

Abstract
Biosimilars are biological products that are highly similar to their biological reference products, notwithstanding minor differences in clinically inactive components. However, unlike generics of small-molecule drugs, biosimilars are not identical to their reference products, since each manufacturer uses unique cell lines and processes, and these lead to slight structural differences between products. Because these structural variations can lead to differences in clinical response, clinical studies demonstrating biosimilarity are required before and robust pharmacovigilance after approval. Although the FDA has not yet issued formal guidance on interchangeable biosimilars, higher standards of similarity will be required in order to achieve an interchangeable designation. In this commentary, we review the differences between generics and biosimilars, describe their respective regulatory approval pathways, discuss interchangeability and substitution, and review substitution of interchangeable biosimilars, focusing on key professional considerations for pharmacists.
The entrance of biosimilars into the U.S. health care system is imminent. Biosimilars are subsequent versions of an original biological product that have the same mechanism of action and are used for the same clinical indication but are not identical to the original product (referred to as the reference product throughout this article). Reference (or innovator) biologics are already on the market, and these are the products to which biosimilars will be compared. According to the U.S. Food and Drug Administration (FDA), biosimilars have no clinically meaningful differences from their reference products in terms of safety, purity, and potency.1 Since 2006, biosimilars have been approved for use in patients in the European Union (EU) and other regions, including Australia, Japan, and Latin America.2 When biosimilars become available in the United States, it will be important for pharmacists and managed care providers to have an understanding of the differences and similarities between small-molecule drugs, biologics, and interchangeable biologics and to be well informed about the scientific and regulatory concepts governing the substitution of biologics.
In this commentary, we (a) examine the differences between traditional small-molecule drugs and biologics; (b) review the U.S. approval pathways for generics and biosimilars; and (c) compare and contrast the substitution of generic versions of small-molecule drugs with the substitution of interchangeable biologics. We also review the evolving U.S. legal framework and FDA statements relevant to the substitution of interchangeable biologics and discuss practical considerations for interchangeable biosimilar substitution that may be of interest to pharmacists.
Differences Between Small-Molecule Drugs and Biologics
The size, complexity, manufacturing, and potential immunogenicity of biologics differentiate them from small-molecule drugs (Figure 1). In general, small-molecule drugs are structurally simple and have a lower likelihood for causing immunogenic responses.3-6 Small-molecule drugs are synthesized using predictable chemical manufacturing processes that allow for the production of equivalent copies of their branded counterparts.3 Biologics are structurally large and complex and are made by living cell systems that are unique to each manufacturer. The product and its variants (chemically modified forms and glycoforms), impurities, and excipients are formulated and combined into a sterile dosage form collectively called the biological drug substance. Because biologics are manufactured using extensive and sophisticated processes, changes in these processes can potentially result in structural alterations that lead to differences in clinical response (efficacy and safety).3 Additionally, biologics are sensitive to storage and handling conditions and have a higher potential for immunogenicity.3,7 The innate differences among manufacturers coupled with sensitivity to conditions result in “similar” but not “identical” copies. This disparity in being “similar” to the reference product is the basis for the fundamental difference between generics, which are identical to the brand medication, and biosimilars. This difference translates into a different regulatory framework (i.e., approval pathway) and practice-related considerations.
FIGURE 1.

Small-Molecule Drugs Versus Biologicsa
Distinct U.S. Approval Pathways for Generics and Biosimilars
In the United States, small-molecule drugs are approved under the Food, Drug, and Cosmetic Act (FDCA) and require a New Drug Application (NDA; Figure 2).8 Their generic counterparts follow an abbreviated approval pathway under the Drug Price Competition and Patent Term Restoration Act of 1984, commonly called the Hatch-Waxman Act (Figure 3). For a small-molecule drug to be approved as a generic of a brand-name small-molecule drug, a manufacturer has to demonstrate to the FDA pharmaceutical equivalence and bioequivalence; together these establish therapeutic equivalence (Figure 3).9 Once granted therapeutic equivalence by the FDA, the safety and efficacy of the generic is inferred from the Reference List Drug or branded counterpart.
FIGURE 2.

U.S. Approval Pathways for Small-Molecule Drugs Versus Biologics
FIGURE 3.

U.S. Approval Pathways for Generic Drugs Versus Biosimilars
In the United States, biologics are generally approved under the Public Health Service Act (PHSA), which requires a Biologics License Application (BLA; Figure 2).10 Biosimilars, however, have an abbreviated pathway for approval under the Biologics Price Competition and Innovation (BPCI) Act of 2009, part of the Patient Protection and Affordable Care Act, and rely for approval, in part, on the prior approval of the reference product.11,12 According to the BPCI Act, manufacturers must show that the biosimilar is “highly similar to the reference product notwithstanding minor differences in clinically inactive components” and that, for the intended condition or conditions of use, there are no clinically meaningful differences in terms of the safety, purity, and potency of the product as compared with the reference product (Figure 3). Applicants must show that the biosimilar is analytically, functionally, pharmacologically, and clinically similar (Figure 3). As in the abbreviated NDA process for generic drugs, a biosimilar sponsor may rely to some extent on the prior findings of safety and efficacy for the reference product. However, the BPCIA Act stipulates that the FDA may still require additional data comparing the safety, efficacy, and immunogenicity of a biosimilar to its reference product to ensure that there are no clinically meaningful differences in these areas.11 Alternatively, the FDA may, at its discretion, waive any of the analytical, animal, or clinical studies otherwise required for a biosimilar.
Interchangeability Designation
In addition to approving biosimilars, the FDA may make an additional determination regarding an “interchangeability” designation. Such a determination requires additional proof above and beyond that needed for a biosimilarity determination, as described in Figure 4.
FIGURE 4.
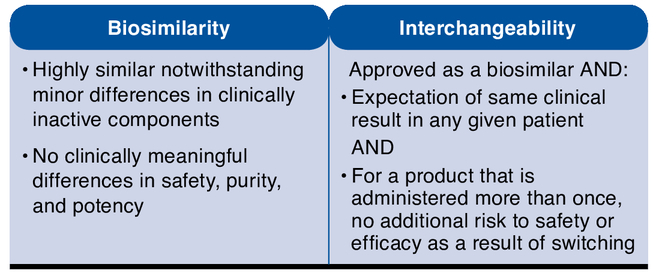
Biosimilarity and the Interchangeability of Biosimilars
The importance of the interchangeability designation is apparent in the language of the BPCI Act, which defines an “interchangeable” biosimilar as a biological product that may be substituted for the reference product without the intervention of the health care provider who prescribed the reference product.11 However, this provision must be evaluated in the context of state pharmacy practice laws that govern the ability of pharmacists to autonomously substitute one product for another.
Given this new regulatory pathway and the impending loss of exclusivity for many branded biological products, there will be many regulatory types of biological products available in the United States.13 As previously described, a biosimilar will be approved through an abbreviated pathway, section 351(k) of the PHSA; biosimilars will be “highly similar” to the reference product in terms of safety and efficacy based on an extensive comparability exercise; and some biosimilars may have an added designation as being “interchangeable” with the reference product. Another regulatory type of biological product is the noninnovator biologic, which is also an intended copy of the reference product; however, noninnovator biologics are approved through section 351(a) of the PHSA, and these products may or may not be explicitly compared with a reference product in terms of their physiochemical or clinical attributes. Yet another regulatory type of biological product is the second-generation “bio-better” product, which has a structural or functional modification or improvement upon a biological molecule in order to achieve a better clinical response. These “bio-better” products are approved through section 351(a) of the PHSA and are clearly distinct products.
Substitution Versus Interchange
According to the American Society of Health-System Pharmacists (ASHP), there are differences between what constitutes a “substitution” and an “interchange.”14 Generic substitution occurs when a pharmacist selects and dispenses a drug product that is identical to the branded product in terms of active ingredient, strength, concentration, dosage form, and administration route. State pharmacy practice laws give pharmacists the authority to make this substitution within reason, and the ASHP guidelines state that the use of generic products deemed by the FDA to be bioequivalent to branded products does not need review by an institution’s pharmacy and therapeutics (P&T) committee. Therapeutic interchange is the practice of exchanging the prescribed medicine with a product that is not identical in the active ingredient, but it exhibits a similar pharmacologic and therapeutic response (i.e., a “therapeutic alternative”).14 This interchange requires an a priori evaluation and authorization by a P&T committee in the form of written protocols or guidelines. Interchange occurs when a pharmacist executes a predefined written protocol when exchanging one product for another, typically within an institution or health system. Therapeutic substitution occurs when a pharmacist dispenses a therapeutic alternative in place of the intended product without a written policy or prior authorization by the provider.14 Substitution practice by a pharmacist does not involve the prescriber at the point of dispensing; the pharmacist exercises professional autonomy as a licensed health care practitioner, and such practice may or may not be legal depending on the authority given by state pharmacy practice laws. Additionally, we utilize the term autonomous substitution to refer to the independent professional practice of substituting one medicinal product for another by a pharmacist without explicit interaction with the prescriber at the point of dispensing.
In the context of this article, we limit our discussion to the substitution of interchangeable biosimilar agents for their corresponding reference products by a community pharmacist. Accordingly, there is no term defined by the ASHP to refer to such practice because biosimilars are not generic products. Thus, throughout our discussion, we refer to this as interchangeable biosimilar substitution.
Substitution in Practice
Although U.S. federal law establishes pathways for the approval of drugs (and thus licensure for sale in the United States), individual state laws regulate the practice of pharmacy.15 This typically includes the practice of substituting one medication for another (e.g., a generic for a branded medication) and any associated requirements for record keeping and communications with patients or prescribers. Before the 1970s, pharmacists in most states were legally obligated to dispense the branded product as written due to antisubstitution laws enacted to prevent the substitution of lower quality, and sometimes counterfeit, medications.15 Substitution in these cases would be considered misbranding.15 As higher quality generic medications became available and the cost of drugs began to rise, these laws were modified, and it eventually became standard practice to routinely conduct autonomous generic substitutions (i.e., substitution at the point of dispensing without informing the prescriber).15 For drug substitutions, there are 2 major concepts that can be applied to traditional small-molecule drugs and to interchangeable biologics: (1) if a prescription has been written that specifies a particular manufacturer (usually through the use of a brand name), then state pharmacy practice laws give pharmacists the ability to substitute the product and (2) if the prescription is written in a manner that does not specify a specific manufacturer, then pharmacists may use professional judgment in product selection.
Generic Substitution of Traditional Small-Molecule Drugs
In 1979, the FDA created the publication “Approved Drug Products with Therapeutic Equivalence Evaluations,” commonly known as the Orange Book, to assist the states in preparing formularies. Currently, the Orange Book is widely used as a reference for determining therapeutic equivalence, although some state laws may not directly reference the Orange Book.16,17 States take various approaches to regulation of the generic substitution of small-molecule drugs, including positive formularies (i.e., substitutions are limited to drugs on a list) and negative formularies (i.e., substitutions are permitted for all drugs except those on a list).16,18 Most states include provisions in their laws and regulations that respect the roles of physicians and patients in determining the best therapy, such as “dispense as written” authority and patient notification and/or counseling on generic substitution. States also regulate record keeping at pharmacies and require records of substitutions. Currently, 38 states and 2 territories have provisions for patients to be notified/informed and/or provide consent for small-molecule drug substitutions.19 Furthermore, some state laws have special provisions for narrow therapeutic index drugs that have either less than a 2-fold difference between the median lethal dose and the median effective dose, or have less than a 2-fold difference between the minimum toxic concentrations and minimum effective concentrations in the blood. Safe and effective use of narrow therapeutic index drugs require careful titration and patient monitoring.20
Since most generic drugs are bioequivalent to their respective reference counterparts, generics may be substituted, with rare exceptions, and autonomous substitution is a standard part of pharmacy practice under specified conditions applicable to specific state law.15 In practice, pharmacists dispense medications following state regulations. Patients may potentially receive the same drug from different manufacturers with the dispensing of each prescription, and in most cases, there are no clinical consequences associated with following this practice.
Many states have laws and regulations mandating communication between pharmacists and physicians in matters associated with the dispensing process. Many state laws and regulations also require pharmacists to interact with prescribers in contexts other than drug substitution. Increased communication was cited by pharmacists as having a positive effect on their relations with patients.21
Considerations for Interchangeable Biosimilar Substitution
Interchangeable biosimilar products are ideal candidates for autonomous substitution (akin to generic substitution), while biosimilars without this designation are not. To date, the FDA has not issued any guidance on interchangeability and indicated that it will “develop standards to ensure that products not deemed interchangeable are not inadvertently substituted for a reference product without the prescriber’s consent.”22 Toward this goal, the FDA recently published the “Lists of Licensed Biological Products with Reference Product Exclusivity and Biosimilarity or Interchangeability Evaluations,” nicknamed the Purple Book, which intends to be analogous to the Orange Book for generic products.23 The publication lists biosimilar agents (and whether they have been deemed to be interchangeable) and their corresponding reference product, as well as information regarding a biologic’s licensure pathway and exclusivity status.
Most of the current state pharmacy laws on autonomous substitution were enacted decades ago and primarily address the substitution of generic small-molecule drugs for branded products and do not explicitly address the issue of interchangeable biosimilar substitution. It is difficult to interpret current state pharmacy laws and determine if current legislation can be readily applied to interchangeable biosimilar substitution. Pharmacists may need to be involved with their respective state legislatures and boards of pharmacy to discuss the need to update such laws and rules to incorporate biosimilars, if necessary. These discussions should consider the following points:
Use of the FDA’s interchangeability criteria to determine whether biosimilar substitution can be applied by pharmacists. The FDA’s Purple Book may be used to guide this requirement.
Prescribers’ ability to “opt-out” of interchangeable biosimilar substitution by exercising “dispense as written” authority as is currently done for generic substitution in all 50 states.
Communication between the prescriber and patient about whether an interchangeable biosimilar substitution occurred in order to facilitate pharmacovigilance and product tracking. For generic drugs, notification to the patient of generic availability is legally required in 38 states and 2 territories.19
Requirements for record keeping of when a biosimilar substitution occurred to ensure adequate product tracking. Record keeping for generic substitution is currently required in all 50 states, with variation in how long records are kept.
It is anticipated that most states will adopt the “interchangeable” designation used by the FDA as one of the considerations to inform decisions regarding the substitution of interchangeable biologics, since this is supported by many professional organizations, including the Academy of Managed Care Pharmacy, American Pharmacists Association, National Association of Chain Drug Stores, National Community Pharmacists Association, and Biotechnology Industry Organization.24-26 Although the state substitution laws do not directly affect hospitals and institutional pharmacies, there are basic underlying principles that bear consideration for these entities. Similarly, legislation regulating interchangeable biosimilar substitution and its potential effect on payers’ ability to make formulary decisions through their reimbursement and/or cost-sharing policies should also be considered. In a staff report by the Bureau of Consumer Protection to the Federal Trade Commission in 1979, 81% of pharmacists reported that increased record keeping did not cause them to substitute less often when generic equivalents of chemical brand drugs were introduced.21 Integral to all discussions is the importance of determining the effect of interchangeable biosimilar substitution upon the ability to trace adverse events accurately and ensure patient safety.
Professional Considerations When Conducting Interchangeable Biosimilar Substitution
When considering interchangeable biosimilar substitution, some guiding questions are useful. The following key questions were based on the current process for conducting generic substitution and are applicable to any pharmacy practice setting:
Does the state pharmacy practice law give pharmacists the authority to autonomously substitute interchangeable biosimilars, or will the prescriber need to be contacted?
What criteria define what products may be substituted for one another?
What indications are allowed to be substituted with the product? Has the product considered for substitution been determined to be interchangeable with the prescribed product for all indications?
Does the state require prescriber and/or patient notification (or counseling)?
Does the state require record keeping of drugs dispensed and any related notifications of dispensing or substitutions?
Is the patient being switched from one product to another and, if so, are there any differences in packaging, excipients, devices, or other issues that require counseling?
What is the scope of state legislation on substitution of interchangeable biosimilars by setting (e.g., retail, mail order, and institutional)?
Implications for Managed Care
One important function and perspective of managed care stakeholders is to promote the use of the most cost-effective interventions. Therefore, managed care stakeholders will be interested in utilizing biosimilars, since these products are expected to be “highly similar” in clinical response and to cost less than their reference products. Current tools such as patient cost sharing and tiering can be powerful strategies to drive utilization towards the less costly but equally safe and effective biologics. When making formulary decisions, it is important for managed care providers to understand that biosimilars are not generics. The reference product can be indicated for multiple conditions. The interchangeable biosimilar does not have to be clinically tested for each of the indications where the reference product has been approved. However, as stated by the FDA draft guidance on biosimilars, “the sponsor will need to provide sufficient scientific justification for extrapolating clinical data to support a determination of biosimilarity for each indication for which licensure is sought.”1 In this situation, the formulary review should consider whether the interchangeable biosimilar is deemed to be acceptable or not acceptable for the nonapproved indications and if it should also be used in medically accepted off-label indications.
Implications for Pharmacovigilance
Accurate attribution of safety signals is important for all biologics so that any long-term and/or rare adverse events, including diminished efficacy, can be identified and so that any changes in the safety profile of a given product over time can be detected and investigated. Accordingly, the European Commission has established special provisions in the 2010 Pharmacovigilance Directive to enhance the traceability of biologics.27 Some of these measures mandate EU member states responsibilities for regulating pharmacy practice and medical record keeping.27
The FDA shares concerns for traceability of biologics and is considering policies within its federal jurisdiction: “The FDA process for biosimilars must include product specific safety monitoring. Pharmaceutical companies may make manufacturing-related changes to biologics periodically throughout their lifecycles, and even small changes could affect safety or efficacy. Tracking adverse events associated with the use of reference and biosimilar products will be difficult if the specific product or manufacturer cannot be readily identified. Appropriate strategies must be developed to ensure the implementation of robust, modern pharmacovigilance programs for biologics.”22 State boards of pharmacy can support FDA’s public health mandate by recognizing that biologics merit practices that ensure enhanced traceability.
Cases have been reported where unintended clinical consequences occurred when noncomparable biologics were treated like small-molecule generics in practice.28,29 Specifically, the inappropriate switching of noncomparable biologics along with the lack of clinical data comparisons between the products purporting to be therapeutically interchangeable resulted in increased incidences of certain adverse events.28,29
In a health care environment that includes highly similar biological products developed by different manufacturers, it is important to note that pharmacovigilance is strengthened when all members of a patient’s health care team—including the pharmacist and the prescriber—share access to the same medication history for a patient for all biological products. If a patient is switched between products without physician-pharmacist communication, it may be challenging to attribute adverse events or loss of efficacy to the applicable biologic in a timely fashion.
Moreover, attribution may be particularly problematic in cases where immune reactions develop slowly. Physician knowledge of the actual biologic dispensed is important, since adverse events, including diminished efficacy, may occur weeks or months after treatment, and patients may be more likely to contact their physicians in the event of a change in their health condition. An example of this dynamic has been published in a case study of severe immunogenicity in a European patient with kidney disease.30 The immunogenicity developed in conjunction with therapeutic switches among 3 biologics.30 In this case, the prescriber directed the therapeutic switches, but the diagnosis and treatment of the immune reaction were nonetheless impeded by the latency of the onset and the intervening shifts in therapy.30
Because the data source for some pharmacovigilance programs may be based on records obtained from physician offices, physician-pharmacist communication regarding the substitution of interchangeable biologics will foster long-term safety monitoring that could otherwise be confounded by inaccurate medical records. An emerging tactic to accomplish appropriate tracking and complete medical records is through the advancement and adoption of interoperable electronic medical records, where pharmacists and physicians share access to a patient’s medication history. Some pharmacovigilance initiatives and outcomes research projects may rely on the retrospective review of claims or electronic health record data. Accurate logging of the actual biologic that the patient receives is important. This may occur through billing codes, National Drug Code numbers, the nonproprietary name, or some combination thereof.
Conclusions
The BPCI Act of 2009 sets forth standards that must be met for products to be approved as biosimilar by the FDA. The BPCI Act also sets forth higher standards that must be met for biosimilar products to be approved as interchangeable. Real-world experience with biosimilars emphasizes the need for adequate clinical studies and comprehensive pharmacovigilance programs. It is important for pharmacists to be at the forefront of dispensing medicines and to stay apprised of their respective state laws for information regarding the autonomous substitution of interchangeable biologics. Unlike substituting generics for small-molecule drugs, where there is no expected clinical difference between products from one manufacturer and another, interchangeable biosimilar substitution requires systems and processes that ensure appropriate tracking and complete medical records at pharmacies and at physician offices. Pharmacists will play a key role in ensuring the maintenance of accurate and complete medical records for pharmacovigilance purposes and in notifying physicians if an interchangeable biosimilar substitution was performed. Pharmacists have the unique opportunity to engage patients in discussions regarding the biologics that they receive. The clinical judgment of pharmacists is paramount in ensuring the safe and effective use of biologics.
ACKNOWLEDGMENTS
The authors thanks Micah Robinson, PhD, and Jenilyn Virrey, PhD, of Amgen Inc. for medical writing assistance with the manuscript.
Funding Statement
This study was funded by Amgen Inc. Li has received honoraria for serving on advisory committees for Amgen Inc. and Hospira and for service on the speaker’s bureau for Pfizer. Ramanan and Green are employees of, and own stock in, Amgen Inc.
REFERENCES
- 1.U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research, Center for Biologics Evaluation and Research. Scientific considerations in demonstrating biosimilarity to a reference product: guidance for industry. April 2015. Available at: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291128.pdf. Accessed June 1, 2015.
- 2.European Medicines Agency. Omnitrope (somatropin). Authorisation details. Available at: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/000607/human_med_000946.jsp&mid=WC0b01ac058001d124. Accessed June 1, 2015.
- 3.Genazzani AA, Biggio G, Caputi AP, et al. . Biosimilar drugs: concerns and opportunities. BioDrugs. 2007;21(6):351-56. [DOI] [PubMed] [Google Scholar]
- 4.Prugnaud JL. Similarity of biotechnology-derived medicinal products: specific problems and new regulatory framework. Br J Clin Pharmacol. 2008;65(4):619-20. Available at: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2291378/. Accessed June 1, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crommelin DJ, Storm G, Verrijk R, de Leede L, Jiskoot W, Hennink WE.. Shifting paradigms: biopharmaceuticals versus low molecular weight drugs. Int J Pharm. 2003;266(1-2):3-16. [DOI] [PubMed] [Google Scholar]
- 6.Gottlieb S. Biosimilars: policy, clinical, and regulatory considerations. Am J Health Syst Pharm. 2008;65(14 Suppl 6):S2-S8 [DOI] [PubMed] [Google Scholar]
- 7.Roger SD. Biosimilars: How similar or dissimilar are they? Nephrology (Carlton). 2006;11(4):341-46. [DOI] [PubMed] [Google Scholar]
- 8.Federal Food, Drug, and Cosmetic Act. 21 U.S.C., chapter 9. Available at: http://www.fda.gov/RegulatoryInformation/Legislation/FederalFoodDrugandCosmeticActFDCAct/default.htm. Accessed June 1, 2015.
- 9.U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research. Guidance for industry: bioavailability and bioequivalence studies for orally administered drug products—general considerations. Revision March 1, 2003. Available at: http://www.fda.gov/ohrms/dockets/ac/03/briefing/3995B1_07_GFI-BioAvail-BioEquiv.pdf. Accessed June 1, 2015.
- 10.Public Health Service Act. 42 U.S.C., chapter 6A. Available at: http://www.fda.gov/RegulatoryInformation/Legislation/ucm148717.htm. Accessed June 1, 2015.
- 11.Biologics Price Competition and Innovation Act. Public Law 111-148, section 7001-7003. Available at: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/UCM216146.pdf. Accessed June 1, 2015.
- 12.Patient Protection and Affordable Care Act. Public Law 111-148. Available at: http://www.gpo.gov/fdsys/pkg/BILLS-111hr3590enr/pdf/BILLS-111hr3590enr.pdf. Accessed June 1, 2015.
- 13.Lucio SD, Stevenson JG, Hoffman JM.. Biosimilars: implications for health-system pharmacists. Am J Health Syst Pharm. 2013;70(22):2004-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tyler LS, Cole SW, May JR, et al. . ASHP guidelines on the pharmacy and therapeutics committee and the formulary system. Am J Health Syst Pharm. 2008;65(13):1272-83. [DOI] [PubMed] [Google Scholar]
- 15.Abood, RR. Pharmacy Practice and the Law. 6th ed. Sudbury, MA: Jones & Bartlett Publishers; 2010. [Google Scholar]
- 16.Food U.S. and Administration Drug. Orange Book preface. Available at: http://www.fda.gov/drugs/developmentapprovalprocess/ucm079068.htm. Accessed June 1, 2015.
- 17.State regulations on generic substitution. Pharmacist’s Letter/Prescriber’s Letter. 2006;22(9):220901. Available at: http://pharmacistsletter.therapeu-ticresearch.com/pl/ArticlePDF.aspx?dd=220901&segment=1186. Accessed June 1, 2015. [Google Scholar]
- 18.Vivian JC. Generic-substitution laws. US Pharm. 2008;33(6):30-34. [Google Scholar]
- 19.National Association of Boards of Pharmacy. Generic drug substitution requires pharmacist attention to ensure compliance with state laws and regulations. National Association of Boards of Pharmacy Newsletter. 2013;42(6):135-36,140. Available at: http://www.nabp.net/system/redactor_assets/documents/618/Final_June-July_2013_Newsletter.pdf. Accessed June 15, 2015. [Google Scholar]
- 20.U.S. Food and Drug Administration. Code of Federal Regulations, Title 21 Section 320.33(c). Available at: http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=320.33. Accessed June 1, 2015.
- 21.U.S. Federal Trade Commission, Bureau of Consumer Protection. Drug Product Selection: Staff Report to the Federal Trade Commission. Ann Arbor, MI: University of Michigan Library; 1979. [Google Scholar]
- 22.Kozlowski S, Woodcock J, Midthun K, Behrman Sherman RB.. Developing the nation’s biosimilars program. N Engl J Med. 2011;365(5): 385-88. Available at: http://www.nejm.org/doi/full/10.1056/NEJMp1107285. Accessed June 1, 2015. [DOI] [PubMed] [Google Scholar]
- 23.U.S. Food and Drug Administration, Center for Drug Evauation Research. List of licensed biological products with (1) reference product exclusivity and (2) biosimilarity or interchangeability evaluations to date. Available at: http://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/Biosimilars/UCM411909.pdf. Accessed June 1, 2015.
- 24.Biotechnology Industry Organization. BIO principles on patient safety in the substitution of biologic products. January 24, 2013. Available at: http://www.bio.org/advocacy/letters/bio-principles-patient-safety-substitution-biologic-products. Accessed June 1, 2015.
- 25.Academy of Managed Care Pharmacy. Where we stand: biosimilar drug therapies. April 2012. Available at: http://www.amcp.org/WorkArea/DownloadAsset.aspx?id=18693. Accessed June 15, 2015.
- 26.American Pharmacists Association, National Association of Chain Drug Stores, National Community Pharmacists Association. Letter to U.S. Food and Drug Administration, Division of Dockets Management (HFA-305). May 25, 2012. Available at: http://www.ncpanet.org/pdf/leg/may12/joint_biosimilar_letter.pdf. Accessed June 1, 2015.
- 27.European Commission. Directive 2010/84/EU of the European Parliament and of the Council of 15 December 2010 amending, as regards pharmacovigilance, directive 2001/83/EC on the Community code relating to medicinal products for human use. 2010 OJ L348/74. Available at: http://ec.europa.eu/health/files/eudralex/vol-1/dir_2010_84/dir_2010_84_en.pdf. Accessed June 1, 2015.
- 28.Rosendaal FR, Nieuwenhuis HK, van den Berg HMet al. . A sudden increase in factor VIII inhibitor development in multitransfused hemophilia A patients in The Netherlands. Dutch Hemophilia Study Group. Blood. 1993;81(8):2180-86. Available at: http://www.bloodjournal.org/content/81/8/2180.long. Accessed June 1, 2015. [PubMed] [Google Scholar]
- 29.Praditpornsilpa K, Tiranathanagul K, Kupatawintu P, et al. . Biosimilar recombinant human erythropoietin induces the production of neutralizing antibodies. Kidney Int. 2011;80(1):88-92. [DOI] [PubMed] [Google Scholar]
- 30.Wieser C, Rosenkranz AR.. Pure red cell aplasia after treatment of renal anaemia with epoetin theta. Clin Kidney J. 2013;6(5):539-42. [DOI] [PMC free article] [PubMed] [Google Scholar]