

Abstract
Heparanase (Hpa1) is expressed by tumor cells and cells of the tumor microenvironment and functions extracellularly to remodel the extracellular matrix (ECM) and regulate the bioavailability of ECM‐bound factors, augmenting, among other effects, gene transcription, autophagy, exosome formation, and heparan sulfate (HS) turnover. Much of the impact of heparanase on tumor progression is related to its function in mediating tumor‐host crosstalk, priming the tumor microenvironment to better support tumor growth, metastasis, and chemoresistance. The enzyme appears to fulfill some normal functions associated, for example, with vesicular traffic, lysosomal‐based secretion, autophagy, HS turnover, and gene transcription. It activates cells of the innate immune system, promotes the formation of exosomes and autophagosomes, and stimulates signal transduction pathways via enzymatic and nonenzymatic activities. These effects dynamically impact multiple regulatory pathways that together drive tumor growth, dissemination, and drug resistance as well as inflammatory responses. The emerging premise is that heparanase expressed by tumor cells, immune cells, endothelial cells, and other cells of the tumor microenvironment is a key regulator of the aggressive phenotype of cancer, an important contributor to the poor outcome of cancer patients and a valid target for therapy. So far, however, antiheparanase‐based therapy has not been implemented in the clinic. Unlike heparanase, heparanase‐2 (Hpa2), a close homolog of heparanase (Hpa1), does not undergo proteolytic processing and hence lacks intrinsic HS‐degrading activity, the hallmark of heparanase. Hpa2 retains the capacity to bind heparin/HS and exhibits an even higher affinity towards HS than heparanase, thus competing for HS binding and inhibiting heparanase enzymatic activity. It appears that Hpa2 functions as a natural inhibitor of Hpa1 regulates the expression of selected genes that maintain tissue hemostasis and normal function, and plays a protective role against cancer and inflammation, together emphasizing the significance of maintaining a proper balance between Hpa1 and Hpa2.
Keywords: heparanase, heparanase 2, heparan sulfate, signal transduction
Abbreviations
- ECM
extracellular matrix
- EGFR
epidermal growth factor receptor
- Hpa1
heparinase
- Hpa2
heparanase‐2
- HS
heparan sulfate
- HSPGs
heparan sulfate proteoglycans
- VEGF
vascular endothelial growth factor
BACKGROUND
The heparanase (HPSE) messenger RNA (mRNA) encodes a 65 kDa proenzyme that is cleaved by cathepsin L into 8 and 50 kDa subunits that noncovalently associate to form the active enzyme. Structurally, heparanase is composed of a TIM‐barrel fold that contains the enzyme's active site and a flexible C‐terminus domain required for the secretion and signaling function of the protein. 1 , 2 Heparanase belongs to the wider class of enzymes known as ‘retaining glycosidases,’ which catalyze hydrolytic cleavage of glycosidic bonds with net retention of anomeric stereochemistry. 3 It employs a conserved ‘double displacement mechanism,’ involving two key catalytic amino acid residues—a nucleophile (Glu343) and a general acid/base proton donor (Glu225)—and transient formation of a covalent enzyme–substrate intermediate during the catalytic cycle. 3 The heparin/heparan sulfate (HS)‐binding domains (HBD1, HBD2) are situated close to the active site micropocked fold. 1 , 3 Relevant observations are referred to in Table 1.
Table 1.
Key observations.
| Selected key observations (Heparanase) |
| 1975—Endoglucuronidase is responsible for cleavage of heparin by mastocytoma cells. 4 , 5 |
| 1982—Purification and characterization of HS‐degrading endoglycosidase in platelets. 6 , 7 |
| 1983—HS‐degrading endoglycosidase is associated with cancer metastasis. 8 , 9 |
| 1983—Present. Heparanase promotes the pathogenesis of solid and hematological malignancies. 10 , 11 , 12 , 13 |
| 1983—Present. High levels of heparanase correlate with decreased survival of cancer patients. 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23 |
| 1984—Activated T lymphocytes produce a matrix‐degrading HS endoglycosidase. 24 |
| 1987–1989—Experimental metastasis and autoimmunity are attenuated by heparanase‐inhibiting species of heparin. 25 , 26 , 27 , 28 |
| 1986—Heparanase regulates the bioavailability of HS‐bound growth factors, chemokines, and cytokines. 29 , 30 , 31 |
| 1999—Heparanase (HPSE) gene cloning, expression, and function in tumor progression. 32 , 33 , 34 , 35 , 36 |
| 1999–2003—Latent heparanase is a heterodimer composed of 8 and 50 kDa subunits connected by a 6 kDa linker peptide. 37 , 38 , 39 , 40 |
| 1999—PI‐88 (= Muparfostat), SST0001 (= Roneparstat), and PG545 (= Pixatomid) attenuate tumor growth and angiogenesis in preclinical models. 41 , 42 , 43 , 44 |
| 2000—Heparanase catalytic mechanism involves a proton donor (Glu‐225) and nucleophile (Glu‐343). 1 |
| 2002—Transcriptional activity of the HPSE gene promoter. 45 , 46 , 47 , 48 , 49 , 50 |
| 2002—HPSE gene silencing inhibits tumor angiogenesis and metastasis. 51 , 52 |
| 2004—The two subunits of the latent enzyme are connected by a 6 kDa linker peptide that obstructs access to the active site. 37 |
| 2004—HPSE overexpressing and null mice unravel physiological functions of the enzyme. 53 , 54 , 55 |
| 2004—Cellular uptake of heparanase is mediated by cell membrane HS. 56 |
| 2004—Processing and activation of latent heparanase occur in lysosomes (79) and involves cleavage of the linker segment by cathepsin L. 57 , 58 |
| 2005—Heparanase is involved in the pathogenesis of sepsis, amyloidosis, colitis, pancreatitis, and tissue fibrosis 59 , 60 , 61 , 62 , 63 |
| 2005—Identification of heparin/HS‐binding domains of heparanase. 64 |
| 2006—Heparanase regulates VEGF gene expression via activation of Src family members. 65 |
| 2006—Heparanase is involved in coagulation and the pathogenesis of thrombosis and atherosclerosis. 66 , 67 , 68 , 69 , 70 , 71 |
| 2009—Heparanase signaling is mediated by its carboxy‐terminal domain. 72 |
| 2011—Nuclear heparanase enhances histone acetyltransferase activity and promotes gene expression. 73 |
| 2012—Heparanase is involved in the pathogenesis of diabetes, diabetic nephropathy, diabetic cardiomyopathy, and kidney dysfunction. 74 , 75 , 76 , 77 , 78 |
| 2012—Nuclear heparanase interacts with key chromatin‐modifying enzymes and regulates gene transcription. 79 |
| 2012—Heparanase augments Akt, EGFR, and STAT phosphorylation. 80 , 81 |
| 2013—Heparanase regulates secretion, composition, and function of exosomes. 82 , 83 , 84 , 85 , 86 |
| 2014—Crystallization, structural characterization, and substrate recognition of human heparanase. 3 , 87 |
| 2014—Heparanase mediates the crosstalk between cells and the tumor microenvironment. 10 , 11 , 12 , 13 , 88 |
| 2014—Heparanase is a key mediator of macrophage activation and polarization. 89 , 90 |
| 2014—HS degradation fragments trigger expression of proinflammatory cytokines through TLR‐4 activation. 89 |
| 2014—Present—Examination of antiheparanase therapies in clinical trials. 41 , 91 , 92 , 93 , 94 |
| 2015—Present. Heparanase is involved in the pathogenesis viral infection. 95 , 96 , 97 , 98 |
| 2015—Heparanase enhances tumorigenicity by promoting autophagy. 99 |
| 2017—Heparanase plays a critical role in NK‐ and T‐cell recruitment into tumors. 100 |
| 2021—Heparanase is involved in sensing DNA damage. 96 |
| Key observations (Hpa2) |
| 2000—HPSE2 gene cloning and characterization. 101 |
| 2010—HPSE2 is mutated in urofacial syndrome, a rare congenital disease featuring urological defects and inverted facial expression. 102 , 103 , 104 |
| 2010—Hpa2 interacts with HS with high affinity and inhibits heparanase activity. 105 |
| 2015—Overexpression of HPSE2 attenuates tumor growth, whereas HPSE2 gene silencing promotes tumorigenesis. 106 , 107 , 108 , 109 , 110 , 111 , 112 , 113 , 114 , 115 |
| 2016—Hpa2 protects against sepsis, endotoxemia, renal inflammation, and Covid‐19. 116 , 117 , 118 |
| 2018—Hpa2 functions as a natural inhibitor of Hpa1, emphasizing the significance of a proper Hpa1/Hpa2 ratio in tissue hemostasis. 116 , 117 , 118 |
| 2022—Hpa2‐KO homozygosity is embryonic lethal, indicating an essential involvement of Hpa2 in embryonic development (unpublished). |
| 2022—Hpa2‐KO mice are highly susceptible to aggressive cancer and inflammation (unpublished), emphasizing the protective function of host‐derived Hpa2 and encouraging the development of Hpa2‐based therapy. |
Abbreviations: Akt, protein kinase B; EGFR, epidermal growth factor receptor; Hpa2, heparanase‐2; HS, heparan sulfate; KO, knockout; NK, natural killer; STAT, signal transducer and activator of transcription; TLR‐4; toll‐like receptor 4; VEGF, vascular endothelial growth factor.
Heparan sulfate proteoglycans (HSPGs) are a fundamental class of extracellular matrix (ECM) constituents, comprised of pericellular or extracellular core proteins conjugated to one or more chains of the glycosaminoglycan polysaccharide HS. 119 HSPGs mediate myriad biological processes, including signal transduction, developmental patterning, 120 cell adhesion, 121 barrier formation, 122 endocytosis 123 and viral entry. 95 , 124 These processes largely depend upon the HS polysaccharides adorning the core protein, whose heterogeneous structure allows interaction with multiple partners. Given their heterogeneity and versatility, HSPGs serve as important functional components of the cell surface, glycocalyx, and ECM. Hence, cleavage of HS by heparanase affects a diverse and expanding repertoire of physiological and pathological processes. The enzyme appears to fulfill some normal functions, associated, for example, with vesicular traffic, lysosomal‐based secretion, autophagy, tissue remodeling, HS turnover, and gene transcription. 10 , 11 , 12 , 13 It activates cells of the innate immune system, promotes the formation of exosomes and autophagosomes, and stimulates signal transduction pathways via enzymatic and nonenzymatic activities. 10 , 11 , 12 , 13 , 82 These effects dynamically impact multiple regulatory pathways that together drive tumor growth, dissemination, and drug resistance as well as inflammatory responses. 2 , 11 , 12 , 13 A key venue by which heparanase accomplishes its multiple effects on cells and tissues is by regulating the bioavailability of HS‐bound growth factors, chemokines, and cytokines. In this way, heparanase mediates tumor‐host crosstalk and promotes basic cellular processes that together orchestrate tissue remodeling. 82 Among the proteins sequestered by the ECM are typical proangiogenic mediators such as platelet‐derived growth factor, hepatocyte growth factor (HGF), basic fibroblast growth factor, heparin‐binding epithelial growth factor, and vascular endothelial growth factor A (VEGF‐A). 29 , 30 , 125 , 126 Release of these proteins by heparanase contributes to the strong proangiogenic response observed in preclinical models and clinical settings. 13 , 31 , 127 , 128 , 129 , 130
HISTORY
Activity capable of cleaving macromolecular heparin at a limited number of sites was first reported in mastocytoma cells. 4 Soon thereafter, Höök et al. 131 reported an endoglycosidase activity that degrades HS glycosaminoglycans into oligosaccharides. Attempts to purify the enzyme yielded some misleading results, culminating, nearly 10 years later, in purification of the platelet enzyme 7 and cloning of a single human heparanase complementary DNA sequence, independently by four groups. 32 , 33 , 34 , 35 Given the structural role of HSPGs in the assembly of the ECM and basement membrane, it was hypothesized that HS‐degrading activity will loosen the ECM, thus promoting cell dissemination. Indeed, early on, heparanase activity was found to correlate with the metastatic potential of tumor cells, 8 , 9 , 132 a correlation that still directs and guides heparanase research. Studies performed before cloning of the HPSE gene contributed immensely to our understanding of key features in the biology of the enzyme, its mode of action, and involvement in cancer metastasis and inflammation. 133 , 134 Soon after cloning of the HPSE gene and the development of antiheparanase antibodies and probes, many studies examined its expression in human tumors compared with the adjacent normal tissue. Immunohistochemistry, in situ hybridization, real‐time‐polymerase chain reaction and enzymatic activity analyses revealed that heparanase is upregulated in essentially all human tumors examined. 2 , 11 , 12 , 13 , 14 , 128 , 130 , 135 In contrast, the normal‐looking tissue adjacent to the malignant lesion expresses little or no detectable levels of heparanase, indicating that fibroblasts and epithelial cells do not normally express the enzyme. The molecular mechanisms underlying heparanase induction in tumor cells are not entirely clear but involve epigenetic alterations (i.e., DNA methylation), hormones, oncogenes, and transcriptional/posttranscriptional regulation by elements (3ʹ‐UTR, enhancer, insulator) that activate or suppress the HPSE promoter. 12 , 45 , 136 Selected observations are referred to in Table 1.
Clinically, patients that were diagnosed as heparanase‐positive exhibited a significantly higher rate of local and distant metastases as well as reduced postoperative survival, compared with patients that were diagnosed as heparanase‐negative. 15 , 16 , 17 , 137 These and more recent studies 18 , 19 , 20 , 21 , 22 , 23 provide strong clinical support for the prometastatic function of heparanase. Subsequent studies provided compelling evidence that ties heparanase levels with all steps of tumor formation including tumor initiation, angiogenesis, growth, metastasis, and chemoresistance. 22 , 88 , 99 , 138 , 139 , 140 , 141 Heparanase not only enhances tumor cell dissemination but also accelerates the growth of the primary tumor. We and others have shown that heparanase induces the expression of VEGF‐A 65 and VEGF‐C, 142 , 143 leading to increased blood and lymph vessel density. Subsequent studies revealed that heparanase downregulates the expression of tumor suppressors (i.e., CXCL10 139 ) and induces the transcription of proangiogenic (i.e., cyclooxygenase–2, matrix metalloproteinase–9), prothrombotic (i.e., tissue factor [TF]), proinflammatory (i.e., tumor necrosis factor‐α, interleukin [IL]‐1, IL‐6, macrophage inflammatory protein–2), profibrotic (i.e., transforming growth factor‐β), mitogenic (i.e., HGF), osteolytic (receptor activator of nuclear factor kappa beta) and other genes, 66 , 89 , 90 , 128 , 142 , 144 , 145 , 146 thus significantly expanding its functional repertoire and mode of action in promoting tissue inflammation and aggressive tumor behavior. 96 Several excellent up‐to‐date reviews describe basic and translational aspects of heparanase. 11 , 12 , 96 This review is aimed at further increasing awareness of this multifaceted protein, highlighting the significance of the Hpa1–heparanase‐2 (Hpa2) axis and addressing obstacles in implementing antiheparanase therapies.
NONENZYMATIC ACTIVITIES
Years before the resolution of the heparanase crystal structure, Fux et al. 72 predicted the structure of enzymatically active, single‐chain, heparanase enzyme, in which the linker segment was replaced by three glycine–serine repeats (GS3), resulting in a constitutively active enzyme. 37 The structure clearly illustrated a C‐terminus (═C‐domain) fold positioned next to the TIM‐barrel structure. 72 Remarkably, protein kinase B (Akt) phosphorylation was stimulated by cells overexpressing the C‐domain (amino acids 413–543), while the TIM‐barrel protein variant yielded no Akt activation, 72 indicating a nonenzymatic signaling function of heparanase mediated by the C‐domain. Notably, Akt phosphorylation was best enhanced in cells transfected with a mini gene comprising a segment of the 8 kDa subunit (Gln36–Ser55) linked to the C‐domain sequence. 72 These findings further indicate that the C‐domain is indeed a valid functional domain responsible for Akt phosphorylation. The cellular consequences of C‐domain overexpression were best revealed by monitoring tumor xenograft growth. Notably, tumor xenografts produced by C‐domain‐transfected glioma cells appeared comparable to those produced by cells transfected with the full‐length heparanase. 72 While signaling through HS clustering appears straightforward in its rationale, HS‐independent signaling by heparanase requires a mediator, possibly in the form of cell surface receptor(s). Binding studies performed with wild‐type CHO‐KI cells and their HS‐deficient CHO‐745 counterpart cells reinforced the notion that while HSPGs serve as low affinity, high abundant binding sites, heparanase also associates with high affinity, low abundant cell surface receptor(s). 72 Notably, Wood and Hulett 147 have reported that the 300 kDa cation‐independent mannose 6‐phosphate receptor (CD222) can bind enzymatically active heparanase and may serve as a heparanase receptor (Figure 1).
Figure 1.
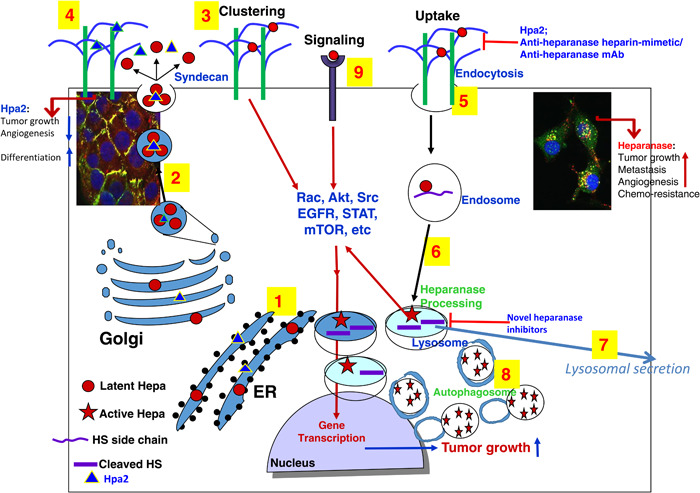
Schematic presentation of heparanase and Hpa2 biosynthesis and trafficking. Pre–proheparanase (red circles) and Hpa2 (blue triangles) are first targeted to the ER lumen via their own signal peptides (1). The proteins are then shuttled to the Golgi apparatus and are subsequently secreted via vesicles that bud from the Golgi (2). Once secreted, heparanase rapidly interacts with syndecans, resulting in their clustering and signaling (3), followed by rapid endocytosis of the heparanase–syndecan complex (5) that accumulates in late endosomes (6). Hpa2 interacts with cell membrane HSPG (i.e., syndecans) with higher affinity but unlike heparanase, is not subjected to uptake but rather remains on the cell membrane for a relatively long period of time (4 and left inset). Accumulation of Hpa2 in the extracellular compartment is enhanced by heparin or anti‐Hpa2 monoclonal antibody. Heparanase uptake is inhibited by heparin/heparin mimetics, antiheparanase monoclonal antibodies, or Hpa2, resulting in extracellular accumulation of the latent enzyme (5). Conversion of endosomes to lysosomes (6) results in heparanase processing and activation (primarily by cathepsin L) awaiting secretion (7). Typically, heparanase appears in perinuclear lysosomes (right inset), promoting autophagy (8) and tumor growth, metastasis, angiogenesis, and chemoresistance due to its enzymatic and signaling (9) functions. Hpa2, on the other hand, attenuates tumor growth and vascularity. Novel heparanase inhibitors are expected to target extracellular latent (signaling) and active heparanase as well as the intracellular, lysosomal, enzyme. ER, endoplasmic reticulum; Hpa2, heparanase‐2; HSPG, heparan sulfate proteoglycan.
Another way used to distinguish between enzymatic and nonenzymatic functions of heparanase applied a double mutant protein devoid of heparanase enzymatic activity due to point mutations (Glu225–Ala = proton donor, Glu343–Ala = nucleophile) in the enzyme' active site. 1 This inactive form of heparanase was found to exert epidermal growth factor receptor (EGFR) phosphorylation and Akt phosphorylation. 80 It was also reported that enzymatically quiescent heparanase augmented T‐cell interactions with VCAM‐1 and ECM components. 148 Notably, heparanase enhances platelet adhesive capacity and thrombogenicity 149 and also supports the clustering of circulating tumor cells 150 thereby contributing to the metastatic cascade, largely independent of its enzymatic activity. Likewise, the upregulation of HGF, MCP‐1, and TF expression in response to heparanase was independent of its enzyme activity. 145 , 151 , 152 Moreover, heparanase enhances the phosphorylation of selected signaling molecules including Akt, Src, and EGFR, which in turn facilitates STAT3 phosphorylation, in a manner that requires secretion but not enzymatic activity of heparanase, evident by being mediated by the enzyme C‐terminus domain and by the inactive double mutant protein 72 , 80 , 81 (Table 1). A later observation of integrin‐dependent phosphoinositide 3‐kinase (PI3K)/Akt activation in response to heparanase further highlighted the nonenzymatic activity of heparanase in promoting signal transduction. 153 Notably, the ability of heparanase to activate PI3K/Akt in a nonenzymatic manner, essentially bypassing PTEN signaling, is evidence of its ability to counter tumor‐suppressive mechanisms. 11 , 153 The results suggest that in certain cells, heparanase activates SRC family kinase in an enzymatically independent manner which proceeds to stimulate EGFR. EGFR can then activate downstream pathways such as PI3K–Akt and mitogen‐activated protein kinase–extracellular signal‐regulated kinase, possibly resulting in phosphorylation of signal transducer and activator of transcription 3 (STAT3) which then drives tumorigenic responses. 10 , 128
SIGNAL TRANSDUCTION
Heparanase interacts with syndecans by virtue of their HS content and the typical high affinity that exists between the enzyme and its substrate. This high‐affinity interaction directs the clustering of syndecans followed by rapid and efficient uptake of heparanase 56 , 154 (Figure 1). Mechanistically, syndecan clustering by heparanase or the KKDC peptide, corresponding to the heparin‐binding domain of heparanase, 64 enhance cell adhesion and spreading, associated with PKC, Src, and Rac1 activation, 155 molecular determinants shown to be induced by syndecans. 156 , 157 , 158 Cell adhesion represents a nonenzymatic signaling function of heparanase in its simplest term. 159 , 160 Heparanase was also noted to elicit signaling in a manner that does not involve HS. Signaling is considered to be HS‐independent if it occurs in HS‐deficient cells (i.e., CHO‐745) or in the presence of heparin, as demonstrated for enhanced Akt phosphorylation by heparanase. 161 Heparin, a potent competitive inhibitor of heparanase enzymatic activity, when added together with heparanase, augmented, rather than attenuated, Akt phosphorylation, 161 critically implying that heparanase enzymatic activity is not required for Akt activation. Importantly, in xenograft models, heparanase overexpression resulted in tumors bigger in size 45 , 162 coupled with increased Akt phosphorylation. 45 , 72 Adversely, heparanase gene silencing was associated with reduced Akt phosphorylation. 140 , 163 Related studies revealed that heparanase stimulates the phosphorylation of STAT3, STAT5, Src, EGFR, Erk, and the insulin receptor and also activates G‐protein receptor signaling, 65 , 80 , 81 , 164 all function to promote tumorigenesis (Figure 1 and Table 1).
DNA DAMAGE
Associations between heparanase and various pathologies, including inflammation and cancer metastasis, have historically been attributed to the cleavage of HS chains at the cell surface and basement membrane; however, as discussed above, heparanase may possess unique roles arising independently of its enzymatic active site. For example, while the enzymatic activity of heparanase appears to enable viral release through splitting of HS residues at the cell surface, heparanase was reported to also regulate gene expression and trigger proviral signaling through some distinct nonenzymatic activity. 97 It appears that heparanase acts beyond its established endoglycosidase activity as a potent regulator of the signal transduction phase of cellular defense. In this respect, it was reported that cells lacking heparanase display enhanced sensitivity to DNA damage‐induced death 96 and are intrinsically resistant to herpes simplex virus‐1 infection. 96 Moreover, the interferon system is constitutively enhanced in the absence of heparanase and deletion of heparanase protects against cellular infiltration and associated inflammation. 96 , 97 , 165 Repeat immunopurifications of heparanase showed robust binding of proteins heavily implicated in DNA damage sensing and repair, suggesting that heparanase plays a significant role as a regulator of DNA damage response signals. 96
NUCLEAR HEPARANASE
Nuclear HS inhibits histone acetyltransferases (HATs), and thereby gene transcription. 73 Heparanase contains two potential nuclear localization sequences, and enzymatically active heparanase has been found in the chromatin compartment of the nucleus where it colocalizes with RNA polymerase II and positively controls the transcription of genes important for T cells' immune function. 79 Likewise, by entering the nucleus and degrading nuclear syndecan‐1, heparanase mediates HAT activation and transcription of genes associated with an aggressive tumor phenotype. 73 Conversely, nuclear heparanase binds nonspecifically to DNA and competes for binding with nuclear factor‐κB (NF‐κB), thus preventing transcription of NF‐κB target genes and acting as a tumor suppressor. 166 At the molecular level, nuclear heparanase appears, among other effects, to regulate histone 3 lysine 4 methylation by influencing the recruitment of demethylases to transcriptionally active genes. 79 Together it appears that nuclear heparanase promotes chromatin remodeling that opens its conformation allowing access to promotors of genes that affect cancer progression. 167 , 168 In‐depth research is still needed to better elucidate the mode of heparanase nuclear translocation and transcriptional activity. 73 , 79
HEPARANASE‐INHIBITING COMPOUNDS
To date, only four compounds have progressed to clinical trials. 10 , 41 , 91 , 92 , 93 , 129 These four “best‐in‐class” inhibitors are all polyanionic oligo‐/polysaccharides that mimic physicochemical properties of the natural heparanase inhibitor heparin, a glycosaminoglycan related to HS. The heparin‐like properties of these inhibitors, along with their structural heterogeneity, likely produce unwanted pleiotropic effects (e.g., anticoagulation, growth factor binding, poor pharmacokinetics) that complicate their clinical use. The active site of heparanase has proven challenging for small‐molecule pharmacological intervention, given its extensive interaction surface evolved to bind large HS polysaccharides. 169 Such challenging sites are often well targeted by mechanism‐based covalent inhibitors, which may still react with an enzyme despite weak or transient initial binding. Indeed, HS‐configured pseudodisaccharide, designed to bind selectively to the heparanase active site and react with its essential catalytic nucleophile, was recently synthesized. 169 This nanomolar, mechanism‐based, irreversible heparanase inhibitor markedly reduced cancer aggression in in cellulo and in vivo cancer models. 169 Recently, chemoenzymatic synthesis of sulfur‐linked sugar polymers was reported to yield potent competitive heparanase inhibitors. 170 Importantly, heparanase knockout (KO) mice exhibit no obvious immunological and other deficits, 53 implying that inhibition of heparanase will cause minimal side effects in cancer patients.
Dual and apparently antagonistic effects of antiheparanase therapy should be considered. 11 For example, it was reported that heparanase plays a critical role in natural killer (NK) cell invasion into tumors. 100 It was shown that cytokine and immune checkpoint blockade immunotherapy for metastases were compromised when NK cells lacked heparanase. 100 Likewise, it was found that in contrast to freshly isolated T lymphocytes, HPSE mRNA is downregulated in in vitro‐expanded T cells. This may explain the reduced ability of cultured CAR‐T cells to penetrate stroma‐rich solid tumors compared with lymphoid tissues. Indeed, engineering the CAR‐T cells to express HPSE resulted in their improved capacity to degrade the ECM, which promoted tumor T cell infiltration and antitumor activity. 171 It was suggested that the use of this strategy might enhance the activity of CAR‐T cells in individuals with stroma‐rich solid tumors. These results should be considered when systemically treating cancer patients with heparanase inhibitors since the potential adverse effect on NK and CAR‐T cells on cell infiltration might limit the antitumor activity of the inhibitors. 171 It appears that targeting the tumor microenvironment by heparanase inhibitors enhances the antitumor activity of approved therapies, further providing a strong rationale for applying antiheparanase therapy in combination with conventional anticancer drugs. 141 Remarkably, heparanase inhibitors were effective even when the xenografted tumor cells were devoid of heparanase, emphasizing the significance of heparanase contributed by cells residing in the tumor microenvironment. 88
HPA2—A NEW PLAYER IN THE HEPARANASE FIELD
HPSE2, the gene encoding Hpa2, was cloned soon after the cloning of heparanase, based on sequence homology. 101 Hpa2 gained attention when it was found that the HPSE2 gene is mutated in a human disease called urofacial syndrome (UFS). 102 , 103 UFS is a rare autosomal recessive congenital disease, featuring a combination of urological defects and an inverted facial expression attributed to peripheral neuropathy. 104 Notably, Hpa2 lacks intrinsic HS‐degrading activity, the hallmark of heparanase, 105 yet it retains the capacity to interact with HS. 105 Hpa2 exhibits an even higher affinity towards heparin and HS than heparanase, 105 thus competing for HS binding and thereby inhibiting heparanase enzymatic activity. 105 Exogenously added Hpa2 binds to HS and clusters syndecan‐1 and 4. Surprisingly, however, unlike heparanase, it fails to get internalized and remains on the cell surface 105 (Figure 2). Hpa2 regulates selected genes that promote normal differentiation, endoplasmic reticulum (ER) stress, fibrosis, and apoptosis, resulting in antitumor, antiangiogenic, and anti‐inflammatory effects. 2 , 10 , 106 , 116 , 129 Interestingly, stress conditions induce the expression of Hpa2, thus establishing a feedback loop by which Hpa2 enhances ER stress which, in turn, induces Hpa2 expression 107 , 108 , 109 (Figure 2). Unlike the intense research effort devoted to exploring the significance of heparanase in cancer progression, very little attention was given to Hpa2. Recent evidence indicates, nonetheless, that Hpa2 expressed by both the tumor cells and the host tumor microenvironment functions as a tumor suppressor. Clinically, it was reported that, unlike heparanase, Hpa2 expression is readily detected in normal epithelium of the bladder, breast, cervical, gastric, and ovarian tissues, whereas its expression is substantially decreased in the resulting carcinomas, 10 , 108 , 109 , 110 , 111 , 112 expression pattern that is typical of a tumor suppressor. Furthermore, patients that retain high levels of Hpa2 survived longer than patients bearing Hpa2‐low tumors. 105 , 107 , 108 , 109 , 113 Experimentally, overexpression of Hpa2 attenuated the growth of tumor xenografts, whereas Hpa2 gene silencing resulted in bigger tumors. 106 , 107 , 108 , 109 , 111 , 114 , 115 This is best demonstrated by gene editing of Hpa2 in pharyngeal FaDu cells applying the CRISPR technology. Notably, Hpa2‐null cells produced bigger tumors vs control cells, whereas the rescue of Hpa2 in the null cells resulted in smaller tumors. 114 Collectively, these results support the notion that Hpa2 functions as a tumor suppressor (Figure 2 and Table 1). Heparanase and Hpa2 not only exhibit opposite functions in terms of tumor growth but also in terms of the underlying mechanism. For example, while heparanase induces VEGF‐A and VEGF‐C 142 expression and promotes angiogenesis, Hpa2 attenuates the expression of VEGF‐A and VEGF‐C and decreases tumor vascularity 106 ; whereas heparanase attenuates cell differentiation and promotes epithelial‐to‐mesenchymal transition (EMT), 172 , 173 Hpa2 increases cell differentiation. 106 , 111 This mirrored functionality suggests that Hpa2 exerts these properties in part by modulating heparanase, as demonstrated by a significant decrease in heparanase activity in sarcoma cells overexpressing Hpa2. 108
Figure 2.
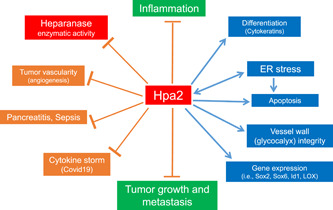
Protective effects of Hpa2 against cancer, inflammation, and tissue damage. Hpa2 functions in heparanase‐dependent and independent manners, and affects gene expression (i.e., Sox2, Id1, Lox), tumor vascularity, ER stress, and cell apoptosis, resulting in cancer suppression. 2 , 106 , 107 , 114 , 129 Hpa2 levels are elevated in response to stress conditions, generating a vicious cycle that contributes to attenuation of tumor progression. Hpa2 exerts a protective effect in models of sepsis, endotoxemia, diabetes‐induced renal inflammation, pancreatitis, and in patients with Covid‐19, attributed, in part, to inhibition of heparanase‐mediated damage to the integrity of the glycocalyx. 116 , 117 , 118 Covid‐19, coronavirus disease 2019; ER, endoplasmic reticulum; Hpa2, heparanase‐2.
CONCLUSIONS AND PERSPECTIVES
Among other aspects, heparanase research reinforced the significance of the ECM in the control of cell proliferation and differentiation. 174 , 175 It led to important and often unexpected observations in diverse normal and pathological processes including, wound healing, 176 angiogenesis, 31 autophagy, 99 signal transduction, 72 , 97 , 128 , 153 protein trafficking, 177 lysosomal secretion, 129 , 178 blood coagulation, 67 , 151 EMT, 173 , 179 glycocalyx remodeling, 116 , 180 activation of immune cells, 24 , 59 , 89 , 90 exosome formation, 82 , 83 , 84 drug resistance, 13 , 181 gene transcription, 73 , 74 and other key biological features. 11 , 12 , 97 While most studies emphasize the involvement of heparanase in cancer and inflammation, other pathologies were investigated. Among these are diabetes, 74 , 75 diabetic complications (i.e., diabetic nephropathy, diabetic cardiomyopathy), 76 , 77 kidney dysfunction, 182 fibrosis, 78 , 183 inflammatory disorders (i.e., neuroinflammation, pancreatitis, ulcerative colitis, arthritis, sepsis), 59 , 60 , 184 , 185 amyloidosis, 61 atherosclerosis, 66 , 68 , 69 , 70 viral diseases, 96 , 97 and other pathologies. 11 , 12 , 97 Heparanase accomplishes all these by exerting both enzymatic and nonenzymatic functions that are mostly HS‐dependent yet in some cases are HS‐independent. 10 , 161 The enzyme is expressed by tumor cells and cells of the tumor microenvironment and functions extracellularly to remodel the ECM and regulate the bioavailability of HS‐bound factors, as well as intracellularly (i.e., lysosome, nucleus) 10 (Figure 1). Unraveling these and other aspects of heparanase biology (e.g., mode of heparanase nuclear translocation and transcriptional activity, activation and polarization of macrophages, 59 , 73 , 79 , 89 , 90 , 186 is ongoing and is critical to our understanding of its multiple functions in health and disease.
Unfortunately, antiheparanase‐based therapy has not yet been implemented in the clinic. Notably, all antiheparanase compounds that were or are being examined in clinical trials are heparin/HS‐like saccharides. 10 , 169 While these compounds were very successful in numerous mouse models, 18 , 141 , 163 unforeseen adverse effects are documented and ascribed primarily to the poor pharmacokinetics, heterogeneous nature, nonspecific, and pleiotropic effects of those HS mimetics. Development of more specific oligosaccharides, small molecules and neutralizing monoclonal antibodies is ongoing. 88 , 169 , 170 , 187 Of the four compounds examined in clinical trials, PG545 (Pixatimod) appears the most potent and promising, likely due to its lipophilic moiety and superior pharmacokinetic properties. 92 The specificity of this compound is, nonetheless, questionable, because it exerts also heparanase‐independent functions and attenuates the growth of tumor xenografts produced by heparanase‐negative lymphoma cells. 188 Second‐generation heparanase inhibitors, possibly in the form of disaccharides that covalently bind to the enzyme active site, 169 should target also its intracellular activities and hence better neutralize all aspects of heparanase function. Heparanase is a multifaceted protein having both enzymatic and nonenzymatic activities. To the best of our knowledge, all heparanase inhibitors under development are predominately targeting the enzymatic activity of heparanase. Therefore, a main question raised in the development of antiheparanase inhibitors is whether the enzymatic activity of heparanase is the critical determinant of its protumorigenic and prometastasis effects, given the fact that species of heparanase (i.e., C‐terminus domain, active site double‐mutant, T5 splice variant) lacking enzymatic activity still promotes tumor progression. 10 , 72 , 189 , 190
Based on the housekeeping nature of its gene promoter, we suggest that heparanase is expressed at low levels by all cells, modulating autophagy and possibly other functions of the lysosome. According to this notion, heparanase function in the lysosome may not be less important than its function extracellularly. This may turn relevant in platelets, neutrophils, lymphocytes, and macrophages that show relatively high levels of heparanase expression and activity. 11 , 12 , 33 , 90 , 191 Beyond serving as a cellular recycling center, recent evidence suggests that the lysosome is involved in homeostasis, generating building blocks for cell growth, mitogenic signaling, angiogenesis, metastasis, and activation of transcriptional programs, 192 , 193 repertoire that closely resembles those of heparanase. The PI3‐kinase/Akt/mammalian target of rapamycin (mTOR) is highly implicated in the regulation of cell metabolism, protein homeostasis, and cell growth due, in part, to the localization of mTOR at the lysosome membrane which is required for its activation. 194 , 195 Indeed, Akt is the most common kinase activated by heparanase, 72 , 80 , 140 , 153 , 161 , 163 and its instrumental role in the regulation of mTOR would likely convey to the lysosome. 194 , 195 Research is needed to further resolve the significance of heparanase in modulating lysosomal function in normal and diseased cells.
Unlike heparanase, Hpa2 does not undergo proteolytic processing and hence lacks intrinsic HS‐degrading activity, the hallmark of heparanase. Our working hypothesis is that Hpa2 functions as a natural inhibitor of heparanase, playing a protective role in maintaining tissue hemostasis and normal function and regulating to a large extent the balance between disease progression and suppression (Figure 2). Hpa2 functions in heparanase activity‐ and HS‐dependent and independent manners, and regulates the expression of selected genes that affect tumor vascularity, tumor fibrosis, cell differentiation and apoptosis, resulting in cancer suppression. 10 , 129 A protective effect of Hpa2 was also demonstrated in models of sepsis, endotoxemia, and diabetes‐induced renal inflammation, 116 , 117 as well as in patients with Covid‐19 118 (Figure 2), encouraging the development of Hpa2‐based therapies. This was attributed, in part, to inhibition of Hpa1‐mediated endothelial injury and damage to the glycocalyx integrity, emphasizing the physiological significance of maintaining a proper Hpa1/Hpa2 ratio. 116 Applying the CRISPR/Cas9 technology we have generated constitutive Hpa2‐KO mice. Unexpectedly, we found that Hpa2‐KO homozygosity is embryonic lethal, indicating an essential involvement of Hpa2 in embryonic development (unpublished). We have recently generated conditional Hpa2‐KO mice and found that these mice are highly susceptible to aggressive cancer and inflammation (unpublished), encouraging further research on the protective function of host‐derived Hpa2.
AUTHOR CONTRIBUTIONS
Israel Vlodavsky: Conceptualization; writing—review & editing; supervision; funding acquisition. Yasmin Kayal: Investigation. Maram Hilwi: Investigation. Soaad Soboh: Investigation. Ralph D. Sanderson: Writing—review & editing; conceptualization; funding acquisition. Neta Ilan: Writing—review & editing; supervision; conceptualization.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest
ETHICS STATEMENT
Not applicable.
ACKNOWLEDGMENTS
These studies were generously supported by research grants awarded by the National Institutes of Health (R01CA211752; Ralph D. Sanderson and Israel Vlodavsky); the Israel Science Foundation (ISF‐1021/19); the DKFZ‐MOST cancer research program; the US‐Israel binational Science Foundation (BSF‐2021059; Israel Vlodavsky and Ralph D. Sanderson); The Israel Cancer Association (ICA), and the Technion Integrated Cancer Center (TICC) Rubinstein scholarship (to Yasmin Kayal).
Vlodavsky I., Kayal Y., Hilwi M., Soboh S., Sanderson R. D., Ilan N., Proteoglycan Res 2023, 1, e6. 10.1002/pgr2.6
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- 1. Hulett M. D., Hornby J. R., Ohms S. J., Zuegg J., Freeman C., Gready J. E., Parish C. R., Biochemistry 2000, 39(51), 15659. [DOI] [PubMed] [Google Scholar]
- 2. Karamanos N. K., Piperigkou Z., Passi A., Götte M., Rousselle P., Vlodavsky I., Trends Mol. Med. 2021, 27(10), 1000. [DOI] [PubMed] [Google Scholar]
- 3. Wu L., Viola C. M., Brzozowski A. M., Davies G. J., Nat. Struct. Mol. Biol. 2015, 22(12), 1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ogren S., Lindahl U., J. Biol. Chem. 1975, 250(7), 2690. [PubMed] [Google Scholar]
- 5. Ögren S., Lindahl U., Biochem. J. 1971, 125(4), 1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Oosta G. M., Favreau L. V., Beeler D. L., Rosenberg R. D., J. Biol. Chem. 1982, 257(19), 11249. [PubMed] [Google Scholar]
- 7. Freeman C., Parish R. C., Biochem. J. 1998, 330(Pt 3), 1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nakajima M., Irimura T., DiFerrante D., DiFerrante N., Nicolson G. L., Science 1983, 220, 611. [DOI] [PubMed] [Google Scholar]
- 9. Vlodavsky I., Fuks Z., Bar‐Ner M., Ariav Y., Schirrmacher V., Cancer Res. 1983, 43(6), 2704. [PubMed] [Google Scholar]
- 10. Ilan N., Bhattacharya U., Barash U., Boyango I., Yanku Y., Gross‐Cohen M., Vlodavsky I., Adv. Exp. Med. Biol. 2020, 1221, 253. [DOI] [PubMed] [Google Scholar]
- 11. Jayatilleke K. M., Hulett M. D., J. Transl. Med. 2020, 18(1), 453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mayfosh A. J., Nguyen T. K., Hulett M. D., Int. J. Mol. Sci. 2021, 22(20), 11096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vlodavsky I., Singh P., Boyango I., Gutter‐Kapon L., Elkin M., Sanderson R. D., Ilan N., Drug Resistance Updates 2016, 29, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vlodavsky I., Beckhove P., Lerner I., Pisano C., Meirovitz A., Ilan N., Elkin M., Cancer Microenviron. 2012, 5(2), 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Takaoka M., Naomoto Y., Ohkawa T., Uetsuka H., Shirakawa Y., Uno F., Fujiwara T., Gunduz M., Nagatsuka H., Nakajima M., Tanaka N., Haisa M., Lab. Invest. 2003, 83(5), 613. [DOI] [PubMed] [Google Scholar]
- 16. Rohloff J., Zinke J., Schoppmeyer K., Tannapfel A., Witzigmann H., Mössner J., Wittekind C., Caca K., Br. J. Cancer 2002, 86(8), 1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sato T., Yamaguchi A., Goi T., Hirono Y., Takeuchi K., Katayama K., Matsukawa S., J. Surg. Oncol. 2004, 87(4), 174. [DOI] [PubMed] [Google Scholar]
- 18. Barash U., Lapidot M., Zohar Y., Loomis C., Moreira A., Feld S., Goparaju C., Yang H., Hammond E., Zhang G., Li J. P., Ilan N., Nagler A., Pass H. I., Vlodavsky I., JNCI, J. Natl. Cancer Inst. 2018, 110(10), 1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hu B., Wang Q., Shi Y., Lu S., Qu H., Wang L., Cui J., Oncol. Lett. 2017, 13(5), 3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kundu S., Xiong A., Spyrou A., Wicher G., Marinescu V. D., Edqvist P. D., Zhang L., Essand M., Dimberg A., Smits A., Ilan N., Vlodavsky I., Li J. P., Forsberg‐Nilsson K., Mol. Cancer Res. 2016, 14(12), 1243. [DOI] [PubMed] [Google Scholar]
- 21. Sun X., Zhang G., Nian J., Yu M., Chen S., Zhang Y., Yang G., Yang L., Cheng P., Yan C., Ma Y., Meng H., Wang X., Li J. P., Oncotarget 2017, 8(8), 43521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vornicova O., Naroditsky I., Boyango I., Shachar S. S., Mashiach T., Ilan N., Vlodavsky I., Bar‐Sela G., Oncotarget 2018, 9(5), 6238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wu X., Yu J., Gao G., Wang X., Liu Y., Zhu S., Gong Z., PLoS One 2015, 10(11), e0143009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Naparstek Y., Cohen I. R., Fuks Z., Vlodavsky I., Nature 1984, 310(5974), 241. [DOI] [PubMed] [Google Scholar]
- 25. Bar‐Ner M., Eldor A., Wasserman L., Matzner Y., Cohen I., Fuks Z., Vlodavsky I., Blood 1987, 70(2), 551. [PubMed] [Google Scholar]
- 26. Parish C. R., Coombe D. R., Jakobsen K. B., Bennett F. A., Underwood P. A., Int. J. Cancer 1987, 40(4), 511. [DOI] [PubMed] [Google Scholar]
- 27. Willenborg D. O., Parish C. R., J. Immunol. 1988, 140(10), 3401. [PubMed] [Google Scholar]
- 28. Lider O., Baharav E., Mekori Y. A., Miller T., Naparstek Y., Vlodavsky I., Cohen I. R., J. Clin. Invest. 1989, 83(3), 752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Folkman J., Klagsbrun M., Sasse J., Wadzinski M., Ingber D., Vlodavsky I., Am. J. Pathol. 1988, 130(2), 393. [PMC free article] [PubMed] [Google Scholar]
- 30. Vlodavsky I., Folkman J., Sullivan R., Fridman R., Ishai‐Michaeli R., Sasse J., Klagsbrun M., Proc. Natl. Acad. Sci. U. S. A. 1987, 84(8), 2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Elkin M., Ilan N., Ishai‐Michaeli R., Friedmann Y., Papo O., Pecker I., Vlodavsky I., FASEB J. 2001, 15(9), 1661. [DOI] [PubMed] [Google Scholar]
- 32. Hulett M. D., Freeman C., Hamdorf B. J., Baker R. T., Harris M. J., Parish C. R., Nat. Med. 1999, 5(7), 803. [DOI] [PubMed] [Google Scholar]
- 33. Kussie P. H., Hulmes J. D., Ludwig D. L., Patel S., Navarro E. C., Seddon A. P., Giorgio N. A., Bohlen P., Biochem. Biophys. Res. Commun. 1999, 261(1), 183. [DOI] [PubMed] [Google Scholar]
- 34. Toyoshima M., Nakajima M., J. Biol. Chem. 1999, 274(34), 24153. [DOI] [PubMed] [Google Scholar]
- 35. Vlodavsky I., Friedmann Y., Elkin M., Aingorn H., Atzmon R., Ishai‐Michaeli R., Bitan M., Pappo O., Peretz T., Michal I., Spector L., Pecker I., Nat. Med. 1999, 5(7), 793. [DOI] [PubMed] [Google Scholar]
- 36. Miao H. Q., Navarro E., Patel S., Sargent D., Koo H., Wan H., Plata A., Zhou Q., Ludwig D., Bohlen P., Kussie P., Protein Expression Purif. 2002, 26(3), 425. [DOI] [PubMed] [Google Scholar]
- 37. Nardella C., Lahm A., Pallaoro M., Brunetti M., Vannini A., Steinkühler C., Biochemistry 2004, 43(7), 1862. [DOI] [PubMed] [Google Scholar]
- 38. Fairbanks M. B., Mildner A. M., Leone J. W., Cavey G. S., Mathews W. R., Drong R. F., Slightom J. L., Bienkowski M. J., Smith C. W., Bannow C. A., Heinrikson R. L., J. Biol. Chem. 1999, 274(42), 29587. [DOI] [PubMed] [Google Scholar]
- 39. McKenzie E., Young K., Hircock M., Bennett J., Bhaman M., Felix R., Turner P., Stamps A., McMillan D., Saville G., Ng S., Mason S., Snell D., Schofield D., Gong H., Townsend R., Gallagher J., Page M., Parekh R., Stubberfield C., Biochem. J. 2003, 373(Pt 2), 423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Levy‐Adam F., Miao H. Q., Heinrikson R. L., Vlodavsky I., Ilan N., Biochem. Biophys. Res. Commun. 2003, 308(4), 885. [DOI] [PubMed] [Google Scholar]
- 41. Liu C. J., World J. Gastroenterol. 2014, 20(32), 11384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dredge K., Hammond E., Handley P., Gonda T. J., Smith M. T., Vincent C., Brandt R., Ferro V., Bytheway I., Br. J. Cancer 2011, 104(4), 635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ritchie J. P., Ramani V. C., Ren Y., Naggi A., Torri G., Casu B., Penco S., Pisano C., Carminati P., Tortoreto M., Zunino F., Vlodavsky I., Sanderson R. D., Yang Y., Clin. Cancer Res. 2011, 17(6), 1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Parish C. R., Freeman C., Brown K. J., Francis D. J., Cowden W. B., Cancer Res. 1999, 59(14), 3433. [PubMed] [Google Scholar]
- 45. Arvatz G., Barash U., Nativ O., Ilan N., Vlodavsky I., FASEB J. 2011, 24(12), 4969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jiang P., Kumar A., Parrillo J. E., Dempsey L. A., Platt J. L., Prinz R. A., Xu X., J. Biol. Chem. 2002, 277(11), 8989. [DOI] [PubMed] [Google Scholar]
- 47. Shteper P. J., Zcharia E., Ashhab Y., Peretz T., Vlodavsky I., Ben‐Yehuda D., Oncogene 2003, 22(49), 7737. [DOI] [PubMed] [Google Scholar]
- 48. Ogishima T., Shiina H., Breault J. E., Terashima M., Honda S., Enokida H., Urakami S., Tokizane T., Kawakami T., Ribeiro‐Filho L. A., Fujime M., Kane C. J., Carroll P. R., Igawa M., Dahiya R., Oncogene 2005, 24(45), 6765. [DOI] [PubMed] [Google Scholar]
- 49. de Mestre A. M., Rao S., Hornby J. R., Soe‐Htwe T., Khachigian L. M., Hulett M. D., J. Biol. Chem. 2005, 280(42), 35136. [DOI] [PubMed] [Google Scholar]
- 50. Baraz L., Haupt Y., Elkin M., Peretz T., Vlodavsky I., Oncogene 2006, 25(28), 3939. [DOI] [PubMed] [Google Scholar]
- 51. Uno F., Fujiwara T., Takata Y., Ohtani S., Katsuda K., Takaoka M., Ohkawa T., Naomoto Y., Nakajima M., Tanaka N., Cancer Res. 2001, 61(21), 7855. [PubMed] [Google Scholar]
- 52. Edovitsky E., Elkin M., Zcharia E., Peretz T., Vlodavsky I., J. Natl. Cancer Inst. 2004, 96, 1219. [DOI] [PubMed] [Google Scholar]
- 53. Zcharia E., Jia J., Zhang X., Baraz L., Lindahl U., Peretz T., Vlodavsky I., Li J. P., PLoS One 2009, 4(4), e5181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zcharia E., Metzger S., Chajek‐Shaul T., Aingorn H., Elkin M., Friedmann Y., Weinstein T., Li J. P., Lindahl U., Vlodavsky I., FASEB J. 2004, 18(2), 252. [DOI] [PubMed] [Google Scholar]
- 55. Escobar Galvis M. L., Jia J., Zhang X., Jastrebova N., Spillmann D., Gottfridsson E., van Kuppevelt T. H., Zcharia E., Vlodavsky I., Lindahl U., Li J. P., Nat. Chem. Biol. 2007, 3(12), 773. [DOI] [PubMed] [Google Scholar]
- 56. Gingis‐Velitski S., Zetser A., Kaplan V., Ben‐Zaken O., Cohen E., Levy‐Adam F., Bashenko Y., Flugelman M. Y., Vlodavsky I., Ilan N., J. Biol. Chem. 2004, 279(42), 44084. [DOI] [PubMed] [Google Scholar]
- 57. Abboud‐Jarrous G., Atzmon R., Peretz T., Palermo C., Gadea B. B., Joyce J. A., Vlodavsky I., J. Biol. Chem. 2008, 283(26), 18167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Abboud‐Jarrous G., Rangini‐Guetta Z., Aingorn H., Atzmon R., Elgavish S., Peretz T., Vlodavsky I., J. Biol. Chem. 2005, 280(14), 13568. [DOI] [PubMed] [Google Scholar]
- 59. Lerner I., Hermano E., Zcharia E., Rodkin D., Bulvik R., Doviner V., Rubinstein A. M., Ishai‐Michaeli R., Atzmon R., Sherman Y., Meirovitz A., Peretz T., Vlodavsky I., Elkin M., J. Clin. Invest. 2011, 121(5), 1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Schmidt E. P., Yang Y., Janssen W. J., Gandjeva A., Perez M. J., Barthel L., Zemans R. L., Bowman J. C., Koyanagi D. E., Yunt Z. X., Smith L. P., Cheng S. S., Overdier K. H., Thompson K. R., Geraci M. W., Douglas I. S., Pearse D. B., Tuder R. M., Nat. Med. 2012, 18, 1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Li J. P., Galvis M. L. E., Gong F., Zhang X., Zcharia E., Metzger S., Vlodavsky I., Kisilevsky R., Lindahl U., Proc. Natl. Acad. Sci. U. S. A. 2005, 102(18), 6473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Masola V., Gambaro G., Onisto M., Adv. Exp. Med. Biol. 2020, 1221, 669. [DOI] [PubMed] [Google Scholar]
- 63. Khamaysi I., Hamo‐Giladi D. B., Abassi Z., Adv. Exp. Med. Biol. 2020, 1221, 703. [DOI] [PubMed] [Google Scholar]
- 64. Levy‐Adam F., Abboud‐Jarrous G., Guerrini M., Beccati D., Vlodavsky I., Ilan N., J. Biol. Chem. 2005, 280(21), 20457. [DOI] [PubMed] [Google Scholar]
- 65. Zetser A., Bashenko Y., Edovitsky E., Levy‐Adam F., Vlodavsky I., Ilan N., Cancer Res. 2006, 66(3), 1455. [DOI] [PubMed] [Google Scholar]
- 66. Blich M., Golan A., Arvatz G., Sebbag A., Shafat I., Sabo E., Cohen‐Kaplan V., Petcherski S., Avniel‐Polak S., Eitan A., Hammerman H., Aronson D., Axelman E., Ilan N., Nussbaum G., Vlodavsky I., Arterioscler., Thromb., Vasc. Biol. 2013, 33, e56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Nadir Y., Brenner B., Gingis‐Velitski S., Levy‐Adam F., Ilan N., Zcharia E., Nadir E., Vlodavsky I., Thromb. Haemostasis 2008, 99(1), 133. [PubMed] [Google Scholar]
- 68. Osterholm C., Folkersen L., Lengquist M., Ponten F., Renne T., Li J., Hedin U., Atherosclerosis 2012, 226(1), 67. [DOI] [PubMed] [Google Scholar]
- 69. Vlodavsky I., Blich M., Li J. P., Sanderson R. D., Ilan N., Matrix Biol. 2013, 32(5), 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Nguyen T. K., Paone S., Chan E., Poon I. K. H., Baxter A. A., Thomas S. R., Hulett M. D., Cells 2022, 11(20), 3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Nadir Y., Adv. Exp. Med. Biol. 2020, 1221, 771. [DOI] [PubMed] [Google Scholar]
- 72. Fux L., Feibish N., Cohen‐Kaplan V., Gingis‐Velitski S., Feld S., Geffen C., Vlodavsky I., Ilan N., Cancer Res. 2009, 69(5), 1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Purushothaman A., Hurst D. R., Pisano C., Mizumoto S., Sugahara K., Sanderson R. D., J. Biol. Chem. 2011, 286(35), 30377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Parish C. R., Freeman C., Ziolkowski A. F., He Y. Q., Sutcliffe E. L., Zafar A., Rao S., Simeonovic C. J., Matrix Biol. 2013, 32(5), 228. [DOI] [PubMed] [Google Scholar]
- 75. Ziolkowski A. F., Popp S. K., Freeman C., Parish C. R., Simeonovic C. J., J. Clin. Invest. 2012, 122(1), 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Gil N., Goldberg R., Neuman T., Garsen M., Zcharia E., Rubinstein A. M., van Kuppevelt T., Meirovitz A., Pisano C., Li J. P., van der Vlag J., Vlodavsky I., Elkin M., Diabetes 2012, 61(1), 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wan A., Rodrigues B., Cardiovasc. Res. 2016, 111(3), 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Masola V., Zaza G., Onisto M., Lupo A., Gambaro G., J. Transl. Med. 2015, 13, 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. He Y. Q., Sutcliffe E. L., Bunting K. L., Li J., Goodall K. J., Poon I. K. A., Hulett M. D., Freeman C., Zafar A., McInnes R. L., Taya T., Parish C. R., Rao S., Transcription 2012, 3(3), 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Cohen‐Kaplan V., Doweck I., Naroditsky I., Vlodavsky I., Ilan N., Cancer Res. 2008, 68(24), 10077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Cohen‐Kaplan V., Jrbashyan J., Yanir Y., Naroditsky I., Ben‐Izhak O., Ilan N., Doweck I., Vlodavsky I., J. Biol. Chem. 2012, 287(9), 6668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Sanderson R. D., Bandari S. K., Vlodavsky I., Matrix Biol. 2019, 75‐76, 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Roucourt B., Meeussen S., Bao J., Zimmermann P., David G., Cell Res. 2015, 25(4), 412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Thompson C. A., Purushothaman A., Ramani V. C., Vlodavsky I., Sanderson R. D., J. Biol. Chem. 2013, 288(14), 10093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. David G., Zimmermann P., Mol. Cell. Oncol. 2016, 3(3), e1047556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Bandari S. K., Purushothaman A., Ramani V. C., Brinkley G. J., Chandrashekar D. S., Varambally S., Mobley J. A., Zhang Y., Brown E. E., Vlodavsky I., Sanderson R. D., Matrix Biol. 2017, 65, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Wu L., Jiang J., Jin Y., Kallemeijn W. W., Kuo C. L., Artola M., Dai W., van Elk C., van Eijk M., van der Marel G. A., Codée J. D. C., Florea B. I., Aerts J. M. F. G., Overkleeft H. S., Davies G. J., Nat. Chem. Biol. 2017, 13(8), 867. [DOI] [PubMed] [Google Scholar]
- 88. Weissmann M., Arvatz G., Horowitz N., Feld S., Naroditsky I., Zhang Y., Ng M., Hammond E., Nevo E., Vlodavsky I., Ilan N., Proc. Natl. Acad. Sci. U. S. A. 2016, 113(3), 704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Goodall K. J., Poon I. K. H., Phipps S., Hulett M. D., PLoS One 2014, 9(10), e109596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Gutter‐Kapon L., Alishekevitz D., Shaked Y., Li J. P., Aronheim A., Ilan N., Vlodavsky I., Proc. Natl. Acad. Sci. U. S. A. 2016, 113(48), E7808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Galli M., Chatterjee M., Grasso M., Specchia G., Magen H., Einsele H., Celeghini I., Barbieri P., Paoletti D., Pace S., Sanderson R. D., Rambaldi A., Nagler A., Haematologica 2018, 103(10), e469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Lemech C., Dredge K., Bampton D., Hammond E., Clouston A., Waterhouse N. J., Stanley A. C., Leveque‐El Mouttie L., Chojnowski G. M., Haydon A., Pavlakis N., Burge M., Brown M. P., Goldstein D., J. Immunother. Cancer 2023, 11(1), e006136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Noseda A., Barbieri P., Adv. Exp. Med. Biol. 2020, 1221, 523. [DOI] [PubMed] [Google Scholar]
- 94. Dredge K., Brennan T. V., Hammond E., Lickliter J. D., Lin L., Bampton D., Handley P., Lankesheer F., Morrish G., Yang Y., Brown M. P., Millwar M., Br. J. Cancer 2018, 118(8), 1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Shukla D., Liu J., Blaiklock P., Shworak N. W., Bai X., Esko J. D., Cohen G. H., Eisenberg R. J., Rosenberg R. D., Spear P. G., Cell 1999, 99(1), 13. [DOI] [PubMed] [Google Scholar]
- 96. Agelidis A., Suryawanshi R. K., Patil C. D., Campeau A., Gonzalez D. J., Shukla D., iScience 2021, 24(3), 102242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Koganti R., Suryawanshi R., Shukla D., Cell. Mol. Life Sci. 2020, 77(24), 5059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Hadigal S. R., Agelidis A. M., Karasneh G. A., Antoine T. E., Yakoub A. M., Ramani V. C., Djalilian A. R., Sanderson R. D., Shukla D., Nat. Commun. 2015, 6, 6985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Shteingauz A., Boyango I., Naroditsky I., Hammond E., Gruber M., Doweck I., Ilan N., Vlodavsky I., Cancer Res. 2015, 75(18), 3946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Putz E. M., Mayfosh A. J., Kos K., Barkauskas D. S., Nakamura K., Town L., Goodall K. J., Yee D. Y., Poon I. K. H., Baschuk N., Souza‐Fonseca‐Guimaraes F., Hulett M. D., Smyth M. J., J. Clin. Invest. 2017, 127(7), 2777. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 101. McKenzie E., Tyson K., Stamps A., Smith P., Turner P., Barry R., Hircock M., Patel S., Barry E., Stubberfield C., Terrett J., Page M., Biochem. Biophys. Res. Commun. 2000, 276(3), 1170. [DOI] [PubMed] [Google Scholar]
- 102. Daly S. B., Urquhart J. E., Hilton E., McKenzie E. A., Kammerer R. A., Lewis M., Kerr B., Stuart H., Donnai D., Long D. A., Burgu B., Aydogdu O., Derbent M., Garcia‐Minaur S., Reardon W., Gener B., Shalev S., Smith R., Woolf A. S., Black G. C., Newman W. G., Am. J. Hum. Genet. 2010, 86(6), 963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Pang J., Zhang S., Yang P., Hawkins‐Lee B., Zhong J., Zhang Y., Ochoa B., Agundez J. A. G., Voelckel M. A., Gu W., Xiong W. C., Mei L., She J. X., Wang C. Y., Am. J. Hum. Genet. 2010, 86(6), 957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Guo C., Kaneko S., Sun Y., Huang Y., Vlodavsky I., Li X., Li Z. R., Li X., Hum. Mol. Genet. 2015, 24(7), 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Levy‐Adam F., Feld S., Cohen‐Kaplan V., Shteingauz A., Gross M., Arvatz G., Naroditsky I., Ilan N., Doweck I., Vlodavsky I., J. Biol. Chem. 2010, 285(36), 28010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Gross‐Cohen M., Feld S., Doweck I., Neufeld G., Hasson P., Arvatz G., Barash U., Naroditsky I., Ilan N., Vlodavsky I., Cancer Res. 2016, 76(9), 2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Kayal Y., Singh P., Naroditsky I., Ilan N., Vlodavsky I., Matrix Biol. 2021, 98, 21. [DOI] [PubMed] [Google Scholar]
- 108. Knani I., Singh P., Gross‐Cohen M., Aviram S., Ilan N., Sanderson R. D., Aronheim A., Vlodavsky I., Matrix Biol. 2022, 105, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Liu J., Knani I., Gross‐Cohen M., Hu J., Wang S., Tang L., Ilan N., Yang S., Vlodavsky I., Neoplasia 2021, 23(9), 966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Fu J., Khaybullin R., Zhang Y., Xia A., Qi X., BMC Cancer 2015, 15, 473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Gross‐Cohen M., Feld S., Naroditsky I., Nativ O., Ilan N., Vlodavsky I., Oncotarget 2016, 7(16), 22556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Wu B., Liu G., Jin Y., Yang T., Zhang D., Ding L., Zhou F., Pan Y., Wei Y., Front. Oncol. 2020, 10, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Zhang X., Xu S., Tan Q., Liu L., Cancer Epidemiol. 2013, 37(6), 1010. [DOI] [PubMed] [Google Scholar]
- 114. Gross‐Cohen M., Yanku Y., Kessler O., Barash U., Boyango I., Cid‐Arregui A., Neufeld G., Ilan N., Vlodavsky I., Matrix Biol. 2021, 99, 58. [DOI] [PubMed] [Google Scholar]
- 115. Knani I., Yanku Y., Gross‐Cohen M., Ilan N., Vlodavsky I., Matrix Biol. 2022, 113, 22. [DOI] [PubMed] [Google Scholar]
- 116. Pape T., Hunkemöller A. M., Kümpers P., Haller H., David S., Stahl K., Matrix Biol. Plus 2021, 12, 100095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Kiyan Y., Tkachuk S., Kurselis K., Shushakova N., Stahl K., Dawodu D., Kiyan R., Chichkov B., Haller H., Sci. Rep. 2019, 9(1), 13591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Stahl K., Gronski P. A., Kiyan Y., Seeliger B., Bertram A., Pape T., Welte T., Hoeper M. M., Haller H., David S., Am. J. Respir. Crit. Care Med. 2020, 202(8), 1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Sarrazin S., Lamanna W. C., Esko J. D., Cold Spring Harbor Perspect. Biol. 2011, 3(7), a004952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Poulain F. E., Yost H. J., Development 2015, 142(20), 3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Chronopoulos A., Thorpe S. D., Cortes E., Lachowski D., Rice A. J., Mykuliak V. V., Róg T., Lee D. A., Hytönen V. P., del Río Hernández A. E., Nat. Mater. 2020, 19(6), 669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Bode L., Salvestrini C., Park P. W., Li J. P., Esko J. D., Yamaguchi Y., Murch S., Freeze H. H., J. Clin. Invest. 2008, 118(1), 229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Christianson H. C., Belting M., Matrix Biol. 2014, 35, 51. [DOI] [PubMed] [Google Scholar]
- 124. Clausen T. M., Sandoval D. R., Spliid C. B., Pihl J., Perrett H. R., Painter C. D., Narayanan A., Majowicz S. A., Kwong E. M., McVicar R. N., Thacker B. E., Glass C. A., Yang Z., Torres J. L., Golden G. J., Bartels P. L., Porell R. N., Garretson A. F., Laubach L., Feldman J., Yin X., Pu Y., Hauser B. M., Caradonna T. M., Kellman B. P., Martino C., Gordts P. L. S. M., Chanda S. K., Schmidt A. G., Godula K., Leibel S. L., Jose J., Corbett K. D., Ward A. B., Carlin A. F., Esko J. D., Cell 2020, 183(4), 1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Billings P. C., Pacifici M., Connect. Tissue Res. 2015, 56(4), 272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Lindahl U., Couchman J., Kimata K., Esko J. D., in Essentials of Glycobiology (Eds: Varki A., Cummings R. D., Esko J. D., Stanley P., Hart G. W., Aebi M., Darvill A. G., Kinoshita T., Packer N. H., Prestegard J. H., Schnaar R. L., Seeberger P. H.), Cold Spring Harbor, New York: 2015, p. 207. [PubMed] [Google Scholar]
- 127. Barash U., Cohen‐Kaplan V., Dowek I., Sanderson R. D., Ilan N., Vlodavsky I., FEBS J. 2010, 277(19), 3890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Ilan N., Elkin M., Vlodavsky I., Int. J. Biochem. Cell. Biol. 2006, 38(12), 2018. [DOI] [PubMed] [Google Scholar]
- 129. Vlodavsky I., Gross‐Cohen M., Weissmann M., Ilan N., Sanderson R. D., Trends Biochem. Sci. 2018, 43(1), 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Vreys V., David G., J. Cell. Mol. Med. 2007, 11(3), 427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Höök M., Wasteson Å., Oldberg Å., Biochem. Biophys. Res. Commun. 1975, 67(4), 1422. [DOI] [PubMed] [Google Scholar]
- 132. Nakajima M., Irimura T., Di Ferrante N., Nicolson G. L., J. Biol. Chem. 1984, 259(4), 2283. [PubMed] [Google Scholar]
- 133. Bar‐Ner M., Kramer M. D., Schirrmacher V., Ishai‐Michaeli R., Fuks Z., Vlodavsky I., Int. J. Cancer 1985, 35(4), 483. [DOI] [PubMed] [Google Scholar]
- 134. Bar‐Ner M., Mayer M., Schirrmacher V., Vlodavsky I., J. Cell. Physiol. 1986, 128(2), 299. [DOI] [PubMed] [Google Scholar]
- 135. Rivara S., Milazzo F. M., Giannini G., Future Med. Chem. 2016, 8(6), 647. [DOI] [PubMed] [Google Scholar]
- 136. Ostrovsky O., Grushchenko‐Polaq A. H., Beider K., Mayorov M., Canaani J., Shimoni A., Vlodavsky I., Nagler A., Oncogenesis 2018, 7(6), 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Gohji K., Okamoto M., Kitazawa S., Toyoshima M., Dong J., Katsuoka Y., Nakajima M., J. Urol. 2001, 166(4), 1286. [PubMed] [Google Scholar]
- 138. Arvatz G., Shafat I., Levy‐Adam F., Ilan N., Vlodavsky I., Cancer Metastasis Rev. 2011, 30(2), 253. [DOI] [PubMed] [Google Scholar]
- 139. Barash U., Zohar Y., Wildbaum G., Beider K., Nagler A., Karin N., Ilan N., Vlodavsky I., Leukemia 2014, 28(11), 2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Boyango I., Barash U., Naroditsky I., Li J. P., Hammond E., Ilan N., Vlodavsky I., Cancer Res. 2014, 74, 4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Ramani V. C., Zhan F., He J., Barbieri P., Noseda A., Tricot G., Sanderson R. D., Oncotarget 2016, 7, 1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Cohen‐Kaplan V., Naroditsky I., Zetser A., Ilan N., Vlodavsky I., Doweck I., Int. J. Cancer 2008, 123(11), 2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Lv B., Zhang B., Hu X. Y., Zeng Q. D., Oncol. Lett. 2016, 11(2), 1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Nadir Y., Brenner B., Zetser A., Ilan N., Shafat I., Zcharia E., Goldshmidt O., Vlodavsky I., J. Thromb. Haemostasis 2006, 4(11), 2443. [DOI] [PubMed] [Google Scholar]
- 145. Ramani V. C., Yang Y., Ren Y., Nan L., Sanderson R. D., J. Biol. Chem. 2011, 286(8), 6490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Yang Y., Ren Y., Ramani V. C., Nan L., Suva L. J., Sanderson R. D., Cancer Res. 2010, 70(21), 8329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Wood R. J., Hulett M. D., J. Biol. Chem. 2008, 283(7), 4165. [DOI] [PubMed] [Google Scholar]
- 148. Sotnikov I., Hershkoviz R., Grabovsky V., Ilan N., Cahalon L., Vlodavsky I., Alon R., Lider O., J. Immunol. 2004, 172(9), 5185. [DOI] [PubMed] [Google Scholar]
- 149. Cui H., Tan Y., Österholm C., Zhang X., Hedin U., Vlodavsky I., Li J. P., Oncotarget 2016, 7(26), 39486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Wei R., Sun D., Yang H., Yan J., Zhang X., Zheng X., Fu X., Geng M., Huang X., Ding J., Acta Pharmacol. Sin. 2018, 39(8), 1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Nadir Y., Brenner B., Fux L., Shafat I., Attias J., Vlodavsky I., Haematologica 2010, 95(11), 1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Tsunekawa N., Higashi N., Kogane Y., Waki M., Shida H., Nishimura Y., Adachi H., Nakajima M., Irimura T., Biochem. Biophys. Res. Commun. 2016, 469(4), 878. [DOI] [PubMed] [Google Scholar]
- 153. Riaz A., Ilan N., Vlodavsky I., Li J. P., Johansson S., J. Biol. Chem. 2013, 288(17), 12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Zetser A., Levy‐Adam F., Kaplan V., Gingis‐Velitski S., Bashenko Y., Schubert S., Flugelman M. Y., Vlodavsky I., Ilan N., J. Cell Sci. 2004, 117(11), 2249. [DOI] [PubMed] [Google Scholar]
- 155. Levy‐Adam F., Feld S., Suss‐Toby E., Vlodavsky I., Ilan N., PLoS One 2008, 3(6), e2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Bass M. D., Roach K. A., Morgan M. R., Mostafavi‐Pour Z., Schoen T., Muramatsu T., Mayer U., Ballestrem C., Spatz J. P., Humphries M. J., J. Cell Biol. 2007, 177(3), 527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Tkachenko E., Elfenbein A., Tirziu D., Simons M., Circ. Res. 2006, 98(11), 1398. [DOI] [PubMed] [Google Scholar]
- 158. Woods A., Couchman J. R., Curr. Opin. Cell Biol. 2001, 13(5), 578. [DOI] [PubMed] [Google Scholar]
- 159. Goldshmidt O., Zcharia E., Cohen M., Aingorn H., Cohen I., Nadav L., Katz B. Z., Geiger B., Vlodavsky I., FASEB J. 2003, 17, 1015. [DOI] [PubMed] [Google Scholar]
- 160. Fux L., Ilan N., Sanderson R. D., Vlodavsky I., Trends Biochem. Sci. 2009, 34(10), 511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Gingis‐Velitski S., Zetser A., Flugelman M. Y., Vlodavsky I., Ilan N., J. Biol. Chem. 2004, 279(22), 23536. [DOI] [PubMed] [Google Scholar]
- 162. Cohen I., Pappo O., Elkin M., San T., Bar‐Shavit R., Hazan R., Peretz T., Vlodavsky I., Abramovitch R., Int. J. Cancer 2006, 118(7), 1609. [DOI] [PubMed] [Google Scholar]
- 163. Spyrou A., Kundu S., Haseeb L., Yu D., Olofsson T., Dredge K., Hammond E., Barash U., Vlodavsky I., Forsberg‐Nilsson K., Mol. Cancer Ther. 2017, 16(8), 1705. [DOI] [PubMed] [Google Scholar]
- 164. Purushothaman A., Babitz S. K., Sanderson R. D., J. Biol. Chem. 2012, 287(49), 41288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Agelidis A., Turturice B. A., Suryawanshi R. K., Yadavalli T., Jaishankar D., Ames J., Hopkins J., Koujah L., Patil C. D., Hadigal S. R., Kyzar E. J., Campeau A., Wozniak J. M., Gonzalez D. J., Vlodavsky I., Li J., Perkins D. L., Finn P. W., Shukla D., JCI Insight 2021, 6(7), e144255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Yang Y., Gorzelanny C., Bauer A. T., Halter N., Komljenovic D., Bäuerle T., Borsig L., Roblek M., Schneider S. W., Oncogene 2015, 34(47), 5832. [DOI] [PubMed] [Google Scholar]
- 167. Amin R., Tripathi K., Sanderson R. D., Cells 2020, 9(9), 2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Li H., Zhang H., Wenz A., Kang Z., Wang H., Vlodavsky I., Chen X., Li J., Cells 2023, 12(6), 891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. de Boer C., Armstrong Z., Lit V. A. J., Barash U., Ruijgrok G., Boyango I., Weitzenberg M. M., Schröder S. P., Sarris A. J. C., Meeuwenoord N. J., Bule P., Kayal Y., Ilan N., Codée J. D. C., Vlodavsky I., Overkleeft H. S., Davies G. J., Wu L., Proc. Natl. Acad. Sci. U. S. A. 2022, 119(31), e2203167119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. He P., Zhang X., Xia K., Green D. E., Baytas S., Xu Y., Pham T., Liu J., Zhang F., Almond A., Linhardt R. J., DeAngelis P. L., Nat. Commun. 2022, 13(1), 7438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Caruana I., Savoldo B., Hoyos V., Weber G., Liu H., Kim E. S., Ittmann M. M., Marchetti D., Dotti G., Nat. Med. 2015, 21, 524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Masola V., Zaza G., Granata S., Gambaro G., Onisto M., Lupo A., J. Transl. Med. 2013, 11, 292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Masola V., Franchi M., Zaza G., Atsina F. M., Gambaro G., Onisto M., Front. Oncol. 2022, 12, 918419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Gospodarowicz D., Delgado D., Vlodavsky I., Proc. Natl. Acad. Sci. U. S. A. 1980, 77(7), 4094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Vlodavsky I., Lui G., Gospodarowicz D., Cell 1980, 19(3), 607. [DOI] [PubMed] [Google Scholar]
- 176. Zcharia E., Zilka R., Yaar A., Yacoby‐Zeevi O., Zetser A., Metzger S., Sarid R., Naggi A., Casu B., Ilan N., Vlodavsky I., Abramovitch R., FASEB J. 2005, 19(2), 211. [DOI] [PubMed] [Google Scholar]
- 177. Nadav L., Eldor A., Yacoby‐Zeevi O., Zamir E., Pecker I., Ilan N., Geiger B., Vlodavsky I., Katz B. Z., J. Cell Sci. 2002, 115(Pt 10), 2179. [DOI] [PubMed] [Google Scholar]
- 178. Shafat I., Vlodavsky I., Ilan N., J. Biol. Chem. 2006, 281(33), 23804. [DOI] [PubMed] [Google Scholar]
- 179. Masola V., Zaza G., Gambaro G., Onisto M., Bellin G., Vischini G., Khamaysi I., Hassan A., Hamoud S., Nativ O., N. Heyman S., Lupo A., Vlodavsky I., Abassi Z., PLoS One 2016, 11(7), e0160074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Rangarajan S., Richter J. R., Richter R. P., Bandari S. K., Tripathi K., Vlodavsky I., Sanderson R. D., J. Histochem. Cytochem. 2020, 68(12), 823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Sanderson R. D., Elkin M., Rapraeger A. C., Ilan N., Vlodavsky I., FEBS. J. 2017, 284(1), 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Rabelink T. J., van den Berg B. M., Garsen M., Wang G., Elkin M., van der Vlag J., Nat. Rev. Nephrol. 2017, 13(4), 201. [DOI] [PubMed] [Google Scholar]
- 183. Masola V., Zaza G., Secchi M. F., Gambaro G., Lupo A., Onisto M., Biochim. Biophys. Acta, Mol. Cell Res. 2014, 1843(9), 2122. [DOI] [PubMed] [Google Scholar]
- 184. Li J., Vlodavsky I., Thromb. Haemostasis 2009, 102(5), 823. [DOI] [PubMed] [Google Scholar]
- 185. Zhang X., Wang B., Li J. P., Matrix Biol. 2014, 35, 174. [DOI] [PubMed] [Google Scholar]
- 186. Hermano E., Meirovitz A., Meir K., Nussbaum G., Appelbaum L., Peretz T., Elkin M., JNCI, J. Natl. Cancer Inst. 2014, 106(12), dju332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Giannini G., Battistuzzi G., Rivara S., Adv. Exp. Med. Biol. 2020, 1221, 567. [DOI] [PubMed] [Google Scholar]
- 188. Weissmann M., Bhattacharya U., Feld S., Hammond E., Ilan N., Vlodavsky I., Matrix Biol. 2019, 77, 58. [DOI] [PubMed] [Google Scholar]
- 189. Barash U., Cohen‐Kaplan V., Arvatz G., Gingis‐Velitski S., Levy‐Adam F., Nativ O., Shemesh R., Ayalon‐Sofer M., Ilan N., Vlodavsky I., FASEB J. 2010, 24(4), 1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Barash U., Spyrou A., Liu P., Vlodavsky E., Zhu C., Luo J., Su D., Ilan N., Forsberg‐Nilsson K., Vlodavsky I., Yang X., Int. J. Cancer 2019, 145(6), 1596. [DOI] [PubMed] [Google Scholar]
- 191. Mestre A. M., Soe‐Htwe T., Sutcliffe E. L., Rao S., Pagler E. B., Hornby J. R., Hulett M. D., Immunol. Cell Biol. 2007, 85(3), 205. [DOI] [PubMed] [Google Scholar]
- 192. Davidson S. M., Vander Heiden M. G., Annu. Rev. Pharmacol. Toxicol. 2017, 57, 481. [DOI] [PubMed] [Google Scholar]
- 193. Lamming D. W., Bar‐Peled L., Traffic 2019, 20(1), 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Carroll B., Dunlop E. A., Biochem. J. 2017, 474(9), 1453. [DOI] [PubMed] [Google Scholar]
- 195. Saxton R. A., Sabatini D. M., Cell 2017, 168(6), 960. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.