

Abstract
Immune checkpoint inhibitors currently serve an important role in prolonging patients' overall survival. However, the prognostic signatures of immune checkpoint inhibitors in colorectal cancer (CRC) remain uncertain and more knowledge on the genetic characteristics of colorectal cancer is needed. Patients with CRC from The Cancer Genome Atlas were classified into high-immunity group and low-immunity group based on median scores from single-sample gene set enrichment analysis using the GSVA package. We explored immune status by immune scores, stromal scores and tumor purity scores in ESTIMATE package and surveyed the difference of immune cells distribution with CIBERSORT package. Eighteen genes were selected using the LASSO Cox regression method and a prognostic risk model was constructed. Compared with patients in the low-risk group, those in the high-risk group had a significantly shorter survival time. For assessment of the prognostic validity of the risk model, receiver operating characteristic curves with areas under the curve of 0.769, 0.774 and 0.771 for 1, 3 and 5 years respectively. Differences in molecular mechanisms between high- and low-risk groups were analyzed using the clusterProfiler package. Tumor Immune Dysfunction and Exclusion data were downloaded and analyzed. The top 5 enriched pathways in the high-risk group involved ‘calcium signaling’, ‘dilated cardiomyopathy’, ‘extracellular matrix receptor interaction’, ‘hypertrophic cardiomyopathy’ and ‘neuroactive ligand receptor interaction’. HAMP was identified as a hub gene, which was highly expressed in tumor samples. The results of the present study indicate that the prognostic model based on both immune-related genes and HAMP has the potential to support personalized treatment.
Keywords: colorectal cancer, HAMP, prognostic value, immune-related signatures
Introduction
In 2020, 1.9 million new cases and 935,000 deaths from colorectal cancer (CRC) were reported in the United States, and CRC had the third highest incidence and the second highest mortality rate among all cancers in the country (1). Furthermore, the incidence of colorectal cancer before the age of 50 has increased by 1–4% per year in numerous countries (2). CRC patients without metastasis have a good 5-year overall survival (OS) rate at >84.0% (3). For CRC patients with metastasis, the 5-year OS is <15% (4,5). The properties of the tumor microenvironment are strongly linked to the occurrence, development, metastasis, recurrence and treatment resistance of CRC (6).
Currently, the main treatments for CRC include surgery, targeted therapy, radiotherapy, chemotherapy and immunotherapy (7,8). Surgery, chemotherapy and radiotherapy are the preferred treatments because immunotherapy is generally less effective (9,10). However, among CRC patients receiving immunotherapy, patients with microsatellite instability tumors were reported to have shown greater therapeutic effects than patients with microsatellite stable tumors (11,12). The Tumor-Node-Metastasis (TNM) staging system is widely used to evaluate the prognosis of CRC patients (13). However, this system is insufficient for evaluating the effect of treatment in patients receiving immunotherapy and for making treatment decisions. The development of next-generation sequencing will help to elucidate the biological molecular mechanisms underlying colorectal cancer, and further contribute to the development of personalized treatment (14,15). Therefore, new biomarkers for predicting the prognosis of CRC patients need to be identified.
Recent studies have reported biomarkers that can guide systemic therapy in colorectal cancer. For example, insensitivity to cetuximab or panitumumab is associated with mutations of the KRAS and NRAS gene exons 2, 3 and 4, and the presence of these mutations precludes the use of these drugs (16–20). Furthermore, patients with CRC and the BRAF V600E mutation have a worse prognosis (21). The addition of cetuximab to first-line treatment also showed no better OS benefit when compared with treatment without cetuximab, and is additionally associated with increased toxicity of the treatment (22). Immune checkpoint inhibitors have attracted great attention owing to their unique clinical therapeutic effects. According to a phase II clinical trial, the immune-related objective response rate (ORR) was 40% in the DNA mismatch repair (dMMR) colorectal cancer group and 0% in the microsatellite instability-high/mismatch repair-deficient (MSI-H/dMMR) group, respectively (12). An open-label study (KEYNOTE-164) reported that the ORR was 33% in dMMR/MSI-H patients receiving pembrolizumab therapy regardless of whether they received first-line or second-line therapy (23).
Immune-related genes and immune-infiltrating cells undoubtedly serve an indispensable role in the tumor microenvironment and their roles in CRC are worth exploring. A comprehensive analysis of immune cells and immune-related genes is needed to further elucidate the underlying mechanisms of immune resistance and immune response in the context of CRC (24,25).
Materials and methods
Data download and processing
Level 3 RNA sequencing data (such as TPM or FPKM data), high throughput sequencing-counts transcriptome data and clinical information for colon adenocarcinoma (COAD) and rectum adenocarcinoma (READ) were downloaded from TCGA database (https://portal.gdc.cancer.gov/). After processing, data on 659 COADREAD patients (51 normal patients and 608 patients with colorectal cancer) with both gene expression profile and clinical information were included for subsequent analysis. The gene expression profiles were normalized using the DEseq2 package (Bioconductor version 3.14) (26). The clinical features of the patients were presented in Table I. Gene expression data and clinical data were downloaded from the National Center for Biotechnology Information Gene Expression Omnibus (GEO) under the accession number GSE87211.
Table I.
Patient clinical information and features (n=608).
| Group | n | Proportion (%) |
|---|---|---|
| Age | ||
| <60 | 177 | 29.1 |
| ≥60 | 431 | 70.9 |
| Sex | ||
| Female | 280 | 46.1 |
| Male | 328 | 53.9 |
| M stage | ||
| M0 | 450 | 74.0 |
| M1 | 85 | 14.0 |
| MX | 63 | 10.4 |
| NA | 10 | 1.6 |
| N stage | ||
| N0 | 345 | 56.7 |
| N1 | 147 | 24.2 |
| N2 | 112 | 18.4 |
| NA | 2 | 0.3 |
| NX | 2 | 0.3 |
| T stage | ||
| T1 | 20 | 3.3 |
| T2 | 106 | 17.4 |
| T3 | 414 | 68.1 |
| T4 | 65 | 10.7 |
| Missing | 3 | 0.5 |
| Pathologic stage | ||
| I | 106 | 17.4 |
| II | 220 | 36.2 |
| III | 175 | 28.8 |
| IV | 86 | 14.1 |
| NA | 21 | 3.5 |
T, tumor; N, node; M, metastasis; NA, not available.
ssGSEA and cluster analysis
A total of 29 immune gene sets were collected from the literature (27). Three R packages (GSVA, GSEABase and limma; all Bioconductor version 3.14) were used to obtain the ssGSEA algorithm results and estimate the scores of immunological cells infiltrating in all TCGA samples (28). All COADREAD patients were divided into a high immune cluster (Immunity_H) or a low immune cluster (Immunity_L) based on the median ssGSEA score (29,30). Rtsne [version 0.16, CRAN-Package Rtsne (r-project.org)] package was used to generate the t-SNE result and evaluated the distribution between the two groups (31).
Immunity and immune cell type distribution analyses
ESTIMATE analysis, performed using the ESTIMATE package (version 1.0.13, ESTIMATE: R Package; mdanderson.org), was used to calculate the tumor purity, stromal score and immune score of each CORDREAD patient (32). CIBERSORT (R version 1.03; http://cibersort.stanford.edu) was used to calculate the distributions of 22 types of immune cells (33). Wilcoxon's rank-sum test was used to compare the scores between the high- and low-immunity groups.
Construction of the immune-related gene prognostic risk signature
Immune-related genes associated with OS of COADREAD patients were screened using univariate Cox regression analysis using the survival (version 3.2–13; http://cran.r-project.org/src/contrib/Archive/survival/survival_3.2–13.tar.gz) package in R. Then, a LASSO regression model was constructed using the glmnet (version 4.1–4; http://cran.r-project.org/src/contrib/Archive/glmnet/glmnet_4.1–4.tar.gz) package based on the results of univariate Cox regression, and the COADREAD patients were divided into high-risk group and low-risk groups according to the median risk score. The risk signature in the GSE87211 cohort was then validated, and survival and survminer (version 0.4.9; http://cran.r-project.org/web/packages/survminer/index.html) packages were used to construct a Kaplan-Meier curve between the high- and low-risk groups. The sensitivity and specificity of the prognostic signature were evaluated using a receiver-operating characteristic (ROC) curve.
GSEA analysis
The signaling pathway differences were analyzed using functional enrichment analysis by the clusterProfiler package (version 4.4.4; Bioconductor-clusterProfiler) (34,35). The dataset named c2.cp.kegg.v7.5.1.symbols.gmt was download from the GSEA website (https://www.gsea-msigdb.org/gsea/msigdb/collections.jsp#C2) (36).
Immune checkpoint inhibitors response prediction
The COADREAD gene expression data were uploaded to the TIDE website (http://tide.dfci.harvard.edu/) and then the prediction response results were visualized using a violin plot. Wilcoxon rank-sum test was used to compare the scores between the high and low-immunity groups.
Exploration of immune-related hub genes
The STRING online tool (https://string-db.org/cgi/input?sessionId=bojBmgAExQGs&input_page_show_search=on) was used to explore protein-protein interaction pairs with a combined score of >0.15 and the cytoHubba (version 0.1) app in Cytoscape (version 3.8.0) was used to calculate the node scores (37). The expression of the hub genes and the relationship between the hub genes and OS were analyzed in TCGA cohort. In the present study, hub genes were considered to be those with a high Clustering Coefficient, with a threshold value of 1, as presented in Table II.
Table II.
Hub genes with high Clustering Coefficient.
| Node name | Clustering Coefficient |
|---|---|
| NUMBL | 0.00 |
| PMCH | 0.00 |
| MC1R | 0.00 |
| VAV2 | 1.00 |
| HAMP | 1.00 |
| IL20RB | 0.00 |
| SEMA5B | 1.00 |
| CX3CL1 | 0.67 |
| CD1B | 0.00 |
| CD1A | 0.00 |
| NRG1 | 0.40 |
| PPARGC1A | 0.47 |
| EPO | 0.33 |
| ANGPTL4 | 1.00 |
Hub gene validation using reverse transcription-quantitative PCR (RT-qPCR)
To further validate the expression of hub genes in the present study, tissue was collected from 3 CRC patients who had not undergone any treatment. The transcription level of the hub gene in the colorectal cancer tissue and adjacent tissues was assessed. Total RNA was extracted from tissues using TRNzol (cat. no. DP424; Tiangen Biotech Co., Ltd.), and cDNA was synthesized using the Evo M-MLV RT premix kit (cat. no. AG11601; Accurate Biotechnology; Hunan Aikerui Biological Engineering Co., Ltd.) according to the manufacturer's instructions. RT-qPCR was performed using the SYBR Green Real-time PCR Master Mix kit (cat. no. 11201ES08; Shanghai Yeasen Biotechnology Co., Ltd.). The following conditions apply to the reaction: An initial 5 min at 95°C and denaturation at 95°C for 15 sec, followed by 40 cycles of annealing at 60°C for 30 sec and extension at 72°C for 30 sec. Relative mRNA expression levels were calculated using the 2−ΔΔCq method (38) and assessed statistically using Student's t-test. The primer sequences used were as follows: HAMP forward (F), 5′-CTCCTTCGCCTCTGGAACAT-3′ and reverse (R), 5′-AGTGGCTCTGTTTTCCCACA-3′; and GAPDH F, 5′-GAAGATGGTGATGGGATTTC-3′ and R, 5′-GAAGGTGAAGGTCGG-3′.
Statistical Analyses
All statistical analysis was performed using R 4.1.0 (https://mirrors.tuna.tsinghua.edu.cn/CRAN/src/base/R-4/R-4.1.0.tar.gz). The relationship between high- and low-risk scores and OS were evaluated using univariate and multivariate Cox analyses. The sensitivity and specificity of high- and low-risk groups and OS were examined by ROC analysis. P<0.05 was considered to indicate a statistically significant difference.
Results
Construction of colorectal cancer groups based on ssGSEA
Data for patients with CRC were obtained from TCGA database, and the distribution of 22 types of immune cells in these patients was analyzed using the ssGSEA algorithm. CRC patients were divided into high-immunity and low-immunity groups according to consensus cluster analysis based on the median ssGSEA scores. The t-SNE method preliminarily evaluated the distribution between the two groups, and the high- and low-immunity groups were well differentiated in colorectal cancer samples (Fig. 1A). A significant difference in OS was observed between the high-immunity group and low-immunity group using the KM curve (Fig. 1B; P=0.026). Stromal, immune and ESTIMATE scores were calculated using ESTIMATE analysis. The Wilcoxon rank-sum test method was used to assess the statistical significance and the violin plot demonstrated significantly higher scores in the high-immunity group compared with the low-immunity group (Fig. 1C; P<2×10−16). In particular, CIBERSORT analysis demonstrated significantly higher CD8+ T cell levels in the high-immunity group compared with the low-immunity group (Fig. 1D; P=0.0019). The heatmap demonstrated that in the high-immune group, the stromal, immune and ESTIMATE scores had a similar trend to immune cell expression (Fig. 1E).
Figure 1.

Classification immune landscape of high- and low-immunity groups in colorectal cancer. (A) The distribution of colorectal patients in high- and low-immunity groups. (B) The Kaplan-Meier curves predict the overall survival in the high- and low-immunity groups. (C) The immune scores, stromal scores and ESTIMATE scores for the high- and low-immunity groups. (D) Distribution of 22 types of immune cells in the high- and low-immunity groups. (E) Heatmap of ESTIMATE scores and immune gene sets for high- and low-immunity groups. *P<0.05, **P<0.01 and ***P<0.001. tSNE, T-distributed stochastic neighbor embedding; TME, tumor microenvironment; H, high; L, low; CD, cluster of differentiation; NK, natural killer; aDCs, activated dendritic cell; pDCs, plasmacytoid dendritic cell; Th, helper T cells; Tfh, follicular helper T cells; iDCs, immature dendritic cells; APC, antigen-presenting cell; HLA, human leukocyte antigen; CCR, chemokine receptor; DCs, dendritic cell; TIL, tumor infiltrating lymphocyte.
Exploration of differentially expressed genes between high- and low-immunity groups
Differentially expressed genes (DEGs) between the high- and low-immunity groups were assessed (Fig. 2A). Immune-related genes were downloaded from two websites, ImmPort (https://www.immport.org/home) and innateDB (https://www.innatedb.com/). Among the DEGs, 1,436 genes were immune system related (Fig. 2B), with 1,195 being upregulated and 241 being downregulated (Fig. 2C).
Figure 2.

Evaluation of DEGs in the high- and low-immunity groups. (A) Volcano plot and (B) Venn of the DEGs in the high- and low-immune groups and immune-related genes from ImmPort and innateDB databases. (C) Heatmap of the 1195 upregulated genes and 241 downregulated genes. DEGs, differentially expressed genes; H, high; L, low.
Construction of immunity-related prognostic signature
To construct a risk model, 111 mRNAs related to OS were identified using the univariate analysis (Fig. 3A). Among the identified mRNAs, 18 genes were used to build a risk model by LASSO Cox regression analysis. The risk score was calculated using the coefficients of the 18 genes (Fig. 3B and C) as follows: risk score=USP7 × (−0.0153034090477698) + VAV2 × 0.0655011160044214 + CX3CL1 × 0.00124855618096817 + NUMBL × 0.0548798283592552 + PPARGC1A × (−0.052320825824987) + ANGPTL4 × 0.00461031918855942 + MC1R × 0.165908532485693 + MID2 × 0.115738969177335 + NRG1 × (−0.108632261933379) + SEMA5B × 0.00929691428939524 + IL20RB × 0.238904701764134 + RBP7 × 0.127007108169451 + CD1A × (−0.0254834618450881) + PTH1R × 0.0610074708594601 + HAMP × 0.00478998991273158 + CD1B × (−0.150298945645048) + PMCH × (−0.0339401840839164) + EPO × 0.00100587552113195. The coefficient values of the 18 genes was calculated by glmnet package using the coef function. Using the median risk scores, the CRC patients were divided into high-risk and low-risk groups. Patients' mortality and risk score distributions were plotted (Fig. 3D and E) and a higher death rate was demonstrated in patients in the high-risk group. Compared with patients in the low-risk group, those in the high-risk group had a significantly shorter overall survival time (P<0.001; Fig. 3F). For assessment of the prognostic validity of the risk score, ROC curves were generated, the area under the curve (AUC) values of 0.769, 0.774 and 0.771 for 1, 3, and 5 year survival rates respectively, indicated that the risk model was valid (Fig. 3G).
Figure 3.

Construction of the risk signature. (A) Univariate analysis identified 111 genes related to overall survival. (B and C) The minimum criteria and coefficients were calculated by LASSO regression analysis. (D and E) Distributions of the risk score and mortality status. (F) Clinical outcome of patients with colorectal cancer in the high-risk and low-risk groups. (G) Receiver operating characteristic curve of the predictive efficiency for 1, 3 and 5 year survival. AUC, area under the curve.
Validation of the immunity-related risk signature
To better understand the value of the immunity-related risk signature, a GEO cohort (GSE87211) with a survival time <9 years was used to construct ROC curves and for KM analysis. Consistent with the trend demonstrated in the TCGA cohort, the OS in the high-risk group was shorter and the risk scores were higher compared with the low-risk group (Fig. 4A and B). The patients in the high-risk group in the GSE87211 dataset demonstrated significantly shorter OS compared with those in the low-risk group (P=0.034; Fig. 4C). ROC analysis was performed to test the stability and robustness of the immunity-related risk model. The AUC values were 0.673, 0.620, and 0.617 for 1, 3, and 5 year OS, respectively (Fig. 4D).
Figure 4.

Validation of the risk signature. (A and B) Mortality rate was associated with high risk scores. (C) The validation cohort demonstrated distinctly different survival curves between the high- and low-risk groups. (D) Receiver operating characteristic curves predicted the risk model efficiency in the validation cohort. AUC, area under the curve.
Relationship between clinical features and prognosis of CRC
To evaluate the correlation between the clinical features and the prognosis of CRC, univariate Cox regression analysis was performed, which demonstrated that age, T stage, N stage, M stage and risk score were significantly associated with OS (P<0.001; Fig. 5A). More importantly, multivariate Cox analysis demonstrated that risk score could be an independent prognostic risk factor (P<0.001; Fig. 5B).
Figure 5.

Univariate and multivariate Cox analyses for clinical features and risk model. (A) Univariate analysis results. (B) Multivariate analysis results. T, tumor; N, node; M, metastasis.
Functional enrichment analysis by GSEA and clinical characteristics between high- and low-risk groups
GSEA analysis indicated that the extracellular matrix (ECM) receptor interaction was enriched in the high-risk group (Fig. 6A), which directly or indirectly controls cellular activities. Other enriched pathways in the high-risk group involved calcium signaling, dilated cardiomyopathy, hypertrophic cardiomyopathy and neuroactive ligand receptor interaction (Fig. 6A). The low-risk group demonstrated enrichment of pathways such as chemokine signaling pathway or cytokine-cytokine receptor interaction (Fig. 6B). The correlation between risk score and clinical characteristics was analyzed and there were significant differences in T, N and M stages between the high- and low-risk groups, but there were no differences in age and sex (Fig. 6C).
Figure 6.

Functional and clinical characteristics between high- and low-risk groups. Top 5 enrichment pathways in the (A) high-risk group and the (B) low-risk group. (C) Heatmap for risk group and clinical characteristics. ***P<0.001. T, tumor; N, node; M, metastasis.
Potential role of risk signature in predicting immune checkpoint blockade responses
To evaluate the clinical function of the risk model, the TIDE method was used to compare the differences in TIDE, dysfunction and exclusion scores between high- and low-risk groups. Patients in the high-risk group had higher scores for all measures (P=8.3×10−5, P=9.5×10−5 and P=1.1×10−8, respectively; Fig. 7). These findings suggested that the poor immune response of patients in the high-risk group was due to immune dysfunction and immune rejection.
Figure 7.
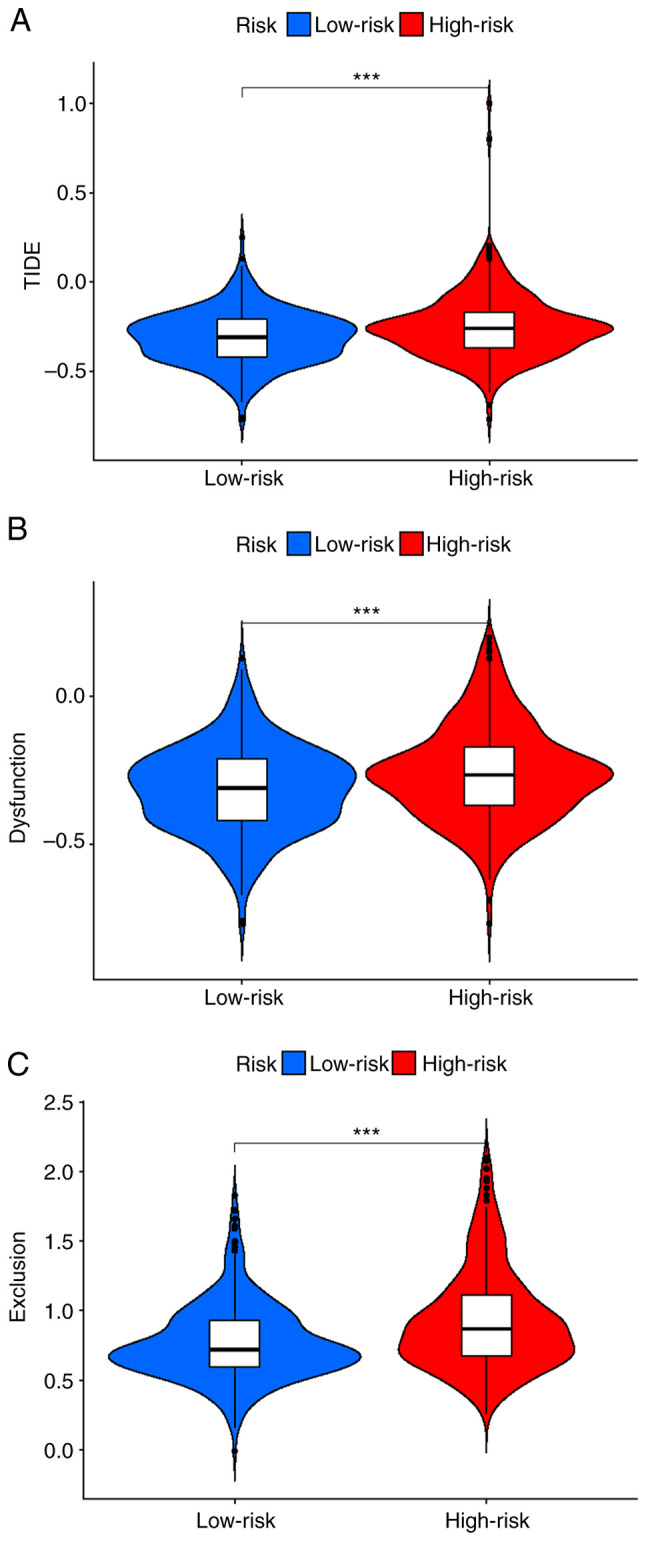
Prediction of immune checkpoint blockade responses in the high- and low-risk groups. (A) TIDE score, (B) dysfunction score and (C) exclusion score in the high- and low-risk groups. ***P<0.001.
Evaluation of the hub gene
An analysis of the 18 genes was performed using the STRING website (https://string-db.org/) to evaluate the hub gene among the risk signatures. The minimum required interaction score was set at 0.15, which resulted in a file containing interactions among all degrees of nodes for 14 genes. cytoHubba was used to further assess hub objects among the complex interactions. The genes were sorted according to clustering coefficient, and VAV2, HAMP, SEMA5B and ANGPTL4 were indicated as the hub genes (Table II). The expression differences of these four genes and survival were further analyzed and no significant differences in the expression of ANGPTL4 between the tumor group and the normal group were demonstrated (data not shown). Furthermore, no significant difference in survival was demonstrated between the high and low expression groups with regard to SEMA5B and VAV2 expression (data not shown). Therefore, HAMP demonstrated significant differences and was selected for subsequent analysis. In both the unpaired and the paired groups, significant differences were demonstrated in the mRNA expression levels of HAMP (P<0.05; Fig. 8A and B). In the tumor group, the expression of HAMP was significantly correlated with tumor pathological stage, T stage and N stage; however, no significant difference was demonstrated for M stage (Fig. 8C-F). The mRNA expression of HAMP was also positively correlated with PDCD1 (PD1) and CD274 (PD-L1) (Fig. 8G-H). The relationship between HAMP expression and 22 types of immune cells was assessed, which demonstrated that HAMP was mainly expressed in CD8+ T cells, NK cells, monocytes and macrophages (cells with importance in immune therapy) (Fig. 8I). After removing duplicated data and data missing clinical information, it was demonstrated that high expression of HAMP was significantly associated with shorter OS (P=0.004), shorter progression free interval (P=0.015) and shorter disease specific survival (P<0.001) (Fig. 8J-M) (39). The ROC curve was used to evaluate the predictive performance of HAMP, the AUC was 0.743, which indicated good predictive performance (Fig. 8N). To evaluate the role of HAMP in tumor immunity, the correlations between HAMP expression and stromal score, immune score and ESTIMATE score were analyzed. The results demonstrated that HAMP expression was significantly positively correlated with all three scores (Fig. 8O-Q). To validate the expression of HAMP, qPCR was performed in colorectal cancer and adjacent tissues, which demonstrated a similar pattern of expression (P=0.0031; Fig. 8R).
Figure 8.

Evaluation of the hub gene and validation with reverse transcription-quantitative PCR. (A and B) The expression of HAMP in unpaired and paired groups. (C-F) The relationship between HAMP expression and clinical features. (G and H) The relationship between HAMP expression and PDCD1/CD274. (I) Different distributions of tumor-infiltrating cells in high/low HAMP expression groups. The Kaplan-Meier curves for (J) overall survival, (K) progression free interval and (L) disease specific survival in The Cancer Genome Atlas datasets and (M) overall survival in GSE17537 for patients between groups with high and low HAMP expression. (N) The receiver operating characteristic curve analysis of HAMP expression. (O-Q) The relationship between HAMP expression and stromal scores, immune scores, and ESTIMATE scores. (R) Validation of HAMP expression in three colorectal cancer specimens and three matched normal adjacent tissues (n=3). *P<0.05, **P<0.01 and ***P<0.001. HAMP, hepcidin antimicrobial peptide; TPM, transcripts per million; CD, cluster of differentiation; NK, natural killer; AUC, area under the curve; CI, confidence interval; T, tumor; N, node; M, metastasis.
Discussion
CRC is the third most common type of malignant tumor and the second most common cause of tumor-related death in the United States (1). Targeted therapy for CRC is still ineffective for patients with advanced tumors (40). PD1 and PD-L1 are the main indicators used to guide the use of immune checkpoint inhibitors (41). A small number of patients with MSI-H/dMMR have a durable response to immune checkpoint inhibitor therapy, which can effectively prolong the OS of patients with advanced CRC (42,43). However, the results in patients with MSI-H/dMMR cannot be generalized to the entire patient population receiving immune checkpoint inhibitor therapy. As the importance of the immune microenvironment in tumor progression is increasingly recognized, there is a critical need to elucidate the molecular pathogenesis of colorectal cancer and to identify reliable prognostic biomarkers based on the immune landscape (44).
In the present study, transcriptomic and clinical information were downloaded from TCGA database and patients with CRC were divided into high- and low-immunity groups based on ssGSEA scores. Previous research reported a pan-cancer tumor inflammation signature and divided patients into high- and low-immune groups, and that patients with renal clear cell carcinoma (45), melanoma (46), lung tumors (47) and head and neck tumors (48) in the high-immune group were more likely to be sensitive to immune checkpoint inhibitors. However, this model has been reported to have limited predictive ability for colorectal cancer (49), because tumor cells could rapidly proliferate by transitioning from an immune homeostasis state to an immune escape state. Therefore, patients in the immune elimination and immune editing phases have higher immunity and better prognosis (50). The antitumor role of the immune system can be summarized as preventing pro-inflammatory effects, protecting the host from viral infection and killing tumor cells (51).
The present study demonstrated that the stromal score, immune score and ESTIMATE score were higher in the high-immune group. By performing CIBERSORT analyses, it was demonstrated that the levels of CD8+ T cells and macrophage M1 cells in the high-immune group were significantly increased compared with the low-immune group. As key components of the adaptive immune system, CD8+ T cells serve important roles in immune defense against intracellular pathogens such as viruses and bacteria, and tumors (52,53). In general, cytotoxic T cells serve an important antitumor role through interferon-γ, tumor necrosis factor-α, interleukin-2 and interleukin-17 (54,55). An increase in T-cell infiltration is more likely with high-immune status (56). Similarly, an increase in CD8+ T cells was demonstrated in the high-immune group in the present study. Tumors contain a large amount of macrophages, and M1 macrophages have anti-tumor and pro-inflammatory effects (57,58). Elevated CD8+ T cell infiltration has been reported to be associated with better prognosis in colorectal cancer (59), improved OS in oral squamous cell carcinoma (59) and disease free survival in laryngeal carcinoma (60–62). High macrophage infiltration is also associated with improved prognosis in colorectal cancer (63). These reports together with the results of the present study explain why colorectal cancer patients in the high-immune group have a better prognosis. By analyzing the DEGs of the high- and low-immune groups, and the intersecting immune-related genes from the ImmPort and innateDB immune databases, a risk model was constructed for the identified intersecting genes. Risk scores and clinical characteristics were combined to evaluate the model's effectiveness, and the ROC curve demonstrated the effectiveness of the model. Univariate and multivariate analyses demonstrated that the model had a better prognostic result than the TNM classification system. The risk model was also validated using external GEO data (GSE87211). Functional enrichment analysis of the high- and low-risk groups was performed and the results demonstrated that the genes in the high-risk group were mainly enriched in the ECM receptor signaling pathway. To evaluate the predictive effect of the model for immune checkpoint inhibitors, the TIDE algorithm was used, which demonstrated that the high-risk group had a high score of immune dysfunction and immune rejection, which may indicate a poor response to immunotherapy (24,25).
Finally, a hub gene, HAMP, was identified using a protein-protein interaction network. HAMP is involved in iron homeostasis and ferroptosis. Ferroptosis, an intracellular iron-dependent form of cell death, serves a key role in tumor suppression and is clearly associated with resistance to cancer therapy (64–66). Ferroptosis is associated with T-cell immunity and cancer immunotherapy, and inhibition of ferroptosis may lead to resistance to immune checkpoint inhibitor therapy (67). Immune checkpoint inhibitor therapy has clear clinical benefits in inducing long lasting responses, but drug resistance remains a formidable challenge (68).
HAMP encodes hepcidin antimicrobial peptide (HAMP), a pro-peptide of 84 amino acids. HAMP is cleaved into mature peptides of 20, 22 and 25 amino acids, undergoing enzymatic digestion (69). Its product, hepcidin, serves an important role in regulating macrophage iron storage and intestinal iron absorption (70).
Hepcidin, as an acute-phase protein, participates in innate immune reactions in an interleukin-6 dependent manner (71). Hepcidin in conventional dendritic cells can promote mucosal repair in a nutritional immunity manner (72). Macrophages serve an important role in regulating iron levels with hepcidin, which in turn influences inflammation, infection and possibly cancer, and overexpression of hepcidin is linked with cancer development and prognosis (73).
HAMP affects iron homeostasis, inflammation and oxidative regulation through the mTOR, JAK/STAT and BMP/SMAD signaling pathways (74–76). Hepcidin serves an important role in the occurrence, development and metastasis of liver cancer. Iron sensing is dysregulated in patients with liver cancer, which in turn leads to the dysregulation of hepcidin. As such, hepcidin may serve as a drug therapy target in liver cancer (77–79). Hepcidin is also involved in breast cancers, promoting proliferation, invasion and metastasis (80). In prostate cancer, hepcidin dysregulation contributes to the development and progression of the cancer (81,82). Serum hepcidin levels are significantly correlated with lymph node metastasis status and T stage in non-small cell lung cancer (83). In colorectal cancer, patients with adequate iron have superior outcomes and increased response to therapy (84). If hepcidin is deficient, tumor number, burden and size are diminished (85–87). A previous in vitro study reported that hepcidin promotes growth in the colorectal cancer cell line HT-29 cell; however, similar results were not reported in other colorectal cell types (87). A previous immunohistochemistry study reported that the positive rate of hepcidin in CRC tissues was significantly higher than that in adjacent tissues (88). In the present study, differences in HAMP expression between colorectal cancer and normal tissues were demonstrated. Furthermore, in terms of clinical features, HAMP expression was higher in patients with advanced clinical features (such as T1 and T2 vs. T3 and T4, and N0 vs. N1 and N2). Colorectal cancer patients were divided into groups based on high and expression of HAMP, using the median value of HAMP expression. Patients with high HAMP expression had a worse prognosis. Furthermore, in the high HAMP expression group, CD8+ T cell and macrophage M1 cell levels were significantly increased compared with the low HAMP expression group. Finally, qPCR was performed using tissue samples from colorectal cancer patients to verify the differences in expression levels. Based on the results of the present study, HAMP could be further used to identify target molecules for subsequent studies and as possible treatment candidates.
The present study demonstrated for the first time, to the best of our knowledge, the role of HAMP in the immune microenvironment of colorectal cancer, combining the current immune microenvironment with the ferroptosis hotspot. The present study has certain limitations. These research findings are preliminary and there are still mechanisms needing further elaboration. Firstly, only the GEO database was used to verify the model, and prospective clinical trial results are needed in the future. Secondly, more clinical samples and in vivo and in vitro experiments are needed to verify the role of HAMP in colorectal cancer. Thirdly, the present study only analyzed the risk model and the correlation between HAMP and immune cells and immunotherapy. More clinical samples are needed to evaluate the roles in the model and HAMP in immunotherapy in the future. The mechanism of HAMP in the progression of colorectal cancer still needs to be further elucidated, and clinical data and molecular experiments are needed to verify these results.
The present study constructed an immune-related prognostic model, and then identified the key gene HAMP, which linked the immune microenvironment with ferroptosis. Risk models and HAMP may provide evidence for colorectal cancer prognosis and drug selection. The finding of HAMP gene as a hub gene is significant and it is important to perform further experiments to elucidate how this gene is related to colorectal cancer. Furthermore, whether modified HAMP could change tumor environment and allow more people to benefit from immunotherapy or reverse immunotherapy resistance should be studied further in future.
Acknowledgements
Not applicable.
Funding Statement
This work was funded by the Fundamental Research Funds for the Central Universities (grant no. 21JNQN14); the Technology Development Cultivation of Program Southern Medical University (grant no. KJ20161121); Guangdong Sci-tech Commissioner (grant no. 20211800500322); the National Natural Science Foundation of China (grant no. 81803877); the Dongguan City Social Science and Technology Development (Key) Project (grant no. 202050715001207); and the Fundamental and Applied Basic Research Fund of Guangdong Province (grant no. 2020B1515120063).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
HW, YW and XZ designed the study. HD collected the data from databases and performed analyses. HW and HD organized and arranged all the figures. SR and JC performed the experiments and the formal analysis. YZ and MD collected tissue samples. HW and XZ prepared and wrote the original draft of the manuscript. HW, HD, YW and XZ reviewed and edited the manuscript. All authors read and approved the final version of the manuscript. HW and HD confirm the authenticity of all the raw data.
Ethics approval and consent to participate
This study was approved by the Ethics Committee of The Affiliated Dongguan People's Hospital of Southern Medical University (approval no. KYKT2021-018) and informed consents was provided by the patients. The study was conducted in accordance with the Declaration of Helsinki.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Torre LA, Soerjomataram I, Hayes RB, Bray F, Weber TK, Jemal A. Global patterns and trends in colorectal cancer incidence in young adults. Gut. 2019;68:2179–2185. doi: 10.1136/gutjnl-2019-319511. [DOI] [PubMed] [Google Scholar]
- 3.Kim MJ, Jeong SY, Choi SJ, Ryoo SB, Park JW, Park KJ, Oh JH, Kang SB, Park HC, Heo SC, Park JG. Survival paradox between stage IIB/C (T4N0) and stage IIIA (T1-2N1) colon cancer. Ann Surg Oncol. 2015;22:505–512. doi: 10.1245/s10434-014-3982-1. [DOI] [PubMed] [Google Scholar]
- 4.Almatroudi A. The incidence rate of colorectal cancer in Saudi Arabia: An observational descriptive epidemiological analysis. Int J Gen Med. 2020;13:977–990. doi: 10.2147/IJGM.S277272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gan GL, Liu J, Chen WJ, Ye QQ, Xu Y, Wu HT, Li W. The diverse roles of the mucin gene cluster located on chromosome 11p15.5 in colorectal cancer. Front Cell Dev Biol. 2020;8:514. doi: 10.3389/fcell.2020.00514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sasidharan Nair V, Saleh R, Taha RZ, Toor SM, Murshed K, Ahmed AA, Kurer MA, Abu Nada M, Al Ejeh F, Elkord E. Differential gene expression of tumor-infiltrating CD4+ T cells in advanced versus early stage colorectal cancer and identification of a gene signature of poor prognosis. Oncoimmunology. 2020;9:1825178. doi: 10.1080/2162402X.2020.1825178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ihnát P, Vávra P, Zonča P. Treatment strategies for colorectal carcinoma with synchronous liver metastases: Which way to go. World J Gastroenterol. 2015;21:7014–7021. doi: 10.3748/wjg.v21.i22.7014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mármol I, Sánchez-de-Diego C, Pradilla Dieste A, Cerrada E, Rodriguez Yoldi MJ. Colorectal carcinoma: A general overview and future perspectives in colorectal cancer. Int J Mol Sci. 2017;18:197. doi: 10.3390/ijms18010197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ciardiello D, Vitiello PP, Cardone C, Martini G, Troiani T, Martinelli E, Ciardiello F. Immunotherapy of colorectal cancer: Challenges for therapeutic efficacy. Cancer Treat Rev. 2019;76:22–32. doi: 10.1016/j.ctrv.2019.04.003. [DOI] [PubMed] [Google Scholar]
- 10.Cunningham D, Atkin W, Lenz HJ, Lynch HT, Minsky B, Nordlinger B, Starling N. Colorectal cancer. Lancet. 2010;375:1030–1047. doi: 10.1016/S0140-6736(10)60353-4. [DOI] [PubMed] [Google Scholar]
- 11.de Weger VA, Turksma AW, Voorham QJ, Euler Z, Bril H, van den Eertwegh AJ, Bloemena E, Pinedo HM, Vermorken JB, van Tinteren H, et al. Clinical effects of adjuvant active specific immunotherapy differ between patients with microsatellite-stable and microsatellite-instable colon cancer. Clin Cancer Res. 2012;18:882–889. doi: 10.1158/1078-0432.CCR-11-1716. [DOI] [PubMed] [Google Scholar]
- 12.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372:2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Benson AB, Venook AP, Al-Hawary MM, Azad N, Chen YJ, Ciombor KK, Cohen S, Cooper HS, Deming D, Garrido-Laguna I, et al. Rectal cancer, version 2.2022, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2022;20:1139–1167. doi: 10.6004/jnccn.2022.0051. [DOI] [PubMed] [Google Scholar]
- 14.Koncina E, Haan S, Rauh S, Letellier E. Prognostic and predictive molecular biomarkers for colorectal cancer: Updates and challenges. Cancers (Basel) 2020;12:319. doi: 10.3390/cancers12020319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vacante M, Borzì AM, Basile F, Biondi A. Biomarkers in colorectal cancer: Current clinical utility and future perspectives. World J Clin Cases. 2018;6:869–881. doi: 10.12998/wjcc.v6.i15.869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, Juan T, Sikorski R, Suggs S, Radinsky R, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:1626–1634. doi: 10.1200/JCO.2007.14.7116. [DOI] [PubMed] [Google Scholar]
- 17.Bokemeyer C, Bondarenko I, Makhson A, Hartmann JT, Aparicio J, de Braud F, Donea S, Ludwig H, Schuch G, Stroh C, et al. Fluorouracil, leucovorin, and oxaliplatin with and without cetuximab in the first-line treatment of metastatic colorectal cancer. J Clin Oncol. 2009;27:663–671. doi: 10.1200/JCO.2008.20.8397. [DOI] [PubMed] [Google Scholar]
- 18.Douillard J, Oliner KS, Siena S, Tabernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham D, Jassem J, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med. 2013;369:1023–1034. doi: 10.1056/NEJMoa1305275. [DOI] [PubMed] [Google Scholar]
- 19.Karapetis CS, Khambata-Ford S, Jonker DJ, O'Callaghan CJ, Tu D, Tebbutt NC, Simes RJ, Chalchal H, Shapiro JD, Robitaille S, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359:1757–1765. doi: 10.1056/NEJMoa0804385. [DOI] [PubMed] [Google Scholar]
- 20.Sorich MJ, Wiese MD, Rowland A, Kichenadasse G, McKinnon RA, Karapetis CS. Extended RAS mutations and anti-EGFR monoclonal antibody survival benefit in metastatic colorectal cancer: A meta-analysis of randomized, controlled trials. Ann Oncol. 2015;26:13–21. doi: 10.1093/annonc/mdu378. [DOI] [PubMed] [Google Scholar]
- 21.Van Cutsem E, Köhne CH, Láng I, Folprecht G, Nowacki MP, Cascinu S, Shchepotin I, Maurel J, Cunningham D, Tejpar S, et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: Updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J Clin Oncol. 2011;29:2011–2019. doi: 10.1200/JCO.2010.33.5091. [DOI] [PubMed] [Google Scholar]
- 22.Maughan TS, Adams RA, Smith CG, Meade AM, Seymour MT, Wilson RH, Idziaszczyk S, Harris R, Fisher D, Kenny SL, et al. Addition of cetuximab to oxaliplatin-based first-line combination chemotherapy for treatment of advanced colorectal cancer: Results of the randomised phase 3 MRC COIN trial. Lancet. 2011;377:2103–2114. doi: 10.1016/S0140-6736(11)60613-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Le DT, Kim TW, Van Cutsem E, Geva R, Jäger D, Hara H, Burge M, O'Neil B, Kavan P, Yoshino T, et al. Phase II open-label study of pembrolizumab in treatment-refractory, microsatellite instability-high/mismatch repair-deficient metastatic colorectal cancer: KEYNOTE-164. J Clin Oncol. 2020;38:11–19. doi: 10.1200/JCO.19.02107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fu J, Li K, Zhang W, Wan C, Zhang J, Jiang P, Liu XS. Large-scale public data reuse to model immunotherapy response and resistance. Genome Med. 2020;12:21. doi: 10.1186/s13073-020-0721-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X, Li Z, Traugh N, Bu X, Li B, et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med. 2018;24:1550–1558. doi: 10.1038/s41591-018-0136-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang X, Duanmu J, Fu X, Li T, Jiang Q. Analyzing and validating the prognostic value and mechanism of colon cancer immune microenvironment. J Transl Med. 2020;18:324. doi: 10.1186/s12967-020-02491-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li L, Zhang W, Qiu J, Zhang W, Lu M, Wang J, Jin Y, Xi Q. Stem cell-associated signatures help to predict diagnosis and prognosis in ovarian serous cystadenocarcinoma. Stem Cells Int. 2023;2023:4500561. doi: 10.1155/2023/4500561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hänzelmann S, Castelo R, Guinney J. GSVA: Gene set variation analysis for microarray and RNA-Seq data. BMC Bioinformatics. 2013;14:7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Finotello F, Mayer C, Plattner C, Laschober G, Rieder D, Hackl H, Krogsdam A, Loncova Z, Posch W, Wilflingseder D, et al. Molecular and pharmacological modulators of the tumor immune contexture revealed by deconvolution of RNA-seq data. Genome Med. 2019;11:34. doi: 10.1186/s13073-019-0638-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yoshihara K, Shahmoradgoli M, Martínez E, Vegesna R, Kim H, Torres-Garcia W, Treviño V, Shen H, Laird PW, Levine DA, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun. 2013;4:2612. doi: 10.1038/ncomms3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, Alizadeh AA. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12:453–457. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z, Feng T, Zhou L, Tang W, Zhan L, et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation (Camb) 2021;2:100141. doi: 10.1016/j.xinn.2021.100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu G, Wang LG, Han Y, He QY. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chin CH, Chen SH, Wu HH, Ho CW, Ko MT, Lin CY. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst Biol. 2014;8((Suppl 4)):S11. doi: 10.1186/1752-0509-8-S4-S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 39.Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, Kovatich AJ, Benz CC, Levine DA, Lee AV, et al. An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell. 2018;173:400–416.e11. doi: 10.1016/j.cell.2018.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tang YL, Li DD, Duan JY, Sheng LM, Wang X. Resistance to targeted therapy in metastatic colorectal cancer: Current status and new developments. World J Gastroenterol. 2023;29:926–948. doi: 10.3748/wjg.v29.i6.926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seliger B. Basis of PD1/PD-L1 Therapies. J Clin Med. 2019;8:2168. doi: 10.3390/jcm8122168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pitt JM, Vétizou M, Daillère R, Roberti MP, Yamazaki T, Routy B, Lepage P, Boneca IG, Chamaillard M, Kroemer G, Zitvogel L. Resistance mechanisms to immune-checkpoint blockade in cancer: Tumor-intrinsic and -extrinsic factors. Immunity. 2016;44:1255–1269. doi: 10.1016/j.immuni.2016.06.001. [DOI] [PubMed] [Google Scholar]
- 43.Sun C, Mezzadra R, Schumacher TN. Regulation and function of the PD-L1 checkpoint. Immunity. 2018;48:434–452. doi: 10.1016/j.immuni.2018.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541:321–330. doi: 10.1038/nature21349. [DOI] [PubMed] [Google Scholar]
- 45.Horn L, Spigel DR, Vokes EE, Holgado E, Ready N, Steins M, Poddubskaya E, Borghaei H, Felip E, Paz-Ares L, et al. Nivolumab versus docetaxel in previously treated patients with advanced non-small-cell lung cancer: Two-Year outcomes from two randomized, open-label, phase III trials (CheckMate 017 and CheckMate 057) J Clin Oncol. 2017;35:3924–3933. doi: 10.1200/JCO.2017.74.3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Topalian SL, Sznol M, McDermott DF, Kluger HM, Carvajal RD, Sharfman WH, Brahmer JR, Lawrence DP, Atkins MB, Powderly JD, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. 2014;32:1020–1030. doi: 10.1200/JCO.2013.53.0105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reck M, Rodríguez-Abreu D, Robinson AG, Hui R, Csőszi T, Fülöp A, Gottfried M, Peled N, Tafreshi A, Cuffe S, et al. Pembrolizumab versus chemotherapy for PD-L1-Positive non-small-cell lung cancer. N Engl J Med. 2016;375:1823–1833. doi: 10.1056/NEJMoa1606774. [DOI] [PubMed] [Google Scholar]
- 48.Ferris RL, Blumenschein G, Jr, Fayette J, Guigay J, Colevas AD, Licitra L, Harrington K, Kasper S, Vokes EE, Even C, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med. 2016;375:1856–1867. doi: 10.1056/NEJMoa1602252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Danaher P, Warren S, Lu R, Samayoa J, Sullivan A, Pekker I, Wallden B, Marincola FM, Cesano A. Pan-cancer adaptive immune resistance as defined by the Tumor Inflammation Signature (TIS): Results from The Cancer Genome Atlas (TCGA) J Immunother Cancer. 2018;6:63. doi: 10.1186/s40425-018-0367-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Koebel CM, Vermi W, Swann JB, Zerafa N, Rodig SJ, Old LJ, Smyth MJ, Schreiber RD. Adaptive immunity maintains occult cancer in an equilibrium state. Nature. 2007;450:903–907. doi: 10.1038/nature06309. [DOI] [PubMed] [Google Scholar]
- 51.Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: From immunosurveillance to tumor escape. Nat Immunol. 2002;3:991–998. doi: 10.1038/ni1102-991. [DOI] [PubMed] [Google Scholar]
- 52.Pagès F, Berger A, Camus M, Sanchez-Cabo F, Costes A, Molidor R, Mlecnik B, Kirilovsky A, Nilsson M, Damotte D, et al. Effector memory T cells, early metastasis, and survival in colorectal cancer. N Engl J Med. 2005;353:2654–2666. doi: 10.1056/NEJMoa051424. [DOI] [PubMed] [Google Scholar]
- 53.Zhang N, Bevan MJ. CD8(+) T cells: Foot soldiers of the immune system. Immunity. 2011;35:161–168. doi: 10.1016/j.immuni.2011.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Alspach E, Lussier DM, Schreiber RD. Interferon γ and its important roles in promoting and inhibiting spontaneous and therapeutic cancer immunity. Cold Spring Harb Perspect Biol. 2019;11:a028480. doi: 10.1101/cshperspect.a028480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu Y, Zhou N, Zhou L, Wang J, Zhou Y, Zhang T, Fang Y, Deng J, Gao Y, Liang X, et al. IL-2 regulates tumor-reactive CD8+ T cell exhaustion by activating the aryl hydrocarbon receptor. Nat Immunol. 2021;22:358–369. doi: 10.1038/s41590-020-00850-9. [DOI] [PubMed] [Google Scholar]
- 56.Zhang J, Endres S, Kobold S. Enhancing tumor T cell infiltration to enable cancer immunotherapy. Immunotherapy. 2019;11:201–213. doi: 10.2217/imt-2018-0111. [DOI] [PubMed] [Google Scholar]
- 57.Franklin RA, Liao W, Sarkar A, Kim MV, Bivona MR, Liu K, Pamer EG, Li MO. The cellular and molecular origin of tumor-associated macrophages. Science. 2014;344:921–925. doi: 10.1126/science.1252510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wu D, Liu X, Mu J, Yang J, Wu F, Zhou H. Therapeutic approaches targeting proteins in tumor-associated macrophages and their applications in cancers. Biomolecules. 2022;12:392. doi: 10.3390/biom12030392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dahlin AM, Henriksson ML, Van Guelpen B, Stenling R, Oberg A, Rutegård J, Palmqvist R. Colorectal cancer prognosis depends on T-cell infiltration and molecular characteristics of the tumor. Mod Pathol. 2011;24:671–682. doi: 10.1038/modpathol.2010.234. [DOI] [PubMed] [Google Scholar]
- 60.Badoual C, Hans S, Merillon N, Van Ryswick C, Ravel P, Benhamouda N, Levionnois E, Nizard M, Si-Mohamed A, Besnier N, et al. PD-1-expressing tumor-infiltrating T cells are a favorable prognostic biomarker in HPV-associated head and neck cancer. Cancer Res. 2013;73:128–138. doi: 10.1158/0008-5472.CAN-12-2606. [DOI] [PubMed] [Google Scholar]
- 61.Vassilakopoulou M, Avgeris M, Velcheti V, Kotoula V, Rampias T, Chatzopoulos K, Perisanidis C, Kontos CK, Giotakis AI, Scorilas A, et al. Evaluation of PD-L1 expression and associated tumor-infiltrating lymphocytes in laryngeal squamous cell carcinoma. Clin Cancer Res. 2016;22:704–713. doi: 10.1158/1078-0432.CCR-15-1543. [DOI] [PubMed] [Google Scholar]
- 62.Wang J, Wang S, Song X, Zeng W, Wang S, Chen F, Ding H. The prognostic value of systemic and local inflammation in patients with laryngeal squamous cell carcinoma. Onco Targets Ther. 2016;9:7177–7185. doi: 10.2147/OTT.S113307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Forssell J, Oberg A, Henriksson ML, Stenling R, Jung A, Palmqvist R. High macrophage infiltration along the tumor front correlates with improved survival in colon cancer. Clin Cancer Res. 2007;13:1472–1479. doi: 10.1158/1078-0432.CCR-06-2073. [DOI] [PubMed] [Google Scholar]
- 64.Jung M, Mertens C, Tomat E, Brüne B. Iron as a central player and promising target in cancer progression. Int J Mol Sci. 2019;20:273. doi: 10.3390/ijms20020273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Torti SV, Torti FM. Iron and cancer: More ore to be mined. Nat Rev Cancer. 2013;13:342–355. doi: 10.1038/nrc3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang Y, Yu L, Ding J, Chen Y. Iron metabolism in cancer. Int J Mol Sci. 2018;20:95. doi: 10.3390/ijms20010095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jiang Z, Lim SO, Yan M, Hsu JL, Yao J, Wei Y, Chang SS, Yamaguchi H, Lee HH, Ke B, et al. TYRO3 induces anti-PD-1/PD-L1 therapy resistance by limiting innate immunity and tumoral ferroptosis. J Clin Invest. 2021;131:e139434. doi: 10.1172/JCI139434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168:707–723. doi: 10.1016/j.cell.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jacolot S, Le Gac G, Scotet V, Quere I, Mura C, Ferec C. HAMPas a modifier gene that increases the phenotypic expression of the HFEpC282Y homozygous genotype. Blood. 2004;103:2835–2840. doi: 10.1182/blood-2003-10-3366. [DOI] [PubMed] [Google Scholar]
- 70.Casu C, Nemeth E, Rivella S. Hepcidin agonists as therapeutic tools. Blood. 2018;131:1790–1794. doi: 10.1182/blood-2017-11-737411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Armitage AE, Eddowes LA, Gileadi U, Cole S, Spottiswoode N, Selvakumar TA, Ho LP, Townsend AR, Drakesmith H. Hepcidin regulation by innate immune and infectious stimuli. Blood. 2011;118:4129–4139. doi: 10.1182/blood-2011-04-351957. [DOI] [PubMed] [Google Scholar]
- 72.Bessman NJ, Mathieu JRR, Renassia C, Zhou L, Fung TC, Fernandez KC, Austin C, Moeller JB, Zumerle S, Louis S, et al. Dendritic cell-derived hepcidin sequesters iron from the microbiota to promote mucosal healing. Science. 2020;368:186–189. doi: 10.1126/science.aau6481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vyoral D, Petrák J. Hepcidin: A direct link between iron metabolism and immunity. Int J Biochem Cell Biol. 2005;37:1768–1773. doi: 10.1016/j.biocel.2005.02.023. [DOI] [PubMed] [Google Scholar]
- 74.Mleczko-Sanecka K, Roche F, da Silva AR, Call D, D'Alessio F, Ragab A, Lapinski PE, Ummanni R, Korf U, Oakes C, et al. Unbiased RNAi screen for hepcidin regulators links hepcidin suppression to proliferative Ras/RAF and nutrient-dependent mTOR signaling. Blood. 2014;123:1574–1585. doi: 10.1182/blood-2013-07-515957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ren F, Yang Y, Wu K, Zhao T, Shi Y, Song M, Li J. The effects of dandelion polysaccharides on iron metabolism by regulating hepcidin via JAK/STAT signaling pathway. Oxid Med Cell Longev. 2021;2021:7184760. doi: 10.1155/2021/7184760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Saad HKM, Abd Rahman AA, Ab Ghani AS, Taib WRW, Ismail I, Johan MF, Al-Wajeeh AS, Al-Jamal HAN. Activation of STAT and SMAD signaling induces hepcidin re-expression as a therapeutic target for β-Thalassemia patients. Biomedicines. 2022;10:189. doi: 10.3390/biomedicines10010189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Joachim JH, Mehta KJ. Hepcidin in hepatocellular carcinoma. Br J Cancer. 2022;127:185–192. doi: 10.1038/s41416-022-01753-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kessler SM, Barghash A, Laggai S, Helms V, Kiemer AK. Hepatic hepcidin expression is decreased in cirrhosis and HCC. J Hepatol. 2015;62:977–979. doi: 10.1016/j.jhep.2014.10.046. [DOI] [PubMed] [Google Scholar]
- 79.Maegdefrau U, Arndt S, Kivorski G, Hellerbrand C, Bosserhoff A. Downregulation of hemojuvelin prevents inhibitory effects of bone morphogenetic proteins on iron metabolism in hepatocellular carcinoma. Lab Invest. 2011;91:1615–1623. doi: 10.1038/labinvest.2011.123. [DOI] [PubMed] [Google Scholar]
- 80.Scimeca M, Bonanno E. New highlight in breast cancer development: The key role of hepcidin and iron metabolism. Ann Transl Med. 2018;6((Suppl 1)):S56. doi: 10.21037/atm.2018.10.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tesfay L, Clausen KA, Kim JW, Hegde P, Wang X, Miller LD, Deng Z, Blanchette N, Arvedson T, Miranti CK, et al. Hepcidin regulation in prostate and its disruption in prostate cancer. Cancer Res. 2015;75:2254–2263. doi: 10.1158/0008-5472.CAN-14-2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhao B, Li R, Cheng G, Li Z, Zhang Z, Li J, Zhang G, Bi C, Hu C, Yang L, et al. Role of hepcidin and iron metabolism in the onset of prostate cancer. Oncol Lett. 2018;15:9953–9958. doi: 10.3892/ol.2018.8544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chen Q, Wang L, Ma Y, Wu X, Jin L, Yu F. Increased hepcidin expression in non-small cell lung cancer tissue and serum is associated with clinical stage. Thorac Cancer. 2014;5:14–24. doi: 10.1111/1759-7714.12046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Phipps O, Brookes MJ, Al-Hassi HO. Iron deficiency, immunology, and colorectal cancer. Nutr Rev. 2021;79:88–97. doi: 10.1093/nutrit/nuaa040. [DOI] [PubMed] [Google Scholar]
- 85.Colorectal cancer cells ectopically express hepcidin to sequester iron. Cancer Discov. 2021;11:OF2. doi: 10.1158/2159-8290.CD-RW2021-092. [DOI] [PubMed] [Google Scholar]
- 86.Schwartz AJ, Goyert JW, Solanki S, Kerk SA, Chen B, Castillo C, Hsu PP, Do BT, Singhal R, Dame MK, et al. Hepcidin sequesters iron to sustain nucleotide metabolism and mitochondrial function in colorectal cancer epithelial cells. Nat Metab. 2021;3:969–982. doi: 10.1038/s42255-021-00406-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sornjai W, Nguyen Van Long F, Pion N, Pasquer A, Saurin JC, Marcel V, Diaz JJ, Mertani HC, Smith DR. Iron and hepcidin mediate human colorectal cancer cell growth. Chem Biol Interact. 2020;319:109021. doi: 10.1016/j.cbi.2020.109021. [DOI] [PubMed] [Google Scholar]
- 88.Xiang-Tao P. Expression of hepcidin and neogenin in colorectal cancer. Open Med (Wars) 2017;12:184–188. doi: 10.1515/med-2017-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.