

Abstract
Diabetes mellitus (DM) is a risk factor for pancreatic ductal adenocarcinoma (PDAC) that promotes the promoter methylation of CDH1. It is still unclear whether DM can exert other epigenetic effects, such as altering microRNA (miR) expression, in PDAC. The expression of miR‐100‐5p is known to be changed in DM patients and can suppress the expression of E‐cadherin. In this study, the correlation between DM status and dual epigenetic changes was evaluated in PDAC specimens from patients who underwent radical surgical resection. A total of 132 consecutive patients with PDAC were clinicopathologically evaluated. E‐cadherin and nuclear β‐catenin expression was measured using immunohistochemistry. DNA and miRs were extracted from the main tumor site on formalin‐fixed paraffin‐embedded tissue sections. TaqMan miR assays were applied to assess miR‐100‐5p expression. Bisulfite modification was conducted on the extracted DNA, which was then subjected to methylation‐specific polymerase chain reaction. Immunohistochemistry revealed that decreased E‐cadherin expression and increased nuclear β‐catenin expression were significantly associated with DM and poor tumor cell differentiation. The presence of long‐duration DM (≥3 years) was a significant factor contributing to CDH1 promoter methylation (p < 0.01), while miR‐100‐5p expression was proportionally correlated with the preoperative HbA1c level (R = 0.34, p < 0.01), but not the duration of DM. The subjects with high miR‐100‐5p expression and CDH1 promoter methylation showed the highest level of vessel invasion and prevalence of tumor size ≥30 mm. PDAC subjects with dual epigenetic changes showed poorer overall survival (OS) than those with a single epigenetic change. miR‐100‐5p expression ≥4.13 and CDH1 promoter methylation independently predicted poor OS and disease‐free survival (DFS) in the multivariate analysis. OS and DFS worsened in DM subjects with both HbA1c ≥ 6.5% and DM duration ≥3 years. Thus, DM is associated with two modes of epigenetic change by independent mechanisms and worsens prognosis.
Keywords: diabetes mellitus, pancreatic ductal adenocarcinoma, promoter methylation, microRNA, E‐cadherin
Introduction
Pancreatic ductal adenocarcinoma (PDAC) remains an intractable cancer with a poor prognosis. The 5‐year survival rate of PDAC is low at approximately 10% [1]. Systemic treatment in addition to local control is clearly essential to curing PDAC, while surgical resection of PDAC is still a predominant treatment option. Genetic studies have identified mutations of four key genes, including KRAS, TP53, SMAD4, and CDKN2A, as responsible for the development of PDAC [2, 3, 4, 5, 6]. However, targeting these genetic variations has yet to produce a useful therapeutic strategy to combat PDAC. Epigenetic changes are also involved in the tumorigenesis of PDAC [7]. Interventions related to epigenetic changes are expected to lead to the development of new therapeutic strategies for PDAC.
Diabetes mellitus (DM) has long been known to be a risk factor for the development of PDAC [8, 9, 10]. DM elicits epigenetic changes, such as promoter methylation or microRNA (miR) expression changes, related to epithelial–mesenchymal transition (EMT) [11, 12, 13, 14]. Loss of E‐cadherin leads to nuclear translocation of β‐catenin, poor prognosis, and the development of resistance to chemotherapy in PDAC patients via induction of EMT [15, 16, 17, 18]. Regarding PDAC associated with DM, our previous studies reported that a long duration of DM (long‐DM; 3 years and more) exacerbates the frequency of CDH1 promoter methylation, stellate cell activation, and EMT [10, 19], which worsens the prognosis and enhances the malignant behavior of PDAC. Nevertheless, it has yet to be clarified how DM is implicated in the comprehensive epigenetic changes, including miR expression and promoter methylation of CDH1 in PDAC.
miRs are small, noncoding endogenous RNAs that mainly function as negative regulators of gene expression. miR‐100, also referred to as miR‐100‐5p, is upregulated in PDAC tissue and promotes the EMT of PDAC cells, resulting in disease progression [20, 21, 22, 23, 24]. Moreover, in diabetic subjects, the expression of miR‐100 in blood exosomes of pre‐type 2 DM subjects was increased compared to that in those of nondiabetic subjects [25], while the expression in T cells and serum of type 1 DM subjects and visceral fatty tissue in type 2 DM was decreased [26, 27, 28]. Therefore, it is still unknown how miR‐100 is involved in the progression of PDAC associated with DM.
Herein, we focused on the involvement and association of epigenetic changes in E‐cadherin, including promoter methylation and miR‐100‐5p expression in PDAC complicated with DM. Our study also provides novel therapeutic options for PDAC complicated with DM.
Materials and methods
Case selection
A total of 132 consecutive subjects who underwent surgical resection for PDAC were evaluated between 2014 and 2019 in Hirosaki University Hospital, and patients with unresectable disease and distant metastasis at diagnosis were excluded. Clinicopathological information was obtained from the medical records. The HbA1c value at a single time point (within 1 month before surgery) was used for clinicopathological evaluation, because it was proportionally correlated with average HbA1c values at multiple points (3–12 months before surgery; supplementary material, Figure S1). DM was confirmed from the medical records and was diagnosed based on the criteria proposed by the Japan Diabetes Society [29]. According to our previous reports, long‐DM was defined as DM lasting 3 years or more, and DM lasting less than 3 years was defined as short‐duration DM (short‐DM) [10, 19]. This study was approved by the ethical committee of Hirosaki University Graduate School of Medicine (approval number: 2020‐143).
Histopathological assessment
Screening of pathological findings was performed with H&E sections in each subject. The pathological diagnosis of PDAC was re‐evaluated according to the 2019 WHO classification of tumors of the digestive system and graded based on the Union for International Cancer Control tumor‐node‐metastasis classification of malignant tumors (eighth edition) by two pathologists (HM and KK) [30]. Histologic grade was divided into three categories of well‐differentiated carcinoma (well), moderately differentiated adenocarcinoma (mod), and poorly differentiated adenocarcinoma (poor) based on the degrees of tubular formation, mucin production, and mitoses. The highest grade in the sections represented the histological grade of the individuals regardless of the proportion. Venous invasion was assessed on tumor sections stained with Elastica‐van Gieson. Lymphatic invasion was evaluated on immunostained sections for podoplanin (clone D2‐40, 1:4, Nichirei Bioscience Inc., Tokyo, Japan). The degree of invasion into venules and lymph vessels was graded as 0 (none), 1 (1–3 sites), 2 (4–6 sites), and 3 (>6 sites) within 10 high‐power fields.
Immunohistochemical analysis
For immunohistochemistry, the standard streptavidin–biotin technique was applied to the sections using the Benchmark Ultra Automated Slide Preparation system (Ventana Medical Systems, Inc., Tucson, AZ, USA). Antibodies against E‐cadherin (clone NCH‐38, 1:100 dilution, Agilent Technologies, Santa Clara, CA, USA) and β‐catenin (clone 17C2, 1:100 dilution, Leica Biosystems, Deer Park, IL, USA) were used. Negative control stains were performed by omitting the primary antibodies or substituting nonimmune rabbit or swine sera. Normal pancreatic tissue in each section was used as a positive control. E‐cadherin expression patterns were classified into three categories according to the definition by Saito et al: loss of expression, <5% of the tumor cells were stained; reduced expression, 5–49% of the tumor cells were stained; and preserved expression, 50% or more tumor cells were stained [10]. Cases showing reduced or lost E‐cadherin expression were defined as ‘low E‐cadherin expression’ cases. For β‐catenin expression, a total of 300 cancer cells were counted in each section. The number of carcinoma cells showing nuclear staining for β‐catenin was assessed in relation to the total number of carcinoma cells and expressed as a percentage regardless of the staining intensity (referred to as the β‐catenin index) [31].
Evaluation of CDH1 promoter methylation by methylation‐specific polymerase chain reaction
Methylation‐specific PCR (MS‐PCR) was carried out according to a previous protocol [10, 32]. Tissue samples included in this study had >60% viable cells and <20% necrosis. Tumor areas without hemorrhage or severe inflammation and areas adjacent to the tumor (6 cm2 on average) were selected for MS‐PCR evaluation. DNA was extracted from formalin‐fixed paraffin‐embedded (FFPE) tissue sections (10 μm) following the protocol of the DNA extraction kit for FFPE (Qiagen K.K., Tokyo, Japan). Subsequently, bisulfite modification was conducted on the extracted samples with a commercially available kit (EpiTect Fast Bisulfite Conversion Kits, Qiagen K.K.), and they were then subjected to MS‐PCR with identical primer sequences to Herman et al using a Taq DNA polymerase designed for bisulfite PCR (EpiTaq™ HS, TAKARA BIO INC., Shiga, Japan) [33]. The amplicons were analyzed by electrophoresis on a 3% agarose gel. A positive methylated band indicated high rates of methylation of the CpG region.
Analysis of miR‐100‐5p expression by real‐time PCR
FFPE tumor tissue samples with 40–60% cancer cells were eligible for miR analysis. Total RNA enriched in the miR fraction was purified by using the miRNeasy FFPE kit isolation system following the manufacturer's protocols (Qiagen K.K.). The RNA samples with OD260/OD280 ratios ranging between 1.8 and 2.0 were considered to be of good quality. Reverse transcription and quantitative PCR for miR‐100‐5p and the endogenous control RNU6B were conducted by using TaqMan MicroRNA Assays (Thermo Fisher Scientific, Waltham, MA, USA). Real‐time PCR measurements with specific TaqMan primers for each miR were performed in duplicate on the ABI PRISM 7000 system (Applied Biosystems, Foster City, CA, USA), determining a mean Ct value for each sample. The relative expression of miR‐100‐5p was calculated using the comparative Ct method [34, 35].
Statistical analysis
All statistical analyses were conducted using the free statistics application EZR (ver. 1.55, https://www.jichi.ac.jp/saitama-sct/SaitamaHP.files/statmed.html). Disease‐free survival (DFS) was defined as the time elapsed between surgical resection and tumor recurrence. Overall survival (OS) was calculated as the time between surgery and death from all causes. Continuous variables were compared with Student's t test or the Mann–Whitney U test. Categorical variables were compared by chi‐square analysis or the Mann–Whitney U test, where appropriate. Comparisons of average values between two groups were analyzed by the nonparametric Mann–Whitney U test. For multiple comparisons, the Z test with Bonferroni adjustment was used. Survival curves were calculated with Kaplan–Meier analysis, and p values were determined by the log‐rank test for censored survival data. Multivariate survival analysis was performed by the Cox proportional hazard model. All tests were two‐tailed, and a p value of <0.05 was considered statistically significant.
Results
Clinical and pathological characteristics
There were no significant differences in sex, age, smoker proportion, tumor size, T stage, N stage, histologic grade, lymph vessel and venous invasion (ly and v factors), nerve invasion (ne factor), INF type, curative resection, neoadjuvant chemotherapy ratio, postadjuvant chemotherapy ratio, resectability or CA19‐9 level between the control and DM groups (supplementary material, Table S1). Body mass index (BMI) was higher in DM patients than in non‐DM patients (23.5 versus 21.8, p < 0.01). In terms of the location of the tumor, the pancreatic head was more common in control patients (75%) than in DM patients (54%; p < 0.01). Pancreatico‐duodenectomy was performed more often in control patients (72%) than in DM patients (51%; p < 0.05).
E‐cadherin protein expression and nuclear translocation of β‐catenin in PDAC were decreased in patients who also had DM
In H&E sections of nonneoplastic areas in each group, ducts were composed of typical monolayered epithelial cells (Figure 1A). In the adjacent tumor area, the invasive growth of carcinoma cells with irregular ductal structures mixed with dense fibrous stroma was observed in all PDAC lesions (Figure 1B ). Carcinoma cells exhibiting diffuse or solid growth were diagnosed as poorly differentiated carcinoma (Figure 1C). Immunohistochemical analysis revealed that E‐cadherin was expressed on the cell membrane of nonneoplastic ductal cells, acinar cells, and tumor cells (Figure 1D,E). E‐cadherin expression was low around the cell membrane in poorly differentiated PDAC samples (Figure 1F). β‐Catenin was localized in the cell membrane of non‐DM and short‐DM subjects, while nuclear and cytosolic expression of β‐catenin was more evident in long‐DM subjects (Figure 1G–I). Semiquantitative evaluation of cancerous tissues revealed that the prevalence of low E‐cadherin expression was significantly increased in poorly differentiated PDAC (versus moderately differentiated to well‐differentiated PDAC; p < 0.05; Figure 1J). Low E‐cadherin was significantly more common in the DM group than in the non‐DM group (57% versus 33%, p < 0.01; Figure 1K). Low E‐cadherin expression was significantly more common in subjects with high levels of HbA1c (≥6.5%) and long‐DM (p < 0.05; Figure 1L,M ). The β‐catenin index was significantly increased in poorly differentiated PDAC (versus moderately differentiated to well differentiated PDAC; p < 0.01; Figure 1N), the DM group (versus the non‐DM group; p < 0.01; Figure 1O), and patients with high levels of HbA1c and long‐DM (p < 0.01; Figure 1P,Q ).
Figure 1.
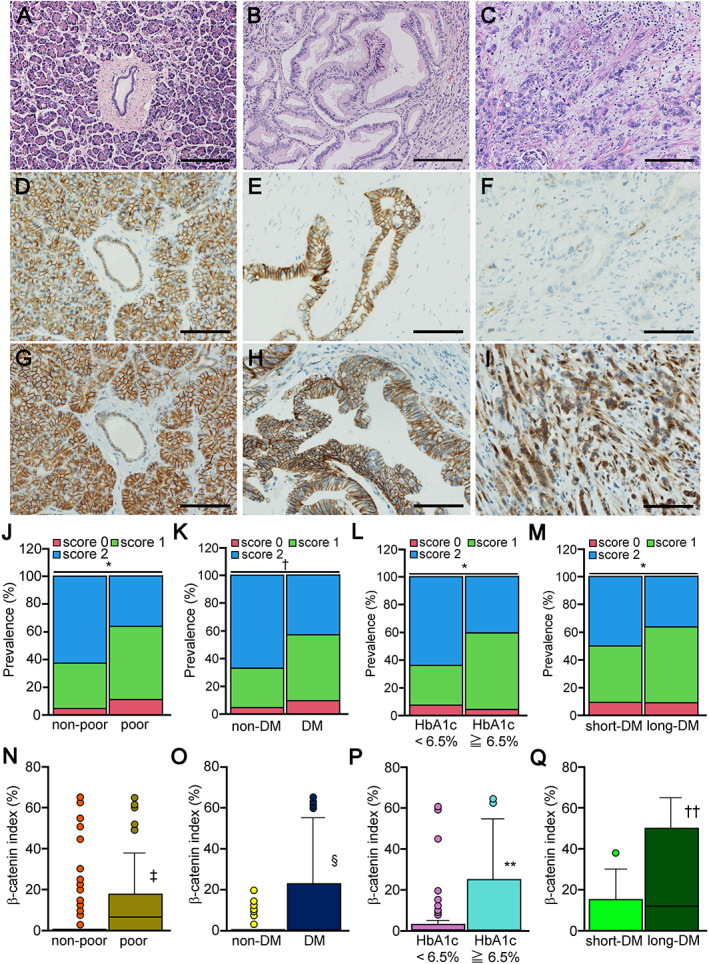
Histology of pancreatic ductal adenocarcinoma and E‐cadherin expression. (A) In H&E sections, the epithelial cells in the duct showed no atypia and monolayered arrays in the nonneoplastic area. (B) PDAC cells exhibited an irregular ductal pattern with stromal reaction and were diagnosed as well or moderately differentiated adenocarcinoma. (C) PDAC cells exhibiting diffuse or solid growth were diagnosed as poorly differentiated carcinoma. (D) Immunohistochemical analysis revealed that E‐cadherin was strongly expressed on the cell membrane of nonneoplastic ductal cells. (E) Moderately differentiated PDAC cells showed robust E‐cadherin expression on the cell membrane, while (F) there was no E‐cadherin expression in poorly differentiated PDAC cells, judged as E‐cadherin loss. β‐Catenin was robustly expressed on the cell membrane of (G) nonneoplastic ductal cells and (H) moderately differentiated PDAC cells, while nuclear expression of β‐catenin was evident in (I) poorly differentiated PDAC cells. Semiquantitative evaluation of cancerous tissues revealed that (J) low E‐cadherin expression was significantly increased in poorly differentiated compared to non‐poorly differentiated tumors. The prevalence of low E‐cadherin subjects was significantly higher (K) in the DM group than in the non‐DM group, (L) the HbA1c ≥ 6.5% group than in the HbA1c < 6.5% group, and (M) the long‐DM group than in the short‐DM group. The β‐catenin index was significantly higher in (N) poorly differentiated tumors compared with non‐poorly differentiated tumors, the (O) the DM group versus the non‐DM group, (P) the HbA1c ≥ 6.5% group versus the HbA1c < 6.5% group, and (Q) the long‐DM group versus the short‐DM group. Significance: *p < 0.05, † p < 0.01 (chi‐square test), ‡ p < 0.01 versus non‐poor, § p < 0.01 versus non‐DM, **p < 0.01 versus HbA1c < 6.5%, and †† p < 0.01 versus long‐DM. Scale bars are 200 μm (A–C) and 100 μm (D–I).
The frequency of CDH1 promoter methylation was increased in patients with long‐DM
The frequency of CDH1 promoter methylation in PDAC evaluated by MS‐PCR was significantly increased in the DM group compared to the control group (56.9% versus 43.1%, p < 0.05; Table 1). When the DM groups were compared with the remaining subjects, no increase in the frequency was observed in those with short‐DM (43.8% versus 56.3%, p = 0.69), while the frequency was significantly increased in PDAC tissues in those with long‐DM (69.7% versus 30.3%, p < 0.01). In contrast, HbA1c level had no impact on the prevalence of CDH1 promoter methylation. DM treatment [diet therapy, metformin, dipeptidyl peptidase 4 (DPP4) inhibitor, and insulin] did not influence the prevalence. The prevalence of CDH1 promoter methylation was significantly increased in the E‐cadherin low group (39.0% versus 61.0%, p < 0.01).
Table 1.
CDH1 promoter methylation analysis.
| Methylation− (cases) | Methylation+ (cases) | P value | |
|---|---|---|---|
| Age ≧ 65 years | 57.7% (60/104) | 42.3% (44/104) | 0.054 |
| Male | 51.6% (33/64) | 48.4% (31/64) | |
| Female | 54.4% (37/68) | 45.6% (31/68) | 0.862 |
| BMI ≧ 25 kg/m2 | 48.6% (17/35) | 51.4% (18/35) | 0.559 |
| DM | 43.1% (28/65) | 56.9% (37/65) | <0.05 |
| Short‐DM | 56.3% (18/32) | 43.8% (14/32) | 0.690 |
| Long‐DM | 30.3% (10/33) | 69.7% (23/33) | <0.01 |
| HbA1c ≧ 6.5% | 42.2% (19/45) | 57.8% (26/45) | 0.098 |
| Diet therapy | 45.5% (10/22) | 54.5% (12/22) | 0.488 |
| Metformin user | 37.5% (3/8) | 62.5% (5/8) | 0.474 |
| DPP4 inhibitor user | 41.2% (14/34) | 58.8% (20/34) | 0.116 |
| Insulin user | 36.4% (4/11) | 63.6% (7/11) | 0.347 |
| Smoker | 46.4% (32/69) | 53.6% (37/69) | 0.120 |
| E‐cadherin, low | 39.0% (23/59) | 61.0% (36/59) | <0.01 |
The proportion of patients with long‐DM was significantly higher among the subjects with CDH1 promoter methylation than among those without CDH1 promoter methylation (p < 0.05; Figure 2A). The DM subjects with CDH1 promoter methylation showed a significantly higher β‐catenin index than those without DM and DM subjects without CDH1 promoter methylation (p < 0.01 and p < 0.05, respectively; Figure 2B). The HbA1c level in DM subjects was significantly higher than that in non‐DM subjects regardless of the presence of CDH1 promoter methylation (p < 0.01, respectively; Figure 2C).
Figure 2.
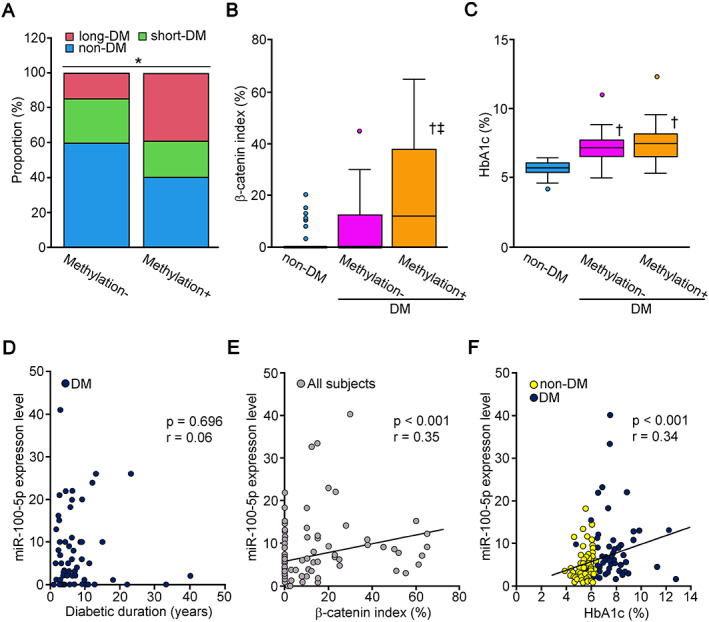
Correlation between epigenetic changes and diabetes‐related measurements. (A) The prevalence of a long duration of DM (≥3 years) was significantly higher in PDAC subjects with CDH1 promoter methylation than in PDAC subjects without CDH1 promoter methylation. (B) The β‐catenin index was significantly higher in DM subjects with CDH1 promoter methylation than in non‐DM subjects and DM subjects without CDH1 promoter methylation. (C) The HbA1c level was significantly higher in DM subjects than in non‐DM subjects regardless of the presence of CDH1 promoter methylation. The HbA1c level was comparable between the subjects with CDH1 promoter methylation − and +. (D) The duration of DM did not correlate significantly with the expression level of miR‐100‐5p (r = 0.06, p = 0.70). (E) The expression level of miR‐100‐5p was proportionally correlated with the β‐catenin index in all subjects (r = 0.35, p < 0.001). (F) The expression level of miR‐100‐5p was proportionally correlated with the HbA1c level in subjects with PDAC complicated with DM (r = 0.34, p < 0.001). Significance: *p < 0.05 (chi‐square test), † p < 0.01 versus non‐DM, ‡ p < 0.05 versus DM methylation−.
miR‐100‐5p expression was significantly correlated with preoperative HbA1c levels
miR‐100‐5p expression was higher in PDAC subjects with DM than in those without DM (expression value: 3.5 versus 6.0, p < 0.001; Table 2). In contrast to the effect on CDH1 promoter methylation, long‐DM had a marginal impact on miR‐100‐5p expression (6.0 versus 3.7, p = 0.06); rather, short‐DM significantly increased miR‐100‐5p expression (5.9 versus 4.0, p < 0.001). miR‐100‐5p expression was higher in the high HbA1c group than in the low HbA1c group (6.0 versus 3.7, p < 0.001). The subjects treated with the DPP4 inhibitor showed increased expression compared to nontreated subjects (6.0 versus 3.9, p < 0.01). The subjects exhibiting low expression of E‐cadherin showed higher expression of miR‐100‐5p than those with high E‐cadherin expression (6.0 versus 3.9, p < 0.001). miR‐100‐5p expression was not influenced by CDH1 promoter methylation (5.2 versus 3.9, p = 0.17).
Table 2.
miR‐100‐5p analysis.
| No. | miR‐100‐5p median (25–75% IQR) | P value | ||
|---|---|---|---|---|
| Age (year) | <65 | 29 | 3.8 (3.1–7.0) | |
| ≧65 | 103 | 4.3 (2.9–7.6) | 0.724 | |
| Sex | Male | 65 | 3.9 (2.9–6.0) | |
| Female | 67 | 5.1 (3.0–9.0) | 0.124 | |
| BMI (kg/m2) | <25 | 97 | 4.2 (2.9–7.4) | |
| ≧25 | 35 | 4.1 (3.1–6.6) | 0.920 | |
| DM | − | 67 | 3.5 (2.4–5.4) | |
| + | 65 | 6.0 (3.6–9.2) | <0.001 | |
| Short‐DM | − | 99 | 4.0 (2.7–6.7) | |
| + | 33 | 5.9 (3.8–10.8) | <0.001 | |
| Long‐DM | − | 98 | 3.7 (2.7–9.0) | |
| + | 34 | 6.0 (3.3–9.0) | 0.061 | |
| HbA1c (%) | <6.5 | 81 | 3.7 (2.7–6.3) | |
| ≧6.5 | 51 | 6.0 (4.1–9.4) | <0.001 | |
| Diet therapy | − | 22 | 4.1 (2.9–6.9) | |
| + | 110 | 6.0 (3.8–10.5) | 0.089 | |
| Metformin user | − | 124 | 4.1 (2.9–7.3) | |
| + | 8 | 6.5 (4.7–9.1) | 0.198 | |
| DPP4 inhibitor user | − | 97 | 3.9 (2.7–6.9) | |
| + | 35 | 6.0 (3.7–8.9) | <0.01 | |
| Insulin user | − | 119 | 4.1 (2.9–7.4) | |
| + | 13 | 6.0 (3.5–7.1) | 0.450 | |
| Smoker | − | 63 | 3.8 (2.5–7.1) | |
| + | 69 | 4.3 (3.2–7.6) | 0.469 | |
| E‐cadherin expression | High | 83 | 3.9 (2.7–5.5) | |
| Low | 59 | 6.0 (2.7–9.2) | <0.001 | |
| CDH1 promoter methylation | − | 68 | 3.9 (2.7–6.4) | |
| + | 64 | 5.2 (3.2–8.3) | 0.174 |
IQR, interquartile range.
In the bivariate correlation analysis, there was no correlation between DM duration and miR‐100‐5p expression (r = 0.06, p = 0.70; Figure 2D). The expression level of miR‐100‐5p was proportionally correlated with the β‐catenin index (r = 0.35, p < 0.001; Figure 2E). The expression level of miR‐100‐5p was significantly correlated with preoperative HbA1c value (r = 0.34, p < 0.001; Figure 2F).
Clinicopathological features associated with multiple epigenetic changes in subjects with PDAC associated with DM
The proportion of subjects with high CA19‐9 (≥200 U/ml) was significantly higher in the groups showing a single epigenetic change (S‐epi) and dual epigenetic changes (D‐epi) than in the group with no epigenetic changes (N‐epi; p < 0.01 versus N‐epi, respectively; Table 3). The proportion of males was significantly higher in the D‐epi group than in the N‐epi group (p < 0.05). The prevalence of poorly differentiated carcinoma (poor), ly2‐3, and low E‐cadherin expression gradually increased along with the number of epigenetic changes in the subjects with PDAC complicated with DM. The prevalence of tumor size ≥30 mm and N1, borderline resectable, v2‐3, and ne2‐3 tumors was significantly higher in the D‐epi group than in the N‐epi and S‐epi groups.
Table 3.
Clinicopathological characteristics related to epigenetic changes.
| Degree of epigenetic change | N‐epi | S‐epi | D‐epi |
|---|---|---|---|
| Age ≧ 65 years | 69.2% (9/13) | 82.8% (24/29)* | 80.0% (20/25) |
| Male | 30.8% (4/13) | 44.8% (13/29) | 48.0% (12/25)* |
| Female | 69.2% (9/13) | 55.2% (16/29) | 52.0% (13/25) |
| CA19‐9 ≧ 200 U/ml | 15.4% (2/13) | 35.5% (10/29) † | 36.0% (9/25) † |
| Tumor size ≧ 30 mm | 38.5% (5/13) | 37.9% (11/29) | 60.0% (15/25) † , ‡ |
| N1 | 46.2% (6/13) | 51.7% (15/29) | 68.0% (17/25) † , § |
| Borderline resectable | 15.4% (2/13) | 10.3% (3/29) | 32.0% (8/25) † , ‡ |
| Poorly differentiated | 7.7% (1/13) | 20.7% (6/29)* | 52.0% (13/25) † , ‡ |
| v2‐3 | 69.2% (9/13) | 72.4% (21/29) | 96.0% (24/25) † , ‡ |
| ly2‐3 | 46.2% (6/13) | 62.1% (18/29)* | 80.0% (20/25) † , ‡ |
| ne2‐3 | 84.6% (11/13) | 89.7% (26/29) | 100% (25/25) † , ‡ |
| E‐cadherin expression, low | 30.8% (4/13) | 55.2% (16/29) † | 72.0% (18/25) † , § |
D‐epi, subjects showing dual epigenetic change; N‐epi, subjects showing non‐epigenetic change; S‐epi, subjects showing single epigenetic change.
P < 0.05 versus N‐epi.
P < 0.01 versus N‐epi.
P < 0.01 versus S‐epi.
P < 0.05 versus S‐epi.
Epigenetic status was associated with short OS and DFS in PDAC
Univariate analysis of OS showed that tumor size ≥30 mm (p < 0.05), N1 (p < 0.05), CA19‐9 ≥ 200 U/ml (p < 0.01), adjuvant chemotherapy (p < 0.01), HbA1c ≥ 6.5% (p < 0.05), long‐DM (p < 0.05), miR‐100‐5p ≧ 4.13 (p < 0.05), and CDH1 promoter methylation (p < 0.01) were significant risk factors for shorter survival (Table 4). Tumor size ≥30 mm (p < 0.05), adjuvant chemotherapy (p < 0.01), miR‐100‐5p ≧ 4.13, and CDH1 promoter methylation (p < 0.05) were independent predictors of OS in the multivariate analysis (Table 5). Univariate analysis of DFS showed that tumor size ≥30 mm (p < 0.05), N1 (p < 0.001), CA19‐9 ≥ 200 U/ml (p < 0.01), adjuvant chemotherapy (p < 0.001), HbA1c ≥ 6.5% (p < 0.05), miR‐100‐5p ≥ 4.13 (p < 0.05), and CDH1 promoter methylation (p < 0.01) were significant risk factors for shorter survival (supplementary material, Table S2). Tumor size ≥30 mm (p < 0.05), CA19‐9 ≥ 200 U/ml (p < 0.05), adjuvant chemotherapy (p < 0.01), miR‐100‐5p ≧ 4.13 and CDH1 promoter methylation (p < 0.05) were independent factors for DFS in multivariate analysis (supplementary material, Table S3). The Kaplan–Meier survival curves clearly indicated that prognosis in terms of OS worsened as the number of epigenetic modifications increased (Figure 3A). The number of epigenetic modifications was indirectly associated with worsened DFS (p < 0.05, N‐epi versus S‐epi and p < 0.01 N‐epi versus D‐epi; Figure 3B). OS and DFS in the subjects with both high‐HbA1c and long‐DM were significantly worse than those in the remaining diabetic subjects (others; p < 0.01 and p < 0.05, respectively; Figure 3C,D).
Table 4.
Univariate analysis (OS).
| Factor | Median OS (date) | P value |
|---|---|---|
| Age (years): <65 versus ≧65 | 943 versus 979 | 0.528 |
| Male versus female | 711 versus 1,166 | 0.111 |
| Location: body‐tail versus head | 1,166 versus 943 | 0.52 |
| Tumor size (mm): <30 versus ≧30 | 1,010 versus 711 | <0.05 |
| T1–2 versus T3–4 | 979 versus 847 | 0.216 |
| N: N0 versus N1 | 1,184 versus 646 | <0.05 |
| CA19‐9 (U/ml): <200 versus ≧200 | 1,097 versus 602 | <0.01 |
| Adjuvant chemotherapy: (−) versus (+) | 537 versus 1,163 | <0.01 |
| HbA1c (%): <6.5 versus ≧6.5 | 1,166 versus 742 | <0.05 |
| DM: (−) versus (+) | 1,184 versus 847 | 0.074 |
| Long‐DM: (−) versus (+) | 1,066 versus 632 | <0.05 |
| miR‐100‐5p expression: <4.13 versus ≧4.13 | 1,184 versus 703 | <0.05 |
| CDH1 promoter methylation: (−) versus (+) | 1,001 versus 602 | <0.01 |
Table 5.
Multivariate analysis (OS).
| Factor | Risk ratio | 95% CI | P value |
|---|---|---|---|
| Tumor size (mm) ≧30 | 1.73 | 1.06–2.81 | <0.05 |
| N: N0 versus N1 | 1.20 | 0.72–2.00 | 0.48 |
| CA19‐9 (U/ml) ≧200 | 1.43 | 0.87–2.34 | 0.17 |
| Adjuvant chemotherapy (+) | 0.37 | 0.21–0.65 | <0.01 |
| miR‐100‐5p expression ≧4.13 | 1.80 | 1.12–2.92 | <0.05 |
| CDH1 promoter methylation (+) | 1.64 | 1.02–2.66 | <0.05 |
Figure 3.
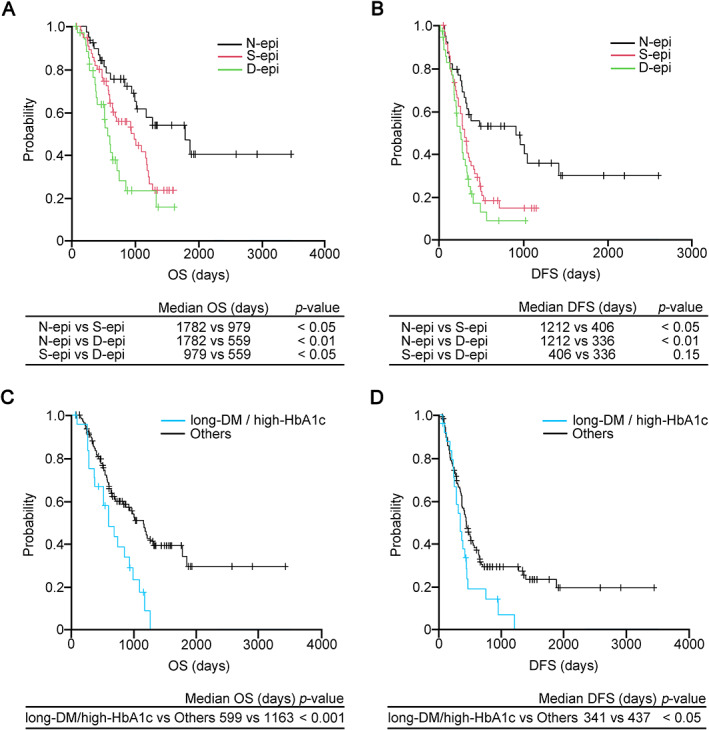
Survival curves based on OS and DFS. (A) The OS of the S‐epi group (red line) was significantly lower than that of the N‐epi group (black line). The survival rate of the D‐epi group (green line) was significantly lower than that of the S‐epi group (p < 0.05). (B) In terms of DFS, the survival rates of the S‐epi and D‐epi groups were significantly lower than that of the N‐epi group (p < 0.05 and p < 0.01, respectively), while the survival rate of the S‐epi group was comparable to that of the D‐epi group. The group exhibiting both long‐DM and high HbA1c showed significantly worse (C) OS and (D) DFS in subjects with PDAC associated with DM compared to the remaining diabetic subjects (others; p < 0.001 and p < 0.05, respectively). D‐epi, subjects showing dual epigenetic change; N‐epi, subjects showing no epigenetic change; S‐epi, subjects showing a single epigenetic change.
Discussion
In this study, we first found that multiple epigenetic processes were associated with the development of PDAC associated with DM. CDH1 promoter methylation was associated with the long‐DM, while the prevalence did not correlate with HbA1c level. Moreover, the expression of miR‐100‐5p correlated positively with preoperative HbA1c but not with the duration of DM. Both variables were strongly correlated with suppressed expression of E‐cadherin in PDAC subjects. The decrease in E‐cadherin expression was accompanied by an increase in the β‐catenin index. PDAC subjects with double positivity for CDH1 promoter methylation and high expression of miR‐100‐5p showed a significantly greater prevalence of tumor size ≥30 mm and N1, borderline resectable, poorly differentiated, v2‐3, ly2‐3, ne2‐3, and E‐cadherin expression‐low tumors than subjects with negative or single‐positive tumors. The OS of double‐positive subjects was significantly worse than that of negative or single‐positive subjects. The subjects with a long‐DM and high HbA1c showed significantly worse OS and DFS than the remaining subjects with PDAC associated with DM.
miR‐100 expression differs depending on DM state and tissue type [25, 26, 27, 28]. This suggests that miR‐100‐5p expression widely varies in different diabetic states and that E‐cadherin expression can be altered in patients with a relatively short duration of DM depending on HbA1c levels. E‐cadherin expression mediated by miR‐100‐5p may be reversible and preventable with glycemic control in PDAC patients with DM.
On the other hand, as shown in our previous study, a long‐DM was associated with a high prevalence of CDH1 promoter methylation but not high HbA1c levels in PDAC subjects. Although the precise mechanism is still speculative, prolonged metabolic disorder can cause epigenetic alterations in diabetic patients [36]. It is generally known that the prevalence of diabetic complications can increase along with diabetic progression [37]. The pathophysiology of diabetic complications is chronic inflammation, the production of advanced glycation end products and activation of their signaling, increased levels of oxidative stress, and tissue ischemia exacerbated by micro‐ and macroangiopathy, which include some of the triggers of promoter methylation [38, 39, 40, 41, 42]. Furthermore, the duration of diabetes changes the intratumoral composition of inflammatory cells in PDAC, which may increase the level of CDH1 promoter methylation [43]. Thus, additional insults evoked by long‐DM may be involved in promoter methylation in PDAC. These findings may suggest the possibility that the regulation of CDH1 promoter methylation in PDAC patients with long‐DM is more difficult than repression of miR‐100‐5p expression, which varies only with HbA1c levels.
E‐cadherin downregulation has been described as a major requirement for aggressive malignant behavior such as invasion eliciting EMT [44]. Interestingly, dual epigenetic changes in E‐cadherin significantly worsened OS compared to a single epigenetic change in PDAC. This suggests that the number of epigenetic changes is not highly involved in PDAC tumorigenesis itself; rather, these changes may increase tumor aggressiveness and subsequent chemoresistance. It is known that the degree of methyl base addition in CpG islands of CDH1 promoter can vary between cases [45]. Therefore, high expression of miR‐100‐5p would complementarily suppress E‐cadherin expression and induce robust EMT, leading to poor OS in PDAC cell patients with dual epigenetic changes.
Surgery is the only potentially curative treatment for PDAC. However, due to its early metastatic nature, only up to 20% of patients are candidates for initial resection of PDAC [46]. Therefore, there is an urgent need to develop new methods for PDAC diagnosis and recurrence monitoring. Liquid biopsy is a noninvasive cancer detection method involving the detection of microvesicles and exosomes containing nucleic acids released into body fluids from tumors [47, 48, 49]. High expression of serum exosomal miR‐17‐5p and miR‐21 is observed in PDAC patients in association with metastasis and advanced stages [50]. These findings suggest that miR‐100‐5p liquid biopsy may be similarly applied to early detection of PDAC or postoperative recurrence monitoring in diabetic patients, especially those with poor glycemic control.
Diabetes therapy can influence the progression of PDAC [51]. Metformin can suppress tumor progression, while the effects of DPP4 inhibitors and insulin are still controversial [51, 52, 53, 54]. In our study, treatment with metformin and a DPP4 inhibitor had no effects on the frequency of CDH1 hypermethylation despite their hypoglycemic and anti‐inflammatory effects. Although the reason is unclear, this might be ascribed to a statistical power issue due to the relatively small number of the subjects in this study. On the other hand, DPP4 inhibitor significantly increased the expression of miR‐100‐5p in PDAC. DPP4 inhibitors can modify various types of miRNA expression via activation of glucagon‐like peptide‐1 (GLP‐1) signaling independent of glycemic improvement [55]. This suggests that miR‐100‐5p expression can be regulated by GLP‐1 signaling in PDAC cells, which may lead to the progression of PDAC. Nevertheless, in the univariate analysis, patients treated with DPP4 inhibitor showed no inferior prognosis compared to patients not treated with DPP4 inhibitor. Because DPP4 inhibitors have moderate efficacy in controlling glucose levels and reduce HbA1c on average by approximately 0.6–0.8%, the improvement in blood glucose levels due to DPP4 inhibitor treatment may counteract the adverse prognostic effect elicited by the increase in miR‐100‐5p expression [56].
Our results suggest that strict glycemic control is necessary to suppress the expression of miR‐100‐5p in PDAC patients with DM. Because it is still unclear whether fasting blood glucose or glucose spikes affect miR‐100‐5p expression, further detailed investigation of blood glucose changes and miR‐100‐5p expression is required. In the case of PDAC with long‐DM, concomitant administration of a demethylating agent such as azacytidine is expected to restore CDH1 gene expression by demethylating the promoter. Since azacytidine inevitably causes adverse events such as nausea, vomiting, and myelosuppression, DM history is considered useful in terms of patient selection for demethylating therapy [57].
There are several limitations to this study. First, only FFPE specimens were used. Since alteration of DNA during tissue processing is known to occur, the results may be confounded by technical artifacts. Although the quality of the samples was confirmed by immunostaining based on the reproducibility of the results in this study, confirmation of the results using fresh samples may be warranted in future investigations. Another drawback of this study may be that the prevalence of PDAC associated with DM was not directly evaluated because of the retrospective nature of the data. Future prospective studies will be necessary to confirm our results. Our analysis was also limited to CDH1 promoter methylation and miR‐100‐5p, and it is impossible to speculate the whole scenario related to the implication of promoter methylation and miR expression in the development of PDAC based only on the findings for these two genes. Finally, it was not possible to evaluate epigenetic changes depending on the site of cancer tissue. Because the degrees of inflammatory cell infiltration and ischemia are different between the periphery and the center of cancer tissue, epigenetic traits may vary depending on the site. Future evaluation applying microdissection is required to address this.
Nevertheless, we believe that our study has revealed a novel association between E‐cadherin downregulation, which can be mediated by multiple epigenetic changes, and DM in PDAC. Our results reconfirm the importance of optimal blood glucose control in DM. Future studies are expected to explore the possibility of developing new effective therapy applying demethylating agents for PDAC associated with DM.
Author contributions statement
YH and HM conceived and designed the study. YH, HM and KH developed the methodology and wrote, reviewed and revised the paper. YH, KY, TY, AI, YT, TS, HK, KM, KK and KI acquired, analyzed and interpreted data and performed statistical analysis. HM and KH provided technical and material support. All authors read and approved the final paper.
Ethics approval and consent to participate
This study was approved by the ethical committee of Hirosaki University Graduate School of Medicine (#2020‐143). This study was performed in accordance with the Declaration of Helsinki.
Supporting information
Figure S1. Preoperative HbA1c levels (within 1 month) showed a significant proportional correlation with average HbA1c levels 3–12 months before surgery in PDAC subjects
Table S1. Clinicopathological characteristics of 132 subjects
Table S2. Univariate analysis (disease‐free survival)
Table S3. Multivariate analysis (disease‐free survival)
Acknowledgements
The technical assistance of Ms. Saori Ogasawara, Ms. Misato Sakamoto, Ms. Hiroko Mori, Ms. Saeko Osanai, and Ms. Rumiko Imai was greatly appreciated. This study was in part supported by KAKENHI (Grants‐in‐Aid for Scientific Research) from the Japanese Ministry of Education, Culture, Sports, Science and Technology to YH (#20K17636).
No conflicts of interest were declared.
Data availability statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author upon reasonable request.
References
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics 2017. CA Cancer J Clin 2017; 67: 7–30. [DOI] [PubMed] [Google Scholar]
- 2. Carnevale J, Ashworth A. Assessing the significance of BRCA1 and BRCA2 mutations in pancreatic cancer. J Clin Oncol 2015; 33: 3080–3081. [DOI] [PubMed] [Google Scholar]
- 3. Maurice D, Pierreux CE, Howell M, et al. Loss of Smad4 function in pancreatic tumors: C‐terminal truncation leads to decreased stability. J Biol Chem 2001; 276: 43175–43181. [DOI] [PubMed] [Google Scholar]
- 4. Collins MA, Bednar F, Zhang Y, et al. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest 2012; 122: 639–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ying H, Kimmelman AC, Lyssiotis CA, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012; 149: 656–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Redston MS, Caldas C, Seymour AB, et al. p53 mutations in pancreatic carcinoma and evidence of common involvement of homocopolymer tracts in DNA microdeletions. Cancer Res 1994; 54: 3025–3033. [PubMed] [Google Scholar]
- 7. Hong SM, Park JY, Hruban RH, et al. Molecular signatures of pancreatic cancer. Arch Pathol Lab Med 2011; 135: 716–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Huxley R, Ansary‐Moghaddam A, Berrington de González A, et al. Type‐II diabetes and pancreatic cancer: a meta‐analysis of 36 studies. Br J Cancer 2005; 92: 2076–2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kasuga M, Ueki K, Tajima N, et al. Report of the Japan Diabetes Society/Japanese Cancer Association Joint Committee on Diabetes and Cancer. Cancer Sci 2013; 104: 965–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Saito T, Mizukami H, Umetsu S, et al. Worsened outcome in patients with pancreatic ductal carcinoma on long‐term diabetes: association with E‐cadherin1 (CDH1) promoter methylation. Sci Rep 2017; 7: 18056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Srivastava SP, Goodwin JE. Cancer biology and prevention in diabetes. Cell 2020; 9: 1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Babu M, Durga Devi T, Mäkinen P, et al. Differential promoter methylation of macrophage genes is associated with impaired vascular growth in ischemic muscles of hyperlipidemic and type 2 diabetic mice: genome‐wide promoter methylation study. Circ Res 2015; 117: 289–299. [DOI] [PubMed] [Google Scholar]
- 13. Volkmar M, Dedeurwaerder S, Cunha DA, et al. DNA methylation profiling identifies epigenetic dysregulation in pancreatic islets from type 2 diabetic patients. EMBO J 2012; 31: 1405–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nilsson E, Matte A, Perfilyev A, et al. Epigenetic alterations in human liver from subjects with type 2 diabetes in parallel with reduced folate levels. J Clin Endocrinol Metab 2015; 100: E1491–E1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Brabletz T, Jung A, Reu S, et al. Variable beta‐catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc Natl Acad Sci U S A 2001; 98: 10356–10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Weadick B, Nayak D, Persaud AK, et al. EMT‐induced gemcitabine resistance in pancreatic cancer involves the functional loss of equilibrative nucleoside transporter 1. Mol Cancer Ther 2021; 20: 410–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Garrido VT, Banerjee S. E‐cadherin: an enigma in pancreatic diseases. Cell Mol Gastroenterol Hepatol 2020; 9: 191–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kaneta Y, Sato T, Hikiba Y, et al. Loss of pancreatic E‐cadherin causes pancreatitis‐like changes and contributes to carcinogenesis. Cell Mol Gastroenterol Hepatol 2020; 9: 105–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Uchida C, Mizukami H, Hara Y, et al. Diabetes in humans activates pancreatic stellate cells via RAGE in pancreatic ductal adenocarcinoma. Int J Mol Sci 2021; 22: 11716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ottaviani S, Stebbing J, Frampton AE, et al. TGF‐β induces miR‐100 and miR‐125b but blocks let‐7a through LIN28B controlling PDAC progression. Nat Commun 2018; 9: 1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Panarelli NC, Chen YT, Zhou XK, et al. MicroRNA expression aids the preoperative diagnosis of pancreatic ductal adenocarcinoma. Pancreas 2012; 41: 685–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dhayat SA, Mardin WA, Seggewiß J, et al. MicroRNA profiling implies new markers of gemcitabine chemoresistance in mutant p53 pancreatic ductal adenocarcinoma. PLoS One 2015; 10: e0143755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang Y, Zhong Y, Sun K, et al. Identification of exosome miRNAs in bronchial epithelial cells after PM2.5 chronic exposure. Ecotoxicol Environ Saf 2021; 215: 112127. [DOI] [PubMed] [Google Scholar]
- 24. Ding Y, Mei W, Zheng Z, et al. Exosomes secreted from human umbilical cord mesenchymal stem cells promote pancreatic ductal adenocarcinoma growth by transferring miR‐100‐5p. Tissue Cell 2021; 73: 101623. [DOI] [PubMed] [Google Scholar]
- 25. Sardu C, Modugno P, Castellano G, et al. Atherosclerotic plaque fissuration and clinical outcomes in pre‐diabetics vs. normoglycemics patients affected by asymptomatic significant carotid artery stenosis at 2 years of follow‐up: role of microRNAs modulation: the ATIMIR study. Biomedicine 2021; 9: 401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hezova R, Slaby O, Faltejskova P, et al. microRNA‐342, microRNA‐191 and microRNA‐510 are differentially expressed in T regulatory cells of type 1 diabetic patients. Cell Immunol 2010; 260: 70–74. [DOI] [PubMed] [Google Scholar]
- 27. Erener S, Marwaha A, Tan R, et al. Profiling of circulating microRNAs in children with recent onset of type 1 diabetes. JCI Insight 2017; 2: e89656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pek SL, Sum CF, Lin MX, et al. Circulating and visceral adipose miR‐100 is down‐regulated in patients with obesity and type 2 diabetes. Mol Cell Endocrinol 2016; 427: 112–123. [DOI] [PubMed] [Google Scholar]
- 29. Committee of the Japan Diabetes Society on the Diagnostic Criteria of Diabetes Mellitus , Seino Y, Nanjo K, et al. Report of the committee on the classification and diagnostic criteria of diabetes mellitus. J Diabetes Investig 2010; 1: 212–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hruban RH, Maitra A, Adsay NV, et al. WHO classification of tumours of the digestive system. In: Fifth Edition IARC WHO Classification of Tumors. World Health Organization: Lyon, No 5, 2019; 322–332. [Google Scholar]
- 31. Yanai K, Nakamura M, Akiyoshi T, et al. Crosstalk of hedgehog and Wnt pathways in gastric cancer. Cancer Lett 2008; 263: 145–156. [DOI] [PubMed] [Google Scholar]
- 32. Umetsu S, Mizukami H, Saito T, et al. Diabetes, an independent poor prognostic factor of non‐B non‐C hepatocellular carcinoma, correlates with dihydropyrimidinase‐like 3 promoter methylation. Sci Rep 2020; 10: 1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Herman JG, Graff JR, Myöhänen S, et al. Methylation‐specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci U S A 1996; 93: 9821–9826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods 2001; 25: 402–408. [DOI] [PubMed] [Google Scholar]
- 35. Greither T, Grochola LF, Udelnow A, et al. Elevated expression of microRNAs 155, 203, 210 and 222 in pancreatic tumors is associated with poorer survival. Int J Cancer 2010; 126: 73–80. [DOI] [PubMed] [Google Scholar]
- 36. Davison GW, Irwin RE, Walsh CP. The metabolic‐epigenetic nexus in type 2 diabetes mellitus. Free Radic Biol Med 2021; 170: 194–206. [DOI] [PubMed] [Google Scholar]
- 37. Diabetes Control and Complications Trial Research Group , Nathan DM, Genuth S, et al. The effect of intensive treatment of diabetes on the development and progression of long‐term complications in insulin‐dependent diabetes mellitus. N Engl J Med 1993; 329: 977–986. [DOI] [PubMed] [Google Scholar]
- 38. Mizukami H, Osonoi S. Pathogenesis and molecular treatment strategies of diabetic neuropathy collateral glucose‐utilizing pathways in diabetic polyneuropathy. Int J Mol Sci 2020; 22: 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Damiani LA, Yingling CM, Leng S, et al. Carcinogen‐induced gene promoter hypermethylation is mediated by DNMT1 and causal for transformation of immortalized bronchial epithelial cells. Cancer Res 2008; 68: 9005–9014. [DOI] [PubMed] [Google Scholar]
- 40. Zheng DL, Zhang L, Cheng N, et al. Epigenetic modification induced by hepatitis B virus X protein via interaction with de novo DNA methyltransferase DNMT3A. J Hepatol 2009; 50: 377–387. [DOI] [PubMed] [Google Scholar]
- 41. Chiba T, Marusawa H, Ushijima T. Inflammation‐associated cancer development in digestive organs: mechanisms and roles for genetic and epigenetic modulation. Gastroenterology 2012; 143: 550–563. [DOI] [PubMed] [Google Scholar]
- 42. Ma SC, Zhang HP, Kong FQ, et al. Integration of gene expression and DNA methylation profiles provides a molecular subtype for risk assessment in atherosclerosis. Mol Med Rep 2016; 13: 4791–4799. [DOI] [PubMed] [Google Scholar]
- 43. Pan X, Mizukami H, Hara Y, et al. Diabetes mellitus impacts on expression of DNA mismatch repair protein PMS2 and tumor microenvironment in pancreatic ductal adenocarcinoma. J Diabetes Investig 2023; 14: 132–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hong SM, Li A, Olino K, et al. Loss of E‐cadherin expression and outcome among patients with resectable pancreatic adenocarcinomas. Mod Pathol 2011; 24: 1237–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shimizu K, Hanaoka M, Kato A, et al. Reduced expression of the E‐cadherin gene and its aberrant DNA methylation in hamster pancreatic tumors. Biochem Biophys Res Commun 2005; 336: 49–53. [DOI] [PubMed] [Google Scholar]
- 46. Gillen S, Schuster T, Meyer Zum Büschenfelde C, et al. Preoperative/neoadjuvant therapy in pancreatic cancer: a systematic review and meta‐analysis of response and resection percentages. PLoS Med 2010; 7: e1000267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Joosse SA, Pantel K. Tumor‐educated platelets as liquid biopsy in cancer patients. Cancer Cell 2015; 28: 552–554. [DOI] [PubMed] [Google Scholar]
- 48. Bagcchi S. Urine test can detect early stage pancreatic cancer. Lancet Oncol 2015; 16: e431. [DOI] [PubMed] [Google Scholar]
- 49. Schwarzenbach H, Nishida N, Calin GA, et al. Clinical relevance of circulating cell‐free microRNAs in cancer. Nat Rev Clin Oncol 2014; 11: 145–156. [DOI] [PubMed] [Google Scholar]
- 50. Que R, Ding G, Chen J, et al. Analysis of serum exosomal microRNAs and clinicopathologic features of patients with pancreatic adenocarcinoma. World J Surg Oncol 2013; 11: 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Feng YH, Velazquez‐Torres G, Gully C, et al. The impact of type 2 diabetes and antidiabetic drugs on cancer cell growth. J Cell Mol Med 2011; 15: 825–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stoian AP, Sachinidis A, Stoica RA, et al. The efficacy and safety of dipeptidyl peptidase‐4 inhibitors compared to other oral glucose‐lowering medications in the treatment of type 2 diabetes. Metabolism 2020; 109: 154295. [DOI] [PubMed] [Google Scholar]
- 53.Buse JB, Bethel MA, Green JB, et al. TECOS Study Group. Pancreatic Safety of Sitagliptin in the TECOS Study. Diabetes Care 2017; 40: 164–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fisher WE, Boros LG, Schirmer WJ. Insulin promotes pancreatic cancer: evidence for endocrine influence on exocrine pancreatic tumors. J Surg Res 1996; 63: 310–313. [DOI] [PubMed] [Google Scholar]
- 55.Radbakhsh S, Sathyapalan T, Banach M, et al. Incretins and microRNAs: Interactions and physiological relevance. Pharmacol Res 2020; 153: 104662. [DOI] [PubMed] [Google Scholar]
- 56.Cahn A, Raz I. Emerging gliptins for type 2 diabetes. Expert Opin Emerg Drugs 2013; 18: 245–258. [DOI] [PubMed] [Google Scholar]
- 57.Kaminskas E, Farrell AT, Wang YC, et al. FDA drug approval summary: azacitidine (5‐azacytidine, Vidaza) for injectable suspension. Oncologist 2005; 10: 176–182. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Preoperative HbA1c levels (within 1 month) showed a significant proportional correlation with average HbA1c levels 3–12 months before surgery in PDAC subjects
Table S1. Clinicopathological characteristics of 132 subjects
Table S2. Univariate analysis (disease‐free survival)
Table S3. Multivariate analysis (disease‐free survival)
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author upon reasonable request.