

Abstract
The protein kinase PINK1 and ubiquitin ligase Parkin promote removal of damaged mitochondria via a feed‐forward mechanism involving ubiquitin (Ub) phosphorylation (pUb), Parkin activation, and ubiquitylation of mitochondrial outer membrane proteins to support the recruitment of mitophagy receptors. The ubiquitin ligase substrate receptor FBXO7/PARK15 is mutated in an early‐onset parkinsonian–pyramidal syndrome. Previous studies have proposed a role for FBXO7 in promoting Parkin‐dependent mitophagy. Here, we systematically examine the involvement of FBXO7 in depolarization and mtUPR‐dependent mitophagy in the well‐established HeLa and induced‐neurons cell systems. We find that FBXO7−/− cells have no demonstrable defect in: (i) kinetics of pUb accumulation, (ii) pUb puncta on mitochondria by super‐resolution imaging, (iii) recruitment of Parkin and autophagy machinery to damaged mitochondria, (iv) mitophagic flux, and (v) mitochondrial clearance as quantified by global proteomics. Moreover, global proteomics of neurogenesis in the absence of FBXO7 reveals no obvious alterations in mitochondria or other organelles. These results argue against a general role for FBXO7 in Parkin‐dependent mitophagy and point to the need for additional studies to define how FBXO7 mutations promote parkinsonian–pyramidal syndrome.
Keywords: FBXO7, iNeurons, mitophagy, proteomics, ubiquitin ligase
Subject Categories: Autophagy & Cell Death, Neuroscience, Post-translational Modifications & Proteolysis
FBXO7/PARK15 appears to be dispensable for chemically induced PINK1/Parkin‐mediated mitophagy in various cell culture and iNeuron systems.
Introduction
Organelle quality control underlies cellular health and is often defective in disease and pathological states. Arguably, the best understood such quality control pathway is mitophagy, wherein damaged or supernumerary mitochondria are targeted for removal from the cell via selective autophagy (Pickrell & Youle, 2015; McWilliams & Muqit, 2017; Harper et al, 2018; Goodall et al, 2022). In this process, individual organelles are marked for sequestration within a double‐membrane vesicle called an autophagosome, which then fuses with a lysosome to facilitate the degradation of the organelle by resident lysosomal hydrolases (Stolz et al, 2014; Dikic & Elazar, 2018). Multiple forms of mitophagy have been reported, which differ in the types of regulatory mechanisms involved in marking the organelle for degradation, but these fall into two primary types—ubiquitin‐dependent and ubiquitin‐independent (Pickrell & Youle, 2015; Harper et al, 2018). Our understanding of ubiquitin‐dependent mitophagy has been advanced by the discovery of a signaling pathway composed of the PINK1 protein kinase and the Parkin (also called PRKN) ubiquitin (Ub) E3 ligase, which marks damaged mitochondria for elimination (Narendra et al, 2008). Parkin and PINK1 are both mutated in early‐onset recessive forms of Parkinson's disease and understanding how these enzymes work has been a major focus of the field (Pickrell & Youle, 2015; Ng et al, 2021).
In healthy mitochondria, PINK1 is imported into the mitochondrial translocon and rapidly processed for degradation (Jin et al, 2010; Yamano & Youle, 2013). In response to mitochondrial damage—such as depolarization, accumulation of mitochondrial misfolded proteins, or defects in mitochondrial fusion—PINK1 is stabilized on the mitochondrial outer membrane (MOM) in association with the translocon (Lazarou et al, 2012), where it can phosphorylate Ser65 on Ub already conjugated to proteins on the MOM (Kane et al, 2014; Koyano et al, 2014; Ordureau et al, 2014; Kazlauskaite et al, 2015; Wauer et al, 2015). Accumulation of phospho S65‐Ub (referred to henceforth as pUb) on the MOM promotes recruitment of Parkin—a pUb‐binding protein—thereby facilitating phosphorylation of Parkin on S65 of its N‐terminal Ub‐like (UBL) domain and activation of its Ub ligase activity (Kane et al, 2014; Ordureau et al, 2014; Kazlauskaite et al, 2015; Wauer et al, 2015; Gladkova et al, 2018; Sauve et al, 2018). Parkin then ubiquitylates numerous MOM proteins, resulting in both the accumulation of additional pUb and further MOM protein ubiquitylation (Sarraf et al, 2013; Bingol et al, 2014; Ordureau et al, 2020; Antico et al, 2021). MOM ubiquitylation then promotes recruitment of selective autophagy cargo receptors including OPTN, CALCOCO2 (also called NDP52), and SQSTM1 (also called p62) to facilitate the assembly of an autophagosome around the ubiquitylated organelle (Wong & Holzbaur, 2014; Heo et al, 2015; Lazarou et al, 2015; Evans & Holzbaur, 2020). Thus, PINK1, Parkin, and pUb function in a positive feedback loop to promote selective ubiquitylation and elimination of the damaged organelle (Goodall et al, 2022). Removal of damaged mitochondria may be an important aspect of neuronal protection in both neurodegenerative diseases and in aging, and understanding how the PINK1/Parkin pathway functions in this regard is a central goal of the field (Fang et al, 2019; Fleming et al, 2022).
Previous work (Burchell et al, 2013) concluded that FBXO7, the product of an early‐onset parkinsonian–pyramidal syndrome risk gene also designated as PARK15 (Di Fonzo et al, 2009; Paisan‐Ruiz et al, 2010; Houlden & Singleton, 2012), is a positive modulator of Parkin‐dependent mitophagy (Appendix Fig S1A). FBXO7 is a member of the F‐box family of proteins, which assemble with SKP1, CUL1, and RBX1 to form a modular SCF Ub ligase complex wherein the F‐box protein binds substrates (Jin et al, 2004). FBXO7 is characterized by an N‐terminal UBL domain, a central FP domain with structural similarity to a domain in the proteasome inhibitory factor PI31/PSMF1, followed by the F‐box motif, and a C‐terminal proline‐rich region (Kirk et al, 2008). Patient mutations in FBXO7 are found in multiple regions of the protein (Di Fonzo et al, 2009; Paisan‐Ruiz et al, 2010; Houlden & Singleton, 2012). Initial studies on FBXO7 proposed a functional interaction with the PINK1/Parkin pathway and concluded that FBXO7 functions as a positive regulator of mitophagy through direct interaction with Parkin and with PINK1 (Burchell et al, 2013). Depletion of FBXO7 by siRNA in cell lines was reported to reduce depolarization‐dependent loss in mitochondria based on Western Blotting of matrix proteins and likewise to reduce depolarization‐dependent ubiquitylation of MFN1, a substrate of Parkin (Burchell et al, 2013). However, studies that further substantiate a functional link between FBXO7 and PINK1/Parkin‐dependent mitophagy are lacking, although FBXO7 depletion has recently been reported to alter mitochondrial dynamics, mitochondrial ubiquitination threshold, or PINK1 levels (Huang et al, 2020; Liu et al, 2020; preprint: Al Rawi et al, 2022; preprint: Sanchez‐Martinez et al, 2022).
Given the strong genetic association of FBXO7/PARK15 with Parkinson's Disease (Houlden & Singleton, 2012) and major advances in our understanding of Parkin‐dependent mitophagy (Goodall et al, 2022) since initial links between FBXO7 and mitophagy were reported, we set out to elucidate how FBXO7 might function within the positive feedback loop to promote mitophagy (Appendix Fig S1A). Relevant advances include the identification of pUb as a sentinel response to PINK1/Parkin activation (Kane et al, 2014; Koyano et al, 2014; Ordureau et al, 2014; Kazlauskaite et al, 2015; Wauer et al, 2015), the development of highly quantitative mitophagic flux assays using a mitochondrially localized Keima fluorescent reporter protein (Katayama et al, 2011; Lazarou et al, 2015), and the development of genetically tractable iNeuron systems for functional analysis of the Parkin pathway in neuronal cell types (Ordureau et al, 2018, 2020). Unexpectedly, we find no clear evidence of a role for FBXO7 in the kinetics of depolarization‐dependent pUb accumulation, or mitophagic flux in either iNeurons or the conventional HeLa cell used extensively to examine PINK1/Parkin‐dependent mitophagy (Narendra et al, 2008). Moreover, global proteomics of FBXO7−/− HeLa cells undergoing mitophagy revealed no defect in mitochondrial elimination by autophagy. Taken together with previous studies that did not identify Parkin–FBXO7 interactions during mitophagy (Sarraf et al, 2013), these data indicate that FBXO7 does not play a general role as a positive modulator of PINK1/Parkin‐dependent mitophagy in HeLa and iNeuron systems. This work suggests that elucidation of FBXO7 biochemical functions is needed to understand how it might contribute to suppression of early‐onset parkinsonian–pyramidal syndrome.
Results
A tool kit for functional analysis of cells lacking FBXO7
To test if FBXO7 is a pivotal amplifier of the PINK1/Parkin pathway, we employed CRISPR‐Cas9‐based gene editing to generate knockouts for FBXO7 in HeLa HFT/TO‐PRKN cells (Ordureau et al, 2015) as well as HEK293T cells and confirmed insertion of a frameshift mutation by sequencing and Western Blotting (Appendix Fig S1B–F). HeLa HFT/TO‐PRKN cells contain a DOX‐inducible PRKN cassette allowing regulated expression of Parkin to facilitate an analysis of mitophagic flux in response to mitochondrial damage (Ordureau et al, 2015). Three independent HeLa HFT/TO‐PRKN FBXO7−/− clones were identified (clones 27, 41, and 45; Appendix Fig S1C and D). Two independent HEK293T cell lines lacking FBXO7 were transduced with a lentivirus for stable expression of GFP‐Parkin to facilitate pathway activation (Appendix Fig S1E and F). In parallel, we generated two independent human ES cell clones lacking FBXO7 via CRISPR‐Cas9 (clones 52 and 89) (Appendix Fig S1G–I). Here, an ES cell line harboring a DOX‐inducible NGN2 gene was used, allowing for conversion of these cells to cortical‐like iNeurons over a 2 week or greater time course (Zhang et al, 2013; Ordureau et al, 2020). We have previously demonstrated that iNeurons derived from these cells exhibit PINK1/Parkin‐dependent mitophagic flux in response to mitochondrial damage (Ordureau et al, 2020). Both clones displayed normal karyotypes and the absence of FBXO7 was verified using MiSeq sequencing (Appendix Fig S1H and I).
Robust depolarization‐dependent Ub phosphorylation by PINK1 in cells lacking FBXO7
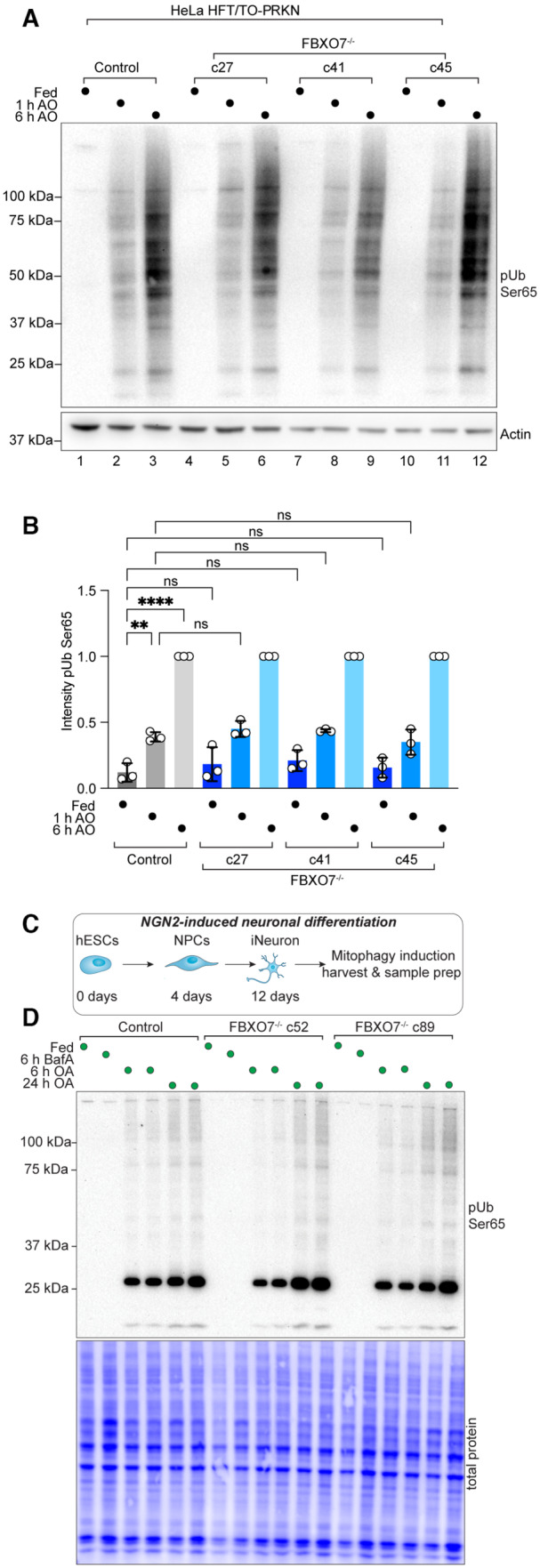
The earliest known event in Parkin‐dependent mitophagy is accumulation of PINK1 on the mitochondrial outer membrane followed by rapid S65 phosphorylation of pre‐existing Ub linked with mitochondrial proteins (Appendix Fig S1A), as has been worked out in detail using the well‐validated HeLa cell system with ectopically expressed Parkin (Narendra et al, 2010; Lazarou et al, 2012; Yamano & Youle, 2013; Kane et al, 2014). In the presence of Parkin, Ub phosphorylation is enhanced as a result of increased Ub conjugation to mitochondrial proteins, thereby providing additional substrate molecules for phosphorylation by PINK1 (Kane et al, 2014; Koyano et al, 2014; Ordureau et al, 2014). To examine pUb accumulation in the context of HeLa cells lacking FBXO7, we induced Parkin expression (16 h) and then depolarized mitochondria for 1 or 6 h with antimycin A/Oligomycin A (AO), which inhibit Complex III and Complex IV activity in the electron transport chain, respectively. As expected, control HeLa cells display time‐dependent accumulation of pUb as measured by Western Blotting, but the absence of FBXO7 had no impact on the extent of pUb accumulation, as assessed in all three FBXO7−/− cell lines in biological triplicate analyses (Fig 1A and B). In addition, we used CCCP (Carbonyl cyanide m‐chlorophenyl hydrazone) as an alternative to induce mitophagy and found pUb levels in either HeLa and HEK293T cells were similar in control and FBXO7−/− cells (Appendix Fig S1J and K).
Figure 1. Robust depolarization‐dependent Ub phosphorylation by PINK1 cells lacking FBXO7.
-
AWestern Blot for pUb on HeLa control and FBXO7−/− cells after treatment with AO for indicated times.
-
BQuantification of pUb signal relative to loading. Western Blots run in triplicates from three independent clones. Error bars depict SD. Data from three biological replicates. One‐way ANOVA with multiple comparisons. **P = 0.0011; ****P < 0.0001.
-
CScheme depicting production of iNeurons for biochemical analysis.
-
DWestern Blot for pUb on extracts from day 12 iNeuron control and FBXO7−/− cells after treatment with BafA or AO for indicated times.
Similarly, we assessed depolarization‐dependent pUb accumulation in the iNeuron system. control or FBXO7−/− ES cells were converted to day 12 iNeurons and depolarized with AO in duplicate (Fig 1C). pUb was detected by Western Blotting at 6 and 24 h and was indistinguishable between WT and two independent FBXO7−/− clones (Fig 1D). Taken together, these data do not support a universal role for FBXO7 in promoting the earliest step in mitophagy signaling—PINK1‐dependent Ub phosphorylation—in the HeLa cell system or iNeurons.
Super‐resolution imaging of pUb formation independent of FBXO7
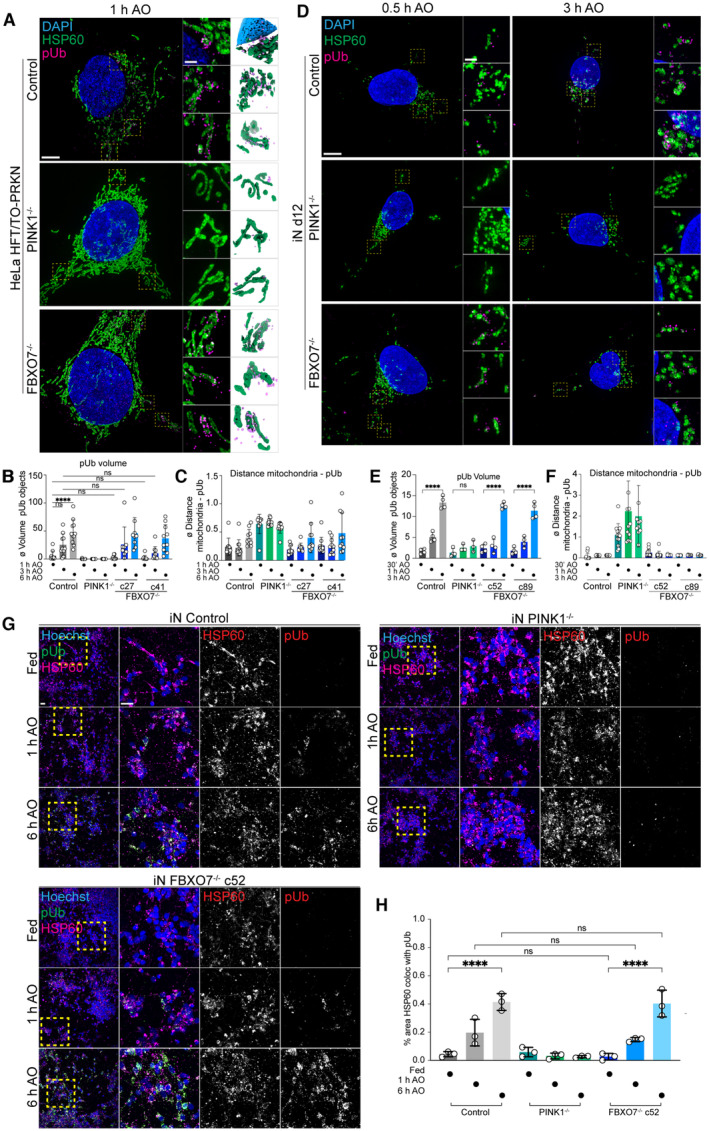
While depolarization‐dependent Ub phosphorylation was not impaired, we considered the possibility that the organization of pUb on damaged mitochondria might be altered in cells lacking FBXO7. To our knowledge, super‐resolution microscopy has not been used to examine the kinetics and spatial properties of pUb accumulation in response to depolarization. We, therefore, optimized conditions to monitor pUb via volumetric 3D‐SIM super‐resolution microscopy in both HeLa and iNeuron systems (see Materials and Methods). After AO treatment of HeLa cells for 1, 3, or 6 h, the mitochondria matrix protein HSP60 and pUb were examined by super‐resolution imaging. In control cells at 1 h, fragmented mitochondria were often proximal to pUb foci with an average volume of 5.71 μm3, which were not observed in cells lacking PINK1 (Figs 2A–C and EV1). The volume of pUb increased ~ 8‐fold over 6 h (47.88 μm3), consistent with the known feed‐forward response (Figs 2B and EV1). At 6 h of AO treatment, we observed a near complete coating of mitochondrial fragments with pUb, as quantified by the significant increase in pUb object volume (Figs 2B and C and EV1). Consistent with analysis of pUb by Western Blotting, two FBXO7−/− clones did not show any significant differences in pUb recruitment, volume, or overall morphological differences compared with the control cells, indicating that FBXO7 is not required for the feed‐forward amplification of PINK1/Parkin mitophagy after AO‐treatment (Fig 2A–C). Analogous super‐resolution experiments in day 12 iNeurons with 0.5, 1, and 3 h of AO revealed similar results, with no obvious effect of FBXO7 deletion on pUb recruitment, volume, or overall morphology when compared with WT iNeurons (Fig 2D–F; Appendix Fig S2A). As expected (Ordureau et al, 2018, 2020), iNeurons lacking PINK1 did not accumulate pUb (Fig 2D and E). These results were also confirmed in iNeurons treated with AO for 1 or 6 h using conventional immunofluorescence and image analysis of HSP60 and pUb co‐localization (Fig 2G and H). To ensure that kinetic effects were not obscured by overt depolarization, we reduced the concentrations of AO used by ~ 10‐fold, but again no defect in pUb recruitment to mitochondria was observed (Appendix Fig S2B and C). These data indicate that the absence of FBXO7 does not alter the ability of PINK1‐dependent pUb foci to accumulate on damaged mitochondria in either HeLa or iNeuron systems.
Figure 2. Super‐resolution imaging of pUb formation independent of FBXO7.
-
A3D‐SIM images of HeLa control, PINK1−/− and FBXO7−/− cell lines after AO‐induced mitophagy. Cells were stained for nuclear DNA (DAPI), mitochondria (HSP60) and pUb. Zoom‐ins of regions of interested are enlarged in the middle panel. 3D‐surface renderings of insets are shown on the right. Scale bar = 5 or 1 μm.
-
B, CEvaluation of 3D‐SIM images from HeLa datasets. The changes in pUb volume and minimal distances between mitochondria and pUb after mitophagy‐induction are plotted. Error bars depict SD from 8 to 14 measured cells per condition. Two‐way ANOVA with multiple comparisons; ****P < 0.0001.
-
D3D‐SIM images of iN day 12 control, PINK1−/− and FBXO7−/− cell lines after AO‐induced mitophagy. Cells were stained for nuclear DNA (DAPI), mitochondria (HSP60) and pUb. Zoom‐ins of regions of interest are enlarged in the middle panel. 3D‐surface renderings of insets are shown on the right. Scale bar = 5 μm or 1 μm.
-
E, FEvaluation of 3D‐SIM images from iNeuron datasets. The changes in pUb volume and minimal distances between mitochondria and pUb after mitophagy‐induction are plotted. Error bars depict SD from 7 to 14 measured cells per condition. One‐way ANOVA with multiple comparisons, ****P < 0.0001.
-
GConfocal images of iNeuron d12 Control, PINK1−/− and FBXO7−/− cell lines after AO‐induced mitophagy. Cells were stained for nuclear DNA (Hoechst33342), mitochondria (HSP60) and pUb. Scale bar = 10 μm and 5 μm.
-
HEvaluation of pUb volume after mitophagy induction. Error bars depict SD from three biological replicates. One‐way ANOVA with multiple comparisons; ****P < 0.0001.
Figure EV1. Super‐resolution pUb detection in HeLa cells in response to mitochondrial depolarization.
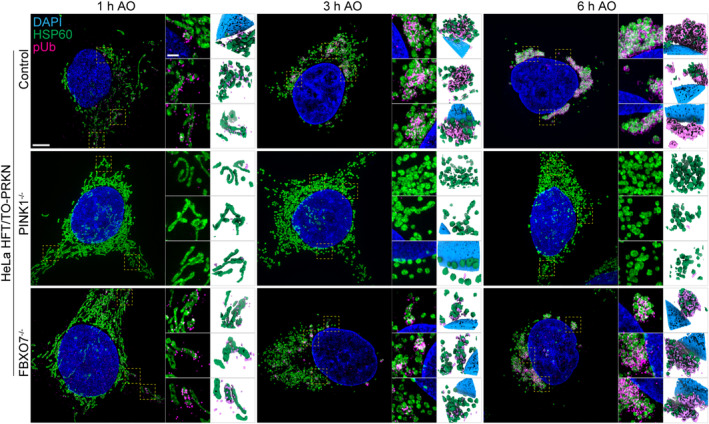
3D‐SIM images of HeLa control, PINK1−/− and FBXO7−/− cell lines after 1 h, 3 h, or 6 h, AO‐induced mitophagy (related to Fig 2A and B). Cells were stained for nuclear DNA (DAPI), mitochondria (HSP60) and pUb. Note that 1 h AO panel is reused from Fig 2A for side‐by‐side comparison. Scale bar = 5 μm or 1 μm.
Parkin and autophagy regulator recruitment in FBXO7 −/− cells
Parkin recruitment depends upon the accumulation of pUb on damaged mitochondria (Appendix Fig S1A; Okatsu et al, 2015). Given that the existing model for FBXO7 in Parkin‐dependent mitophagy posited that Parkin and FBXO7 physically interact and that FBXO7 promotes Parkin function (Burchell et al, 2013), we directly tested whether FBXO7 is required for Parkin recruitment to mitochondria using the HeLa cell system. Control or FBXO7−/− cells were stably transduced with a lentivirus expressing GFP‐Parkin and subjected to imaging with or without depolarization with AO (Fig EV2A). We used CellProfiler (Stirling et al, 2021) to analyze the characteristics of GFP‐Parkin and mitochondria and their relationship to each other. By segmenting out cytosolic and mitochondrially localized Parkin, we could quantify alterations in Parkin abundance within the mitochondrial mask as a measure of Parkin‐translocation (Fig EV2B). Our expectation was that a requirement for FBXO7 in Parkin recruitment would result in reduced Parkin intensity within the mitochondrial mask in response to depolarization. First, we tested whether mitochondrial morphology (mitochondrial integrated density [==mtIntDen] as proxy, stained with HSP60) is changing after AO‐treatment on a single‐cell level. Under fed conditions, control and three FBXO7−/− clones displayed comparable mtIntDen. Depolarization by AO led to the expected mitochondrial fragmentation and clustering, thus causing an increase in local HSP60 signal and a right‐shift in mtIntDen (Fig EV2C, top panel). This shift was also observed to a similar extent in FBXO7−/− cells. Thus, both control and FBXO7−/− cells robustly respond to AO‐induced mitophagy. Next, we tested if Parkin intensities are increasing on mitochondria on an “organelle object” level. As expected, mtIntDen did shift to the right after AO‐treatment, in line with previous experiments. Parkin signal was comparable between the genotypes under fed conditions, and increased in intensity after AO‐treatment, consistent with Parkin accumulation inside the mitochondrial area (Fig EV2C, lower panel). This Parkin shift was observed in both control and FBXO7−/− cell lines, arguing that FBXO7 is not universally required for Parkin recruitment to damaged mitochondria.
Figure EV2. Parkin and autophagy regulator recruitment in FBXO7−/− cells.
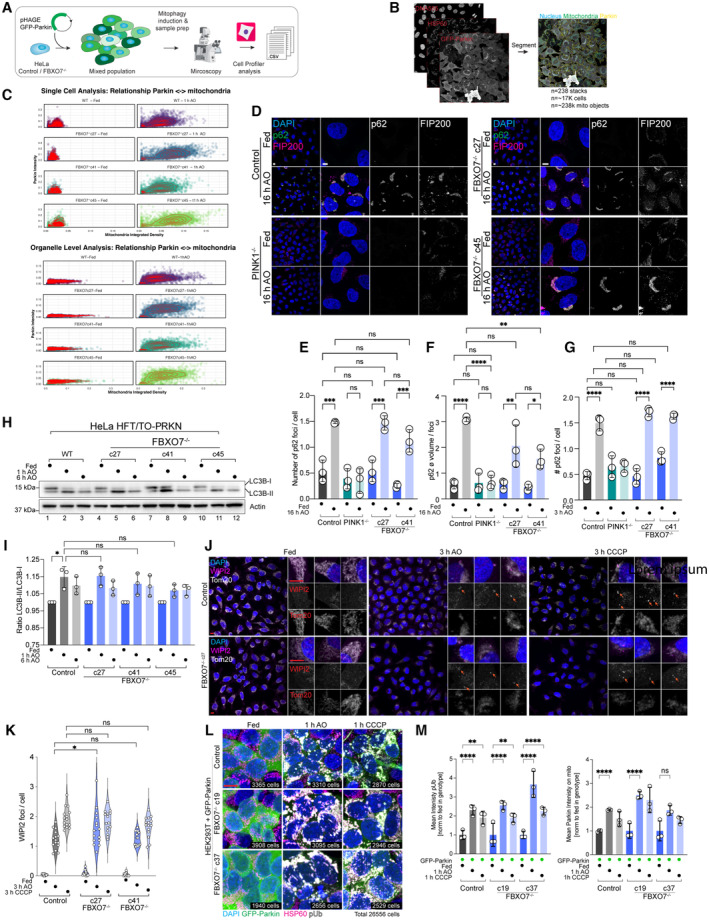
-
AOverview of methods for analysis of Parkin recruitment to damaged mitochondria.
-
BCells were imaged for GFP‐Parkin, nuclei, and mitochondria and segmented to facilitate analysis of Parkin recruitment to mitochondria in single cells/mitochondrial organelles.
-
CStd mitochondrial signal vs. Parkin intensity and Std Parkin vs. mitochondria with or without 1 h AO‐induced mitophagy are shown. Data from three replicates, including a total of 238 image stacks, containing 16,888 cells and 237,848 mitochondrial objects.
-
DRecruitment of p62 to mitochondria (not stained) in control or FBXO7−/− HeLa cells with or without treatment with AO (16 h) was examined by confocal imaging. Scale bar = 10 and 5 μm.
-
E, FQuantification of cells in panel (D). Assays were performed in biological triplicate with 4 image stacks taken per repeat. n = 6,712 cells. Error bars depict SD from triplicate experiments from two independent clones. One‐way ANOVA with multiple comparisons; ***P = 0.0002, *P = 0.0355, **P = 0.0029, ****P < 0.0001.
-
GQuantification of number of p62 foci normalized per cell in control, PINK1−/− and FBXO7−/− cells with or without 3 h AO treatment. Assays were performed in biological triplicate with 4 image stacks taken per repeat. n = 12,646 cells. Error bars depict SD from triplicate experiments from two independent clones. One‐way ANOVA with multiple comparisons; ****P < 0.0001.
-
HControl or FBXO7−/− HeLa cells expressing Parkin were either left untreated or incubated with AO for 1 or 6 h and extracts subjected to Western Blotting with α‐LC3B and α‐Actin as a loading control.
-
IThe ratio of LC3B lipidation (LC3B‐II/LC3B‐I) was quantified from triplicate experiments from three independent clones. One‐way ANOVA with multiple comparisons. *P = 0.0104.
-
JImmunofluorescence staining of WIPI2 (magenta) and Tom20 (gray) in HeLa Control and FBXO7−/− cells after mitophagy induction. Arrows depict WIPI2 foci on the mitochondrial staining. Scale bar = 10 μm (overview) and 10 μm (insets).
-
KEvaluation of mitochondrially localized WIPI2 foci per cell based on images shown in (J). Data based on 15 image stacks from three technical replicates of two independent clones. One‐way ANOVA with multiple comparisons. *P = 0.0331.
-
LImmunofluorescence staining of HEK293T Control and FBXO7−/− cells expressing GFP‐Parkin for HSP60 (mitochondria, magenta), pUb (gray) and GFP (Parkin, green) after mitophagy induction for indicated times. Scale bar = 10 μm. Number of individual cells analyzed per condition is indicted in the right bottom of each image.
-
MAnalysis of pUb intensity (left) and Parkin intensity (right) in the mitochondrial mask. Error bars depict SEM from three biological replicates from two independent clones. 2‐way ANOVA with multiple comparisons; **P = 0.001, ****P < 0.0001.
Mitochondrial ubiquitylation by Parkin promotes recruitment of downstream autophagy machinery, including the FIP200‐ULK1 complex and Ub‐binding autophagy adaptors, ultimately leading to the formation of an autophagosome (Heo et al, 2015; Lazarou et al, 2015; Ravenhill et al, 2019; Vargas et al, 2019). Lipidation of LC3 is associated with autophagosome formation and is thought to contribute to recruitment of autophagy receptors (Chang et al, 2021). Depolarization of Parkin‐expressing HeLa cells resulted in recruitment of both p62 and FIP200 to mitochondria in a PINK1‐depenent manner, as revealed by immunostaining and confocal imaging (Fig EV2D). However, FBXO7−/− cells were as proficient as control cells in recruitment of p62 or FIP200 after 3 h or 16 h or AO‐induced mitophagy (Fig EV2D–G). Previous studies using siRNAs targeting FBXO7 also reported that LC3 lipidation in response to mitochondrial depolarization is defective compared with non‐depleted cells (Burchell et al, 2013). However, all three FBXO7−/− HeLa cell clones underwent depolarization‐dependent LC3 lipidation to similar extents, consistent with a functional Parkin pathway (Fig EV2H and I). Taken together, these data indicate that FBXO7 is not universally required to promote either Parkin‐dependent recruitment to damaged mitochondria or subsequent steps that depend upon Ub chain assembly on mitochondria such as recruitment of autophagy machinery and initiation of LC3 lipidation.
To probe for early mitophagy initiation sites, we treated HeLa control and FBXO7−/− cells with 3 h AO or CCCP and stain for the mitochondrial protein Tom20 and WIPI2 (WD repeat domain phosphoinositide‐interacting protein 2; Katayama et al, 2011; Lazarou et al, 2015). Loss of FBXO7 did not lead to a loss of mitophagy initiation, as WIPI2 foci were detectable in Control and FBXO7−/− cell lines (Fig EV2J). Evaluation of WIPI2 foci normalized per cell found no difference in foci number between Control and two FBXO7−/− clones after 3 h CCCP treatment and a significant difference in only one of the two clones when treated with AO (Fig EV2J and K). Next, we repeated our GFP‐Parkin and pUb translocation measurement after mitophagy induction in HEK293T Control and FBXO7−/− cells stably expressing GFP‐Parkin. Both independent knockout clones displayed a robust response to 1 h AO or CCCP, with both Parkin and pUb accumulating on mitochondria (Fig EV2L). Quantifying the average pUb intensity showed a significant increase in signal in Control and both knockout clones, supporting the notion that FBXO7 is not essential for PINK1/Parkin mitophagy (Fig EV2M). Taken together, our data from HeLa and HEK293T cell lines suggest that FBXO7 is not essential for the initiation and propagation of mitophagy.
Interaction network analysis of FBXO7 −/−
In addition, we have not been able to reproduce a previously reported association between overexpressed FBXO7 and PINK1 (Huang et al, 2020) in either HEK293T or HCT116 cells. Briefly, as part of our BioPlex project (Huttlin et al, 2021), we ectopically expressed PINK1‐HA and performed interaction proteomics. While we identified the known protein kinase chaperone HSP90 subunits in association with PINK1, we did not detect FBXO7 (Fig EV3A; Dataset EV1). Likewise, FBXO7 associated reciprocally with members of the CUL1‐SKP1‐RBX1, the COP9/Signalosome, and both core particle and regulatory subunits of the proteasome in HEK293T and/or HCT116 cells, as expected (Bader et al, 2011; Vingill et al, 2016; Liu et al, 2019), but neither PINK or Parkin were detected (Fig EV3B; Dataset EV2) (Huttlin et al, 2021) (see Materials and Methods). These results are consistent with our previous Parkin interaction proteomics experiments that failed to demonstrate an interaction with FBXO7 during mitophagy induction (Sarraf et al, 2013). Among FBXO7 interacting proteins is PSMF1 (also called PI31). We found that purified SKP1–FBXO7 complexes (with or without CUL1 N‐terminal domain [NTD]) can directly associate with PSMF1, as examined using size exclusion chromatography and analysis of fractions by SDS–PAGE (Fig EV3C and D). To our knowledge, this is the first demonstration that FBXO7 assembled with SKP1/CUL1 can bind directly to PSMF1.
Figure EV3. Interaction analysis of FBXO7.
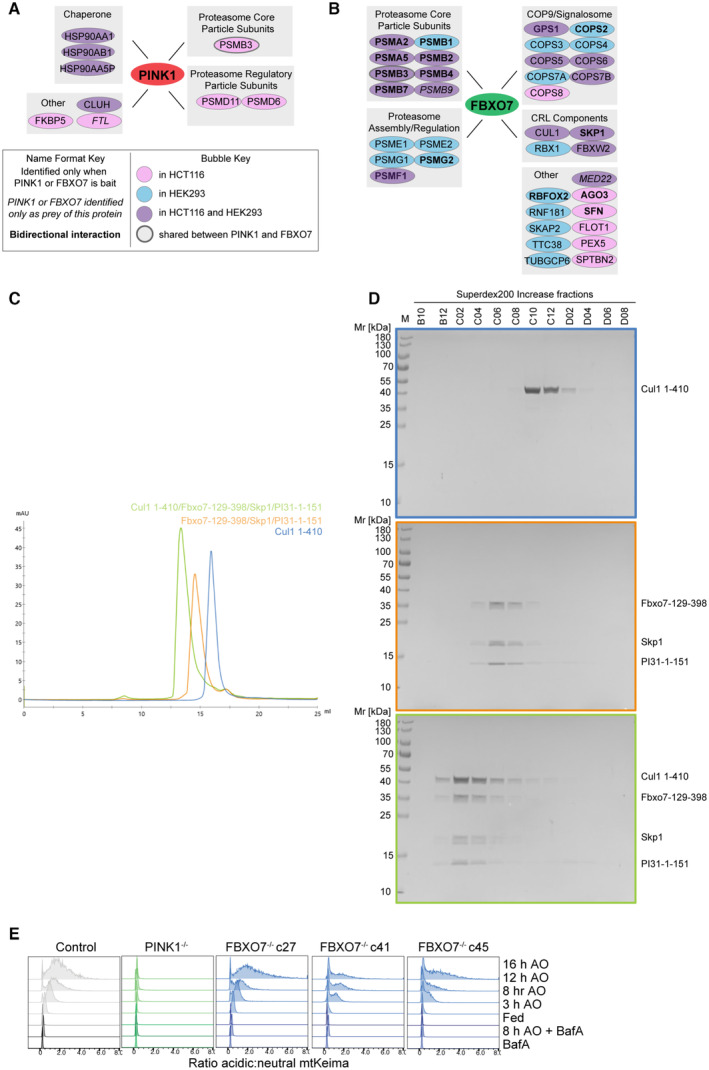
-
AInteraction network analysis of PINK1 (left) and FBXO7 (right) based on interaction proteomics data from our BioPlex Interactome (Huttlin et al, 2021) (see Materials and Methods for details). Figure key can be found beneath panel.
-
BSummary of major interactions observed for FBXO7 in the context of the relevant protein complexes.
-
CSize‐exclusion chromatography traces of indicated FBXO7 complexe on Superdex 200 Increase 10/300 GL.
-
DSDS–PAGE gel analysis of fractions of SKP1‐FBXO7 complexes. Box colors match traces from (C).
-
ERidgeline plots of mtKeima‐shift analysis by flow cytometry in HeLa control, PINK1−/− and FBXO7−/− cell lines treated with AO at the indicated times. All lines are normalized to the BafA sample.
Mitophagic flux in FBXO7 −/− cells
A significant advance has been the development of mitophagy reporters, which cumulatively measure flux of mitochondrial turnover through to the final stages of mitophagy: the fusion of mitochondria‐laden autophagosomes with lysosomes (Lazarou et al, 2015). mKeima is a fluorescent protein that undergoes a pH‐dependent Stokes shift, and delivery of a Keima‐tagged cargo from the cytoplasm to the acidic interior of the lysosome can be monitored by measuring the ratio of emission at 620 nm with maximal excitation at 440 or 586 nm under neutral or acidic conditions, respectively, by flow cytometry or microscopy (Fig 3A). To monitor differences in mitophagic flux upon deletion of FBXO7, we employed mtKeima, an mKeima protein targeted to the mitochondria via a COX8‐matrix targeting sequence (Ordureau et al, 2020).
Figure 3. Mitophagic flux in iNeurons and HeLa cells lacking FBXO7.
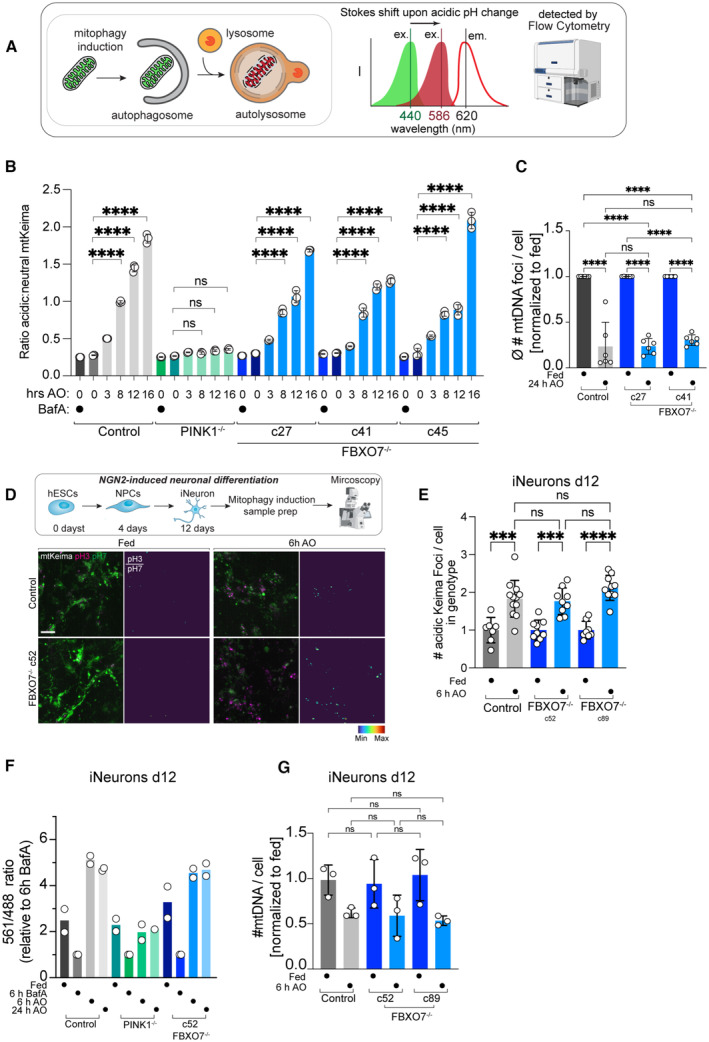
-
AScheme depicting mitophagic flux assay using mtKeima. See text for details.
-
BMean Acidic:Neutral mtKeima per‐cell ratios measured by flow cytometry for HeLa cells expressing Parkin ndicating the number of hours treated with AO (Antimycin A [5 μM] and Oligomycin [10 μM]); or 3 h with 25 nM BafilomycinA (BafA). Error bars depict SD from biological triplicate measurements from three independent clones. Two‐way ANOVA with multiple comparisons; ****P < 0.0001.
-
CBar graph showing the number of mtDNA puncta/cell with or without treatment with AO (6 h) in HeLa control or FBXO7−/− cells. Error bars depict SD from triplicate technical replicates from two independent clones. One‐way ANOVA with multiple comparisons; ****P < 0.0001.
-
DIndicated hESCs expressing mtKeima were differentiated to iNeurons (day 12) and were subjected to image analysis. Scale bar, 5 μm.
-
EThe number of red‐shifted Keima foci per cell is plotted, where each dot represents the average foci‐number per image stack, originating from 7 to 12 image stacks of iNeuron differentiations. Error bars represent SD. One‐way ANOVA with multiple comparisons; ***P = 0.0001, ****P < 0.0001.
-
FmtKeima mitophagy flux readout of indicated iNeuron genotypes. Cells were treated with AO for either 1 or 6 h and mitophagic flux measured by flow cytometry (> 10,000 cells). Pooled data from biological replicates is shown, normalized to 6 h BafA treated cells.
-
GBar graph showing the number of mtDNA puncta/cell with or without treatment with AO (6 h) in control or FBXO7−/− day 12 iNeurons. Error bars depict SD from biological triplicate measurements from two independent clones and three differentiations. One‐way ANOVA with multiple comparisons.
In Parkin‐expressing HeLa cells, the ratio of acidic‐Keima (measured with excitation at 561 nm and emission 620 nm) to neutral Keima (ex. 405 nm, em. 603 nm) increases as cells are treated with AO (Figs 3B and EV3E). This mitophagic flux is PINK1‐dependent as deletion of PINK1 abolishes this shift (Figs 3B and EV3E). As expected, there is little flux without depolarization with AO, and inhibition of the lysosomal V‐ATPase with BafilomycinA1 (BafA) restores the mtKeima acidic:neutral ratio to levels similar to that observed in fed cells (Fig EV3E). In three FBXO7−/− clones, AO‐dependent mitophagic flux is similar to control cells with increasing flux observed with increasing length of AO treatment (Figs 3B and EV3E). We validated the mtKeima flux results using the mitochondrial DNA clearance assay (Heo et al, 2015; Lazarou et al, 2015). Staining for mtDNA foci per cell after 24 h AO treatment in control and FBXO7−/− HeLa cells showed a significant decrease in mtDNA number in all samples (Fig 3C). The remaining mtDNA levels were indistinguishable between control and FBXO7−/− cells (Fig 3C).
We also measured mtKeima foci and flux in control and FBXO7−/− iNeurons at day 12 of differentiation (Fig 3D–G). As with HeLa cells, red‐shifted mtKeima foci increased ~ 2‐fold after 6 h AO treatment in control and FBXO7−/− cells (Fig 3D and E). Using flow cytometry, mitophagic flux (normalized to BafA) after 6 and 24 h AO treatment was comparable between control and FBXO7−/− iNeurons, and was absent in PINK1−/− cells (Fig 3F). Additionally, we investigated mtDNA turnover in d12 control and two independent FBXO7−/− iNeuron clones. Within 6 h of depolarization of iNeurons, we observed a reduction in the number of mitochondrial DNA/cell, and the effect was independent of the presence or absence of FBXO7 (Fig 3G). Taken together, these data indicate that FBXO7 is not universally required for depolarization‐dependent mitophagic flux in either the HeLa or iNeuron systems.
Proteomic analysis of HeLa cells lacking FBXO7 during mitophagy
As an alternative to mtKeima for measuring mitophagic flux, we examined the total proteome of depolarized HeLa cells with the expectation that in control HeLa cells, mitochondrial turnover would result in bulk loss of the mitochondrial proteome in depolarized cells. Thus, control or FBXO7−/− HeLa cells expressing Parkin were depolarized for 16 h in triplicate and total cell extracts subjected to 18‐plex Tandem Mass Tagging (TMT)‐based proteomics (Fig 4A; Dataset EV3 and EV4) (Li et al, 2021). Replicates were highly correlated, with correlation coefficients greater that 0.95 and Principal Component Analysis (PCA) revealed tight clustering of replicates, with the major feature separating the samples being AO‐treatment (Appendix Fig S3A and B). Through hierarchical clustering (Appendix Fig S3C), we identified major alterations in the abundance of mitochondria in response to depolarization (Fig 4B; Appendix Fig S3D–I). Indeed, the majority of proteins annotated as mitochondrial were found to have reduced levels in control cells in response to depolarization, as indicated in the overlap of mitochondrial proteins on the volcano plots for the ~ 8,000 proteins quantified (leftward skew of red dots in Fig 4B, left panel). Importantly, the patterns of protein abundance were indistinguishable from control for two independent FBXO7−/− cell lines, with a leftward skew of mitochondrial proteins in the volcano plot (Fig 4B and C). In similar fashion, ~ 4.3% of detected mitochondrial proteins were significantly more abundant after AO treatment across all genotypes, compared with ~ 67.9% of significantly decreased proteins. The plurality (49%) of proteins with significantly decreased abundance was shared between both knockout clones and the KO, supporting a robust mitophagy response even in the absence of FBXO7 (Appendix Fig S3J and K). When calculating the difference (delta = ∆ log2 FC [16 h AO/Fed]) between FBXO7−/− and Control for each detected protein and compare the ranked abundance of mitochondrial vs. all other proteins, mitochondrially localized proteins are evenly distributed across the whole range, arguing that the proteome of both Control and FBXO7−/− cells remodel in similar fashion after mitophagy treatment (Appendix Fig S3L, green dots).
Figure 4. Proteomic analysis of HeLa cells lacking FBXO7 during mitophagy.
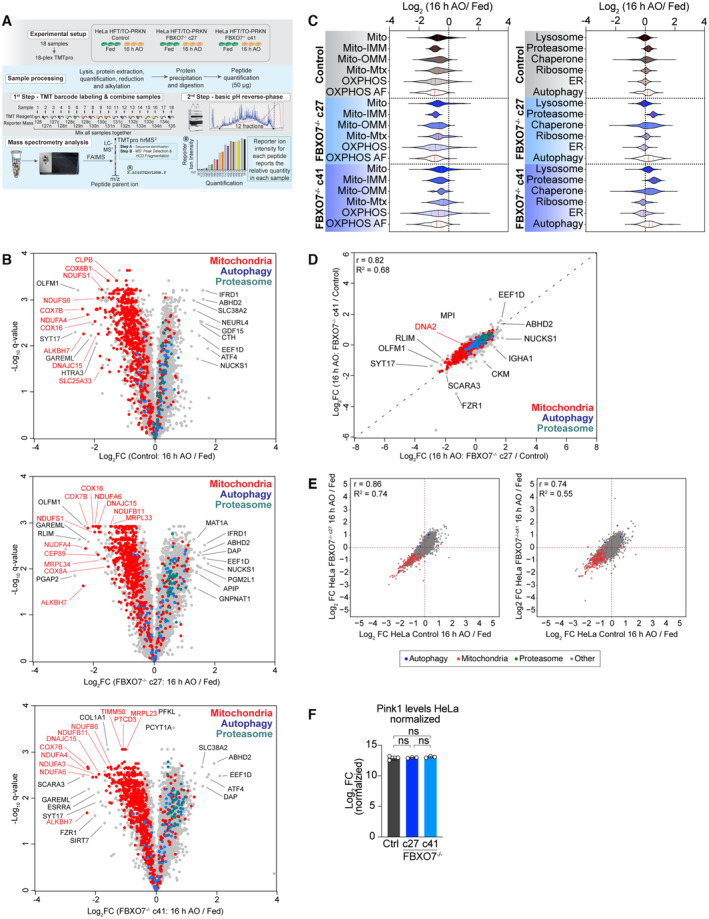
-
AWorkflow for analysis of total protein abundance in HeLa cells expressing Parkin with and without depolarization with AO (16 h). Cell extracts from triplicates were digested with trypsin prior to 18‐plex TMT labeling and analysis by mass spectrometry.
-
BVolcano plots [Log2 FC (16 h AO/Fed) versus ‐Log10(q‐value)] for control or FBXO7−/− cell lines with or without treated with AO (16 h). Red dots, mitochondrial proteins; blue dots, autophagy proteins; green dots, proteasome proteins; gray dots, remainder of the proteome.
-
CViolin plots [Log2 (16 h AO/Fed)] of control or FBXO7−/− cells depicting alterations in the abundance of mitochondrial protein (left panel) or specific organelles or protein complexes (right panel). Red line in violin plot depicts the median, dotted red lines depict the 25 and 75 quartiles. AF, assembly factors; ER, endoplasmic reticulum; IMM, inner mitochondrial membrane; Mtx, mitochondrial matrix; OMM, outer mitochondrial membrane; OXPHOS, Oxidative Phosphorylation.
-
DCorrelation plot of c27 and c41 FBXO7−/− clones. Log2FC (16 h AO for each clone relative to control cells) is plotted.
-
ECorrelation plots [Log2 FC (16 h AO/Fed)] of the proteome of FBXO7−/−c27 or c41 clones against control. Red dots, mitochondrial proteins; blue dots, autophagy proteins; gray dots, remainder of the proteome.
-
FPINK1 levels in control cells and in two FBXO7−/− clones were measured by TMT‐proteomics in fed cells, from biological triplicates from two independent clones. Error bars depict SD.
Proteins with altered abundance were enriched for mitochondrial matrix, inner membrane, outer membrane and oxidative phosphorylation categories, consistent with autophagy of entire organelles (Fig 4C). The behavior of other organelles and large protein complexes were similar for both control and FBXO7−/− cells (Fig 4C). Moreover, both FBXO7 clones displayed similar behavior to each other and to the control, as indicated by correlation analysis (Pearson's R = 0.82, 0.86, and 0.74, respectively; Fig 4D and E). These results are consistent with the absence of a discernible alteration in depolarization‐dependent mitophagic flux in cells lacking FBXO7. A previous report proposed that depletion of FBXO7 resulted in accumulation of PINK1 proteins levels under basal conditions (Huang et al, 2020), but in our experiments, PINK1 levels in the two FBXO7−/− clones were unaffected based on TMT intensities (Fig 4F).
Proteomic analysis of human ES cells lacking FBXO7 during neurogenesis
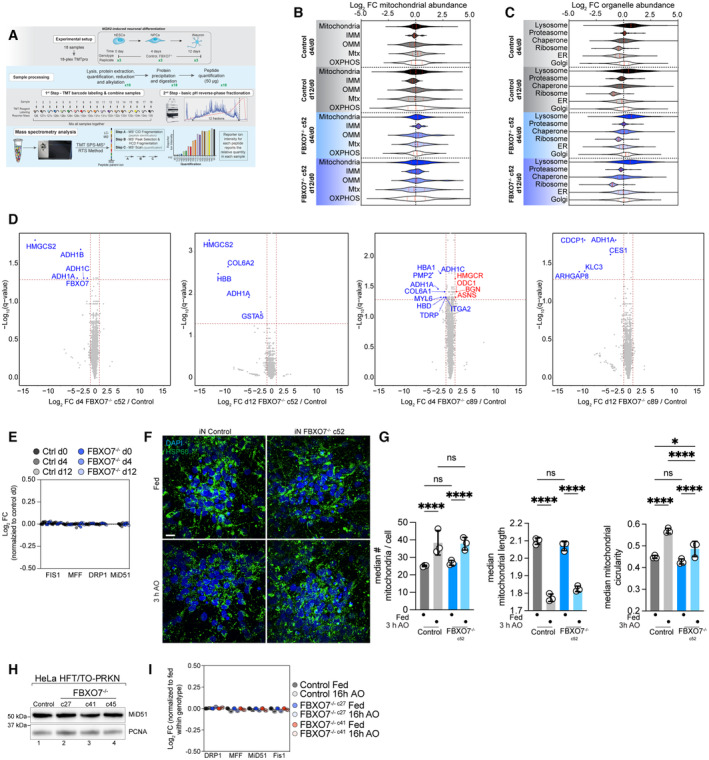
As an initial unbiased approach to examining FBXO7 function in iNeurons, we performed 18‐plex TMT proteomics on control and two clones of FBXO7−/− cells at days 0, 4, and 12 of differentiation (Fig 5A; Dataset [Link], [Link]). We have previously demonstrated dramatic remodeling of mitochondria around day 4 of differentiation to support a switch from glycolysis to oxidative phosphorylation, which is accompanied by BNIP3L‐dependent mitophagy of a fraction of mitochondria (Ordureau et al, 2021). From the ~ 6,000 proteins quantified across all replicates and conditions, we found strong correspondence of control and FBXO7−/− cells, with PC1 being driven by differentiation (Appendix Fig S4A and B). FBXO7 itself was expressed at comparable levels across the time course but was not detected in FBXO7−/− iNeurons, as expected (Appendix Fig S4C). Remarkably, the abundance of the proteome was largely immune to the loss of FBXO7 at both days 4 and 12 of differentiation (Fig 5B–D; Appendix Fig S4D). Moreover, the abundance of mitochondrial proteins (including inner and out membrane and OXPHOS components) were unchanged relative to control cells at day 4 or 12 of differentiation (Fig 5B; Appendix Fig S4D). Correlation analysis of Control and FBXO7−/− cells [iNeuron d12 / hESC d0] did show high similarity between the proteomes, as well as for subsets of mtUPR or autophagy markers (Appendix Fig S4E and F).
Figure 5. Proteomic analysis of human ES cells lacking FBXO7 during neurogenesis.
-
AWorkflow for analysis of total protein abundance in ES cells undergoing NGN2‐driven neurogenesis with or without FBXO7. Cell extracts were digested with trypsin prior to 18‐plex TMT labeling and analysis by mass spectrometry.
-
BLog2 FC for the indicated mitochondrial protein‐groups in control or FBXO7−/− cells at days 0, 4, or 12 during neurogenesis is shown in violin plots.
-
CLog2 FC for the indicated cellular organelle proteins in control or FBXO7−/− cells at days 0, 4, or 12 during neurogenesis is shown in violin plots.
-
DVolcano plots [Log2 FC (FBXO7−/−/Control) versus ‐Log10(q‐value)] for FBXO7−/− (c52 and c89) and control cells at day 4 (left panel) or day 12 (right panel) of differentiation. Proteins showing decreased or increased abundance are shown as blue or red dots.
-
ERelative abundance of DNM1L (Drp1), MFF, and FIS1 in control or FBXO7−/− cells at days 0, 4, or 12 during neurogenesis.
-
F, GMitochondrial morphology in iNeurons (iN) was assessed using confocal imaging after staining cells with HSP60 to detect mitochondria and DAPI to identify nuclei, in either fed cells or cells treated with AO (3 h) (left panel). The number of median # of mitochondria/cell, median mitochondrial length, and median mitochondrial circularity is shown. Error bars depict SD from triplicates from three differentiations (9‐12 image stacks each), as shown in panel I left, center, and right, respectively. Two‐way ANOVA with multiple comparisons; *P = 0.0416, ****P < 0.0001. Scale bar = 10 μm.
-
HWestern Blot analysis on HeLa control and FBXO7−/− whole cell lysate, probed for the mitochondrial fission adapter MiD51.
-
IRelative abundance of DNM1L, MFF, MiD51, and FIS1 in WT (Control) or FBXO7−/− HeLa cells either in the fed state or after 16 h AO as determined from the proteomics data in Fig 5A and B.
Recent reports have directly linked L250P mutations in the dimerization domain of FBXO7 to MiD49/MiD51, a core adapter protein involved in mitochondrial fission (preprint: Al Rawi et al, 2022). Indeed, DNM1L‐mediated mitochondrial fission has been previously indicated in the efficient scission of damaged parts of mitochondria and protection of the remaining healthy mitochondrial network (Burman et al, 2017). Cells expressing FBXO7L250P were found to have reduced levels of proteasome subunits as well (preprint: Al Rawi et al, 2022). Our proteomic data from iNeurons indicates no alterations in the abundance of proteasome subunits or mitochondrial fission and fusion machinery (DNM1L, MFF, and MiD51) in iNeurons lacking FBXO7 (Fig 5C and E). To test if loss of FBXO7 causes in morphological changes in the mitochondrial network, we stained day 12 iNeurons with for the matrix marker HSP60 prior to image analysis (Fig 5F and G). However, we observed no significant alterations in the number of mitochondria/cell or the mean mitochondrial length in control or FBXO7−/− iNeurons. The mean mitochondrial circularity was slightly reduced in FBXO7−/− iNeurons treated with AO for 3 h (Fig 5F). Analogous analyses of mitochondria and fission/fusion proteins also did not reveal significant alterations in response to loss of FBXO7 in two independent clones of HeLa cells (Fig 5H and I). However, there was a ~ 10% decrease in the abundance of proteasome subunits in HeLa cells lacking FBXO7 in the absence of depolarization, but an increase after mitophagy induction (Appendix Fig S3E; Fig 4C). If this response is a compensatory mechanism of the knockout cell line to cope with cytotoxic insults is an intriguing idea. Last, we examined whether FBXO7−/− is required for the maintenance of the mitochondrial organelle pool during differentiation. Previous work has shown significant metabolic rewiring during the differentiation from hESC to iNeurons, including a switch from glycolysis to oxidative phosphorylation as the cells main energy source (Ordureau et al, 2021). Since this switch is accompanied by increased mitophagy, we set out to see if the absence of FBXO7 would lead to accumulation of mitochondria and/or impair differentiation. FBXO7−/− cells did not display obvious differences in the abundance of mitochondrial proteins, when normalized to day 0 (Fig 5B; Appendix Fig S4D). Likewise, abundance of other organelles was unchanged (Fig 5C). Finally, neither differentiation markers or autophagy proteins displayed obvious changes in control versus the two FBXO7−/− clones as measured by TMT‐proteomics and displayed using a T 2‐statstic (Ordureau et al, 2021; Appendix Fig S5A–F).
FBXO7−/− are susceptible to mtUPR
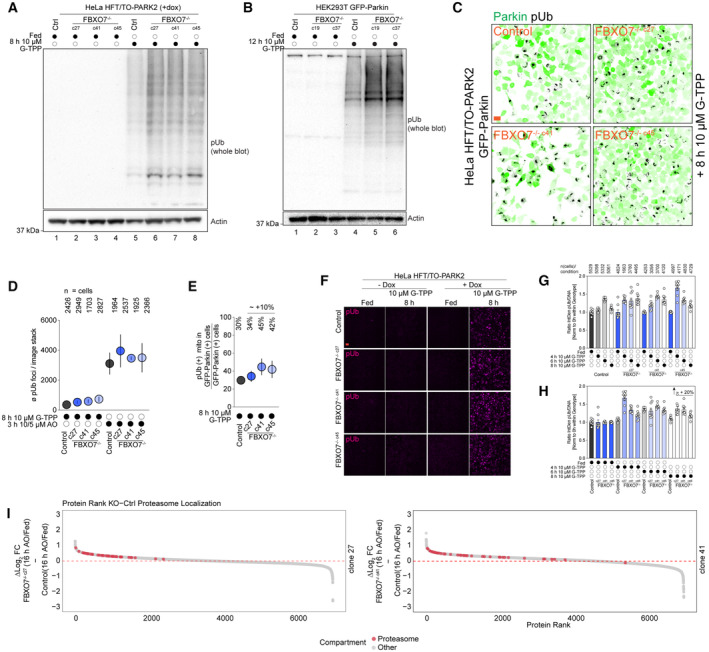
Last, we investigated the role of FBXO7 in response to the mitochondrial unfolding response (mtUPR) via treatment of the mitochondrial HSP90 inhibitor Gamitrinib‐triphenylphosphonium (G‐TPP), which is also known to induce protein misfolding in the matrix and activate the PINK1/Parkin pathway (Munch & Harper, 2016; Fiesel et al, 2017; Kataura et al, 2023). We treated HeLa or HEK293T Control and FBXO7−/− cells expressing Parkin with 10 μM G‐TPP (8 or 12 h) and examined pUb accumulation using Western Blot (Fig EV4A and B). Cells lacking FBXO7 still responded to depolarization by activation of pUb accumulation, indicating that FBXO7 is dispensable for PINK1/Parkin activation in response to mtUPR. Interestingly, loss of FBXO7 did show increased pUb signal in all knockout clones, suggesting that loss of FBXO7 may prime cells for enhances mtUPR signaling to the PINK1/Parkin pathway. As an orthogonal approach, GFP‐Parking expressing HeLa Control and FBXO7−/− were treated with G‐TPP, and image analysis used to examine the co‐incidence of pUb and GFP‐Parkin with mitochondria (Fig EV4C). The average number of pUb after G‐TPP was ~ 4–5 times lower compared to AO‐treated cells, in line with the lower acute mitotoxic nature of the drug (Fig EV4D). However, we observed a ~ 10% increase in pUb foci in FBXO7−/− cells expressing GFP‐Parkin when normalized to all Parkin expressing cells (Fig EV4E).
Figure EV4. Analysis of FBXO7−/− response to G‐TPP.
-
AWestern Blot for pUb of HeLa Control and FBXO7−/− cells after mitophagy induction using G‐TPP for indicated times.
-
BWestern Blot for pUb of HEK293T Control and FBXO7−/− cells after mitophagy induction using G‐TPP for indicated times.
-
CImmunofluorescence images of HeLa Control and FBXO7−/− cells after G‐TPP treatment. Staining for Parkin (GFP, green) and pUb (gray) are shown. Scale bars = 10 μm.
-
D, EEvaluation of mitophagic response to G‐TPP in Hela Control and FBXO7−/− cells, based on images from (C). Ø (average) number of pUb per image stack are plotted. Number of analyzed cells for each condition is indicted in (D). Error bars depict SEM. (E) Ratio of pUb‐positive mitochondria in GFP‐Parkin‐positive cells divided by total number of GFP‐Parkin‐positive cells as proxy for mitophagy induction rate is plotted. Ø (average) % numbers for each genotype are indicted above the plot. Error bars depict SEM.
-
FImmunofluorescence images of HeLa Control and FBXO7−/− cells after G‐TPP timecourse treatment ± doxycycline (= Parkin expression). Staining for pUb (magenta) is shown. Scale bars = 25 μm.
-
G, HQuantification of pUb/DNA integrated density normalized to 0 h treatment within genotype. Panel (H) depicts the measurements sorted by treatment time. N (cells) analyzed per condition is indicated at the bottom of the graph. Error bars depict SEM.
-
IRanked protein abundance of ∆ log2 (KO[16 h AO/Fed]—Control [16 h AO/Fed]) for FBXO7−/− clone 27 (top) and clone 41 (bottom). Proteasomal annotated proteins are depicted red, all other detected proteins in gray.
Next, we tested if Parkin is required for the FBXO7‐mediated changes in pUb recruitment after G‐TPP treatment. HeLa HFT/TO‐PARK2 Control and FBXO7−/− cells were treated with G‐TPP without and with the addition of doxycycline, thus controlling for Parkin expression. Immunofluorescence staining for pUb revealed the absence of signal in cells lacking Parkin [(−) dox samples], while Parkin‐expressing cells responded to HSP90 inhibition with pUb foci appearing after 8 h of treatment (Fig EV4F). Analysis of pUb intensity over G‐TPP treatment‐timecourse suggested potential small differences in pUb kinetics, especially during early states of G‐TPP‐mediated stress (4–6 h of treatment), albeit variability between FBXO7−/− clones was observed (Fig EV4G and H). After 8 h of G‐TPP, the normalized ratio of pUb/DNA intensities were on average 20% higher across the knockouts compared to control cells, consistent with Western Blotting.
Discussion
Despite strong genetic evidence linking mutations in FBXO7 with parkinsonian–pyramidal syndrome (Di Fonzo et al, 2009; Paisan‐Ruiz et al, 2010; Houlden & Singleton, 2012), our understanding of the cellular roles of FBXO7 are limited. An early study reported biochemical links between FBXO7 and PINK1/Parkin‐dependent mitophagy in fibroblasts and SH‐SY5Y cells, including interaction of overexpressed FBXO7 with both PINK1 and Parkin (Burchell et al, 2013). Using siRNA to deplete FBXO7, it was also concluded that FBXO7 promotes Parkin's ability to ubiquitylate the outer mitochondrial membrane substrate MFN1 and also promotes clearance of mitochondria by autophagy. These biochemical and physiological results led to the conclusion that FBXO7 functions as a biochemical amplifier of PINK1/Parkin‐dependent mitophagy (Burchell et al, 2013). However, follow‐up studies have been limited, and the field has experienced dramatic advances in the understanding of the biochemical roles of Parkin and PINK1 in promoting mitochondrial clearance by autophagy, thereby providing the tools to examine potential roles of FBXO7 in the pathway.
With these tools, we set out to define where in the pathway FBXO7 might operate to promote mitophagy. However, in both the HeLa cells with overexpressed Parkin and iNeurons with a fully endogenous PINK1/Parkin pathway, we failed to validate an essential role for FBXO7 in any of several steps in the pathway, including pUb accumulation, Parkin recruitment to the mitochondrial outer membrane, recruitment of autophagy machinery, mitophagic flux as measured by mtKeima, and mitochondrial proteome degradation. The finding that deletion of FBXO7 has no obvious effect on the pathway with either endogenous or overexpressed Parkin in iNeurons and HeLa cells indicates that FBXO7 does not play a general or universally required role in the pathway. A previous study largely based on overexpression concluded that FBXO7 may bind and regulate PINK1 levels, with < 2‐fold increases in PINK1 by Western Blotting in FBXO7‐depleted cells (Huang et al, 2020). However, our quantitative proteomics experiments (Fig 4) indicated that deletion of FBXO7 has no effect on PINK1 abundance, consistent with the finding that pUb accumulation in response to depolarization is unaffected in FBXO7−/− cells. Moreover, our previous interaction proteomics experiments did not identify either PINK1 in association with overexpressed FBXO7 (Huttlin et al, 2021) or FBXO7 in association with overexpressed Parkin (Sarraf et al, 2013). Thus, our functional and biochemical results do not support a biochemical linkage between FBXO7 and the PINK1/Parkin pathway. A recent preprint (preprint: Sanchez‐Martinez et al, 2022) examining the Drosophila FBXO7 ortholog—nutcracker (ntc)—reported a ~ 50% reduction in basal mitolysosome number in ntc mutant larva, but this effect appears to be independent of Pink1 and Parkin. Under basal conditions using Keima reporters, we did not observe a reduction in basal flux in FBXO7−/− HeLa cells or iNeurons.
In order to broadly examine FBXO7−/− iNeurons for pathways that are affected, we performed global proteomic analysis during neurogenesis in vitro. However, we did not observe any alterations in pathways linked with mitochondria, or other quality control pathways, although a reduction in the abundance of alcohol dehydrogenase enzymes among a small number of other proteins, was observed. Whether these signatures are related to FBXO7 function remains to be examined. While a recent study linked the L250P mutation in the dimerization domain of FBXO7 with alterations in proteins associated with mitochondrial fusion and fission (preprint: Al Rawi et al, 2022), we did not observe alterations in either the abundance of these proteins (MiD51, MFF, FIS1) nor did we observe changes in mitochondrial morphology in either HeLa cells or iNeurons lacking FBXO7. However, based on the G‐TPP results, we cannot exclude the possibility that stress response pathways distinct from those examined here, and possibly in specific cell types, may employ FBXO7 to promote mitochondrial health.
Previous studies demonstrated that ntc associates with components of the core particle of the proteasome, as well as with DmPI31 (Bader et al, 2011). Association of human FBXO7 with proteasome components and PSMF1/PI31 have also been observed in both focused studies and in our previous interaction proteomic studies (Vingill et al, 2016; Huttlin et al, 2021; preprint: Al Rawi et al, 2022). In this context, PI31/DmPI31 and FBXO7/ntc have been proposed to functionally link proteasome trafficking via dynein motors in neuronal processes (Liu et al, 2019). Of note, in our HeLa proteomics, proteasome proteins appeared to be overrepresented in FBXO7−/− proteomes after mitophagy treatment (Fig EV4I). Our interaction proteomics analysis has also confirmed association of FBXO7 with PSMF1, CRL, COP9/Signalosome, and proteasome core subunits, and demonstrated that association of FBXO7‐SKP1‐CUL1NTD with PSMF1 is a direct interaction (Fig EV3D and E). Further studies are required to elucidate the relationship between FBXO7 and parkinsonian–pyramidal syndrome, to understand any functional relationships between FBXO7 and other Parkinson's disease risk alleles, and to examine whether FBXO7's physical association with the proteasome is linked with disease.
Materials and Methods
All details and catalog numbers can be found in the Reagents and Tools table. Protocols associated with this work can be found on protocols.io (dx.doi.org/10.17504/protocols.io.kxygx99pwg8j/v2).
Reagents and Tools table
| Reagent/resource | Reference or source | Identifier or catalog number |
|---|---|---|
| Experimental models | ||
| Rosetta™(DE3) pLysS Competent Cells | Novagen | 70956 |
| https://www.emdmillipore.com/US/en/product/RosettaDE3-Competent-Cells-Novagen,EMD_BIO-70954 | ||
| H9 ES cells | WiCell, Madison WI | WA09 |
| RRID: CVCL_9773 | ||
| H9 ES cells + AAVS‐NGN2 | Ordureau et al (2020) | RRID: CVCL_C4EK |
| H9 ES PINK1−/− + AAVS‐NGN2 | Ordureau et al (2020) | RRID: CVCL_C4EM |
| H9 ES FBXO7−/− + AAVS‐NGN2 | This paper | RRID: CVCL_C4EN |
| HeLa HFT/TO‐PRKN | Ordureau et al (2014) | RRID: CVCL_C4EQ |
| HeLa HFT/TO‐PRKN PINK1−/− | Ordureau et al (2014) | RRID: CVCL_C4ES |
| HeLa HFT/TO‐PRKN FBXO7−/− | This paper | RRID: CVCL_C4ER |
| HEK293T | ATCC | RRID:CVCL_0063 |
| HEK293T FBXO7−/− | This paper | RRID: CVCL_C7U0 |
| Recombinant DNA | ||
| pET‐NLS‐Cas9‐6xHis | Zuris et al (2015); Addgene | Addgene #62934 |
| RRID:Addgene_62934 | ||
| gRNA_AAVS1‐T2 | Addgene | Addgene #41818 |
| RRID:Addgene_41818 | ||
| pAAVS1‐TRE3G‐NGN2 | Ordureau et al (2020) | N/A |
| px459 | Addgene | Addgene #62988; RRID:Addgene_62988 |
| hCAS9 plasmid | Addgene | Addgene #41815 |
| RRID:Addgene_41815 | ||
| pAC150‐PBLHL‐4xHS‐EF1a – mtx‐mKeimaXL | Ordureau et al (2020) | Dest vector: Addgene #48234 |
| RRID:Addgene_48234 | ||
| pCMV‐hyPBase – hyperactive piggyBac transposase | Yusa et al (2011) | https://www.sanger.ac.uk/form/Sanger_CloneRequests |
| psPAX2 | Addgene | Addgene #12260 |
| RRID:Addgene_12260 | ||
| pMD2.G | Addgene | Addgene #12259 |
| RRID:Addgene_12259 | ||
| pHAGE‐mt‐mKeima | Vargas et al (2019) | Addgene #131626 |
| RRID:Addgene_131626 | ||
| pHAGE‐GFP‐Parkin | Heo et al (2015) | N/A |
| pGEX4T1‐TEV‐HsFbxo7‐129‐398/HsSkp1 | This study | |
| pRSF1b‐His8‐TEV‐HsPI31‐1‐151 | This study | |
| pGEX4T1‐TEV‐HsCul1‐1‐410 | Hopf et al (2022) | https://pubmed.ncbi.nlm.nih.gov/35982156/ |
| Antibodies | ||
| Anti‐HSP60 | Abcam | ab128567 |
| RRID:AB_11145464 | ||
| Anti‐FBXO7 (D8L4E) | Cell Signaling Technology | 57981S |
| RRID:AB_2799536 | ||
| Anti‐Tomm20 | Abcam | ab186735 |
| RRID:AB_2889972 | ||
| Anti‐Phospho‐Ser65‐Ubiquitin | Cell Signaling Technology | 70973S |
| RRID:AB_2799795 | ||
| Anti‐beta‐Actin | Santa‐Cruz | sc‐69879 |
| RRID:AB_1119529 | ||
| Anti‐SQSTM | Abnova | H00008878‐M01 |
| RRID:AB_437085 | ||
| Anti‐FIP200 (D10D11) | Cell Signaling Technology | 12436S |
| RRID:AB_2797913 | ||
| Anti‐LC3B | Cell Signaling Technology | 2775S |
| RRID:AB_915950 | ||
| Anti‐DNA | Progen | 61014 |
| RRID:AB_1541137 | ||
| Anti‐PCNA (PC10) | Santa‐Cruz | Sc‐56; RRID:AB_628110 |
| Anti‐MiD51 / MIEF2 | ProteinTech | 20164‐1‐AP |
| RRID:AB_10639522 | ||
| Anti‐GFP (chicken) | Thermo Fisher Scientific | A10262 |
| RRID:AB_2534023 | ||
| Anti‐WIPI2 | BioRad | MCA5780GA |
| RRID:AB_10845951 | ||
| Anti‐Rabbit IgG (H+L), HRP Conjugate | Promega | W4011; RRID:AB_430833 |
| Anti‐Mouse IgG (H+L), HRP Conjugate | Promega | W4021; RRID:AB_430834 |
| Goat anti‐mouse Alexa488 | Thermo Fisher Scientific | A‐11001 |
| RRID:AB_2534069 | ||
| Goat anti‐chicken Alexa488 | Thermo Fisher Scientific | A11039 |
| RRID: AB_2534096 | ||
| Goat anti‐rabbit Alexa568 | Thermo Fisher Scientific | A‐11011 |
| RRID:AB_143157 | ||
| Goat anti‐rabbit Alexa647 | Thermo Fisher Scientific | A27040 |
| RRID:AB_2536101 | ||
| Oligonucleotides and sequence‐based reagents | ||
| Primers for 5′ AAVS junction PCR | 5′‐ctctaacgctgccgtctctc‐3′ and 5′‐tgggcttgtactcggtcatc‐3′ | N/A |
| Primers for 3′ AAVS junction PCR | 5′‐cacacaacatacgagccgga‐3′ and 5′‐accccgaagagtgagtttgc‐3′ | N/A |
| Primers for locus PCR add MiSeq Primers FBXO7 | 5′‐ tcctttttcagttagggcctagt‐3′ and 5′‐ atgtctgtgttttgcttgcagt‐3′ | N/A |
| FBXO7 for deletion sgRNA | ACCGATTCACTACAGAGCATGGG | N/A |
| Chemicals, enzymes and other reagents | ||
| Oligomycin A | Sigma‐Aldrich | 75351 |
| Antimycin A | Sigma‐Aldrich | A8674 |
| Bafilomycin A | Sigma‐Aldrich | B1793‐10mg |
| Gamitrinib TPP hexafluorophosphate (G‐TPP) | MedChemExpress | HY‐102007A |
| Dithiothreitol (DTT) | Gold Biotechnology | DTT25 |
| Trypsin | Promega | V511C |
| Lys‐C | Wako Chemicals | 129‐02541 |
| Urea | Sigma | U5378 |
| Formic Acid | Sigma‐Aldrich | 94318 |
| EPPS | Sigma‐Aldrich | E9502 |
| 2‐Chloroacetamide | Sigma‐Aldrich | C0267 |
| Adenosine 5′ triphosphate, disodium, trihydrate (ATP) | Thermo‐Fisher Scientific | 10326943 |
| TCEP | Gold Biotechnology | TCEP2 |
| PhosSTOP phosphatase inhibitor cocktail | Roche | 4906845001 |
| Recombinant SpCas9 | Zuris et al (2015); This paper | N/A |
| 100 × 21 mm Dish, Nunclon Delta | Thermo Fisher Scientific | 172931 |
| Nunc Cell‐Culture Treated 6‐well | Thermo Fisher Scientific | 140685 |
| Nunc Cell‐Culture Treated 12‐well | Thermo Fisher Scientific | 150628 |
| Corning Matrigel Matrix, Growth Factor Reduced | Corning | 354230 |
| Geltrex LDEV‐Free Reduced Growth Factor Basement Membrane Matrix | Thermo Fisher Scientific | A1413202 |
| DMEM/F12 | Thermo Fisher Scientific | 11330057 |
| Neurobasal | Thermo Fisher Scientific | 21103049 |
| NEAA | Life Technologies | 11140050 |
| GlutaMax | Life Technologies | 35050061 |
| N‐2 Supplement (100X) | Thermo Fisher Scientific | 17502048 |
| Neurotrophin‐3 | Peprotech | 450‐03 |
| Brain‐derived neurotrophic factor (BDNF) | Peprotech | 450‐02 |
| Cultrex 3D Culture Matrix Laminin I | R&D Systems | 3446‐005‐01 |
| B27 | Thermo Fisher Scientific | 17504001 |
| B‐27 Supplement (50X), minus antioxidants | Thermo Fisher Scientific | 10889038 |
| Y‐27632 Dihydrochloride (ROCK inhibitor) | PeproTech | 1293823 |
| Accutase | StemCell | 7920 |
| FGF3 | in‐house | N/A |
| Insulin Human | Sigma‐Aldrich | I9278‐5ML |
| TGF‐beta | PeproTech | 100‐21C |
| holo‐Transferrin human | Sigma‐Aldrich | T0665 |
| Sodium Bicarbonate | Sigma‐Aldrich | S5761‐500G |
| Sodium selenite | Sigma‐Aldrich | S5261‐10G |
| Doxycycline | Sigma‐Aldrich | D9891 |
| Puromycin | Gold Biotechnology | P‐600‐500 |
| Hygromycin B | Thermo Fisher Scientific | 10687010 |
| UltraPure 0.5M EDTA, pH 8.0 | Thermo Fisher Scientific | 15575020 |
| DMEM, High Glucose, Pyruvate | Thermo Fisher Scientific | 11995‐073 |
| Penicillin‐Streptomycin (10,000 U/mL) | Thermo Fisher Scientific | 15140163 |
| GlutaMAX | Thermo Fisher Scientific | 35050061 |
| MEM NEAA | Thermo Fisher Scientific | 11140050 |
| Trypsin‐EDTA | Sigma | T4049‐100ML |
| PBS | Corning | 21‐031‐CV |
| OptiMEM I reduced serum media | Thermo Fisher Scientific | 31985062 |
| Cover Glasses | VWR | 16004‐308 |
| Marienfeld Precision cover glasses thickness No. 1.5H (tol. ± 5 μm) 18 × 18 mm | VWR | 107032 |
| 6 Well glass bottom plate with high performance #1.5 cover glass | Cellvis | P06‐1.5H‐N |
| 12 Well glass bottom plate with high performance #1.5 cover glass | Cellvis | P12‐1.5H‐N |
| 24 Well glass bottom plate with high performance #1.5 cover glass | Cellvis | P24‐1.5H‐N |
| 96 Well glass bottom plate with high performance #1.5 cover glass | Cellvis | P96‐1.5H‐N |
| 16% Paraformaldehyde, Elecron‐Mircoscopy Grade | Electron Microscopy Science | 15710 |
| Paraformaldehyde 3% Glutaraldehyde 0.35% in 0.1M Sodium Cacodylate, pH 7.4 | Electron Microscopy Science | 15949‐50 |
| DAPI (4',6‐Diamidino‐2‐Phenylindole, Dihydrochloride) | Thermo Fisher Scientific | D1306 |
| Hoechst33342 | Thermo Fisher Scientific | H1399 |
| DNA SPY555 | Spirochrome | N/A |
| Vectashield | Vector Laboratories | H‐1000‐10 |
| FluoroBrite DMEM | Thermo Fisher Scientific | A1896701 |
| Falcon® 5mL Round Bottom Polystyrene Test Tube, with Cell Strainer Snap Cap, 25/Pack, 500/Case | Corning | 352235 |
| NuPAGE 8%, Bis‐Tris, 1.0 mm, Midi Protein Gel, 12+2‐well | Thermo Fisher Scientific | WG1001BOX |
| NuPAGE Novex® 4–12% Bis‐Tris Midi Protein Gels, 20 well | Thermo Fisher Scientific | WG1402BOX |
| NuPAGE Novex® 4–12% Bis‐Tris Midi Protein Gels, 26 well | Thermo Fisher Scientific | WG1403BOX |
| NuPAGE™ LDS Sample Buffer (4X) | Thermo Fisher Scientific | NP0008 |
| Precision Plus Protein™ Kaleidoscope™ Prestained Protein Standards | BioRad | 1610395 |
| Revert 700 Total Protein Stain Kits for Western Blot Normalization | LI‐COR | 926‐11016 |
| Immobilon‐P Membrane, PVDF, 0.45 μm, 26.5 cm × 3.75 m roll | EMD Millipore | IPVH00010 |
| Criterion™ Cell/Plate Blotter System | BioRad | 1656024 |
| Lipofectaime LTX | Thermo Fisher Scientific | 15338100 |
| cOmplete, EDTA‐free Protease Inhibitor Cocktail | Milipore Sigma | 11873580001 |
| FastBreak Buffer | Promega Inc | V8571 |
| GeneArt Precision gRNA Synthesis Kit | Thermo Fisher Scientific | A29377 |
| Pierce™ High pH Reversed‐Phase Peptide Fractionation Kit | Thermo Fisher Scientific | 84868 |
| Pierce™ BCA Protein Assay Kit | Thermo Fisher Scientific | 23227 |
| TMTpro 18plex Label Reagent | Thermo Fisher Scientific | A52045 |
| Software | ||
| Prism | GraphPad, v9.4.1 (Build 458) | https://www.graphpad.com/scientific-software/prism/ |
| RRID:SCR_002798 | ||
| MassPike, In‐house mass spectrometry data analysis software | Huttlin et al (2010) | N/A |
| Monocle | Rad et al (2021) | RRID:SCR_018685 |
| SEQUEST | Eng et al (1994) | https://proteomicsresource.washington.edu/protocols06/sequest.php |
| Perseus (v1.6.5) | Tyanova et al (2016) | http://www.perseus-framework.org |
| RRID:SCR_015753 | ||
| MitoCharta | Rath et al (2021) | MitoCarta (RRID:SCR_018165) |
| FiJi | ImageJ 1.53t30 | https://imagej.net/Fiji |
| RRID:SCR_002285 | ||
| CellProfiler (v.4.2.1) | Stirling et al (2021) | https://cellprofiler.org |
| RRID:SCR_007358 | ||
| ImageStudioLite | v5.2.5 | https://www.licor.com/bio/image-studio-lite/ |
| RRID:SCR_013715 | ||
| FlowJo | v10.7.2 | https://www.flowjo.com |
| RRID:SCR_008520 | ||
| NIS Elements | 5.21.3 (Build 1489) | https://www.microscope.healthcare.nikon.com/products/software/nis-elements |
| RRID:SCR_014329 | ||
| Imaris | Oxford Instruments, v9.7 | RRID:SCR_007370 |
| Illustrator (v26.2.1) | Adobe Inc. | RRID:SCR_010279 |
| Excel (v.4.2.0) | Microsoft Inc. | RRID:SCR_016137 |
| R (4.2.0)/RStudio (2022.07.01 Build 554) | https://www.rstudio.com/ | |
| RRID:SCR_000432 | ||
| library(tibble) | v3.1.8 | https://cran.r-project.org/web/packages/tibble/tibble.pdf |
| library(ggVennDiagram) | v1.2.0 | https://cran.r-project.org/web/packages/ggVennDiagram/readme/README.html#:~:text=ggVennDiagram%20maps%20the%20fill%20color,%3Dsample(genes%2C350)) |
| library(stringr) | v1.4.0 | RRID:SCR_022813 |
| library(dplyr) | v1.0.9 | RRID:SCR_016708 |
| library(ggsci) | v2.9 | https://github.com/nanxstats/ggsci |
| library(ggrepel) | v0.9.1 | RRID:SCR_017393 |
| library(ggplot2) | v3.3.6 | RRID:SCR_014601 |
| library(tidyverse) | v1.3.2 | RRID:SCR_019186 |
| library(timecourse) | V1.68.0 | RRID:SCR_000077 |
| UCSF ChimeraX | version 1.4.dev202201050842 (2022‐01‐05) | https://www.cgl.ucsf.edu/chimerax/ |
| RRID:SCR_015872 | ||
| Snapgene (v.5.1.7) | Dotmatics | https://snapgene.com |
| RRID:SCR_015052 | ||
| Other | ||
| Orbitrap Fusion Lumos Mass Spectrometer | ThermoFisher Scientific | IQLAAEGAAPFADBMBHQ |
| RRID:SCR_020562 | ||
| Orbitrap Eclipse Tribrid Mass Spectrometer | ThermoFisher Scientific | FSN04‐10000 |
| RRID:SCR_020559 | ||
| Easy‐nLC 1200 | ThermoFisher Scientific | LC140 |
| Aeris™ 2.6 μm PEPTIDE XB‐C18 100 Å 250 × 4.6 mm | Phenomenex | 00G‐4505‐E0 |
| Sep‐Pak C18 1cc Vac Cartridge, 50 mg | Waters | WAT054960 |
| SOLA HRP SPE Cartridge, 10 mg | Thermo Fisher Scientific | 60109‐001 |
| Empore™ SPE Disks C18 | 3M Bioanalytical Technologies | 2215 |
| Neon™ Transfection System | Thermo Fisher Scientific | MPK5000 |
| ChemiDoc MP imaging system | BioRad | 12003154 |
| RRID:SCR_019037 | ||
| Nikon Ti w/ Yokagawa CSU‐W1 spinning disk confocal | Nikon Corporation | https://www.microscope.healthcare.nikon.com/products/confocal-microscopes/csu-series/csu-w1 |
| Nikon Ti w/ Yokagawa CSU‐X1 spinning disk confocal | Nikon Corporation | https://www.microscope.healthcare.nikon.com/products/confocal-microscopes/csu-series/csu-x1 |
| DeltaVision OMX V4 | Applied Precision (GE Healthcare) | https://sim.hms.harvard.edu/tech-specs/ |
| Symphony A5 | BD Biosciences | https://www.bdbiosciences.com/en-us/instruments/research-instruments/research-cell-analyzers/facsymphony |
| Attune NxT | Thermo Fisher Scientific | https://www.thermofisher.com/ca/en/home/life-science/cell-analysis/flow-cytometry/flow-cytometers/attune-nxt-flow-cytometer.html |
| RRID:SCR_019590 | ||
| ÄKTApure system | GE Healthcare/Cytiva | https://www.cytivalifesciences.com/en/us/shop/chromatography/chromatography-systems/akta-pure-p-05844 |
| Superdex 200 Increase 10/300 GL column | Cytiva | https://www.cytivalifesciences.com/en/us/shop/chromatography/prepacked-columns/size-exclusion/hiload-superdex-200-pg-preparative-size-exclusion-chromatography-columns-p-06283?s_kwcid=AL!14612!3!614128999325!b!!g!!superdex%20200&dtid=ppc_google_17817864916_143232689001&ps_kw=superdex%20200&extcmp=cy22498-se-paid-hiload-superdex-200-sec-columns&adgrp=&gclid=EAIaIQobChMImrT2j_aD_gIVfWxvBB1AYgKqEAAYASAAEgKTbPD_BwE |
| Cat no: 28989336 | ||
Methods and Protocols
Cloning and plasmid generation
The construction of lentiviral expression constructs used in this study have been previously described: pHAGE‐mt‐mKeima in (Heo et al, 2015), PB‐mt‐mKeimaXL (Ordureau et al, 2021) and pHAGE‐GFP‐Parkin in (Ordureau et al, 2018).
Cell culture, generation of lentiviral stable cell lines
HeLa Flip‐In T‐Rex (HFT) expressing doxycycline‐inducible WT‐Parkin (HeLa HFT/TO‐PRKN‐WT) cells (Heo et al, 2015) and HEK293T were maintained in Dulbecco's modified Eagle's medium (DMEM), supplemented with 10% vol/vol fetal bovine serum (FBS), 5% vol/vol penicillin–streptomycin (P/S), 5% vol/vol GlutaMAX and 5% vol/vol Non‐essential amino acids (NEAA) at 37°C, 5% O2. Unless otherwise noted, we refer to independently grown and handled cultures as biological replicates to distinguish from assays performed on identical samples (i.e. technical replicates).
Stable cell lines were generated using lentivirus generated from HEK293T cells. pHAGE‐mt‐mKeima or pHAGE‐eGFP‐Parkin were co‐transfected in HEK293T cells together with the lentiviral vectors pSpax2 and pMD2.1 using Lipofectaime LTX reagent (Thermo Fisher Scientific, 15338100), according to manufacturer's instructions. Virus was harvested, filtered (0.45 μm) and added with 8 μg/ml polybrene and target cells infected for 18 h. Antibiotic selection was performed using 1 μg/ml puromycin or FACS sorting and cells verified using Western Blotting or fluorescence microscopy.
If not stated otherwise, Parkin expression in HeLa HFT/TO‐PRKN was induced using 2 μg/ml doxycycline o/n, and subsequent mitophagy induced using 5 μM Antimycin A/10 μM Oligomycin A (==AO), 1 μM CCCP or 10 μM G‐TPP for the indicated times.
H9 ES cells harboring the mitochondrial mt‐mKeima flux reporter (Ordureau et al, 2020) were generated by electroporation of 1 × 106 cells with 2.5 μg of pAC150‐PiggyBac‐matrix‐mKeimaXL along with 2.5 μg of pCMV‐HypBAC‐PiggyBac‐Helper, as described (Ordureau et al, 2020). The cells were selected and maintained in E8 medium supplemented with 50 μg/ml Hygromycin and Hygromycin was kept in the medium during differentiation to iNeurons. Mitophagy was induced with either 0.5 or 5 μM Antimycin A or 0.5 or 10 μM Oligomycin A.
Gene‐Editing and iNeuron differentiation
Generation of HeLa HFT cells lacking FBXO7−/− was facilitated using CRISPR/Cas9 with target sites determined using CHOPCHOP (Labun et al, 2019). Guide RNAs were ligated into the px459 plasmid (Addgene plasmid # 62988) and cells transfected using Lipofectaime LTX reagent (Thermo Fisher Scientific, 15338100), according to manufacturer's instructions. Two days post‐transfection, single, GFP positive cells were sorted into 96‐well dishes containing 300 μl full growth medium (composition as described above). Single cells were allowed to grow into colonies, then duplicated for MiSeq analysis and maintenance. Knockout candidates were confirmed by Western blot on whole cell lysates. The sgRNAs were generated using GeneArt Precision gRNA Synthesis Kit (Thermo Fisher Scientific) according to the manufacturer's instruction and purified using RNeasy Mini Kit (Qiagen). The sgRNA target sequence: ACCGATTCACTACAGAGCAT.
The PINK1−/− H9 cells used here, wherein sequences in exon 1 were deleted using CRISPR‐Cas9 to create a null allele, were reported previously (Ordureau et al, 2018). To generate FBXO7−/− H9 ES or HeLa cells, 0.6 μg sgRNA was incubated with 3 μg SpCas9 protein for 10 min at room temperature and electroporated into 2 × 105 control H9 cells using Neon transfection system (Thermo Fisher Scientific). Mutants were identified by Illumina MiSeq and further confirmed by Western blotting. For introduction of TRE3G‐NGN2 into the AAVS1 site, a donor plasmid pAAVS1‐TRE3G‐NGN2 was generated by replacing the EGFP sequence with N‐terminal flag‐tagged human NGN2 cDNA sequence in plasmid pAAVS1‐TRE3G‐EGFP (Addgene plasmid # 52343). Five micrograms of pAAVS1‐TRE3G‐NGN2, 2.5 μg hCas9 (Addgene plasmid # 41815), and 2.5 μg gRNA_AAVS1‐T2 (Addgene plasmid # 41818) were electroporated into 1 × 106 H9 cells. The cells were treated with 0.25 μg/ml Puromycin for 7 days and surviving colonies were expanded and subjected to genotyping.
Human ES cells (H9, WiCell Institute) were cultured in E8 medium (Chen et al, 2011) on Matrigel‐coated tissue culture plates with daily medium change. Cells were passaged every 4–5 days with 0.5 mM EDTA in 1× DPBS (Thermo Fisher Scientific). SpCas9 and AsCas12a/AsCpf1 expression plasmids pET‐NLS‐Cas9‐6xHis (Addgene plasmid # 62934) and modified pDEST‐his‐AsCpf1‐EC, generated by deleting the MBP sequence from plasmid pDEST‐hisMBP‐AsCpf1‐EC (Addgene plasmid # 79007), were transformed into Rosetta™(DE3)pLysS Competent Cells (Novagen), respectively, for expression. SpCas9 and AsCas12a/AsCpf1 proteins were purified as described elsewhere (Zuris et al, 2015; Hur et al, 2016). Briefly, cells expressing SpCas9 [0.5 mM isopropylthio‐β‐galactoside, 14‐h induction] were lysed in FastBreak buffer (Promega, Inc) and the NaCl concentration adjusted to 500 mM. Extracts were centrifuged at 38,000 g for 10 min at 4°C and the supernatant incubated with Ni‐NTA resin for 1 h. The resin was washed extensively with 50 mM Tris pH8.0, 500 mM NaCl, 10% glycerol, 20 mM imidazole, and 2 mM TCEP prior to elution with this buffer supplemented with 400 mM imidazole. Proteins were diluted two volumes/volume in PBS and fractionated on a Heparin‐Sepharose column using a 0.1–1.0 M NaCl gradient. Cas9‐containing fractions were stored in PBS, 20% glycerol, 2 mM TCEP at −80°C. AsCpf1 expression was induced similarly, and cells pelleted by centrifugation. Cells were lysed by sonication in 50 mM HEPES pH7, 200 mM NaCl, 5 mM MgCl2, 1 mM DTT, and 10 mM imidazole supplemented with lysozyme (1 mg/ml) and protease inhibitors (Roche complete, EDTA‐free). After centrifugation (16,000 g for 30 min), the supernatant was incubated with Ni‐NTA resin, the resin washed with 2 M NaCl, and bound proteins eluted with 250 mM imidazole, and buffer exchanged into lysis buffer lacking MgCl2 and imidazole prior to storage at −80°C.
For human ES cell conversion to iNeurons, cells were expanded and plated at 2 × 104/cm2 on Matrigel‐coated tissue plates in DMEM/F12 supplemented with 1× N2, 1× NEAA (Thermo Fisher Scientific), human brain‐derived neurotrophic factor (BDNF, 10 ng/ml, PeproTech), human Neurotrophin‐3 (NT‐3, 10 ng/ml, PeproTech), mouse laminin (0.2 μg/ml, Cultrex), Y‐27632 (10 μM, PeproTech) and Doxycycline (2 μg/ml, Alfa Aesar) on day 0. On day 1, Y‐27632 was withdrawn. On day 2, medium was replaced with Neurobasal medium supplemented with 1× B27 and 1× Glutamax (Thermo Fisher Scientific) containing BDNF, NT‐3 and 1 μg/ml Doxycycline. Starting on day 4, half of the medium was replaced every other day thereafter. On day 7, the cells were treated with Accutase (Thermo Fisher Scientific) and plated at 3–4 × 104/cm2 on Matrigel‐coated tissue plates. Doxycycline was withdrawn on day 10.
Western blotting
At the indicated times, ES cells, iNeurons, or HeLa cells were washed on ice in 1× PBS, harvested and pellet wash with 1× PBS and resuspended in 8 M urea buffer (8 M urea, 150 mM TRIS pH 7.4, 50 mM NaCl, PhosSTOP phosphatase inhibitor cocktail). Resuspended cell lysates were sonicated for 10 s and debris pelleted at 13,000 rpm for 10 min. Protein concentration was determined by BCA assay according to manufacturer's instructions (Thermo Fisher Scientific, 23227). Indicated amounts of proteins were resuspended in 1× LDS + 100 mM DTT and boiled for 10 min at 85°C. Equal amount of protein and volume were loaded run on 4–20% Bis‐Tris, 8% Tris‐NuPAGE gels for 5 min at 100 V, 5 min at 150 V and then run at 200 V for the required time. Gels were transferred via wet transfer system onto PDVF membranes for Western Blotting. Chemiluminescence and colorimetric images were acquired using a BioRad ChemiDoc MP imaging system. Images from Western Blots were exported and analyzed using Image Lab and ImageJ/FiJi (Schindelin et al, 2012).
Expression, purification and analysis of SKP1‐FBXO7 complexes
All constructs were prepared utilizing standard molecular biological techniques and verified by Sanger sequencing. The cDNAs coding for HsFbxo7‐129‐398 and Skp1 preceding a second RBS were cloned into pGEX4T1 as previously described for other substrate receptor/Skp1 complexes (Schulman et al, 2000) (pGEX4T1‐TEV‐HsFbxo7‐129‐398/HsSkp1). The cDNA coding for HsPI31‐1‐151 (with N‐terminal TEV cleavable His8‐tag) was cloned into pRSF1b (pRSF1b‐His8‐TEV‐HsPI31‐1‐151). For co‐expression of Fbxo7/Skp1/PI31 complexes both plasmids were co‐transformed into E. coli BL21 Rosetta (DE3). Cultures were grown in Terrific Broth medium, and at an OD600 of 0.8 expression was induced with 0.5 mM IPTG, and E. coli were further cultured at 18°C for 16 h. Fbxo7/Skp1/PI31 complexes were purified by sequential standard GST and His‐affinity chromatography. Affinity tags were cleaved by incubation with TEV protease at 4°C for 16 h, and the complexes were further purified by preparative size exclusion chromatography (SEC) in buffer A (25 mM HEPES pH7.5 (KOH), 150 mM NaCl, 1 mM DTT) on a Superdex 200 Increase 10/300 GL column. Fractions of interest were pooled, aliquoted, snap frozen in liquid N2, and stored at −80°C until further usage. HsCul1‐1‐410 was expressed as GST‐fusion protein and purified as described previously (Hopf et al, 2022).
Analytical size exclusion chromatography (SEC)
Analytical SEC was carried out on an ÄKTApure system (GE Healthcare) equipped with a Superdex 200 Increase 10/300 GL column (Cytiva), in buffer A (25 mM HEPES pH7.5 (KOH), 150 mM NaCl, 1 mM DTT). In brief, samples (100 μl at a concentration of 45 μM) of HsCul1‐1‐410, HsFbxo7‐129‐398/HsSkp1/HsPI31‐1‐151, or HsFbxo7‐129‐398/HsSkp1/HsPI31‐1‐151 + HsCul1‐410 (preincubated at 37°C for 10 min before loading) were applied to the column with a flow rate of 1 ml/min, UV absorbance was recorded at 280 nm, fractions of 200 μl were collected, and analyzed by SDS–PAGE.
Proteomics
Proteomics—General sample preparation
Sample preparation of proteomic analysis of whole‐cell extract from HeLa, hESC, NPC, and iNeurons was performed according to previously published studies (Ordureau et al, 2020, 2021). Cells were harvested on ice and plates were washed twice with 1× PBS and detached in 1× PBS using cell scraper. After pelleting at 2,000 rpm for 5 min at 4°C, cells were washed 2× with 1× PBS and resuspended in 8 M urea buffer (composition stated above). After sonification for 10 s, resuspended cells were pelleted for 10 min at 13,000 rpm. Protein concentration was determined using BCA kit (Thermo Fisher Scientific, 23227).
Unless otherwise noted, proteomics and data analysis was performed as described (Ordureau et al, 2020, 2021). Briefly, protein extracts (100 μg) were subjected to disulfide bond reduction with 5 mM TCEP (room temperature, 10 min) and alkylation with 25 mM chloroacetamide (room temperature, 20 min). Methanol–chloroform precipitation was performed prior to protease digestion. In brief, four parts of neat methanol were added to each sample and vortexed, one part chloroform was then added to the sample and vortexed, and finally three parts water was added to the sample and vortexed. The sample was centrifuged at 6,000 rpm for 2 min at room temperature and subsequently washed twice with 100% methanol. Samples were resuspended in 100 mM EPPS pH8.5 containing 0.1% RapiGest and digested at 37°C for 8 h with Trypsin at a 100:1 protein‐to‐protease ratio. Samples were acidified with 1% formic acid for 15 min and subjected to C18 solid‐phase extraction (SPE) (Sep‐Pak, Waters).
Proteomics—Quantitative proteomics using TMT
Tandem mass tag labeling of each sample (50 mg peptide input) was performed by adding 5 μl of the 25 ng/ml stock of TMTpro reagent along with acetonitrile to achieve a final acetonitrile concentration of approximately 30% (v/v). Following incubation at room temperature for 1 h, the reaction was quenched with hydroxylamine to a final concentration of 0.5% (v/v) for 15 min. The TMTpro‐ labeled samples were pooled together at a 1:1 ratio. The sample was vacuum centrifuged to near dryness, and following reconstitution in 1% FA, samples were desalted using C18 SPE (50 mg, Sep‐Pak, Waters), according to manufacturer protocol.
Dried TMTpro‐labeled peptides (~ 300 μg) were resuspended in 10 mM NH4HCO3 pH 8.0 and fractionated using basic pH reverse phase HPLC (Wang et al, 2011). Briefly, samples were offline fractionated over a 90 min run, into 96 fractions by high pH reverse‐phase HPLC (Agilent LC1260) through an Aeris peptide xb‐c18 column (Phenomenex; 250 mm × 3.6 mm) with mobile phase A containing 5% acetonitrile and 10 mM NH4HCO3 in LC‐MS grade H2O, and mobile phase B containing 90% acetonitrile and 10 mM NH4HCO3 in LC‐MS grade H2O (both pH 8.0). The 96 resulting fractions were then pooled in a non‐continuous manner into 24 fractions (as outlined in figure S5 of Paulo et al, 2016) and sets of 12 fractions (even or odd numbers) were used for subsequent mass spectrometry analysis. Fractions were vacuum centrifuged to near dryness. Each consolidated fraction was desalted via StageTip, dried again via vacuum centrifugation, and reconstituted in 5% acetonitrile, 1% formic acid for LC‐MS/MS processing.
TMT proteomics samples were subjected to analysis using a Orbitrap Fusion Lumos Tribrid mass spectrometer (Thermo Fisher Scientific, San Jose, CA) online with Proxeon EASY‐nLC1200 liquid chromatography (Thermo Scientific). Peptides were resuspended in 5% ACN/5% FA and 10% of the samples were loaded on a 35 cm analytical column (100 mm inner diameter) packed in‐house with Accurcore150 resin (150 Å, 2.6 mm, Thermo Fisher Scientific, San Jose, CA) for LC‐MS analysis. Peptide separation was performed with a gradient of acetonitrile (ACN, 0.1% FA) from 3 to 13% (0–83 min) and 13 to 28% (80–83 min) during a 90 min run.
For the HeLa TMTpro proteomic samples, LC‐hrMS/MS was combined with three optimized CV parameters on the FAIMS Pro Interface to reduced precursor ion interference (Schweppe et al, 2019). Data‐dependent acquisition (DDA) was performed by selecting the most abundant precursors from each CV's (−40/−60/−80) MS1 scans for hrMS/MS over a 1–1.5 s duty cycle (1 s/1.5 s/1 s respectively). The MS1 scan parameters include a 400–1,600 m/z mass range at 60,000 resolution (at 200 Th) with 4 × 105 automated gain control (AGC) (100%), and a maximum injection time (max IT) of 50 ms. Precursors (z = 2–5) were isolated with 0.7 Th (quadrupole), fragmented with high‐energy C‐trap dissociation (HCD) at 36 normalized collision energy (NCE), and subjected to hrMS/MS on the Orbitrap at 50,000 resolution (at 200 Th), 120 ms max IT, fixed first mass 110 Th, and 1.0 × 105 AGC (200%). Precursors were placed on 90 s dynamic exclusion (± 10 ppm) to prevent redundant sampling.
In the iNeuron TMTpro experiments, the same FAIMS and MS1 parameters were implemented (with 1.25 s duty cycle/CV for DDA) with the Multi‐Notch SPS‐MS3 acquisition method (McAlister et al, 2014), to further reduce ion interference in TMT reporter quantification (Paulo et al, 2016). Most abundant precursors (with 120 s dynamic exclusion ± 10 ppm) were selected from MS1 scans, isolated using the quadrupole (0.6 Th isolation), fragmented with collision‐induced dissociation (CID) at 35 NCE, and subjected to MS/MS in the ion trap (turbo scan speed, 35 ms max IT, 1.0 × 104 AGC). Using Real Time Search analysis software (Erickson et al, 2019; Schweppe et al, 2020), a synchronous‐precursor‐selection (SPS) API‐MS3 scan was collected on the top 10 most intense b‐ or y‐ions from the matched peptide identification (determined by an online search of its respective MS/MS scan). MS3 scans were performed on the Orbitrap (AGC 2.0 × 105; NCE 55; max IT 250 ms, 50,000 resolution at 200 Th). To increase quantitative sampling (MS3 scans) of proteins during each mass spectrometry injection, a 2 peptide per protein per sample closeout was set. This ensures no more than two peptide‐spectrum matches per protein (that pass quality filters) are subjected to MS3 scans, reducing redundant protein MS sampling and potentially increasing proteome depth (Schweppe et al, 2020).
Proteomics data analysis
Mass spectrometry raw data were converted to mzXML and monoisotopic peaks were reassigned using Monocle (Rad et al, 2021). Mass spectra were database searched using Sequest algorithm (2019.01 rev. 5; Eng et al, 1994) against the Human Reference Proteome Uniprot database appended with sequences of common contaminates and reverse sequences of proteins as decoys, for target‐decoy competition (Elias & Gygi, 2007). Sequest search parameters include: 50 ppm precursor tolerance, 0.9 Da product ion tolerance, trypsin endopeptidase specificity (C‐terminal to [KR], 2 max missed cleavages), static modifications on peptide N‐terminus and lysines with TMTpro tags (+304.207 Da) and carbamidomethylation on cysteines (57.021 Da), and variable modification of oxidation on methionines (+15.995 Da). Peptide‐spectrum matches were filtered at 2% false discovery rate (FDR) using linear discriminant analysis (Huttlin et al, 2010), based on XCorr, DeltaCn, missed cleavages, peptide length, precursor mass accurracy, fraction of matched product ions, charge state, and number of modifications per peptide (additionally restricting PSM Xcorr > 1 and peptide length > 6). Following a 2% protein FDR filter (Savitski et al, 2015), PSMs reporter ion intensities were quantified (most intense centroid within 0.003 Da of theoretical TMT reporter mass), filtered based on a precursor isolation specificity > 0.5, and filtered by a summed signal‐to‐noise across TMT channels > 100.
Protein quantification was performed via the summation (weighted average) of its constituent PSMs' reporter intensities and TMT channels were normalized for protein input to total TMT channel intensity across all quantified PSMs (adjusted to median total TMT intensity for the TMT channels) (Plubell et al, 2017). Log2 normalized summed protein reporter intensities were compared using a Student's t‐test and P‐values were corrected for multiple hypotheses using the Benjamini–Hochberg adjustment (Benjamini & Hochberg, 1995). Resultant q‐values and mean log2 fold changes between conditions were used to generate volcano plots. Hotelling T 2 analysis was performed using the normalized summed protein reporter ion intensities and the timecourse (v. 1.66.0; Tai, 2022) package in R.
The annotation list for the subcellular localization of organellar protein markers was derived from previously published high confidence HeLa dataset (Itzhak et al, 2016; “high” & “very high” confidence) and additional manual entries (Ordureau et al, 2021). MitoCharta 3.0 (Rath et al, 2021) was used for mitochondrial annotation, whereas annotations for ribosome and autophagy components were used from previous studies (An et al, 2020). Transcription factor and neuronal development markers were based on previously published databases and publications (Lambert et al, 2018; Ordureau et al, 2021). Figures were generated using a combination of Excel, R (v.4.2.0) in RStudio (2022.07.01 Build 554), Perseus (v1.6.5; Tyanova & Cox, 2018), GraphPad Prism (v9.1.0), and Adobe Illustrator (26.2.1).
The Dataset [Link], [Link] contains the quantified proteins as well as associated TMT reporter ratio to control channels used for quantitative analysis. The TMT reporter channel assignment used for this study are added to each table.
Interaction proteomic analysis
For interaction analysis of FBXO7 and PINK1, interaction proteomics data from the Bioplex Interactome (Huttlin et al, 2021) was extracted for HEK293T and HCT116 samples (Dataset EV1 and EV2). The number of replicates in which a peptide‐spectral match (PSM) mapped to PINK1's or FBXO7's the Bioplex 3.0 interactomes was plotted via a network diagram in R (packages to add from R include; igraph 1.3.1, tidygraph 1.2.1, and ggraph 2.0.5).
Microscopy
Macros and pipelines used in this work can be found on GitHub (https://github.com/harperlaboratory/FBXO7.git) or Zenodo (https://zenodo.org/record/7803294).
Live‐cell confocal microscopy for mitophagic flux analysis over differentiation (mt‐mKeimaXL )
For quantitative Keima‐flux analysis (Ordureau et al, 2020), hESC were seeded on day 4 of differentiation into 6‐well 1.5 high‐performance glass bottom plates (Cellvis, P06‐1.5H‐N) and further differentiated in the vessel for the indicated times until reaching appropriate confluency and imaged. iNeurons were imaged using a Yokogawa CSU‐W1 spinning disk confocal on a Nikon Eclipse Ti‐E motorized microscope. The system is equipped with a Tokai Hit stage top incubator and imaging was performed at 37°C, 5% CO2 and 95% humidity under a Nikon Plan Apo 60×/1.40 NA immersion oil objective lens. For ratiometic imaging, mtKeimaXL were excited in sequential manner with a Nikon LUN‐F XL solid state laser combiner ([laser line—laser power]: 445–80 mW, 561–65 mW) using a Semrock Di01‐T445/515/561 dichroic mirror. Fluorescence emissions were collected through a Chroma ET605/52 m [for 445 nm] and a 568 Chroma ET605/52 m [for 561 nm], filters, respectively (Chroma Technologies). Confocal images were acquired with a Hamamatsu ORCA—Fusion BT CMOS camera (6.5 μm2 photodiode, 16‐bit) camera and NIS‐Elements image acquisition software. Consistent laser intensity and exposure time were applied to all the samples, and brightness and contrast were adjusted equally by applying the same minimum and maximum display values in ImageJ/FiJi (Schindelin et al, 2012). Image quantification was performed in ImageJ/FiJi using custom‐written batch‐macros.
In brief, raw confocal images of mitochondrial targeted mt‐mKeimaXL were divided [ex:561 nm/ex:445 nm] resulting in a ratiometic image of only acidic Keima‐puncta. These signals were subjected to background subtraction (rolling kernel size 25, sliding paraboloid) and converted to binary objects. The “Analyze Particles…” command (pixel size exclusion: 0.5‐∞, exclude edge objects) was used to measure foci‐abundance and other morphological parameters. Results for each image‐stack saved as .csv files, together with the original ratiometic .tiff file for QC purposes. Unless stated otherwise all images represent z‐projections. Statistical analysis and plotting of microscopy data was performed in Prism (v9.1.0, GraphPad).
Immunocytochemical analysis
iNeurons or HeLa cells were fixed with warm 4% paraformaldehyde (Electron Microscopy Science, #15710, purified, EM grade) in PBS at 37°C for 30 min and permeabilized with 0.5% Triton X‐100 in PBS for 15 min at room temperature. After three washes with 0.02% Tween20 in PBS (PBST), cells were blocked for 10 min in 3% BSA‐1× PBS at room temperature and washed again three times in PBST. Cells were incubated for 3 h in primary antibodies in 3% BSA‐1× PBS and washed three times with PBST. Secondary antibodies (Thermo Scientific, 1:400 in 3% BSA‐1× PBS) where applied for 1 h at room temperature. To stain nuclei, Hoechst33342 (1:10,000) was added for 5 min to cells in PBST and final three washes performed before mounting in Vectashield (Vector Laboratories, H‐1000‐10). Primary and secondary antibodies used in this study can be found in the Reagents and Tools table.
Fixed‐cell microscopy—General acquisition parameters
Immunofluorescently labeled Hela or iNeurons (antibodies indicated in figures and figure legends and details in Reagents and Tools table) were imaged at room temperature using a Yokogawa CSU‐W1 spinning disk confocal on a Nikon Eclipse Ti‐E motorized microscope equipped with a Nikon Plan Apochromat 40×/0.40 NA air‐objective lens, Nikon Apochromat 60×/1.42 NA oil‐objective lens and a Plan Apochromat 100×/1.45 NA oil‐objective lens. Signals of 405/488/568/647 fluorophores were excited in sequential manner with a Nikon LUN‐F XL solid state laser combiner ([laser line—laser power]: 405 nm–80 mW, 488 nm–80 mW, 561 nm–65 mW, 640 nm–60 mW) using a Semrock Di01‐T405/488/568/647 dichroic mirror. Fluorescence emissions were collected with Chroma ET455/50 m [405 nm], 488 Chroma ET525/50 m [488 nm], 568 Chroma ET605/52 m [561 nm], 633 Chroma ET705/72 m [640 nm] filters, respectively (Chroma Technologies). Confocal images were acquired with a Hamamatsu ORCA‐Fusion BT CMOS camera (6.5 μm2 photodiode, 16‐bit) camera and NIS‐Elements image acquisition software. Consistent laser intensity and exposure time were applied to all the samples, and brightness and contrast were adjusted equally by applying the same minimum and maximum display values in ImageJ/FiJi (Schindelin et al, 2012).
Microscopy‐based mitochondrial morphology measurements in iNeurons
For quantitative measurement of mitochondrial morphology in d12 iNeurons under fed or mitophagy conditions, HeLa cells were seeded on 6‐well 1.5 high‐performance glass bottom plates (Cellvis, P06‐1.5H‐N) and mitophagy induced for the indicated time durations. Alternatively, hESC were seeded on day 4 of differentiation into 6‐well 1.5 high‐performance glass bottom plates (Cellvis, P06‐1.5H‐N) and differentiated in the vessel into iNeurons and mitophagy induced for the indicated time durations. Cells were fixed and stained as described above. Z‐stacks were acquired with a Nikon Plan Apo 100×/1.45 NA oil‐objective lens and with the parameters stated above. Image quantification was performed in ImageJ/FiJi using custom‐written batch‐macros.
Statistical analysis and plotting of microscopy data was performed in Prism (v9.1.0, GraphPad). Primary and secondary antibodies used in this study can be found in the Reagents and Tools table.
Microscopy‐based mtDNA turnover measurements in HeLa and iNeurons
For quantitative measurement of mtDNA turnover after AO‐induced mitophagy, HeLa cells were seeded on 6‐well 1.5 high‐performance glass bottom plates (Cellvis, P06‐1.5H‐N) and mitophagy induced for the indicated time durations. Alternatively, hESC were seeded on day 4 of differentiation into 6‐well 1.5 high‐performance glass bottom plates (Cellvis, P06‐1.5H‐N) and differentiated in the vessel into iNeurons and mitophagy induced for the indicated time durations. Cells were fixed and stained as described above, but primary antibody incubation with aDNA (1:200) was performed overnight. Z‐stacks were acquired with a Nikon Plan Apo 100×/1.45 NA oil‐objective lens and with the parameters stated above. Image quantification was performed in ImageJ/FiJi using custom‐written batch‐macros. In brief, both aDNA and nuclear signals were converted to binary files and the nuclear signal subtracted from the aDNA signal, resulting in an image stack containing only the mtDNA intensities. The “Analyze Particles…” command (pixel size exclusion: 0.05–3, exclude edge objects) was used to measure morphological features and results for each image‐stack saved as .csv files, together with the analyzed binary‐mask overlay .tiff file for QC purposes. Number of mtDNA signals were normalized to cell number found in the same image stack. Statistical analysis and plotting of microscopy data was performed in Prism (v9.1.0, GraphPad). Primary and secondary antibodies used in this study can be found in the Reagents and Tools table.
Microscopy‐based measurements of p62 recruitment in HeLa
For quantitative measurement of p62 recruitment to mitochondria, HeLa control and knockout cells were seed in 1.5 high‐performance glass bottom plates and doxycycline added over night to induce Parkin expression. Mitophagy was induced using Oligomycin A/Antimycin A for 16 h in full DMEM in presence of doxycycline. Cells were fixed and stained as stated above and imaged using a Nikon Plan Apo 60×/1.42 NA air‐objective lens and with the parameters stated above. 8 μM z‐stacks were taken for each selected field of view.
Image quantification was performed in ImageJ/FiJi using custom‐written batch‐macros. The p62 channel was filtered (Gaussian Blue, sigma = 2) and converted into binary files using the Intermodes thresholding method. p62 spots were counted using the “Analyze Particles…” function (pizel size exclusion: 0.1–30, exclude edge objects) and results for each image‐stack saved as .csv files, together with the analyzed binary‐mask overlay .tiff file for QC purposes. The DAPI channel was used to count nuclei for per cell normalization. Statistical analysis and plotting of microscopy data was performed in Prism (v9.1.0, GraphPad). Primary and secondary antibodies used in this study can be found in the Reagents and Tools table.
Microscopy‐based pUb‐coverage measurements of mitochondria in iNeurons
For quantitative measurement of pUb coverage over mitochondria AO‐induced mitophagy, hESC were seeded on day 4 of differentiation into 6‐well 1.5 high‐performance glass bottom plates (Cellvis, P06‐1.5H‐N) and differentiated in the vessel into iNeurons and mitophagy induced for the indicated time durations. Cells were fixed and stained as described above, using anti‐HSP60 to label mitochondria and anti‐pUb (Ser65) to label pUb. 3–5 10 μm thick z‐stacks per replicate per sample were acquired using a Nikon Plan Apo 60×/1.42 NA air‐objective lens and with the parameters stated above.
Image quantification was performed in ImageJ/FiJi using custom‐written batch‐macros. In brief, mitochondrial signal was filtered (Gaussian Blur, sigma = 2), converted into binary files and holes in the resulting mask filled. pUb channel was thresholded into a binary file (Triangle method); these masks were measured using the “Analyze Particles…” command (pixel size exclusion: 0.5‐∞, exclude edge objects) and results for each image‐stack saved as .csv files, together with the analyzed binary‐mask overlay .tiff file for QC purposes. % of mitochondrial pUb coverage was calculated and normalized to [t] = 6 h AO. Statistical analysis and plotting of microscopy data was performed in Prism (v9.1.0, GraphPad). Primary and secondary antibodies used in this study can be found in the Reagents and Tools table.
Microscopy‐based evaluation of Parkin translocation and mitophagy in FBXO7 −/− cell lines
For quantitative measurement of Parkin translocation kinetics in FBXO7−/− cell lines, stable HeLa or HEK293T cell lines expressing GFP‐Parkin were created. WT and knockout cell lines were seeded into 12, 24 or 96‐well 1.5 high‐performance glass bottom plates (Cellvis, P12/24/96‐1.5H‐N) 2 days prior to experimental manipulation. Mitophagy was induced as described above using Antimycin A/Oligomycin A or CCCP for 1 h. Treated and control (fed) cells were fixed as described above, stained for mitochondria (HSP60), Parkin (GFP), and DNA (SpyDNA‐555) and 8 μm z‐stacks were acquired with a Nikon Plan Apo 60×/142 NA oil‐objective lens and with the parameters stated above. HEK293T cells were stained for mitochondria (HSP60), Parkin (GFP), and DNA (Hoechst) and 8 μm z‐stacks were acquired with a Nikon Plan Apochromat 40×/0.40 NA air‐objective lens, using the HCA‐module in NIS‐Elements.
For quantitate single‐cell analysis, a total of ~ 240 (HeLa) or 135 (HEK293T) randomly xy‐marked stacks were acquired, and MIPs used for subsequent analysis. CellProfiler (Stirling et al, 2021) was used for the quantitative analysis of single‐cells and mitochondrial objects (segmentation and analysis pipelines can be found on GitHub). Plotting of microscopy data was performed in Prism (v9.1.0, GraphPad) and R using the following libraries: tidyverse, dplyr, tibble, viridis, ggplot2, ggridges, ggsci. Primary and secondary antibodies used in this study can be found in the Reagents and Tools table.
Microscopy‐based evaluation pUb recruitment after G‐TPP treatment in HeLa FBXO7 −/− cell lines
For quantitative measurement of pUb recruitment to mitochondria after G‐TPP treatment, HeLa cell lines were seeded into 96‐well 1.5 high‐performance glass bottom plates (Cellvis, 96‐1.5H‐N) 2 days prior to experimental manipulation. Cells were treated for the indicated times and fixed and immunofluorescently labeled as described above. Cells were stained for pUb, Parkin (GFP) and DNA (Hoechst33342) and 8 μm z‐stacks were acquired with a Nikon Plan Apochromat 40×/0.40 NA air‐objective lens, using the HCA‐module in NIS‐Elements.
9 stacks of randomly selected field of views were taken and MIPs used for analysis using CellProfiler. Plotting of microscopy data was performed in Prism (v9.1.0, GraphPad). Primary and secondary antibodies used in this study can be found in the Reagents and Tools table.
Immunocytochemical sample preparation for 3D‐SIM
Sample preparation and SIM acquisition guidelines from (Kraus et al, 2017) were used as a basis for the super‐resolution analysis of pUb spreading after mitophagy induction. iNeurons or HeLa cells were seeded on 18 × 18 mm Marienfeld Precision cover glasses thickness No. 1.5H (tol. ± 5 μm). Cover glasses were coated, if necessary, for hESC/iNeuron culture as described above. After experimental manipulation, cells were fixed with warm paraformaldehyde 3% Glutaraldehyde 0.35% in 0.1 M Sodium Cacodylate, pH 7.4 (Electron Microscopy Science) at 37°C for 30 min and permeabilized with 0.5% Triton X‐100 in PBS for 15 min at room temperature. After three washes with 0.02% Tween20 in PBS (PBST), cells were blocked for 10 min in 3% BSA‐1×PBS at room temperature and washed again three times in PBST. iNeurons were incubated overnight in primary antibodies in 3% BSA‐1×PBS and washed three times with PBST. Secondary antibodies (Thermo Scientific, 1:400 in 3% BSA‐1×PBS) where applied for 1 h at room temperature. To stain nuclei, DAPI was added for 5 min to cells in PBST, washed three times for 5 min in 1×PBST before a post‐fixation in 4% paraformaldehyde was performed. After 2 washes in PBST, coverslips were washed once in 1×PBS and mounted in Vectashield (Vector Laboratories, H‐1000‐10) on glass slides. Primary and secondary antibodies used in this study can be found in the Reagents and Tools table.
3D‐SIM microscopy—acquisition parameters
3D‐SIM microscopy was performed on a DeltaVision OMX v4 using an Olympus 60×/1.42 NA Plan Apo oil objective (Olympus, Japan). The instrument is equipped with 405, 445, 488, 514, 568 and 642 nm laser lines (all ≥ 100 mW) and images were recorded on a front‐illuminated sCMOS (PCO Photonics, USA) in 512x512px image size mode, 1× binning, 125 nm z‐stepping and with 15 raw images taken per z‐plane (5 phase‐shifts, 3 angles). Raw image data was computationally reconstructed using CUDA‐accelerated 3D‐SIM reconstruction code (https://github.com/scopetools/cudasirecon) based on (Gustafsson et al, 2008). Optimal optical transfer function (OTF) was determined via an in‐house build software, developed by Talley Lambert from the NIC/CBMF (GitHub: https://github.com/tlambert03/otfsearch, all channels were registered to the 528 nm output channel, Wiener filter: 0.002, background: 90).
3D‐SIM microscopy—pUb—mitochondria 3D renderings & analysis
3D renderings of 3D‐SIM images were performed in UCSF Chimera X (https://www.cgl.ucsf.edu/chimerax/) using 32‐bit .tiff image stacks. Image channels were sequentially imported and visualized as surfaces using the “Volume Viewer” tool. Surface representations were cleaned up using the “High dust” command, based on size filtering thresholding.
3D‐analysis of HeLa and iNeuron datasets from 3D‐SIM datasets was performed using Imaris (Oxford Instruments, v9.7). After converting all multi‐color .tiff to native .ims files, import into Imaris Arena and global background subtraction, mitochondrial and pUb objects were segmented from seeds (XY starting diameter: 0.08 μm == pixel size of images), segmented based on automatic thresholding with local background subtraction and splitting of touching objects (0.4 μm). Objects were piped into Imaris Vantage module for further analysis. In Vantage, nearest neighbor distances of pUb to pUb and between pUb and mitochondria, as well as volume of segmented objects were computed. This pipeline was tested on WT control cells and then applied for batch processing on all other genotype to allow for unbiased segmentation and analysis. On average, 10 cells were analyzed per genotype per condition (121 total) for HeLa cells, and 12 cells (139 total) for iNeurons.
Unless stated otherwise, all images depicted in figures are maximum‐intensity projections.
Flow cytometry‐based measurement of mitophagic flux
HeLa HFT/TO‐PRKN cells of the indicated genotype (control, PINK1−/−, FBXO7−/−) expressing mtKeima (Heo et al, 2015) were seeded into 12‐well dishes. Upon reaching 60% confluency, 2 μg/ml doxycycline was added to induce Parkin expression for at least 8 h before depolarization of mitochondria. Following Parkin induction, cells were treated with Antimycin A (5 μM) and Oligomycin (10 μM) for the indicated number of hours. Bafilomycin A (25 nM, Sigma‐Aldrich, B1793)‐treated samples were treated for 3 h. Control cells were fed 3 h before experimental manipulation. To harvest cells for analysis, each well was washed with 1 ml PBS then treated with 100 μl of 0.25% trypsin for 3 min at room temperature, resuspended in 300 μl of DMEM with 10% FBS. 200 μl of each sample was transferred to a flat‐bottom 96‐well plate for analysis by flow cytometry.
hESCs were seeded at day 4 into coated 6‐well dishes and differentiated. At day 12, mitophagy was induced with Antimycin A (5 μM) and Oligomycin (10 μM) or Bafilomycin A (25 nM, Sigma‐Aldrich, B1793) for the indicated number of hours. Control cells were fed 2 h before experimental manipulation. Cells were dissociated from the wells using Accutase and resuspended in 600 μl ND2 medium +10 μM Y27632 and filtered through a cell strainer cap tube (Corning, 352235).
Analysis of a population of at least 10,000 cells was performed on an Attune NxT (Thermo Fisher Scientific) detecting neutral mtKeima signal with excitation at 445 nm and emission 603 nm with a 48 nm bandpass and acidic mtKeima with 561 nm excitation and emission 620 nm and a 15 nm band pass. mtKeima ratio was analyzed as previously described (An & Harper, 2018). Briefly, acidic:neutral Keima ratios were measured by gating a population of well‐behaved single cells using forward and side scatter, followed by calculation of acidic:neutral mtKeima ratio on a per‐cell basis in FlowJo Software (FlowJo, LLC).
Quantification and statistical analysis
Unless stated otherwise all quantitative experiments were performed in triplicate and average with SEM or SD as indicated in legends reported. All the numerical data used in plots can be found in Dataset EV9.
Author contributions
Felix Kraus: Conceptualization; resources; data curation; formal analysis; validation; investigation; visualization; methodology; writing – original draft; project administration; writing – review and editing. Ellen A Goodall: Conceptualization; data curation; formal analysis; validation; investigation; visualization; writing – original draft; writing – review and editing. Ian R Smith: Resources; data curation; software; formal analysis; validation; investigation; visualization; methodology; writing – original draft; writing – review and editing. Yizhi Jiang: Resources; validation; investigation; methodology. Julia C Paoli: Resources; validation; project administration; writing – review and editing. Frank Adolf: Investigation; methodology. Jiuchun Zhang: Resources. Joao A Paulo: Resources; software; investigation; methodology. Brenda A Schulman: Supervision; funding acquisition; project administration. J Wade Harper: Conceptualization; data curation; formal analysis; supervision; funding acquisition; writing – original draft; project administration; writing – review and editing.
Disclosure and competing interests statement
J.W.H. is a consultant and founder of Caraway Therapeutics and a founding scientific advisory board member of Interline Therapeutics. B.A.S. is an EMBO council member. B.A.S. is a member of the scientific advisory boards of Interline Therapeutics and BioTheryX, and a co‐inventor of intellectual property related to DCN1 inhibitors licensed to Cinsano. No other authors declare a conflict of interest.
Supporting information
Appendix S1
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Dataset EV6
Dataset EV7
Dataset EV8
Dataset EV9
PDF+
Acknowledgements
This work was supported by Aligning Science Across Parkinson's (ASAP) (J.W.H. and B.A.S.), the NIH (R01 NS083524 to J.W.H. and RO1 GM132129 to J.A.P.), The Max Planck Society (B.A.S.), and the Harvard Medical School Cell Biology Initiative for Molecular Trafficking and Neurodegeneration. Michael J Fox Foundation administers the grant ASAP‐000282 on behalf of ASAP and itself. For the purpose of open access, the author has applied a CC‐BY public copyright license to the Author Accepted Manuscript (AAM) version arising from this submission. We thank Laura Pontano Vaites for help with the BioPlex 3.0 interactome analysis. We thank Jennifer Waters, Talley Lambert, Federico Gasparoli and Rylie Walsh in the Nikon Imaging Center and the Cell Biology Microscopy Facility (CBMF) at Harvard Medical School for microscopy support and technical support.
EMBO reports (2023) 24: e56399
Data availability
Proteomic data and analysis files part of this study are deposited at ProteomeXchange Consortium by the PRIDE partner (Deutsch et al, 2020; Perez‐Riverol et al, 2022). The PRIDE project identification number is PXD037797. Macros and pipelines used in this work can be found on GitHub (https://github.com/harperlaboratory/FBXO7.git) or Zenodo (https://zenodo.org/record/7803294). Raw files associated with this work are deposited on Zenodo (https://zenodo.org/record/7803294).
References
- Al Rawi S, Simpson L, McDonald NQ, Chernuha V, Elpeleg O, Zeviani M, Barker RA, Spiegel R, Laman H (2022) Study of an FBXO7 patient mutation reveals Fbxo7 and PI31 co‐regulate proteasomes and mitochondria. bioRxiv 10.1101/2021.12.22.473884 [PREPRINT] [DOI] [PubMed]
- An H, Harper JW (2018) Systematic analysis of ribophagy in human cells reveals bystander flux during selective autophagy. Nat Cell Biol 20: 135–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- An H, Ordureau A, Korner M, Paulo JA, Harper JW (2020) Systematic quantitative analysis of ribosome inventory during nutrient stress. Nature 583: 303–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antico O, Ordureau A, Stevens M, Singh F, Nirujogi RS, Gierlinski M, Barini E, Rickwood ML, Prescott A, Toth R et al (2021) Global ubiquitylation analysis of mitochondria in primary neurons identifies endogenous Parkin targets following activation of PINK1. Sci Adv 7: eabj0722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader M, Benjamin S, Wapinski OL, Smith DM, Goldberg AL, Steller H (2011) A conserved F box regulatory complex controls proteasome activity in drosophila. Cell 145: 371–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B 57: 289–300 [Google Scholar]
- Bingol B, Tea JS, Phu L, Reichelt M, Bakalarski CE, Song Q, Foreman O, Kirkpatrick DS, Sheng M (2014) The mitochondrial deubiquitinase USP30 opposes parkin‐mediated mitophagy. Nature 510: 370–375 [DOI] [PubMed] [Google Scholar]
- Burchell VS, Nelson DE, Sanchez‐Martinez A, Delgado‐Camprubi M, Ivatt RM, Pogson JH, Randle SJ, Wray S, Lewis PA, Houlden H et al (2013) The Parkinson's disease‐linked proteins Fbxo7 and Parkin interact to mediate mitophagy. Nat Neurosci 16: 1257–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burman JL, Pickles S, Wang C, Sekine S, Vargas JNS, Zhang Z, Youle AM, Nezich CL, Wu X, Hammer JA et al (2017) Mitochondrial fission facilitates the selective mitophagy of protein aggregates. J Cell Biol 216: 3231–3247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C, Shi X, Jensen LE, Yokom AL, Fracchiolla D, Martens S, Hurley JH (2021) Reconstitution of cargo‐induced LC3 lipidation in mammalian selective autophagy. Sci Adv 7: eabg4922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Gulbranson DR, Hou Z, Bolin JM, Ruotti V, Probasco MD, Smuga‐Otto K, Howden SE, Diol NR, Propson NE et al (2011) Chemically defined conditions for human iPSC derivation and culture. Nat Methods 8: 424–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch EW, Bandeira N, Sharma V, Perez‐Riverol Y, Carver JJ, Kundu DJ, Garcia‐Seisdedos D, Jarnuczak AF, Hewapathirana S, Pullman BS et al (2020) The ProteomeXchange consortium in 2020: enabling ‘big data’ approaches in proteomics. Nucleic Acids Res 48: D1145–D1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Fonzo A, Dekker MC, Montagna P, Baruzzi A, Yonova EH, Correia Guedes L, Szczerbinska A, Zhao T, Dubbel‐Hulsman LO, Wouters CH et al (2009) FBXO7 mutations cause autosomal recessive, early‐onset parkinsonian‐pyramidal syndrome. Neurology 72: 240–245 [DOI] [PubMed] [Google Scholar]
- Dikic I, Elazar Z (2018) Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol 19: 349–364 [DOI] [PubMed] [Google Scholar]
- Elias JE, Gygi SP (2007) Target‐decoy search strategy for increased confidence in large‐scale protein identifications by mass spectrometry. Nat Methods 4: 207–214 [DOI] [PubMed] [Google Scholar]
- Eng JK, McCormack AL, Yates JR (1994) An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom 5: 976–989 [DOI] [PubMed] [Google Scholar]
- Erickson BK, Mintseris J, Schweppe DK, Navarrete‐Perea J, Erickson AR, Nusinow DP, Paulo JA, Gygi SP (2019) Active instrument engagement combined with a real‐time database search for improved performance of sample multiplexing workflows. J Proteome Res 18: 1299–1306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans CS, Holzbaur EL (2020) Degradation of engulfed mitochondria is rate‐limiting in Optineurin‐mediated mitophagy in neurons. Elife 9: e50260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang EF, Hou Y, Palikaras K, Adriaanse BA, Kerr JS, Yang B, Lautrup S, Hasan‐Olive MM, Caponio D, Dan X et al (2019) Mitophagy inhibits amyloid‐beta and tau pathology and reverses cognitive deficits in models of Alzheimer's disease. Nat Neurosci 22: 401–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiesel FC, James ED, Hudec R, Springer W (2017) Mitochondrial targeted HSP90 inhibitor Gamitrinib‐TPP (G‐TPP) induces PINK1/Parkin‐dependent mitophagy. Oncotarget 8: 106233–106248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming A, Bourdenx M, Fujimaki M, Karabiyik C, Krause GJ, Lopez A, Martin‐Segura A, Puri C, Scrivo A, Skidmore J et al (2022) The different autophagy degradation pathways and neurodegeneration. Neuron 110: 935–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladkova C, Maslen SL, Skehel JM, Komander D (2018) Mechanism of parkin activation by PINK1. Nature 559: 410–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodall EA, Kraus F, Harper JW (2022) Mechanisms underlying ubiquitin‐driven selective mitochondrial and bacterial autophagy. Mol Cell 82: 1501–1513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson MG, Shao L, Carlton PM, Wang CJ, Golubovskaya IN, Cande WZ, Agard DA, Sedat JW (2008) Three‐dimensional resolution doubling in wide‐field fluorescence microscopy by structured illumination. Biophys J 94: 4957–4970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper JW, Ordureau A, Heo JM (2018) Building and decoding ubiquitin chains for mitophagy. Nat Rev Mol Cell Biol 19: 93–108 [DOI] [PubMed] [Google Scholar]
- Heo JM, Ordureau A, Paulo JA, Rinehart J, Harper JW (2015) The PINK1‐PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote Mitophagy. Mol Cell 60: 7–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopf LVM, Baek K, Klügel M, von Gronau S, Xiong Y, Schulman BA (2022) Structure of CRL7FBXW8 reveals coupling with CUL1–RBX1/ROC1 for multi-cullin-RING E3-catalyzed ubiquitin ligation. Nat Struct Mol Biol 29: 854–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houlden H, Singleton AB (2012) The genetics and neuropathology of Parkinson's disease. Acta Neuropathol 124: 325–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang T, Fang L, He R, Weng H, Chen X, Ye Q, Qu D (2020) Fbxo7 and Pink1 play a reciprocal role in regulating their protein levels. Aging (Albany NY) 13: 77–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur JK, Kim K, Been KW, Baek G, Ye S, Hur JW, Ryu SM, Lee YS, Kim JS (2016) Targeted mutagenesis in mice by electroporation of Cpf1 ribonucleoproteins. Nat Biotechnol 34: 807–808 [DOI] [PubMed] [Google Scholar]
- Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villen J, Haas W, Sowa ME, Gygi SP (2010) A tissue‐specific atlas of mouse protein phosphorylation and expression. Cell 143: 1174–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin EL, Bruckner RJ, Navarrete‐Perea J, Cannon JR, Baltier K, Gebreab F, Gygi MP, Thornock A, Zarraga G, Tam S et al (2021) Dual proteome‐scale networks reveal cell‐specific remodeling of the human interactome. Cell 184: 3022–3040.e28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itzhak DN, Tyanova S, Cox J, Borner GH (2016) Global, quantitative and dynamic mapping of protein subcellular localization. Elife 5: e16950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Cardozo T, Lovering RC, Elledge SJ, Pagano M, Harper JW (2004) Systematic analysis and nomenclature of mammalian F‐box proteins. Genes Dev 18: 2573–2580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ (2010) Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol 191: 933–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, Banerjee S, Youle RJ (2014) PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol 205: 143–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kataura T, Otten EG, Rabanal‐Ruiz Y, Adriaenssens E, Urselli F, Scialo F, Fan L, Smith GR, Dawson WM, Chen X et al (2023) NDP52 acts as a redox sensor in PINK1/Parkin‐mediated mitophagy. EMBO J 42: e111372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama H, Kogure T, Mizushima N, Yoshimori T, Miyawaki A (2011) A sensitive and quantitative technique for detecting autophagic events based on lysosomal delivery. Chem Biol 18: 1042–1052 [DOI] [PubMed] [Google Scholar]
- Kazlauskaite A, Martinez‐Torres RJ, Wilkie S, Kumar A, Peltier J, Gonzalez A, Johnson C, Zhang J, Hope AG, Peggie M et al (2015) Binding to serine 65‐phosphorylated ubiquitin primes Parkin for optimal PINK1‐dependent phosphorylation and activation. EMBO Rep 16: 939–954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirk R, Laman H, Knowles PP, Murray‐Rust J, Lomonosov M, Meziane el K, McDonald NQ (2008) Structure of a conserved dimerization domain within the F‐box protein Fbxo7 and the PI31 proteasome inhibitor. J Biol Chem 283: 22325–22335 [DOI] [PubMed] [Google Scholar]
- Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T et al (2014) Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510: 162–166 [DOI] [PubMed] [Google Scholar]
- Kraus F, Miron E, Demmerle J, Chitiashvili T, Budco A, Alle Q, Matsuda A, Leonhardt H, Schermelleh L, Markaki Y (2017) Quantitative 3D structured illumination microscopy of nuclear structures. Nat Protoc 12: 1011–1028 [DOI] [PubMed] [Google Scholar]
- Labun K, Montague TG, Krause M, Torres Cleuren YN, Tjeldnes H, Valen E (2019) CHOPCHOP v3: expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res 47: W171–W174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert SA, Jolma A, Campitelli LF, Das PK, Yin Y, Albu M, Chen X, Taipale J, Hughes TR, Weirauch MT (2018) The human transcription factors. Cell 172: 650–665 [DOI] [PubMed] [Google Scholar]
- Lazarou M, Jin SM, Kane LA, Youle RJ (2012) Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev Cell 22: 320–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ (2015) The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524: 309–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Cai Z, Bomgarden RD, Pike I, Kuhn K, Rogers JC, Roberts TM, Gygi SP, Paulo JA (2021) TMTpro‐18plex: the expanded and complete set of TMTpro reagents for sample multiplexing. J Proteome Res 20: 2964–2972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Jones S, Minis A, Rodriguez J, Molina H, Steller H (2019) PI31 is an adaptor protein for proteasome transport in axons and required for synaptic development. Dev Cell 50: 509–524.e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Lear TB, Verma M, Wang KZ, Otero PA, McKelvey AC, Dunn SR, Steer E, Bateman NW, Wu C et al (2020) Chemical inhibition of FBXO7 reduces inflammation and confers neuroprotection by stabilizing the mitochondrial kinase PINK1. JCI Insight 5: e131834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlister GC, Nusinow DP, Jedrychowski MP, Wuhr M, Huttlin EL, Erickson BK, Rad R, Haas W, Gygi SP (2014) MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal Chem 86: 7150–7158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWilliams TG, Muqit MM (2017) PINK1 and Parkin: emerging themes in mitochondrial homeostasis. Curr Opin Cell Biol 45: 83–91 [DOI] [PubMed] [Google Scholar]
- Munch C, Harper JW (2016) Mitochondrial unfolded protein response controls matrix pre‐RNA processing and translation. Nature 534: 710–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, Youle RJ (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183: 795–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ (2010) PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 8: e1000298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng MYW, Wai T, Simonsen A (2021) Quality control of the mitochondrion. Dev Cell 56: 881–905 [DOI] [PubMed] [Google Scholar]
- Okatsu K, Koyano F, Kimura M, Kosako H, Saeki Y, Tanaka K, Matsuda N (2015) Phosphorylated ubiquitin chain is the genuine Parkin receptor. J Cell Biol 209: 111–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordureau A, Sarraf SA, Duda DM, Heo JM, Jedrychowski MP, Sviderskiy VO, Olszewski JL, Koerber JT, Xie T, Beausoleil SA et al (2014) Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol Cell 56: 360–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordureau A, Heo JM, Duda DM, Paulo JA, Olszewski JL, Yanishevski D, Rinehart J, Schulman BA, Harper JW (2015) Defining roles of PARKIN and ubiquitin phosphorylation by PINK1 in mitochondrial quality control using a ubiquitin replacement strategy. Proc Natl Acad Sci USA 112: 6637–6642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordureau A, Paulo JA, Zhang W, Ahfeldt T, Zhang J, Cohn EF, Hou Z, Heo JM, Rubin LL, Sidhu SS et al (2018) Dynamics of PARKIN‐dependent mitochondrial ubiquitylation in induced neurons and model systems revealed by digital snapshot proteomics. Mol Cell 70: 211–227.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordureau A, Paulo JA, Zhang J, An H, Swatek KN, Cannon JR, Wan Q, Komander D, Harper JW (2020) Global landscape and dynamics of Parkin and USP30‐dependent Ubiquitylomes in iNeurons during mitophagic signaling. Mol Cell 77: 1124–1142.e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordureau A, Kraus F, Zhang J, An H, Park S, Ahfeldt T, Paulo JA, Harper JW (2021) Temporal proteomics during neurogenesis reveals large‐scale proteome and organelle remodeling via selective autophagy. Mol Cell 81: 5082–5098.e11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paisan‐Ruiz C, Guevara R, Federoff M, Hanagasi H, Sina F, Elahi E, Schneider SA, Schwingenschuh P, Bajaj N, Emre M et al (2010) Early‐onset L‐dopa‐responsive parkinsonism with pyramidal signs due to ATP13A2, PLA2G6, FBXO7 and spatacsin mutations. Mov Disord 25: 1791–1800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulo JA, O'Connell JD, Gygi SP (2016) A triple knockout (TKO) proteomics standard for diagnosing ion interference in isobaric labeling experiments. J Am Soc Mass Spectrom 27: 1620–1625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez‐Riverol Y, Bai J, Bandla C, Garcia‐Seisdedos D, Hewapathirana S, Kamatchinathan S, Kundu DJ, Prakash A, Frericks‐Zipper A, Eisenacher M et al (2022) The PRIDE database resources in 2022: a hub for mass spectrometry‐based proteomics evidences. Nucleic Acids Res 50: D543–D552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell AM, Youle RJ (2015) The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson's disease. Neuron 85: 257–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plubell DL, Wilmarth PA, Zhao Y, Fenton AM, Minnier J, Reddy AP, Klimek J, Yang X, David LL, Pamir N (2017) Extended multiplexing of tandem mass tags (TMT) labeling reveals age and high fat diet specific proteome changes in mouse Epididymal adipose tissue. Mol Cell Proteomics 16: 873–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rad R, Li J, Mintseris J, O'Connell J, Gygi SP, Schweppe DK (2021) Improved monoisotopic mass estimation for deeper proteome coverage. J Proteome Res 20: 591–598 [DOI] [PubMed] [Google Scholar]
- Rath S, Sharma R, Gupta R, Ast T, Chan C, Durham TJ, Goodman RP, Grabarek Z, Haas ME, Hung WHW et al (2021) MitoCarta3.0: an updated mitochondrial proteome now with sub‐organelle localization and pathway annotations. Nucleic Acids Res 49: D1541–D1547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravenhill BJ, Boyle KB, von Muhlinen N, Ellison CJ, Masson GR, Otten EG, Foeglein A, Williams R, Randow F (2019) The cargo receptor NDP52 initiates selective autophagy by recruiting the ULK complex to cytosol‐invading bacteria. Mol Cell 74: 320–329.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez‐Martinez A, Martinez A, Whitworth AJ (2022) FBXO7/ntc and USP30 antagonistically set the basal ubiquitination threshold for mitophagy. bioRxiv 10.1101/2022.10.10.511602 [PREPRINT] [DOI] [PMC free article] [PubMed]
- Sarraf SA, Raman M, Guarani‐Pereira V, Sowa ME, Huttlin EL, Gygi SP, Harper JW (2013) Landscape of the PARKIN‐dependent ubiquitylome in response to mitochondrial depolarization. Nature 496: 372–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauve V, Sung G, Soya N, Kozlov G, Blaimschein N, Miotto LS, Trempe JF, Lukacs GL, Gehring K (2018) Mechanism of parkin activation by phosphorylation. Nat Struct Mol Biol 25: 623–630 [DOI] [PubMed] [Google Scholar]
- Savitski MM, Wilhelm M, Hahne H, Kuster B, Bantscheff M (2015) A scalable approach for protein false discovery rate estimation in large proteomic data sets. Mol Cell Proteomics 14: 2394–2404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda‐Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B et al (2012) Fiji: an open‐source platform for biological‐image analysis. Nat Methods 9: 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweppe DK, Prasad S, Belford MW, Navarrete‐Perea J, Bailey DJ, Huguet R, Jedrychowski MP, Rad R, McAlister G, Abbatiello SE et al (2019) Characterization and optimization of multiplexed quantitative analyses using high‐field asymmetric‐waveform ion mobility mass spectrometry. Anal Chem 91: 4010–4016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweppe DK, Eng JK, Yu Q, Bailey D, Rad R, Navarrete‐Perea J, Huttlin EL, Erickson BK, Paulo JA, Gygi SP (2020) Full‐featured, real‐time database searching platform enables fast and accurate multiplexed quantitative proteomics. J Proteome Res 19: 2026–2034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirling DR, Swain‐Bowden MJ, Lucas AM, Carpenter AE, Cimini BA, Goodman A (2021) CellProfiler 4: improvements in speed, utility and usability. BMC Bioinformatics 22: 433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolz A, Ernst A, Dikic I (2014) Cargo recognition and trafficking in selective autophagy. Nat Cell Biol 16: 495–501 [DOI] [PubMed] [Google Scholar]
- Tai YC (2022) timecourse: statistical analysis for developmental microarray time course data. R package version 1.68.0. http://wwwbioconductororg
- Tyanova S, Cox J (2018) Perseus: a bioinformatics platform for integrative analysis of proteomics data in cancer research. Methods Mol Biol 1711: 133–148 [DOI] [PubMed] [Google Scholar]
- Tyanova S, Temu T, Cox J (2016) The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat Protoc 11: 2301–2319 [DOI] [PubMed] [Google Scholar]
- Vargas JNS, Wang C, Bunker E, Hao L, Maric D, Schiavo G, Randow F, Youle RJ (2019) Spatiotemporal control of ULK1 activation by NDP52 and TBK1 during selective autophagy. Mol Cell 74: 347–362.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vingill S, Brockelt D, Lancelin C, Tatenhorst L, Dontcheva G, Preisinger C, Schwedhelm‐Domeyer N, Joseph S, Mitkovski M, Goebbels S et al (2016) Loss of FBXO7 (PARK15) results in reduced proteasome activity and models a parkinsonism‐like phenotype in mice. EMBO J 35: 2008–2025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Yang F, Gritsenko MA, Wang Y, Clauss T, Liu T, Shen Y, Monroe ME, Lopez‐Ferrer D, Reno T et al (2011) Reversed‐phase chromatography with multiple fraction concatenation strategy for proteome profiling of human MCF10A cells. Proteomics 11: 2019–2026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wauer T, Simicek M, Schubert A, Komander D (2015) Mechanism of phospho‐ubiquitin‐induced PARKIN activation. Nature 524: 370–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YC, Holzbaur EL (2014) Optineurin is an autophagy receptor for damaged mitochondria in parkin‐mediated mitophagy that is disrupted by an ALS‐linked mutation. Proc Natl Acad Sci USA 111: E4439–E4448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamano K, Youle RJ (2013) PINK1 is degraded through the N‐end rule pathway. Autophagy 9: 1758–1769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusa K, Zhou L, Li MA, Bradley A, Craig NL (2011) A hyperactive piggyBac transposase for mammalian applications. Proc Natl Acad Sci USA 108: 1531–1536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Pak C, Han Y, Ahlenius H, Zhang Z, Chanda S, Marro S, Patzke C, Acuna C, Covy J et al (2013) Rapid single‐step induction of functional neurons from human pluripotent stem cells. Neuron 78: 785–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuris JA, Thompson DB, Shu Y, Guilinger JP, Bessen JL, Hu JH, Maeder ML, Joung JK, Chen ZY, Liu DR (2015) Cationic lipid‐mediated delivery of proteins enables efficient protein‐based genome editing in vitro and in vivo . Nat Biotechnol 33: 73–80 [DOI] [PMC free article] [PubMed] [Google Scholar]