

Abstract
Misfolded Aβ is involved in the progression of Alzheimer's disease (AD). However, the role of its polymorphic variants or conformational strains in AD pathogenesis is not fully understood. Here, we study the seeding properties of two structurally defined synthetic misfolded Aβ strains (termed 2F and 3F) using in vitro and in vivo assays. We show that 2F and 3F strains differ in their biochemical properties, including resistance to proteolysis, binding to strain‐specific dyes, and in vitro seeding. Injection of these strains into a transgenic mouse model produces different pathological features, namely different rates of aggregation, formation of different plaque types, tropism to specific brain regions, differential recruitment of Aβ40/Aβ42 peptides, and induction of microglial and astroglial responses. Importantly, the aggregates induced by 2F and 3F are structurally different as determined by ssNMR. Our study analyzes the biological properties of purified Aβ polymorphs that have been characterized at the atomic resolution level and provides relevant information on the pathological significance of misfolded Aβ strains.
Keywords: amyloid‐beta, animal models, prion, protein conformation, strains
Subject Categories: Molecular Biology of Disease, Neuroscience, Structural Biology
Two structurally defined Aβ conformational variants differ in their biochemical properties in vitro and induce different pathological features in a transgenic mouse model.
Introduction
Alzheimer's disease (AD) is clinically diverse (Devi & Scheltens, 2018). Several reports suggest that patient‐specific pathological traits are responsible for AD clinical variation (Thal et al, 2015; Zhang et al, 2016; Dujardin et al, 2020; Duran‐Aniotz et al, 2021). Amyloid‐β (Aβ) deposition, an early and perhaps the most characteristic feature of AD pathology, accumulates in the brain in a variety of arrangements (types of deposits) and displays tropisms towards different anatomical, tissue and intra‐ or extra‐cellular locations (Knobloch et al, 2007; Thal et al, 2008, 2015; Duran‐Aniotz et al, 2021). This pathological variability is similar to that observed in other diseases associated with the accumulation of misfolded proteins (Prusiner & Hsiao, 1994; Irwin, 2016; Soto & Pritzkow, 2018). The most classical example of variation at this level can be found in prion diseases. There, different conformations of the misfolded prion protein (PrPSc), which is the main component of the prion infectious agent, are thought to be the cause for the diverse clinical and pathological manifestations observed in these neuropathies (Morales et al, 2007; Morales, 2017). Interestingly, several prion strains can be generated from prion proteins with identical primary structure. In the same line, several reports suggest that variabilities in synucleinopathies (e.g., Parkinson's disease [PD] and multiple system atrophy [MSA]), and tauopathies (Alzheimer's disease, Pick's disease, progressive supranuclear palsy, and others) are also caused by variations in the misfolded conformation of α‐synuclein and tau, respectively (Shahnawaz et al, 2020; Shi et al, 2021).
Conformational prion strains can be faithfully propagated in vitro and in animal models (Castilla et al, 2008). Similarly, misfolded Aβ, tau, and α‐synuclein “strains” also propagate their conformations in appropriate systems (Petkova et al, 2005; Clavaguera et al, 2013; Watts et al, 2014; Shahnawaz et al, 2020). In early in vitro experiments, synthetic Aβ40 polymorphs were shown to serially propagate while maintaining their unique strain‐specific properties as characterized by solid‐state nuclear magnetic resonance (ssNMR; Petkova et al, 2005). The aggregates described in this report were later termed “2F” or “3F” due to their approximate rotational symmetry around the fibril growth axis in structural models and their characteristically different mass‐per‐length values (Tycko, 2014). Although generated under laboratory‐controlled conditions, these aggregates provided initial evidence on the conformational plasticity of misfolded Aβ. Later reports using animal models of brain amyloidosis showed that inoculation of possibly diverse misfolded Aβ strains differentially propagated in these systems (Heilbronner et al, 2013; Stöhr et al, 2014; Watts et al, 2014). Interestingly, brains from AD patients that experienced rapid and slow clinical decline have been linked to different populations of misfolded Aβ conformers as resolved by the conformation‐dependent immunoassay (Cohen et al, 2015). On the other hand, changes on misfolded Aβ conformation may explain why some individuals carrying large quantities of Aβ deposits in their brains do not manifest clinical signs (Zolochevska & Taglialatela, 2016). Additional evidence highlighting the role of misfolded Aβ strains in AD pathology has been provided at the structural level, as specific Aβ structures (as resolved by ssNMR) have been linked to different AD types (Lu et al, 2013; Qiang et al, 2017). Despite of all this information, the pathological relevance that different conformations of misfolded Aβ exert in AD is still unclear. In this study, we investigated the biological changes induced by the prion‐like propagation of the structurally defined 2F and 3F synthetic Aβ aggregates upon injection in the brain of transgenic mice expressing human amyloid precursor protein (APP). Considering this, the results presented here are the first to describe the pathological features generated by the prion‐like spreading of Aβ polymorphs that have been characterized at the atomic resolution level. Along these lines, our findings enable to relate the structure of Aβ aggregates to the neuropathological alterations produced in the brain in vivo.
Results
2F and 3F aggregates display different structural and biochemical features
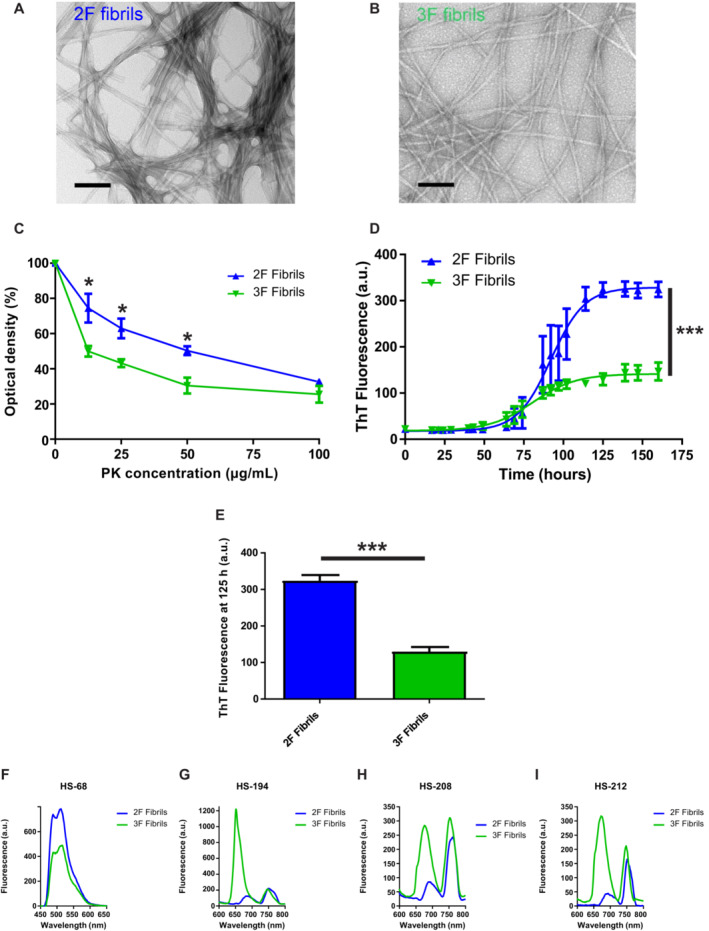
To properly model the pathological features of conformational variants of misfolded Aβ, we used the previously described 2F and 3F Aβ fibrils due to their relative homogeneity and clear structural differences indicated by electron microscopy and ssNMR (Petkova et al, 2005; Tycko, 2014). Importantly, the structural motifs of these particular Aβ morphotypes have been extensively characterized (Petkova et al, 2005). Although previous articles have extensively described the molecular structures of these fibrils, they have not addressed their biological effects (Petkova et al, 2005; Tycko, 2014). Our initial efforts were focused to analyze the biochemical properties of these aggregates using similar tools used to characterize prion strains (Morales et al, 2007; Castilla et al, 2008; Morales, 2017). First, we visualized these aggregates using transmission electron microscopy (TEM, Fig 1A and B). TEM images displayed gross differences in the morphological features of these aggregates, similarly as previously described (Petkova et al, 2005). Then, these aggregates were tested for their resistance to proteolytic degradation using increasing concentrations of proteinase K (PK). Results show that 2F and 3F fibrils were distinguishable at this level. Specifically, 2F fibrils displayed a higher resistance to proteolytic digestion compared to their 3F counterpart (Fig 1C). Additionally, we tested the in vitro seeding activity of these aggregates using the protein misfolding cyclic amplification (PMCA) technique modified for Aβ aggregates. The PMCA technology was originally developed to replicate infectious prions in an accelerated manner and in cell‐free conditions (Saborio et al, 2001; Morales et al, 2012). Further modifications on PMCA allowed this technique to be implemented for their use to amplify misfolded Aβ (Salvadores et al, 2014), α‐synuclein (Shahnawaz et al, 2020), and tau (Saijo et al, 2019) aggregates. Importantly, the PMCA technique has been shown to faithfully replicate the conformation and biological properties of infectious prions (Castilla et al, 2008) and distinguish between conformational variants of α‐synuclein associated with PD or MSA (Shahnawaz et al, 2020). Our Aβ‐PMCA assay using 2F and 3F as seeds provided us with relevant information supporting the differential pathological activity of misfolded Aβ strains. Importantly, the Aβ aggregates generated by 2F and 3F seeding displayed different fluorescence readings at plateau (Fig 1D and E). This result further supports the differential molecular structural and seeding properties of 2F and 3F, as it suggests differential accessibility and reactivity of thioflavin T (ThT) to the Aβ aggregates generated by seeding (Fig 1D). Importantly, similar outcomes have been reported for Aβ fibril polymorphs (Qiang et al, 2011) and α‐synuclein‐PMCA products seeded with MSA‐ and PD‐derived specimens (Shahnawaz et al, 2020).
Figure 1. Biochemical properties of 2F and 3F fibrils.
-
A, BTransmission electron micrographs of negatively stained 2F (A) and 3F (B) fibrils before sonication and injection into mice. The two fibril polymorphs were grown in vitro from synthetic Aβ40 and show the morphological differences originally reported by Petkova et al (2005). Fibrils are negatively stained with uranyl acetate. Bars represent 100 nm.
-
CPK resistance profile of 2F and 3F fibrils. Values are expressed as the mean densitometric value at each PK concentration (technical triplicates) ± standard error. Statistical analyses were performed by student's t‐test (*P < 0.05).
-
DSeeding assays using 100 pM of 2F (blue) and 3F (green) fibrils as seeds as described in Materials and Methods. Curves were generated by measuring ThT emission values at different time points. Reaction kinetics were statistically different as assessed by using the multiple comparisons method to compare pairs of curves (***P < 0.0001). Data in graphs is represented as averages depicting standard errors of five technical replicates.
-
EMean fluorescence (quintuplicates) of the seeding reaction in (E) at 125 h. Statistical differences were assessed by student's t‐test (***P < 0.0001). Data in graphs is represented as averages depicting standard errors of five technical replicates.
-
F–I2F and 3F fibrils were tested for their reactivity against a variety of luminescent conjugated thiophenes able to discriminate among conformational variants of misfolded proteins. Stock solutions of LCOs (1.5 mM) were diluted in distilled water to 15 μM and added to the wells to a final concentration of 0.3 μM. All wells contained 10 μM of either 2F or 3F fibrils. The samples were incubated at 37°C, and the emission spectrum of each probe was collected after 30 min by exciting the samples at 440 nm (HS‐68) or 535 nm (HS‐194, HS‐208, and HS‐212).
Source data are available online for this figure.
To further confirm structural differences, we tested 2F and 3F fibrils with a battery of thiophene‐based ligands, including luminescent conjugated oligothiophenes (LCOs). LCOs have great affinities for amyloids and have been previously shown to discriminate between prion strains (Magnusson et al, 2014) and different types of misfolded Aβ deposits (Rasmussen et al, 2017). Here, we tested nine different LCOs (Fig 1F–I and Appendix Figs S1 and S2). Several of these molecules were able to discriminate between 2F and 3F fibrils (Fig 1F–I) while some others revealed similar spectra when both fibrils were used (Appendix Fig S2). The most pronounced spectral difference was observed for the structurally related analogs, HS‐194, HS‐208, and HS‐212 (Fig 1F–I and Appendix Fig S1), suggesting that ligands with a distinct chemical composition display differential binding modes to 2F and 3F fibrils. In contrast, the two LCOs, HS‐169 and HS‐199, previously used to distinguish α‐synuclein PMCA products seeded with MSA and PD seeds (Shahnawaz et al, 2020) display comparable spectra for both aggregated species. Altogether, these results further support the differences between 2F and 3F fibrils at the biochemical and seeding activity levels, complementing previous structural characterizations (Petkova et al, 2005; Tycko, 2014). In addition, these results suggest that a similar battery of assays used to discriminate between prion strains can be used to distinguish Aβ strains.
Synthetic Aβ misfolded strains induce diverse pathological outcomes in the brains of seeding‐susceptible transgenic mice
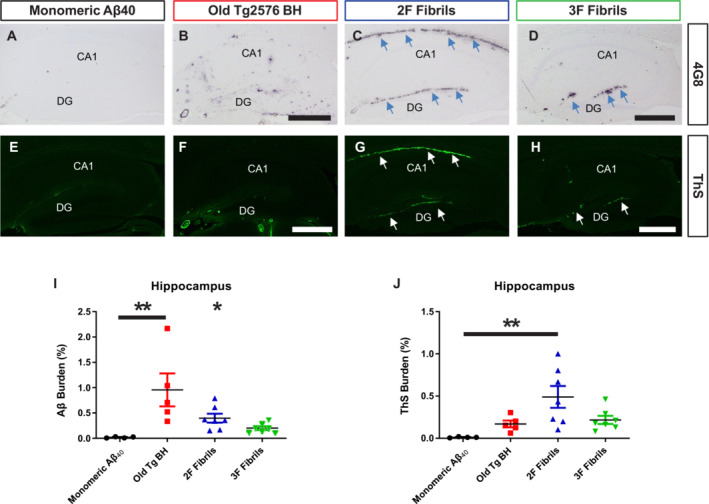
Currently, the pathological relevance of Aβ polymorphic variants is not clear. To explore this, we injected 2F and 3F fibrils in both brain hemispheres (hippocampus) of 50 days old Tg2576 mice. Controls included animals injected with either monomeric Aβ40 peptides (negative control) and brain extracts from old Tg2576 mice with proven seeding activities (positive control). Animals were sacrificed and brains collected at 300 days old (250 days post‐inoculation), time when nontreated mice display only small amount of Aβ deposits (Morales et al, 2021). Collected brains were separated in both hemispheres, keeping one frozen for biochemical analyses while the other was preserved for histopathological assessments (Appendix Fig S3). First, we analyzed the histopathological features of experimental and control mice. We observed pronounced differences, specifically at the injection site (hippocampus). Mice treated with monomeric Aβ40 peptides displayed a modest amount of deposits, similar to what has been described for untreated mice at the same age (Morales et al, 2015, 2021; Fig 2A). On the contrary, the group of mice injected with the brain of an old Tg2576 animal showed abundant diffuse amyloid deposition across all hippocampal layers (Fig 2B) in agreement with previous reports using the same model (Morales et al, 2015). Interestingly, mice injected with 2F and 3F aggregates displayed a substantially different pattern of amyloid deposition compared to mice injected with the in vivo (Tg2576) generated seeds. 2F induced abundant amyloid lesions in the hippocampus, mostly in the dentate gyrus and alveus (Fig 2C). Interestingly, 3F seeded aggregation was restricted to the dentate gyrus (Fig 2D). We also observed differences on the aggregates seeded by the different Aβ strains in terms of their reactivity against thioflavin S (ThS, a dye known to bind fibrillar and compact Aβ deposits). For animals treated with monomeric Aβ, reactivity against ThS was negligible (Fig 2E). Interestingly, amyloid deposits seeded by the Tg2576‐derived seeds were poorly reactive against this dye (Fig 2F). On the contrary, amyloid deposits in 2F‐ (Fig 2G) and 3F‐ (Fig 2H) treated mice were reactive against ThS. Quantification of Aβ burden showed a significant increase in the hippocampi of mice injected with Tg2576 seeds (Old Tg BH) when compared to all other groups (Fig 2I, **P < 0.001). However, ThS only reached a statistically significant increase in mice injected with the 2F fibrils (Fig 2J, **P < 0.001). An exhaustive analysis of the mice's brains demonstrated that while animals treated with the Old Tg BH induced Aβ deposition in other brain regions (cortex and caudate nucleus/putamen Appendix Fig S4), 2F and 3F deposits were restricted to the hippocampus and the lateral ventricle (Appendix Fig S5). In the latter brain region, 2F seeds induced strong amyloidosis (Appendix Fig S5). 3F seeds also showed aggregation in the lateral ventricle compared to the positive and negative control groups, although this increase was not significantly different compared to the pathology induced by the Old Tg2576 seeds (Appendix Fig S5). Consistent with the data obtained in the hippocampus, the amyloidosis promoted by the Old Tg2576 seeds in the lateral ventricle was ThS negative (Fig 2). Importantly, the patterns of seeded amyloid deposition are unlikely to be generated from the needle injury as (i) the needle track lesion and associated seeded amyloid deposition runs horizontally from the cortex through the hippocampus and (ii) the patterns of amyloid deposition in the different groups were inoculum‐specific and reproducible (Appendix Fig S6).
Figure 2. Amyloid pathology seeded by 2F and 3F fibrils, and brains extracts from old Tg2576 mice.
-
A–HRepresentative images of hippocampi from mice treated with 2F‐, 3F‐, and Tg2576‐ derived seeds after visualization for Aβ deposits using the 4G8 antibody (A–D) or ThS (E–H). CA, cornu ammonis 1; DG, dentate gyrus. Scale bar in (B), (D), (F), and (H) represents 500 μm and applies to all panels. Arrows represent regions of interest (alveus, DG).
-
I, JAβ (I) and ThS (J) burden quantification in hippocampus. Statistical analyses were performed using one‐way ANOVA (*P < 0.05, **P < 0.001). Data in graphs is represented as averages depicting standard errors. Measurements considered five tissue slices per animal, and 4–7 animals were included per group.
Source data are available online for this figure.
To further explore 2F and 3F seeding activity properties, additional groups of mice treated with these aggregates were sacrificed 100 days after exposure. In line with the data presented above, 2F aggregates displayed higher, although no significant quantities of PBS‐insoluble Aβ compared to mice treated with the 3F fibrils (Appendix Fig S7). Histological analyses displayed deposition in the alveus and dentate gyrus of 2F‐treated mice, suggesting that accumulation in these areas occurs way before the 300 days old experimental endpoint. It is relevant to note that some mice treated with 3F fibrils and Old Tg2576 seeds also displayed Aβ deposition in the alveus (one animal per group, Appendix Figs S5 and S7). However, these animals were not representative of the tropism observed in all other subjects within the same groups.
Aβ pathology induced by 2F and 3F aggregates recruits different Aβ peptides and result in differential reactivity to amyloid binding dyes
Another difference found for mice induced by 2F, 3F, and Tg2576‐derived seeds was observed on their differential abilities to recruit Aβ40 and Aβ42 peptides. We first focused our analyses on the dentate gyrus, the area of the hippocampus where the three seeds induced amyloidosis. Amyloid deposits primed by Tg2576 seeds in this brain region were composed by both Aβ peptides. In fact, Aβ40 and Aβ42 seemed to coexist in these specific aggregates (Appendix Fig S8A). 2F‐ and 3F‐induced deposits in the dentate gyrus also recruited both Aβ peptides but with a predominance of the Aβ40 version (Appendix Fig S8A). Interestingly, while some deposits displayed colocalization of both Aβ40 and Aβ42, others appeared to be exclusively formed by Aβ40. This phenomenon was more pronounced in the 3F group where plaques were mostly composed by Aβ40 (Appendix Fig S8A). Additional differences were observed in the alveus (Appendix Fig S8B). While the extensive amyloidosis exerted by 2F seeds in this region was mostly composed by Aβ40, the few deposits induced by the Old Tg2576 seeds in this area were mostly formed by Aβ42 (Appendix Fig S8B). Mice treated with monomeric Aβ40 and 3F seeds generated negligible quantities of amyloidosis in this brain region, as discussed above.
Luminescent conjugated oligothiophenes have been previously shown to be useful in differentiating between prion strains (Magnusson et al, 2014) and diverse Aβ arrangements (Rasmussen et al, 2017). In this study, we confirmed the use of these amyloid binding dyes to differentiate between Aβ strains. Based on the data presented in Fig 1, we used dyes HS‐68 and HS‐194 in brain tissue to explore their binding properties in the in vivo seeded aggregates (Appendix Fig S9). In the dentate gyrus, we observed that deposits induced by the Old Tg2576 and 2F seeds were, in their vast majority, reactive to both dyes. In contrast, 3F‐induced deposits were strongly reactive to HS‐194 and poorly reactive against HS‐68 (Appendix Fig S9A). Interestingly, 2F‐induced amyloidosis in the alveus was reactive almost exclusively to HS‐68, consistent with the in vitro results presented in Fig 1. These data are relevant as they suggest that different brain regions play a relevant role in the differential propagation of Aβ strains, similarly as it has been shown for prion strains (Morales et al, 2007; Morales, 2017). These data also verify that ligands with different chemical compositions, such as HS‐68 and HS‐94, can aid in the identification of different aggregated Aβ species.
The specific properties of amyloid deposits induced by 2F, 3F, and Tg2576‐derived seeds in Tg2576 mice are summarized in Table 1.
Table 1.
Specific properties of hippocampal Aβ deposits induced by old Tg2576‐derived seeds, 2F, or 3F fibrils.
| Amyloid deposits | Aβ40 and Aβ42 content | ThS reactivity | LCO reactivity | ||
|---|---|---|---|---|---|
| DG | Alveus | ||||
| Old Tg BH | Widespread | Aβ40 and Aβ42 | N/A | Negative | HS‐68/HS‐194 |
| 2F Fibrils | DG and Alveus | Aβ40 and Aβ42 | Aβ40 | Positive | HS‐68/HS‐194 |
| 3F Fibrils | DG | Aβ40 and Aβ42 | N/A | Positive | HS‐194 |
Glial activation induced by 2F‐ and 3F‐seeded aggregates is Aβ strain dependent
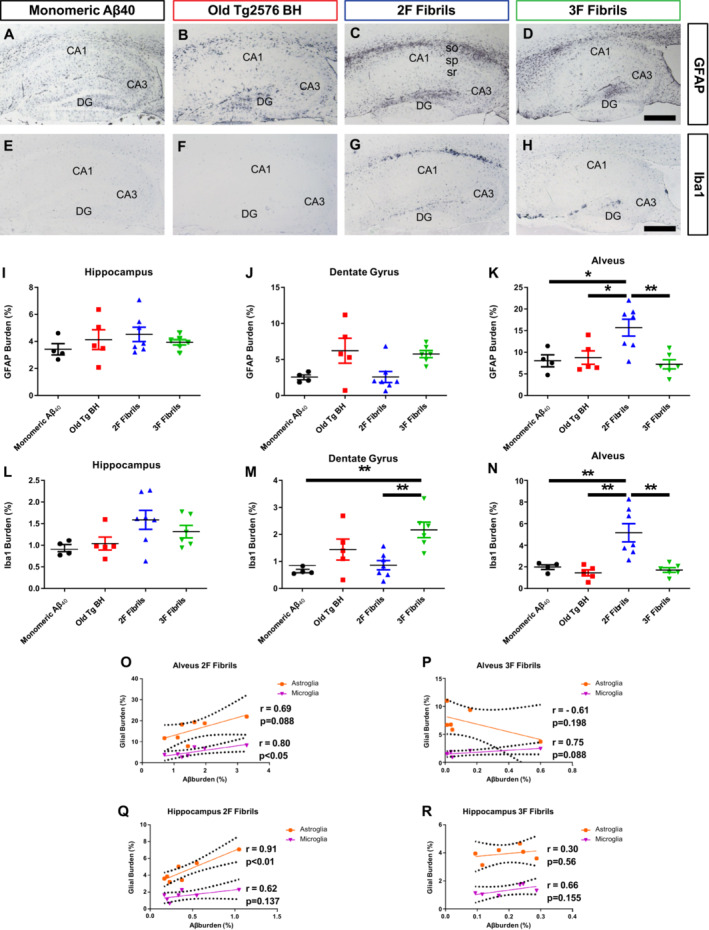
To further explore the biological differences in the brain produced by injection of distinct Aβ strains, we studied differential glial responses. For that purpose, astroglial (Fig 3A–D) and microglial (Fig 3E–H) markers in control and experimental groups were measured. Astroglial reactivity, evaluated through immunolabeling for the glial fibrillary acidic protein (GFAP, Fig 3A–D), showed no differences between experimental and control groups when burden staining was measured in the whole hippocampus (Fig 3I). A similar outcome was found in the dentate gyrus, albeit 3F aggregates seeded predominantly in that area (Fig 3J). The alveus of 2F‐treated mice displayed a significantly increase of GFAP signal compared to other groups (Fig 3K, 46% of increase compared to 3F injected animals, **P < 0.001). This effect is likely induced by the specific Aβ aggregates being deposited in these animals. Changes in microglial response were assessed by evaluating the ionized calcium binding adaptor molecule 1 (Iba‐1) signals in the same brain regions (Fig 3E–H). In analogy to the GFAP data, no differences were found when considering Iba‐1 staining across the whole hippocampus (Fig 3L). However, differences in microglial activity were found in specific areas. In agreement with amyloid deposition, mice induced by 3F seeds displayed significantly higher levels of Iba‐1 reactivity in the dentate gyrus compared to other groups (Fig 3M, 21.27% of increase compared with 2F fibrils, **P < 0.001), with the exception of the mice treated with the Old Tg2576 brain extract (Fig 3M). In the same line, 2F aggregates induced higher Iba‐1 reactivity in the alveus (Fig 3N, 33.13% of increase compared with 2F fibrils, **P < 0.001). These nonlinear Aβ/glial activation responses were better appreciated when correlations between these parameters were done (Fig 3O–R).
Figure 3. Glial activation in the brain of mice treated with 2F‐, 3F‐, and Tg2576‐derived Aβ seeds.
-
A–HRepresentative pictures of hippocampi from mice treated with different injectate after for micro‐ and astro‐glial associated signals using anti‐GFAP (A–D) and anti‐Iba‐1 (E–H) antibodies. Scale bar in (D) and (H) represents 500 μm and applies to all panels. CA1‐3, cornu ammonis 1–3; DG, dentate gyrus; so, striatum oriens; sp, striatum pyramydale; sr, striatum radiatum.
-
I–NAnti‐GFAP burden in hippocampus (I), dentate gyrus (J), and alveus (K) for all animal groups was quantified for image analysis. Similarly, anti‐Iba‐1 burdens for all experimental and control groups were calculated in hippocampus (L), dentate gyrus (M), and alveus (N). Statistical analyses were performed by one‐way ANOVA (*P < 0.05, **P < 0.001). Data in graphs is represented as averages depicting standard errors. Measurements considered five tissue slices per animal, and 4–7 animals were included per group.
-
O–RCorrelations between Aβ burden and glial burden were calculated in the alveus for mice injected with 2F (O) and 3F (P) fibrils. In the same way, the relationship between glial activation and Aβ burden in the hippocampus was calculated for mice treated with 2F (Q) and 3F (R) aggregates. Statistical analyses were performed by Pearson correlation coefficients test in which P < 0.05 was considered significant.
Source data are available online for this figure.
Next, we explored in more detail the effect that the different seeded aggregates exerted over alveus and dentate gyrus glial cells by epifluorescence microscopy analysis of double immunofluorescent 4G8/GFAP (Fig 4) and 4G8/Iba1 (Fig 5) labeling. In the dentate gyrus (Fig 4A a1–16), all Aβ aggregates induced astroglial reactivity in a similar manner, regardless of the plaque type/morphology. In the alveus region (Fig 4B b1–16), GFAP signal was highly detected in 2F inoculated mice (Fig 4B b9–12) as only 2F aggregates were strongly deposited in this region. Importantly, the few diffuse Aβ deposits induced by the Tg2576 seeds area did not result in strong astroglial reaction surrounding them (Fig 4 b1–4). Mice injected with monomeric Aβ displayed considerably less astroglial reactivity in both alveus and dentate gyrus when compared to all other groups (Fig 4A a5–8 and B b5–b8). For microglial reaction in the dentate gyrus (Fig 5A a1–16), the strongest Iba‐1 positive signal was detected around 3F seeded plaques (Fig 5A a13–16). The microglial response of seeded aggregates induced by 2F and the Tg2576 seeds was not as strong and few Iba1‐positive cells were clustered around plaques, regardless of plaque size. In the alveus (Fig 5B b1–16), the microglial response against 2F seeded aggregates (Fig 5B b9–12) appeared weaker compared to what was found in the dentate gyrus (Fig 5A a9–12), further suggesting that different Aβ arrangements induce differential glial responses in a hippocampal region‐specific manner. These spatial variations were supported by correlation analyses (Appendix Fig S10). Both microglial and astroglial reactivity correlated positively with amyloid burden in the dentate gyrus for 3F fibrils, whereas Tg2576 seeds showed positive correlation with microgliosis and 2F with astroglial reaction.
Figure 4. Astroglial reactivity to Aβ deposits seeded by 2F‐, 3F‐ and Tg2576‐derived aggregates.
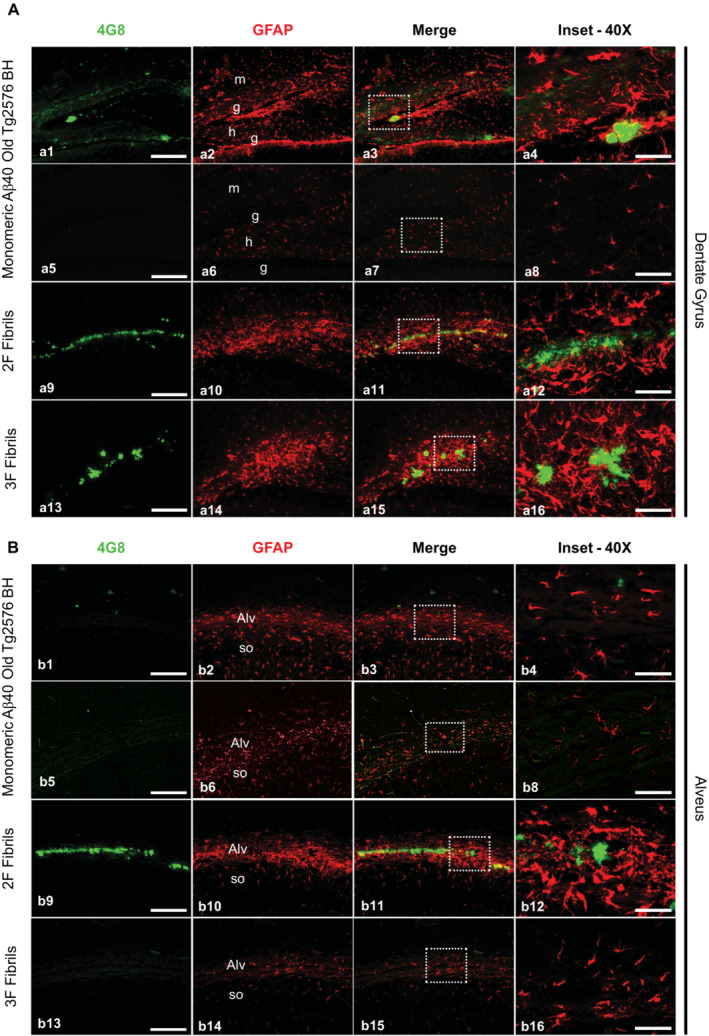
- Panels showing representative epifluorescence microscopy images of the dentate of mice treated with experimental and control materials and immunostained with both 4G8 (green) and anti‐GFAP (red) antibodies. The third column of panels from left to right depict merged images. Right panels are insets obtained from the merged images (white punctuated squares).
- A similar analysis as explained in (A) was performed for the alveus. Images were arranged in the same manner as described above.
Data information: Scale bars at the left panels represent 200 μm and are applicable to the pictures labeled as “4G8,” “GFAP,” and “Merge.” Scale bars on the right images (inset) represent 50 μm. Alv, alveus; g, granular layer; h, hilar region; m, molecular layer; so, striatum oriens. One tissue slice per animal was used for these analyses.
Source data are available online for this figure.
Figure 5. Microglial reactivity to Aβ aggregates seeded by 2F‐, 3F‐ and Tg2576‐derived aggregates.
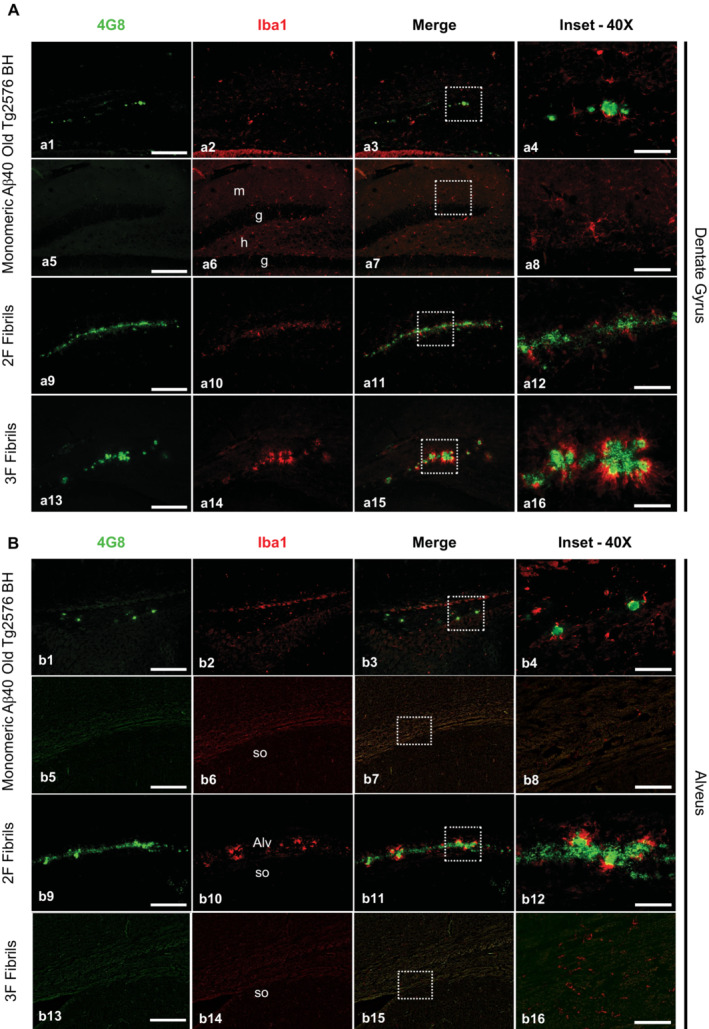
- Panels showing representative pictures of the dentate gyrus of mice treated with experimental and control Aβ seeds and stained with both 4G8 (green) and anti‐Iba‐1 (red) antibodies. The third column of panels from left to right depict merged images. Right panels are insets obtained from the merged images (white punctuated squares).
- A similar analysis as explained in (A) was performed for the alveus. Images were arranged in the same manner as described above.
Data information: Scale bars at the left panels represent 200 μm and are applicable to the pictures labeled as “4G8”, “Iba‐1” and “Merge”. Scale bars on the right images represent 50 μm. Alv, alveus; g, granular layer; h, hilar region; m, molecular layer; so, striatum oriens. One tissue slice per animal was used for these analyses.
Source data are available online for this figure.
To evaluate whether these aggregates induced distinct inflammatory profiles, we selected and measured the concentrations of 23 key cytokines related to inflammation in brain extracts using a multiplex immunoassay (Appendix Fig S11). The cytokines' levels were significantly different between groups. Mice inoculated with the Tg2576‐derived seeds displayed greater cytokine alterations, with IL‐1a, IL‐6, IL‐5, lL‐9, IL‐13, MIP‐1a, MIP‐1B, RANTES, TNF‐α and INF‐γ levels significantly increased. On the contrary, IL‐2, IL‐3, and IL‐17 showed lower levels in mice treated with Tg2576 seeds when compared to control animals challenged with monomeric Aβ. Moreover, Tg2576 seeds induced higher levels of IL‐1b, IL‐6, IL‐5, IL‐9, IL‐13, and INF‐γ than 2F and 3F aggregates. Despite these differences, these three distinct Aβ strains also exhibited a common cytokine signature with significant elevations of CCL3 and INF‐γ and reduced IL‐2, IL‐3 and IL‐17 when comparing with the monomeric Aβ injected group. Levels of the anti‐inflammatory cytokines IL‐4 and IL‐10 were unchanged regardless of the injectate used. 2F fibrils induced a proinflammatory profile (IL‐1a, CCL3, CCL5, and INF‐γ) that slightly differed from 3F fibrils (CCL3 and INF‐γ).
Vascular amyloid deposition in mice treated with in vivo‐derived and synthetic Aβ fibrils
We and others have shown that Aβ deposition induced by seeding promoted the formation of vascular amyloidosis in animal models that naturally display negligible levels (Eisele et al, 2010; Morales et al, 2021). Moreover, a previous report from our group shows that AD brain extracts displaying amyloid pathology in diverse arrangements differentially seed Aβ deposits in brain vessels of APP/PS1 mice (Duran‐Aniotz et al, 2021). Here, we measured whether seeding to brain vessels was differentially induced by synthetic or in vivo derived seeds. Analyses were restricted to meningeal vessels as parenchymal cerebral amyloid angiopathy (CAA) was absent in all mice included in this study. A significant increase was only observed for mice treated with the Tg2576 seeds when compared to the animals treated with either monomeric Aβ40 and 3F fibrils (Appendix Fig S12, *P < 0.05). 2F‐treated mice did not display any significant difference at this level when compared to any other group.
Amyloid deposits induced by different seeds contain Aβ fibrils with different molecular structures
We used ssNMR to test whether amyloid deposits induced in mouse brain tissue by injection of 2F‐, 3F‐, or Tg2576‐derived seeds contains fibrils with different molecular structures. Since ssNMR requires milligram‐scale quantities of fibrils and isotopic labeling, fibrils in mouse brain homogenates were amplified by seeded growth in vitro (see Materials and Methods and Appendix Fig S13A), as previously demonstrated in studies of Aβ fibrils from human brain tissue (Lu et al, 2013; Qiang et al, 2017). Aβ40 fibrils for ssNMR were 15N,13C‐labeled at all nitrogen and carbon sites in eight residues, namely F19, V24, G25, S26, A30, I31, L34, and M35. NMR frequencies (i.e., chemical shifts) of these residues are sensitive to structural variations among Aβ polymorphs, allowing the one‐dimensional (1D) and two‐dimensional (2D) ssNMR spectra to be used as structural fingerprints (Lu et al, 2013; Qiang et al, 2017).
Figure 6A shows 1D 13C ssNMR spectra of Aβ40 fibrils grown from amyloid‐containing extracts of brain homogenates from mice that had been treated with 2F‐, 3F‐, or Tg2576‐derived seeds. These spectra show clear differences, especially in the aliphatic signal region (13C chemical shifts in the 0–70 ppm range). Clear differences in crosspeak positions and crosspeak shapes are also observed in 2D 13C‐13C and 15N‐13C ssNMR spectra, in which signals can be assigned to specific amino acids (Fig 6B and C). All labeled residues show differences in 13C and 15N chemical shifts. Two sets of crosspeaks are resolved for F19, G25, and A30 in all three samples and for I31, S26, and L34 in the sample derived from Tg2576‐treated mice. Thus, each brain‐derived fibril sample contains at least two distinct polymorphs.
Figure 6. Characterization of brain‐seeded Aβ40 fibrils by ssNMR.
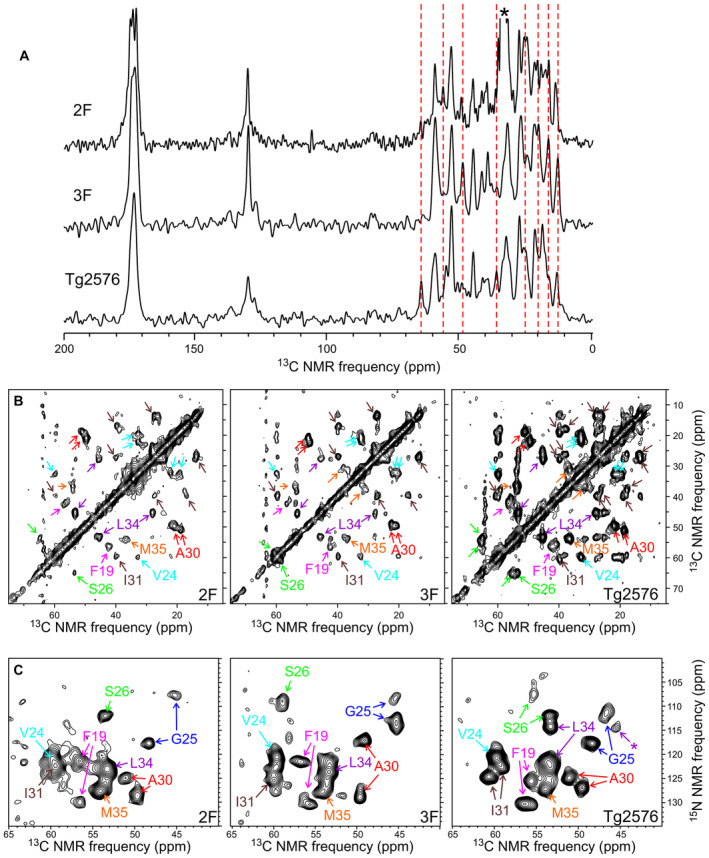
- 1D 13C ssNMR spectra of fibrils that were prepared in vitro by seeded growth from amyloid‐containing extracts of mouse brain homogenates. Mice had been treated with 2F‐, 3F‐, or Tg2576‐derived seeds. Aβ40 was 15N,13C‐labeled at F19, V24, G25, S26, A30, I31, L34, and M35. Vertical dashed lines indicate some of the ssNMR peaks that vary among the three samples. Asterisk indicates a peak from residual sodium dodecyl sulfate in the 2F sample.
- Aliphatic regions of 2D 13C‐13C ssNMR spectra of the same samples, with crosspeaks signals assigned to the labeled residues.
- 2D 15N‐13C ssNMR spectra of the same samples, showing intraresidue crosspeaks between chemical shifts of backbone nitrogen and α‐carbon sites.
Data information: Asterisk indicates a crosspeak to the L34 β‐carbon. Differences in peak positions in panel (A) and differences in crosspeak positions and shapes in panels (B) and (C) indicate differences in molecular structures of the brain‐seeded fibrils. One technical replicate per group was used in this study.
Superpositions of the 2D 15N‐13C ssNMR spectra from the three samples are shown in Appendix Fig S13B. Based on the presence or absence of common crosspeaks in these superpositions, we conclude that Aβ40 fibrils derived from 2F‐ and Tg2576‐treated mice may have one polymorph in common but differ in the identity of their second prevalent polymorph. The most prevalent polymorphs derived from 3F‐treated mice are distinct from those derived from 2F‐ and Tg2576‐treated mice. Moreover, one of the prevalent polymorphs in the Tg2576‐derived sample may closely resemble the predominant Aβ40 polymorph derived from cortical tissue of typical AD patients (Qiang et al, 2017; Ghosh et al, 2021), based on overlap of 15N‐13C crosspeaks (green X's in Appendix Fig S13B).
Aβ40 fibrils derived from 2F‐ and 3F‐treated mice do not have the same molecular structures as the original 2F and 3F fibrils that were used as seeds to induce amyloid deposition in the mouse brains. Appendix Fig S13C shows that crosspeak positions in 2D 15N‐13C ssNMR spectra of the brain‐derived samples do not agree with crosspeak positions in similar spectra of 2F and 3F fibrils with the same isotopic labeling pattern (orange and cyan X's). This observation may indicate that Aβ fibril structures evolve or adapt as they propagate in brain tissue, presumably due to the pronounced differences in chemical, physical, and biological environments between a mouse brain and the simple phosphate buffer used to grow the original 2F and 3F fibrils. That Aβ fibril structures can evolve during multiple rounds of self‐propagation in a new environment has been amply demonstrated in vitro (Qiang et al, 2011). Nonetheless, even though molecular structures of synthetic Aβ seeds do not propagate faithfully in mouse brains, data in Fig 6 and Appendix Fig S13 show that different molecular structures exist within amyloid deposits that are induced by structurally distinct seeds.
Discussion
Misfolded Aβ has been reported to exist in different conformations, similarly as described for other misfolded proteins including prions (Morales, 2017), tau (Dujardin et al, 2020), and α‐synuclein (Shahnawaz et al, 2020). In all these cases, conformational variability can be linked with different clinical or pathological manifestations. Whether different Aβ conformations are at the root of clinical variability in AD patients is still unknown. If this proves to be correct, Aβ strain variation should be considered when designing diagnostic and therapeutic strategies directed to identify and modify amyloid pathology.
In this work, we describe the seeding properties of two synthetic misfolded Aβ strains using in vitro and in vivo systems. The original seeds used in this study (termed 2F and 3F) have been extensively characterized for their structural features (Petkova et al, 2005; Tycko, 2014), although their biological properties have not been described. To our knowledge, this is the first study analyzing the biological properties of purified Aβ strains that have been characterized at the atomic resolution level. These Aβ polymorphs were prepared in vitro using somewhat different fibril growth conditions (quiescent growth for 3F, gentle agitation during growth for 2F). The preparation and characterization of these fibrils have been previously described (Petkova et al, 2005) although the “2F, 3F” nomenclature was not used at that moment. Previous work from the Tycko lab shows that these polymorphs exhibit different morphologies in TEM images and different 13C and 15N chemical shifts in solid state NMR spectra. Thus, they contain different molecular structures. These aggregates transmit their conformational features by seeding (Petkova et al, 2005), mirroring the strain specific in vitro replication extensively described for infectious prions (Castilla et al, 2008). Here, we describe that the distinct structural motifs observed in 2F and 3F fibrils are linked with different biochemical features such as their differential resistance to proteolytic digestion and their in vitro seeding activities. Remarkably, these aggregates displayed strain‐specific affinities to dyes previously shown to differentiate conformational variants of other misfolded proteins such as α‐synuclein (Shahnawaz et al, 2020) and prion proteins (Magnusson et al, 2014). Moreover, 2F and 3F fibrils induced the formation of easily distinguishable pathological features in treated mice (Table 1). The seeding properties of 2F and 3F fibrils in vivo resulted in remarkably different pathological features, namely different rates of aggregation, formation of different plaque types, tropism to specific brain regions, differential recruitment of Aβ40/Aβ42 peptides, and induction of microglial and astroglial responses. Importantly, the seeded amyloidosis and linked pathological features induced by synthetic peptides were easily distinguishable to the ones generated by in vivo‐derived seeds (collected from an aged Tg2576 mouse). The structural differences between the aggregates generated by the three seeds used in this study was confirmed by different means, including the reactivity of seeded aggregates to a variety of dyes able to bind misfolded protein structures, and ssNMR. It is important to note that in all cases, the properties of the seeded aggregates did not match the ones recorded for the original seeds. This outcome was expected as we believe that seeded pathology in treated animals is likely the result of both, the seeds administered and the endogenous aggregates that are normally displayed by Tg2576 mice as they age. The fact that different ssNMR results were obtained with human brain tissue and mouse brain tissue (and different results were obtained with human brain tissue from rapidly progressing AD cases vs. typical long‐duration AD cases, as reported by Qiang et al (2017)) supports the idea that in vitro amplification does not preferentially select a single polymorph (or strain or conformer). It is possible that in vitro amplification alters the relative populations of different polymorphs to some extent, but we have designed our amplification protocol to minimize this effect. In particular, we confirm by TEM that abundant long fibrils develop within 4 h after the initial seeding step, and we avoid multiple rounds of seeded fibril growth in our protocol for preparing solid state NMR samples (see Materials and Methods). An alternative explanation is that the accelerated induction of amyloid pathology through seeding is responsible to alter the conformation of the resulting aggregates, analogous to what is observed when different protein concentrations are used in in vitro aggregation assays, or the type and size of aggregates displayed in mouse models carrying the same mutations on APP but producing different concentrations of Aβ (Hsiao et al, 1996; Jankowsky et al, 2004; Oakley et al, 2006). It is important to mention that the aggregates induced by Tg2576 seeds did not induce the same pathology observed spontaneously in older Tg2576 transgenic mice (Morales et al, 2015, 2021). Specifically, the natural amyloid pathology in aged Tg2576 mice is characterized by compact Aβ deposits in the cortex and hippocampus that strongly react against ThS and displaying limited CAA. On the contrary, pathology induced by Tg2576 seeds show diffuse aggregates poorly reactive against ThS, mild CAA, and tropism directed to the injection site (Morales et al, 2015). In addition, the glial response toward naturally versus induced aggregates is different, being more prominent in aged, not seeded subjects (Appendix Fig S14). Interestingly, the pathology observed in animals treated with the Tg2576‐derived aggregates is similar as the one described for humans potentially exposed to biologically active Aβ seeds (Gomez‐Gutierrez & Morales, 2020). Whether the mechanisms of exogenous versus endogenous seeding lead to different pathologies should be carefully explored as they could unveil the etiology of certain cases of brain amyloidosis.
An important contribution of this work is to describe the pathological significance of strain‐specific replication of Aβ misfolding in vivo. Our results show that Aβ aggregates with known differences in molecular structural properties induce amyloid deposition in specific brain regions, mimicking the extensively described strain‐specific tropism of infectious prions (Morales et al, 2007; Morales, 2017). Specifically, our results show that while Tg2576‐derived seeds induce diffuse amyloid deposition across the hippocampus, synthetic Aβ aggregates promoted amyloidosis prominently in the alveus (2F) and dentate gyrus (2F and 3F). Along the same line, amyloid deposition was also induced in other brain regions in a Aβ strain‐specific fashion. Particularly, the lateral ventricle (2F) and meningeal blood vessels (Old Tg2576 BH) displayed these pathological differences. Although connected, these anatomical structures are away from the injection site. This, and the fact that each injectate induced a well‐defined response in this pathological feature, supports the idea of strain‐specific spreading of Aβ misfolding. Importantly, the amyloid deposition in meningeal vessels presented here reproduce our previous findings in Tg2576 mice intra‐cerebrally injected with Tg2576‐derived seeds (Morales et al, 2015, 2021).
We also show that the induction of amyloid pathology by distinct Aβ seeds resulted in different glial responses. Our interpretation of this data is that 2F‐, 3F‐, and Tg2576‐Aβ seeded aggregates represents unique populations of misfolded proteins inducing specific biological responses. Though a more comprehensive inflammatory characterization of the Aβ seed‐specific inflammatory profiles is necessary, our multiplex analysis support our initial conclusions. This data demonstrates a predominance of proinflammatory cytokines expression in mice receiving the Tg2576‐derived seeds. This indicates that neuroinflammation is exacerbated by this specific Aβ strain compared to mice challenged with the other seeds. 2F and 3F fibrils elicited a moderate proinflammatory signature characterized by INF‐γ and CCL3 production, with 2F increasing also IL‐1a and CCL5 production. These findings indicate the existence of differential glial responses to the seed strain used as inocula. Cytokines can be produced by microglia, astroglia and neurons, however the exact sources for each cytokine in the different seeded mice need to be further investigated. Moreover, the biological impact of the Aβ strains in the innate immune response may be underestimated by the complexity of microglial and astroglial responses in the disease context, with the coexistence of multiple functional phenotypes including spatial and temporal variations (Escartin et al, 2021). Interestingly, the three seeds (Tg2576, 2F, and 3F) induced similar CCL3 upregulation, a chemokine involved in chemotaxis and shown to contribute to T‐cell recruitment to the brain (Shechter et al, 2013). Moreover, these seeds also induced IFN‐γ elevation, an essential element of the neuroinflammatory network involved in leukocyte trafficking (Kunis et al, 2013). Future research in this line may help to link specific conformational motifs on misfolded Aβ with strain‐specific inflammatory profiles. It is important to consider that future experiments specifically focusing on the behavioral, synaptic and neurotoxic aspects of naturally occurring and seeded pathology should be conducted to further evaluate the biological significance of strain‐specific spreading of misfolded Aβ.
Another interesting finding in this study is the strain‐specific recruitment of Aβ peptides into amyloid deposits. While synthetic Aβ seeds (2F and 3F) had a preference to recruit Aβ40, the in vivo derived seeds displayed a higher affinity for Aβ42. Interestingly, we observed that while some aggregates were composed by both Aβ40 and Aβ42 peptides, some others were exclusively formed by one specific Aβ peptide. Moreover, we observed that the population of aggregates formed by different patterns of Aβ40 and Aβ42 varied depending on the brain region within a single brain, suggesting that the different recruitment of Aβ peptides is just partially due to the seeds administered. This conclusion is further supported by the brain region‐specific reactivity of amyloid plaques to LCOs.
One limitation of this study involves the use of synthetic Aβ seeds. As judged by their seeding activity in animal models (Stöhr et al, 2014 and this report), the seeding titers of misfolded synthetic Aβ are considerably lower compared to their in vivo‐derived counterparts (Morales et al, 2015). In addition, 2F and 3F fibrils used in this study were developed under lab‐controlled conditions using chemically synthesized Aβ40. On the contrary, it is very likely that Aβ aggregates in AD brains are composed by a multitude of different structural arrangements, similarly to what has been hypothesized for infectious prions (Polymenidou et al, 2005; Collinge & Clarke, 2007). In that sense, the use of these particular synthetic aggregates may be advantageous, as they are expected to contain either limited or even a unique set of structural motifs. This homogeneity may help to properly characterize the contribution of specific Aβ strains in AD pathology. In addition, the well‐characterized structural properties of these fibrils (Petkova et al, 2005) may allow to make structure/pathology predictions. Regardless of the advantages expressed above, it is uncertain whether the synthetic Aβ strains used in this study represent naturally occurring aggregates present in human diseased brains. This obviously limits the potential impact of the results and conclusions presented in this article. However, we observed that HS‐194 and HS‐68 LCOs recognized amyloid deposits in post‐mortem brains from AD and nondemented individuals (Appendix Fig S15), suggesting that seeded aggregates in experimental rodents share common structural motifs with the amyloid deposits present in human brains. Based in their LCOs reactivities, aggregates from humans seem heterogenous as they react to dyes in different patterns. Specifically, while some parenchymal deposits may be recognized by either one, or both LCOs (Appendix Fig S15A–C and J–L), others appears to be recognized by a single dye (Appendix Fig S15G–I). In addition, the proportion of LCO binding appears to be different, depending of the type (vascular/parenchymal) and morphology of the aggregates. Interestingly, the pattern of LCO binding was different for amyloid deposits in the AD and nondemented brains, suggesting that these clinically diverse cases accumulate different proportions of Aβ strains. Nevertheless, we acknowledge that the previously discussed evidence is indirect. Along these lines, future studies must confirm or discard 2F and 3F as surrogates of AD‐relevant Aβ strains, as well as the applicability of our conclusions to AD pathophysiology.
In conclusion, our results provide useful information on the pathological significance of misfolded Aβ strains. Future studies using relevant (patient isolated) Aβ propagons are necessary to confirm or rule out a putative role of misfolded strains in the clinical variability observed across AD patients.
Materials and Methods
Preparation of synthetic Aβ aggregates and monomeric Aβ
Aβ40 peptides with isotopic labeling of specific residues were synthesized at a 0.1 mmol scale by standard solid phase methods with Fmoc chemistry, using a Biotage Initator+ Alstra synthesizer, Fmoc‐Val‐NovaSyn‐TGA resins (Sigma‐Aldrich), and DIC/OxymaPure activation. All Fmoc‐amino acids were double‐coupled with a fivefold excess at 75°C, except that Fmoc‐His(Boc)‐OH was double‐coupled at 50°C. Each labeled amino acid was single coupled with a threefold excess, followed by single coupling with a fivefold excess of the corresponding unlabeled amino acid. Acetic anhydride capping was performed before deprotection steps, which was performed with 20% piperidine in DMF at room temperature. Cleavage from the resin was performed with a standard cocktail, including tetrabutylammonium bromide in the final 15 min of cleavage to reduce any oxidized methionine at M35. After precipitation and washing with cold methyl t‐butyl ether, crude peptides were purified by reverse‐phase high‐performance liquid chromatography, using a Zorbax 300SB‐C3 column (Agilent Technologies). Purified peptides were lyophilized and stored at −25°C. Final purity was determined to be greater than 95% by liquid chromatography/mass spectrometry.
For fibril growth in vitro, 3–5 mg of lyophilized Aβ40 were first dissolved in DMSO at approximately 5 mM peptide concentration. Aliquots of DMSO‐solubilized peptide were then diluted rapidly into 10 mM sodium phosphate buffer, pH 7.4, containing 0.1% w/v NaN3 to produce final Aβ40 concentrations of 100 μM. Solutions in phosphate buffer were incubated at room temperature in 15 mL or 50 mL polypropylene tubes, either quiescently to produce 3F fibrils or with orbital mixing (approximately 60 rotations per minute) to produce 2F fibrils (Petkova et al, 2005). Seeds of previously prepared 2F or 3F fibrils (i.e., sonicated fibril fragments with typical lengths of 50–150 nm, approximate 1:30 ratio of Aβ40 in seeds to DMSO‐solubilized Aβ40) were added to the solutions before incubation, to accelerate fibril growth and ensure formation of the desired polymorphs. Solutions were incubated for several days. Fibril formation was verified by negative‐stain TEM (Fig 1A).
2F and 3F fibrils for injection into mouse brains were prepared from Aβ40 with 13C labels at V18 carbonyl, A30 β‐carbon, and G33 α‐carbon sites. Additional 2F and 3F fibrils for ssNMR measurements, which were used for the comparisons in Appendix Fig S13C, were prepared from Aβ40 with uniform 15N,13C labeling of F19, V24, G25, S26, A30, I31, L34, and M35.
Biochemical characterization of the synthetic Aβ aggregates
The structural features of 2F and 3F fibrils have been previously characterized (Petkova et al, 2005; Tycko, 2014). Additional structural characterization presented in this article included analyses by transmission electron microscopy (TEM, see above). Both fibrils preparations, at 0.1 mg/ml, were treated with different concentrations of proteinase K (PK, 100, 50, 25, and 12.5 μg/ml) for 1 h at 37°C with gentle agitation (600 rpm in an Eppendorf thermomixer). Digestion products were analyzed by silver staining after separation in NuPage Bis‐Tris 12% gels. The monomeric band of Aβ40 was analyzed using the WCIF ImageJ software (National Institutes of Health, Bethesda, MD, USA).
In vitro aggregation assay
2F and 3F synthetic Aβ aggregates were further characterized for their seeding potential in vitro using the Aβ version of the protein misfolding cyclic amplification (Aβ‐PMCA) technique (Salvadores et al, 2014). In summary, purified, aggregate‐free Aβ40 (1 μM) was incubated in 100 mM Tris–HCl buffer, pH 7.4, and 5 μM ThT, at 20°C in opaque 96‐well plates with cycles of 1‐min cyclic agitation (500 rpm) every 30 min. Samples were incubated in the presence of 100 pM of either 2F or 3F fibrils. Protein aggregation was monitored by ThT fluorescence measured at 485 nm after excitation at 435 nm using a plate spectrofluorometer. All graphs were modeled by using a Boltzmann sigmoidal equation.
Synthesis of thiophene‐based ligands and LCOs
p‐FTAA, h‐FTAA, HS‐68, HS‐167, HS‐169, HS‐194, and HS‐199 were synthesized as described previously (Åslund et al, 2009; Klingstedt et al, 2011, 2015; Shirani et al, 2015, 2017; Shahnawaz et al, 2020). HS‐208 and HS‐212 were synthesized as shown in Fig 7A.
Figure 7. Supplemental schemes for the synthesis of thiophene‐based ligands and LCOs.
- General procedure (G1) for Suzuki coupling to generate compounds 4a and 4b.
- General procedure (G2) for te synthesis of HS‐208 and HS‐212 from the dimer building blocks 4a and 4b.
General procedure for Suzuki coupling (G1)
A mixture of the (2) or (3), and (1), K2CO3 (3 equiv./bromine), in 1,4‐dioxane/methanol (8: 2, 8 ml/mmol, degassed) and PEPPS‐IPr (5 mol %) was heated to 80°C until the LC–MS indicated completion of reaction. After cooling to room temperature, pH was adjusted to 4 by 1 M HCl and the residue was extracted with DCM (3 × 30 ml/mmol), washed with water (3 × 30 ml/mmol) and brine (30 ml). The combined organic phase was dried over MgSO4 and the solvent was evaporated. The crude product was subjected to column chromatography with appropriate solvent to give desired products 4a‐4b (Fig 7B).
General procedure for condensation reactions (G2)
A few drops of pyridine were added to a cold solution of the 4a or 4b (1 equiv) and the corresponding 2‐methyl‐3‐alkylbenzothiazolium salt 5 (1 equiv.) in anhydrous MeOH. The mixture was refluxed until completion of the reaction (monitored by HPLC). The solvent was evaporated in vacuo to provide a dark red solid, which was crystallized from appropriate solvent. The red solid was collected by filtration, washed with cold MeOH and dried in vacuum to afford desired final products in good purity and yield.
4a
General procedure of Suzuki coupling (G1) was applied starting with 5‐formyl‐2‐thiopheneboronic acid (1) (102 mg, 0.657 mmol) and 5‐Bromo‐1,3‐benzodioxole (2) (120 mg, 0.597 mmol). The residue was subjected to column chromatography using [heptane/EtOAc (8:1 → 5:1)] to give 4a (73 mg, 53%) as yellow solid. IR (neat) 1,644, 1,499, 1,436, 1,358, 1,252, 1,224, 1,104, 1,051, 1,031, 931, 843, 815, 785, 755 cm−1. 1H NMR (500 MHz, CDCl3) δ 9.86 (s, 1H), 7.70 (d, J = 3.9 Hz, 1H), 7.27 (d, J = 3.9 Hz, 1H), 7.19 (dd, J = 8.1, 1.9 Hz, 1H), 7.12 (d, J = 1.8 Hz, 1H), 6.86 (d, J = 8.1 Hz, 1H), 6.03 (s, 2H). 13C NMR (126 MHz, CDCl3) δ 182.8, 154.5, 149.0, 148.6, 141.6, 137.7, 127.5, 123.6, 121.0, 109.1, 106.9, 101.8. LCMS (ESI): m/z calcd for C12H8O3S (M + H) 232.3 found: 233.6.
4b
General procedure of Suzuki coupling (G1) was applied starting with 5‐formyl‐2‐thiopheneboronic acid (1) (175 mg, 1.15 mmol) and 5‐bromo‐2,1,3‐benzothiadiazole (3) (0.2 mg, 0.930 mmol). The residue was subjected to column chromatography using [DCM/MeOH (1%)] to give 4b (162 mg, 75%) as yellow solid. IR (neat) 1,653, 1,477, 1,448, 1,426, 1,223, 1,189, 1,146, 1,064, 845, 800, 758 cm−1. 1H NMR (500 MHz, CDCl3) δ 9.95 (s, 1H), 8.32 (dd, J = 1.8, 0.8 Hz, 1H), 8.07 (dd, J = 9.1, 0.8 Hz, 1H), 7.91 (dd, J = 9.1, 1.8 Hz, 1H), 7.81 (d, J = 4.0 Hz, 1H), 7.59 (d, J = 3.9 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 182.9, 155.1, 154.9, 152.1, 144.0, 137.4, 134.4, 128.6, 126.0, 122.5, 118.5. LCMS (ESI): m/z calcd for C11H6N2OS2 (M + H) 246.3 found: 247.2.
HS‐208
General procedure of condensation reactions (G1) was applied with 5 (100 mg, 0.328 mmol) and 4a (76 mg, 0.328 mmol). The residue was purified by crystallization from MeOH/THF (80:20) to give stilbene HS‐208 (110 mg, 65%) as deep red solid. IR (neat) 1,596, 1,584, 1,506, 1,448, 1,419, 1,332, 1,275, 1,241, 1,209, 1,139, 1,067, 1,034, 926, 863, 799, 753 cm−1. 1H NMR (500 MHz, DMSO‐d6) δ 8.46–8.39 (m, 2H), 8.26 (d, J = 8.5 Hz, 1H), 7.96 (d, J = 4.2 Hz, 1H), 7.85 (ddd, J = 8.5, 7.3, 1.2 Hz, 1H), 7.79–7.74 (m, 1H), 7.69 (d, J = 4.0 Hz, 1H), 7.64 (d, J = 15.4 Hz, 1H), 7.41 (d, J = 1.9 Hz, 1H), 7.30 (dd, J = 8.1, 1.9 Hz, 1H), 7.05 (d, J = 8.1 Hz, 1H), 6.12 (s, 2H), 4.91 (q, J = 7.2 Hz, 2H), 1.46 (t, J = 7.2 Hz, 3H). 13C NMR (126 MHz, DMSO‐d6) δ 170.7, 151.5, 148.5, 148.3, 141.4, 140.9, 137.4, 137.3, 129.4, 128.2, 128.0, 126.8, 125.4, 124.3, 120.4, 116.4, 110.3, 109.1, 106.0, 101.8, 44.3, 14.1. LCMS (ESI): m/z calcd for C22H18NO2S2 (M + H) 392.5 found: 392.7.
HS‐212
General procedure of condensation reactions (G1) was applied with 5 (62 mg, 0.203 mmol) and 4b (50 mg, 0.203 mmol). The residue was purified by crystallization from MeOH to give stilbene HS‐212 (70 mg, 65%) as deep red solid. IR (neat) 1,594, 1,581, 1,510, 1,421, 1,334, 1,261, 1,169, 962, 850, 822, 810, 760, 722 cm−1. 1H NMR (500 MHz, DMSO‐d6) δ 8.49 (d, J = 14.6 Hz, 2H), 8.43 (d, J = 8.0 Hz, 1H), 8.28 (d, J = 8.5 Hz, 1H), 8.21 (d, J = 9.1 Hz, 1H), 8.15 (dd, J = 9.2, 1.6 Hz, 1H), 8.09 (q, J = 4.1 Hz, 2H), 7.86 (t, J = 7.7 Hz, 1H), 7.78 (dd, J = 15.2, 5.8 Hz, 2H), 4.95 (q, J = 6.9 Hz, 2H), 1.49 (t, J = 7.2 Hz, 3H). 13C NMR (126 MHz, DMSO‐d6) δ 170.6, 154.5, 154.1, 148.7, 140.9, 140.8, 139.9, 136.6, 133.9, 129.5, 128.5, 128.3, 128.3, 128.2, 124.4, 122.2, 116.9, 116.5, 111.9, 44.5, 14.2. LCMS (ESI): m/z calcd for C21H16N3S3 (M + H) 406.6 found: 406.6.
In vitro reactivity of Aβ strains to luminescent conjugated oligothiophenes (LCOs)
2F and 3F fibrils were resuspended at 1 mg/ml (vide infra) and diluted to 100 μM in PBS. These fibrils were further diluted at a 1:10 ratio (10 μM) in PBS in a 96‐well microtiter plate. Stock solutions of LCOs at 1.5 mM, were diluted in distilled water to 15 μM and added to the wells containing the 2F and 3F fibrils to a final concentration of 0.3 μM. The samples were incubated at 37°C and the emission spectrum of each probe was collected after 30 min by exciting the samples at 450 nm (p‐FTAA), 465 nm (h‐FTAA), 440 nm (HS‐68), 525 nm (HS‐167), 535 nm (HS‐169, HS‐194, HS‐208, and HS‐212), or 545 nm (HS‐199).
Preparation of inocula
(i) A brain from an aged (15 months old) Tg2576 mouse (frontal cortex) was homogenized at 10% w/v using a glass homogenizer in Phosphate Buffer Saline (PBS, MP Biomedicals, Santa Ana, CA, USA) containing a cocktail of protease inhibitors (Roche Diagnostics). (ii) Both 2F and 3F fibrils were pelleted by ultracentrifugation at 100,000 g, resuspended in PBS at 1 mg/ml concentration, and sonicated for 30 s right before injection into animals. The resulting inocula, both from brains and the synthetic fibrils, was stored at −80°C until used for animal injection.
Mice
Tg2576 transgenic mice were used in this study. These mice express the human APP gene harboring the Swedish mutation and start developing cerebral Aβ deposits at approximately 8 months old (Hsiao et al, 1996; Morales et al, 2015, 2021). Mice were housed in standard conditions (22°C, 12 h dark/light cycles, food and water ad libitum) as groups of no more than five animals per cage. All animal procedures described in this work complied with the regulations of the Institutional Animal Care and Use Committee of The University of Texas Health Science Center at Houston (UTHealth, protocol number AWC‐21‐0065) and approved by an Animal Welfare Committee. We used 5–10 animals (sex randomized) per experimental group.
Animal procedures
Fifty days old Tg2576 mice were intracerebrally (i.c.) injected with a 10% w/v brain homogenate from an aged Tg2576 mouse or synthetic Aβ (2F or 3F fibrils, or monomeric protein). Injections were performed stereotaxically in the hippocampus (both hemispheres) of anesthetized animals using the following coordinates from bregma: anteroposterior: −1.8 mm; medio‐lateral: ±1.8 mm; dorso‐ventral: −1.8 mm. Ten microlitres of the brain homogenate or synthetic Aβ (1 mg/ml) were administered per injection using a Hamilton syringe. Mice were sacrificed by CO2 inhalation at either 100‐ or 250‐days post injection (dpi). Brain halves were frozen to be analyzed by protein‐based multiplex immunoassay for Aβ40, Aβ42, and cytokines levels using commercially available kits (Miliplex from Milipore for Aβ40 and Aβ42, and Bio‐Plex Pro Mouse Cytokine 23‐plex Assay from Bio‐Rad). Opposite brain halves were fixed by immersion in formalin and saved for further immunohistochemical (IHC) studies.
Serial extraction and multiplex immunoassay measurement of aqueous insoluble Aβ and cytokine panel
Two hundred microlitres of 10% w/v brain homogenates were ultra‐centrifuged at 100,000 g for 1 h at 4°C using a L100K Beckman–Coulter ultracentrifuge (Beckman–Coulter). Supernatants (S1) were snap frozen in liquid nitrogen and pellets were resuspended in 200 μl of 70% v/v formic acid. Samples were sonicated and centrifuged again, in the same conditions, for 30 min. Supernatants (S2) were diluted 25 times in 1 M Tris buffer (pH 11) and snap frozen in liquid nitrogen. Resulting samples were stored at −80°C until used (next day) for measuring Aβ concentration using Aβ40/Aβ42 specific protein‐based multiplex immunoassay kits (Miliplex, Milipore). S1 samples were used to measure a panel of 23 cytokines (Bio‐Plex Pro Mouse Cytokine 23‐plex Assay, Bio‐Rad). The protocol provided by the kits' manufacturers was followed for measuring Aβ and cytokines. Extractions and multiplex measurements were performed in duplicates.
Human samples
Human autopsied fixed brains hemispheres from AD patients and non‐demented individuals were obtained from the National Disease Research Interchange (NDRI, USA) in a de‐identified manner. Fixed hemispheres were dissected and temporal cortices from one AD patient and one non‐demented individual were used for LCO staining (see below). The AD sample corresponded to a 66‐year‐old female AD that was histologically scored for Tau and Aβ pathology as a Braak V‐VI and Thal IV, respectively. The non‐demented brain tissue was obtained from a 84‐year‐old female that was scored as a Braak III‐IV and Thal IV. Research on human samples was performed following The Code of Ethics of the World Medical Association (Declaration of Helsinki). Brain tissues were manipulated following the universal precautions for working with human samples and as directed by the Institutional Review Board (IRB) and the Biosafety Office of The University of Texas Health Science Center at Houston.
Histological studies
Fixed mouse brains and human temporal cortices were dehydrated in graded ethanol for paraffin embedding. Then, paraffin‐embedded brains were serially sliced in 10 μm thick sagittal sections. Five slices per animal were used for the histological studies. In short, sections were deparaffinized and Thioflavin S (ThS) staining was performed by incubating tissue slices with a ThS (Sigma, St. Louis, MO, USA) solution (0.1% w/v in 50% ethanol) for 15 min. After incubation, sections were washed for 2 min in 80% ethanol, dehydrated, cleared in xylene, and cover slipped with DPX mounting medium (Innogenex, San Ramon, CA, USA). For immunohistochemistry, after deparaffinization, the endogenous peroxidase activity was blocked with a solution containing 10% methanol and 3% H2O2 in PBS, for 30 min. Then, brain sections were incubated in 85% formic acid for 5 min to enhance antigen retrieval. Sections were incubated overnight with the mouse 4G8 antibody (Covance), the rabbit Iba‐1 antibody (Wako) or the rabbit GFAP antibody (Abcam), diluted 1:1,000 in PBS with 0.02% Triton‐X100 (Sigma). After washing with PBS, sections were incubated for 1 h with an HRP‐linked secondary anti‐mouse antibody (GE Healthcare) or HRP‐linked secondary anti‐rabbit antibody (Sigma Aldrich) at a 1:500 dilution. Peroxidase reaction was visualized using a DAB Kit (Vector Labs) following the manufacturer's instructions. Finally, sections were dehydrated in graded ethanol, cleared in xylene, and cover slipped as described previously (Duran‐Aniotz et al, 2021; Morales et al, 2021). For double immunofluorescence staining, deparaffinized sections were consecutively incubated with anti‐4G8 and anti‐Iba‐1 or anti‐GFAP antibodies. After primary antibody incubations, sections were washed with PBS and incubated for 1.5 h with anti‐mouse Alexa Fluor‐488 and anti‐rabbit Alexa Fluor‐594 at a 1:500 dilution. Then, sections were washed and cover slipped using FluorSave Mounting medium (Millipore Sigma). For LCOs staining of mouse and human brain sections, paraffin‐embedded tissues were deparaffinized and incubated with LCOs at 300 nM diluted in PBS for 30 min at room temperature. Sections were rinsed with PBS and cover slipped with VECTASHIELD® Antifade Mounting Medium with DAPI (Vector Labs).
Image analyses of brain slices
Immunostained sections, as well as those stained with ThS and LCOs, were visualized in a dual (bright field and epifluorescence) microscope (DMI6000B microscope from Leica Microsystems, Germany). For quantification, image analysis was performed using the ImageJ software (National Institutes of Health). Aβ burden was defined as the 4G8 antibody labeled area in each slice per total area analyzed and expressed as percentages. Quantification in the lateral ventricle was performed by analyzing plaques that surround the lateral ventricles and giving a visual score ranging from 0 (No plaques) to 10 (plaques found in the tissue with more plaques). Given the subjectivity of this measurement, this was performed blinded by 2 independent researchers (RG‐G and KD). Quantification of CAA was performed by counting the total amount of meningeal vessels and the amount of 4G8‐positive meningeal vessels following this formula: % 4G8 positive vessels = (Total number of 4G8 positive vessels) / (Total number of vessels).
Preparation of brain‐seeded fibrils for solid‐state nuclear magnetic resonance
Amyloid‐containing extracts were prepared from mouse brain homogenates by following a protocol developed previously for ssNMR studies of Aβ fibrils from human cortical tissue (Lu et al, 2013; Qiang et al, 2017). Briefly, frozen homogenates were thawed, added to 5 mL of buffer A (10 mM Tris–HCl, pH 7.5, 0.25 M sucrose, 3 mM EDTA, 0.1% w/v NaN3) with one half tablet of Roche Complete Protease Inhibitor, then rotated end‐over‐end overnight at 4°C. After increasing the sucrose concentration to 1.2 M, the mixtures were centrifuged at 220,000 g for 60 min at 4°C. The resulting pellets were resuspended in 11 ml of buffer B (10 mM Tris–HCl, pH 7.5, 1.9 M sucrose, 3 mM EDTA, 0.1% w/v NaN3) and centrifuged again with the same conditions. Top layers were resuspended in 10 ml of Tris–HCl buffer (50 mM, pH 8.0) and spun at 11,000 g for 15 min at 4°C. Pellets were resuspended in 10 ml of Tris–HCl buffer with 2 mM CaCl2 and 10 μg/ml of DNAse I, then incubated at room temperature for 60 min with orbital mixing. After pelleting again, pellets were resuspended in 4.5 ml of buffer C (10 mM Tris–HCl, pH 7.5, 1.3 M sucrose, 3 mM EDTA, 0.1% w/v NaN3, 1% w/v SDS), then centrifuged at 350,000 g for 30 min at 4°C. Pellets were washed twice by resuspension in deionized water followed by centrifugation.
Fibrils for ssNMR measurements were then prepared by resuspending the pelleted extracts from mouse brain homogenates in 0.5 ml of 10 mM sodium phosphate buffer, pH 7.4, containing 0.1% w/v NaN3. Suspensions were sonicated vigorously for 20 min (Branson S‐259A sonifier with tapered 1/8" microtip horn, lowest power, 10% duty factor) to break amyloid into short fragments. Aliquots of isotopically labeled Aβ40, solubilized in DMSO and containing 0.22 mg of the peptide to produce [Aβ40] = 100 μM, were then added to the sonicated suspensions and mixed by brief vortexing. TEM images were recorded after 4 h of quiescent incubation at room temperature (Appendix Fig S11A, top row of images). Observation of abundant long fibrils confirmed seeded fibril growth, as control experiments without brain extract produced no detectable fibrils after 4 h incubation. Sample volumes were then increased to 2.5 ml by addition of the same buffer and DMSO‐solubilized, isotopically labeled Aβ40 was added to restore the total peptide concentration to 100 μm (1.1 mg total peptide). Samples were then incubated at room temperature for an additional 4‐5 days, after which TEM images were recorded again (Appendix Fig S11B, bottom row). To maximize conversion of soluble Aβ40 to fibrils, samples were given a 10 s burst of sonication once per hour for the first 24 h (Qsonica model Q55 sonifier, 1/8" horn, power level 20, controlled by an Omega PTC‐16 timer). Aside from these brief sonication bursts, incubation was quiescent.
Finally, fibrils were pelleted, resuspended in deionized water, pelleted again, and lyophilized. Lyophilized material (containing ≤ 1 mg of isotopically labeled fibrils in a larger quantity of nonfibrillar material from the brain tissue) was then packed into 1.8 mm‐diameter magic‐angle spinning (MAS) rotors for ssNMR measurements and fully rehydrated by addition of 10 mM sodium phosphate buffer to the rotor.
Transmission electron microscopy
TEM images of 2F and 3F fibrils and fibrils grown from brain homogenates were obtained with an FEI Morgagni microscope, operating at 80 keV, equipped with an Advantage HR camera (Advanced Microscopy Techniques). Each fibril sample was diluted by a factor of 5–10 in deionized water, then applied as a 10 μl aliquot to a glow‐discharged grid (carbon film supported by lacey carbon on 300 mesh copper), allowed to adsorb for approximately 120 s, blotted, rinsed twice with deionized water, blotted, stained with a 10 μl aliquot of 2% w/v uranyl acetate for 15–30 s, blotted, and dried in air.
Solid‐state nuclear magnetic resonance
ssNMR data spectra were acquired at 17.5T (187.5 MHz 13C NMR frequency, 75.6 MHz 15N NMR frequency), using a Varian InfinityPlus spectrometer and a 1.8 mm magic‐angle spinning (MAS) probe obtained from the laboratory of Dr. Ago Samoson (Tallinn University of Technology, Estonia). MAS frequencies were 17.00 kHz. Sample temperatures were 25° ± 1°C. All measurements used standard cross‐polarization techniques for 1H‐13C, 1H‐15N, and 15N‐13C polarization transfers and two‐pulse phase‐modulated 1H decoupling with a 100 kHz radio‐frequency (rf) field amplitude (Bennett et al, 1998). 2D 13C‐13C spectra used 2.82 ms mixing periods with finite‐pulse rf‐driven recoupling (Bennett et al, 1998; Ishii, 2001), using 20.0 μs 13C π pulses at a carrier frequency of 32 ppm to maximize polarization transfers among aliphatic 13C sites. 2D 13C‐13C spectra of brain‐derived fibrils in Fig 6B were obtained in 2.5–4.4 days, using maximum t1 values of 4.00 ms, t1 increments of 20.0 μs, and recycle delays of 1.0–1.3 s. 2D 15N‐13C spectra in Fig 6C were obtained in 2.0–3.5 days, using maximum t1 values of 7.35 ms, t1 increments of 49.0 μs, and 1.0 s recycle delays. Similar conditions were used to obtain 2D spectra of synthetic 2F and 3F fibrils (grown in vitro, not seeded with brain extract), which were used for the comparisons in Appendix Fig S11C.
Data were processed with nmrPipe software (Delaglio et al, 1995). Pure Gaussian apodization functions were used, corresponding to 0.8 and 1.3 ppm line‐broadening in 13C and 15N dimensions, respectively, without artificial resolution enhancement. 13C and 15N chemical shifts are relative to 4,4‐dimethyl‐4‐silapentane‐1‐sulfonic acid (DSS) and liquid ammonia, respectively, based on an external standard of 1‐13C‐L‐alanine powder at 179.65 ppm relative to DSS. Contour levels in plots of 2D 13C‐13C and 2D 15N‐13C spectra increase by successive factors of 1.4 and 1.2, respectively.
Quantification and statistical analysis
According to the data distribution, Student's t‐test or Mann–Whitney U‐test were used to compare Aβ burden in injected and noninjected animals. The values are expressed as means ± standard deviation. Data was analyzed using the Graph Pad prism software. Statistical differences were considered significant for values of P < 0.05. All analyses were performed in a blinded manner by different investigators.
Author contributions
Ruben Gomez‐Gutierrez: Data curation; formal analysis. Ujjayini Ghosh: Data curation; formal analysis; investigation. Wai‐Ming Yau: Investigation. Nazaret Gamez: Data curation; formal analysis; investigation. Katherine Do: Data curation; investigation. Carlos Kramm: Investigation. Hamid Shirani: Resources. Laura Vegas‐Gomez: Investigation. Jonathan Schulz: Investigation. Ines Moreno‐Gonzalez: Data curation; formal analysis; investigation. Antonia Gutierrez: Formal analysis. K Peter R Nilsson: Resources. Robert Tycko: Conceptualization; data curation; formal analysis. Claudio Soto: Conceptualization. Rodrigo Morales: Conceptualization; methodology; writing – original draft; project administration.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix S1
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Acknowledgments
This work was supported by grants from the National Institutes of Health (R56AG061878 and RF1AG059321) to RM and CS, the Alzheimer's Association (AARGD‐18‐566576 to RM), the Health Institute Carlos II (ISCIII) of Spain co‐financed by FEDER funds from the European Union (grant PI21/00915 to AG), and the Junta de Andalucía Consejería de Economía y Conocimiento of Spain co‐financed by Programa Operativo FEDER 2014‐2020 (grants UMA18‐FEDERJA‐211 and P18‐RT‐2233 to AG). Work by UG, WMY, and RT was supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health (project Z01‐DK029061‐14). We acknowledge the use of tissues procured by the National Disease Research Interchange (NDRI) with support from NIH grant U42OD11158.
EMBO reports (2023) 24: e57003
Contributor Information
Robert Tycko, Email: robertt@niddk.nih.gov.
Claudio Soto, Email: claudio.soto@uth.tmc.edu.
Rodrigo Morales, Email: rodrigo.moralesloyola@uth.tmc.edu.
Data availability
The solid state NMR data included in this paper are available at https://data.mendeley.com/datasets/96nvvpz9y8/1 (DOI: 10.17632/96nvvpz9y8.1).
References
- Åslund A, Sigurdson CJ, Klingstedt T, Grathwohl S, Bolmont T, Dickstein DL, Glimsdal E, Prokop S, Lindgren M, Konradsson P et al (2009) Novel Pentameric Thiophene derivatives for in vitro and in vivo optical imaging of a plethora of protein aggregates in cerebral Amyloidoses. ACS Chem Biol 4: 673–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett AE, Rienstra CM, Auger M, Lakshmi KV, Griffin RG (1998) Heteronuclear decoupling in rotating solids. J Chem Phys 103: 6951–6958 [Google Scholar]
- Castilla J, Morales R, Saá P, Barria M, Gambetti P, Soto C (2008) Cell‐free propagation of prion strains. EMBO J 27: 2557–2566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clavaguera F, Akatsu H, Fraser G, Crowther RA, Frank S, Hench J, Probst A, Winkler DT, Reichwald J, Staufenbiel M et al (2013) Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci U S A 110: 9535–9540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen ML, Kim C, Haldiman T, ElHag M, Mehndiratta P, Pichet T, Lissemore F, Shea M, Cohen Y, Chen W et al (2015) Rapidly progressive Alzheimer's disease features distinct structures of amyloid‐β. Brain 138: 1009–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collinge J, Clarke AR (2007) A general model of prion strains and their pathogenicity. Science 318: 930–936 [DOI] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6: 277–293 [DOI] [PubMed] [Google Scholar]
- Devi G, Scheltens P (2018) Heterogeneity of Alzheimer's disease: consequence for drug trials? Alzheimers Res Ther 10: 122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dujardin S, Commins C, Lathuiliere A, Beerepoot P, Fernandes AR, Kamath TV, De Los Santos MB, Klickstein N, Corjuc DL, Corjuc BT et al (2020) Tau molecular diversity contributes to clinical heterogeneity in Alzheimer's disease. Nat Med 26: 1256–1263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran‐Aniotz C, Moreno‐Gonzalez I, Gamez N, Perez‐Urrutia N, Vegas‐Gomez L, Soto C, Morales R (2021) Amyloid pathology arrangements in Alzheimer's disease brains modulate in vivo seeding capability. Acta Neuropathol Commun 9: 56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisele YS, Obermüller U, Heilbronner G, Baumann F, Kaeser SA, Wolburg H, Walker LC, Staufenbiel M, Heikenwalder M, Jucker M (2010) Peripherally applied Aβ‐containing inoculates induce cerebral β‐amyloidosis. Science 330: 980–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escartin C, Galea E, Lakatos A, O'Callaghan JP, Petzold GC, Serrano‐Pozo A, Steinhäuser C, Volterra A, Carmignoto G, Agarwal A et al (2021) Reactive astrocyte nomenclature, definitions, and future directions. Nat Neurosci 24: 312–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh U, Thurber KR, Yau W‐M, Tycko R (2021) Molecular structure of a prevalent amyloid‐β fibril polymorph from Alzheimer's disease brain tissue. Proc Natl Acad Sci U S A 118: e2023089118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez‐Gutierrez R, Morales R (2020) The prion‐like phenomenon in Alzheimer's disease: evidence of pathology transmission in humans. PLoS Pathog 16: e1009004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilbronner G, Eisele YS, Langer F, Kaeser SA, Novotny R, Nagarathinam A, Aslund A, Hammarström P, Nilsson KPR, Jucker M (2013) Seeded strain‐like transmission of β‐amyloid morphotypes in APP transgenic mice. EMBO Rep 14: 1017–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G (1996) Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 274: 99–102 [DOI] [PubMed] [Google Scholar]
- Irwin DJ (2016) Tauopathies as clinicopathological entities. Parkinsonism Relat Disord 22: S29–S33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii Y (2001) 13C–13C dipolar recoupling under very fast magic angle spinning in solid‐state nuclear magnetic resonance: applications to distance measurements, spectral assignments, and high‐throughput secondary‐structure determination. J Chem Phys 114: 8473–8483 [Google Scholar]
- Jankowsky JL, Fadale DJ, Anderson J, Xu GM, Gonzales V, Jenkins NA, Copeland NG, Lee MK, Younkin LH, Wagner SL et al (2004) Mutant presenilins specifically elevate the levels of the 42 residue β‐amyloid peptide in vivo: evidence for augmentation of a 42‐specific γ secretase. Hum Mol Genet 13: 159–170 [DOI] [PubMed] [Google Scholar]
- Klingstedt T, Åslund A, Simon RA, Johansson LBG, Mason JJ, Nyström S, Hammarström P, Nilsson KPR (2011) Synthesis of a library of oligothiophenes and their utilization as fluorescent ligands for spectral assignment of protein aggregates. Org Biomol Chem 9: 8356–8370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingstedt T, Shirani H, Mahler J, Wegenast‐Braun BM, Nyström S, Goedert M, Jucker M, Nilsson KPR (2015) Distinct spacing between anionic groups: an essential chemical determinant for achieving Thiophene‐based ligands to distinguish β‐amyloid or tau polymorphic aggregates. Chemistry 21: 9072–9082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knobloch M, Konietzko U, Krebs DC, Nitsch RM (2007) Intracellular Abeta and cognitive deficits precede beta‐amyloid deposition in transgenic arcAbeta mice. Neurobiol Aging 28: 1297–1306 [DOI] [PubMed] [Google Scholar]
- Kunis G, Baruch K, Rosenzweig N, Kertser A, Miller O, Berkutzki T, Schwartz M (2013) IFN‐γ‐dependent activation of the brain's choroid plexus for CNS immune surveillance and repair. Brain 136: 3427–3440 [DOI] [PubMed] [Google Scholar]
- Lu J‐X, Qiang W, Yau W‐M, Schwieters CD, Meredith SC, Tycko R (2013) Molecular structure of β‐amyloid fibrils in Alzheimer's disease brain tissue. Cell 154: 1257–1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnusson K, Simon R, Sjölander D, Sigurdson CJ, Hammarström P, Nilsson KPR (2014) Multimodal fluorescence microscopy of prion strain specific PrP deposits stained by thiophene‐based amyloid ligands. Prion 8: 319–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales R (2017) Prion strains in mammals: different conformations leading to disease. PLoS Pathog 13: e1006323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales R, Abid K, Soto C (2007) The prion strain phenomenon: molecular basis and unprecedented features. Biochim Biophys Acta 1772: 681–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales R, Duran‐Aniotz C, Diaz‐Espinoza R, Camacho MV, Soto C (2012) Protein misfolding cyclic amplification of infectious prions. Nat Protoc 7: 1397–1409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales R, Bravo‐Alegria J, Duran‐Aniotz C, Soto C (2015) Titration of biologically active amyloid‐β seeds in a transgenic mouse model of Alzheimer's disease. Sci Rep 5: 9349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales R, Bravo‐Alegria J, Moreno‐Gonzalez I, Duran‐Aniotz C, Gamez N, Edwards G, Soto C (2021) Transmission of cerebral amyloid pathology by peripheral administration of misfolded Aβ aggregates. Mol Psychiatry 26: 5690–5701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet‐Bongaarts A, Ohno M, Disterhoft J, Van Eldik L et al (2006) Intraneuronal beta‐amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci 26: 10129–10140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkova AT, Leapman RD, Guo Z, Yau W‐M, Mattson MP, Tycko R (2005) Self‐propagating, molecular‐level polymorphism in Alzheimer's beta‐amyloid fibrils. Science 307: 262–265 [DOI] [PubMed] [Google Scholar]
- Polymenidou M, Stoeck K, Glatzel M, Vey M, Bellon A, Aguzzi A (2005) Coexistence of multiple PrPSc types in individuals with Creutzfeldt‐Jakob disease. Lancet Neurol 4: 805–814 [DOI] [PubMed] [Google Scholar]
- Prusiner SB, Hsiao KK (1994) Human prion diseases. Ann Neurol 35: 385–395 [DOI] [PubMed] [Google Scholar]
- Qiang W, Yau W‐M, Tycko R (2011) Structural evolution of Iowa mutant β‐amyloid fibrils from polymorphic to homogeneous states under repeated seeded growth. J Am Chem Soc 133: 4018–4029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiang W, Yau W‐M, Lu J‐X, Collinge J, Tycko R (2017) Structural variation in amyloid‐β fibrils from Alzheimer's disease clinical subtypes. Nature 541: 217–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen J, Mahler J, Beschorner N, Kaeser SA, Häsler LM, Baumann F, Nyström S, Portelius E, Blennow K, Lashley T et al (2017) Amyloid polymorphisms constitute distinct clouds of conformational variants in different etiological subtypes of Alzheimer's disease. Proc Natl Acad Sci U S A 114: 13018–13023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saborio GP, Permanne B, Soto C (2001) Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 411: 810–813 [DOI] [PubMed] [Google Scholar]
- Saijo E, Groveman BR, Kraus A, Metrick M, Orrù CD, Hughson AG, Caughey B (2019) Ultrasensitive RT‐QuIC seed amplification assays for disease‐associated tau, α‐Synuclein, and prion aggregates. Methods Mol Biol 1873: 19–37 [DOI] [PubMed] [Google Scholar]
- Salvadores N, Shahnawaz M, Scarpini E, Tagliavini F, Soto C (2014) Detection of misfolded Aβ oligomers for sensitive biochemical diagnosis of Alzheimer's disease. Cell Rep 7: 261–268 [DOI] [PubMed] [Google Scholar]
- Shahnawaz M, Mukherjee A, Pritzkow S, Mendez N, Rabadia P, Liu X, Hu B, Schmeichel A, Singer W, Wu G et al (2020) Discriminating α‐synuclein strains in Parkinson's disease and multiple system atrophy. Nature 578: 273–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shechter R, Miller O, Yovel G, Rosenzweig N, London A, Ruckh J, Kim K‐W, Klein E, Kalchenko V, Bendel P et al (2013) Recruitment of beneficial M2 macrophages to injured spinal cord is orchestrated by remote brain choroid plexus. Immunity 38: 555–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Zhang W, Yang Y, Murzin AG, Falcon B, Kotecha A, van Beers M, Tarutani A, Kametani F, Garringer HJ et al (2021) Structure‐based classification of tauopathies. Nature 598: 359–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirani H, Linares M, Sigurdson CJ, Lindgren M, Norman P, Nilsson KPR (2015) A palette of fluorescent Thiophene‐based ligands for the identification of protein aggregates. Chemistry 21: 15133–15137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirani H, Appelqvist H, Bäck M, Klingstedt T, Cairns NJ, Nilsson KPR (2017) Synthesis of Thiophene‐based optical ligands that selectively detect tau pathology in Alzheimer's disease. Chemistry 23: 17127–17135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto C, Pritzkow S (2018) Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat Neurosci 21: 1332–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stöhr J, Condello C, Watts JC, Bloch L, Oehler A, Nick M, DeArmond SJ, Giles K, DeGrado WF, Prusiner SB (2014) Distinct synthetic Aβ prion strains producing different amyloid deposits in bigenic mice. Proc Natl Acad Sci U S A 111: 10329–10334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thal DR, Griffin WST, Braak H (2008) Parenchymal and vascular Abeta‐deposition and its effects on the degeneration of neurons and cognition in Alzheimer's disease. J Cell Mol Med 12: 1848–1862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thal DR, Walter J, Saido TC, Fändrich M (2015) Neuropathology and biochemistry of Aβ and its aggregates in Alzheimer's disease. Acta Neuropathol 129: 167–182 [DOI] [PubMed] [Google Scholar]
- Tycko R (2014) Physical and structural basis for polymorphism in amyloid fibrils. Protein Sci 23: 1528–1539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts JC, Condello C, Stohr J, Oehler A, Lee J, DeArmond SJ, Lannfelt L, Ingelsson M, Giles K, Prusiner SB (2014) Serial propagation of distinct strains of a prions from Alzheimer's disease patients. Proc Natl Acad Sci U S A 111: 10323–10328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Mormino EC, Sun N, Sperling RA, Sabuncu MR, Yeo BTT (2016) Bayesian model reveals latent atrophy factors with dissociable cognitive trajectories in Alzheimer's disease. Proc Natl Acad Sci U S A 113: E6535–E6544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolochevska O, Taglialatela G (2016) Non‐demented individuals with Alzheimer's disease neuropathology: resistance to cognitive decline may reveal new treatment strategies. Curr Pharm Des 22: 4063–4068 [DOI] [PMC free article] [PubMed] [Google Scholar]