

Abstract
BACKGROUND:
The Biologics Price Competition and Innovation Act, introduced as part of the Affordable Care Act, directed the FDA to create an approval pathway for biologic products shown to be biosimilar or interchangeable with an FDA-approved innovator drug. These biosimilars will not be chemically identical to the reference agent. Investigational studies conducted with biosimilar agents will likely provide limited real-world evidence of their effectiveness and safety. How do we best monitor effectiveness and safety of biosimilar products once approved by the FDA and used more extensively by patients?
OBJECTIVE:
To determine the feasibility of developing a distributed research network that will use health insurance plan and health delivery system data to detect biosimilar safety and effectiveness signals early and be able to answer important managed care pharmacy questions from both the government and managed care organizations.
METHODS:
Twenty-one members of the AMCP Task Force on Biosimilar Collective Intelligence Systems met November 12, 2013, to discuss issues involved in designing this consortium and to explore next steps.
RESULTS:
The task force concluded that a managed care biosimilars research consortium would be of significant value. Task force members agreed that it is best to use a distributed research network structurally similar to existing DARTNet, HMO Research Network, and Mini-Sentinel consortia. However, for some surveillance projects that it undertakes, the task force recognizes it may need supplemental data from managed care and other sources (i.e., a “hybrid” structure model).
CONCLUSIONS:
The task force believes that AMCP is well positioned to lead the biosimilar-monitoring effort and that the next step to developing a biosimilar-innovator collective intelligence system is to convene an advisory council to address organizational governance.
What is already known about this subject
The FDA’s implementation of the 351(k) pathway for the approval of biosimilars is pending.
Biosimilar agents will not be exact copies of the innovator or reference product, raising issues of interchangeability and equivalent outcomes.
With limited real-world data available upon approval, managed care pharmacy can assure the public that safety and effectiveness will be monitored by initiating active surveillance methods to monitor biosimilars versus innovator products using a managed care data consortium.
Surveillance systems based on distributed research networks have demonstrated ability to answer public health questions through the data collection capabilities of the partner organizations (e.g., payers and providers).
What this study adds
The need will exist for a distributed research network-based surveillance system dedicated to monitoring biosimilar drugs and their innovator products for clinical and safety outcomes.
This surveillance system should be a hybrid model built on the foundation established by existing initiatives but utilizing additional data sources.
On November 12, 2013, the Academy of Managed Care Pharmacy (AMCP) convened a multidisciplinary task force to identify a system to monitor the safety and effectiveness of biosimilar products after U.S. Food and Drug Administration (FDA) approval and provide consumers and providers with greater confidence in biosimilars. The AMCP Task Force on Biosimilar Collective Intelligence Systems included individuals from health insurance plans, health delivery systems, pharmacy benefit management companies (PBMs), specialty pharmacies and the pharmaceutical industry, along with executives and policymakers from professional and trade associations, patient advocacy, government agencies, and academia. A full list of task force members is shown at the end of this article.
The purpose of this task force was to determine the feasibility of launching a managed care biosimilars collective intelligence system to monitor the safety and efficacy of biosimilars, with reference to the data available for the innovator specialty drug, using available technologies, data sources, and observational research methods.
The Biologics Price Competition and Innovation Act, introduced as part of the Affordable Care Act,1 directed the FDA to create an abbreviated approval pathway for biologic products shown to be biosimilar or interchangeable with an FDA-approved biologic innovator or reference drug. It is expected that the FDA’s section 351(k) approval pathway for biosimilars will require that investigational studies of the biosimilar demonstrate pharmacokinetics and pharmacodynamics comparable with the innovator product, as well as yield clinical outcomes that approximate those seen with the original agent.2 Using the “totality of evidence” approach, a sponsor is expected to demonstrate physicochemically, pharmacokinetically, pharmacodynamically, biologically, and clinically that there are no clinically meaningful differences between the biosimilar and the reference product.
However, biosimilars are not exact copies of the approved branded medical product. They may be manufactured through different means, and the final structure of the resulting compound may be similar to, but not exactly the same as, the innovator product. This can result in differences that are subtle and are difficult to predict. It is not yet clear whether the FDA will designate all biosimilars as interchangeable with the innovator product. In addition, because clinical data requirements remain unknown, the FDA has stated that it is unlikely any biosimilars will be designated as interchangeable medicines in the short term.
Like all new drug entries, the premarketing investigational studies required for biosimilar approval will provide limited evidence regarding the biosimilar’s safety and effectiveness in real-world practice. For biosimilars, the investigational studies will likely be carried out in even more carefully specified populations and controlled conditions, which suggests a strong case for active postmarketing surveillance by the managed care pharmacy community. Specifically, questions may still linger regarding the long-term safety of the product and whether it is as effective as the reference agent in populations with various comorbid conditions. In addition, the duration of the investigational trials may be inadequate to identify potentially important safety signals that may arise.
Specialty Drug Trends
The task force agreed that specialty products are generally defined by the complexity in storage and distribution, biologic processes necessary in their manufacturing, need for more careful patient monitoring, and their relatively high cost. At a commonly accepted definition of at least $1,200 per month (and even a threshold of at least $600 per month), the issue of affordability is very real.3,4 Specialty pharmaceutical spending in the United States is expanding rapidly, with double-digit increases each year. One PBM documented an 18.4% annual increase in specialty drug expenditures, while the money spent on traditional drugs dropped by 2.4%.3 Express Scripts estimates that by 2016, 7 of the top 10 drugs in terms of sales will be biologics.5 Artemetrx calculates that by 2018, specialty drug spending in the United States is expected to surpass spending for traditional drug products.6
This trend is fueled both by the number of biologic agents in the pipeline and the number being approved by the FDA. According to the Pharmaceutical Research and Manufacturers of America, more than 600 specialty drugs are in the mid- to late-stage pipeline, with oncology products dominating the landscape.7 By October 31, 2013, the FDA had approved 25 specialty drugs and 17 traditional pharmaceuticals, which was more than in 2012 and in any year since 1998.8
The introduction of biosimilars may help blunt the impact to pharmacy and medical budgets. An analysis by Express Scripts found that the introduction of biosimilars for 11 biologic agents could save the U.S. health system more than $250 billion over the next decade.5 This analysis is based on assumptions of a certain level of savings per prescription and does not consider the potential costs of any new adverse drug events (ADEs) resulting from the use of new biosimilars. Although it is not known how ADEs may affect possible savings associated with biosimilars, public health experience (e.g., rofecoxib and the higher risk of myocardial infarction and stroke9) suggests that the cost of treating any ADEs discovered after approval could be substantial. This provides a strong argument for close clinical monitoring of biosimilar products to identify and minimize associated negative clinical and financial effects.
The Need for Postapproval Biosimiar Monitoring
The FDA has the authority to implement the 351(k) approval pathway, which is expected to be operational in the near future. However, existence of a formal approval pathway will not solve the need for a biologics effectiveness or safety-monitoring system. A monitoring system can collect and continually analyze new data for both the innovator and biosimilar as more experience is gained with newly approved biosimilars, searching for potential safety signals using coding and information technology infrastructure that exists today (e.g., National Drug Code [NDC] numbering; International Classification of Diseases, Ninth Revision, Clinical Modification [ICD-9-CM]; Healthcare Common Procedure Coding System [HCPCS], and National Council for Prescription Drug Programs [NCPDP] standards for electronic data interchange).5 The monitoring system could complement postmarketing safety-monitoring requirements that might be implemented by the FDA (e.g., Risk Evaluation & Mitigation Strategy [REMS] or Elements to Assure Safe Use [ETASU] programs). For example, European regulators have required biosimilar manufacturers to develop postapproval risk-management plans.
The system can be constructed using available tools. Health plans’ and insurers’ administrative processes already capture claims and differentiate drug exposures, diagnoses, and procedures. Consider this analogous example: Multiple manufacturers produce the growth hormone somatropin, which is marketed under different brand names but has 1 U.S. Adopted Name (USAN) designation.5 (Somatropin is not considered a biosimilar, as the pathway did not exist when it was approved by the FDA.) Modern data systems of individual health plans and insurers gather claims and utilization information and evaluate the use of various somatropin products, despite the potentially confusing mix of manufacturers.5
The need for biosimilar monitoring grows out of our understanding of today’s FDA review and approval process, and the experience gained with biologic utilization to date. Payers have little comparative effectiveness information at the time of approval for any new product or molecular entity. The safety profile of innovator products is the sum of experience gained over many years of use (and clinical trials), including off-label use. For the most part, systems in place to report ADEs are passive in nature (e.g., FDA Adverse Event Reporting System). Biosimilars are not exact replicas of the innovator or reference product; therefore, they may have ADE risks that differ from those seen with the innovator product in addition to ADE risks that can be similar to those seen with the innovator product. Unfortunately, an efficient and reliable system for actively monitoring and uncovering what we don’t know does not yet exist. An active monitoring system that achieves this crucial objective could be of great benefit to patients, health care providers, drug makers, federal regulators, and the health care delivery system in general.
A Sample of Approvals to Come?
In 2012, the FDA approved Teva Pharmaceuticals’ new form of filgrastim, called tbo-filgrastim. Teva filed a conventional new biologic license application because the new biosimilar approval pathway was not available. The drug was approved but without the full labeling carried by the innovator product.10
The European Medicines Agency (EMA) approved tbo-filgrastim as a biosimilar in 2008. In fact, Europe is well ahead of the United States in the acceptance and use of biosimilars overall. Biosimilars for erythropoiesis-stimulating agents were approved by European regulators in 2006.11 Table 1 lists the biosimilar agents that have been approved by the EMA’s Committee for Medicinal Products for Human Use since employing its pathway to approval in 2005.4,12
TABLE 1.
Biosimilar Products Approved by the European Medicine Agency 12
| Name | Nonproprietary Name | Indications | Date of Approval |
|---|---|---|---|
| Abseamed | Epoetin alfa | Anemia, cancer, chronic kidney failure | 8/28/2007 |
| Binocrit | Epoetin alfa | Anemia, chronic kidney failure | 8/28/2007 |
| Epoetin Alfa Hexal | Epoetin alfa | Anemia, cancer, chronic kidney failure | 8/28/2007 |
| Retacrit | Epoetin zeta | Anemia, autologous blood transfusion, cancer, chronic kidney failure | 12/18/2007 |
| Silapo | Epoetin zeta | Anemia, autologous blood transfusion, cancer, chronic kidney failure | 12/18/2007 |
| Biograstim | Filgrastim | Cancer, hematopoietic stem cell transplantation, neutropenia | 9/15/2008 |
| Filgrastim Hexal | Filgrastim | Cancer, hematopoietic stem cell tnsplantation, neutropenia | 2/6/2009 |
| Grastofil | Filgrastim | Neutropenia | 10/18/2013 |
| Ratiograstim | Filgrastim | Cancer, hematopoietic stem cell tansplantation, neutropenia | 9/15/2008 |
| Nivestim | Filgrastim | Cancer, hematopoietic stem cell transplantation, neutropenia | 6/8/2010 |
| Tevagrastim | Filgrastim | Cancer, hematopoietic stem cell transplantation, neutropenia | 9/15/2008 |
| Zarzio | Filgrastim | Cancer, hematopoietic stem cell transplantation, neutropenia | 2/6/2009 |
| Ovaleap | Follitropin alfa | Anovulation | 9/27/2013 |
| Inflectra | Infliximab | Psoriatic arthritis, rheumatoid arthritis, ulcerative colitis, Crohn’s disease, psoriasis, akylosing spondylitis | 9/10/2013 |
| Remsima | Infliximab | Psoriatic arthritis, rheumatoid arthritis, ulcerative colitis, Crohn’s disease, psoriasis, akylosing spondylitis | 9/10/2013 |
| Omnitrope | Somatropin | Pituitary dwarfism, Prader-Willi syndrome, Turner syndrome | 4/12/2006 |
Note: The medications listed in this table are not all distinct molecules. For example, there are only 2 biosimilar epoetins (epoetin alfa, marketed as Abseamed, Binocrit, and Epoetin Alfa Hexal, and epoetin zeta, marketed as Retacrit and Silapo).
Surveillance Systems and Distributed Research Networks
Biologics are often used to treat rare diseases in small patient populations. Therefore, capturing significant safety signals associated with the use of biosimilar and innovator products will require surveillance systems that rely on large health care networks, perhaps a multisite system, to provide the breadth of information necessary to detect rare outcomes and determine whether the outcomes are associated with use of the biologic above and beyond the occurrence in the underlying population in which it is used.
Managing, coordinating, and integrating the clinical and administrative data inputs from varied geographic locations, organization types, lines of business, and different information technology systems suggests that the “distributed research network” (DRN) model may be an excellent guide for a new surveillance initiative (Table 2).
TABLE 2.
Distributed Research Networks: Sample Organizations
|
Such a system requires inclusion of standardized data held and maintained behind the individual contributors’ firewalls. The data partners must control the system’s use and when (and how) the data are transferred to the DRN coordinating center. 13 DRNs typically require that only the minimum necessary information be transferred for analysis.
When doing multisite, sequential analysis through a DRN, the role of a central coordinating center is critical, not only for data analysis but also for coordination among the network partners.13 For example, a unique computer program must be developed for each query addressed through the DRN and provided by the coordinating center to each of the data partners to ensure consistent data analysis. These programs may not be modified by the contributing data partner.13
The FDA’s Mini-Sentinel
In 2009, the Mini-Sentinel program was implemented under the FDA’s public health authority. The agency utilizes a coordinating center (located at the Harvard Pilgrim Health Care Institute) to coordinate its data requests and reports. Mini-Sentinel enables the FDA to rapidly issue queries and implement assessments related to medical product safety surveillance.13 In its fifth year, the Mini-Sentinel program, through its DRN, has answered questions regarding the following:
What types of people (e.g., age groups, gender, conditions/diseases) are taking specific drugs
Whether the implementation of black box warnings changed patterns of care
The relationship between medication use and outcomes (prospective and retrospective analyses)14,15
To date, most of Mini-Sentinel’s activity has involved signal refinement and signal evaluation and obtaining rapid response to specific questions from the FDA (Figure 1).
FIGURE 1.
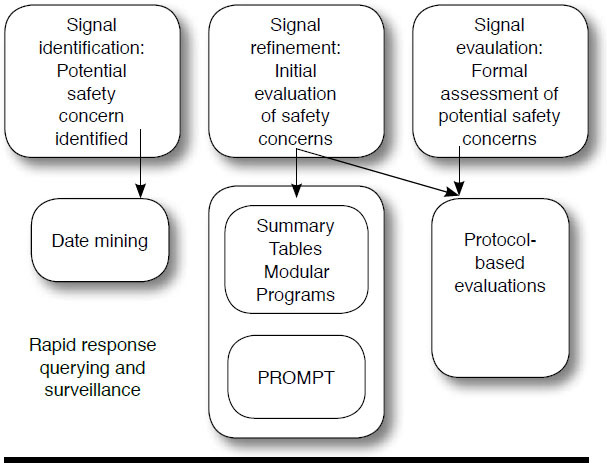
Mini-Sentinel Postmarket Safety Surveillance Activities13
The distributed data system of Mini-Sentinel uses administrative and clinical data captured by the Mini-Sentinel data partners. The Mini-Sentinel Coordinating Center sends questions to Mini-Sentinel’s partners, who run the queries on their own data (which have been formatted into the Mini-Sentinel Common Data model16 that standardizes data table formats at all participating sites), and return back to the Coordinating Center tabular data results (e.g., counts of medication exposures and outcomes). Participating Mini-Sentinel data partners refresh their distributed database tables periodically. Each time data are refreshed by the data partners, the data tables are subjected to 4 levels of checking and evaluation (i.e., more than 1,400 total checks per refresh; Figure 2).13
FIGURE 2.

Mini-Sentinel’s Steps for Data Quality Assurance and Evaluation13
Mini-Sentinel uses a variety of methods to query the distributed data. Queries can use validated “modular” programs or summary tables for routine requests, or new analytic programs can be developed for cases in which the prespecified programs are not appropriate.17 As previously mentioned, to preserve the integrity and consistency of the analysis, programs (queries) cannot be changed by the data partners. The programming, testing, and certification process is time consuming, but it is a necessary step to ensure that Mini-Sentinel can accurately address each query (Figure 3).13 To illustrate, among Mini-Sentinel’s data and scientific partners, there are 10 different computing environments (e.g., Windows, Linux, Unix) and multiple versions of the SAS software (e.g., 9.2, 9.3, or 9.4, in addition to different versions of each release). Each of the data partners has a unique local hardware system. Every time a program is written, it must be tested to ensure that it runs flawlessly across all of these environments.13 Current Mini-Sentinel data partners include Aetna, HealthCore (Wellpoint), HMO Research Network, Humana, Kaiser Permanente, Optum, and the Vanderbilt School of Medicine.13
FIGURE 3.

Mini-Sentinel New Program Development
New Electronic Clinical Data Infrastructure
One hundred million dollars was invested by the Agency for Healthcare Research and Quality (AHRQ) to create a new multipurpose, electronic clinical data infrastructure and to advance the methods to create and use this infrastructure. It achieved this by funding 4 interrelated grant programs to advance methods in this field:
Prospective outcome systems using patient-specific electronic data to compare tests and therapies (PROSPECT; 6 grants)
Scalable distributed research networks (3 grants)
Enhanced registries for quality improvement and comparative effectiveness research (2 grants)
Electronic Data Methods (EDM) Forum (1 grant)18
The EDM Forum helped advance the methods and share lessons learned in creating and using the infrastructure. The AHRQ project will focus on 4 domains: (1) analytic methods, (2) clinical informatics, (3) governance, and (4) learning health system.
The objectives of the EDM Forum and the other electronic data infrastructure projects are listed in Table 3. The EDM Forum convenes investigators and stakeholders to understand, prioritize, and tackle challenges in building and using electronic data infrastructure for diverse purposes. It conducts collaborative methods projects and creates a variety of products to disseminate knowledge and inform a broad audience.
TABLE 3.
Objectives of AHRQ’s Electronic Data Infrastructure Projects
|
AHRQ = Agency for Healthcare Research and Quality.
Study results from the EDM Forum are disseminated through a number of different venues:
Community web portal (http://www.edm-forum.org)
Online repository (http://repository.academyhealth.org), which contains links to publications, issue briefs, archived webinars, symposia materials
eGEMs (http://repository.academyhealth.org/egems)—a new open-access journal
Supplements to other journals (e.g., Medical Care July 2012 and August 2013)18,19
EDM Forum monthly newsletter (https://ahedm.wufoo.com/embed/z7p9r7/)
Twitter and RSS feeds (https://ahedm.wufoo.com/embed/z7p9r7/)
eGEMS, published with AcademyHealth, emphasizes clinical informatics, analytics, and governance issues relevant to research and to building the learning health system. It includes a review of data governance programs and pathways for success in multisite research and a governance toolkit for multisite research.20
Analytic Approaches to Surveillance
Three types of active surveillance approaches exist, when searching for ADEs: (1) epidemiologic study of an existing safety signal after a specified time or exposure, (2) sequential analysis of accumulating data based on a potential safety signal observed with the same or a similar medical product, and (3) data mining for identifying potential (unspecified) safety signals.13 Monitoring for safety signals in biosimilar use is an exercise in sequential analysis of accumulating data, in which one starts reviewing data on both the innovator and biosimilar as early as possible. Over time, more observational information is added to the surveillance database. In sequential analysis, data are extracted, manipulated, summarized, and analyzed continuously as more information accumulates to search for safety signals. This form of surveillance requires specific data analysis methodology (e.g., sequential probability ratio testing) as the same data are being subjected to repeated statistical testing.13,21
When applied to drug-specific issues, additional adjusters must be addressed in the data collection and analysis. These adjusters include the following22:
Determining patterns of drug use; i.e., whether drug use is associated with new therapy (incident use), chronic treatment, or intermittent use
Accommodating misclassification of exposure (e.g., nonadherence, prior drug use, concomitant drug use)
Evaluating the effect of comorbidities on outcomes
Brown et al. (2007)22 applied these methods retrospectively to simulate how ADEs would be flagged in a DRN, using several significant historical examples. Figure 4 illustrates how this surveillance model would have identified an increased incidence of acute myocardial infarction (the safety signal) after 34 months of exposure for patients taking rofecoxib compared with naproxen. The safety signal is raised when the observed event line in Figure 4 diverges significantly from the expected number of events until it crosses the relative risk line. This does not, however, prove that the drug therapy is responsible for the outcome. It simply identifies the signal, allowing one to then investigate causality.
FIGURE 4.
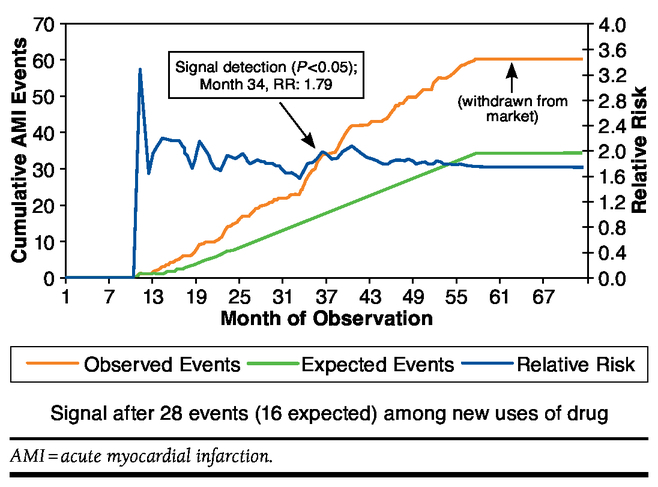
Observed and Expected Events for Rofecoxib Versus Naproxen Users: 2000-2005 22
Critical Success Factors in Working with Distributed Research Networks
Multisite, electronic health record-based data networks have tremendous potential for improving patient outcomes and advancing scientific knowledge but need upfront buy-in from multiple decision makers and stakeholders. This means many resources are required to set it up for optimal use, with the upfront efforts yielding long-term benefits.18
Another key to successfully coordinating (and participating) in these collaborative networks is to develop trust and respect, with all DRN partners working towards a common mission. To gain that trust, each partner must be transparent and frank regarding the potential and limitations of its organization’s data.23
When entering into these collaborations, no one partner can dominate decision making. The collaborating partners may have different governance and corporate philosophies, and it may be necessary to obtain contract approval in different countries (e.g., pharmaceutical company parents based in Europe). Even agreeing on the right research question takes patience, understanding, and time.23
Project classification is an important consideration for the new DRN’s data partners. For example, data collection and dissemination can be conducted under the umbrella of research or as a public health surveillance initiative. Under the former, privacy issues, human subjects’ protection in research, and other obstacles may be challenging. Under the public health umbrella, a simpler approval process may prevail when de-identified data are used (i.e., the use of third-party data that have not been de-identified will require approval of the third party).
The time frame for obtaining the institutional review board approval and accessing charts for public health surveillance, writing the research protocol, and completing the background work necessary for the launch of a surveillance project should be taken into account.13 Furthermore, communicating important information to all key stakeholders, including the public, must also be considered. A protocol around how the public is notified of a clinically important finding (e.g., a significant safety signal) would be developed, and this issue includes the notification of all key stakeholders. It is possible that notifications could be efficiently communicated through a central, respected source, such as the FDA.
Task Force Consensus Decision Making
Fine-Tuning the Pharmacovigilance Mission
When the FDA approves a new product, it sometimes may make the decision based on limited available evidence. For these products, including biosimilars, postmarketing surveillance will be critical to ensuring the long-term safety and efficacy of a new product when used widely after approval. One example of how the FDA is managing the risks of drug approval based on limited evidence is through the REMS program.
Pharmacovigilance through active surveillance networks will be critical to this effort. Although some coordinated DRNs equipped to handle pharmacovigilance are operational (e.g., HMO Research Network, Mini-Sentinel), others are still developing (e.g., PCORnet DRN, NIH Health Care Systems Collaboratory DRN). In addition, the FDA’s biosimilar approval pathway may not require the same investigational clinical trial prerequisites as the innovator product, resulting in a perceived greater need for more monitoring of real-world outcomes associated with these agents and the innovator to confirm efficacy and safety.
The AMCP task force decided that the appropriate role of a new biosimilar surveillance system would be to monitor the long-term safety and effectiveness of these products along with the innovator products. The task force also considered that some populations may not be well represented in investigational clinical trials of biosimilars, and monitoring the effectiveness and safety of the biosimilar and the innovator in these specific populations would be an important objective of the new DRN-based consortium. The results may also have important implications for the interchangeability of biosimilars and the innovator biologics. Task force members also considered that such a DRN might also be used to monitor safety signals and effectiveness for previously approved biologics including new biosimilars.
Use an Existing Consortium or Build a New System?
The panel concluded that existing consortia do not publish sufficient quantities of information, and the time span between data evaluation and publication can be lengthy. The task force believes that unless managed care motivates the effort, this will not change. Several managed care organizations have contributed significant resources to develop an infrastructure that makes active DRN surveillance possible. Managed care pharmacy must marshal resources to realize important public health benefits inherent in monitoring biosimilar and innovator safety and effectiveness.
The task force considered whether an alternative approach might better support its ability to conduct pharmacovigilance (i.e., help it to evolve more quickly to meet the required task). A few of the issues considered in the question of whether to “buy or build” are discussed here:
The Need to Better Capture ADEs.
The probability that a practitioner will report an ADE is relatively low, unless it is a life-threatening or mortal event. This may relate to clinicians’ questions as to whether a particular outcome directly results from an ADE or the lack of convenient ADE-reporting systems. Processes should be in place or other sources should be available to capture fuller information on ADE occurrences. The task force recommended that in addition to an active DRN, the research construct should incorporate secondary data sources either residing within health plan and insurers’ medical and claims records or data mining social media outlets for reports of ADEs (e.g., Facebook, disease-oriented discussion groups).
The task force acknowledged that part of the surveillance challenge may be in “asking the right question” for data collection and analysis. The information-gathering capabilities of managed care plans are prodigious, but only by constructing an unambiguous query can the precise data point be found for analysis. For biosimilar agents, this may mean trying to identify an unexpected, new ADE—not simply ADEs that have been observed with the innovator agent. An active sequential approach with the biosimilar and innovator reduces the likelihood that the unexpected will be missed. Furthermore, the safety signals may only be relevant when a product, biosimilar or originator, is used for unapproved (i.e., off-label) indications. This may be another significant value of the collection and analysis of observational, real-world data.
The Capability to Investigate Broader Questions.
The AMCP task force considered the possibility of setting additional goals for the Biosimilar Collective Intelligence System. For example, consider a biosimilar tumor necrosis factor (TNF)-alpha inhibitor:
Will the efforts of the surveillance system be able to identify whether the biosimilar TNF-alpha inhibitor exhibits a better side-effect profile compared with the innovator product and other members of the class?
Will the surveillance system be able to evaluate whether this new TNF-alpha inhibitor is subject to dose escalation?
Will the surveillance system be able to contribute information on the health economics associated with these products?
The task force stated that the question asked in the first bullet is of fundamental importance to the mission of the new surveillance network. It acknowledged that, though important, the second and third bullets are examples of questions that can be addressed by individual payers’ research groups and might not require the scope of this public health postmarketing drug surveillance effort. In particular, researching economic questions, such as cost drivers of a particular episode of care, requires an in-depth understanding of contracting and other factors that influence costs, which vary significantly across health plans. The task force agreed that the stated public health mission, to actively monitor the safety and effectiveness of biosimilars, should be the focus of the surveillance effort.
Ask Questions of Interest to Managed Care.
Although the main objectives of this new collaborative drug surveillance program may overlap those of Mini-Sentinel, the questions chosen for evaluation in Mini-Sentinel are chosen by the FDA alone. A new consortium would be able to address questions chosen by other qualified parties and by managed care. For example, does the FDA ask the same safety questions that managed care may ask? Different organizational priorities may result in different questions and issues to be evaluated. For example, managed care might want to understand longitudinal utilization of growth hormones with respect to changes in children’s growth curves over a specified period. Fundamentally, managed care has a need to expedite understanding of biosimilars, especially regarding how their safety and effectiveness compare with the originator products.
The task force believes that existing DRNs have much to offer this drug surveillance effort, and leveraging the work of networks such as Mini-Sentinel would be preferable to a doit-yourself approach. To illustrate the breadth of its existing network, Mini-Sentinel collects data from 130 million unique individuals; this represents over 40 million current lives for which data are being accumulated.13
Efforts must still be taken to improve the existing data sets at the source, the task force acknowledged, with consideration given especially to accurate product identification (e.g., NDC vs. HCPC codes) and lab results in particular. These issues, however, are shortcomings of most payer-provided data. In addition, for biosimilar drug research, access to hospital data as well as infusion center data might be needed to answer some questions. These considerations favor the hybrid approach, where a core set of information exists in the DRN that can be supplemented by additional data from existing partners or from external sources (e.g., a patient registry, specialty pharmacy provider, or other resource) based on the specific query. A hybrid approach allows for other data, including ADEs reported to MedWatch and ADEs reported in journals’ reader correspondence or case reports. Although it would be challenging to merge these data sources into the larger feed and evaluation pathway, they can contribute important information to the biosimilar surveillance effort.
Technical Challenges: Accuracy, Transparency, Reliability, and Accountability
Sequential Analysis and Data Coordination.
The Mini-Sentinel program and the HMO Research Network may be considered models for how any new surveillance consortium approaches real-time data collection and analysis. Prospective, sequential analysis typically requires each data partner to execute an analytic program after each data refresh. That is, under sequential analysis, the analytic program is run once at time point A to obtain an observed and expected count for the innovator and biosimilar, and each time the data is refreshed, the same program runs again, and those data are added to generate a cumulative count. No real coordination is needed in terms of when programs must refresh. Coordination of refresh times may be necessary to evaluate the effectiveness of a new flu vaccine, for example, but this is not the norm. This type of sequential analysis requires adjustment for multiple statistical tests.
In existing DRNs, the rate at which data are refreshed depends upon the question asked. With the new DRN, it is possible that data refreshes can occur weekly, depending on the public health question being addressed.
Claims Coding to Comprehensively Identify Biosimilar Use.
Biosimilar products such as filgrastim, which are covered on the medical benefit of commercial plans, typically receive a “miscellaneous” J code upon approval. The medical benefit databases frequently lack data on the specific product and dosage used.
A coding question to be answered is whether the nonproprietary name of each biosimilar agent will be unique. The FDA is currently considering several options, including keeping the same USAN for the biosimilar and innovator product, having completely different USANs (which could complicate the use of interchangeable products), or using a prefix or suffix to the originator’s USAN to differentiate the products (e.g., tbo-filgrastim [even though this is not technically a biosimilar, based on the FDA’s approval processes]).
For the purposes of drug surveillance and data evaluation, the easiest scenario would be for each biosimilar to have a unique HCPCS code, which is the case today with the 2 currently available forms of epoetin alfa, Epogen and Procrit. Additionally, similar vaccine preparations are also assigned unique HCPCS codes. Ironically, biologic products may not be completely identical from one manufactured batch to another. Although this may not have any effect on clinical safety or effectiveness. a unique J code assigned to a biosimilar product, for example, does not imply that all of the lots of that agent are homogeneous.
A months-long delay in assigning a permanent HCPCS code to a new biosimilar could hinder surveillance efforts during this very early period of utilization. Yet, other challenges need to be overcome with the use of HCPCS codes only:
Dose cannot be identified through the HCPCS code in claims databases
Indication or off-label use cannot be identified through the HCPCS code alone (e.g., biosimilar TNF-alpha inhibitor approved only for use in rheumatoid arthritis will likely be used in Crohn’s disease and perhaps psoriasis—these patient populations may have very different risk profiles).
Furthermore, one cannot accurately infer dosage through drug quantity information.
If the dosage or off-label use cannot be identified from the HCPCS code, is this code the best option available for surveillance purposes? Coupling this information with electronic medical record data may provide a more complete picture, but this process is associated with its own challenges. Ideally, one would want to identify the product, the manufacturer, the lot number, the dosage, and the indication. The AMCP task force considered the feasibility of using NDC data, if it were widely available in the medical claims records, to help fill the gaps. Inclusion of NDC is not presently required for reimbursement, and until NDC coding is required for claim reimbursement, there will be significant hurdles. Some analyses, however, suggest that several payer databases have access to NDC-level data that can be mapped in a DRN.24
A Beta-Test in Preparation for the First Biosimilar Approval?
The approval of tbo-filgrastim, which is technically not a biosimilar approved through the FDA 351(k) pathway, represents an opportunity to test a multisite drug surveillance consortium’s systems, data queries, and statistical analyses. If some payers viewed this new product as a biosimilar, using it instead of, or in addition to, the innovator version of filgrastim, this scenario could prepare the consortium’s drug surveillance program for the approval of the first FDA-approved biosimilar agent. In fact, if 1 of the health plans in the data consortium implements a full switch to tbo-filgrastim, and another delays the full switch for 6 months, this could provide the foundation of a natural experiment: to compare outcomes in the 2 populations.
Developing an Action Plan for a Biosimilar Collective Intelligence System
Value Proposition
The AMCP task force believes that having a resource to collect and analyze biosimilar and innovator safety and effectiveness data derived from managed care populations in a longitudinal manner is of considerable value. Ad hoc safety questions also arise on a regular basis, which could be addressed through the network.
The overall value to managed care and to U.S. health care in general will be in the early detection of safety signals for biosimilar products and, perhaps, other biologics. The early detection of safety signals may result in direct and indirect societal savings, including mortality, ADE management, lost productivity, and use of health resources.
For participating partners in the distributed research network, the cost of providing data for any 1 project (i.e., to answer any 1 public health question) should not be exclusively borne by the managed care plans. Whereas the managed care plans will benefit from the results provided by a DRN, a methodology for paying data partner expenses must be considered.
Choosing a Foundational Program on Which to Base the New Drug Surveillance System
The task force believes that it would make sense to establish which of the established DRNs can provide the best foundation for the new drug surveillance consortium. This may be addressed by a future advisory council through a request for proposal.
Potential Clients for the Drug Surveillance System
The task force agreed that any data partner should have the ability to submit a query to the consortium for analysis. They also agreed that the FDA should have access to the consortium’s DRN to help answer questions regarding ADEs or other critical drug outcome questions.
Whereas the FDA and managed care organizations are expected to be the primary clients for information generated from the surveillance consortium, the manufacturing sector may want to utilize the biosimilar consortium for product safety tracking and other quality questions. However, this may raise credibility and conflict-of-interest issues. These issues will need to be explicitly addressed in the consortium’s charter and policies before manufacturers could submit independent questions for research through the consortium’s governing structure.
Integral to any DRN is the understanding that individual data partners may decide against researching a particular query because of a potential perceived conflict. The task force agreed that the biosimilar consortium governance would follow existing consortia’s leads in addressing this issue.
Consortium Participants
Because the task force members favored a hybrid model for data collection and analysis, they would first consider inviting existing FDA’s Mini-Sentinel program data partners. Other managed care organizations, data warehouses, and governmental institutions may be asked to participate, subject to additional data needed to yield an optimally inclusive information set for collection and analysis.
Funding Model
Mini-Sentinel currently costs the FDA $12 million per year for data infrastructure and routine querying. Mini-Sentinel partners can use their data in Mini-Sentinel format for other purposes, such as the proposed network. Since this partnership model—with its program development, common data model, and data quality assurance—is established, developing a coordinating center similar to Mini-Sentinel’s model may cost considerably less.
The funding model will help define the scope of the surveillance consortium’s activities. The task force discussed 2 parallel streams of funding: (1) a government stream and (2) a science stream. The source of funding is also affected by whether the work done by the consortium is considered public health surveillance (as in Mini-Sentinel) or research. The answer to this question has implications for whether the consortium can quickly answer ad hoc questions or whether a research protocol will first need to be developed, with possible institutional review board review and approval.
The task force wondered whether AMCP should serve as a potential funding source, as well as play a key role in helping to organize the surveillance network. Funding through AMCP would come from solicited sponsors and patrons, and AMCP would serve as the fiscal intermediary. Furthermore, the task force maintained that possible seed funding for the project may be obtained through conference or planning grants from the AHRQ or the National Institutes of Health.
Funding may be more topic-specific. Not everyone will be interested in a question asked by one payer or on a particular aspect of therapy. Therefore, individual query analysis may be funded by the interested health plan.
Creation of an Advisory Council
The next step in the process is to appoint an advisory council (Table 4) to address governance issues, data partner contracts, and additional foundational questions, informed by the task force’s findings.
TABLE 4.
Proposed Advisory Council Representatives
|
CMS = Centers for Medicare & Medicaid Services; FDA = U.S. Food and Drug Administration.
The governance discussions are the first priority. In the FDA’s Mini-Sentinel program, fully 12 months were needed to finalize its governance. An additional 6 months were required to address contracting issues.13 Mini-Sentinel’s contracts are public and could be leveraged for this new consortium. The task force also considered that time to develop a governance structure could be shortened by considering the surveillance program as a pilot program, establishing a strong governance foundation later for implementing the fully evolved consortium.
Conclusions
As the FDA continues to complete its section 351(k) pathway for the evaluation and approval of biosimilar agents, a mechanism should be developed to help ensure that these agents and their innovators are monitored for safety and effectiveness over the long term, in far more patients than available in an investigational clinical phase 2 or 3 trial.
The primary mission of surveillance consortia, such as Mini-Sentinel, the proposed biosimilar drug surveillance consortium, and others, is to gather data and conduct analysis to address public health issues (e.g., monitor the long-term safety and effectiveness of new biosimilar agents and identify safety signals that require further evaluation). A secondary important mission is to answer ad hoc queries involving these topics that are of importance to managed care organizations and the health system to improve care and care efficiency.
The AMCP gathered a task force to begin the planning process for the Biosimilar Collective Intelligence System, a distributed research network of health plans, PBMs, and academia to achieve these goals. AMCP will take a leading role in the next step of development—the organization of an advisory council to address the governance of the Biosimilar Collective Intelligence System and partner contracting.
ACKNOWLEDGMENTS
The AMCP task force would like to express its thanks to Stanton R. Mehr of SM Health Communications for his assistance in reporting on the deliberations of the task force and in producing this manuscript.
Authors
TASK FORCE MEMBERS: MIkE BALDzIkI, CRCM, Executive Vice President, Industry Relations & Advocacy, Armada; PHIL BALL, PhD, Director, Biologics Alliance Management and Government Affairs, Actavis, Inc.; JEFF BROWN, PhD, Therapeutics Research and Infectious Disease Epidemiology, Department of Population Medicine, Harvard Pilgrim Health Care Institute/Harvard Medical School (speaker); HUNGCHING CHAN, MPH, Vice President, Medical Informatics, OptumInsights; T. CRAIG CHEETHAM, Researcher, Pharmacy Analytical Services, Kaiser Permanente; THOMAS CONN, Chief Strategy Officer, Armada; GREGORY W. DANIEL, PhD, MPH, RPH, Fellow, Economic Studies and Managing Director, Brookings Engelberg Center for Health Care Reform; MARK HENDRICKSON, Director for Sciences and Regulatory Affairs, Generic Pharmaceutical Association; LUTZ HILBRICH, MD, MBA, Global Head Integrated Program Management Biosimilars, Boehringer Ingelheim; AYANNA JOHNSON, MS, Health Policy Associate, National Consumers League; STEVEN B. MILLER, MD, Senior Vice President & Chief Medical Officer, Express Scripts, Inc. (speaker); TOM MOORE, Director, US Value and Access, Amgen, Inc.; BRENDA MOTHERAL, RPh, MBA, PhD, President, Artemetrx (moderator); SARAH A. PRIDDY, PhD, Director of Research, Humana Comprehensive Health Insights (speaker); MARSHA A. RAEBEL, PharmD, Senior Investigator, Kaiser Permanente Colorado Institute for Health Research; GURVANEET RANDHAWA, MD, MPH, Medical Officer, Senior Advisor on Clinical Genomics & Personalized Medicine, Center for Outcomes and Evidence (COE), Agency for Healthcare Research and Quality (speaker); PENNY SURRATT, RN, MBA, Manager, Channel Strategies—Specialty Products, Humana; CHERYL WALRAVEN, PhD, Informatics Manager, FDA Mini-Sentinel Project, Aetna; T. JEFF WHITE, PharmD, MS, Director, Drug Evaluation and Clinical Analytics, Clinical Pharmacy Policy, Wellpoint; and YI ZHENG, Vice President and Chief Pharmacy Officer, Aetna.
AMCP STAFF: KEVIN BRUNS, Senior Director of Communications; MARY JO CARDEN, RPh, JD, Director, Regulatory Affairs; CHARLIE DRAGOVICH, Director, Business Development; BERNADETTE EICHELBERGER, PharmD, Director, Pharmacy Affairs; EDITH ROSATO, RPh, IOM, Chief Executive Officer; and TODD SEGA, PharmD, Manager, Pharmacy Affairs.
AUTHOR CORRESPONDENCE: Bernadette Eichelberger, PharmD, Director, Pharmacy Affairs, Academy of Managed Care Pharmacy, 100 N. Pitt St., Ste. 400, Alexandria, VA 22314. E-mail: beichelberger@amcp.org.
REFERENCES
- 1.Patient Protection and Affordable Care Act. Public Law 111-148: 124 Statute 119. 111th Congress. March 23, 2010. Congress.gov. Available at: http://beta.congress.gov/111/plaws/publ148/PLAW-111publ148.pdf. Accessed April 21, 2014.
- 2.U.S. Food and Drug Administration. Guidance for industry: scientific consideration in demonstrating biosimilarity to a reference product. February 2012. Available at: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291128.pdf. Accessed April 21, 2014.
- 3.Express Scripts Inc. The 2013 drug trend report. April 2014. Available at: http://lab.express-scripts.com/~/media/pdfs/drug%20trend%20report/express%20scripts%202013%20drug%20trend%20report.ashx. Accessed April 21, 2014.
- 4.Dalgaard K, Evers M, de Silva JS.. Biosimilars seven years on: where are we and what’s next? McKinsey and Company. February 2013. Available at: http://www.mckinsey.com/Search.aspx?q=Biosimilars&l=Insights%20%26%20Publications. Accessed April 21, 2014.
- 5.Miller S. Defining biosimilars and their effect on healthcare. Presented at: Meeting of the AMCP Task Force on Biosimilar Collective Intelligence System: Utilizing Data Consortiums to Prove Safety and Effectiveness of Biosimilars; Alexandria, VA; November 12, 2013. [Google Scholar]
- 6.Motheral B. Specialty drug trend across the medical and pharmacy benefit. Artemetrix. 2013. Available at: http://www.artemetrx.com/docs/ARTEMETRX_Specialty_Trend_Rpt.pdf. Accessed April 21, 2014.
- 7.Pharmaceutical Research and Manufacturers of America. Medicines in development: biologics. 2013. Available at: http://www.phrma.org/sites/default/files/pdf/biologics2013.pdf. Accessed April 21, 2014.
- 8.CenterWatch. FDA approved drugs. November 2013. Available at: http://www.centerwatch.com/drug-information/fda-approvals/. Accessed April 21, 2014.
- 9.Atchison JW, Herndon CM, Rusie E.. NSAIDs for musculoskeletal pain management: current perspectives and novel strategies to improve safety. J Manag Care Pharm. 2013;19(9 Suppl A):S1-S19. Available at: http://www.amcp.org/WorkArea/DownloadAsset.aspx?id=17383. [PubMed] [Google Scholar]
- 10.Food U.S. and Administration Drug. FDA approves new treatment for severe neutropenia in certain cancer patients. News and Events. August 29, 2012. Available at: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm317392.htm. Accessed April 21, 2014.
- 11.Mikhail A, Farouk M.. Epoetin biosimilars in Europe: five years on. Adv Ther. 2013;30(1):28-40. [DOI] [PubMed] [Google Scholar]
- 12.European Medicines Agency. Biosimilar medicines. November 19, 2013. Available at: http://www.ema.europa.eu/ema/index.jsp?curl=pages/special_topics/document_listing/document_listing_000318.jsp&mid=WC0b01ac0580281bf0. Accessed April 21, 2014.
- 13.Brown J. Reviewing current landscape of existing data consortiums: how they are being used, what they uncover, how they function—the Mini-Sentinel example. Presented at: Meeting of the AMCP Task Force on Biosimilar Collective Intelligence System: Utilizing Data Consortiums to Prove Safety and Effectiveness of Biosimilars; Alexandria, VA; November 12, 2013. [Google Scholar]
- 14.Southworth MR, Reichman ME, Unger ER.. Dabigatran and postmarketing reports of bleeding. N Engl J Med. 2013;368(14):1272-74. [DOI] [PubMed] [Google Scholar]
- 15.Food U.S. and Administration Drug. FDA drug safety communication: FDA approves label changes to include intestinal problems (sprue-like enteropathy) linked to blood pressure medicine olmesartan medoxomil. July 3, 2013. Available at: http://www.fda.gov/downloads/Drugs/DrugSafety/UCM359496.pdf. Accessed April 21, 2014.
- 16.Mini-Sentinel. Overview and description of the common data model v4.0. 2014. Available at: http://www.mini-sentinel.org/work_products/Data_Activities/Mini-Sentinel_Common-Data-Model.pdf. Accessed April 21, 2014.
- 17.Curtis LH, Weiner MG, Boudreau DM, et al. Design considerations, architecture, and use of the Mini-Sentinel distributed data system. Pharmacoepidemiol Drug Saf. 2012;21(Suppl 1):S23-S31. [DOI] [PubMed] [Google Scholar]
- 18.Marsolo K. Approaches to facilitate institutional review board approval of multicenter research studies. Med Care. 2012;50(Suppl):S77-S81. [DOI] [PubMed] [Google Scholar]
- 19.Hersh WR, Weiner MG, Embi PJ, et al. Caveats for the use of operational electronic health record data in comparative effectiveness research. Med Care. 2013;51(8 Suppl 3):S30-S37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Randhawa G. The EDM Forum approach to overcome traditional barriers in using electronic data sources for CER and QI. Presented at: Meeting of the AMCP Task Force on Biosimilar Collective Intelligence System: Utilizing Data Consortiums to Prove Safety and Effectiveness of Biosimilars; Alexandria, VA; November 12, 2013. [Google Scholar]
- 21.Davis RL, Kolczak M, Lewis E, et al. Active surveillance of vaccine safety: a system to detect early signs of adverse events. Epidemiology. 2005;16(3):336-41. [DOI] [PubMed] [Google Scholar]
- 22.Brown JS, Kulldorff M, Chan KA, et al. Early detection of adverse drug events within population-based health networks: application of sequential testing methods. Pharmacoepidemiol Drug Safety. 2007;16(12):1274-84. [DOI] [PubMed] [Google Scholar]
- 23.Priddy SA. Distributed data networks: meeting MCO knowledge management concerns—security, privacy, and access. Presented at: Meeting of the AMCP Task Force on Biosimilar Collective Intelligence System: Utilizing Data Consortiums to Prove Safety and Effectiveness of Biosimilars; Alexandria, VA; November 12, 2013. [Google Scholar]
- 24.Curtis JR, Xie F, Chen R, et al. Identifying newly approved medications in Medicare claims data: a case study using tocilizumab. Pharmacoepidemiol Drug Saf. 2013;22(11):1214-21. [DOI] [PMC free article] [PubMed] [Google Scholar]