

Abstract
MutLα is essential for human DNA mismatch repair (MMR). It harbors a latent endonuclease, is responsible for recruitment of process associated proteins and is relevant for strand discrimination. Recently, we demonstrated that the MMR function of MutLα is regulated by phosphorylation of MLH1 at serine (S) 477. In the current study, we focused on S87 located in the ATPase domain of MLH1 and on S446, S456 and S477 located in its linker region. We analysed the phosphorylation-dependent impact of these amino acids on DNA binding, MMR ability and thermal stability of MutLα. We were able to demonstrate that phosphorylation at S87 of MLH1 inhibits DNA binding of MutLα. In addition, we detected that its MMR function seems to be regulated predominantly via phosphorylation of serines in the linker domain, which are also partially involved in the regulation of DNA binding. Furthermore, we found that the thermal stability of MutLα decreased in relation to its phosphorylation status implying that complete phosphorylation might lead to instability and degradation of MLH1. In summary, we showed here, for the first time, a phosphorylation-dependent regulation of DNA binding of MutLα and hypothesized that this might significantly impact its functional regulation during MMR in vivo.
Subject terms: Cell biology, Molecular biology
Introduction
Different DNA repair mechanisms are responsible for the maintenance of the DNA information and are required to safeguard cellular functions and identities in all organisms. The major role of the DNA mismatch repair (MMR) is the correction of base–base mismatches and insertion-deletion loops, which occur during replication. In humans, the mismatch recognition can be initiated by two heterodimeric MutS complexes: The MSH2/MSH6 (MutSα) complex is primarily responsible for recognition of single base–base and small insertion deletion loops; the MSH2/MSH3 (MutSβ) complex is mainly responsible for the recognition of insertion-deletion loops containing up to 16 additional nucleotides in one strand1,2. After MMR initiation, the MLH1/PMS2 (MutLα) complex interacts with and binds to the MutS complex. MutLα plays a very important role in 5' as well as in 3' directed MMR, has an endonuclease function3–6, and is required for the recruitment of several other proteins involved in further steps of the process2,7.
Deficiency of MMR results in a massive increase of the mutation rate in affected cells and is especially associated with the development of colorectal cancer (CRC), where loss of MMR plays a role in up to one fifth of cases8. However, MMR deficiency is also frequently involved in other types of cancer9.
In sporadic CRCs, loss of MMR is detected in 12–17% of cases and is due to hypermethylation of the MLH1 promoter, which avoids MLH1 gene transcription10. In the hereditary Lynch syndrome, which accounts for approximately 3% to 5% of all CRC cases, germline mutations in the MLH1 gene are detectable in around 50% of patients whereas one-third of the detected alterations are non-synonymous, non-truncating variants, or missense variants, in the coding region11,12. In up to 3% of patients with Lynch syndrome, constitutional hypermethylation of the MLH1 promoter has been described, especially in patients with unclear family history13.
In addition to the lack of MLH1 caused by promoter hypermethylation or gene mutation, MLH1 function can also be limited by post-translational modifications. We previously showed that MLH1 can be phosphorylated by casein kinase 2 α (CK2α) at amino acid position serine 477 (S477) and we identified that phosphorylation of MLH1 at position S477 can switch off MMR activity in vitro14. Very recently, we were able to demonstrate in vivo, that CK2α is frequently overexpressed in CRCs and that a high nuclear/cytoplasmic expression of CK2α is responsible for enhanced phosphorylation of MLH1 and increased tumor mutation rates in these tumors15. Moreover, affected patients demonstrated a significantly reduced 5-year survival outcome15. Thus, we were able to show very clearly that post-translational modification of MLH1 by phosphorylation can also cause a loss or at least a marked restriction of MMR function, a condition that significantly drives tumor progression.
Furthermore, by mass spectrometry of MLH1, we were able to detect one serine located in the ATPase domain, serine 87 (S87), and two serines located in the linker region, serine 446 (S446) and serine 456 (S456), which were phosphorylatable in MLH1, albeit to a lesser extent14. The relevance of phosphorylation of MLH1 at S87, S446 and S456 is completely unknown so far. Therefore, the current study, we analyzed the influence of MLH1 phosphorylation at amino acid position S87, S446, S456 alone and in combination with S477 on the expression level, the DNA binding ability and the repair efficiency of MutLα.
Materials and methods
Cells
HEK293T cells, obtained from Dr. Kurt Ballmer (Paul Scherrer Institute, Villigen, Switzerland), were grown in DMEM with 10% Fetal Calf Serum and 1% Penicillin/Streptomycin. As previously described, MLH1 is not expressed in HEK293T16.
Antibodies
Anti-MLH1 (G168-728) and anti-PMS2 (A16-4) were purchased from Pharmingen (BD Biosciences, Heidelberg, Germany). Anti-beta Actin (Clone AC-15) was from Sigma-Aldrich (Munich, Germany). Anti-phospho-AKT-substrate (23C8D2), used for the detection of phosphorylation of MLH1 at amino acid position serine 477 and hereinafter referred to as anti-p-MLH1, was obtained from Cell Signaling (New England Biolabs GmbH, Frankfurt, Germany).
Anti-fluorescence-labeled anti-mouse IRDye680LT and anti-fluorescence-labeled anti-rabbit IRDye800CW were from LI-COR (LI-COR Biosciences GmbH, Bad Homburg, Germany).
Plasmids
The pcDNA3.1 + /PMS2 expression plasmid was described earlier17. The pEBG-2 T/MLH1 vector was generated by cloning the cDNA of MLH1 from the pcDNA3.1 + plasmid into the pEBG-2 T vector (a gift from David Baltimore (Addgene plasmid # 22227; http://n2t.net/addgene:22227; RRID:Addgene_22227;18) via the BamHI restriction site. This placed MLH1 downstream of the coding sequence for Glutathione-S-Transferase (GST).
Further pEBG-2 T plasmids containing MLH1-variants (MLH1S87A, MLH1S446A, MLH1S456A, MLH1S87A/S477A, MLH1S446A/S477A and MLH1S456A/S477A) were subsequently prepared by site-directed mutagenesis using appropriate primers (Table 1) with the Q5 Site-Directed Mutagenesis Kit (New England Biolabs GmbH, Frankfurt, Germany). All resulting plasmids were confirmed by direct sequencing.
Table 1.
Primers used for site directed mutagenesis.
| MLH1 variant | Forward primer | Reverse primer |
|---|---|---|
| MLH1 S477A | 5′- CATCGGGAAGATGCTGATGTGGAAATGG-3′ | 5′- CCATTTCCACATCAGCATCTTCCCGATG -3′ |
| MLH1 S87A | 5′- ACTAGTAAACTGCAGGCCTTTGAGGATTTAGCCA -3′ | 5′- TGGCTAAATCCTCAAAGGCCTGCAGTTTACTAGT -3′ |
| MLH1 S446A | 5′- GCTGCCAAAAATCAGGCCTTGGAGGGGGATACAA -3′ | 5′- TTGTATCCCCCTCCAAGGCCTGATTTTTGGCAGC -3′ |
| MLH1 S456A | 5′- ACAACAAAGGGGACTGCCGAAATGTCAGAGAAGA -3′ | 5′- TCTTCTCTGACATTTCGGCAGTCCCCTTTGTTGT -3′ |
The pEGFP_C1 plasmid, negative control plasmid for transfection control and negative control for MMR assay, was purchased from Clontech Laboratories (Clontech Laboratories, Mountain View, United States of America).
The pEBG-2 T empty vector containing only GST was a used as a negative control plasmid for the electrophoretic mobility shift assay (EMSA).
Cell transfection and treatment
HEK293T at 50–70% confluence were transiently a) co-transfected with pEGB-2 T/MLH1 and pcDNA3.1 + /PMS2 as positive controls, b) co-transfected with pEGB-2 T constructs containing different MLH1 variants (pEGB-2 T/MLH1S87A, pEGB-2 T/MLH1S446A, pEGB-2 T/MLH1S456A, pEGB-2 T/MLH1S477A, pEGB-2 T/MLH1S87A/S477A, pEGB-2 T/MLH1S446A/S477A, pEGB-2 T/MLH1S456A/S477A) and pcDNA3.1 + /PMS2 and c) single transfected with pEGFP_C1 or empty pEGB-2 T as negative controls. For the generation of protein-extracts used in the MMR-assay, transfection of HEK293T cells was carried out in ø100mm dishes using 5 µg plasmid DNA and 2 µL/mL of the cationic polymer polyethylenimine (PEI) (Polysciences, Warrington, PA, USA; stock solution 1 mg/mL). For immunoprecipitation experiments, transfection of HEK293T cells was performed in ø100mm dishes as well, 5 µg Plasmid, 2 µL/mL PEI. For generation and overexpression of GST-tagged MutLα, HEK293T cells were co-transfected in cell culture dishes (ø145mm) using 12.5 µg plasmid DNA and 4.375 µL/mL of PEI. At 48 h post-transfection, cells were directly harvested or treated 8 h with Calyculin (50 nM) or DMSO as a control if indicated.
Finally, cells were harvested, protein extracts were generated and used for immunoprecipitation and analyzed by Western blotting or used as required. All experiments were performed at least three times.
Protein extraction
Denatured protein extracts of transiently transfected HEK293T cells, used for Western blotting, were prepared as described earlier15. In brief, cells were isolated by re-suspending the cells directly in medium and centrifuging (3000 g, 3 min, 4 °C). The cells were washed in 1 mL PBS and centrifuged again (3000 g, 3 min, 4 °C). The supernatant was discarded and cells were solved in lysis reagent (CelLytic™ M Lysispuffer Sigma-Aldrich (St. Louis, Missouri, USA)) combined with cOmplete™ Protease Inhibitor Cocktail Roche (Roche Pharma, Basel, Switzerland)), incubated on ice for 5 min, lysed by ultrasound for 10 s at 35% power, and centrifuged (14,000 g, 10 min, 4 °C). The supernatant (whole protein extract) was transferred to a new Eppendorf tube and stored at −20°C until use.
Whole cell extracts used for the MMR assay were generated as described before19. In brief, native protein extracts were generated by re-suspending PBS-washed cells in 2 times their packed cell volume of ice-cold hypotonic buffer (10 mM HEPES (pH 7.6), 5 mM MgCl2, 10 mM NaCl, 10 mM NaF, 0.1 mM EDTA, supplemented with 0.2 mM PMSF, 0.5 mM DTT directly before use). This suspension was carefully homogenized, incubated on ice for 5 min and snap-frozen in liquid nitrogen for 2 min for lysis. Afterwards, the cell suspension was thawed on ice and after 1 h, an identical volume of hypertonic buffer (10 mM HEPES (pH 7.6), 5 mM MgCl2, 830 mM NaCl, 10 mM NaF, 0.1 mM EDTA, 34% (v/v) glycerol, supplemented with 0.2 mM PMSF and 0.5 mM DTT directly before use) was added. The suspension was rocked on ice for 30 min with gentle agitation and centrifuged (14,000g, 10 min, 4 °C). The supernatant (native cell extract) was transferred to a new Eppendorf tube. The protein concentration was determined according to Bradford before the samples were frozen in liquid nitrogen and stored in aliquots at −80°C until further use.
Generation of nuclear extracts
10 cell culture dishes (ø145mm) of untransfected HEK293T cells were harvested, washed with PBS, pelleted and re-suspended in three times their packed cell volume of ice-cold hypotonic buffer (20 mM HEPES pH 7.6, 5 mM KCl, 0.5 mM MgCl2, supplemented with 0.2 mM PMSF, 0.5 mM DTT directly before use) and lysed with Dounce pestle B until lysis was sufficient. After centrifugation (10,000 g, 2 min, 4 °C), the cytoplasmic supernatant was again removed and centrifuged (12,000 g, 3 min, 4 °C) for removal of all cytoplasmic residuals. The pellet was re-suspended in 0.4 times the pellet volume of re-suspension-buffer (20 mM HEPES–KOH pH 7.6, 10% sucrose, supplemented with 0.2 mM PMSF and 1 mM DTT directly before use) with cOmplete™ Protease Inhibitor Cocktail Roche (Roche Pharma, Basel, Switzerland) and 0.4 times the pellet volume of high-salt buffer (50 mM HEPES–KOH pH 7.6, 10% sucrose, 840 mM KCl) was added under agitation. Extraction was performed for 30 min and extracted nuclei were removed by centrifugation (21,000 g, 30 min, 4 °C). Finally, the supernatant was dialyzed 3 h against 100 times the volume of dialysis buffer (25 mM HEPES pH 7.6, 100 mM KCl, 0.1 mM EDTA, supplemented with 0.2 mM PMSF and 0.5 mM DTT directly before use). The extract was centrifuged (21,000 g, 10 min, 4 °C), snap-frozen in liquid nitrogen and stored in aliquots at −80 °C.
Immunoprecipitation
Immunoprecipitations were carried out as described in Wessbecher et al.14 using 500 µg of whole cell extract from MutLα wild type (wt) or MutLα variant overexpressing HEK293T cells in a total volume of 1000 µl precipitation buffer (50 mM HEPES–KOH (pH 7.6), 100 mM NaCl, 0.5 mM EDTA, 0.2 mM PMSF, 0.5 mM DTT, 1% Triton X-100) with 2 µg of anti-MLH1 (G168-728). After one hour of agitated incubation at 4 °C, 20 µl protein G sepharose (Santa Cruz Biotechnology, Heidelberg, Germany) were added and incubation continued for 3 h. Precipitates were extensively washed in cold precipitation buffer using SigmaPrep™ spin columns (Sigma, Munich, Germany). Success of washing was always confirmed by running samples without antibody in parallel. The SpinColumns were boiled in SDS-PAGE sample buffer for 5 min, followed by centrifugation (10,000 g, 1 min) and proteins were separated on 10% polyacrylamide gels, followed by Western blotting on nitrocellulose membranes and antibody detection using standard procedures.
Western blotting
Proteins were separated on 10% polyacrylamide gels, followed by Western blotting on nitrocellulose membranes and antibody detection using standard procedures. Nitrocellulose membranes on which immunoprecipitated proteins were blotted were cut between 50 and 75 kDa, only the upper part of the membrane was incubated with antibody. This prevented hybridisation of the antibody of interest to the antibody used for precipitation.
Fluorescent-labeled secondary antibodies (anti-mouse 680 LT from LiCor Bioscience, anti-mouse 800 CW from LiCor Bioscience, anti-rabbit 680 LT from LiCor Bioscience) were used to detect signals in a FLA-9000 scanner (Fujifilm, Tokyo, Japan). If indicated, the band intensity of the protein expression was quantified using the Multi Gauge V3.2 program (Fujifilm, Tokyo, Japan).
The amount of p-MLH1S477 was detected after immunoprecipitation and quantified in correlation to total MLH1 levels as previously described14 using Multi Gauge V3.2. p-MLH1S477 levels were calculated in relation to MLH1 set to 100%. Thereafter, all p-MLH1S477 level were normalized to the p-MLH1S477 level of the MLH1 wt. All experiments were performed at least three times.
Generation of GST-tagged recombinant MutLα
GST-MLH1 and GST-MLH1 variants were coexpressed with PMS2 wt in 7 cell culture dishes (ø145 mm) of HEK293T cells as detailed above. Cells were harvested after 48 h in lysis buffer (50 mM Trizma pH 7.4, 0.27 M Sucrose, 1 mM Na-Ortho-Vanadate, 1 mM EDTA, 1 mM EGTA,10 mM Na-ß-glycerolphosphate, 50 mM NaF, 5 mM Na-pyrophosphate, 1% Triton-x-100) and centrifuged (4000 rpm, 15 min, 4 °C). The Supernatant was incubated (4 °C) with 0.5 ml Glutathione-sepharose 4B (GE Healthcare, Chicago, U.S.A) pre-washed twice with one volume of buffer A (50 mM Trizma pH 7.4, 0.1 mM EGTA, 0.1% β-Mercaptoethanol) and once with one volume of lysis buffer. After 2 h, the Glutathione-sepharose-suspension was washed 4 times with 10 ml lysis buffer with 500 mM NaCl, 8 times with 10 ml buffer A, and once with 10 ml buffer A plus 0.26 M Sucrose. Thereafter, 500 µl buffer A plus 0.26 M Sucrose and 80 mM glutathione was added per 500 µl Glutathione-sepharose and this suspension was incubated again for 2 h. Finally, the Glutathione-sepharose was pelleted (2500 rpm, 2 min, 4 °C) and used for EMSA, since a large amount of GST-tagged MutLα was still bound. In addition, the supernatant was loaded on SigmaPrep columns (Sigma Aldrich, Munich, Germany) and GST-tagged MutLα was eluted by centrifugation (3000 rpm, 5 min, 4 °C). The elution was repeated with 250 µl buffer A plus 0.26 M Sucrose and 80 mM glutathione. Recombinant GST-tagged MutLα was frozen with liquid nitrogen and stored at -80 °C until use.
Synthesis of GT-DNA-substrate
Generation of GT-DNA-substrate was carried out using pUC19 CPDC and pUC19 CPD20 plasmid-constructs on which the MMR assay of this manuscript is based on. Both vectors only differ in one base, which enabled us to generate a suitable DNA substrate for the EMSA: a double stranded DNA fragment carrying a G/T mismatch. First, amplification of the 101 bp pUC19 CPDC-depending fragment was performed using 100 ng pUC19 CPDC vector together with a 5′-P-tagged forward primer and a 5′-Atto680-tagged reverse primer (Table 2), the pUC19 CPD -depending fragment was amplified using 100 ng of pUC19 CPD together with a 5′-Atto680-tagged forward primer and a 5′-P-tagged reverse primer (Table 2). 10 µl of PCR fragments were treated with 0.25U of exonuclease λ (M0262S (5U/µl), New England Biolabs GmbH, Frankfurt, Germany) for 10 min at 37 °C followed by adding further 0.5U exonuclease λ and incubating again for 10 min at 37 °C to catalyze the removal of nucleotides from the 5’-phosphorylated double-stranded DNA, respectively. Resulting single stranded DNA fragments were purified, mixed 1/1, boiled at 95 °C for 5 min, and cooled down at room temperature. The mixture was centrifuged (14,000 rpm, 5 min, RT) and the supernatant was transferred to a new test tube. 10 µl of this supernatant were digested with 5U of exonuclease I (M0293S (20U/µl), New England Biolabs GmbH, Frankfurt, Germany) and incubated for 1 h at 37 °C to remove residual abundant nucleotides from linear single-stranded DNA in the 3' to 5' direction. Finally, the GT-DNA substrate was precipitated with sodium acetate 3 M and ethanol, washed with 70% ethanol and re-suspended in H2O.
Table 2.
Primers used for generation of GT-DNA-substrate.
Electrophoretic mobility shift assay
To analyze binding of GST-tagged MutLα wt and variants to DNA, a) 10 µl of Glutathione-sepharose-bound protein samples or b) 10 µl dephosphorylated Glutathione-sepharose-bound protein samples pretreated with 10 units of CIP (at 37 °C for 1 h), were first combined with 1.5 µl 10 mM ATP in 20 mM HEPES pH 7.5, 40 µg/ml BSA, 1 mM DTT, 100 mM NaCl, mixed with 95 ng GT-DNA-substrate and incubated on ice. After 5 min, 1 µl of 8% (w/v) sucrose and 2 µl of 75% glycerol were added to each sample. Now, the electrophoretic mobility shift assay (EMSA) was carried out in 4% non-denaturing polyacrylamide gels. After a first gel run for 30 min (150 V with ice-cold 0.5xTBE-buffer (pH 7.5)) without the addition of any probe, samples were applied and electrophoresed for 2 h at 48 V (in a dark room). Thereafter, gel-included glass plates were placed in a plastic bag and GT-DNA-substrate detection was performed at 685 nm using a FLA-9000 scanner (Fujifilm, Tokyo, Japan) and quantified using the Multi Gauge V3.2 program (Fujifilm, Tokyo, Japan). Subsequently, the DNA bound protein-amount was determined via Western blotting and compared to a known concentration of a control sample analyzed in parallel. All experiments were performed at least three times.
Analysis of MMR activity
The analysis of the MMR activity of the MutLα variants was determined in vitro as described19,21. Briefly, 2.5 μg protein extracts of transfected HEK293T cells were mixed with 50 μg nuclear extract of HEK293T cells and 35 ng of DNA substrate containing an intact AseI restriction site, a G-T mismatch in between an (not digestible) EcoRV restriction site and a 3′ single-strand nick at a distance of 83 bp to guide the MMR direction. If the tested heterodimer is fully qualified for mismatch repair, then the G-T mismatch will be corrected and the EcoRV restriction site will be restored. After incubation at 37 °C (20 min) the reaction was stopped, following by purification and digestion of the DNA substrate with AseI (5 units/15 µl) and EcoRV (10 units/15 µl). The restriction fragments were separated in agarose gels, analyzed using GelDoc XR plus detection and band intensities were quantified using Image Lab version 3.0 (Bio-Rad). If the substrate is efficiently repaired the AseI and EcoRV digestion will result in three bands: a 2.0 kb band corresponding to singly AseI cut (uncorrected and in excess added) DNA substrate, a 1.2 kb and a 0.8 kb band caused by the successfully corrected and restricted EcoRV restriction site. The repair efficiency (e) was calculated as: (e) = intensity of bands of repaired substrate/intensity of all bands of substrate. This result is independent of the amount of DNA recovered through plasmid purification. The (e) of MutLα variants was analyzed in direct comparison with MutLα wt that had been produced in parallel and calculated as (e)relative = (e)variant/(e)wild-type × 100.
In addition, it should be mentioned that a small amount of Calyculin is abundant in the MMR assay during the protocol when adding whole protein extracts of Calyculin treated cells. To exclude that this amount of Calyculin has any influence, an additional control experiment was performed in the current study. The amount of Calyculin present in the protein extract was calculated and was added separately and co-incubated according to the MMR assay protocol. In brief, 2.5 μg protein extracts of untreated MutLα wt expressing HEK293T cells, 50 μg nuclear extract of HEK293T cells, 35 ng of DNA substrate and 0.815 nM Calyculin were mixed and incubated. In parallel, the same reaction mixture without the addition of Calyculin was prepared and incubated. The repair efficacy of assays with Calyculin supplementation was compared to the efficacy of assays without Calyculin. All experiments were performed at least three times.
Nano differential scanning fluorimetry
Protein stabilities of recombinant GST-tagged MutLα variants were compared to the protein stability of recombinant GST-tagged MutLα wt as previously described22 on the basis of thermal stability, using nano-differential scanning fluorimetry (nanoDSF), a modified version of differential scanning fluorimetry, working with a Prometheus NT.48 (NanoTemper) with standard capillaries (NanoTemper).
This label-free technique uses the intrinsic fluorescence of the aromatic amino acids tryptophan and tyrosine to determine protein folding and stability. The fluorescence is excited at 280 nm and is detected at 330 nm and 350 nm. nanoDSF determines the apparent melting temperature (Tm), where half of the protein is unfolded, and onset temperature (Tonset) of thermal unfolding by measuring the ratio of the fluorescence intensity at 330 nm and 350 nm as a function of temperature. The derivation of F350/330 is used for data analysis. The protein samples were set up to a concentration of 1.2 mg/mL for every measurement, loaded into capillaries by capillary force action and placed into the instrument. The capillaries were heated from 20 to 80 °C with a heating rate of 2 °C /min and the changes in the fluorescence ratio (F350/F330) were monitored. For each experiment, a capillary containing only buffer A (50 mM Trizma pH 7.4, 0.1 mM EGTA, 0.1% β-Mercaptoethanol) plus 0.26 M Sucrose and 80 mM glutathione was used to calculate the blank value.
Statistical analysis
Unpaired two-tailed T-tests for normality, followed by Welch correction for uneven variations, and the Mann–Whitney-U test for non-normally distributed data were used to assess statistical significance.
All calculations were analyzed using the software GraphPad Prism 7 for Windows, Version 7.04 (La Jolla, CA, USA). The data shown are means ± SD, unless otherwise stated, the following p-values were considered as statistically significant: * p < 0.05, ** p < 0.005, *** p < 0.005, **** p < 0.0001 unless otherwise stated.
Results
Phosphorylation at amino acid positions S87, S446 or S456 has no influence on phosphorylatability of MLH1 at position S477
We previously detected that MLH1 harbors three additional amino acids, besides S477, that can be phosphorylated. Since it is known that different phosphorylation sites within one protein can influence each other, we first analyzed if phosphorylation of one of these amino acid positions (S87, S446 and S456) impacts the phosphorylatability of MLH1 at position S477. Therefore, non-phosphorylatable MLH1 variants (MLH1S87A, MLH1S446A, MLH1S456A, and MLH1S477A) were co-expressed together with PMS2 wt protein in HEK293T cells for 48 h. To stabilize phosphorylated amino acids, cells were then treated with Calyculin for 8 h, whole cell extracts were isolated, MutLα was immunoprecipitated using anti-MLH1 and the amount of p-MLH1S477 as well as PMS2 was compared via Western blotting (Fig. 1A, Figure S1). Quantification of bands was performed by using Multi Gauge V3.2 and correlated to those of MLH1 wt expressing cells which were set to 100% (Fig. 1B).
Figure 1.
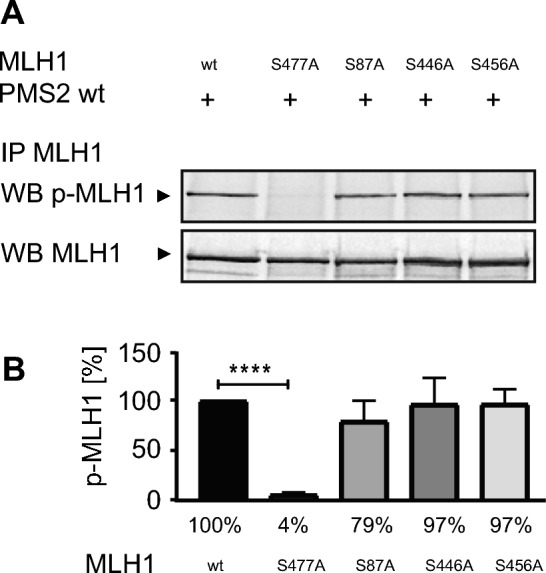
Amino acid positions S87, S446 or S456 have no influence on phosphorylatability of position S477. Using immunoprecipitated MutLα and a specific antibody which recognizes the phospho-S477-motif of MLH1 (A) the expression of p-MLH1S477 was determined in MutLα wt as well as in MLH1S477A/PMS2, MLH1S87A/PMS2, MLH1S446A/PMS2, MLH1S456A/PMS2 variant overexpressing HEK293T cells (shown as cropped blots) and (B) expression levels of five independent experiments were quantified (mean ± S.D.) using Multi Gauge V3.2 program and normalized in relation to the value of MLH1 wt levels. p-MLH1S477 was well detectable in all protein extracts despite in the extract of HEK293T cells overexpressing the non-phosphorylatable MLH1S477A/PMS2 variant. P-values were calculated by paired T-test. **** p < 0.0001; n = 5.
We detected that the phosphorylatability of S87, S446 and S456 did not affect the amount of p-MLH1S477 (Fig. 1A + B). The amount of p-MLH1S477 was comparable in the MLH1S87A/PMS2, the MLH1S446A/PMS2, as well as in the MLH1S456A/PMS2 variant after Calyculin treatment and similar to the amount of p-MLH1S477 of expressed MutLα wt. MLH1S87A/PMS2 showed 79% ± 21%, MLH1S446A/PMS2 showed 96% ± 26% and MLH1S456A/PMS2 showed 96% ± 15% of the p-MLH1S477 amount precipitated from MutLα wt extracts. No p-MLH1S477 could be detected at position S477 using the non-phosphorylatable MLH1S477A /PMS2 variant (4% ± 2%) (Fig. 1B).
Amino acid changes at S87, S446, S456 and S477 do not affect the amount of whole MLH1 expression
To investigate the impact of amino acid changes from serine to alanine at positions S87, S446, S456 and S477 of MLH1 on the amount of protein expression of MutLα, HEK293T cells were transiently cotransfected with pEBG-2 T/MLH1 (or the respective MLH1 variant) and pcDNA3.1 + /PMS2 and the amount of MLH1 and PMS2 for each variant was determined via Western blotting. As shown in Fig. 2 (and Figure S2), MLH1 as well as PMS2 expression of all MutLα variants was slightly increased, but in general very similar compared to the MLH1 and PMS2 expression of MutLα wt and showed no significant differences. In detail, the relative MLH1 expression level of MLH1S87A/PMS2 was 140.5% ± 45.58%, of MLH1S446A/PMS2 it was 136% ± 81.89%, of MLH1S456A/PMS2 it was 187.3% ± 112.8%, of MLH1S477A/PMS2 it was 140.6% ± 91.42%, of MLH1S87A/S477A/PMS2 it was 115.8% ± 86.86%, of MLH1S446A/S477A/PMS2 it was 174.4% ± 91.35% and of MLH1S456A/S477A/PMS2 it was 126.7% ± 82.27%. Thus, the amino acid changes seem to have no influence on the expression level of MLH1 or PMS2.
Figure 2.
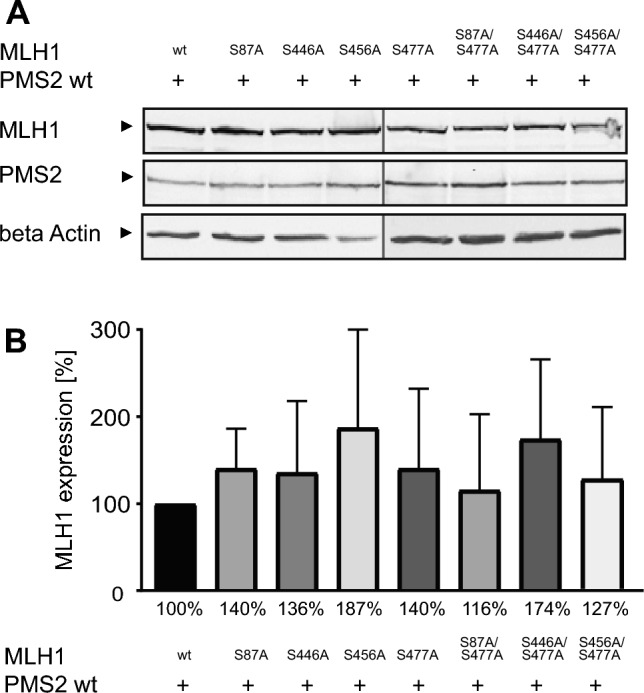
Changes from serine to alanine in phosphorylatable positions of MLH1 have no significant impact on the expression level of MutLα. HEK293T cells were cotransfected with pEBG-2 T/MLH1 or different pEBG-2 T/MLH1 variants and pcDNA3.1 + /PMS2. Protein expression of MLH1 and PMS2 was (A) analyzed via Western blotting (shown as cropped blots) and (B) expression level of MLH1 was quantified using Multi Gauge V3.2 program and normalized in relation to the value of MLH1 wt levels. The expression levels of all MLH1 variants were similar compared to MLH1 wt as well as all corresponding PMS2 levels. The shown percentages are given as rounded numbers in the figure. n = 3.
Amino acid position S87 is of great importance for DNA binding of MutLα
MutLα has been shown to be able to bind to the DNA, which is required, for example, for its endonuclease activity during the MMR3–6. In order to determine the influence of phosphorylation of MLH1 at position S87, S446, S456 and S477 on DNA binding of MutLα an EMSA was performed. For this assay fluorescently labeled double-stranded DNA with a mismatch, recombinant GST tagged MutLα wt and MutLα variants were used. Since the amount of protein bound to the DNA was the same for purified GST-tagged MutLα in comparison to Glutathione-sepharose-bound MutLα (Figure S3A), Glutathione-sepharose-bound protein samples were used for all EMSAs. The different recombinant MutLα variants were subjected to two treatments: Calf intestinal phosphatase (CIP), causing complete dephosphorylation, and treatment with the serine/threonine protein phosphatase inhibitor Calyculin, causing hyperphosphorylation. The untreated recombinant MutLα wt complex was used as a control in all experiments and, after calculating the exact amount of DNA-bound protein via Western blotting in parallel, the DNA-bound amount of MutLα wt was set to 100%, respectively (Fig. 3; Figure S3; Figure S4A-G).
Figure 3.

Phosphorylation of MLH1 inhibits DNA binding of MutLα. The relevance of phosphorylation of MLH1 was determined by using EMSA. The assay was performed using a missense mutation harboring fluorescence labeled DNA substrate and different GST-tagged MutLα variants. The DNA binding of non-phosphorylated (untreated) or phosphorylated (Calyculin treated) MutLα variants was compared to the DNA binding of MutLα wt, respectively. Shown are the results of (A) MutLα wt, (B) MLH1S87A/PMS2, (C) MLH1S446A/PMS2, (D) MLH1S456A/PMS2, (E) MLH1S477A/PMS2, (F) MLH1S87A/S477A/PMS2, (G) MLH1S446A/S477A/PMS2 and (H) MLH1S456A/S477A/PMS2. P-values were calculated by Mann–Whitney-U test. * p < 0.05; ** p < 0.005; n.s. = not significant; n = 3–5.
Hyperphosphorylation of MutLα wt leads to significantly decreased DNA binding while dephosphorylated MutLα binds as similar as the untreated MutLα complex to the DNA (Fig. 3, upper panel, first graph from the left; Figure S3B).
Comparing the DNA binding intensity of MutLα wt with the respective MLH1/PMS2 variants, it becomes clear that phosphorylation of position S87 seems to play the most important role for DNA binding. The Calyculin treated, fully phosphorylated, but at position S87 non-phosphorylatable MLH1S87A/PMS2 variant shows no difference in DNA binding compared to the unphosphorylated MutLα wt (Fig. 3, upper panel, second graph from the left; Figure S4A). Interestingly, the completely dephosphorylated, CIP treated, MLH1S87A/PMS2 variant even binds significantly better to DNA than the MutLα wt.
In the case of the MLH1S87A/S477A/PMS2 variant, only the dephosphorylated form was able to bind to the DNA very well and in a comparable amount as the MutLα wt complex (Fig. 3, lower panel, second graph from the left; Figure S4E). In contrast, Calyculin-treatment, which leads to phosphorylation at position S446 and S456, inhibits binding of MLH1S87A/S477A/PMS2 variant and showed significantly less DNA bound protein (Fig. 3, lower panel, second graph from the left; Figure S4E). However, none of the tested variants bind to DNA with or without phosphorylated serines (Fig. 3, upper panel, third and fourth graph from the left; lower panel, first, third and fourth graph from the left; Figure S4B-D; Figure S4F + G).
Phosphorylation of MLH1 prevents MMR
To investigate the MMR relevance of MLH1 phosphorylation at amino acid positions S87, S446, S456 and S477, an in vitro MMR assay was performed. The analysis was carried out using protein extracts of untreated (Fig. 4A + B) and Calyculin treated (Fig. 4C + D) HEK293T cells which were transiently cotransfected with pEBG-2 T plasmids containing MLH1 wt or the different MLH1-variants and pcDNA3.1 + /PMS2 wt. Protein extract of transiently pEGFP_C1 transfected HEK293T served as a negative control.
Figure 4.
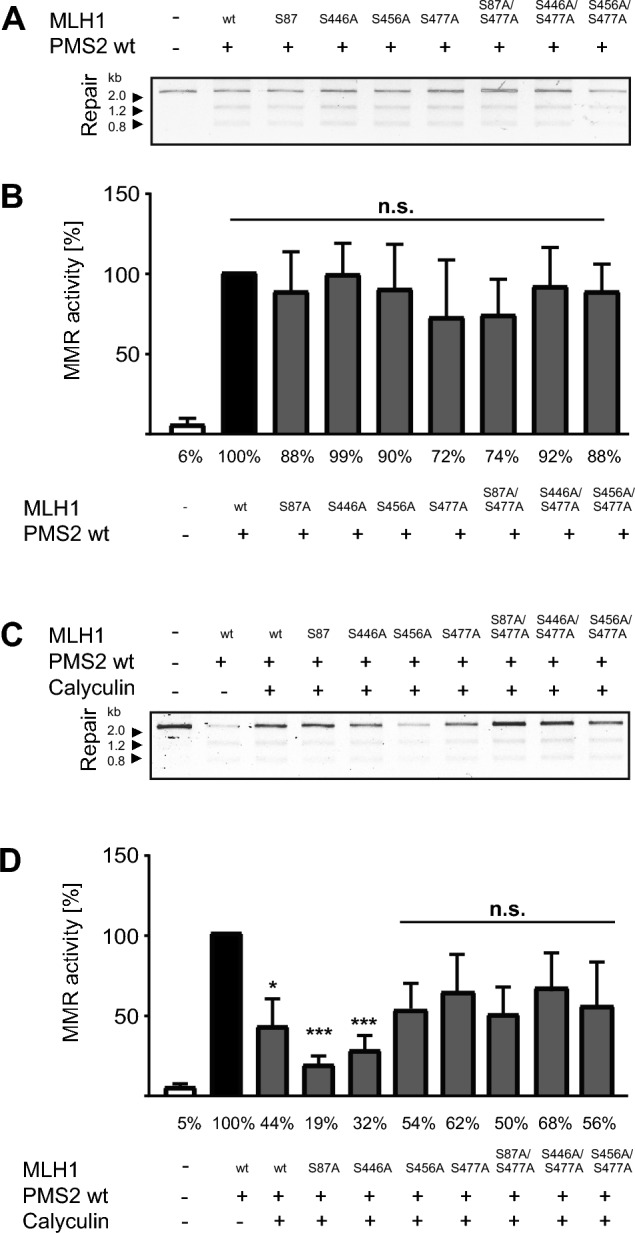
Phosphorylation levels of phosphorylatable amino acids of MLH1 significantly impact the MMR function of MutLα. An in vitro MMR assay was performed to determine the impact of phosphorylation at positions S87, S446, S456 or S477 on the functionality of MutLα. DNA MMR substrate was incubated with differently phosphorylatable MutLα variants and the MMR activity of the (A + B) non-phosphorylated (untreated) and the (C + D) phosphorylated (Calyculin treated) form of these variants was compared to the MMR activity of MutLα wt, repectively. All non-phosphorylated MutLα variants showed MMR activity similar to that of MutLα wt; significant differences were detectable using the phosphorylated forms of these proteins. The shown percentages are given as rounded numbers in the figure. P-values were calculated by unpaired two-tailed t-test, followed by Welch correction and the Mann–Whitney-U test. * p < 0.005; *** p < 0.0001; n.s. = not significant; n = 4–6.
Numerical values were quantified and set in relation to MutLα containing MLH1 wt (positive control) (100 ± 0). The results indicate that all non-phosphorylated MutLα variants were able to repair the DNA substrate in a similar manner as MutLα wt (Fig. 4A + B, Figure S5A). Variant MLH1S87A/PMS2 showed 88.33% ± 24.60% repair activity, MLH1S446A/PMS2 showed 98.98% ± 19.24% repair activity, MLH1S456A/PMS2 showed 89.93% ± 27.64% repair activity, MLH1S477A/PMS2 showed 72.37% ± 35.48% repair activity; MLH1S87A/S477A/PMS2 showed 73.91% ± 22.11% repair activity, MLH1S446A/S477A/PMS2 showed 91.57% ± 24.05% repair activity and MLH1S456A/S477A/PMS2 showed 88.38% ± 17.03% repair activity.
After Calyculin treatment and hyperphosphorylation of the used MutLα variants, a different picture has been drawn. Some phosphorylated protein-complexes showed significantly reduced MMR activity compared to the unphosphorylated MutLα wt while the MMR functionality of other MutLα variants was impaired but still present after Calyculin treatment (Fig. 4C+D, Figure S5B). The reduction of the MMR activity of phosphorylated MutLα wt was significant with 44.27% ± 20.02%, of the phosphorylated MLH1S87A/PMS2 the activity with 19.35% ± 3.91% was the lowest, and of phosphorylated MLH1S446A/PMS2 the MMR activity was also very weak and significantly reduced with 32.10% ± 3.68%. However, for all other variants no significant reduction in MMR activity could be determined. The MMR activity of phosphorylated MLH1S456A/PMS2 was 53.56% ± 16.78%, of phosphorylated MLH1S477A/PMS2 it was 61,98% ± 20.22%, of phosphorylated MLH1S87A/S477A/PMS2 it was 49.38% ± 12.64%, of phosphorylated MLH1S446A/S477A/PMS2 it was 67.82% ± 20.14% and of phosphorylated MLH1S456A/S477A/PMS2 it was 56,46% ± 27.02% in comparison to the untreated MutLα wt control (Fig. 4C + D, Figure S5B).The negative control performed with protein extracts of pEGFP_C1 transfected HEK293T cells demonstrated no MMR activity, with 4.70% ± 2.10% (Fig. 4A + B, Figure S5A) or 5.14% ± 2.80% (Fig. 4C + D, Figure S5B). The supplementation of Calyculin itself, separately added to the assay during incubation with the DNA substrate, was without influence on the MMR efficacy (Figure S6).
Thermostability of MutLα is significantly influenced by the phosphorylation status of MLH1
The influence of phosphorylation of MLH1 at positions S87, S446, S456 and S477 of MLH1 on the general protein stability was determined by nanoDSF using recombinant purified MutLα wt and the different MutLα variants (Fig. 5). As demonstrated in Fig. 5 A + C, all untreated MutLα variants, except those carrying the non-phosphorylatable amino acid S87A (MLH1S87A/PMS2; MLH1S87A/S477A/PMS2), showed very similar thermostability compared to the MutLα wt complex. In contrast, MLH1S87A/PMS2 as well as MLH1S87A/S477A/PMS2 showed impaired thermostability which manifests in a significantly flatter curve (Fig. 5A + C). Looking at the complexes after treatment with Calyculin and therefore hyperphosphorylation, the thermostability of the MutLα variants was very different (Fig. 5B + D). The lowest thermostability (with first derivative of the fluorescence intensity (FI) (Fi350nm/Fi330nm) = peak at 0.0035) was shown by the hyperphosphorylated MutLα wt and the MLH1S477A/PMS2 variant (first derivative (Fi350nm/Fi330nm) = peak at 0.0027). The highest thermostability after treatment with Calyculin was shown by MLH1S456A/PMS2 (first derivative (Fi350nm/Fi330nm) = peak at 0.0064) followed by MLH1S446A/PMS2 (first derivative (Fi350nm/Fi330nm) = peak at 0.0057) and MLH1S87A/PMS2 (first derivative (Fi350nm/Fi330nm) = peak at 0.0053) (Fig. 5B).
Figure 5.

Phosphorylation status of MLH1 influences the thermostability of MutLα. The thermostability of recombinant, unphosphorylated and phosphorylated versions of MutLα wt and different MutLα variants was determined using nano-differential scanning fluorimetry (nanoDSF). Measurements of (A + C) unphosphorylated and (B + D) phosphorylated proteins were performed and data are shown as the first derivative of the fluorescence intensity (Fi) (Fi350nm/Fi330nm).
For the MutLα variants that carried two changes, all variants were found to have higher thermostability than the MutLα wt after treatment with Calyculin (Fig. 5D). The peak of the MutLα double variants were all on very similar levels: MLH1S87A/S477A/PMS2 where the first derivative (Fi350nm/Fi330nm) peak was at 0.0051, MLH1S446A/S477A/PMS2 where the first derivative (Fi350nm/Fi330nm) peak was at 0.0050 and MLH1S456A/S477A/ PMS2 where the first derivative (Fi350nm/Fi330nm) peak was at 0.0059 (Fig. 5D).
The majority of the examined phosphorylation sites are located in the linker region of MLH1
In order to visualize the localization of S87, S446, S456 and S477 in silico, we used Pymol and modelled the biologically active structural form of dimeric MutLα (Fig. 6). While S87 (Fig. 6, red ball) is located in the N-terminal ATPase and MutS interacting domain of MLH1, all other phosphorylatable serines of MLH1 are localized in its flexible linker region (Fig. 6, light blue balls).
Figure 6.

Potential structure of the p-MLH1S87/S446/S456/S477 / PMS2 heterodimer. The biologically active structural form of MLH1 (green) in its heterodimeric form with PMS2 (grey) is shown. The structure is a prediction based on AlphaFold223. S87 is located in the ATPase domain (indicated as red ball) whereas S446, S456 and S477 of MLH1 are located in the flexible linker region and are indicated as light blue balls (shown on the left side). aa: amino acid; NTD: N-terminal domain; CTD: C-terminal domain.
Discussion
The human MMR is a highly complex mechanism for the maintenance of genetic information. Its functionality depends on diverse factors e.g. germline mutations24, promoter hypermethylation25,26 and posttranslational modification14,27–29. Recent mass spectrometry studies of our group showed that MLH1 is phosphorylatable at several sites, albeit with varying intensity (see supplementary data of14). In the present study, we investigated the effect of phosphorylation of MLH1 at amino acid position S87, S446, S456, and S477 in different recombinant MutLα variants and analyzed their protein expression and stability, DNA binding ability and MMR functionality in vitro. We were able to demonstrate for the first time that the ability of MutLα to bind to the DNA seems to be orchestrated via a phosphorylation status-dependent fine tuning and partial interplay between the N-terminal ATPase domain as well as serines located in the linker region of MLH1.
Our major finding is that S87, which is located in the N-terminal ATPase site of MLH1, is most essential for DNA binding of MutLα and that phosphorylation of S87 destroys this ability. The MLH1S87A/PMS2 variant, which is non-phosphorylatable at position S87, showed significantly enhanced binding to the DNA compared to MutLα wt. Interestingly, its DNA binding capacity was not influenced by the phosphorylation of all serine positions in the linker region. In contrast, the phosphorylated form of MLH1S87A/PMS2 showed the worst and most significantly reduced MMR activity compared to all other variants. Furthermore, we were able to detect that phosphorylation of S446 and S456 in the linker region of MutLα variants was enough to avoid the DNA binding as long as S87 as well as S477 were not phosphorylatable.
A general relevance of DNA binding of MutL for the MMR process has been demonstrated by Dortschmann et al. by using yeast Mlh1-Pms1 heterodimer30. Our observation that the linker region of MLH1 also seems to be important for DNA binding as well fits with Plys et al. who showed that the unstructured linker domains of MLH proteins of yeast provided distinct interactions with DNA during MMR31. The observation that DNA binding of MutLα seems to depend on amino acids of the N-terminus as well as partially on the linker region, is closely in line with previously published data for MutLγ, a MutL heterodimer which has been implicated in the processing of meiotic recombination intermediates into crossovers32. In their study, Bouuaert and Keeney mapped DNA contacts of MutLγ and showed that the DNA binding sites of MutLγ for single stranded DNA were located in the N-terminal ATPase domain as well as in the linker region32. Interestingly, the authors additionally demonstrated that most contacts with Holiday junction substrates were solely detectable in the linker region. Since DNA binding in our study was analyzed by using only one DNA substrate, we cannot give any information regarding the DNA binding ability to different substrates. However, this would be worthwhile to investigate more detailed in future studies and might give an explanation for the observation that phosphorylation of serines in the linker region differentially affects the DNA binding of MutLα variants in correlation to the phosphorylation status of S87. Another question is why single amino acid changes in the linker region show this very strong effects on DNA binding even in the unphosphorylated status of most used MutLα variants. We can only speculate about this and assume that the flexibility of the linker region of MutLα allows interactions within the own protein and that changes of single amino acids in the linker can significantly affect protein structure and function, which has been demonstrated for other proteins before33. Of course, we have to emphasize here that the DNA binding assay used in the current study was intended to focus solely on MutLα, therefore, no other proteins were included in this assay. The DNA binding behavior of MLH1 in combination with components of the MMR cannot be answered in the present case and has to be analyzed in detail by future experiments.
Concerning our detection of a phosphorylation-dependent regulation of the N-terminus of MLH1 in which the ATPase is located, one has to point out that the regulation of ATPases by phosphorylation is a common mechanism. A cAMP activated protein kinase phosphorylation site is e.g. responsible for regulating the pumping activity of Na+, K+- and H+, K+-ATPases, either by changing the cellular distribution of the ATPases or by directly altering their kinetic properties34,35. Positions of amino acids which are directly responsible for ATP binding in the ATPase region of MutLα have been already described for yeast36. Based on crystal structures of the N-terminal domain of yeast MutL, two conserved asparagine residues (position 35 in Mlh1 and position 65 in Pms1) have been shown as essential for ATP binding and two Glutamates (position 31 in Mlh1 and position 61 in Pms1) as important for ATP hydrolysis36. Since human and yeast MutLα are predominantly homologous, position S87 of human MLH1 is highly unlikely to be directly involved in ATP binding and phosphorylation at this position rather seems to have an indirect effect.
Considering the ATPase activity of MMR proteins, it is still not finally clarified in which step of the MMR process the respective ATPases are actually involved. It has been detected that, although the interaction of MutLα and MutSα requires ATP, the ATPase activity of MutLα is not essential for the formation of ternary complexes with MutSα, but required for downstream processes37,38. ATP hydrolysis by MutS and MutL is involved in the mismatch-dependent activation of MutL endonuclease activity39 and enables MutL to act as a switch to coordinate MMR40. Furthermore, it has been suggested that the ATPase activity of MutLα is regulated by conformational changes from an ADP bound open to an ATP bound condensed form, thus modulated by adenine nucleotides in the ATPase domain41. These changes have been identified to mediate the interaction of MutLα with other proteins in the MMR pathway and are of importance for the recognition of DNA mismatches by MutSα. As a consequence, MutLα is able to coordinate downstream events that lead to repair41 and the conformational status of MutLα also impacts its interaction with PCNA42 which is assumed to be necessary for strand discrimination43. Therefore, we hypothesize that the phosphorylation of MLH1 at position S87 might induce conformational changes at the catalytic interface of the N-terminus in the ATP binding region in a way that its ATP binding and ATPase function is suppressed, which might prevent the interaction with MutSα as well as the DNA binding and finally the MMR mechanism.
Our differential results concerning phosphorylation-dependent DNA binding and MMR functionality of MutLα lead us to the hypothesis that binding of MutLα to the DNA is regulated most dominantly by phosphorylation of S87 in the N-terminal ATPase domain while MMR activity is rather regulated by phosphorylation of positions in the linker region of MLH1. While the prevention of phosphorylation at position S87 increased DNA binding, which also remained in the case of phosphorylation of amino acids in the linker region, the phosphorylated form of this MLH1S87A/PMS2 variant showed the lowest MMR activity. In contrast, incomplete phosphorylatability of positions in the linker domain, especially in the presence of S477A, allowed MMR activity which is in line with our recently published data14 and underlines the importance of amino acid S477 for the regulation of the MMR. To our surprise, hyperphosphorylated variant MLH1S446A/PMS2 showed a significant reduction of MMR activity. The hypothesis that even minor changes in the linker region could affect protein structure and function, as mentioned above, might also explain this MMR result. However, this needs to be clarified by further experiments.
The thermal stability data of the tested MutLα variants underline our observations of a phosphorylation-dependent regulation and showed that, overall, MutLα is less stable in the phosphorylated state than in the non-phosphorylated state. The stability of the complex is inversely correlated to the number of phosphorylated positions which seems to be in accordance with the loss of DNA binding and MMR function that we identified. Although these differences do not manifest themselves until a non-physiological temperature range is reached, they are nevertheless consistent with Becher et al. who analyzed protein thermal stability variation during the cell cycle and found that protein thermal stability serves as a proxy for enzyme activity, DNA binding, and complex formation44. Discussing a phosphorylation-dependent regulation of MutLα stability, it also seems appropriate to mention, that phosphorylation can promote or inhibit ubiquitination, which in turn can lead to proteasomal protein degradation among other consequences45,46. Thus, it is also conceivable that complete phosphorylation of MutLα is in the end required to degrade the protein complex and remove it from the MMR process.
Finally, it remains to be elucidated why phosphorylation of MLH1 was predominantly detected at position S477 in the mass spectrometry analysis, while all other serines were detected to a significantly lower proportion (S87 (6%), S446 (25%), S456 (5%), S477 (61%))14. We could imagine that other posttranslational modifications, e.g. ubiquitination45,46, the order of addition or removal of post translational modifications in the lifespan of the protein47, or the abundance of special kinases and phosphatases48,49 might significantly impact the phosphorylatability of the investigated positions.
Altogether, one might speculate that the complete detachment of the MutLα complex from the DNA is a process that occurs rather infrequently, whereas the direct acting in the context of MMR is more precisely regulated. Descriptions of the clamp of MutS, which actually slides along the DNA during the entire replication process and only locks at a certain position when an error occurs, would fit this assumption50. However, this hypothesis has to be investigated in more detail in future.
Conclusions
In summary, we hypothesize a phosphorylation-dependent fine tuning of the MutLα complex during the MMR process. While the phosphorylation of serines located in the linker region seems to be responsible for interrupting the ability of MutLα to participate in the MMR process, the phosphorylation of S87, located in the N-terminal ATPase domain, might finally cause the dissociation of MutLα from the MMR complex (Fig. 7).
Figure 7.

Model of phosphorylation-dependent binding, activity and dissociation of MutLα during the MMR process. 1. Initiation of the MMR after binding of MutSα. 2. Starting process and performance of the MMR after MutLα binding and activity. 3. Stop of the MMR process after phosphorylation of MLH1 at positions S446, S456 and S477. 4. Separation of MutLα from the DNA after phosphorylation of MLH1 at positon S87.
Supplementary Information
Acknowledgements
The authors would like to thank Nazzareno Dominelli from the Institute of Molecular Physiology of the Gutenberg University Mainz for his assistance with the nano differential scanning fluorimetry measurements and Sarah Overby for proofreading and constructive criticism of the manuscript. This manuscript is part of the Ph.D. thesis of May-Britt Firnau.
Author contributions
M.-B.F. performed the experiments, generated the figures and co-wrote the manuscript. A.B. conceptualized the idea, supervised the project and wrote the paper. G.P. performed the Pymol analysis and reviewed the manuscript. S.Z. edited the publication. All authors have read and agreed to the published version of the manuscript.
Funding
Open Access funding enabled and organized by Projekt DEAL. This work was supported by Wilhelm Sander Foundation, Grant number: 2015.161.2, and Institutional funds of the University Clinic Frankfurt.
Data availability
All datasets used and/or analysed during the current study are shown in the supplementary data. For any other additional data desired on reasonable request, please contact the corresponding author.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-023-39750-x.
References
- 1.McCulloch SD, Gu L, Li GM. Nick-dependent and -independent processing of large DNA loops in human cells. J. Biol. Chem. 2003;278:50803–50809. doi: 10.1074/jbc.M309025200M309025200[pii]. [DOI] [PubMed] [Google Scholar]
- 2.Kunkel TA, Erie DA. DNA mismatch repair. Annu. Rev. Biochem. 2005;74:681–710. doi: 10.1146/annurev.biochem.74.082803.133243. [DOI] [PubMed] [Google Scholar]
- 3.Kadyrov FA, Dzantiev L, Constantin N, Modrich P. Endonucleolytic function of MutLalpha in human mismatch repair. Cell. 2006;126:297–308. doi: 10.1016/j.cell.2006.05.039. [DOI] [PubMed] [Google Scholar]
- 4.Kadyrov FA, et al. Saccharomyces cerevisiae MutLalpha is a mismatch repair endonuclease. J. Biol. Chem. 2007;282:37181–37190. doi: 10.1074/jbc.M707617200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kadyrova LY, Kadyrov FA. Endonuclease activities of MutLalpha and its homologs in DNA mismatch repair. DNA Repair (Amst) 2016;38:42–49. doi: 10.1016/j.dnarep.2015.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van Oers JM, et al. PMS2 endonuclease activity has distinct biological functions and is essential for genome maintenance. Proc. Natl. Acad. Sci. USA. 2010;107:13384–13389. doi: 10.1073/pnas.1008589107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Modrich P. Mechanisms and biological effects of mismatch repair. Annu. Rev. Genet. 1991;25:229–253. doi: 10.1146/annurev.ge.25.120191.001305. [DOI] [PubMed] [Google Scholar]
- 8.Poynter JN, et al. Molecular characterization of MSI-H colorectal cancer by MLHI promoter methylation, immunohistochemistry, and mismatch repair germline mutation screening. Cancer Epidemiol. Biomark. Prev. 2008;17:3208–3215. doi: 10.1158/1055-9965.EPI-08-0512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hause RJ, Pritchard CC, Shendure J, Salipante SJ. Corrigendum: Classification and characterization of microsatellite instability across 18 cancer types. Nat. Med. 2018;24:525. doi: 10.1038/nm0418-525a. [DOI] [PubMed] [Google Scholar]
- 10.Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peltomaki P, Vasen H. Mutations associated with HNPCC predisposition – Update of ICG-HNPCC/INSiGHT mutation database. Dis. Markers. 2004;20:269–276. doi: 10.1155/2004/305058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thompson BA, et al. Application of a 5-tiered scheme for standardized classification of 2,360 unique mismatch repair gene variants in the InSiGHT locus-specific database. Nat. Genet. 2014;46:107–115. doi: 10.1038/ng.2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ponti G, Manfredini M, Tomasi A, Pellacani G. Muir-Torre Syndrome and founder mismatch repair gene mutations: A long gone historical genetic challenge. Gene. 2016;589:127–132. doi: 10.1016/j.gene.2015.06.078. [DOI] [PubMed] [Google Scholar]
- 14.Wessbecher IM, et al. DNA mismatch repair activity of MutLalpha is regulated by CK2-dependent phosphorylation of MLH1 (S477) Mol. Carcinog. 2018;57:1723–1734. doi: 10.1002/mc.22892. [DOI] [PubMed] [Google Scholar]
- 15.Ulreich K, et al. High expression of casein kinase 2 Alpha Is responsible for enhanced phosphorylation of DNA mismatch repair protein MLH1 and increased tumor mutation rates in colorectal cancer. Cancers Basel. 2022 doi: 10.3390/cancers14061553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Trojan J, et al. Functional analysis of hMLH1 variants and HNPCC-related mutations using a human expression system. Gastroenterology. 2002;122:211–219. doi: 10.1053/gast.2002.30296. [DOI] [PubMed] [Google Scholar]
- 17.Brieger A, et al. Characterization of the nuclear import of human MutLalpha. Mol. Carcinog. 2005;43:51–58. doi: 10.1002/mc.20081. [DOI] [PubMed] [Google Scholar]
- 18.Tanaka M, Gupta R, Mayer BJ. Differential inhibition of signaling pathways by dominant-negative SH2/SH3 adapter proteins. Mol. Cell Biol. 1995;15:6829–6837. doi: 10.1128/MCB.15.12.6829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Plotz G, et al. Mutations in the MutSalpha interaction interface of MLH1 can abolish DNA mismatch repair. Nucleic Acids Res. 2006;34:6574–6586. doi: 10.1093/nar/gkl944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang H, Hays JB. Mismatch repair in human nuclear extracts. Quantitative analyses of excision of nicked circular mismatched DNA substrates, constructed by a new technique employing synthetic oligonucleotides. J. Biol. Chem. 2002;277:26136–26142. doi: 10.1074/jbc.M200357200. [DOI] [PubMed] [Google Scholar]
- 21.Hinrichsen I, et al. Expression defect size among unclassified MLH1 variants determines pathogenicity in Lynch syndrome diagnosis. Clin. Cancer Res. 2013 doi: 10.1158/1078-0432.CCR-12-3299. [DOI] [PubMed] [Google Scholar]
- 22.Dominelli N, Platz F, Heermann R. The insect pathogen photorhabdus luminescens protects plants from phytopathogenic fusarium graminearum via chitin degradation. Appl. Environ. Microbiol. 2022;88:e0064522. doi: 10.1128/aem.00645-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jumper J, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–589. doi: 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Olkinuora A, et al. Molecular basis of mismatch repair protein deficiency in tumors from lynch suspected cases with negative germline test results. Cancers (Basel) 2020 doi: 10.3390/cancers12071853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Loukovaara M, Pasanen A, Butzow R. Mismatch repair protein and MLH1 methylation status as predictors of response to adjuvant therapy in endometrial cancer. Cancer Med. 2021;10:1034–1042. doi: 10.1002/cam4.3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaneko E, et al. MLH1 promoter hypermethylation predicts poorer prognosis in mismatch repair deficiency endometrial carcinomas. J. Gynecol. Oncol. 2021;32:e79. doi: 10.3802/jgo.2021.32.e79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Edelbrock MA, Kaliyaperumal S, Williams KJ. Structural, molecular and cellular functions of MSH2 and MSH6 during DNA mismatch repair, damage signaling and other noncanonical activities. Mutat Res. 2013;743–744:53–66. doi: 10.1016/j.mrfmmm.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wessbecher IM, Brieger A. Phosphorylation meets DNA mismatch repair. DNA Repair (Amst) 2018;72:107–114. doi: 10.1016/j.dnarep.2018.09.001. [DOI] [PubMed] [Google Scholar]
- 29.Li F, Ortega J, Gu L, Li GM. Regulation of mismatch repair by histone code and posttranslational modifications in eukaryotic cells. DNA Repair (Amst) 2016;38:68–74. doi: 10.1016/j.dnarep.2015.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Drotschmann K, et al. DNA binding properties of the yeast Msh2-Msh6 and Mlh1-Pms1 heterodimers. Biol. Chem. 2002;383:969–975. doi: 10.1515/BC.2002.103. [DOI] [PubMed] [Google Scholar]
- 31.Plys AJ, Rogacheva MV, Greene EC, Alani E. The unstructured linker arms of Mlh1-Pms1 are important for interactions with DNA during mismatch repair. J. Mol. Biol. 2012;422:192–203. doi: 10.1016/j.jmb.2012.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Claeys Bouuaert C, Keeney S. Distinct DNA-binding surfaces in the ATPase and linker domains of MutLgamma determine its substrate specificities and exert separable functions in meiotic recombination and mismatch repair. PLoS Genet. 2017;13:e1006722. doi: 10.1371/journal.pgen.1006722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fiorani P, et al. Single mutation in the linker domain confers protein flexibility and camptothecin resistance to human topoisomerase I. J. Biol. Chem. 2003;278:43268–43275. doi: 10.1074/jbc.M303899200. [DOI] [PubMed] [Google Scholar]
- 34.Cornelius F, Mahmmoud YA. Direct activation of gastric H, K-ATPase by N-terminal protein kinase C phosphorylation. Comparison of the acute regulation mechanisms of H, K-ATPase and Na K-ATPase. Biophys. J. 2003;84:1690–1700. doi: 10.1016/S0006-3495(03)74977-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fisone G, et al. Identification of the phosphorylation site for cAMP-dependent protein kinase on Na+, K(+)-ATPase and effects of site-directed mutagenesis. J. Biol. Chem. 1994;269:9368–9373. doi: 10.1016/S0021-9258(17)37117-X. [DOI] [PubMed] [Google Scholar]
- 36.Hall MC, Shcherbakova PV, Kunkel TA. Differential ATP binding and intrinsic ATP hydrolysis by amino-terminal domains of the yeast Mlh1 and Pms1 proteins. J. Biol. Chem. 2002;277:3673–3679. doi: 10.1074/jbc.M106120200. [DOI] [PubMed] [Google Scholar]
- 37.Plotz G, Raedle J, Brieger A, Trojan J, Zeuzem S. N-terminus of hMLH1 confers interaction of hMutLalpha and hMutLbeta with hMutSalpha. Nucleic Acids Res. 2003;31:3217–3226. doi: 10.1093/nar/gkg420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Raschle M, Dufner P, Marra G, Jiricny J. Mutations within the hMLH1 and hPMS2 subunits of the human MutLalpha mismatch repair factor affect its ATPase activity, but not its ability to interact with hMutSalpha. J. Biol. Chem. 2002;277:21810–21820. doi: 10.1074/jbc.M108787200. [DOI] [PubMed] [Google Scholar]
- 39.Shimada A, et al. MutS stimulates the endonuclease activity of MutL in an ATP-hydrolysis-dependent manner. FEBS J. 2013;280:3467–3479. doi: 10.1111/febs.12344. [DOI] [PubMed] [Google Scholar]
- 40.Ban C, Junop M, Yang W. Transformation of MutL by ATP binding and hydrolysis: A switch in DNA mismatch repair. Cell. 1999;97:85–97. doi: 10.1016/S0092-8674(00)80717-5. [DOI] [PubMed] [Google Scholar]
- 41.Sacho EJ, Kadyrov FA, Modrich P, Kunkel TA, Erie DA. Direct visualization of asymmetric adenine-nucleotide-induced conformational changes in MutL alpha. Mol. Cell. 2008;29:112–121. doi: 10.1016/j.molcel.2007.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Genschel J, et al. Interaction of proliferating cell nuclear antigen with PMS2 is required for MutLalpha activation and function in mismatch repair. Proc. Natl. Acad. Sci. USA. 2017;114:4930–4935. doi: 10.1073/pnas.1702561114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pluciennik A, et al. PCNA function in the activation and strand direction of MutLalpha endonuclease in mismatch repair. Proc. Natl. Acad. Sci. USA. 2010;107:16066–16071. doi: 10.1073/pnas.1010662107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Becher I, et al. Pervasive protein thermal stability variation during the cell cycle. Cell. 2018;173:1495–1507. doi: 10.1016/j.cell.2018.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hunter T. The age of crosstalk: Phosphorylation, ubiquitination, and beyond. Mol. Cell. 2007;28:730–738. doi: 10.1016/j.molcel.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 46.Zhao X, Lai G, Tu J, Liu S, Zhao Y. Crosstalk between phosphorylation and ubiquitination is involved in high salt-induced WNK4 expression. Exp. Ther. Med. 2021;21:133. doi: 10.3892/etm.2020.9565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hammaren HM, Geissen EM, Potel CM, Beck M, Savitski MM. Protein-peptide turnover profiling reveals the order of PTM addition and removal during protein maturation. Nat. Commun. 2022;13:7431. doi: 10.1038/s41467-022-35054-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smoly I, Shemesh N, Ziv-Ukelson M, Ben-Zvi A, Yeger-Lotem E. An asymmetrically balanced organization of kinases versus phosphatases across eukaryotes determines their distinct impacts. PLoS Comput Biol. 2017;13:e1005221. doi: 10.1371/journal.pcbi.1005221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Levy ED, Michnick SW, Landry CR. Protein abundance is key to distinguish promiscuous from functional phosphorylation based on evolutionary information. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2012;367:2594–2606. doi: 10.1098/rstb.2012.0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Groothuizen FS, Sixma TK. The conserved molecular machinery in DNA mismatch repair enzyme structures. DNA Repair (Amst) 2015;38:14–23. doi: 10.1016/j.dnarep.2015.11.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All datasets used and/or analysed during the current study are shown in the supplementary data. For any other additional data desired on reasonable request, please contact the corresponding author.