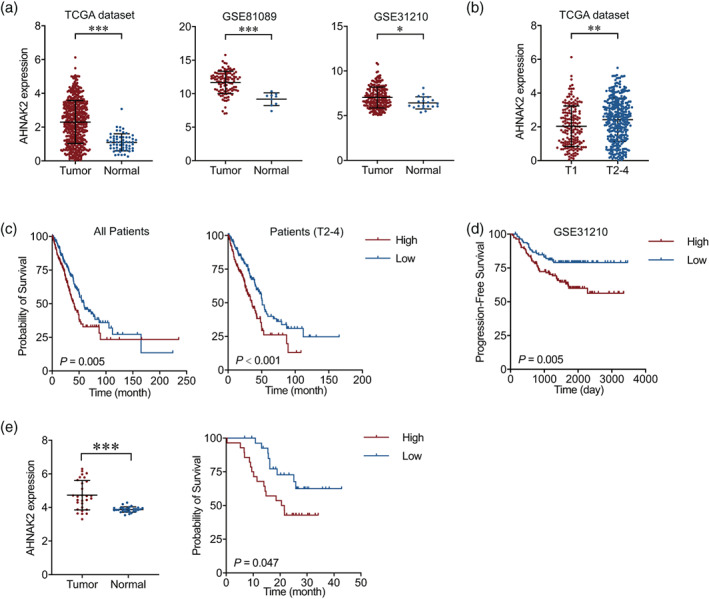
Upregulation of AHNAK2 is correlated with poor prognosis in lung adenocarcinoma (LUAD) patients. (a) mRNA levels of AHNAK2 in LUAD tumor and normal tissues in TCGA (left) and GEO database (middle and right). (b) mRNA levels of AHNAK2 in LUAD tumor samples with different T stages. (c) Kaplan–Meier estimate of LUAD overall survival time based on AHNAK2 mRNA expression levels in all patients (left) and advanced patients (right). (d) Kaplan–Meier estimate of LUAD progression‐free survival time based on AHNAK2 mRNA expression levels. (e) Quantitative real‐time polymerase chain reaction (qRT‐PCR) of AHNAK2 was performed in adenocarcinoma tumor tissues and adjacent normal tissues.
AHNAK2 knockdown suppresses LUAD cells proliferation, migration and invasion
To verify our hypotheses of the function of AHNAK2 in LUAD, two cell lines (NCI‐H1975, NCI‐H1299 and NCI‐A549) were employed to be transfected with sh‐NC, sh‐AHNAK2‐A12 or sh‐AHNAK2‐B1. Then the transfection efficiency was detected with qRT‐PCR, and consequently, NCI‐H1975 and NCI‐A549 were chosen for subsequent experiments with the high expression level of AHNAK2 suppressed remarkably. We performed a CCK‐8 proliferation assay to observe the changes in the proliferation capability of cell lines. As shown in Figure 2a, AHNAK2 knockdown aberrantly suppressed the proliferative capability of H1975 and A549 cells in comparison to the sh‐NC group (Figure 2a). A transwell assay was carried out to explore the effects of AHNAK2 on migration and invasion capacities. We found that low expression of AHNAK2 markedly suppressed the migration and invasion of LUAD cells (Figure 2b, c).
FIGURE 2.
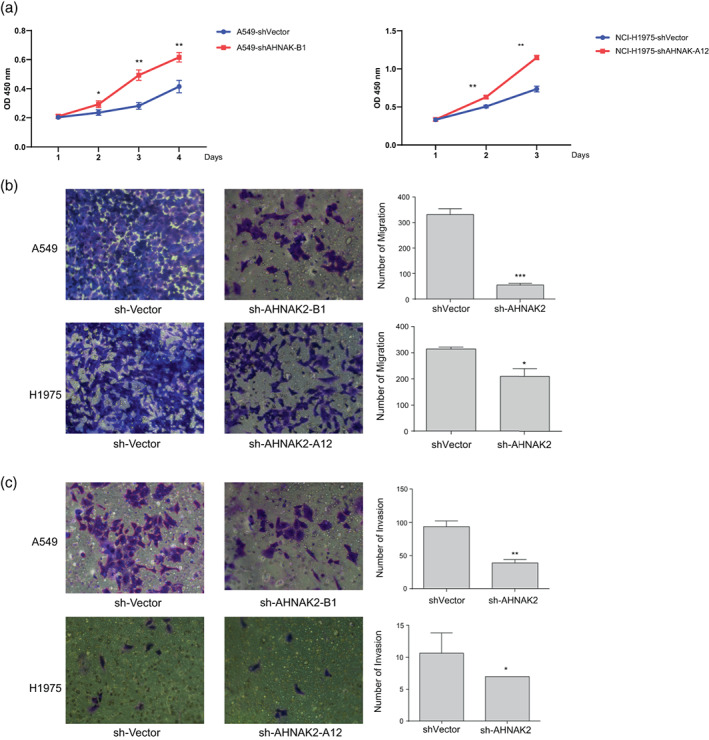
AHNAK2 knockdown suppressed lung adenocarcinoma (LUAD) cell proliferation, migration and invasion. (a) AHNAK2 knockdown aberrantly suppressed the proliferative capability of H1975 and A549 cells. (b) The effect of sh‐AHNAK2 on migration of the two lung adenocarcinoma cells using a transwell assay. (c) The effect of sh‐AHNAK2 on invasion of the two lung adenocarcinoma cells using a transwell assay.
AHNAK2 downregulation suppresses cell cycle in A549 cells
To reveal the promoting effect of AHNAK2 on LUAD cells, RNA sequencing was performed using NCI‐A549 in triplicate (sh‐NC vs. sh‐AHNAK2‐B1). The results of pathway enrichment suggested that AHNAK2 downregulation induced significant changes in DNA replication, NF‐kappa B signaling pathway and cell cycle (Figure 3a, b). It is worth noting that most of regulated genes were enriched to cell cycle G1/S phase transition, chromosome segregation and protein‐DNA complex subunit organization (Figure 3c). Furthermore, our bioinformatic analysis indicated that the expression of CCND1 was positively correlated with AHNAK2 expression and the same correlation appeared between PCNA and RUVBL1 (Figure 3d). To confirm the results of gene ontology (GO) enrichment (biological progress [BP]), we verified the checkpoint proteins of cell cycle using western blot. Unsurprisingly, the expression of cell cycle regulation proteins, including PCNA and CCND1, were downregulated to varying degrees in the experimental group of A549 cells transfected with AHNAK2 shRNA compared with control cells. Meanwhile CDKN1A (P21), as the potent cyclin‐dependent kinase inhibitor, increased when AHNAK2 was suppressed (Figure 3e).
FIGURE 3.
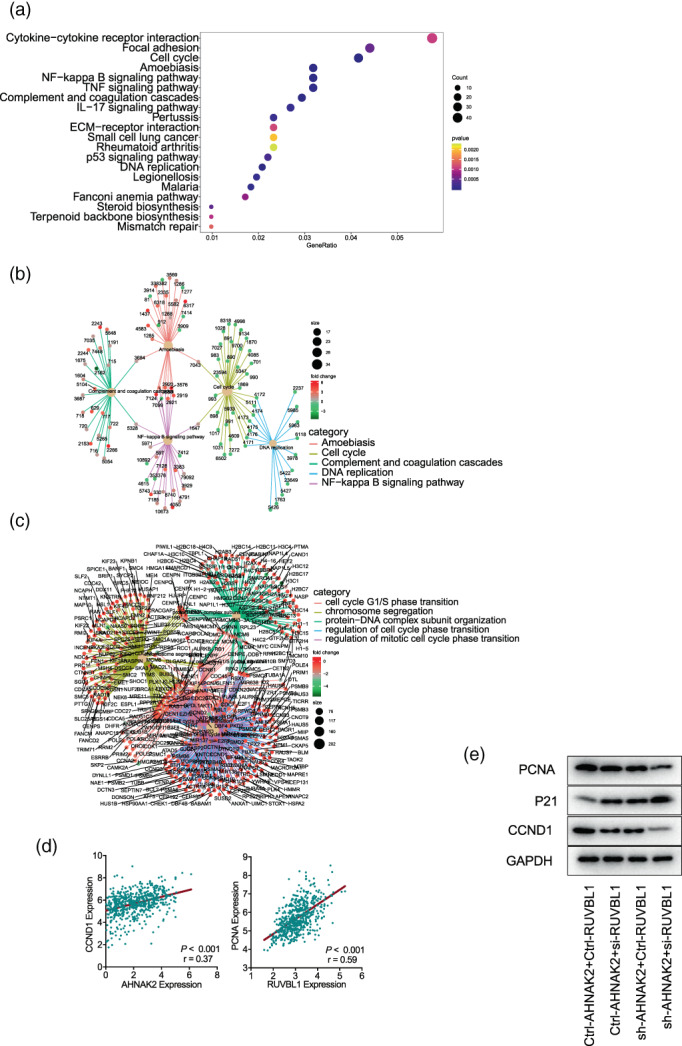
AHNAK2 downregulation suppresses cell cycle in A549 cells. (a, b) Pathway enrichment: differential genes were enriched to focal adhesion, cell cycle, DNA replication, NF‐kappa B signaling pathway and so forth. (c) Gene ontology (GO) enrichment: differential genes were enriched to cell cycle G1/S phase transition, chromosome segregation and protein‐DNA complex subunit organization. (d) Correlation of the mRNA levels of AHNAK2 and CCND1 (left) and RUVBL1 and PCNA (right) in TCGA lung adenocarcinoma (LUAD) tumor samples. (e) PCNA and CCND1 were downregulated in the A549 cells transfected with AHNAK shRNA and RUVBL1 siRNA in sequence, while CDKN1A(P21) expression increased.
AHNAK2 interacts with RUVBL1 and causes G1/S phase cell cycle arrest in A549 cells
Given that AHNAK2 had not been mentioned in previous studies to be associated with cell cycle, we supposed some other proteins were involved in the regulation of cell cycle phase transition. We subsequently performed mass spectrometry analysis to screen for the interacting proteins of AHNAK2. A total of 176 proteins were recognized, and among them, RUVBL1(TIP49a or Pontin) attracted our attention as a cell cycle associated protein and ranks high on the list (Table 3).
TABLE 3.
A total of 176 proteins interacted with AHNAK2 were recognized in mass spectrometry analysis.
| No. | UniProt ID | Protein description | Score | emPAI |
|---|---|---|---|---|
| 1 | Q8IVF2 | Protein AHNAK2 (AHNAK2) | 8339 | 0.23 |
| 2.1 | P07437 | Tubulin beta chain (TUBB) | 4630 | 2.82 |
| 2.2 | P68371 | Tubulin beta‐4B chain (TUBB4B) | 4109 | 2.34 |
| 2.3 | P04350 | Tubulin beta‐4A chain (TUBB4A) | 3508 | 2.15 |
| 3 | Q9BQE3 | Tubulin alpha‐1C chain (TUBA1C) | 2186 | 0.56 |
| 4.1 | P04264 | Keratin, type II cytoskeletal 1 (KRT1) | 2016 | 0.88 |
| 4.2 | P04259 | Keratin, type II cytoskeletal 6B (KRT6B) | 715 | 0.24 |
| 4.3 | P05787 | Keratin, type II cytoskeletal 8 (KRT8) | 525 | 0.2 |
| 4.4 | P19013 | Keratin, type II cytoskeletal 4 (KRT4) | 462 | 0.12 |
| 4.5 | P35908 | Keratin, type II cytoskeletal 2 epidermal (KRT2) | 418 | 0.34 |
| 4.6 | P13647 | Keratin, type II cytoskeletal 5 (GN=KRT5) | 354 | 0.23 |
| 5.1 | P11142 | Heat shock cognate 71 kDa protein (HSPA8) | 962 | 0.5 |
| 5.2 | P0DMV8 | Heat shock 70 kDa protein 1A (HSPA1A) | 772 | 0.32 |
| 5.3 | P11021 | Endoplasmic reticulum chaperone BiP (HSPA5) | 432 | 0.36 |
| 5.4 | P34931 | Heat shock 70 kDa protein 1‐like (HSPA1L) | 329 | 0.15 |
| 6 | P35527 | Keratin, type I cytoskeletal 9 (KRT9) | 824 | 0.36 |
| 7 | P02768 | Albumin (ALB) | 618 | 0.09 |
| 8.1 | P62258 | 14‐3‐3 protein epsilon (YWHAE) | 562 | 0.71 |
| 8.2 | P27348 | 14‐3‐3 protein theta (YWHAQ) | 440 | 0.4 |
| 8.3 | P63104 | 14‐3‐3 protein zeta/delta (YWHAZ) | 438 | 0.4 |
| 8.4 | P61981 | 14‐3‐3 protein gamma (YWHAG) | 429 | 0.39 |
| 9.1 | P63261 | Actin, cytoplasmic 2 (ACTG1) | 507 | 0.25 |
| 9.2 | P62736 | Actin, aortic smooth muscle (ACTA2) | 228 | 0.25 |
| 10 | P49411 | Elongation factor Tu, mitochondrial (TUFM) | 483 | 0.29 |
| 11 | O43175 | D‐3‐phosphoglycerate dehydrogenase (PHGDH) | 429 | 0.18 |
| 12 | P17844 | Probable ATP‐dependent RNA helicase DDX5 (DDX5) | 428 | 0.26 |
| 13.1 | Q06830 | Peroxiredoxin‐1 (PRDX1) | 361 | 0.75 |
| 13.2 | P32119 | Peroxiredoxin‐2 (PRDX2) | 214 | 0.33 |
| 14 | O14980 | Exportin‐1 (XPO1) | 302 | 0.23 |
| 15 | Q92499 | ATP‐dependent RNA helicase DDX1 (DDX1) | 300 | 0.17 |
| 16 | Q9Y265 | RuvB‐like 1 (RUVBL1) | 299 | 0.29 |
| 17 | Q9Y230 | RuvB‐like 2 (RUVBL2) | 270 | 0.21 |
| 18 | P08670 | Vimentin (VIM) | 259 | 0.43 |
| 19 | P62249 | 40S ribosomal protein S16 (RPS16) | 257 | 0.45 |
| 20 | P07355 | Annexin A2 (ANXA2) | 256 | 0.63 |
| 21 | P13645 | Keratin, type I cytoskeletal 10 (KRT10) | 228 | 0.18 |
| 22 | A0A075B6P5 | Immunoglobulin kappa variable 2‐28 (IGKV2‐28) | 201 | 0.26 |
| 23 | P12268 | Inosine‐5′‐monophosphate dehydrogenase 2 (IMPDH2) | 195 | 0.41 |
| 24 | P23396 | 40S ribosomal protein S3 (RPS3) | 193 | 0.26 |
| 25 | Q07021 | Complement component 1 Q subcomponent‐binding protein, mitochondrial (C1QBP) | 182 | 0.22 |
| 26 | Q9Y285 | Phenylalanine – tRNA ligase alpha subunit (FARSA) | 178 | 0.06 |
| 27 | O00148 | ATP‐dependent RNA helicase DDX39A (DDX39A) | 176 | 0.14 |
| 28 | Q96JM3 | Chromosome alignment‐maintaining phosphoprotein 1 (CHAMP1) | 174 | 0.11 |
| 29 | P62826 | GTP‐binding nuclear protein Ran (RAN) | 172 | 0.67 |
| 30 | P63244 | Receptor of activated protein C kinase 1 (RACK1) | 168 | 0.43 |
| 31 | P60903 | Protein S100‐A10 (S100A10) | 168 | 0.3 |
To verify the interactions between AHNAK2 and RUVBL1, Co‐IP assay and flow cytometry were carried out to confirm their collaboration in the cell cycle pathway. The results of the Co‐IP assay demonstrated the specific interactions between AHNAK2 and RUVBL1 derived from A549 cells (Figure 4a).
FIGURE 4.
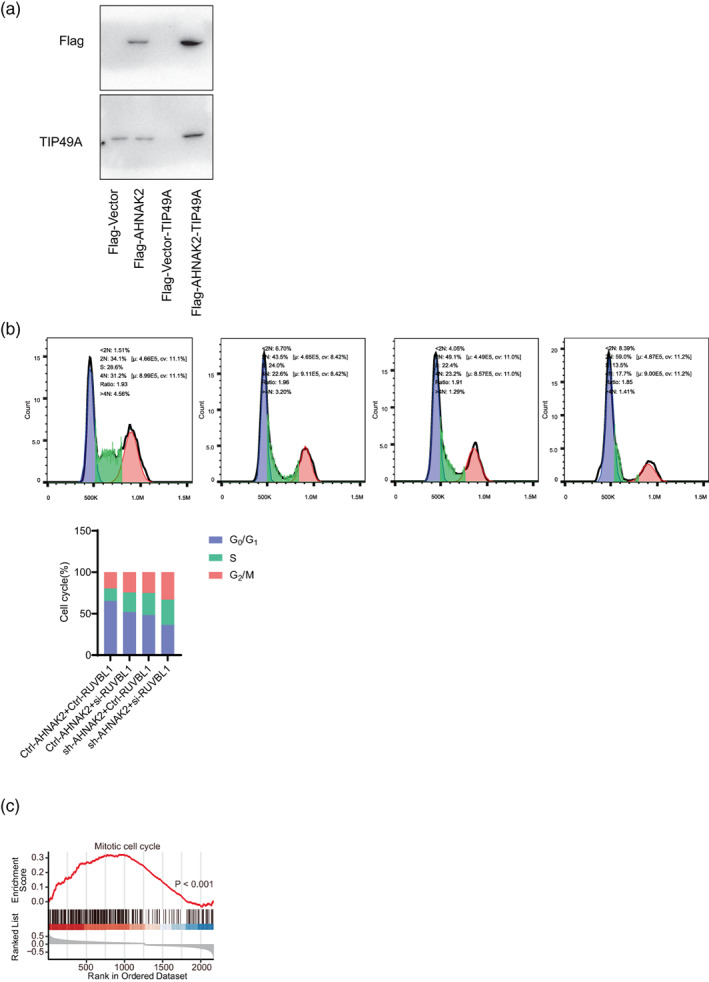
AHNAK2 interacts with RUVBL1 and causes G1/S phase cell cycle arrest in A549 cells. (a) The Co‐immunoprecipitation (Co‐IP) assay demonstrated the specific interactions between AHNAK2 and RUVBL1. Cell cycle analysis. (bi) A549 cells treated with Ctrl‐AHNAK2 and Ctrl‐RUVBL1. (bii) A549 cells with lower expression of AHNAK2 showed cell cycle arrest at the G1/S phase. (biii) H1975 cells with lower expression of RUVBL1 showed cell cycle arrest at the G1/S phase. (biv) H1975 cells treated with sh‐AHNAK2 and si‐RUVBL1 showed cell cycle arrest at the G1/S phase more significantly. (bv) The G1/S cell cycle arrest deteriorated when both AHNAK2 and RUVBL1 were suppressed in sequence. (c) Gene set enrichment analysis (GSEA) revealed that samples with higher AHNAK2 expression exhibited higher pathway activity of “mitotic cell cycle” as compared to lower samples in TCGA datasets.
We first knocked down expression of AHNAK2 and RUVBL1 with siRNA into A549 cells respectively, and then knocked them down simultaneously to observe their cell cycle distributions. A549 cells with a lower expression of AHNAK2 and RUVBL1, respectively showed cell cycle arrest at the G1/S phase, with an accumulation of cells in the G1 and S phases and a decrease in G2 phase cells compared with control cells. In addition, the G1/S cell cycle arrest deteriorated even further when both AHNAK2 and RUVBL1 were suppressed (Figure 4b).
In addition, tubulin β‐4B, tubulin β‐4A and tubulin α‐1C were also detected by mass spectrometry, which were predicted to be involved in microtubule cytoskeleton organization and mitotic cell cycle. Gene set enrichment analysis (GSEA) revealed that samples with higher AHNAK2 expression exhibited higher pathway activity of “mitotic cell cycle” as compared to lower samples in TCGA datasets (Figure 4c). The above results suggested that AHNAK2 probably took part in mitotic progression through α‐tubulin and β‐tubulin.
DISCUSSION
Our interest in AHNAKs can be traced back to a whole exon sequencing test in 2017, that showed as significantly mutated genes, the mutations of AHNAK and AHNAK2 were associated with many tumor pathways, such as PI3K‐Akt, MAPK and the calcium signaling pathway. However, due to the insufficient sample size, no reliable conclusions could be drawn. Then, we found that the high expression of AHNAK2 in LUAD tissues led to poor prognosis in patients with LUAD and a variety of other tumors.
Previous studies have suggested that AHNAK2 plays a tumor promoter role in several tumor diseases. For instance, AHNAK2 is upregulated in CCRC cells and promotes tumorigenesis and progression through hypoxia (HIF1α). 8 The upregulations of AHNAK2 have also been observed in pancreatic ductal adenocarcinoma, breast carcinoma, uveal melanoma and LUAD. 4 , 9 , 11 Recent studies revealed that AHNAK2 promotes migration, invasion, and EMT in LUAD cells via the TGF‐β/Smad3 pathway and MAPK pathway. 17 Furthermore, deleterious AHNAK2 mutations have been reported to be associated with activated CTL effector functions, IFN‐γ signaling, and infiltration of TIILs, which contribute to the activated immune microenvironment. 15 As a large nucleoprotein, the sequence of AHNAKs is approximately 616 kDa, comprising 5795 amino acids and it is because of its high molecular mass many difficulties were introduced into our experiments. Our early attempts at western blot, coimmunoprecipitation and immunofluorescence had previously failed. In our studies, we had to rely on the results of qRT‐PCR to verify the knockout effect and insert a protein tag to perform Co‐IP assays.
RUVBL1 (RuvB Like AAA ATPase 1, also named as INO80H, Pontin, TIP49a) belong to highly conserved ATPases of the AAA+ superfamily and are involved in various cellular processes that are important for oncogenesis. The main functions of RUVBL1 include transcriptional regulation, DNA damage signaling and repair, regulating cell cycle/mitotic progression. 18 , 19 , 20 , 21 As part of TIP60 histone acetyl transferase activity histone acetylation, RUVBL1 regulates gene expression as it typically relaxes chromatin structure allowing the binding of the transcriptional machinery to proper promoter regions. 22 In a previous study, RUVBL1 also had an effect on cell proliferation and caused G1/S phase cell cycle arrest, which were attributed to repression of the AKT/GSK‐3β/cyclin D1 pathway and probably to the activation of IRE1α‐mediated endoplasmic reticulum (ER) stress. 19
In the present study, we observed high expression of AHNAK2 in LUAD, which led to a poor prognosis, especially in patients with stage T2–T4 disease. Based on this clinical observation, we performed proliferation, migration and invasion assays on sh‐AHNAK2‐H1975 and sh‐AHNAK2‐A549 cells compared with control cells.
The results confirmed our hypothesis about the biological functions of AHNAK2 in tumorigenesis that the overexpression of AHANK2 mainly contribute to cell proliferation, migration and invasion. This also was consistent with our observation of independent expression level in different tumor stages.
The results of RNA sequencing in our study showed that the downregulation of AHNAK2 was associated with G1/S phase transition. Therefore, to verify whether AHNAK2 affects cells proliferation through regulating G1/S phase transition in LUAD, we examined the protein levels of PCNA/CCND1/P21 and performed flow cytometry in shAHNAK2‐A549 cells. We found that suppression of AHNAK2 caused G1/S phase cell cycle arrest and induced the decreased expression of PCNA, CCND1, while the expression of P21 increased. Given the lack of evidence that AHNAK2 had an effect on the cell cycle in previous studies, we attempted to find interacting proteins, which might be relevant to the cell cycle or some other upstream pathways, by mass spectrometry analysis. RUVBL1 was screened out and it was verified by coimmunoprecipitation that it definitely interacted with AHNAK2. It has previously been reported to have an effect on the AKT/GSK‐3β/cyclin D1 pathway, HIF1α regulation, mitosis and NF‐κB regulation. 19 , 23 , 24 Therefore, we subsequently explored which target AHNAK2 acts on upstream of the cell cycle pathway, such as MAPK, PI3K‐Akt, NF‐κB and the focal adhesion signaling pathways (Figure 5). We examined Src and FAK, including their phosphorylated forms, in shAHNAK2‐A549 cells compared with control cells although the results were somewhat contradictory to our in vitro experiments (Figure 6).
FIGURE 5.
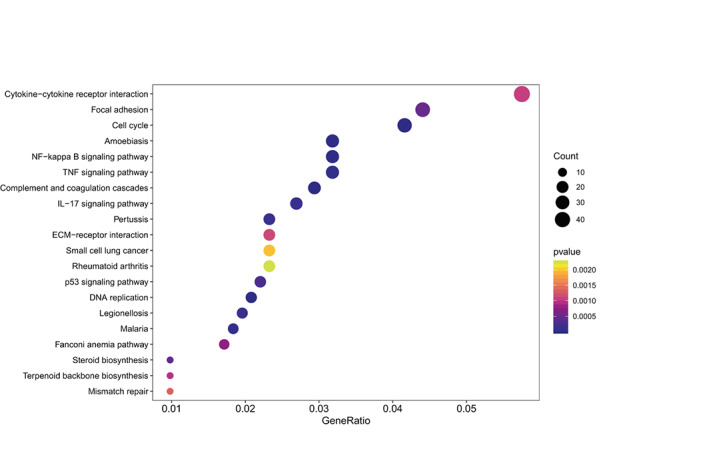
Overrepresentation analysis (ORA) for A549 cells transfected with sh‐AHNAK2 versus sh‐NC.
FIGURE 6.
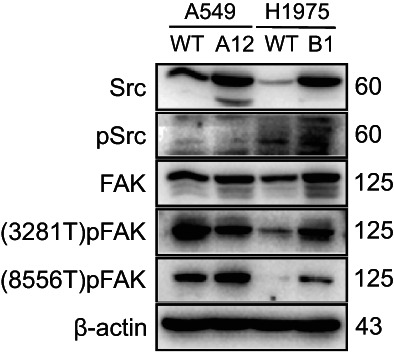
The expression levels of markers in the focal adhesion pathway were not consistent with phenotype.
Furthermore, we performed western blot and flow cytometry for groups of A549 cells, in which AHNAK2 and RUVBL1 were knocked down in sequence. As we speculated, the simultaneous suppression of AHNAK2 and RUVBL1 caused the G1/S phase cell cycle arrest and PCNA/CCND1 downregulation more significantly. Previous studies have reported that RUVBL1 is involved in regulating diverse mitosis and histone acetylation processes in tumor, by colocalizing with both α‐ and γ‐tubulin or play a part in TIP60. 25 , 26 , 27 , 28 In addition, at telophase, RUVBL1 has previously been found to form two foci that colocalized with β‐tubulin at the sides of the cytokinetic furrow. 29 In our present study, the mass spectrometry analysis showed that AHNAK2 also interacted with tubulin‐α (1C) and ‐β (4A, 4B), which were predicted to be involved in microtubule cytoskeleton organization and mitotic cell cycle.
Abnormal gene expression in tumor tissues might be caused by a variety of upstream factors, including genetic changes such as gene mutation and copy number variation, as well as epigenetic changes such as DNA methylation and chromatin conformation. In the TCGA cohort, neither gene mutation nor copy number variation was associated with AHNAK2 expression (Figure S2A, B). Interestingly, the methylation levels of three methylation sites in AHNAK2 gene were significantly negatively correlated with AHNAK2 mRNA expression, and their methylation levels in tumor tissues were significantly lower than in normal tissues (Figure S2C, D). Therefore, we believe that abnormal DNA methylation was an important factor in regulating the abnormal expression of AHNAK2 in LUAD.
In conclusion, this study clarified the oncogenic effect of AHNAK2 on the proliferation of A549 and H1975 cells. Furthermore, the suppression of AHNAK2 was verified to inhibit the cell cycle pathway and caused the G1/S phase cell cycle arrest, which could be attributed to the interaction with RUVBL1. Based on the data of RNA sequencing, there is some ambiguous speculation about the upstream mechanism that needs further experimental verification. Present studies on the biological function of AHNAK2 in various tumors are still lacking and some evidence is unauthentic, on account of experimental difficulties arising from the high molecular weight of AHNAK2. Our study also offers a reliable experimental method for exploring the functional role of AHNAK2 in the LUAD. Further studies will be performed in the future to reveal the upstream pathways and the mechanism of co‐action between AHNAK2 and RUVBL1. In summary, these results demonstrably suggest that AHNAK2 should be paid more attention and considered a promising target to determine the pathogenetic mechanism of LUAD.
AUTHOR CONTRIBUTION
Xin Li: Experimental studies and Writing. Hui Li: Design. Ming‐Ming Shao: Data Analysis and Statistical Analysis. Jin Bai Miao and Yi Li Fu: Manuscript Review. Bin Hu: Validation and Manuscript Review.
CONFLICT OF INTEREST
No potential conflict of interest was reported by the authors.
Supporting information
Figure S1. mRNA levels of AHNAK2 in tumor and normal tissues of multitumor in TCGA database.
Figure S2. Upstream factor of AHNAK2. (A) Somatic mutation of AHNAK2 in TCGA cohort. (B) Copy number variation of AHNAK2 in TCGA cohort. (C) DNA methylation levels of AHNAK2 in TCGA cohort. (D) The correlation of DNA methylation and AHNAK2 mRNA expression.
Li X, Li H, Shao M‐M, Miao J, Fu Y, Hu B. Downregulation of AHNAK2 inhibits cell cycle of lung adenocarcinoma cells by interacting with RUVBL1 . Thorac Cancer. 2023;14(22):2093–2104. 10.1111/1759-7714.14989
REFERENCES
- 1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021. CA Cancer J Clin. 2021;71(1):7–33. [DOI] [PubMed] [Google Scholar]
- 2. Ettinger DS, Wood DE, Aisner DL, Akerley W, Bauman JR, Bharat A, et al. Non‐small cell lung cancer, version 3.2022, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2022;20(5):497–530. [DOI] [PubMed] [Google Scholar]
- 3. Ettinger DS, Wood DE, Aisner DL, Akerley W, Bauman JR, Bharat A, et al. NCCN guidelines insights: non‐small cell lung cancer, version 2.2021. J Natl Compr Canc Netw. 2021;19(3):254–66. [DOI] [PubMed] [Google Scholar]
- 4. Chen B, Wang J, Dai D, Zhou Q, Guo X, Tian Z, et al. AHNAK suppresses tumour proliferation and invasion by targeting multiple pathways in triple‐negative breast cancer. J Exp Clin Cancer Res. 2017;36(1):65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sheppard HM, Feisst V, Chen J, Print C, Dunbar PR. AHNAK is downregulated in melanoma, predicts poor outcome, and may be required for the expression of functional cadherin‐1. Melanoma Res. 2016;26(2):108–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhao Z, Xiao S, Yuan X, Yuan J, Zhang C, Li H, et al. AHNAK as a prognosis factor suppresses the tumor progression in glioma. J Cancer. 2017;8(15):2924–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Park JW, Kim IY, Choi JW, Lim HJ, Shin JH, Kim YN, et al. AHNAK loss in mice promotes type II Pneumocyte hyperplasia and lung tumor development. Mol Cancer Res. 2018;16(8):1287–98. [DOI] [PubMed] [Google Scholar]
- 8. Wang M, Li X, Zhang J, Yang Q, Chen W, Jin W, et al. AHNAK2 is a novel prognostic marker and oncogenic protein for clear cell renal cell carcinoma. Theranostics. 2017;7(5):1100–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lu D, Wang J, Shi X, Yue B, Hao J. AHNAK2 is a potential prognostic biomarker in patients with PDAC. Oncotarget. 2017;8(19):31775–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhou YY, Kang YT, Chen C, Xu FF, Wang HN, Jin R. Combination of TNM staging and pathway based risk score models in patients with gastric cancer. J Cell Biochem. 2018;119(4):3608–17. [DOI] [PubMed] [Google Scholar]
- 11. Li M, Liu Y, Meng Y, Zhu Y. AHNAK nucleoprotein 2 performs a promoting role in the proliferation and migration of uveal melanoma cells. Cancer Biother Radiopharm. 2019;34(10):626–33. [DOI] [PubMed] [Google Scholar]
- 12. Liu G, Guo Z, Zhang Q, Liu Z, Zhu D. AHNAK2 promotes migration, invasion, and epithelial‐mesenchymal transition in lung adenocarcinoma cells via the TGF‐beta/Smad3 pathway. Onco Targets Ther. 2020;13:12893–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang DW et al. Down‐regulation of AHNAK2 inhibits cell proliferation, migration and invasion through inactivating the MAPK pathway in lung adenocarcinoma. Technol Cancer Res Treat. 2020;19:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang S, Lu Y, Qi L, et al. AHNAK2 is associated with poor prognosis and cell migration in lung adenocarcinoma. Biomed Res Int. 2020;2020:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zheng M, Liu J, Bian T, Liu L, Sun H, Zhou H, et al. Correlation between prognostic indicator AHNAK2 and immune infiltrates in lung adenocarcinoma. Int Immunopharmacol. 2021;90:107134. [DOI] [PubMed] [Google Scholar]
- 16. Zardab M, Stasinos K, Grose RP, Kocher HM. The obscure potential of AHNAK2. Cancers (Basel). 2022;14(3):1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lee IH, Sohn M, Lim HJ, Yoon S, Oh H, Shin S, et al. Ahnak functions as a tumor suppressor via modulation of TGFbeta/Smad signaling pathway. Oncogene. 2014;33(38):4675–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gallant P. Control of transcription by Pontin and Reptin. Trends Cell Biol. 2007;17(4):187–92. [DOI] [PubMed] [Google Scholar]
- 19. Yuan XS, Wang ZT, Hu YJ, Bao FC, Yuan P, Zhang C, et al. Downregulation of RUVBL1 inhibits proliferation of lung adenocarcinoma cells by G1/S phase cell cycle arrest via multiple mechanisms. Tumour Biol. 2016;37:16015–27. [DOI] [PubMed] [Google Scholar]
- 20. Grigoletto A, Lestienne P, Rosenbaum J. The multifaceted proteins Reptin and Pontin as major players in cancer. Biochim Biophys Acta. 2011;1815(2):147–57. [DOI] [PubMed] [Google Scholar]
- 21. Wu S, Shi Y, Mulligan P, Gay F, Landry J, Liu H, et al. A YY1‐INO80 complex regulates genomic stability through homologous recombination‐based repair. Nat Struct Mol Biol. 2007;14(12):1165–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. DesJarlais R, Tummino PJ. Role of histone‐modifying enzymes and their complexes in regulation of chromatin biology. Biochemistry. 2016;55(11):1584–99. [DOI] [PubMed] [Google Scholar]
- 23. Perez‐Perri JI, Dengler VL, Audetat KA, Pandey A, Bonner EA, Urh M, et al. The TIP60 complex is a conserved coactivator of HIF1A. Cell Rep. 2016;16(1):37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mao YQ, Houry WA. The role of Pontin and Reptin in cellular physiology and cancer etiology. Front Mol Biosci. 2017;4:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhao LJ, Loewenstein PM, Green M. Ad E1A 243R oncoprotein promotes association of proto‐oncogene product MYC with the NuA4/Tip60 complex via the E1A N‐terminal repression domain. Virology. 2016;499:178–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gartner W, Rossbacher J, Zierhut B, Daneva T, Base W, Weissel M, et al. The ATP‐dependent helicase RUVBL1/TIP49a associates with tubulin during mitosis. Cell Motil Cytoskeleton. 2003;56(2):79–93. [DOI] [PubMed] [Google Scholar]
- 27. Sigala B, Edwards M, Puri T, Tsaneva IR. Relocalization of human chromatin remodeling cofactor TIP48 in mitosis. Exp Cell Res. 2005;310(2):357–69. [DOI] [PubMed] [Google Scholar]
- 28. Ducat D, Kawaguchi SI, Liu H, Yates JR III, Zheng Y. Regulation of microtubule assembly and organization in mitosis by the AAA+ ATPase Pontin. Mol Biol Cell. 2008;19(7):3097–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gentili C, Castor D, Kaden S, Lauterbach D, Gysi M, Steigemann P, et al. Chromosome Missegregation associated with RUVBL1 deficiency. PLoS One. 2015;10(7):e0133576. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. mRNA levels of AHNAK2 in tumor and normal tissues of multitumor in TCGA database.
Figure S2. Upstream factor of AHNAK2. (A) Somatic mutation of AHNAK2 in TCGA cohort. (B) Copy number variation of AHNAK2 in TCGA cohort. (C) DNA methylation levels of AHNAK2 in TCGA cohort. (D) The correlation of DNA methylation and AHNAK2 mRNA expression.