

Abstract
BACKGROUND:
Companion diagnostic tests (CDTs) have emerged as a vital technology in the effective use of an increasing number of targeted drug therapies. Although CDTs can offer a multitude of potential benefits, assessing their value within a health technology appraisal process can be challenging because of a complex array of factors that influence clinical and economic outcomes.
OBJECTIVE:
To develop a user-friendly tool to assist managed care and other health care decision makers in screening companion tests and determining whether an intensive technology review is necessary and, if so, where the review should be focused to improve efficiency.
METHODS:
First, we conducted a systematic literature review of CDT cost-effectiveness studies to identify value drivers. Second, we conducted key informant interviews with a diverse group of stakeholders to elicit feedback and solicit any additional value drivers and identify desirable attributes for an evidence review tool. A draft tool was developed based on this information that captured value drivers, usability features, and had a particular focus on practical use by nonexperts. Finally, the tool was pilot tested with test developers and managed care evidence evaluators to assess face-validity and usability. The tool was also evaluated using several diverse examples of existing companion diagnostics and refined accordingly.
RESULTS:
We identified 65 cost-effectiveness studies of companion diagnostic technologies. The following factors were most commonly identified as value drivers from our literature review: clinical validity of testing; efficacy, safety, and cost of baseline and alternative treatments; cost and mortality of health states; and biomarker prevalence and testing cost. Stakeholders identified the following additional factors that they believed influenced the overall value of a companion test: regulatory status, actionability, utility, and market penetration. These factors were used to maximize the efficiency of the evidence review process. Stakeholders also stated that a tool should be easy to use and time efficient. Cognitive interviews with stakeholders led to minor changes in the draft tool to improve usability and relevance. The final tool consisted of 4 sections: (1) eligibility for review (2 questions), (2) prioritization of review (3 questions), (3) clinical review (3 questions), and (4) economic review (5 questions).
CONCLUSIONS:
Although the evaluation of CDTs can be challenging because of limited evidence and the added complexity of incorporating a diagnostic test into drug treatment decisions, using a pragmatic tool to identify tests that do not need extensive evaluation may improve the efficiency and effectiveness of CDT value assessments.
What is already known about this subject
Managed care formulary staff and other health care decision makers are increasingly faced with the need to assess clinical and economic evidence for companion diagnostics associated with targeted drug therapies.
The evaluation process for companion diagnostics can be challenging because of varying regulatory pathways, a paucity of evidence, and a complex array of factors influencing value.
What this study adds
There are 6 important factors that decision makers should pay attention to: (1) clinical validity and utility of test; (2) efficacy, safety and cost of treatment; (3) severity and cost of disease; (4) biomarker prevalence; (5) test cost; and (6) test result adherence.
The evaluation process can be expedited by considering (1) the regulatory pathway, (2) clinical utility, (3) clinical value, and (4) economic value.
A tool was developed to help decision makers determine if a full technology review is necessary and, if so, what factors are likely to be most influential in the test’s overall value.
Companion diagnostic tests (CDTs) have emerged as a growing and vital technology in the effective use of many drug therapies.1,2 Although CDTs may offer a multitude of potential benefits, such as improved effectiveness, selected patient populations, and improved side-effect profiles, assessing the value of CDTs can be challenging because of the complex array of factors that can influence their value.3-7
Payers and providers are increasingly seeing CDTs in the market, and most are just beginning to search for approaches to developing reimbursement and treatment guidelines.5,8 Some payers rely on third-party technology assessments. While these assessments answer some questions, they are not available for every test, do not address questions of budget impact, and are not personalized for individual plans. Stakeholders are faced with lingering questions such as “How much does this test improve patient outcomes compared with usual care?” “Is this test worth the cost?” and “Are targeted therapies more expensive, or do they save money overall?” Thus, now is an opportune time to provide payers and providers with guidance and tools to address these types of questions.
The Academy of Managed Care Pharmacy (AMCP) recently launched an addendum to its format for the provision of clinical and economic evidence to payers.9 The Companion Diagnostics Addendum provides more detailed guidance to drug and diagnostic manufacturers about how evidence on CDTs should be presented to decision makers. However, practical guidance on how decision makers can efficiently evaluate this evidence is lacking. The overall objective of this study was to develop a tool to facilitate the health technology assessment process for CDTs in a time-efficient manner that is complementary to the AMCP Companion Diagnostics Addendum for assessing a test’s value in the clinical pathway.
Methods
We conducted a systematic literature review of CDT cost-effectiveness studies to identify factors that were drivers of value. Next, we conducted key informant interviews with a diverse group of stakeholders to elicit feedback on identified value drivers, identify novel factors, and elicit desirable attributes for an evidence evaluation tool. We then drafted an evidence evaluation tool based on the findings from the first 2 steps. Finally, we refined the tool based on feedback from cognitive interviews with stakeholders.
For this process, we defined a CDT as a laboratory test that provides differential information on predicted response to medical treatment among patients with the same clinical condition. We did not exclude tests based on analyte (DNA, RNA, or protein) but did exclude tests that provided information exclusively on disease diagnosis or prognosis rather than differential response to treatment.
Phase 1: Literature Review
We updated a previous systematic review by searching the following resources: PubMed, Tufts Cost-Effectiveness Analysis (CEA) Registry, National Institute for Health and Care Excellence (NICE; United Kingdom), and Canadian Agency for Drugs and Techologies in Health (CADITH).10-13 The following strategies were employed:
Broad search terms. The terms pharmacogenetics and costs were used as the first inclusive search. Additional searches with the terms genetic testing and cost-effectiveness were also conducted.
Specific (MeSH) search terms. A search using the following MeSH terms was conducted: genetic screening/economics, genotype, costs and cost analysis, economics, and pharmacogenetics/economics.
Disease-specific search terms. Based on results from the first 2 search strategies, disease-specific terms were used: clopidogrel, CYP2C19 EGFR, warfarin, ACEI, CYP2D6, HLA-B*1502, carbamazepine, CYP2C9, trastuzumab, Oncotype, and 21 gene assay.
Expert recommendations. Opinions from investigators with experience in the field (the authors) were used to identify additional articles that may have been missed by the previous methods.
Cross-reference with other systematic reviews. As a final stage after our inclusion review of identified studies, the same search criteria was employed and limited to systematic reviews to identify other analyses with similar aims. The studies identified as part of these other analyses were then compared with our own in order to identify any missed studies.
All searches were limited to studies with abstracts and written in the English language published after October 2009. Articles published before this date were assumed to be captured under the previous review,10 which included studies up to this date. We employed the following inclusion criteria: (a) the study included measures of clinical effectiveness and costs and (b) the CDT being considered had predictive value for a drug treatment outcome beyond diagnosis of the indicated condition or prognosis for disease trajectory. Studies were excluded that evaluated markers that provided information only on definition of disease (diagnosis) or disease-related events and trajectory irrespective of therapy (prognosis). Studies were also excluded if they did not capture downstream costs and health events of the interventions evaluated, such as cost minimization or cost per case detected evaluations.
Data abstracted from each study included year published, biomarkers, condition, drugs, population, test actionability, results, country setting, payer or societal modeling perspective, and influential factors in one-way sensitivity. The one-way sensitivity analyses from each of these studies, when available, were subsequently abstracted to determine the plausible range of input values used, as well as their relative effects on the summary incremental cost-effectiveness ratio. Each of these variables were then coded into broader categories: adherence; clinical validity; fatality of adverse events; cost of clinical events, treatments, and adverse events; rate of disease progression; disease severity; efficacy of compared treatments; genotypic prevalence; and utilities of health states and treatments (Table 1). From these studies, the top 3 influential factors from one-way sensitivity analyses—those that swayed the final cost-effectiveness metric by the greatest absolute margin—were considered to be key value drivers.
TABLE 1.
Definition of Terms
| • Companion diagnostic test (CDT): A laboratory assay that provides predictive information about a patient’s response to drug therapy. This is in contrast to diagnostic or prognostic tests, which provide information about the disease process rather than response to treatment. This definition includes assays of RNA expression and allows for the potential of multiple marker tests with results interpreted by an algorithm. Assays of nonhuman analytes (e.g., viral genotype) are excluded from this definition. |
| • Value: A quantification of treatment outcomes (benefit vs. harm, life expectancy, quality of life) relative to health care costs. |
| • Value driver: A characteristic of a health care decision (test cost, drug safety, etc.) that, when modified, will substantially affect the overall value of a CDT. |
| • Clinical validity: The ability of a test to predict the clinical outcome of interest.17 |
| • Clinical utility: The ability of a test to lead to improved patient outcomes when used in clinical practice. |
RNA = ribonucleic acid.
Phase 2: Key Stakeholder Interviews
Stakeholder interviews were conducted with payers (those making coverage decisions for tests), test facilitators (those identifying testing opportunities and performing outreach), and test developers (those involved in developing and commercializing new tests). All participants were given a structured interview (see Appendix, available in online article) that focused on perceptions of the value of CDTs as well as preferred approaches to assessing their value. The interview consisted of 4 sections: (1) discussion of interviewee’s perceptions of the market and regulatory environment for companion diagnostic technologies; (2) discussion of interviewee’s perception of key value drivers that effect market uptake and reimbursement of CDTs; (3) review of results from the phase 1 literature review and discussion of preliminary results; and (4) discussion of factors for usability of a decision tool for CDTs and relative advantages of a qualitative framework and quantitative model.
We selected participants based on prominence in the field, with additional participants enrolled via snowball sampling. Whenever possible, we attempted to recruit participants from a variety of geographies. All but 1 interview was conducted by phone. Whenever possible, interviews were conducted by at least 2 of the authors. All interviews were recorded with the participant’s verbal permission, and the recording was abstracted afterwards to identify key themes and quotes. At the conclusion of all interviews, these reports were reviewed by the authors to identify issues that resonated in the interviews within and across stakeholder types (payer, test developers, and facilitators).
Phase 3: Companion Test Assessment Tool Development
Overall Structure.
The results of the systematic review and key informant interviews were synthesized qualitatively to create an ordered series of questions that would identify for the user the most influential parameters for a reimbursement review of CDTs. The tool was primarily designed for individuals within managed care organizations tasked with the clinical and economic review of an emerging companion test. This type of user was assumed to have limited experience in genomics and resource constraints that would preclude full review of all potentially relevant factors. Given this perspective, the framework was designed to increase the efficiency of companion diagnostic review rather than to serve as an exhaustive assessment of a particular test.
Question Integration.
The final sequence of questions was determined by piloting numerous example tests and obtaining stakeholder feedback, with the objective of categorizing tests in a minimum number of steps. The ordered questions were then placed in a visual flow to improve ease of use and highlight the natural order of decisions relevant for efficiently evaluating tests from a managed care perspective. In other words, the tool was designed as a sequential flow of questions. Numerous visual layouts of this sequential ordering of questions were prototyped with the ultimate version decided by its presumed effectiveness in communicating the process to the intended audience.
Tool Evaluation.
In order to evaluate the performance of the tool with a diverse group of CDTs, a categorization of tests was identified that captured the majority of test types (Table 2). This categorization grouped tests by the clinical information they provided (safety or efficacy) and their regulatory status. The primary differentiating factor in this 2×2 framework is whether the CDT is required for drug use in the U.S. Food and Drug Administration (FDA) drug label. We chose FDA requirements as a differentiator because a key difference between required and optional test types lies in the strength of the evidence. In other words, required tests likely will have higher levels of evidence than optional tests and not need as extensive an evaluation. For example, a CDT used in an randomized controlled trial (RCT) of a new drug to identify patients for enrollment in that trial (i.e., an enrichment trial) generally does not require that its clinical validity be assessed because the drug has only been tested in the specified population and, importantly, should only be used in that population. In contrast, for tests that are not required, it is critical to evaluate their clinical validity because some patients would not receive a needed drug based on the test result.
TABLE 2.
Categorization of CDTs Based on Regulatory Status and Clinical Effect
| CDT Regulatory Status | |||
|---|---|---|---|
|
Required
Tests Required in FDA Drug Label |
Optional (Not Required)
Tests Not Required in FDA Drug Label |
||
| Treatment Prediction |
Efficacy
CDTs that predict treatment response |
Examples
|
Examples
|
|
Safety
CDTs that predict the risk of adverse events |
Examples
|
Examples
|
|
ALK = anaplastic lymphoma kinase; CDT = companion diagnostic test; FDA = U.S. Food and Drug Administration; HLA = human leukocyte antigen; NSCLC = non-small cell lung cancer; TPMT = thiopurine s-methyltransferase.
More specifically, tests in the “required” category are included in the FDA label as either an indication for use in the case of efficacy markers in this group or as a contraindication in a black box warning for safety markers. Examples in this category include vemurafenib, which was approved specifically for patients with a BRAF mutation, and HLA screening for abacavir, which was evaluated via randomized trial and has subsequently been included as a contraindication for use in B*5701 carriers due to the rate of hypersensitivity reactions. While these tests generally have robust clinical validation, their cost-effectiveness may still be unknown. Optional tests are generally those that are not required in the drug label, although they may be mentioned.
We also differentiated CDTs based on their intended primary effect—improving efficacy or safety. It is assumed for this categorization that efficacy markers predict better responsiveness to treatment (lower number of disease-related events) in those who are marker-positive, while safety markers predict a worse adverse event profile (greater number of events that are caused by the drug and not related to disease progression) in those who are marker-positive. The developed sequence of questions was applied to a case study from each category of this framework in order to test the assessment tool’s ease of use and validity. In situations where it was not immediately clear which path each of these tests would follow, the question flow was adjusted to allow for more efficient categorization. The tool was also informally evaluated for relevance and ease of use via cognitive interviews with stakeholders from managed care and test developers. Feedback from this interview process was incorporated into future versions to improve efficiency and ease of use of the framework.
Results
Phase 1: Literature Review
A total of 65 studies were identified that met our inclusion criteria and were subsequently abstracted (Table 3). These studies were found from the following sources: Wong et al. (2010) review (29),10 updated broad PubMed search (23), and disease-specific PubMed search (3). No additional studies were found from searches of NICE, CADTH, or the Tufts CEA Registry. A search for comparable systematic reviews identified 3 examples with similar objectives: Carlson et al. (2005), Djalalov et al. (2011), and Vegter et al. (2008).14-16 Comparison of these reviews with the list from our own search identified 4, 2, and 4 additional studies, respectively (Figure 1).
TABLE 3.
Identified Cost-Effectiveness Studies
| Biomarker | Clinical Specialty | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Oncology | Cardiology/Internal Medicine | Infectious Disease | Gastrology | Psychology | Rheumatology | Other | Total | ||||||||||
| OncotypeDx: breast | 8 | 8 | |||||||||||||||
| TPMT | 1 | 3 | 2 | 6 | |||||||||||||
| CYP2C19 | 3 | 1 | 4 | ||||||||||||||
| CYP2C9, VKORC1 | 4 | 4 | |||||||||||||||
| EGFR | 4 | 4 | |||||||||||||||
| HLA-B*5701 | 4 | 4 | |||||||||||||||
| ACE | 1 | 2 | 3 | ||||||||||||||
| HER2 | 3 | 3 | |||||||||||||||
| KRAS | 3 | 3 | |||||||||||||||
| UGT1A1 | 2 | 1 | 1 | 3 | |||||||||||||
| Other | 5 | 5 | 2 | 4 | 1 | 5 | 23 | ||||||||||
| Total | 26 | 13 | 7 | 4 | 4 | 3 | 8 | 65 | |||||||||
| United States | United Kingdom | Canada | Netherlands | Japan | Korea | Other | |||||||||||
| Country setting, n (%) | 28 (43) | 6 (9.2) | 5 (7.7) | 3 (4.6) | 2 (3.1) | 2 (3.1) | 19 (29.2) | 65 | |||||||||
| DNA | RNA | Protein | Protein vs. DNA | ||||||||||||||
| Marker type, n (%) | 50 (76.9) | 8 (12.3) | 3 (4.6) | 4 (6.2) | 65 | ||||||||||||
| Payer | Societal | Not Reported | |||||||||||||||
| Perspective, n (%) | 26 (40) | 38 (58.5) | 1 (1.5) | 65 | |||||||||||||
| $50,000/QALY | $100,000/QALY | ||||||||||||||||
| Cost-effective at willingness-to-pay threshold, n (%) | 46 (70.8) | 56 (86.2) | 65 | ||||||||||||||
| Influence in One-Way Sensitivity, n (%) | |||||||||||||||||
| Clinical Validity of Test | Efficacy, Safety, and Cost of Treatments | Cost, Utility, and Mortality of Health States | Genotypic Prevalence | Test Cost | Adherence | Other | Not Reported | ||||||||||
| Most impactful | 8 (12) | 9 (14) | 1 (2) | 2 (3) | 5 (8) | 1 (2) | 3 (5) | 36 (55) | |||||||||
| Second most impactful | 6 (9) | 9 (14) | 7 (11) | 3 (5) | 2 (3) | 1 (2) | 1 (2) | 36 (55) | |||||||||
| Third most impactful | 1 (2) | 5 (8) | 7 (11) | 2 (3) | 5 (8) | 2 (3) | 7 (11) | 36 (55) | |||||||||
ACE = angiotensin-converting enzyme; CYP2C19 = a clinically important enzyme that metabolizes a wide variety of drugs; DNA = deoxyribonucleic acid; EGFR = epidermal growth factor receptor; HER2 = human epidermal growth factor receptor 2; HLA = human leukocyte antigen; KRAS = a protein that plays an important role in many cell functions; QALY = quality-adjusted life-year; RNA = ribonucleic acid; TPMT = thiopurine s-methyltransferase; UGT1A1 = enzyme involved in converting the toxic form of bilirubin to its nontoxic form.
FIGURE 1.

Literature Search Results
Of the studies selected, 40% were in the oncology setting. Nearly 60% of the studies took a societal perspective (i.e., included costs to the health plan and to the patient), and over 70% of the studies found the CDT-based strategy to be cost-effective at a willingness-to-pay threshold of $50,000 per quality-adjusted life-year. Although all of the studies identified performed some form of sensitivity analysis, only 45% reported results from a one-way sensitivity analysis that allowed for the identification of value drivers. Of the 35 studies that did not report full one-way sensitivity results, 20 performed analyses but did not report full results, and 15 performed only multivariate sensitivity analyses. Among those studies that reported one-way sensitivity analyses from which we could infer value drivers, the most commonly identified value drivers were clinical validity of testing; efficacy, safety, and cost of baseline and alternative treatments; cost and mortality of health states; and biomarker prevalence and testing cost (Figure 2). Although clinical utility is a point considered for all of these models and certainly is one of the key considerations in framing research question and structuring decision trees, it was not identified as one of our value drivers in this phase, since it could not be captured and reported in a single value in a sensitivity analysis.
FIGURE 2.

Identified Value Drivers from Literature Reviewa
Phase 2: Stakeholder Interviews
Between April and June 2013, 12 key informant interviews were conducted. The composition of this group included 5 payers, 3 test facilitators, and 4 test developers. All payers interviewed had direct experience reviewing companion diagnostics for purposes of reimbursement and represented a diverse sample of plan size, geography, and familiarity with genomics. Test developers and facilitators expressed frustration with the reluctance of payers to reimburse novel diagnostics despite their relatively low cost and potentially high impact on medical decisions. Members of these groups without exception expressed hope that payers will begin to accept a “chain of (indirect) evidence” instead of large-scale RCTs that can be cost prohibitive for many developers. A test facilitator explained frustration with payers’ reluctance to accept a chain of evidence: “You could tell [payers] this drug saves lives only if the sun is coming up and they’ll ask you ‘How do I know the sun is coming up? It’s observational and all retrospective.’”
But, hope was also expressed that value-based pricing would be used and incentivize development of diagnostics that are likely to improve patient outcomes in a cost-effective manner. Other key themes identified by test facilitators and developers included the following:
Commoditization of expertise and misalignment of incentives. Developers of FDA-approved diagnostics were concerned that less rigorous regulatory oversight of laboratory-developed tests adversely affects the ability of tests developed in collaboration with the drug development process to adequately compete in the marketplace. This ultimately results in undercutting the market and turning their services into a lower margin commodity.
Companion diagnostics improve confidence and decision impact. Developers emphasized that the value of a test may not be just in terms of predicting an outcome but also in reducing the amount of uncertainty around health care decisions.
Companion diagnostic costs are a “blip on the screen.” Concern was raised that undo scrutiny was being given to testing cost when this expense is relatively modest compared with the drug costs that the test informs.
These views contrasted with those of payers. While some payers expressed optimism about the potential of personalized medicine to improve health care, most mentioned a growing fatigue with the failure of companion diagnostics to deliver on the promise of more efficient health care, as 1 interviewee described: “There has been great promise for 30 years but companion diagnostics still don’t have a lot of impact on clinical practice.”
Interviews with payers also highlighted the following themes related to issues of cost containment in the absence of certainty about the clinical utility of available tests:
A clear focus on budget impact. Payers had a focus on budget impact and expressed a need to see a clear return on investment for the multitude of new diagnostics that are presented to them every year.
Need for a clear value proposition. Payers expressed a demand for concise statements of clinical validity and utility for tests that often are used in practice with little regard for how their results might be used to change treatment decisions.
A fear of “mission creep.” Concern was raised that many diagnostics in the context of personalized medicine seemed to be used as a means for demanding high margins in a narrowly defined condition but that this margin was then maintained as the drug was expanded to other conditions with similar biomarker patterns.
Overall, our key informant interviews with payers affirmed the utility of our research objective. Any tool that is developed should be simple, easy to operate, and should quickly categorize tests based on the level of evidence review complexity. Once categorized, the tool should highlight key operating characteristics for the test: clinical sensitivity and specificity, number needed to test, and decision impact on drug spending. Quality of life metrics were not thought to be as important by payers, who instead preferred to see direct medical impact of test use. A more thorough review of the themes among these groups can be found in the Appendix (available in online article), along with representative quotes.
Stakeholders identified 2 value drivers not consistently captured in the review of health economic evaluations: market penetration of testing and adherence to treatment guidelines. Since cost-effectiveness models are designed to capture the incremental effect of clearly identified alternatives, they often make numerous simplifying assumptions. Two of these assumptions are that (1) the choice being made is between a scenario where everyone who qualifies is tested and a scenario where no one is tested, and (2) in the testing scenario, everyone who is tested will act upon the results of the test. In real-world situations, it is often much more of a hybrid, where only a fraction of patients may be tested, and in those who are tested, some patients will seemingly ignore the clinical recommendation from the test. To capture this feedback, market penetration of testing and adherence to guidelines were added as factors in the development of our tool.
Phase 3: Companion test Assessment Tool (CAT)
Factors identified as important in the literature review and stakeholder interviews were structured in such a way as to allow for categorization of tests with a single final recommendation. We then used extensive prototyping to create a process that categorized a majority of test types using the identified factors in a minimum of steps. Feedback from our stakeholder interviews overwhelmingly emphasized that the tool should expedite and augment the existing process for reimbursement decision making rather than attempting to reenvision the whole process. With this in mind, the tool was designed at first to filter those tests that did not require extensive evaluation and then highlight the factors where evidence review should be focused.
The tool development process resulted in a qualitative tool with 13 questions that is referred to here as the Companion test Assessment Tool (CAT; Figure 3). Visually, the CAT is designed to flow sequentially downward, with tests that necessitate further questions progressing downward, and tests that have been categorized moving off to the right. For each of the sequential questions, users either progress to tangible next steps for how their organizations can handle the review of tests or proceed to further questions based on how they respond. Examples of tests that follow each pathway are also provided to give more context and provide a comparison for the user. The framework is divided into 4 main steps to assist in ease of use and allow for a separation of questions regarding clinical benefit and economic consequences. These steps are (in order) as follows: (1) eligibility review, (2) prioritization, (3) clinical review, and (4) economic review.
FIGURE 3.
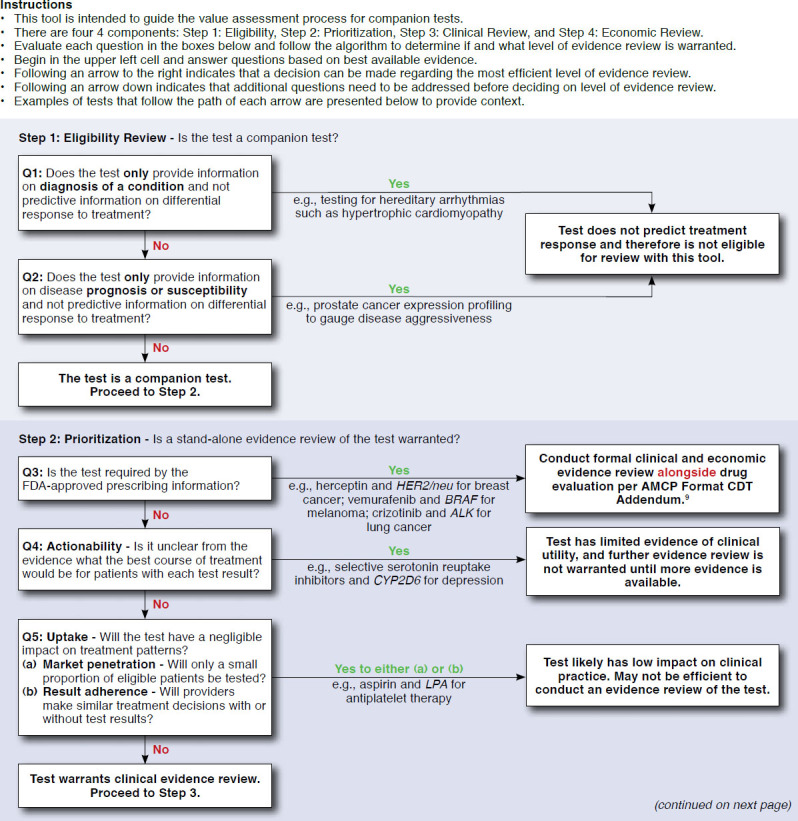
Companion test Assessment Tool (CAT)
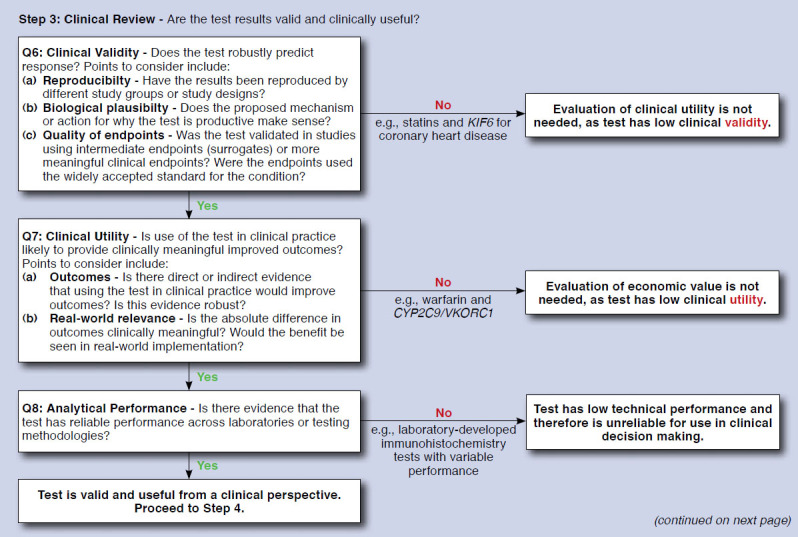
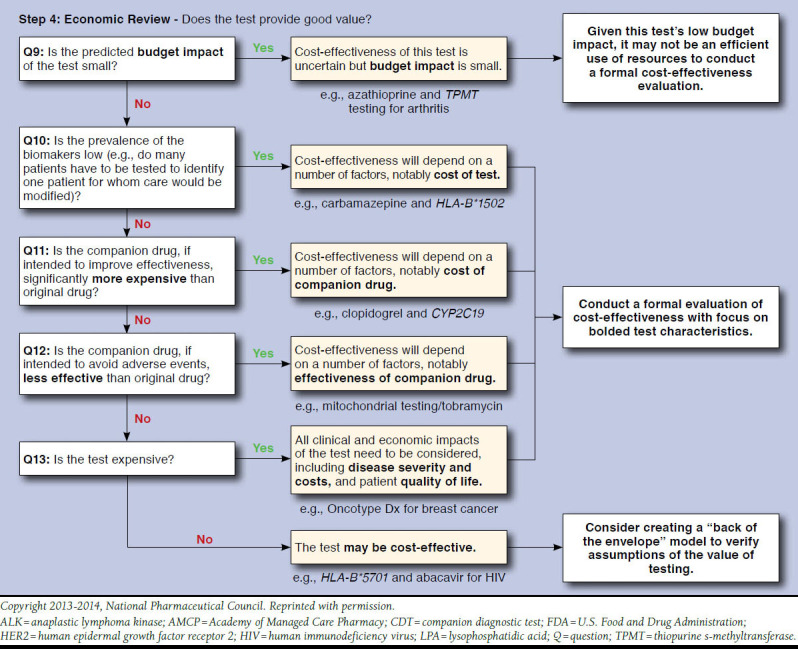
Step 1 includes 2 questions to determine eligibility for use in the CAT. Tests defined as diagnostic or prognostic are filtered out to the right. Tests defined as predictive progress to step 2, with the objective of determining if an evidence review is warranted. Step 2 consists of 3 questions regarding regulatory status and actionability. Tests that progress past step 2 undergo clinical and economic review in steps 3 and 4, respectively. Step 3 includs questions to filter tests based on low levels of evidence. The questions for this step roughly parallel the ACCE framework for genomic tests.17 As opposed to steps 1-3, where the objective is to filter out tests that either do not qualify for use with the tool or lack the strong evidence necessary to warrant extensive review, step 4 contains a middle column that highlights parameters of predicted high influence with all tests recommended for further review. More details on each step are provided as follows:
Step 1: The eligibility review step assesses whether the test under consideration is appropriate to use with the tool. The intent of this section is to exclude tests that are primarily intended for either diagnostic or prognostic purposes rather than the prediction of treatment response. The exclusion is applied because diagnostic and prognostic tests are assumed to have different clinical profiles and value drivers.
Step 2: This step is focused on prioritization of evidence reviews, with the objective of identifying tests that do not warrant extensive or independent review. Tests that are FDA-approved and codeveloped for use with a pharmaceutical company or in clinical guidelines are assumed to not necessitate as rigorous a review process, since they have already been extensively clinically evaluated. Furthermore, evaluation of such tests independently of their companion drug is not feasible, as outlined in the AMCP Companion Diagnostics Addendum.9 Finally, tests that would have negligible levels of clinical impact are also excluded at this step.
Steps 3 and 4: These steps focus on the clinical and economic review of tests, respectively. It was determined based on feedback from stakeholders that managed care decision makers would have varying degrees of comfort with an appraisal based on economics, and a clear distinction was made between these 2 stages of review so that the user could have the option to focus only on the clinical aspects of tests. Questions included in steps 3 and 4 were developed based on value drivers identified in our literature review, as well as in the key informant interviews. Value drivers that were identified in our literature review were subsequently vetted with stakeholders who also provided additional parameters.
Pilot Testing with Stakeholders
All original stakeholders were approached for pilot testing and evaluation of the CAT, with 8 individuals ultimately agreeing to participate: 3 test developers, 2 test facilitators, 2 regional payers, and 1 patient advocate. All those interviewed found strong face validity in the CAT. Recommendations regarding wording and clarity were made and subsequently incorporated to improve ease of use.
Tool Evaluation with Companion Test Examples
The previously developed two-by-two categorization of tests was used to identify the case studies that we would apply to the framework: crizotinib and anaplastic lymphoma kinase (ALK) testing, abacavir and HLA-B*5701 testing, adjuvant chemotherapy and OncotypeDx Breast Cancer Assay, and pharmacogenomically guided warfarin dosing.
Crizotinib and ALK testing is an example of a test that is predictive of the drug’s efficacy and is required by the FDA prior to prescription; therefore, it falls into the upper left quadrant of our categorization. ALK is a tyrosine kinase drug target for several cancers including non-small cell lung cancer (NSCLC).18 Genetic aberrations in ALK are present in roughly 5% of cases of NSCLC and signal a unique molecular profile and subset.19-21 Crizotinib is a protein kinase inhibitor that binds competitively with the adenosine triphosphate-binding pocket of the ALK, cMET, and ROS1 proteins. Crizotinib was approved by the FDA on the basis of an open-label, phase 3 RCT that compared crizotinib with standard chemotherapy. In order to qualify for this trial, patients needed to have NSCLC that was positive for rearrangement in ALK.22 Since crizotinib is an FDA-approved companion diagnostic, this test would be “filtered out” at question 3. At this point, the user is directed to rely on the information in the AMCP Companion Diagnostics Addendum, which is presumed to have the most detailed information.9 The user could potentially also use the questions in step 4 to highlight the most important factors in determining the test’s overall value.
Abacavir and HLA-B*5701 is an example of a test that is predictive of the drug’s safety and required by the FDA prior to prescription. Abacavir is a nucleoside reverse-transcriptase inhibitor that is active against the human immunodeficiency virus. Although abacavir was originally approved by the FDA with no testing requirement, its adoption was limited by a hypersensitivity reaction (HSR) in 5%-8% of patients during the initiation of treatment.23 After an association was found between this HSR and the HLA-B*5701 genotype,24-28 the manufacturer, GlaxoSmithKline, launched a randomized trial, testing the effectiveness of prospective screening.29 This is the only known example of a postmarket RCT sponsored by the manufacturer to test the effectiveness of genotyping in everyday practice. Similar to crizotinib and ALK testing, this test would be filtered out at question 3 and would not necessitate an in-depth review based on its regulatory status and extensive clinical validation. Again, as with the previous example, the user could still use the questions in step 4 to prioritize the economic review of this test.
OncotypeDx is a test that is categorized as “optional-efficacy” and is 1 of only a handful of proprietary tests to successfully implement a value-based pricing scheme and break into routine clinical practice. OncotypeDx is a 21-gene multiplexed assay that combines all results into a recurrence score via algorithm. This test has been shown to have predictive value in identifying women with either node-positive or node-negative breast cancer who are unlikely to respond to adjuvant chemotherapy and so may choose to forgo adjuvant chemotherapy.30,31 This test is eligible for review with our tool, is based on its ability to predict drug response (as well as recurrence risk), and proceeds through clinical review to economic review. OncotypeDx has potentially large budget impact, reasonable prevalence (greater than 20% of women have actionable results), and a relatively high price for a test. Given these characteristics, the test progresses to question 13 with the ultimate recommendation of completing a full economic analysis.
Warfarin pharmacogenomically guided dosing is an example of a test that would be categorized as “optional-safety.” Since its approval by the FDA in 1954, warfarin has become one of the most widely prescribed anticoagulants in the United States.32,33 Warfarin has been shown to reduce this risk of clinical events in patients with thrombotic diathesis but carries numerous limitations, including narrow therapeutic range, wide interpatient variability, and interactions with numerous foods and drugs.34-37 As a result of these limitations, up to half of eligible patients are not prescribed warfarin.38 Recently, several genes have been associated with warfarin response including VKORC1, CYP4F2, and CYP2C9, which affect the reactivity and elimination of warfarin, respectively.39 Within the CAT, this test would proceed to clinical review based on the characteristics of its use. Several previously conducted meta-analyses show that this test has strong clinical validity and is able to provide valid information in dose estimation for patients on warfarin.40-44 So, in reviewing this test, the user would continue past question 6 on clinical validity to question 7 on clinical utility. Recently conducted large randomized trials that compared testing with no testing indicate that use of this test, contrary to preliminary evidence, actually has a small to negligible effect and therefore has low clinical utility and likely would be filtered out at question 7.45,46 Full economic review of this test would not be necessary, since it appears to have low clinical utility.
Discussion
We developed a tool, the CAT, to help evidence evaluators and decision makers more efficiently assess the clinical and economic value of CDTs. This tool highlights the importance of individual test characteristics (clinical validity and utility of test; efficacy, safety, and cost of baseline and alternative treatments; cost and mortality of health states; genotypic prevalence; and testing cost) and market-level forces (market penetration and adherence) in determining the value of a companion diagnostic. Notably, most of these factors can change over time, and use of the CAT may present a means to efficiently reevaluate tests as their evidence and market characteristics change.
Our study has several key implications. First, our literature review highlighted the importance of individual test characteristics in determining the economic value of a test. Second, our stakeholder interviews emphasized how any framework that excluded market characteristics would be inadequate in real-world practice. Our stakeholders identified regulatory status, clear actionability, and market adoption as key characteristics to use in determining if a test requires a full review. A cost-effectiveness model assumes a clear decision after testing and makes simplifying assumptions about the penetration and adherence to results in the testing scenario. While this provides a more clear comparison of clinical pathways, it limits the analysis in that it may ignore valuable market-level characteristics of the test’s use that define its value. Finally, the development of the CAT highlights how these parameters can be sequentially ordered to improve the efficiency of managed care review of CDTs.
Previous frameworks for evaluating companion tests facilitate an exhaustive analysis of relevant factors for the clinical and economic review of tests.7,17 Furthermore, attempts have been made to develop frameworks to assist in the decision making of public payers.8 These tools are limited, however, since they catalogue all factors involved in the coverage decision of a test but do not help the decision maker prioritize to expedite the review of tests—a key objective of the resource-constrained managed care user. For example, the most recent framework by Merlin et al. (2013) contains nearly 80 questions over 20 pages.8 While certainly thorough, this may be too detailed and cumbersome for the typical managed care decision maker to apply in practice.
Limitations
This study has some limitations worth noting. First, this tool is only intended to prioritize elements for review in a test—it does not provide a quantitative assessment of budget impact or clinical outcome for any given test. In order to arrive at a quantitative assessment of value, clinical outcome, or budget impact, a formal modeling exercise would still be necessary. Second, this tool also assumes that payers will be receiving claims for tests from licensed clinical labs with quality systems in place for analytical validity. This is why the ordering is different from previously completed tools, such as the ACCE framework,17 that were completed from a societal perspective and placed analytical validity as a first consideration. It is assumed that payers will have limited insight into laboratory quality and will have to rely on existing regulatory oversight to ensure quality among laboratories.
Conclusions
We developed a tool to assist managed care staff, pharmacy and therapeutics committees, and other health care decision makers in the evaluation process for CDTs. As more CDTs enter the market, it will become increasingly important for those evaluating their use to quickly gain a sense of their value. This tool is designed to be simple to use and to capture the majority of the variability in the value of testing, thereby offering greater efficiency in the CDT evaluation process.
APPENDIX. Detailed Methods and Results
Structured Interview
How do you define companion diagnostics and personalized medicines?
-
What are your current perceptions of the market for companion diagnostics?
-
Can you describe to us:
Have your views on any of the above changed within the past two years?
Who do you see as the “consumer” in the marketplace for companion diagnostics and personalized medicines? Who makes decisions about their use and reimbursement?
Is there a setting, indication, or disease area where you think they will have more value than others?
-
-
How would you define value for a companion diagnostic?
-
Specifically
How would you define the value of a personalized drug versus the test used in conjunction with that drug?
How best can that value be measured?
-
-
What factors drive the value of companion diagnostics?
What factors and types of evidence would or do you consider when evaluating CDx and personalized medicines?
-
Considering implementation of a test in a clinical workflow what are important factors?
~~~~~Review with Interviewee our findings from literature review~~~~~
Having briefly reviewed our findings, is there anything that stands out as missing? Are there any factors that we have highlighted that you don’t feel are necessary?
What type of decision-making tool or framework would be most useful (prompts, if needed: qualitative framework, case studies, excel model)?
How might you use a qualitative checklist or framework? Describe an ideal checklist/framework (prompts: length, time to complete, factors).
-
How might you use a quantitative model?
What variables would you want to be able to adjust to aid in your decision making?
Which outcome measures do you consider most important for your decisions (prompts: test performance [sensitivity/specificity], clinical events, life expectancy, quality of life, cost)?
Would you want to see productivity or quality of life included in a model?
What time horizon do you consider most meaningful for your decision making?
Key Stakeholder Interviews
Test Developers and Facilitators: This combined group represents both those actively engaged in developing and commercializing diagnostics informing drug use (developers) as well as those who offer testing services and interpretation for payers (facilitators). The following key themes were identified:
-
Commoditization of Expertise and Misalignment of Incentives. Overall, test developers expressed concern that in comparing laboratory developed tests (LDTs) with tests that either had received regulatory approval or were proprietary panels with established clinical benefit, payers viewed all companion diagnostics (CDxs) as commodities and reimbursed them as such. This commoditization of expertise also extended to Pharma partners who tended to view their diagnostic partners as vendors rather than partners who should share in the upside of the drug’s commercial success.
“Our value as a diagnostic partner is mostly what can be captured in shepherding the [companion] diagnostic through the regulatory [approval] and commercialization process, not just creating a lab method.” –Test Developer
“Tests are currently undervalued because [payers] are unclear how to reward them monetarily under the current system.” –Test Developer
-
CDxs Improve Confidence and Decision Impact. A theme that emerged from several interviews was that the value of a diagnostic was beyond just its epidemiologic characteristics (sensitivity and specificity) but extended to what degree it improved the confidence and reduced the uncertainty of the health care provider. On the aggregate level this might be measured as a “decision impact”—what percentage of providers follows the recommendation of or base their decisions on a CDx.
“There are not yet many tools for measuring the value of confidence in treatment decisions …. Decision impact is very meaningful, everyone needs to follow recommendations of the test for it to provide value.” –Test Developer
-
Payer Resistance to “Chain of Evidence.” Several test developers and facilitators expressed frustration that payers were not responding to a “chain of evidence” value proposition for diagnostics that were not studied in large-scale randomized trials, which are cost prohibitive for most developers. Developers felt this need is often circumstantial as diagnostics that limit drug use by identifying nonresponders or are clearly cost saving get reimbursed with much less evidence. An example that was repeatedly used was the KRAS test for nonresponse to erlotinib.
“You could tell [payers] this drug saves lives only if the sun is coming up and they’ll ask you ‘How do I know the sun is coming up? It’s observational and all retrospective.’” –Test Facilitator
“Quick uptake of KRAS could be because it could rationalize [cetuximab] even without evidence from an RCT. When it suits, adoption can happen very quickly.” –Test Facilitator
-
CDx Costs Are a “Blip on the Screen.” This term was used several times to express the relative magnitude of test price to the cost of the treatment decisions that CDx inform. Confusion was expressed as to why tests were given such scrutiny given that their cost was relatively modest and their potential impact on future treatment decisions was so great.
“The cost of diagnostics is significantly less than the therapy in many cases. For BCR-ABL, the cost of the test is $100-$400 but annual cost of gleevec is ~$100K…. When weighing the decision of whether to test, the cost of a diagnostic isn’t even a blip on the screen.” –Test Developer
-
Model Framework. Test developers acknowledged that payers responded mostly to budget impact but also expressed optimism that they at least considered quality of life (QoL) factors. Test developers also uniformly expressed a sentiment that at its core each test should start with a rationale for why testing is valuable. It was this clear and concise value proposition they argued that gained traction more than any complex model.
“Payers have gotten sophisticated enough to start caring about QoL.” –Test Developer
“Payers at least review QoL data, although potentially [it’s] not key to decisions.” –Test Developer
“Any test has to start with a qualitative rationale for why testing is valuable. [Test developers] need to first build a compelling narrative. Medicine is currently practiced with experience not data. Quantitative justification comes next.” –Test Facilitator
Payers: This group included both public and private payers that are responsible for making decisions regarding test reimbursement.
-
A Clear Focus on Budget Impact. Payers had a focus on budget impact and expressed a need to see a clear return on investment for the multitude of new diagnostics that are presented to them every year.
“We are concerned with the premium and whether the employer will choose us as their insurer. If we’re not going to be competitive with our premium, we’ll be out of business, and we can’t be competitive by paying for every new thing that comes down the pike.” –Payer
-
Need for a Clear Value Proposition. Payers reported frustration with how many developers came to them for coverage without a clear case for their value propositions. Payers expressed skepticism with observational studies that appeared to be “fishing for associations” without evaluating how the information gained from testing would improve clinical practice.
“There is so much smoke out there around the variety of types of tests that can be useful. I would want to have specific information that would give you real measurable data about how the diagnosis affects the individual.” –Payer
-
A Fear of “Mission Creep.” Concern was voiced that personalized medicine and companion diagnostics are a means sustaining irrationally high drug pricing. This was compared to a “Trojan Horse effect” for pricing. Drug developers would request a high margin because their drug worked in only a narrow subset of patients and sometimes only marginally so. Then drug developers would work to expand use to other diseases with the same biomarker expression while keeping the high pricing from the narrow indication.
“The way CDx are being used by pharmaceutical manufacturers is a justification for irrational product pricing to gouge the market…NSCLC costs around $70K and Xalkori costs around $90K and their justification is that they only use it in 4% of patients, but now they are expanding usage to other diseases with ALK expression without changing pricing.” –Payer
-
Disillusionment. Nearly every payer mentioned a disillusionment with the pace at which personalized medicine and companion diagnostics were delivering on their promise to improve the efficient allocation of health care resources.
“There has been great promise for 30 years but companion diagnostics still don’t have a lot of impact on clinical practice.” –Payer
Model Framework: When asked about what an ideal framework would contain, 2 central themes emerged. First, payers expressed reluctance with using any framework that is administratively burdensome, and nearly all suggested that they look to outside organizations (CMS, Hayes) for guidance decisions in all but a few exceptional cases. Given the complexity of the space, and the relatively low absolute expense, most payers did not find it efficient to complete their own reviews. Second, payers suggested that they would want to see a model that focused on budget impact with no consideration of QoL. Every payer mentioned a concern with “Number Needed to Test” or how many patients they would have to pay for testing before finding an actionable result as the largest factor in their decision making.
“And who’s going to do the work [using this framework]? If we have a list of 15 things and are those all things that we’re going to have to do? Concept is interesting but it depends on how many man hours it will take to implement, that’s why we rely on outside organizations.” –Payer
“The cost of testing has dwarfed drug cost, so the key question is how often does the [companion] diagnostic actually inform drug cost. The most helpful too would be a dossier by the test developer …. [a tool for] bucketing tests would help somewhat.” –Payer
“I’m looking for data gathered prospectively on sensitivity and specificity …. I would just have [test developers] read a 20-year-old textbook on decision analysis, we need more than an association, we need number to test and the characteristics in the population.” –Payer
In summary, these key informant interviews support the need for a tool to set expectations and improve communication of value between test developers, facilitators, and payers. Any tool that is developed should be very simple, easy to operate, and quickly categorize (“bucket”) tests by a concise statement of value. Once categorized, the tool should highlight key operating characteristics for the test: clinical sensitivity and specificity, number needed to test, and decision impact on drug spend. QoL metrics were not thought to be as impactful by payers who instead preferred to see direct medical impact of test use.
REFERENCES
- 1.Zineh I, Gerhard T, Aquilante CL, Beitelshees AL, Beasley BN, Hartzema AG. Availability of pharmacogenomics-based prescribing information in drug package inserts for currently approved drugs. Pharmacogenomics J. 2004;4(6):354-58. [DOI] [PubMed] [Google Scholar]
- 2.Frueh FW, Amur S, Mummaneni P, et al. . Pharmacogenomic biomarker information in drug labels approved by the United States Food and Drug Administration: prevalence of related drug use. Pharmacotherapy. 2008;28(8):992-998. [DOI] [PubMed] [Google Scholar]
- 3.Zineh I, Pacanowski MA. Pharmacogenomics in the assessment of therapeutic risks versus benefits: inside the United States Food and Drug Administration. Pharmacotherapy. 2011;31(8):729-35. [DOI] [PubMed] [Google Scholar]
- 4.Surh LC, Pacanowski MA, Haga SB, et al. . Learning from product labels and label changes: how to build pharmacogenomics into drug-development programs. Pharmacogenomics. 2010;11(12):1637-47. [DOI] [PubMed] [Google Scholar]
- 5.Epstein RS, Frueh FW, Geren D, et al. . Payer perspectives on pharmacogenomics testing and drug development. Pharmacogenomics. 2009;10(1):149-51. [DOI] [PubMed] [Google Scholar]
- 6.Olsen D, Jørgensen JT. Companion diagnostics for targeted cancer drugs - clinical and regulatory aspects. Front Oncol. 2014;4:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Flowers CR, Veenstra D. The role of cost-effectiveness analysis in the era of pharmacogenomics. Pharmacoeconomics. 2004;22(8):481-93. [DOI] [PubMed] [Google Scholar]
- 8.Merlin T, Farah C, Schubert C, Mitchell A, Hiller JE, Ryan P. Assessing personalized medicines in Australia: a national framework for reviewing codependent technologies. Med Decis Making. 2013;33(3):333-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.AMCP Format Executive Committee. The AMCP format for formulary submissions: a format for submission of clinical and economic evidence of pharmaceuticals in support of formulary consideration. Version 3.1. December 2012. Available at: http://amcp.org/practice-resources/amcp-format-formulary-submisions.pdf. Accessed June 29, 2015.
- 10.Wong WB, Carlson JJ, Thariani R, Veenstra DL. Cost effectiveness of pharmacogenomics: a critical and systematic review. Pharmacoeconomics. 2010;28(11):1001-13. [DOI] [PubMed] [Google Scholar]
- 11.Center for the Evaluation of Value and Risk in Health. Cost-Effectiveness Analysis Registry. 2014. Available at: https://research.tufts-nemc.org/cear4/. Accessed June 29, 2015.
- 12.National Institute for Health and Care Excellence. Evidence search. 2014. Available at: https://www.evidence.nhs.uk/. Accessed June 29, 2015.
- 13.Canadian Agency for Drugs and Technologies in Health. Health technology assessment search. 2015. Available at: https://www.cadth.ca/hta. Accessed July 3, 2014.
- 14.Carlson JJ, Henrikson NB, Veenstra DL, Ramsey SD. Economic analyses of human genetics services: a systematic review. Genet Med. 2005;7(8):519-23. [DOI] [PubMed] [Google Scholar]
- 15.Djalalov S, Musa Z, Mendelson M, Siminovitch K, Hoch J. A review of economic evaluations of genetic testing services and interventions (2004-2009). Genet Med. 2011;13(2):89-94. [DOI] [PubMed] [Google Scholar]
- 16.Vegter S, Boersma C, Rozenbaum M, Wilffert B, Navis G, Postma MJ. Pharmacoeconomic evaluations of pharmacogenetic and genomic screening programmes: a systematic review on content and adherence to guidelines. Pharmacoeconomics. 2008;26(7):569-87. [DOI] [PubMed] [Google Scholar]
- 17.Haddow JE, Palomaki GE. ACCE: a model process for evaluating data on emerging genetic tests. In: Khoury M, Little J, Burke W, eds. Human Genome Epidemiology: A Scientific Foundation for Using Genetic Information to Improve Health and Prevent Disease. New York: Oxford University Press; 2003:217-33. [Google Scholar]
- 18.Kwak EL, Bang YJ, Camidge DR, et al. . Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363(18):1693-703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Soda M, Choi YL, Enomoto M, et al. . Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448(7153):561-66. [DOI] [PubMed] [Google Scholar]
- 20.Rikova K, Guo A, Zeng Q, et al. . Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;131(6):1190-203. [DOI] [PubMed] [Google Scholar]
- 21.Camidge DR, Doebele RC. Treating ALK-positive lung cancer—early successes and future challenges. Nat Rev Clin Oncol. 2012;9(5):268-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shaw AT, Kim DW, Nakagawa K, et al. . Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368(25):2385-94. [DOI] [PubMed] [Google Scholar]
- 23.Hetherington S, McGuirk S, Powell G, et al. . Hypersensitivity reactions during therapy with the nucleoside reverse transcriptase inhibitor abacavir. Clin Ther. 2001;23(10):1603-14. [DOI] [PubMed] [Google Scholar]
- 24.Mallal S, Nolan D, Witt C, et al. . Association between presence of HLA-B*5701, HLA-DR7, and HLA-DQ3 and hypersensitivity to HIV-1 reverse-transcriptase inhibitor abacavir. Lancet. 2002;359(9308):727-32. [DOI] [PubMed] [Google Scholar]
- 25.Hetherington S, Hughes AR, Mosteller M, et al. . Genetic variations in HLA-B region and hypersensitivity reactions to abacavir. Lancet. 2002;359(9312):1121-22. [DOI] [PubMed] [Google Scholar]
- 26.Martin AM, Nolan D, Gaudieri S, et al. . Predisposition to abacavir hypersensitivity conferred by HLA-B*5701 and a haplotypic Hsp70-Hom variant. Proc Natl Acad Sci U.S.A. 2004;101(12):4180-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hughes AR, Mosteller M, Bansal AT, et al. . Association of genetic variations in HLA-B region with hypersensitivity to abacavir in some, but not all, populations. Pharmacogenomics. 2004;5(2):203-11. [DOI] [PubMed] [Google Scholar]
- 28.Phillips EJ, Wong GA, Kaul R, et al. . Clinical and immunogenetic correlates of abacavir hypersensitivity. AIDS. 2005;19(9):979-81. [DOI] [PubMed] [Google Scholar]
- 29.Mallal S, Phillips E, Carosi G, et al. . HLA-B*5701 screening for hypersensitivity to abacavir. N Engl J Med. 2008;358(6):568-79. [DOI] [PubMed] [Google Scholar]
- 30.Albain KS, Barlow WE, Shak S, et al. . Prognostic and predictive value of the 21-gene recurrence score assay in postmenopausal women with nodepositive, oestrogen-receptor-positive breast cancer on chemotherapy: a retrospective analysis of a randomised trial. Lancet Oncol. 2010;11(1):55-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tang G, Cuzick J, Costantino JP, et al. . Risk of recurrence and chemotherapy benefit for patients with node-negative, estrogen receptor-positive breast cancer: recurrence score alone and integrated with pathologic and clinical factors. J Clin Oncol. 2011;29(33):4365-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Epstein RS, Moyer TP, Aubert RE, et al. . Warfarin genotyping reduces hospitalization rates results from the MM-WES (Medco-Mayo Warfarin Effectiveness study). J Am Coll Cardiol. 2010;55(25):2804-12. [DOI] [PubMed] [Google Scholar]
- 33.Charland SL, Agatep BC, Epstein RS, et al. . Patient knowledge of pharmacogenetic information improves adherence to statin therapy: results of the additional KIF6 risk offers better adherence to statins (AKROBATS) trial. J Am Coll Cardiol. 2012;59(13 Suppl):E1848. [Google Scholar]
- 34.De Caterina R, Connolly SJ, Pogue J, et al. . Mortality predictors and effects of antithrombotic therapies in atrial fibrillation: insights from ACTIVE-W. Eur Heart J. 2010;31(17):2133-40. [DOI] [PubMed] [Google Scholar]
- 35.Baker WL, Cios DA, Sander SD, Coleman CI. Meta-analysis to assess the quality of warfarin control in atrial fibrillation patients in the United States. J Manag Care Pharm. 2009;15(3):244-52. Available at: http://www.amcp.org/data/jmcp/244-252.pdf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fang MC, Go AS, Chang Y, et al. . Warfarin discontinuation after starting warfarin for atrial fibrillation. Circulation. 2010;3(6):624-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Waldo AL, Becker RC, Tapson VF, Colgan KJ, Committee NS. Hospitalized patients with atrial fibrillation and a high risk of stroke are not being provided with adequate anticoagulation. J Am Coll Cardiol. 2005;46(9):1729-36. [DOI] [PubMed] [Google Scholar]
- 38.Hylek EM, D’Antonio J, Evans-Molina C, Shea C, Henault LE, Regan S. Translating the results of randomized trials into clinical practice: the challenge of warfarin candidacy among hospitalized elderly patients with atrial fibrillation. Stroke. 2006;37(4):1075-80. [DOI] [PubMed] [Google Scholar]
- 39.Flockhart DA, O’Kane D, Williams MS, et al. . Pharmacogenetic testing of CYP2C9 and VKORC1 alleles for warfarin. Genet Med. 2008;10(2):139-50. [DOI] [PubMed] [Google Scholar]
- 40.Yang L, Ge W, Yu F, Zhu H. Impact of VKORC1 gene polymorphism on interindividual and interethnic warfarin dosage requirement—a systematic review and meta analysis. Thromb Res. 2010;125(4):e159-166. [DOI] [PubMed] [Google Scholar]
- 41.Sanderson S, Emery J, Higgins J. CYP2C9 gene variants, drug dose, and bleeding risk in warfarin-treated patients: a HuGEnet systematic review and meta-analysis. Genet Med. 2005;7(2):97-104. [DOI] [PubMed] [Google Scholar]
- 42.Lindh JD, Holm L, Andersson ML, Rane A. Influence of CYP2C9 genotype on warfarin dose requirements—a systematic review and meta-analysis. Eur J Clin Pharmacol. 2009;65(4):365-75. [DOI] [PubMed] [Google Scholar]
- 43.Liang R, Wang C, Zhao H, Huang J, Hu D, Sun Y. Influence of CYP4F2 genotype on warfarin dose requirement—a systematic review and meta-analysis. Thromb Res. 2012;130(1):38-44. [DOI] [PubMed] [Google Scholar]
- 44.Jorgensen AL, FitzGerald RJ, Oyee J, Pirmohamed M, Williamson PR. Influence of CYP2C9 and VKORC1 on patient response to warfarin: a systematic review and meta-analysis. PLoS One. 2012;7(8):e44064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pirmohamed M, Burnside G, Eriksson N, et al. . A randomized trial of genotype-guided dosing of warfarin. N Engl J Med. 2013;369(24):2294-303. [DOI] [PubMed] [Google Scholar]
- 46.Kimmel SE, French B, Kasner SE, et al. . A pharmacogenetic versus a clinical algorithm for warfarin dosing. N Engl J Med. 2013;369(24):2283-93. [DOI] [PMC free article] [PubMed] [Google Scholar]