

Abstract
Possessing only essential genes, a minimal cell can reveal mechanisms and processes that are critical for the persistence and stability of life1,2. Here we report on how an engineered minimal cell3,4 contends with the forces of evolution compared with the Mycoplasma mycoides non-minimal cell from which it was synthetically derived. Mutation rates were the highest among all reported bacteria, but were not affected by genome minimization. Genome streamlining was costly, leading to a decrease in fitness of greater than 50%, but this deficit was regained during 2,000 generations of evolution. Despite selection acting on distinct genetic targets, increases in the maximum growth rate of the synthetic cells were comparable. Moreover, when performance was assessed by relative fitness, the minimal cell evolved 39% faster than the non-minimal cell. The only apparent constraint involved the evolution of cell size. The size of the non-minimal cell increased by 80%, whereas the minimal cell remained the same. This pattern reflected epistatic effects of mutations in ftsZ, which encodes a tubulin-homologue protein that regulates cell division and morphology5,6. Our findings demonstrate that natural selection can rapidly increase the fitness of one of the simplest autonomously growing organisms. Understanding how species with small genomes overcome evolutionary challenges provides critical insights into the persistence of host-associated endosymbionts, the stability of streamlined chassis for biotechnology and the targeted refinement of synthetically engineered cells2,7–9.
Subject terms: Experimental evolution, Bacterial genes, Genome evolution, Synthetic organisms, Molecular evolution
An engineered minimal cell evolves to escape the negative consequences of genome streamlining.
Main
The complexity of a genome is reflected by the number of genes that it contains, a quantity that varies by orders of magnitude across the tree of life. Whereas some obligately endosymbiotic bacteria have fewer than 200 protein-coding genes, many plant and animal genomes contain more than 20,000 genes10–12. In principle, the simplest organism is one that possesses only the minimum number of genes for survival and reproduction in a given environment. Any mutation in such an organism could lethally disrupt one or more cellular functions, placing constraints on evolution, as revealed by the fact that essential proteins change more slowly than those encoded by dispensable genes13,14. Furthermore, organisms with streamlined genomes have fewer targets on which positive selection can act, therefore limiting opportunities for adaptation.
The cell is the simplest independent functional unit of life. However, even unicellular model organisms that are touted for their tractability are complex, possessing thousands of genes and proteins, many of which remain uncharacterized even after decades of in-depth investigation. The quest for the simplest organism has been aided by advances in synthetic biology, which involves the redesign or novel construction of biological parts and modules2,15. Synthetic biology provides a platform for developing powerful simplest-case models through streamlining, whereby non-essential sequences are removed from an organism’s genome1–3,8,16. Guided by such strategies, a minimal cell was constructed with a genome containing only the smallest set of genes required for autonomous cellular life3,4. Although these efforts succeeded in experimentally identifying the genetic requirements for basic cellular processes, such as metabolism and cell division, it remains unclear how a minimal cell will respond to the forces of evolution. On one hand, evolution of a minimal cell could be constrained by the limited raw materials with which natural selection can operate. On the other hand, synthetic streamlining may result in a highly disrupted genome, altering protein interactions and expanding the opportunity for adaption to a new cellular environment.
To gain insights into the dynamics and outcomes of evolution in a minimal cell, we conducted experiments with strains of M. mycoides3,4, which are bacteria belonging to the Mollicutes. The minimal cell (JCVI-syn3B) has a synthetically constructed genome containing a subset of genes found in a corresponding non-minimal strain (JCVI-syn1.0). By reducing the chromosome from 901 to 493 genes, JCVI-syn3B has the smallest genome of any organism that can be grown in pure laboratory culture3,4. With these two strains, we first investigated whether genome streamlining—which included the removal of two DNA-replication genes, eight DNA repair genes and other genes of unknown function—altered the rate and spectrum of new mutations in the minimal cell relative to the non-minimal organism under conditions of relaxed selection. Second, with knowledge of the mutational input, we evaluated whether genome minimization altered the rate and mechanisms of evolution in response to natural selection, as measured using whole-genome sequencing, estimates of population fitness and phenotypic changes in cell size.
Highest recorded mutation rate
Through serial bottlenecking under relaxed selection, we conducted mutation accumulation experiments with populations of M. mycoides (Methods). The number of mutations per nucleotide per generation for the non-minimal cell (3.13 ± 0.12 × 10−8, mean ± s.e.m.) was indistinguishable from that of the minimal cell (3.25 ± 0.16 × 10−8) (t140 = 0.43, P = 0.667; Fig. 1a). These mutation rates, which are the highest recorded for any cellular organism, are consistent with other reports in which organisms with smaller genomes have higher mutation rates17–20. Notably, the mutation rate was not affected by genome minimization that included the elimination of genes involved in replication fidelity (Fig. 1a). Perhaps this is due to the fact that M. mycoides already has an elevated mutation rate. To evaluate the generality of our findings, the effect of genome minimization should be investigated in a microorganism with a lower intrinsic mutation rate. In any case, our data are consistent with predictions from the drift-barrier hypothesis. This theory posits that mutation rates evolve downwards until the selective advantage of another incremental decrease in the mutation rate is small enough to be effectively neutral and outweighed by genetic drift19,20. In other words, populations with a lower effective population size (Ne) experience stronger drift and, therefore, evolve higher mutation rates19. Notably, wild-type M. mycoides is an obligate pathogen and has genomic features (small genome size and low GC content) consistent with it having a low Ne17,18,21,22. Note that mutation-accumulation studies are typically designed to estimate the rate and spectrum of viable mutations. By eliminating redundancy, genome streamlining could alter the contribution of strongly deleterious or lethal mutations that would not be captured in our study.
Fig. 1. The mutation rate and spectrum of the minimal and non-minimal cell.

a–c, The mutation rate (per nucleotide (nt) per generation (gen.)) and spectrum of the minimal and non-minimal cell were estimated from mutation-accumulation experiments. a, Although synthetic M. mycoides has the highest recorded mutation rate (base substitutions and indels), it was not affected by genome minimization. The dark coloured circles represent non-minimal (n = 85) and minimal (n = 57) clones that were sequenced at the end of the experiment. The light coloured areas represent kernel densities of the data. b, The proportions of insertions, deletions and SNMs were also the same for the minimal and non-minimal cells. c, Among SNMs, which accounted for 88% of all mutations, the minimal cell exhibited a stronger A:T bias in its mutation spectrum compared with the non-minimal cell, particularly in the C:G to T:A category. Two-sided χ2 analysis was used for hypothesis testing; ***P = 2.5 × 10−6 (A:T to G:C), ***P = 1.5 × 10−11 (C:G to G:C), ***P = 1.6 × 10−20 (C:G to T:A), ***P = 0.0003 (C:G to A:T); NS, not significant.
Minimization and mutational spectrum
Although the mutation rate was robust to genome streamlining, the types of mutations that arise in a population can still influence evolution. Overall, the composition of mutation types (insertions, deletions and single-nucleotide mutations (SNMs)) was not affected by genome minimization (χ22 = 4.16, P = 0.125; Fig. 1b). However, the composition of SNMs, which constituted the largest category of mutations (88%), differed between the minimal and non-minimal cells (Monte Carlo χ2 = 69.9, P = 1.0 × 10−4). For both cell types, mutations from a G or C nucleotide to an A or T nucleotide occurred at a higher rate compared with mutations in the opposite direction, that is, from A or T to G or C (Fig. 1c; non-minimal cell, χ21 = 3736, P < 2.2 × 10−16; minimal cell, χ21 = 1444, P < 2.2 × 10−16). The magnitude of this A:T bias was affected by genome streamlining (χ21 = 21.8, P = 3.08 × 10−6; Fig. 1c) leading to a 30-fold bias in the non-minimal cells and a 100-fold bias in the minimal cells. The discrepancy is probably due to the deletion of ung, a gene of which the protein product excises misincorporated uracil that can otherwise cause C-to-T mutations23. Its removal from the minimal cell’s genome should elevate A:T mutational bias relative to the non-minimal cell as observed.
Recovery of fitness in a minimal cell
With mutation rates of around 3 × 10−8 per nucleotide per generation and population sizes in excess of 107 individuals, a new mutation would hit every nucleotide in the genome more than 250 times during 2,000 generations of experimental evolution. Thus, neither cell type would be limited by the availability of genetic variation to fuel adaptation. Any differences in the ways the two strains adapt should be driven by alterations in genome content created by synthetic streamlining.
To study natural selection, we passaged replicate populations of M. mycoides for 2,000 generations (Methods), a period during which rapid adaptation is often observed24,25. We then measured fitness, the contribution of a genotype’s offspring to future generations, using two methods26. First, we quantified the maximum growth rate (µmax) of each replicate population every 65–130 generations (Methods). We documented that genome streamlining led to a 57% reduction in µmax, but that this measure of fitness subsequently increased linearly and at comparable rates for the minimal cell (1.71 × 10−5 ± 4.53 × 10−6 per day per generation) and non-minimal cell (1.03 × 10−5 ± 4.53 × 10−6 per day per generation) during the evolution experiment. Using the predicted values from a generalized linear mixed model, the µmax of the non-minimal and minimal cell increased by 17–68% over the course of the experiment (Extended Data Fig. 1 and Extended Data Table 1). Second, we measured relative fitness using head-to-head competition assays with the ancestral (generation 0) and most evolved (generation 2,000) populations (Methods). For the ancestral strains, we determined that genome minimization led to a 53% decrease in fitness (Fig. 2), on par with estimates based on µmax. Despite this major initial cost, the minimal cell rapidly regained fitness. In fact, the competition-based estimates of fitness indicate that the minimal cell adapted 39% more rapidly than the non-minimal cell (t = −2.530, P = 0.032). With the power afforded by our experimental design, the average relative fitness of the evolved minimal cell (0.998) was statistically indistinguishable (t = −0.055, P = 0.957) from that of the ancestral non-minimal cell (1.00). Given this, we conclude that effectively all of the fitness lost to genome streamlining was recovered during 300 days of serial passaging (Fig. 2 and Supplementary Fig. 1). Our findings suggest that a streamlined M. mycoides genome is not inherently crippled and can perform as well as the non-minimized cell after readaptation.
Extended Data Fig. 1. Trajectories of maximum growth rates (µmax) for the minimal cell and non-minimal cell.
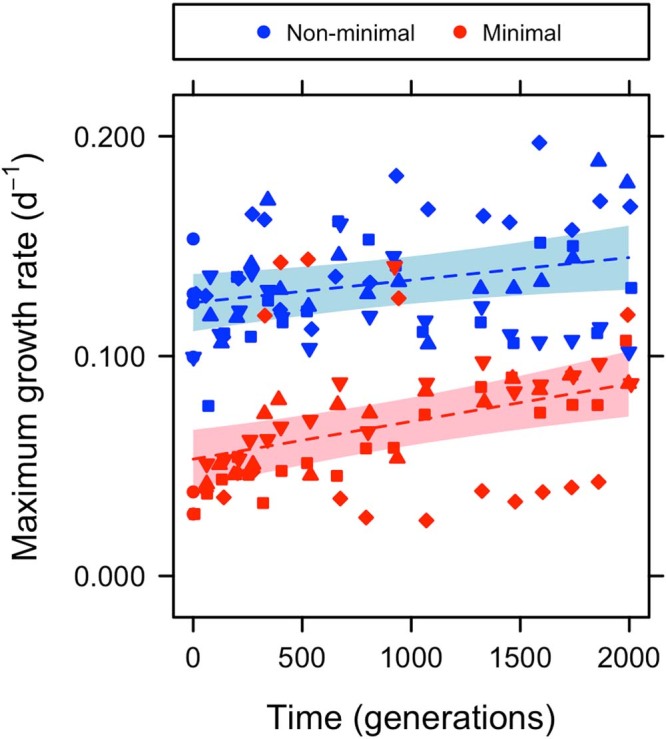
Data (n = 141) were generated from growth-curve assays that were fit using a modified Gompertz equation (see Fig. S5) across 2000 generations of experimental evolution. With these estimates of µmax, we then fit a generalized linear mixed model (GLMM) where time (generation) and cell type (minimal cell vs. non-minimal cell) were treated as fixed effects and replicate evolved populations (n = 8) was treated as a random effect. Based on the intercepts from the GLMM, synthetic streamlining reduced µmax by 57% in the non-evolved ancestors. During subsequent evolution, µmax for both cell types increased at comparable rates over the course of the experiment (see Extended Data Table 1). In the figure, dark-coloured circles represent data from the ancestral populations, while triangles (up- and down-pointing), diamonds, and squares represent data from the replicate evolved populations. Dashed lines and light-coloured regions represent predicted values and 95% confidence intervals, respectively, for the fixed effects (generation and cell type). The conditional R2, which accounts for variance explained by the fixed and random effects, was 0.68. The variance partition coefficient (VPC) of 0.127 indicates that an appreciable portion of the total explained variance in µmax was associated with the random effect of the replicate evolved populations (See Extended Data Table 1). Additional information, including model fits, parameters, summary statistics, and residual plots, can be found in the online Figshare repository.
Extended Data Table 1.
Parameters and summary statistics associated with fitness

We used generalized linear mixed models (GLMM) to explain variation in maximum growth rate (µmax), which was estimated from growth curves for the non-minimal and minimal cells. The cell type (“Cell”) and time (“Generation”) were treated as fixed effects. We included random intercepts for the replicate populations.
Fig. 2. The effect of genome minimization on fitness and adaptation.
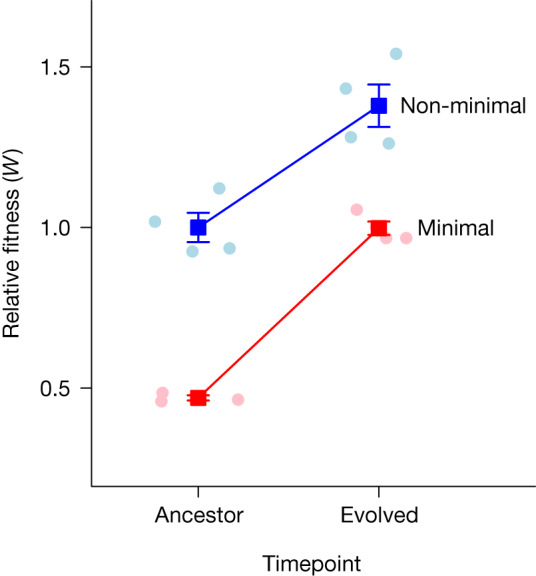
Genome minimization reduced the relative fitness by 50%. However, almost all of this cost was regained over 2,000 generations of evolution. Despite the removal of nearly half of its genome, the minimal cell adapted at a rate comparable to that of the non-minimal cell, which was corroborated by fitness estimates from growth curve experiments (Extended Data Fig. 1 and Extended Data Table 1). The dark coloured symbols represent mean ± s.e.m. As the experiment was initiated with a single clone, error bars for the ancestral timepoint were calculated from technical replicates (n = 4), whereas error bars for evolved populations were calculated from replicate populations (n = 4), both of which are depicted by light coloured symbols. The solid red and blue lines are a visual aid connecting the mean values of the minimal and non-minimal populations, respectively.
On the basis of the fitness dynamics, we conclude that adaptation was not constrained by genome minimization. This interpretation was bolstered by results from population genomic sequencing (Methods). The relative ratio of nonsynonymous to synonymous fixed SNMs (dN/dS) was similar between the two cell types (t6 = 0.81, P = 0.488; Extended Data Fig. 2), consistent with the interpretation that the rates of molecular evolution were comparable even though almost all of the genes in the minimal cell are critical for fitness13,14.
Extended Data Fig. 2. Effect of genome streamlining on the ratio of nonsynonymous to synonymous substitutions.
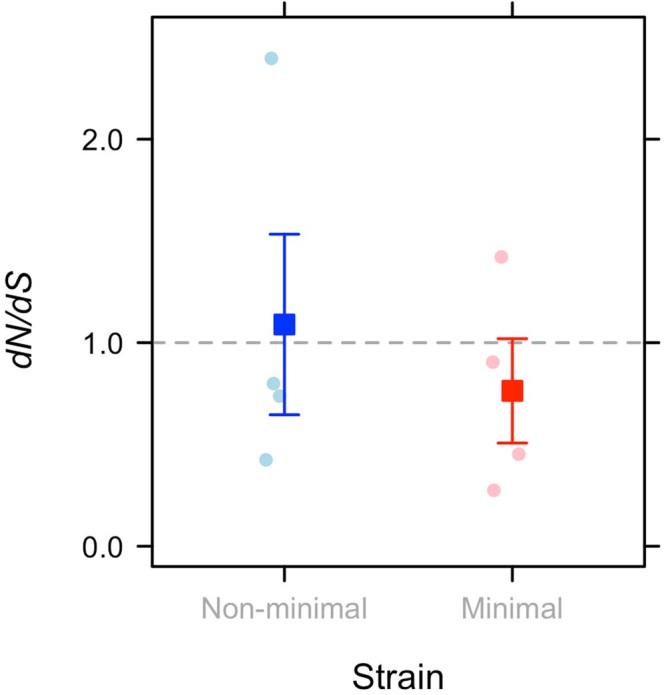
In populations of Mycoplasma mycoides after 2000 generations of evolution, we used the normalized ratio of nonsynonymous to synonymous mutations (dN/dS) as an indicator of natural selection. Values of dN/dS > 1 are associated with positive selection, while values of dN/dS < 1 are associated with the dominance of negative selection and constraint on adaptation. The minimal and non-minimal cell exhibited comparable values of dN/dS (t6 = 0.81, P = 0.488). One of the replicate populations belonging to the non-minimal treatment had an elevated dN/dS (2.06) compared to other replicate populations (mean dN/dS = 0.45). When we removed this potential outlier, there was still no difference in dN/dS between the minimal and non-minimal cell (t5 = −0.25, P = 0.811). Dark-coloured symbols represent the mean ± SEM (n = 4). Light-coloured symbols represent individual values for each replicate population (n = 4). Hypotheses were evaluated with two-sided t-tests.
Divergent mechanisms of adaptation
Using a combination of statistical simulation and reverse genetics, we identified mutations that probably contributed to the observed patterns of adaptation. First, we analysed the gene-by-population matrix for nonsynonymous mutations that arose in the shared set of essential genes during the natural selection experiment (Methods). The two cell types acquired mutations in different sets of essential genes (permutational multivariate analysis of variance (PERMANOVA), F7 = 4.12, P = 0.029; Fig. 3) suggesting that the populations evolved through divergent routes. To examine this hypothesis, we looked for genes that acquired a higher number of nonsynonymous, nonsense and small insertion–deletion (indel) mutations than expected under assumptions of neutrality (Methods). We identified 16 genes in the non-minimal genome and 14 in the minimal genome that were potential targets of positive selection (Extended Data Tables 2–4). Second, we used reverse genetics to experimentally verify that one of the common types of mutation observed in replicate populations of both strains was in fact beneficial (Extended Data Table 5). Using CRISPR editing, we recreated ftsZ C-terminal nonsense mutations by inserting an ftsZ E315* nonsense mutation into the ancestral genomes of the minimized and non-minimized strains (Methods). Head-to-head competition assays with the constructs revealed that this putatively adaptive mutation had a significant effect on Mycoplasma performance that was dependent on genome minimization (two-way analysis of variance (ANOVA), F1,32 = 7.45, P = 0.010). The mutation conferred a 25% fitness advantage in the non-minimal cell and a 14% advantage in the minimal cell (Extended Data Fig. 3).
Fig. 3. The non-minimal cell and minimal cell populations acquired adaptive mutations in different sets of shared genes.
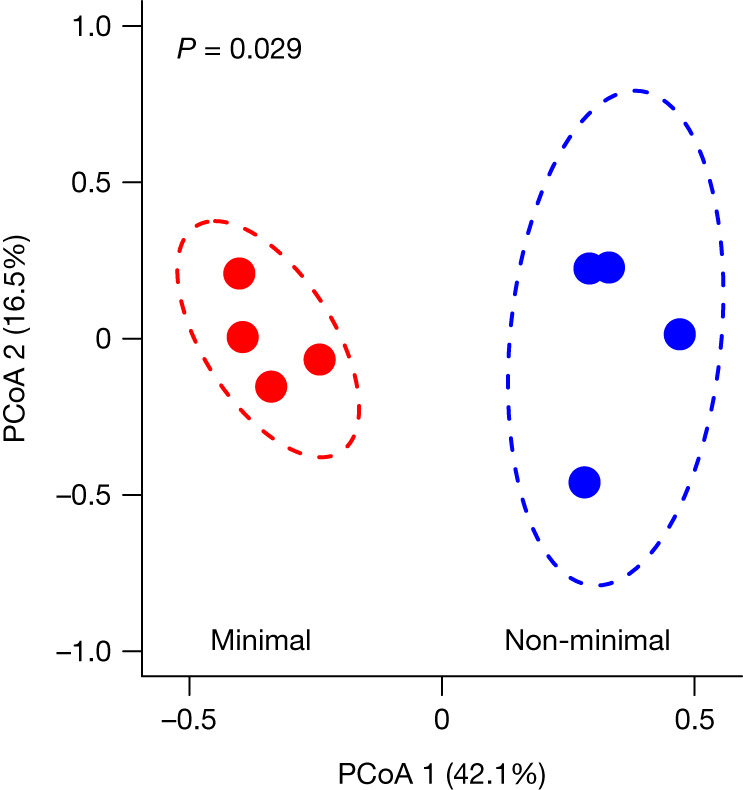
Ordination from a principal coordinates analysis (PCoA) created by a gene-by-population matrix using the Bray–Curtis distance metric after 2,000 generations of evolution (Extended Data Tables 2–4). The dashed lines represent 95% confidence ellipses around replicate populations (n = 4 for each cell type) represented by dark coloured symbols.
Extended Data Table 2.
Mutations only in non-minimal cell that that are putatively under positive selection

Simulations were performed to find genes acquiring more mutations than expected to occur by chance during 2000 generations of experimental evolution. Such mutations are indicative of positive selection. Padj corresponds to significance following Benjamini-Hochberg correction to account for multiple comparisons. Genes are assigned to categories based on the secondary functional classifications4. Note that “Central metabolism” corresponds to “Central carbon metabolism” in the original source4. * = nonessential genes that are absent from the minimal cell, N/A = uncharacterized genes that did not fall into defined category.
Extended Data Table 4.
Mutations found in both non-minimal and minimal cell that are putatively under positive selection

Simulations were performed to find genes acquiring more mutations than expected to occur by chance during 2000 generations of experimental evolution. Such mutations are indicative of positive selection. Padj corresponds to significance following Benjamini-Hochberg correction to account for multiple comparisons. Genes are assigned to categories based on the secondary functional classifications4. In the Padj column, “non” refers to non-minimal cell and “min” refers to the minimal cell. † = nonessential genes that were retained in M. mycoides JCVI-syn3B to facilitate cultivation and robust growth.
Extended Data Table 5.
ftsZ mutations

Mutations observed in ftsZ during adaptive evolution across all replicate populations.
Extended Data Fig. 3. Fitness effects of an ftsZ mutation on populations of Mycoplasma mycoides.
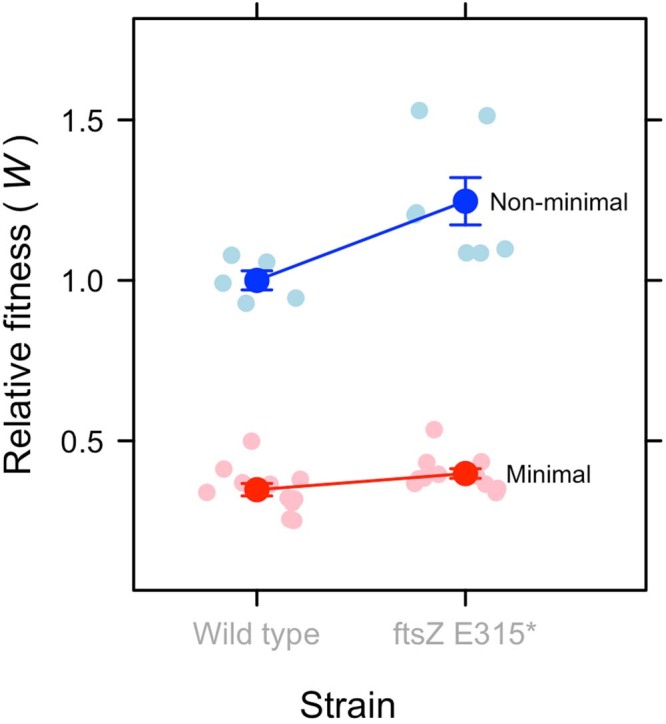
We reengineered the nonsense mutation ftsZ E315* and quantified its effect on relative fitness in both the non-minimal and minimal cells using head-to-head competition assays. The ftsZ E315* nonsense mutation had a significant effect on Mycoplasma cell size that depended on cell type (two-way ANOVA, F1,32 = 7.45, P = 0.010). Compared to the wild type (non-evolved ancestor), the mutation increased relative fitness by 25% in the non-minimal cell and 14% in the minimal cell. Dark-coloured symbols represent the mean ± SEM. Light-coloured symbols represent values for each replicate population. Samples sizes are as follows: wild-type minimal cell, n = 12; ftsZ E315* minimal, n = 12; wild-type non-minimal cell, n = 5; ftsZ E315* non-minimal, n = 5.
Comparative analysis of the genes putatively under positive selection provided insights into the functional consequences of adaptation in the minimal cell. We hypothesized that mutations in genes related to membrane transport would be critical for adaptation because the minimal cell relies on the import and export of metabolites and other biomolecules for metabolism4,27. However, mutations in membrane transport functions were enriched to a similar degree in both cell types (Fisher’s exact test, P = 0.934). Instead, we detected a marginal signal of enrichment for mutations in biosynthetic genes for the minimal cell (Fisher’s exact test, P = 0.090), including those involved in lipid metabolism. Specifically, fakA and clsA (Extended Data Table 3) are considered to be essential for synthesizing cardiolipin and other lipids from free fatty acids4, which are important for the construction of cell membranes and the regulation of cell division. The gene lgt is also critical for membrane construction, encoding the protein that transfers diacylglyceryl moieties to anchor surface lipoproteins in the lipid bilayer4. Thus, metabolic innovations involving lipid synthesis and distribution may be more important for the minimal cell than enhanced acquisition of metabolites that are already present in the growth medium.
Extended Data Table 3.
Mutations only in minimal cell that are putatively under positive selection

Simulations were performed to find genes acquiring more mutations than expected to occur by chance during 2000 generations of experimental evolution. Such mutations are indicative of positive selection. Padj corresponds to significance following Benjamini-Hochberg correction to account for multiple comparisons. Genes are assigned to categories based on the secondary functional classifications4. Note that “Central metabolism” corresponds to “Central carbon metabolism” in the original source4.
To better understand the pattern of evolutionary divergence, we compared mutations that arose in essential and non-essential genes over 2,000 generations specifically within the non-minimal cell. After accounting for the relative numbers of essential and non-essential genes, there was no difference in the number of mutations observed between these two genomic partitions (t3 = 0.646, P = 0.565; Supplementary Table 1). Nor was there any measurable difference in dN/dS between essential and non-essential genes (t3 = 0.91, P = 0.423; Supplementary Fig. 2). Among the genes putatively under positive selection, there was no evidence for bias towards either essential or non-essential genes (χ21 = 0.377, P = 0.539; Extended Data Table 2). We identified 11 deletions in the non-minimal cell, ten of which were at non-essential loci (Supplementary Table 2). Most of these were small (1–3 bp) but three deletions were large (1,483, 1,495 and 7,047 bp). In summary, it appears that essential genes did not disproportionately contribute to the molecular of evolution of the non-minimal cell, although we cannot rule out that epistatic interactions between essential and non-essential genes contributed to new cell phenotypes.
Constraints on the evolution of cell size
The size of single-celled organisms is variable and often linked to fitness in complex ways28–30. In resource-rich environments, cell size tends to be positively correlated with growth rate, one of the most important components of fitness24,29–32. For example, in the first 2,000 generations of a classic long-term evolution experiment with Escherichia coli, cell volume and fitness concomitantly increased by 50% and 30%, respectively24. Although an increase in size can accommodate more macromolecules needed for growth and division, it also decreases a cell’s surface-to-volume ratio, which reduces the efficiency of substrate diffusion. Given these opposing pressures, we evaluated how cell size changed in replicate populations over the course of evolution. Using scanning electron microscopy, we showed that genome streamlining reduced the cell diameter by 31% from 439 ± 0.01 nm to 305 ± 0.01 nm in the ancestral cell types. After 2,000 generation of evolution, the size of the non-minimal cell increased by 85% to 811 ± 0.02 nm (t = 3.77, P = 0.005), which was accompanied by a tenfold increase in volume compared with its ancestor (Fig. 4 and Extended Data Table 6). By contrast, the size of the minimal cell did not appreciably change (0.08 ± 0.05 nm) during evolution (t = 1.51, P = 0.181; Extended Data Fig. 4 and Supplementary Fig. 3).
Fig. 4. The effect of genome minimization on the evolution of cell size.
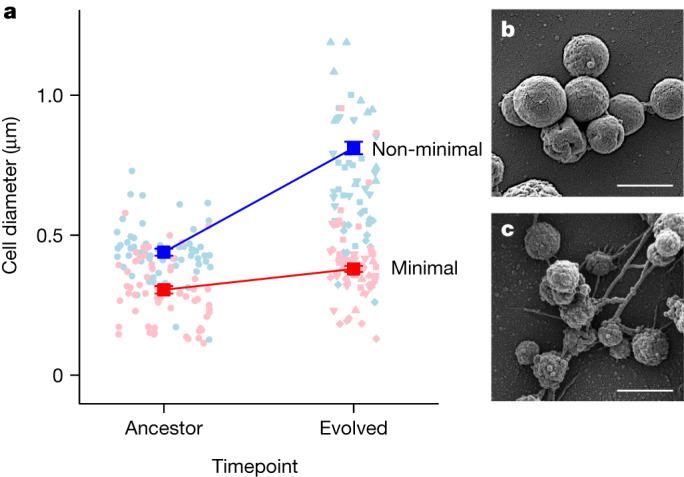
a, Genome minimization was accompanied by a 31% decrease in cell size. Over 2,000 generations of evolution, the size of the non-minimal cells increased by 85% (P = 0.005), whereas the size of the minimal cells remained the same (P = 0.181). Owing to variation associated with replicate evolved populations, there was a marginal effect when directly comparing changes in the size of the minimal and non-minimal cells (P = 0.077; Supplementary Fig. 4). The dark coloured symbols represent the mean ± s.e.m. As the experiment was initiated with a single clone, error bars for the ancestral timepoint were calculated from samples of individuals (n = 62 and n = 75 for the non-minimal and minimal cell, respectively), whereas error bars at the evolved time point were calculated from individuals (n = 285 and n = 181 for the non-minimal and minimal cell, respectively) across replicate populations (n = 4). The light coloured circles represent randomly drawn data (n = 60) corresponding to the diameter of individual cells from the ancestral populations. The light coloured triangles (pointing up and down), diamonds and squares represent randomly drawn data (n = 60) corresponding to the diameter of individual cells from the four replicate evolved populations. The solid red and blue lines are a visual aid connecting the mean values of the minimal and non-minimal populations, respectively. b,c, Scanning electron micrographs obtained from evolved replicate populations of the non-minimal (b) and minimal (c) cells. Scale bars, 1 μm.
Extended Data Table 6.
Evolution of cell size

Diameter and volume measurements of M. mycoides ancestor, evolved, and ftsZ E315* cells observed via scanning electron microscopy. Values represent mean ± SEM.
Extended Data Fig. 4. Cell size of ftsZ mutants compared to wildtype (non-evolved) for the minimal cell and non-minimal cell.
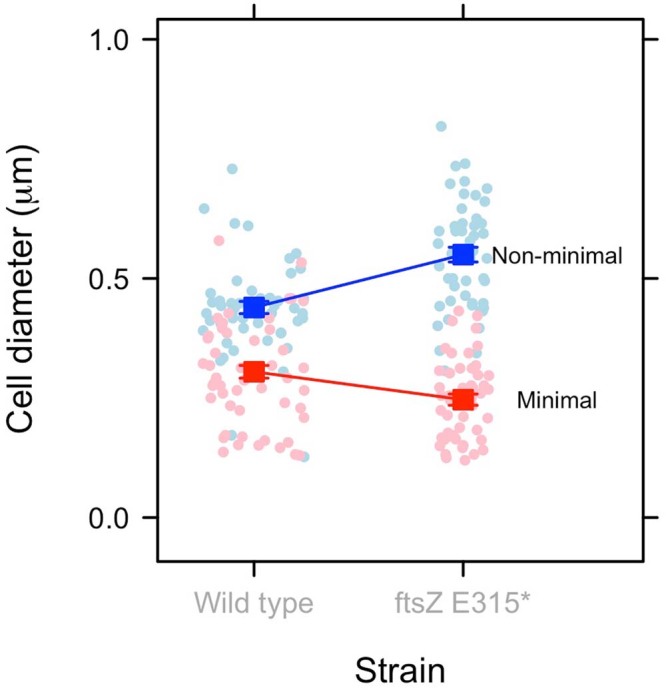
Using scanning electron microscopy, the ftsZ E315* nonsense mutation had a significant effect on Mycoplasma cell size that depended on cell type (two-way ANOVA, F1,241 = 37.9, P < 0.0001). The mutation in the non-minimal cell caused a 25% increase in cell diameter (P < 0.0001) and a corresponding two-fold increase in cell volume. In contrast, the same ftsZ nonsense mutation in the minimal cell led to a 19% decrease in the cell diameter (P = 0.015). Dark-coloured symbols represent mean ± SEM. Light-coloured symbols represent randomly drawn data (n = 60) corresponding to the diameter of individual cells.
While cell size is a complex multigenic trait, previous studies have attributed changes in morphology of the minimal cell to FtsZ33. This protein localizes to the midcell and determines the site of membrane constriction during cell division. Prevalent among diverse lineages of bacteria and archaea5,6, ftsZ is nevertheless non-essential in M. mycoides. However, cells lacking ftsZ exhibit aberrant cell division and morphology3,4,33,34. Thus, along with 18 other non-essential genes, ftsZ was retained in JCVI-syn3B to aid in culture maintenance and stable growth4,33. In our study, ftsZ was consistently mutated over 2,000 generations of evolution and was identified as a target of positive selection in both the minimal and non-minimal cells (Extended Data Table 4 and Extended Data Fig. 3). Introduction of an early termination codon, as was observed in multiple evolved populations, could eliminate the C-terminal region of the protein that is known to interact with membrane-associated products that recruit FtsZ35. The early stop codon could also create a transcriptional polar effect36 that reduces expression of two adjacent downstream genes within a probable polycistronic operon—MMSYN1_0521, an orthologue of cell division protein sepF and MMSYN1_0520, encoding aminopeptidase/esterase/lipase, of the α/β hydrolase superfamily37. Irrespective of mechanism, we demonstrated that mutations in ftsZ had a non-additive effect that contributed to the evolutionary divergence of cell size. We documented that the ftsZ E315* nonsense mutation had a significant effect on Mycoplasma cell size that was dependent on genome minimization (two-way ANOVA, F1,241 = 37.9, P = 3.1 × 10−9). The mutation in the non-minimal cell led to a 25% increase in cell diameter (P = 2.0 × 10−7) and a corresponding twofold increase in cell volume. By contrast, the same ftsZ nonsense mutation in the minimal cell led to a 19% decrease in the cell diameter (P = 0.015; Extended Data Fig. 4), which reduced cell volume by half. Thus, the ftsZ E315* mutation recapitulated nearly 60% of the evolved divergence in cell size, indicating that FtsZ has a central role in the cell size of M. mycoides.
Although changes in ftsZ had opposing effects on the size of the minimal and non-minimal cell, mutations in this gene were beneficial for both strains (Extended Data Fig. 3 and Extended Data Table 4). One adaptive consideration is that the faster-growing non-minimal cell should experience bouts of feast-or-famine conditions. In a serial batch environment, repeated transitions between exponential and stationary growth phases has been shown to select for increased cell size24,28. The observed patterns may also reflect constraints imposed by genome streamlining on the ability of the minimal cell to evolve an adaptive increase in cell size29,30,38,39. With more than 50% of its membrane-transport proteins removed, the minimal cell may have been unable to sequester the resources needed for constructing and maintaining a larger cell3,30,39 under the experimental conditions. Alternatively, cell size could evolve as a fitness-neutral byproduct of selection on other traits, such as DNA-replication rate40. For example, the two strains could have evolved different size trajectories despite similar selection pressures, due to epistatic effects of genome minimization such as those demonstrated using the ftsZE315* mutants (Extended Data Table 6 and Extended Data Fig. 4). In any case, our findings highlight that cell size—a fundamental feature of biological complexity in multicellular and single-celled organisms alike—evolves in a manner that is dependent on the genomic context.
Outlook
We uncovered genes, proteins and traits that are critical for evolutionary performance in the synthetically constructed M. mycoides JCVI-syn3B—a bacterium with the smallest genome of any organism grown in pure culture in the laboratory. In its ancestral state, this working approximation of a minimal cell had significantly reduced fitness. With less than 500 protein-coding genes, M. mycoides JCVI-syn3B had few redundancies when faced with an exceptionally high input of mutations. Despite these challenges, genome reduction did not alter cellular resources in any fundamental way that interfered with the ability to evolve increased fitness. Instead, natural selection during extended laboratory growth outweighed any deleterious effects of genome disruption and drift associated with synthetic streamlining that could have led populations of the minimal cell to extinction.
Our results demonstrate how synthetic biology and engineering can be informed by principles of evolutionary biology and population genetics. While it is now possible to build genomes with desired phenotypes, evolutionary processes represent a powerful but still underdeveloped approach for biological refinement. For example, rapid adaptation of the minimal cell involved selection on distinct targets, 25% of which encoded proteins of unknown function. Future studies combining evolution with a synthetic biology toolset have the potential to improve gene characterization and the mapping of regulatory networks, which may ultimately be used for optimizing stable living systems. Some degree of genome minimization will probably be a common path of development in biotechnology. It would be undesirable if such an approach compromised replication or repair fidelity, owing for example to unexpected cellular changes that might be mutagenic or otherwise interfere with damage maintenance. From an engineering perspective, more studies are needed to evaluate the minimization of other genomes in alternate chassis under different environmental conditions. Nevertheless, if we assume that our findings are somewhat general, it appears that cellular functions are robust to streamlining over time, which is desirable when using minimized cells for biotechnology and bioproduction.
Our findings shed new light on the phenomenon of genome streamlining, which is prevalent in nature, especially among microorganisms that coevolve with hosts in both pathogenic and mutualistic ways, but also among free-living bacteria that dominate the global oceans7,9,41. Both adaptive and neutral theories have been developed to explain why genomes become streamlined42,43. Very few studies have mechanistically investigated how genome streamlining affects subsequent evolution, especially for microorganisms with different phylogenetic backgrounds living in environments with contrasting niches. Despite it reducing the sequence space of possible trajectories, we conclude that streamlining does not constrain fitness evolution and diversification of populations over time. Genome minimization may even create opportunities for evolutionary exploitation of essential genes, which are commonly observed to evolve more slowly13,44.
Methods
Strains and growth conditions
We maintained synthetic M. mycoides JCVI-syn1.0 and synthetic M. mycoides JCVI-syn3B in SP4 medium with KnockOut Serum Replacement (Gibco) substituted for fetal bovine serum (Supplementary Table 3). Cultures of these non-motile bacteria were grown in a dark, static growth chamber at 37 °C. The non-minimal JCVI-syn1.0 strain has been described in detail previously45. The minimal JVCI-syn3B is identical to the strain synthesized in previous studies3 with the following exceptions: JVCI-syn3B possesses a second rRNA operon copy, lacks a gene (MMSYN1_0531) encoding an efflux protein, and has 19 genes that were added back into the minimal genome to render the cell easier to use4,33 (Supplementary Table 4). The strain also contains a landing pad system (cre recombinase and loxP) facilitating genetic manipulation. For competition experiments used to quantify relative fitness, we used a JCVI-syn1.0 strain that expresses mCherry, which enabled us to distinguish it in mixed culture from other strains using flow cytometry and also factor out any costs associated with production of the fluorescent protein (see below).
Mutation accumulation experiment
Overview
Mutation accumulation (MA) experiments are designed to reduce the influence of natural selection through repeated bottlenecks of evolving populations19. When used with microbial populations, this is typically achieved by transferring single colonies, which have undergone single-cell bottlenecks. Before initiating MA experiments, we acclimatized JCVI-syn1.0 and JCVI-syn3B to laboratory conditions by maintaining populations in SP4 liquid medium. We took a clone of each acclimated strain to begin the MA experiment and propagated replicate lineages (n = 87 and n = 57 for JCVI-syn1.0 and JCVI-syn3B, respectively) for 20 to 36 weekly transfers.
Number of generations
To compare rates of mutation across replicates, we normalized all rates as per-generation values. To calculate the number of generations per transfer in the MA, we grew cells on SP4 agar for 1 week and diluted a sample of seventh day colonies into 1 ml of phosphate-buffered saline (pH 7.4). Cells were fixed with 20 μl of 25% glutaraldehyde and stained with 2× SYBR Green, and then counted with a NovoCyte flow cytometer (ACEA Biosciences). We used the dilutions to calculate the number of cells in the original colony, from which we inferred the number of generations (log2[N], where N is the number of cells in the undiluted colony) that must have occurred to reach a colony of that size46, assuming each colony is formed by a single progenitor cell. As the growth rate and other fitness components can decrease during an MA experiment47, we also measured the number of cells per colony during and at the end of the MA, averaging across timepoints to estimate the total number of generations. We then used the number of generations per transfer to estimate the effective population size (Ne) using the harmonic mean method47. Specifically, Ne was approximated as the harmonic mean of the series (20, 21, 22, …, 2f), where f is equal to the number of generations per transfer inferred from the previous step.
Whole-genome sequencing and sequence analysis
We performed DNA extractions from evolved MA cell lines using the DNeasy UltraClean Microbial Kit (Qiagen) according to the manufacturer’s instructions, with the additional step of adding 50 μl of 50 mg ml−1 lysozyme to improve cell lysis. Genomic DNA was sequenced using Illumina MiSeq sequencing to a depth of at least 35× coverage. Library preparation and DNA sequencing were conducted by the Indiana University Bloomington Center for Genomics and Bioinformatics. Whole-genome sequencing reads were quality controlled using cutadapt48 to trim low-quality base pairs and remove residual adapter sequences. We used breseq with the default parameters49,50 to call mutations using the trimmed reads. We only considered fixed mutations for the MA cell lines. We checked for mutations that had arisen in experimental ancestor strains before evolution. Ancestral mutations were removed from the analysis of all evolved MA lines derived from that strain using gdtools49,50. We used the sequencing data to check for contamination or cross-contamination in the evolved cell lines.
Statistical analyses
To compare the mutation rate and spectrum between strains, we used two-sample t-tests for numerical response variables and two-sample χ2 tests with continuity correction for comparing proportions. For comparing proportions to theoretical expectations within a strain, we used one-sample χ2 tests with continuity correction.
Adaptive evolution
Overview
In contrast to the mutation accumulation experiments, we conducted experiments that allowed bacteria to achieve large population sizes to increase the efficacy of natural selection. This involved serial passaging of cells in liquid cultures with limited bottlenecking at each transfer. For example, in our experiment, the minimum population size was 2× 107–4 × 107 for both JCVI-syn1.0 and JCVI-syn3B. We passaged replicate 3 ml liquid cultures of each strain (n = 4 per strain) in 13 mm glass test tubes by 1% (v/v) serial transfer each day for 300 days in a dark, static incubator held at 37 °C. We calculated the number of generations per day as the log2 of the dilution factor, that is, log2[101], the number of binary fissions needed to regenerate the original population size after the 1% (v/v) transfer51. Thus, we estimate that the M. mycoides strains were maintained for 1,997 generations, which, based on other experiments, is long enough for the majority of adaptation to occur51,52.
Measurements of fitness
First, we measured fitness as µmax by conducting growth curves on cells that were isolated at different timepoints during the adaptive evolution experiment (Supplementary Fig. 5). Cryopreserved cells were thawed on ice before preculturing at 37 °C for 24–72 h in 3 ml of SP4 medium in a 13 mm test tube. Before initiating the experiment, we adjusted the start times of precultures to help ensure that cultures from different evolution timepoints were at the same stage of growth. Approximately 6 × 105 cells from turbid precultures were then inoculated into replicate wells of a 96-well plate containing 200 µl of SP4 medium. Separately, each population was incubated in a 96-well plate for 24 h in a BioTek Synergy H1 microplate reader that recorded the absorbance every 15 min at 415 nm. This wavelength is close to a spectral peak for phenol red, a pH indicator that is a component of SP4 medium (Supplementary Table 3). Previous studies have shown that phenol red can be used as proxy for metabolism and growth53 because bacteria like M. mycoides produce organic acids as a byproduct of carbohydrate metabolism4 (Supplementary Fig. 5). With the resulting data, we used maximum likelihood to estimate growth-curve parameters using a modified Gompertz equation54:
where L is the lag time (h), A is the carrying capacity or yield (optical density at 415 nm), µmax is the maximum growth rate (day−1) and b0 is the intercept (Supplementary Fig. 6 and Supplementary Table 5).
Second, we measured relative fitness by competing ancestral and evolved strains against a M. mycoides JCVI-syn1.0 reference strain labelled with mCherry (syn1.0::mCh)26. Cryopreserved cells were used to make precultures in a similar manner to those in the growth curve experiment. Each strain was grown in liquid medium to log phase, and then the labelled and unlabelled strains were simultaneously diluted into a mixed culture in fresh medium. We immediately sampled the axenic cultures or the mixed culture (t0), fixed the cells with 20 μl of cold 25% glutaraldehyde, incubated them at 4 °C for 20 min and then stained the samples with 2× SYBR Green. After 24 h of growth (tf), the mixed culture was sampled and processed again in an identical manner. For samples in the adaptive evolution experiment, we quantified the abundance of each strain using a an LSR II flow cytometer (BD Biosciences) at Indiana University’s Flow Cytometry Core Facility. For measuring the relative fitness of engineered ftsZ mutants, we used the NovoCyte flow cytometer (ACEA Biosciences). While measurements were being made, we vortexed the samples every minute to prevent multiple cells from clumping together and being scored as single events. The purity was assessed during every run using negative controls and axenic controls. We detected 1,800–2,700 events per second and abundances on the order of 1 × 108 cells per ml. With the resulting data, we differentiated cells on the basis of the expression of mCherry. Using NovoExpress, FACSDiva and FCS Express software, we established gates on pure cultures of the non-mCherry-expressing experimental strains and the syn1.0::mCh reference strain (Supplementary Figs. 7 and 8). For the experimental strains, boundaries were established by gating axenic mCherry-negative cells that were positive for only SYBR Green fluorescence. For the reference strain, boundaries were established by gating axenic syn1.0::mCh cells that were positive for SYBR Green and mCherry (Supplementary Fig. 9). In the competition assays used to quantify relative fitness, we applied the axenically established gates to samples that contained a mixture of the reference strain and experimental strain. We obtained the proportion of false-negative mCherry cells by applying the mCherry-negative gate to axenic mCherry-expressing cells; this proportion was then used as a correction factor in mixed populations. Last, we calculated relative fitness as the change in the relative abundance of the strain of interest during the 24 h period of competitive growth versus syn1.0::mCh. Specifically, the relative fitness versus the mCherry reference strain WC is
where N0 represents the initial abundance of the experimental strain, Nf the abundance of the experimental strain after 24 h, and NC0 and NCf are initial and final abundances of the reference strain (syn1.0::mCh), respectively26. We normalized fitness values to be relative to the original M. mycoides JCVI-syn1.0 ancestor strain. In other words, we represent the fitness (W) as , where WJCVI -syn1.0 is the value of WC for M. mycoides JCVI-syn1.0.
Whole-genome sequencing and sequence analysis
DNA extraction, sequencing and bioinformatics were performed according to the same methods as for the mutation accumulation experiment with a few exceptions. Specifically, each replicate population was sequenced to a depth of at least 100× coverage, and polymorphic mutations were included in our analyses. As an indicator of selective pressure, we used the Jukes–Cantor method55 to compute the per-site dN/dS value on the basis of the number of nonsynonymous and synonymous SNMs within each of the evolved replicate populations normalized by the total nonsynonymous and synonymous target sizes. We counted the number of synonymous and nonsynonymous AT to CG, AT to GC, AT to TA, CG to GC, CG to TA and CG to AT sites using the gdtools module of breseq, which is a computational pipeline that identifies mutations from short-read DNA resequencing studies50. We next combined that information with the empirical mutation spectrum from the MA experiment to account for the differing probabilities of each of the six SNM types, and thereby calculate the total expected number of SNMs at nonsynonymous and synonymous sites56. The observed numbers of synonymous and nonsynonymous substitutions were obtained directly from breseq outputs. Synonymous and nonsynonymous polymorphisms were included in the observed count with probability equal to their allele frequency in mapped reads. We added a pseudocount of 1 synonymous substitution for all calculations57 because two of the populations had 0 synonymous substitutions.
To identify mutations possibly contributing to adaptation, we looked for genes that had mutations across two or more replicate populations for each genotype. Mutations in the same gene, arising and increasing in frequency in independent lineages, suggests that that mutation’s rise could be driven by positive selection58. To test this hypothesis, we statistically assessed whether multiply-mutated genes (that is, genes mutated in >1 replicate evolved population) had acquired more mutations than would be expected by chance under the assumption that the mutations were neutral58. To do this, we recorded all of the polymorphic and fixed mutations that were called within genes. Synonymous mutations were excluded. We then used Python59 to simulate the placement of these mutations at random across all genes. The probability of any given gene receiving any given mutation was relativized to the gene’s length and GC content using the known mutation rates of G:C nucleotides and A:T nucleotides from the mutation-accumulation experiment. We repeated this random placement of mutations 100,000 times. In each simulation, we counted the number of mutations received by each gene, with each fixed mutation increasing the count by 1 and each polymorphism increasing the count by an amount equal to its allele frequency. For each multiply-mutated gene from the real adaptation experiment, we calculated the proportion of the 100,000 simulations in which the gene received at least as many mutations as were truly observed and called this proportion the P value. We then used the Benjamini–Hochberg method60,61 to generate corrected P values (Padj) to account for multiple tests with the false-discovery rate set to be α = 0.05 (Extended Data Table 2). As a negative control, we repeated the simulations using only synonymous mutations. This process returned two false-positive significant genes, which was small compared with the 52 significant signatures detected among nonsynonymous mutations, although we also acknowledge that synonymous gene analysis had less power due to the smaller number of synonymous mutations.
Generation of ftsZ E315* mutant cells
This process required mutating the bacterial genomes while they were yeast centromeric plasmids (YCPs) followed by genome transplantation of the mutated genomes. The YCPs were mutated using rounds of CRISPR–Cas9 and yeast homologous recombination that is a modification of a method used previously to mutate M. mycoides strains62.
In the first CRISPR–Cas9 step, the molecule to be mutated was cleaved and the donor DNA comprising sequences from the two flanking genes was recombined with the cut JCVI-syn1.0 or JCVI-syn3B YCP, removing parts of genes of the flanking genes and all of the target gene. The donor DNA had 40 bp overlaps to both genes flanking the target gene and had a 22 bp Mycoplasma gallisepticum 161 CRISPR–Cas9 target sequence with a protospacer adjacent motif (PAM) (5′-GTATAAATACATCCAGGAGTGG-3′) that had no homology elsewhere in JCVI-syn1.0 or JCVI-syn3B. The M. gallisepticum sequence put a new PAM in the genome that was used in the second round of CRISPR–Cas9.
The second round of CRISPR–CAS9 cut the JCVI-syn1.0 or JCVI-syn3B YCP at the new M. gallisepticum PAM. The cut YCP was then recircularized using a donor DNA containing the desired point mutation. The mutagenized regions of the YCPs were PCR amplified and the mutation was confirmed by Sanger sequencing. Correctly mutagenized JCVI-syn1.0 or JCVI-syn3B YCPs were then transplanted into Mycoplasma capricolum recipient cells as reported previously3,59,60,63,64. The mutagenized regions of the transplants were PCR-amplified and sequenced to confirm the presence of the desired mutations.
Microscopy and image analysis
Scanning electron microscopy (SEM) was used to compare changes in the cell size of evolved populations. All of the populations were grown in the same batch of medium and under identical conditions in a single incubator. The start times of cultures were adjusted so that they reached stationary phase at the same time. We centrifuged stationary-phase cultures and resuspended the pellet in 1 ml of phosphate-buffered saline (pH 7.4). The resuspended cells were fixed by adding 20 μl of cold 25% glutaraldehyde and incubating at 4 °C for 20 min. For microscopy observation, fixed cells were concentrated 4× by centrifugation and resuspension. The centrifugation steps were performed at 25 °C for 4 min at 2,000g. SEM was performed at the Indiana University Bloomington Electron Microscopy Center. Fixed cells in PBS were pelleted and resuspended in 100 mM sodium cacodylate buffer (pH 7.2) with 2 mM calcium chloride and 2% sucrose. We coated 12-mm-diameter glass coverslips with 0.1% poly-l-lysine for 5 min, after which coverslips were washed with a few drops of double distilled water. Resuspended cells were added to the coverslip surface and allowed to adhere. After 5 min, the coverslips were washed twice with 100 mM sodium cacodylate buffer (pH 7.2) with 2 mM calcium chloride and 2% sucrose. Next, 300 µl of 2% osmium tetroxide in 100 mM sodium cacodylate buffer (pH 7.2) with 2% sucrose was added to the surface of the coverslips while on ice. After 30 min, the coverslips were washed with double-distilled water. The coverslips were placed into a CPD coverslip holder (Electron Microscopy Sciences, 70193-01). The samples were dehydrated in a graded ethanol series (30%, 50%, 70%, 90%, 95%) while on ice. At room temperature, the coverslips were rinsed three times with 100% ethanol. Each dehydration step lasted for 2 min. Critical-point drying was performed using the Tousimis Samdri 790 critical-point dryer. The dried coverslips were placed on aluminium SEM stubs and sputter-coated using the Safematic CCU-010 with SP-010 Sputter Head with 45 nm of gold/palladium (80%/20%), which is accurate in the Angstrom range. All of the samples were coated simultaneously to minimize variance among samples. We viewed the samples using the FEI Teneo scanning electron microscope at 2.0 kV, 25 pA probe current and 3.0 mm working distance. The T2 detector was used. We calibrated the measurements using line grating replicas (2,160 lines per mm) with 0.261 μm latex spheres (Electron Microscopy Sciences). We analysed the SEM image data using ImageJ65. We used the straight and measure features combined with image scale metadata to measure the vertical diameters of imaged cells that met the following criteria: cells must be round; cells must not have apparent holes or punctures; cells must be completely within the field of view; cells must have an unambiguous perimeter; there must be no suggestion that a cell is currently or has recently undergone binary fission; cells must be ≥0.1 μm across. Each image was processed counterclockwise starting from east. The samples were processed in a randomized order.
Statistical analyses
For the growth-curve experiments, we used a generalized linear mixed model to test for the fixed effects of time (generation) and cell type (minimal versus non-minimal) on growth curve parameters (µmax, lag time, yield) while fitting random intercepts for the replicate evolved populations (Supplementary Table 3). We used variance partition coefficients to estimate the contribution of the replicate populations (random effect) to the total variation explained in the models (Extended Data Fig. 1, Extended Data Table 1, Supplementary Figs. 1 and 2 and Supplementary Table 5). For the adaptative evolution experiment (Figs. 2 and 4), we tested hypotheses using a general linear model (GLM) after subtracting observations of each replicate-evolved population (generation 2,000) from its corresponding ancestor (generation 0). With the intercept term excluded, the GLM tests whether the evolutionary trajectory for each group is different from zero. With the intercept term included, the GLM tests whether the evolutionary trajectories are different among groups. We also used two-way ANOVA with Tukey’s honest significant difference test to test hypotheses about the effects of cell type (minimal versus non-minimal) and ftsZ E315* (wild type versus mutant) on relative fitness and cell size. When necessary, data were log10-transformed to meet statistical assumptions.
We compared the composition of genes acquiring mutations among the evolved replicate populations by first constructing a gene-by-population matrix. Here, each row represented an evolved population and each column represented a gene that had acquired at least one mutation among all of the populations. Each cell of the matrix was filled with the sum value of mutations occurring in that gene in that population, where fixed mutations were valued at 1 and polymorphisms were valued equal to the allele frequency. Only essential genes, shared between JCVI-syn1.0 and JCVI-syn3B, were considered. We used PERMANOVA on the Bray–Curtis distances generated from the gene-by-population matrix to test for the significance of cell type (minimal versus non-minimal) on the composition of mutations using the adonis function in the R package vegan66. For visualization, the Bray–Curtis distances were decomposed into two dimensions using principal coordinate analysis using the cmdscale function.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at 10.1038/s41586-023-06288-x.
Supplementary information
Supplementary Figs. 1–9, Supplementary Tables 1–5 and two citations supporting the experimental and statistical procedures described in the main manuscript.
Delta fitness trajectory data for relative fitness in Supplementary Fig. 1.
dN/dS data for non-minimal cell in Supplementary Fig. 2.
Data for the percentage change in cell size in Supplementary Fig. 3.
Delta size trajectory data for cells in Supplementary Fig. 4.
Growth curve data for Supplementary Fig. 5.
Growth curve data for Supplementary Fig. 6.
Flow cytometry data for Supplementary Fig. 7.
Flow cytometry data for Supplementary Fig. 8.
Flow cytometry data for Supplementary Fig. 9.
Source data
Acknowledgements
We thank B. Stein for assistance with scanning electron microscopy (NIH 1S10OD023501-01); J. French, E. Snider, K. McKenzie and D. Schwartz for assistance in the laboratory; and M. Hahn, M. Behringer, M. Muscarella and C. Hassel for discussions regarding data analysis. We acknowledge financial support from the US National Science Foundation (DEB-1442246, DEB 1934554 and DBI-2022049, to J.T.L.; and MCB-1840301, MCB-1818344, MCB-1840320 and MCB-2221237 to J.I.G., L.S., D.C.M.B. and K.S.W.), US Army Research Office Grant (W911NF-14-1-0411, to J.T.L. and M.L.; and W911NF2210014, to J.T.L.), the National Aeronautics and Space Administration (80NSSC20K0618, to J.T.L.), the National Institutes of Health (R35-GM122566-01 and 2017-202, to M.L.), and the Brazilian Agricultural Research Corporation (21195.002926/2019-98, to D.M.C.B.). Any use of trade, firm or product names is for descriptive purposes only and does not imply endorsement by the US Government.
Extended data figures and tables
Author contributions
J.T.L. and R.Z.M.-R. conceived the study. J.T.L. supervised the project. J.T.L. and R.Z.M.-R. performed administrative tasks. R.Z.M.-R., J.T.L., M.L., J.I.G., K.S.W., B.K.L. and L.S. designed the methodology. R.Z.M.-R., B.K.L., L.S., D.M.C.B. and J.T.L. performed the experiments. J.T.L., R.Z.M.-R. and D.R.S. curated, analysed and visualized the data. J.T.L. and R.Z.M.-R. wrote the paper. All of the authors reviewed, edited and approved the final version of the manuscript.
Peer review
Peer review information
Nature thanks Ilias Tagkopoulos and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Data availability
Data are available at GitHub (https://github.com/LennonLab/MinimalCell), Zenodo (10.5281/zenodo.7953578), Figshare (10.6084/m9.figshare.23119985) and the NCBI Sequence Read Archive (PRJNA743406). Source data are provided with this paper.
Code availability
Computing code for reproduction is available at GitHub (https://github.com/LennonLab/MinimalCell) and Zenodo (10.5281/zenodo.7953578).
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
7/26/2023
A Correction to this paper has been published: 10.1038/s41586-023-06454-1
Extended data
is available for this paper at 10.1038/s41586-023-06288-x.
Supplementary information
The online version contains supplementary material available at 10.1038/s41586-023-06288-x.
References
- 1.Lachance J-C, Rodrigue S, Palsson BO. Minimal cells, maximal knowledge. eLife. 2019;8:e45379. doi: 10.7554/eLife.45379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Glass JI, Merryman C, Wise KS, Hutchison CA, Smith HO. Minimal cells—real and imagined. Cold Spring Harb. Perspect. Biol. 2017;9:a023861. doi: 10.1101/cshperspect.a023861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hutchison CA, et al. Design and synthesis of a minimal bacterial genome. Science. 2016;351:aad6253. doi: 10.1126/science.aad6253. [DOI] [PubMed] [Google Scholar]
- 4.Breuer M, et al. Essential metabolism for a minimal cell. eLife. 2019;8:e36842. doi: 10.7554/eLife.36842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McQuillen R, Xiao J. Insights into the structure, function, and dynamics of the bacterial cytokinetic FtsZ-ring. Annu. Rev. Biophys. 2020;49:309–341. doi: 10.1146/annurev-biophys-121219-081703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liao Y, Ithurbide S, Evenhuis C, Löwe J, Duggin IG. Cell division in the archaeon Haloferax volcanii relies on two FtsZ proteins with distinct functions in division ring assembly and constriction. Nat. Microbiol. 2021;6:594–605. doi: 10.1038/s41564-021-00894-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moran NA, Bennett GM. The tiniest tiny genomes. Annu. Rev. Microbiol. 2014;68:195–215. doi: 10.1146/annurev-micro-091213-112901. [DOI] [PubMed] [Google Scholar]
- 8.Leprince A, van Passel MW, dos Santos VAM. Streamlining genomes: toward the generation of simplified and stabilized microbial systems. Curr. Opin. Biotechnol. 2012;23:651–658. doi: 10.1016/j.copbio.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 9.Giovannoni SJ, et al. Genome streamlining in a cosmopolitan oceanic bacterium. Science. 2005;309:1242–1245. doi: 10.1126/science.1114057. [DOI] [PubMed] [Google Scholar]
- 10.Lynch, M. The Origins of Genome Architecture (Sinauer Associates, 2007).
- 11.Nakabachi A, et al. The 160-kilobase genome of the bacterial endosymbiont Carsonella. Science. 2006;314:267–267. doi: 10.1126/science.1134196. [DOI] [PubMed] [Google Scholar]
- 12.Meyer A, et al. Giant lungfish genome elucidates the conquest of land by vertebrates. Nature. 2021;590:284–289. doi: 10.1038/s41586-021-03198-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hahn MW, Kern AD. Comparative genomics of centrality and essentiality in three eukaryotic protein-interaction networks. Mol. Biol. Evol. 2005;22:803–806. doi: 10.1093/molbev/msi072. [DOI] [PubMed] [Google Scholar]
- 14.Graur, D. & Li, W.-H. Fundamentals of Molecular Evolution (Sinauer Associates, 2000).
- 15.Benner SA, Sismour AM. Synthetic biology. Nat. Rev. Genet. 2005;6:533–543. doi: 10.1038/nrg1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Richardson SM, et al. Design of a synthetic yeast genome. Science. 2017;355:1040–1044. doi: 10.1126/science.aaf4557. [DOI] [PubMed] [Google Scholar]
- 17.Kuo C-H, Ochman H. Deletional bias across the three domains of life. Genome Biol. Evol. 2009;1:145–152. doi: 10.1093/gbe/evp016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Long H, et al. Evolutionary determinants of genome-wide nucleotide composition. Nat. Ecol. Evol. 2018;2:237–240. doi: 10.1038/s41559-017-0425-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lynch M, et al. Genetic drift, selection and the evolution of the mutation rate. Nat. Rev. Genet. 2016;17:704–714. doi: 10.1038/nrg.2016.104. [DOI] [PubMed] [Google Scholar]
- 20.Sung W, Ackerman MS, Miller SF, Doak TG, Lynch M. Drift-barrier hypothesis and mutation-rate evolution. Proc. Natl Acad. Sci. USA. 2012;109:18488–18492. doi: 10.1073/pnas.1216223109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hershberg R. Mutation—the engine of evolution: studying mutation and its role in the evolution of bacteria. Cold Spring Harb. Perspect. Biol. 2015;7:a018077. doi: 10.1101/cshperspect.a018077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moran NA, McLaughlin HJ, Sorek R. The dynamics and time scale of ongoing genomic erosion in symbiotic bacteria. Science. 2009;323:379–382. doi: 10.1126/science.1167140. [DOI] [PubMed] [Google Scholar]
- 23.Lindahl T, Ljungquist S, Siegert W, Nyberg B, Sperens B. DNA N-glycosidases: properties of uracil-DNA glycosidase from Escherichia coli. J. Biol. Chem. 1977;252:3286–3294. doi: 10.1016/S0021-9258(17)40386-3. [DOI] [PubMed] [Google Scholar]
- 24.Vasi F, Travisano M, Lenski RE. Long-term experimental evolution in Escherichia coli. II. Changes in life-history traits during adaptation to a seasonal environment. Am. Nat. 1994;144:432–456. doi: 10.1086/285685. [DOI] [Google Scholar]
- 25.Gifford DR, Schoustra SE, Kassen R. The length of adaptive walks is insensitive to starting fitness in Aspergillus nidulans. Evol. Int. J. Org. Evol. 2011;65:3070–3078. doi: 10.1111/j.1558-5646.2011.01380.x. [DOI] [PubMed] [Google Scholar]
- 26.Wiser MJ, Lenski RE. A comparison of methods to measure fitness in Escherichia coli. PLoS ONE. 2015;10:e0126210. doi: 10.1371/journal.pone.0126210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Antczak M, Michaelis M, Wass MN. Environmental conditions shape the nature of a minimal bacterial genome. Nat. Commun. 2019;10:3100. doi: 10.1038/s41467-019-10837-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gounand I, et al. Size evolution in microorganisms masks trade-offs predicted by the growth rate hypothesis. Proc. R. Soc. B. 2016;283:20162272. doi: 10.1098/rspb.2016.2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mei Z-P, Finkel ZV, Irwin AJ. Light and nutrient availability affect the size-scaling of growth in phytoplankton. J. Theor. Biol. 2009;259:582–588. doi: 10.1016/j.jtbi.2009.04.018. [DOI] [PubMed] [Google Scholar]
- 30.Chien A-C, Hill NS, Levin PA. Cell size control in bacteria. Curr. Biol. 2012;22:R340–R349. doi: 10.1016/j.cub.2012.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mongold JA, Lenski RE. Experimental rejection of a nonadaptive explanation for increased cell size in Escherichia coli. J. Bacteriol. 1996;178:5333–5334. doi: 10.1128/jb.178.17.5333-5334.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hill NS, Buske PJ, Shi Y, Levin PA. A moonlighting enzyme links Escherichia coli cell size with central metabolism. PLoS Genet. 2013;9:e1003663. doi: 10.1371/journal.pgen.1003663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pelletier, J. F. et al. Genetic requirements for cell division in a genomically minimal cell. Cell184, 2430–2440 (2021). [DOI] [PubMed]
- 34.Pelletier JF, Glass JI, Strychalski EA. Cellular mechanics during division of a genomically minimal cell. Trends Cell Biol. 2022;32:900–907. doi: 10.1016/j.tcb.2022.06.009. [DOI] [PubMed] [Google Scholar]
- 35.Cohan MC, Eddelbuettel AMP, Levin PA, Pappu RV. Dissecting the functional contributions of the intrinsically disordered C-terminal tail of Bacillus subtilis FtsZ. J. Mol. Biol. 2020;432:3205–3221. doi: 10.1016/j.jmb.2020.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Graffeuil, A., Uhlin, B. E. & Cisneros, D. A. Polar mutagenesis of bacterial transcriptional units using Cas12a. Microb. Cell Fact.21, 139 (2022). [DOI] [PMC free article] [PubMed]
- 37.Bianchi DM, Pelletier JF, Hutchison CA, Glass JI, Luthey-Schulten Z. Toward the complete functional characterization of a minimal bacterial proteome. J. Phys. Chem. B. 2022;126:6820–6834. doi: 10.1021/acs.jpcb.2c04188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.DeLong JP, Okie JG, Moses ME, Sibly RM, Brown JH. Shifts in metabolic scaling, production, and efficiency across major evolutionary transitions of life. Proc. Natl Acad. Sci. USA. 2010;107:12941–12945. doi: 10.1073/pnas.1007783107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Westfall CS, Levin PA. Bacterial cell size: multifactorial and multifaceted. Annu. Rev. Microbiol. 2017;71:499–517. doi: 10.1146/annurev-micro-090816-093803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Amir A. Is cell size a spandrel? eLife. 2017;6:e22186. doi: 10.7554/eLife.22186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wolf YI, Koonin EV. Genome reduction as the dominant mode of evolution. Bioessays. 2013;35:829–837. doi: 10.1002/bies.201300037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lynch M. Streamlining and simplification of microbial genome architecture. Annu. Rev. Microbiol. 2006;60:327–349. doi: 10.1146/annurev.micro.60.080805.142300. [DOI] [PubMed] [Google Scholar]
- 43.Giovannoni SJ, Cameron Thrash J, Temperton B. Implications of streamlining theory for microbial ecology. ISME J. 2014;8:1553–1565. doi: 10.1038/ismej.2014.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Luo H, Gao F, Lin Y. Evolutionary conservation analysis between the essential and nonessential genes in bacterial genomes. Sci Rep. 2015;5:13210. doi: 10.1038/srep13210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gibson DG, et al. Creation of a bacterial cell controlled by a chemically synthesized genome. Science. 2010;329:52–56. doi: 10.1126/science.1190719. [DOI] [PubMed] [Google Scholar]
- 46.Dillon MM, Sung W, Sebra R, Lynch M, Cooper VS. Genome-wide biases in the rate and molecular spectrum of spontaneous mutations in Vibrio cholerae and Vibrio fischeri. Mol. Biol. Evol. 2017;34:93–109. doi: 10.1093/molbev/msw224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Behringer MG, Hall DW. Genome-wide estimates of mutation rates and spectrum in Schizosaccharomyces pombe indicate CpG sites are highly mutagenic despite the absence of DNA methylation. G3. 2016;6:149–160. doi: 10.1534/g3.115.022129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:10–12. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 49.Barrick JE, et al. Identifying structural variation in haploid microbial genomes from short-read resequencing data using breseq. BMC Genom. 2014;15:1039. doi: 10.1186/1471-2164-15-1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Deatherage DE, Barrick JE. Identification of mutations in laboratory-evolved microbes from next-generation sequencing data using breseq. Methods Mol. Biol. 2014;1151:165–188. doi: 10.1007/978-1-4939-0554-6_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lenski RE, Travisano M. Dynamics of adaptation and diversification: a 10,000-generation experiment with bacterial populations. Proc. Natl Acad. Sci. USA. 1994;91:6808–6814. doi: 10.1073/pnas.91.15.6808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lang, Gregory I, Botstein D, Desai MM. Genetic variation and the fate of beneficial mutations in asexual populations. Genetics. 2011;188:647–661. doi: 10.1534/genetics.111.128942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yus E, et al. Determination of the gene regulatory network of a genome-reduced bacterium highlights alternative regulation independent of transcription factors. Cell Syst. 2019;9:143–158. doi: 10.1016/j.cels.2019.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lennon JT, Khatana SAM, Marston MF, Martiny JBH. Is there a cost of virus resistance in marine cyanobacteria? ISME J. 2007;1:300–312. doi: 10.1038/ismej.2007.37. [DOI] [PubMed] [Google Scholar]
- 55.Yang, Z. Computational Molecular Evolution (Oxford Univ. Press, 2006).
- 56.Ina Y. New methods for estimating the numbers of synonymous and nonsynonymous substitutions. J. Mol. Evol. 1995;40:190–226. doi: 10.1007/BF00167113. [DOI] [PubMed] [Google Scholar]
- 57.Shpak M, Goldberg MM, Cowperthwaite MC. Rapid and convergent evolution in the Glioblastoma multiforme genome. Genomics. 2015;105:159–167. doi: 10.1016/j.ygeno.2014.12.010. [DOI] [PubMed] [Google Scholar]
- 58.Johnson MS, et al. Phenotypic and molecular evolution across 10,000 generations in laboratory budding yeast populations. eLife. 2021;10:e63910. doi: 10.7554/eLife.63910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Van Rossum, G. & Drake, F. L. Python 3 Reference Manual (CreateSpace, 2009).
- 60.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B. 1995;57:289–300. [Google Scholar]
- 61.Benjamini Y, Heller R, Yekutieli D. Selective inference in complex research. Philos. Trans. R. Soc. A. 2009;367:4255–4271. doi: 10.1098/rsta.2009.0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kannan K, et al. One step engineering of the small-subunit ribosomal RNA using CRISPR/Cas9. Sci. Rep. 2016;6:30714. doi: 10.1038/srep30714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lartigue C, et al. Genome transplantation in bacteria: changing one species to another. Science. 2007;317:632–638. doi: 10.1126/science.1144622. [DOI] [PubMed] [Google Scholar]
- 64.Lartigue C, et al. Creating bacterial strains from genomes that have been cloned and engineered in yeast. Science. 2009;325:1693–1696. doi: 10.1126/science.1173759. [DOI] [PubMed] [Google Scholar]
- 65.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Oksanen, J. et al. R package ‘vegan’: community ecology package; version 2.5.7 (2020).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figs. 1–9, Supplementary Tables 1–5 and two citations supporting the experimental and statistical procedures described in the main manuscript.
Delta fitness trajectory data for relative fitness in Supplementary Fig. 1.
dN/dS data for non-minimal cell in Supplementary Fig. 2.
Data for the percentage change in cell size in Supplementary Fig. 3.
Delta size trajectory data for cells in Supplementary Fig. 4.
Growth curve data for Supplementary Fig. 5.
Growth curve data for Supplementary Fig. 6.
Flow cytometry data for Supplementary Fig. 7.
Flow cytometry data for Supplementary Fig. 8.
Flow cytometry data for Supplementary Fig. 9.
Data Availability Statement
Data are available at GitHub (https://github.com/LennonLab/MinimalCell), Zenodo (10.5281/zenodo.7953578), Figshare (10.6084/m9.figshare.23119985) and the NCBI Sequence Read Archive (PRJNA743406). Source data are provided with this paper.
Computing code for reproduction is available at GitHub (https://github.com/LennonLab/MinimalCell) and Zenodo (10.5281/zenodo.7953578).