

Abstract
Glutarimides such as thalidomide, pomalidomide, and lenalidomide are the most frequently used ligands to recruit E3 ubiquitin ligase cereblon (CRBN) for the development of proteolysis targeting chimeras (PROTACs). Due to the rapid and spontaneous racemization of glutarimides, most CRBN-recruiting PROTACs are synthesized as a mixture of racemates or diastereomers. Since the (S)-enantiomer is primarily responsible for binding to CRBN, the existence of the largely inactive (R)-enantiomer complicates the drug development process. Herein, we report that substituted achiral phenyl dihydrouracil (PDHU) can be used as a novel class of CRBN ligands for the development of PROTACs. Although the parent PDHU has minimal binding affinity to CRBN, we found that some substituted PDHUs had comparable binding affinity to lenalidomide. Structural modeling provided further understanding of the molecular interactions between PDHU ligands and CRBN. PDHUs also have greater stability than lenalidomide. Finally, potent BRD4 degraders were developed by employing trisubstituted PDHUs.
Keywords: PROTAC, cereblon, dihydrouracil, thalidomide, lenalidomide
Graphical Abstract
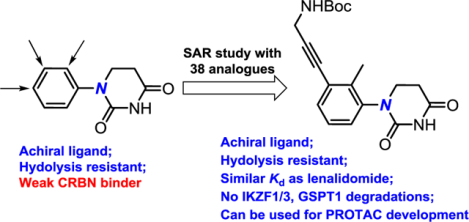
Introduction
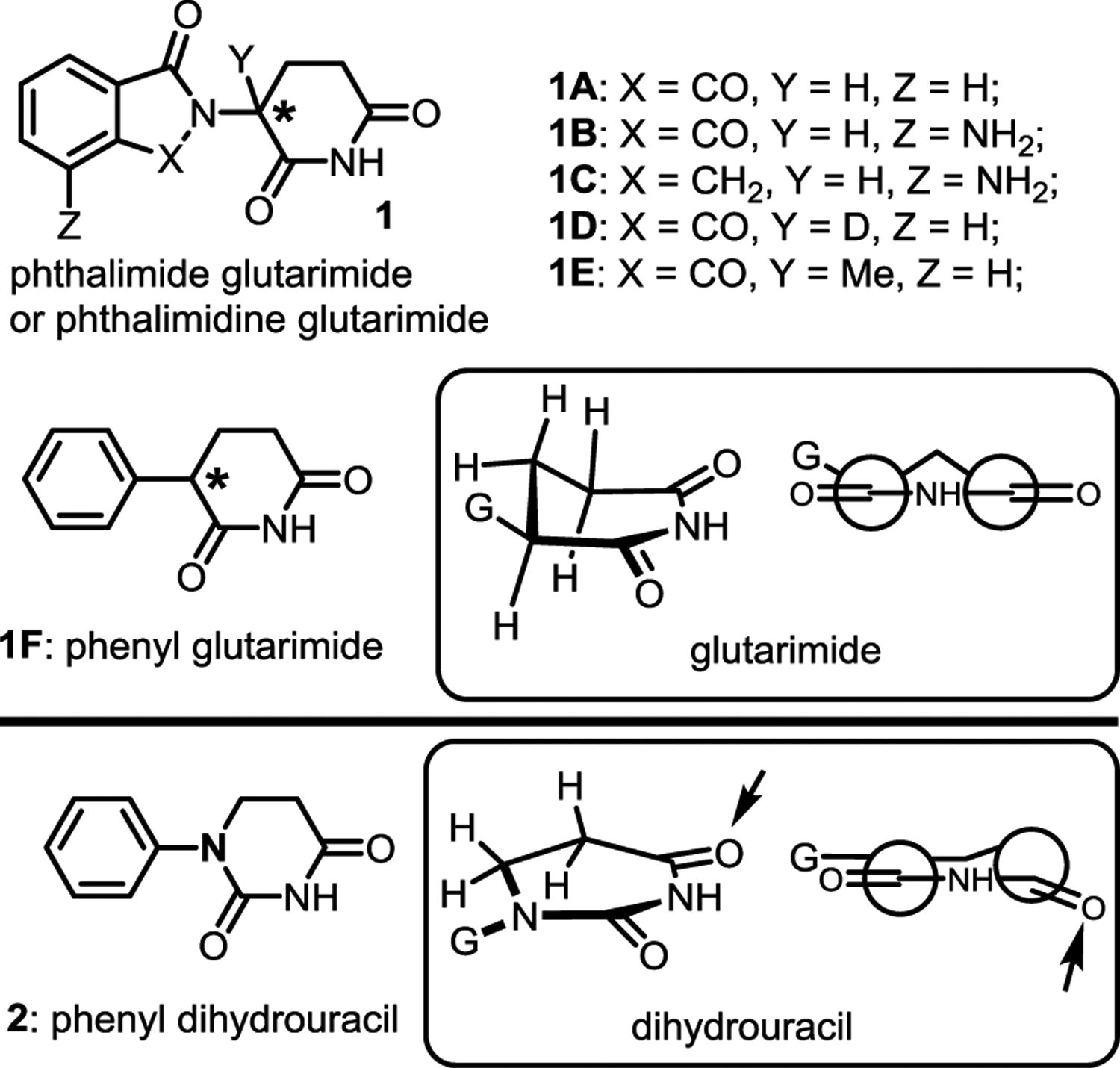
Targeted protein degradation has recently emerged as a promising modality for the development of therapeutics to address numerous unmet medical needs.1–5 Proteolysis targeting chimera (PROTAC) is the most well-known strategy for the degradation of intracellular protein targets.6 PROTACs are bifunctional molecules that are composed of a E3 ubiquitin ligase binding motif, a linker, and a protein of interest (POI) binding motif. PROTACs can serve as a molecular adapter to induce the interactions between the POI and the E3 ligase, which in turn promotes polyubiquitination and subsequent proteasomal degradation of the POI.7 Although there are over 600 E3 ubiquitin ligase complexes in human proteome, few of them have been employed for the development of PROTACs.1 Among them, CRBN is the most frequently utilized E3 ligase due to the low molecular weight and drug-like properties of its glutarimide ligands such as thalidomide 1A, pomalidomide 1B, and lenalidomide 1C (Figure 1).5,8–12 These glutarimides were first applied to the development of PROTACs for targeted protein degradation in 2015.13,14 The first two PROTACs progressed into clinical trials (ARV-110 and ARV-471) both used glutarimide CRBN ligands. However, glutarimides are used as a racemic mixture in almost all reported PROTACs because the two enantiomers can undergo rapid and spontaneous racemization in vitro and in vivo. For example, the racemization half-life of thalidomide is ~2.0 h in human blood and ~5.0 h in-vivo in humans, respectively.15 The binding affinity of the (S)-enantiomeric thalidomide to CRBN is at least 10-fold stronger than the corresponding (R)-enantiomer.16,17 The DDB1-CRBN-thalidomide complex further confirms that only the (S)-enantiomer fits the binding pocket well. The glutarimide moiety is most critical for binding as three hydrogen bonds are formed between the imide motif and CRBN, whereas the phthalimide or the phthalimidine moiety is exposed to the solvent.18
Figure 1.
Comparison of the well-known glutarimides,13,14 the recently reported phenyl glutarimide,28 and the designed achiral phenyl dihydrouracil.
The use of racemic glutarimide as the E3 ligase ligands in PROTACs complicates many aspects of the drug development process and is one of the significant barriers for the therapeutic applications of CRBN-recruiting PROTACs. For example, half of the PROTAC molecules that have poor degradation activity due to ~10-fold weaker binding affinity to CRBN can competitively inhibit the other half of the active portion, because they bear the same POI ligand that can bind to the protein target. In addition, according to the FDA policy updated in 1992, more resources are needed to characterize the pharmacological properties of all isomers including enantiomers or diastereomers and their metabolites during the drug development process.19 To date, various CRBN ligands have been reported by researchers from both industry and academia; some of these reports include attempts to solve the chirality issue.20–24 For example, the racemization of deuterated thalidomide 1D, where the hydrogen is replaced by a deuterium, was about five times slower than thalidomide.25 If an all carbon quaternary center was to be introduced, no racemization would occur. However, when the Y substituent in 1 was replaced by a methyl group, the corresponding compound 1E lost its activity.26,27 In addition to racemization, stability of the glutarimides is another issue. One of the major breakthroughs for the development of CRBN ligands is the design of phenyl glutarimides (PGs) 1F for PROTACs during our study.28 Significantly higher stability was observed after the replacement of the phthalimide or phthalimidine motif in thalidomide, pomalidomide, and lenalidomide by a simple phenyl ring in the PGs. Herein, we report the development of achiral phenyl dihydrouracil (PDHU) 2 as the CRBN ligands for the development of non-racemic PROTACs. Although both PGs and dihydrouracils were described as CRBN ligands in several patents,20 detailed structure-activity relationship (SAR) and properties of the ligands were not disclosed, which prevent their broad applications for the development of PROTACs.
We began our investigation by examining the conformations of simple glutarimide and dihydrouracil (Figure 1) by extracting the relevant X-ray structures 4CI118 and 1R2Z29 from Protein Data Bank. Glutarimide has a half-chair conformation, where five atoms in the imide ring are in the same plane. The remaining methylene group in the ring and the “G” substituent are out of the plane. In contrast, dihydrouracil is much flatter. The G-substituent on the nitrogen is in the same plane as the imide, while the carbonyl group pointed by the arrow and its adjacent methylene group are twisted out of the plane slightly. Before our study, it is not clear how the different orientation of G group and the twisted imide group will impact the binding of PDHU with the CRBN protein complex.
Results and Discussion
The parent PDHU 2 was prepared and its binding with the CRBN-DDB1 complex was compared with positive control lenalidomide 1C using a known fluorescence polarization (FP) assay.18 Lenalidomide was used as the positive control for comparison with PDHUs analogues and 84% of fluorescence probe was displaced by 1 μM of lenalidomide. All analogs were tested at a single concentration of 1 μM three times. As shown in Scheme 1, displacement observed for parent compound 2 was only around 20%, which is much worse than the 84% observed for lenalidomide positive control. This is not surprising based on the conformational analysis as discussed before. To improve the binding affinity, a series of disubstituted PDHUs were prepared. Various substituents were installed at the ortho, meta, and para positions of the phenyl group in PDHUs systematically and compared with the parent compound 2 for relative binding affinity. Interestingly, analogue 3A with an ortho hydroxyl substituent almost completely lost the binding, while analogues 4A and 5A with a meta and para OH enhanced the binding to 57% and 38%, respectively. Methyl, ethyl and chloro substituents on the ortho-position (3B, 3D, 3E) improved the binding to 35%, 29% and 36% from the original 20% for the parent compound 2. Compound 3C with a methoxy substituent on the ortho-position decreased the binding, while placing the MeO on the meta-position (4C) improved the binding to 33%. Compounds 4F and 4K bearing longer substituents on the meta-position further improved the binding to 56% and 44%, respectively. Substituents on the para-positions were also tolerated, though they did not improve the binding as much as those on the meta-positions as shown in 5A and 5K.
Scheme 1.
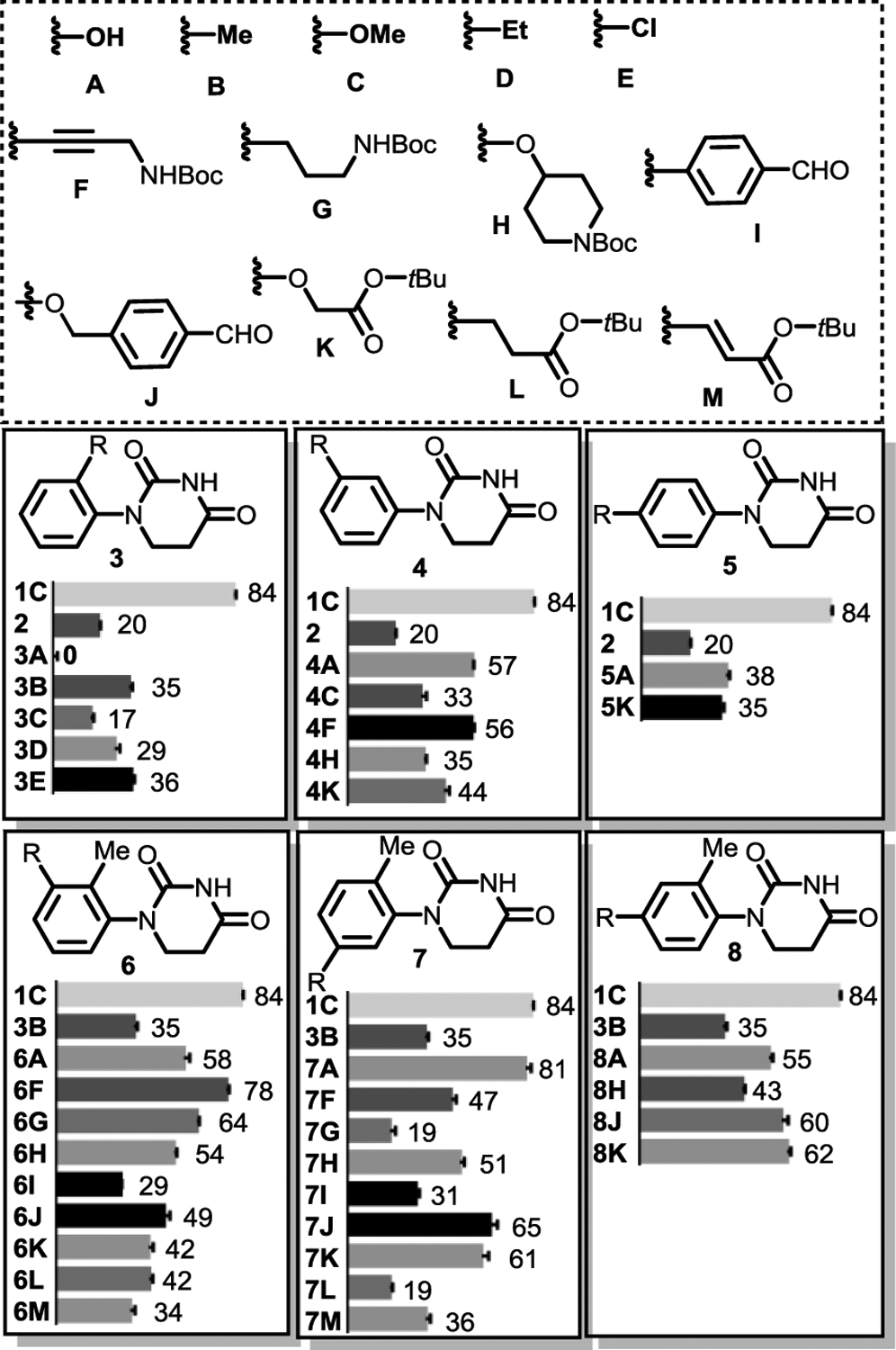
Relative binding affinities of substituted PDHUs in a fluorescent polarization assay. Lenalidomide 1C was used as the positive control and each compound was tested three times.
Based on the structure-activity relationship (SAR) of the disubstituted PDHUs, we designed the second series of trisubstituted PDHUs. Considering the binding affinity observed for disubstituted PDHUs, ligand efficiency, metabolic stability and magic methyl effect,30,31 ortho methyl was selected to explore its synergetic effect of binding improvement with the third substituent. Various 1,2,3-, 1,2,5- and 1,2,4-trisubstituted PDHUs were designed, synthesized, and compared with the lenalidomide positive control and compound 3B with an ortho methyl substituent. We prepared more compounds with meta-substituents on the phenyl group of PDHUs since our results from the disubstituted PDHUs indicated that meta-substituents improved the binding more than the para-substituents. Among different groups, substituent A and F had the strongest synergetic effect. For example, the binding of 6F and 7A were improved to 78% and 81%, respectively. Most other trisubstituted PDHUs also showed better binding affinity than 3B.
It is easiest to install linkers to the phenyl group on PDHUs bearing building blocks F and K for the development of PROTACs. We hypothesize that the tBu group in these two building blocks can be replaced by various linkers for the development of non-racemic PROTACs that can recruit CRBN. With these in mind, we next measured the Kds for lenalidomide 1C, parent PDHU 2, mono substituted PDHU 3B, disubstituted PDHU 4F, and trisubstituted PDUs 6F and 6K for further comparison (Figure 2). The Kd (0.17 μM) we observed in the FP assay for lenalidomide 1C is very close to what was reported in the literature (Kd = 0.18 μM).18 Parent PDHU 2 is 18 times weaker than lenalidomide 1C. The installation of an ortho-Me in 3B increased the affinity about 2.5 times. We were pleased to find that 1,2,3-Trisubstituted PDHU 6F has a comparable Kd to lenalidomide. The difference between 6F and 4F confirms that the ortho- and meta-substituents in 6F both contributed to the binding. Interestingly, the meta-substituent in PDHU 6K does not further increase the binding compared to 3B. We then prepared compounds 9 and 10 by replacing the tBu with an amide functional group to confirm this observation. Similar Kds were observed for compounds 6K, 9 and 10. We also extended the propargyl amide linker in 6F to homopropargyl amide in 11. Surprisingly, the affinity dropped three-fold in this case.
Figure 2.
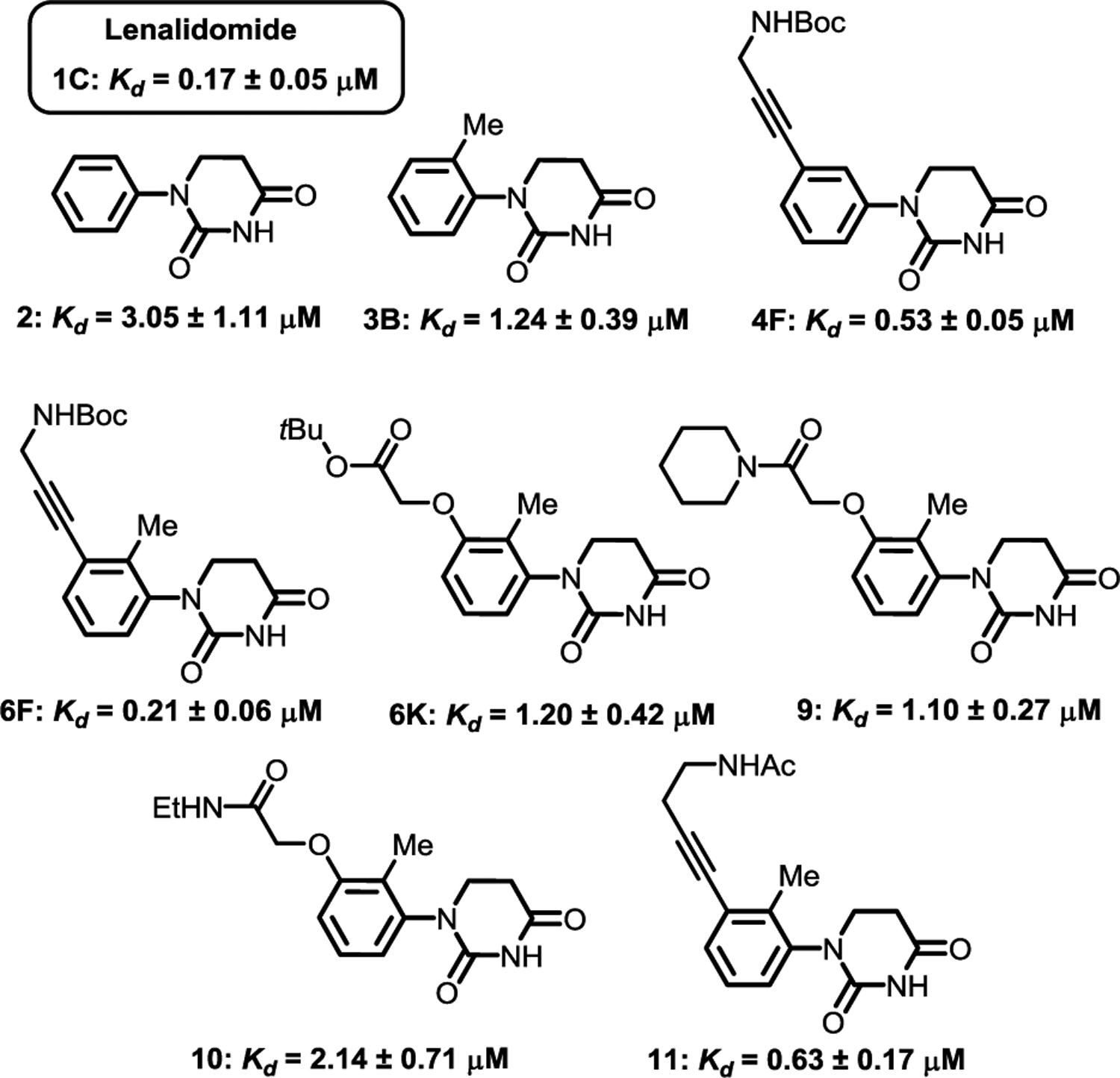
Binding affinities of lenalidomide and selected PDHUs in a fluorescent polarization assay. (Detailed procedures are provided in the SI.)
To establish a binding mode of the achiral ligands and rationalize the observed binding affinity trends, structural modeling was performed for representative structure 6F based on docking followed by Molecular Dynamics (MD) refinement (Figure 3). The refinement allows both the ligand and protein to fully relax since ligand-induced changes may occur on protein side chains. Additionally, ligand binding stabilities can be observed during the refinement process, including filtering out false positive poses. The MD refinement procedure is similar to our previous method used for PROTAC modeling.32 From these results, we observed that the carbamate carbonyl oxygen of 6F form an H-bond with neighboring Histidine 353. The phenyl, 2-methyl, 3-alkyne group and Boc t-butyl all form hydrophobic contacts with the protein. When the rigid alkyne is removed for 6K, 9 and 10, the direct hydrophobic contacts will be diminished and the H-bond will be disturbed. Compound 4F being slightly weaker than 6F is attributed to less hydrophobic space filling lacking the 2-methyl. Compound 11 with a rigid alkyne linker is expected to also form good contacts, but the extended linker length disturbs the H-bond with the carbonyl oxygen. Detailed procedures of the modeling and more images are provided in the SI.
Figure 3.
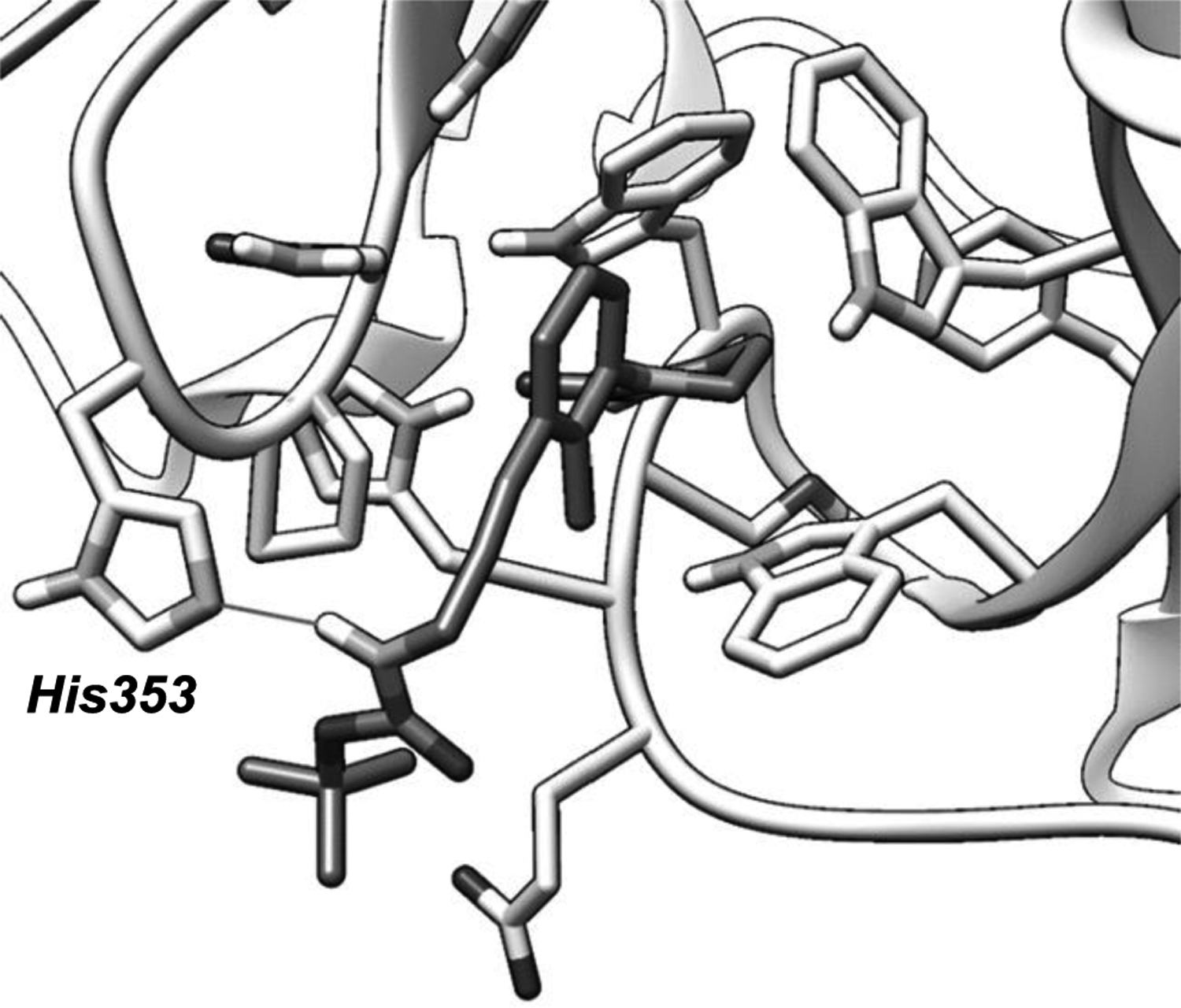
Binding poses of 6F based on computational modeling. The dark sticks are from 6F and a key H-bond between the amide and histidine is marked out with a straight line. Image generated by UCSF Chimera.
We then further evaluated the stability of selected compounds (6F and 9–11) in human liver microsomes (hLM), human plasma (hP) and buffer with different pHs (Table 1). Both compounds 6F and 9–11 showed comparable stability to lenalidomide in hLM. In terms of stability in hP, only 62% of lenalidomide remained after 4h of incubation. In contrast, 97% of 6F and over 80% of 9–11 remained under the same conditions. Under acidic conditions (pH=1.0), all compounds are very stable. However, under neutral conditions that mimic the physiological environment (pH=7.4 buffer), only 39% of lenalidomide was remained after 24 h of incubation at 37°C, while the hydrolysis of 1,2,3-trisubstituted PDHUs 6F and 9–11 was not detected. PDHUs 6F and 9–11 are much more stable than lenalidomide at slightly basic conditions (e.g. pH=8.8). Other than racemization, the ring opening of glutarimides by hydrolysis is a liability of CRBN-recruiting PROTACs.33,34 Our results showed that the dihydrouracil motif has much higher stability and is less prone to hydrolysis. Based on the above data, we can conclude that the achiral CRBN ligands with 1,2,3-trisubstituted PDHUs such as 6F can have similar binding affinity and much better stability profiles compared to lenalidomide.
Table 1.
Stability of lenalidomide and 1,2,3-trisubstituted PDHUs
| Compound | hLM[a] | hP[b] | pH=1.0[c] | pH=7.4[d] | pH=8.8[e] |
|---|---|---|---|---|---|
| Lenalidomide | 89 | 62 | >99 | 39 | 0, 20[f] |
| 6F | 87 | 97 | >99[g] | >99 | 93 |
| 9 | 85 | 81 | >99 | >99 | 95 |
| 10 | 100 | 88 | >99 | >99 | 95 |
| 11 | 90 | 102 | >99 | >99 | 90 |
Percent compound remaining after 0.5 h.
Percent compound remaining after 4.0 h.
Glutarimides and dihydrouracil ring opening hydrolysis, percent compound remaining after 24 h.
Percent compound remaining after 2 h.
Boc was deprotected, dihydrouracil ring opening hydrolysis was not detected by LC-MS. (Detailed procedures are provided in the SI.)
It is well known that the binding of thalidomide, lenalidomide, pomalidomide, their analogues or certain PROTACs bearing the glutarimide motif to CRBN can induce the degradation of neo-substrates, such as IKZF1, IKZF3 and GSPT1.1–5 The neo-substrate degradation activity can be changed by modifying the structure of CRBN ligand, the linking site, the property of the linker, or even the ligand of the POI in PROTACs. We next tested compounds 6F and 9-11 in MV4;11 cells at two different concentrations (10 and 1 μM) for the degradation of IKZF1, IKZF3 and GSPT1. Western blot analysis indicated that these four compounds did not induce the degradation of IKZF1, IKZF3 or GSPT1 (Figure 4A–4C). Although this is encouraging, the selectivity of specific PROTACs bearing the achiral PDHU ligand should be examined thoroughly for the development of any therapeutics as the selectivity depends on the entire structure of the PROTAC.
Figure 4.
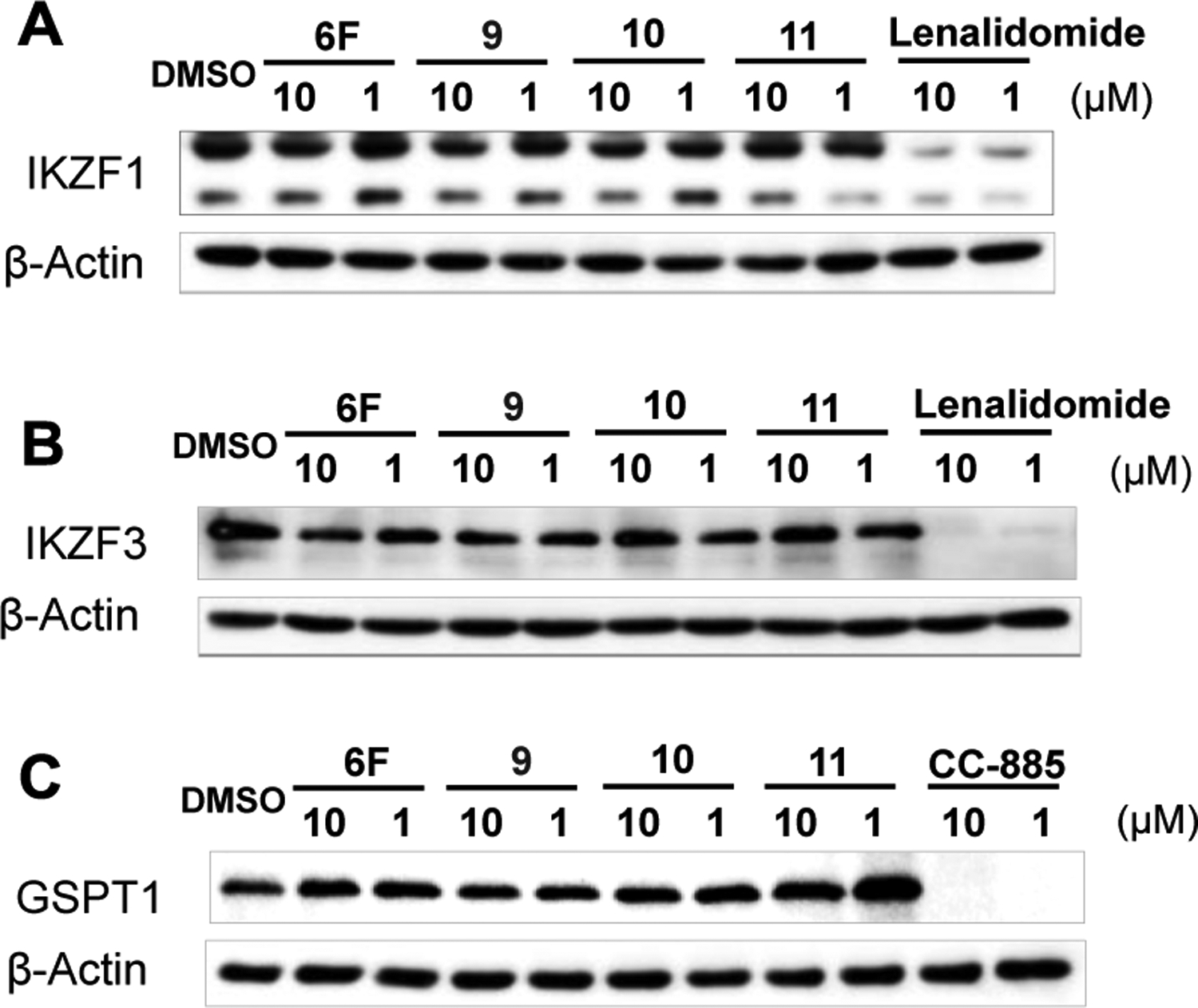
Compounds 6F, 9, 10 and 11 do not induce the degradation of IKZF1, IKZF3 or GSPT1. (A), (B) and (C) Western blots of IKZF1, IKZF3 and GSPT1. MV4;11 cells were treated with 6F, 9, 10, 11, Lenalidomide or CC-885 at 10 and 1 μM for 24 h.
The discovery of the achiral CRBN ligands provides an opportunity to develop various stereochemically well defined PROTACs that can recruit CRBN. To demonstrate the utility of these new CRBN ligands, (e.g., 6F and 9–11), we attached JQ1, a well-known ligand for BRD4, to the 1,2,3-trisubstituted PDHU and prepared potential BRD4 degraders 12A–12C, 13 and 14 bearing several different linkers (Figure 5). These compounds were then tested in four different cell lines at two different concentrations (1.0 and 0.1 μM) for the degradation of BRD4. Western blot analysis indicated that compounds 12B, 13 and 14 could induce the degradation of BRD4 protein in all four cell lines. Compound 12C showed obviously BRD4 degradation activity in MV4;11 and LNCAP, however, 12A bearing the shortest linker did not display activity in all four cell lines (Figure 6A). While compounds 12B, 13 and 14 displayed the most potent activity, compound 13 appeared to be slightly more potent. Our results indicated that the degradation efficiency was related to multiple parameters such as the length and type of linkers, in addition to the binding affinity of the achiral ligand to CRBN. Compound 13 was then selected for further characterization. We first studied the time course and dose response of degrader 13 (Figure 6B–E). Degradation of BRD4 occurred as early as 1 h post-treatment (Figure 6B). Degrader 13 also induced significant degradation of BRD4 at pM concentrations in MV4;11 cells with a DC50 of 22 pM and Dmax of 97% (Figure 6C/D). We also verified the degradation effect of degrader 13 in a Flp-In™-293 cell line that stably expresses e-GFP-tagged BRD4 bromodomain (BD1) fusion and a non-targeted mCherry endogenous control (Figure 6E).35 In the Flp-In™-293 cells, a DC50 of 34 nM and a Dmax of 75% at 10 μM was observed. Interestingly, no obvious “Hook effect”36 was observed with up to 10 μM for degrader 13. The degrader was successful in inducing caspase 3 cleavage, a biomarker for cell apoptosis, at 10 nM to 10 μM concentrations.
Figure 5.
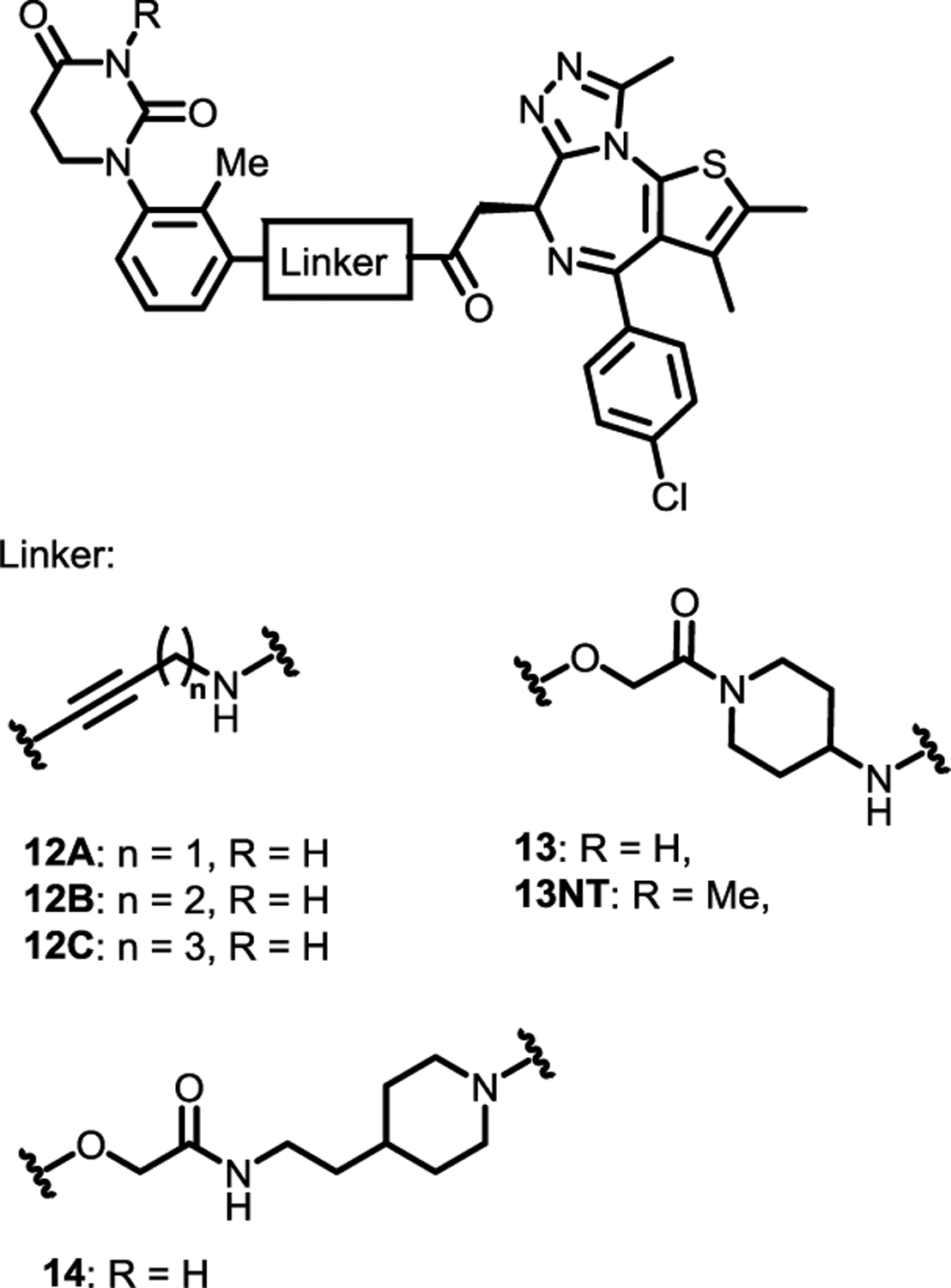
BRD4 degraders with achiral CRBN ligands
Figure 6.
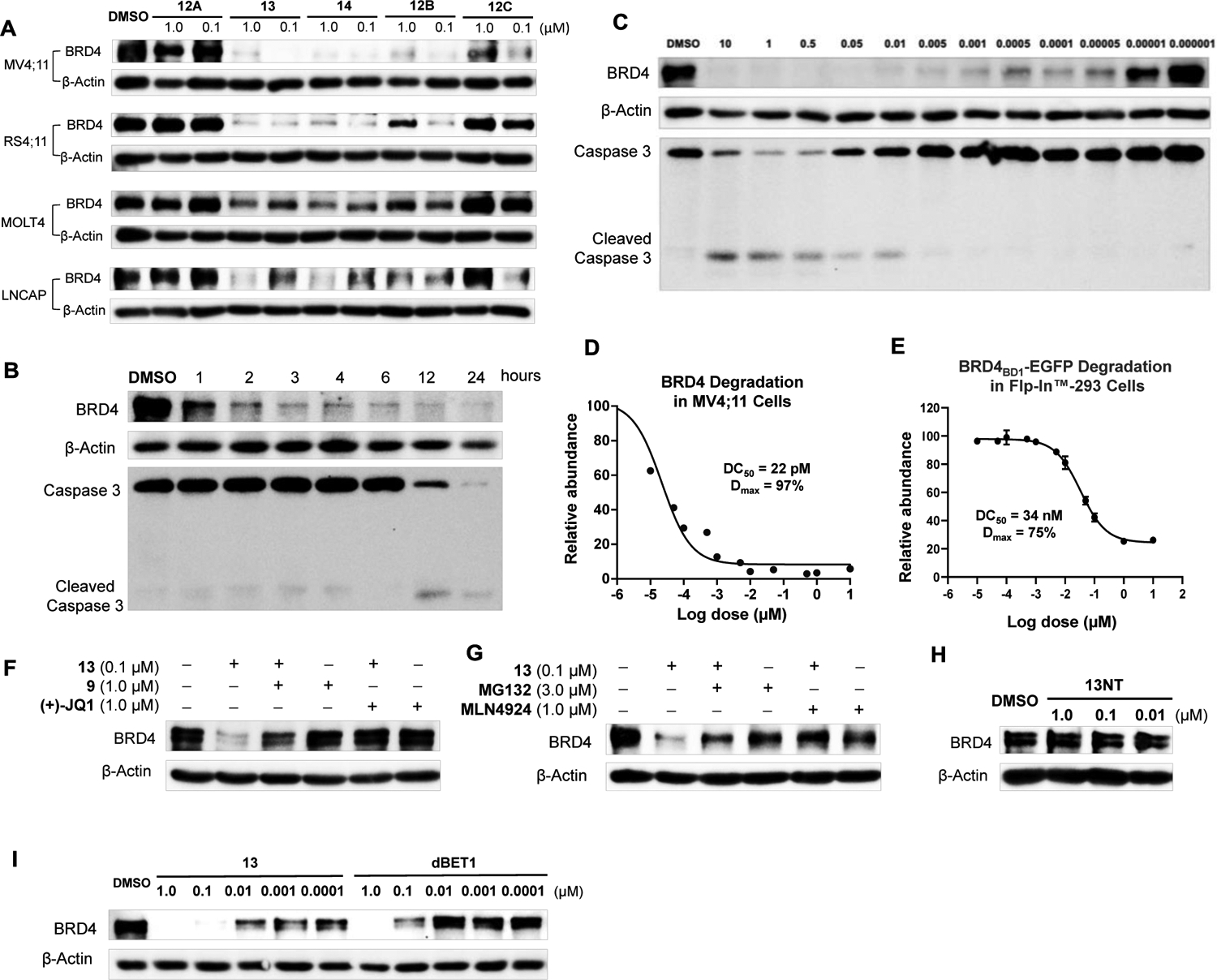
(A) Western blot of BRD4. MV4;11, RS4;11, MOLT4 and LNCaP cells were treated by 12A, 12B, 12C, 13 and 14 at 1.0 and 0.1 μM for 24h. (B) Time course based on Western blot of BRD4, caspase 3 and cleaved caspase 3. MV4;11 cells were treated by 13 with indicated time. (C) and (D) Dose responses based on Western blot of BRD4, caspase 3 and cleaved caspase 3. MV4;11 cells were treated by 13 with indicated concentrations for 24 h. DC50: the concentration where 50% of the protein has been degraded. Dmax: the maximum degradation that can be achieved. (E) Quantitative assessment of degradation using a BRD4BD1 -e-GFP reporter assay. Dose response based on fluorescent signal ratio of e-GFP to mCherry. Flp-In™-293 Cell Line was treated with 13 at indicated concentrations for 24 h. (F), (G) and (H) Confirmation of mechanism of action by Western blot of BRD4. For (F) and (G), MV4;11 cells were pretreated by JQ1, 9, MG132 or MLN4924 for 1 h followed by the treatment of 13 for 3 h. For (H), MV4;11 cells were treated by 13NT at indicated concentrations for 24 h. (I) MV4;11 cells were treated by 13 or dBET1 with indicated concentrations for 24 h.
We next verified the mechanism of action of degrader 13 (Figure 6G–H). Pretreatment of the cells with POI ligand JQ1, achiral CRBN ligand 9, proteasome inhibitor MG132, and neddylation inhibitor MLN492437 abolished the BRD4 degradation induced by 13, suggesting that the degradation involves the engagement of BRD4, CRBN, proteasome, and Cullin-RING E3 ligase complex. We also prepared negative control 13NT, which has an additional methyl group on the imide motif that prevents the binding to CRBN.18 As expected, no degradation activity of BRD4 was observed for 13NT in MV4;11 cells at several different concentrations, further confirming the involvement of CRBN (Figure 6H). Finally, side-by-side comparison of degrader 13 with dBET1 was performed in MV4;11 cells. Degrader 13 showed more potent BRD4 degradation activity than dBET1 (Figure 6I).
We next investigated the functional outcomes of degrader 13 bearing an achiral CRBN ligand including cell proliferation, cell cycle arrest, and cell apoptosis (Figure 7). First, MV4;11 cells were treated by BRD4 degraders 13, POI ligand (+)-JQ1, CRBN ligand 9, and compound 13NT for 72 h. As expected, compound 13 showed the most potent anti-proliferation activity with an IC50 value around 1.1 nM (Figure 7A). POI ligand (+)-JQ1 showed weaker anti-proliferation activity than 13, but higher activity than compound 13NT, which has a very similar structure to 13 but without CRBN recruiting ability. Compound 9, which has the entire CRBN ligand and most parts of the linker, did not show any anti-proliferation activity at up to 100 μM. All these trends are expected and consistent with their mechanism of action. BRD4 degraders 13 could also induce G1 phase arrest, decrease the cell population in S phase, (Figure 7B) and induced cell apoptosis in a dose-dependent manner (Figure 7C and 7D) in MV4;11 cells.
Figure 7.
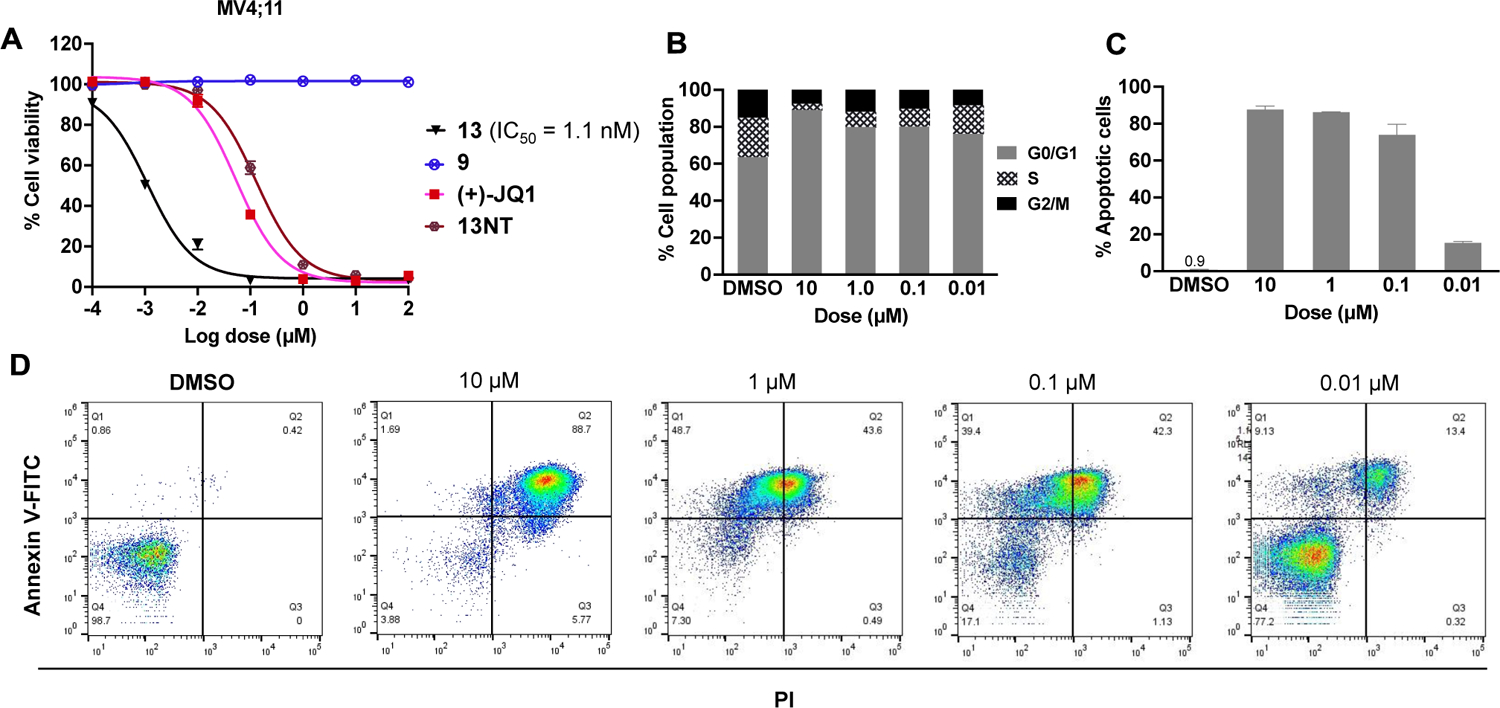
Functional studies of the PROTACs on cell viability, cell cycle progression and cell apoptosis. (A) AlamarBlue™ Cell Viability. MV4;11 cells were treated by 13, 9, (+)-JQ1 and 13NT for 72 h. (B) MV4;11 cells were treated by indicated dosage of 13 for 48 h followed by flow cytofluorimetric cell cycle analysis. (C) and (D) MV4;11 cells were treated by indicated dosage of 13 for 48 h followed by flow cytofluorimetric apoptosis analyses.
Synthesis
Achiral CRBN ligands and BRD4 PROTACs were synthesized as shown in scheme 2. Various substituted PDHUs S2 can be prepared from the corresponding anilines S1 by adapting literature procedures through conjugate addition followed by cyclization (eq 1).38 More substituents can be introduced to the phenyl group of PDHUs by Sonogashira coupling (eq 2), alkylation (eq 3 and eq 6), Suzuki cross coupling (eq 4), or Heck cross-coupling (eq 5). Further manipulations such as reduction (eq 7) and de-protection of Boc group followed by amide formation can afford ligands 6G, 9, and 11, respectively.
Scheme 2.
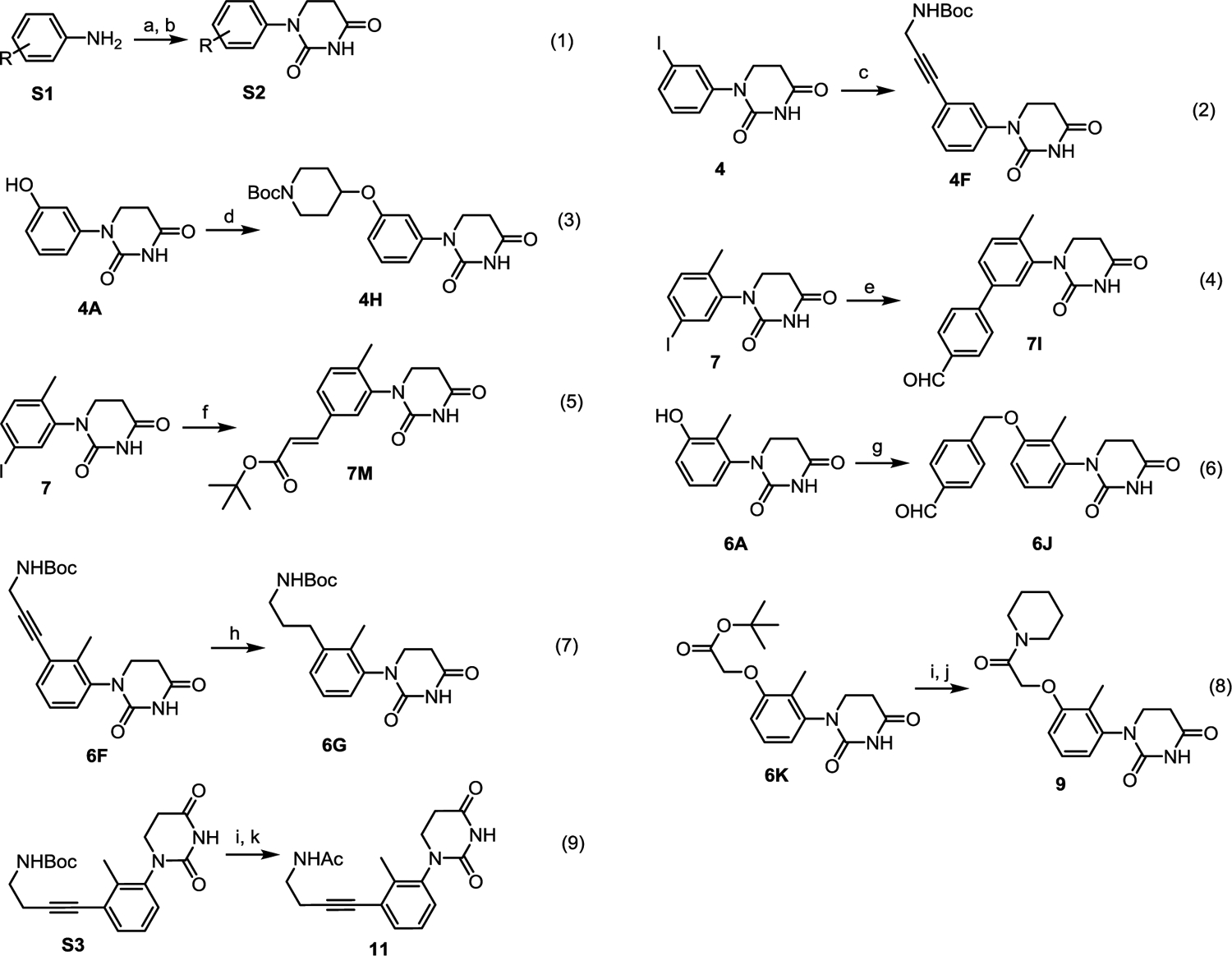
The synthesis of achiral CRBN ligands and BRD4 degraders. Reagents and conditions: (a) Acrylic acid, toluene, 110 °C; (b) Urea, acetic acid, 120 °C, (a) and (b) total yield 33%; (c) N-Boc-propargylamine, Pd(PPh3)2Cl2, CuI, DMF, NEt3, rt, yield 75%; (d) 1-Boc-4-bromopiperidine, K2CO3, Acetonitrile, 110 °C, yield 31%; (e) 4-Formylphenylboronic Acid, Pd(dppf)Cl2, KOAc, DMSO, 90 °C, yield 43%; (f) tert-Butyl acrylate, Pd(OAc)2, P(o-tol)3, NEt3, DMF, 110 °C, yield 61%; (g) 4-(Chloromethyl)benzaldehyde, K2CO3, Acetonitrile, 60 °C, yield 24%; (h) Pd/C, H2, MeOH, rt, yield 95%; (i) TFA/DCM, rt, yield 95%; (j) Piperidine, HATU, DIPEA, DMF, rt, yield 82%; (k) Ac2O, NEt3, DMF, rt, yield 64%.
BRD4 PROTACs bearing achiral CRBN ligands were synthesized as shown in scheme 3. Removal of the Boc protecting group in intermediate S4 followed by amide coupling with JQ1 afforded PROTACs 12A-C. Removal of the tBu protecting group in intermediate S5 followed by coupling with a linker, de-protection, and reaction with JQ1 yielded PROTACs 13, 14 and the negative control 13NT.
Scheme 3.
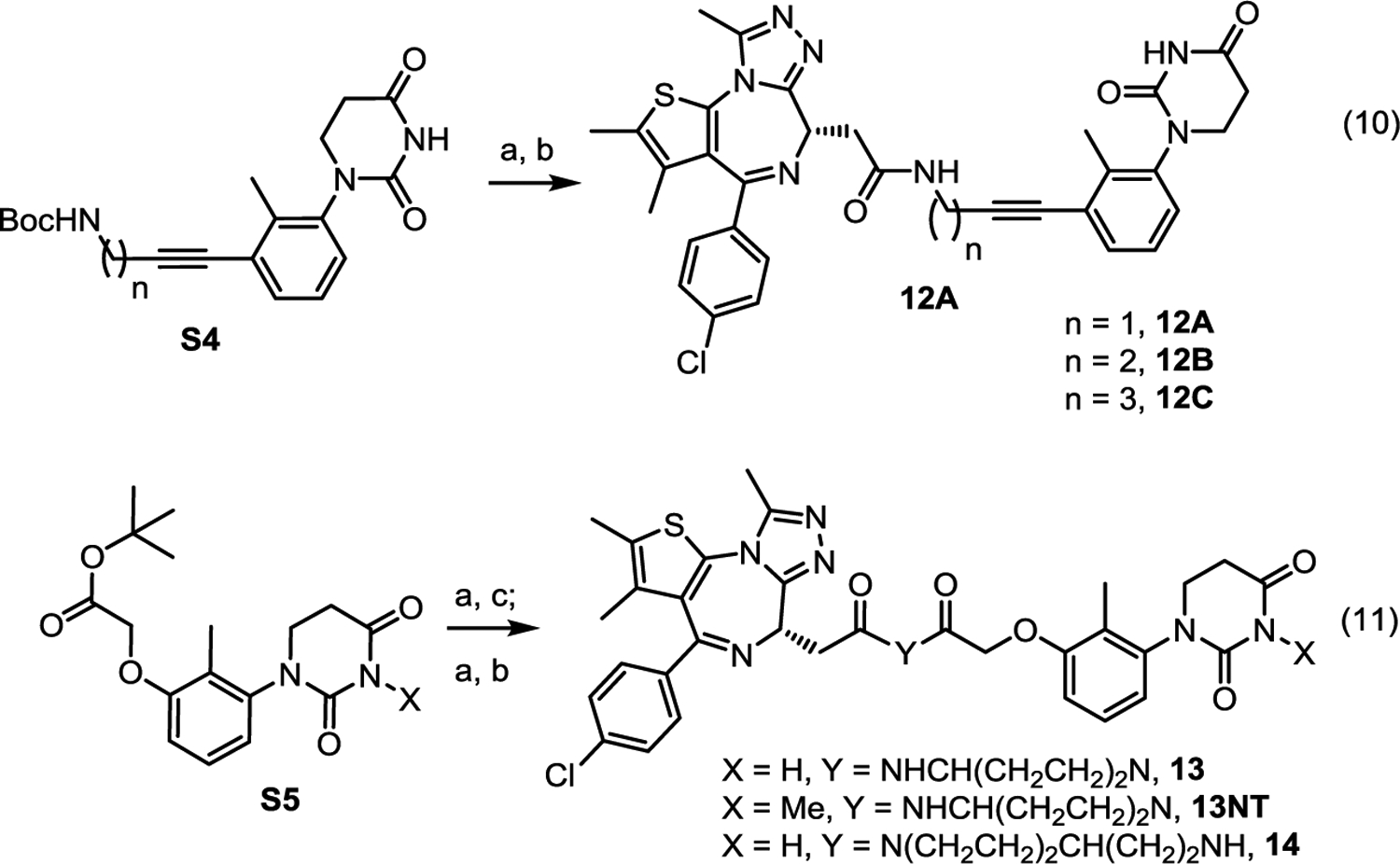
The synthesis of BRD4 degraders using achiral CRBN ligands. Reagents and conditions: (a) TFA/DCM, rt; (b) JQ-1 (carboxylic acid), HATU, DIPEA, DMF, rt, (a) and (b) total yield 86%; (c) 4-(N-Boc-amino)piperidine (or 4-(Aminoethyl)-1-N-Boc-piperidine), HATU, DIPEA, DMF, rt, yield 80%.
Conclusion
Racemization has been a long-standing issue for most CRBN recruiting PROTACs. Due to the racemization between active and inactive enantiomers, CRBN-based PROTACs face not only the issue of half of the PROTAC molecules possessing low E3-ligase binding activity, but also the issue of these low-potency molecules acting as inhibitors to compete with the other half of active PROTACs at the POI binding site. In addition, the half of PROTACs with low E3 ligase binding activity also produce various metabolites that require additional characterization and careful monitoring. These issues significantly complicate the development and manufacturing of PROTACs as therapeutics. Although many molecular glues and PROTACs bearing a racemic CRBN ligand have successfully entered into clinic trials or gained approval, more resources are required to characterize the pharmacological properties of the inactive isomeric compounds according to FDA policy.19 We have developed various substituted PDHUs as a novel class of achiral CRBN ligands and discovered interesting SAR. Although there is only one atom change from PGs to PDHUs, the conformation of dihydrouracil ring in PDHU becomes much flatter than the glutarimide ring in PGs, which results in much weaker binding affinity of the parent PDHU than the parent PG. Through the SAR study of a series of disubstituted and trisubstituted PDHUs, we found that substituted achiral PDHUs could achieve similar binding affinities to lenalidomide, but with much higher stability. Structural modeling studies revealed the additional interactions one can pick up by adding various substituents to the phenyl group, which provides a path for further optimization in the future. Finally, five 1,2,3-trisubstituted PDHU-based BRD4 PROTACs were designed and synthesized. Their remarkable BRD4 degradation activities were verified in multiple cell lines and two orthogonal methods – Western blot analysis of the endogenous BRD4 and fluorescent quantification of BRD4BD1-e-GFP fusion protein, demonstrating that substituted PDHUs can be used for the development of non-racemic CRBN recruiting PROTACs. We anticipate that the readily accessible substituted PDHUs such as 6F and 9 will be widely used for the development of various PROTACs.
Experimental section
Cell Culture
Three human leukemia cell lines (MV4;11, RS4;11, MOLT4) and one prostate cancer cell line (LNCAP) were obtained from ATCC, expanded, and frozen down in cryogenic vials. Cells were thawed and used within 20 passages in the experiments. All cells were cultured in RPMI1640 media (Corning) supplemented with 10% fetal bovine serum (FBS) and 1% Penicillin and Streptomycin in a CO2 incubator (37 °C, with humidified 5% CO2 atmosphere).
AlamarBlue Cell Viability
Cells were seeded in a 96-well plate at a density of 5000–10000 cells in 100 μl media per well and treated with DMSO control or test compounds at indicated doses. After incubated in CO2 incubator for 72 hours, cells were treated with 10 μL AlamarBlue solution (440 mM)). Fluorescence was measured in a plate reader with an excitation wavelength at 530–560 nm and emission wavelength at 590 nm. The fluorescence was normalized to the DMSO-treated cells, and the IC50 was calculated using GraphPad Prism 9 software.
Western Blot Assay
Cells were treated with test compounds at indicated doses, incubated for the indicated time, and then lysed in RIPA plus protease inhibitors. The protein concentration was determined by the BCA assay. Equivalent amounts of protein were loaded and separated in 7.5% SDS-PAGE gels and transferred to PVDF membranes. Membranes were blocked in 5% BSA/TBST solution and then incubated with the appropriate primary antibodies diluted in 5% BSA/TBST in a cold room overnight. After being washed, the membranes were incubated with the appropriate HRP-conjugated secondary antibodies in 2% BSA/TBST for one h at room temperature and then washed again. Bound antibodies were visualized using ECL assay (Bio-Rad), and images were captured using the ChemidocTMMP imaging system (Bio-Rad). All antibodies were purchased from Cell signaling Technology, including Anti-Brd4 (CS#13440), anti-Caspase 3 (CS#9662), Anti-β-Actin (CS#3700), and HRP-conjugated anti-mouse IgG (CS#7076) and HRP-conjugated anti-rabbit IgG (CS#7074).
Flow Cytometry
Cell apoptosis and cell cycle were detected by Attune Flow cytometer (Thermo Fisher). Cells were seeded in a 6-well plate and treated with the indicated concentrations of compounds for 48 h. For samples for cell apoptosis detection, cells were harvested and incubated in Annexin V-FITC and PI reagents each for 15 minutes (Annexin V-FITC apoptosis detection kit, Invitrogen). For the samples for cell cycle detection, cells were fixed in 70% cold ethanol overnight and stained with PI (500μg/ml) for 15 minutes. Flow data were analyzed using Flowjo software.
Cellular Degradation in Flp-In™-293 Cell
BRD4BD1 mammalian pcDNA5/FRT Vector (Ampicillin and Hygromycin B resistant) with MCS-eGFP-P2A-mCherry was obtained from Fischer’s lab.35 Stable cell lines expressing eGFP-protein fusion and mCherry reporter were generated using the Flp-In 293 system according to the manual from the vendor (Thermo Fisher). Plasmid (0.4 μg) and pOG44 (3.6 μg) DNA were pre-incubated in Opti-MEM media with 12 μL FuGene HD and added to Flp-In 293 cells (REF R75007, ThermoFisher) containing DMEM media per well in a 6-well plate format. Cells were propagated after 48 h and transferred into a 10 cm2 plate in DMEM media containing 50 μg/ml of Hygromycin B as a selection marker. Positive colonies were split into a 60 mm dish until confluent and subsequently propagated. eGFP and mCherry signal were confirmed by flow cytometry for assay validation.
Cells were seeded at 5×10^5 per well in 24-well plates one day prior to compound treatment. Titrated compounds were incubated with cells for 24 h followed by detachment and resuspension in PBS with 2% FBS, filtration, and transferred into 5 mL polystyrene tubes for analysis by flow cytometry (Attune, Thermo Fisher). Signal from at least 50,000 events per well was acquired, and the eGFP and mCherry fluorescence monitored. Data were analyzed using FlowJo (FlowJo, LCC). Forward and side scatter outliers were removed by gating. The eGFP signal abundance relative to mCherry was then quantified as a ten-fold amplified ratio using the formula: 10 × eGFP/mCherry. The median fluorescence intensity (MFI) was calculated for eGFP and mCherry for each sample and normalized to the DMSO ratio.
General Information in Synthetic Chemistry
All reactions were conducted under a positive pressure of dry argon in a glassware that had been oven-dried prior to use. Anhydrous solutions of reaction mixtures were transferred via an oven-dried syringe or cannula. All solvents were dried prior to use unless noted otherwise. Thin-layer chromatography (TLC) was performed using precoated silica gel plates. Flash column chromatography was performed with the silica gel. 1H and 13C nuclear magnetic resonance (NMR) spectra were recorded on Bruker 400, 500, 600 MHz and Varian 500 MHz spectrometers. 1H NMR spectra were reported in parts per million (ppm) referenced to 7.26 ppm of CDCl3 or referenced to the center line of a septet at 2.50 ppm of DMSO-d6. Signal splitting patterns were described as singlet (s), doublet (d), triplet (t), quartet (q), quintet (quint), or multiplet (m), with coupling constants (J) in hertz. High-resolution mass spectra (HRMS) were performed on an electron spray injection (ESI) TOF mass spectrometer. The liquid chromatography–mass spectrometry (LC–MS) analysis of final products was processed on an Agilent 1290 Infinity II LC system using a Poroshell 120 EC-C18 column (5 cm × 2.1 mm, 1.9 μm) for chromatographic separation. Purity is >95% as determined by HPLC for all final compounds tested for biological activities.
General procedure for preparation of Phenyl Dihydrouracils
In a 250 mL flask with a magnetic stirring bar, a solution of known 4-hydroxyaniline (4.37 g, 40.0 mmol) in toluene (50 mL, 0.8 M) was added acrylic acid (3.57 mL, 52.0 mmol). The reaction mixture stirred at 110 °C until the staring material disappeared as indicated by TLC, then the toluene was removed by rotavapor. Acetic acid (60 mL) and urea (7.20 g, 120.0 mmol) was then added to the flask and heated to 120 °C for 16 hours. Most of the acetic acid was removed by rotavapor. The residue was then dissolved in water (100 mL) and extracted by ethyl acetate (3×100 mL). The organic layers were combined, dried over Na2SO4, and concentrated under reduced pressure. The solid residue was suspended in ethyl acetate (10 mL) and stirred for 2 hours. The slurry was filtrated and the solid was wash by ethyl acetate (2×5 mL). Compound 5A (2.75 g, yield 33%) was obtain as solid.
General procedure for preparation of 4F, 6F and 7F by Sonogashira Couplings
A 25 mL flask was charge with magnetic stirring bar, compound 4 (316 mg, 1.0 mmol), N-Boc-propargylamine (466 mg, 3.0 mmol), Pd(PPh3)2Cl2 (35 mg, 0.05 mmol) and CuI (10 mg, 0.05 mmol), evacuated and backfilled with argon. Dimethylformamide (DMF) (2.5 mL) and Triethylamine (NEt3) (2.5 mL) were successively added by syringe. The reaction mixture stirred at room temperature until the staring material compound 4 disappeared as indicated by TLC. The mixture was partitioned between ethyl acetate and saturated solution of sodium bicarbonate, the organic layer was washed with brine, dried over Na2SO4, and concentrated in vacuum. The residue was purified by column chromatography on silica to provide 4F (260 mg, yield 75%) as white solid.
General procedure for preparation of 4H, 6H, 7H and 8H
A 35 mL pressure tube was charge with magnetic stirring bar, compound 4A (412 mg, 2.0 mmol). Next 1-Boc-4-bromopiperidine (1585 mg, 6.0 mmol), K2CO3 (910 mg, 6.6 mmol) and acetonitrile (10 mL) was added. Then the reaction mixture was stirred at 110 °C for 18 hours. The mixture was partitioned between ethyl acetate and water, the organic layer was washed with brine, dried over Na2SO4, and concentrated in vacuum. The residue was purified by column chromatography on silica to provide 4H (241 mg, yield 31%) as white solid.
General procedure for preparation of 6I and 7I by Suzuki Couplings
A 25 mL flask was charge with magnetic stirring bar, compound 7 (330 mg, 1.0 mmol), 4-Formylphenylboronic acid (300 mg, 2.0 mmol), Pd(dppf)Cl2 (36 mg, 0.05 mmol) and KOAc (295 mg, 3.0 mmol), evacuated and backfilled with argon. Dimethyl sulfoxide (DMSO) (5.0 mL) was added by syringe. The reaction mixture stirred at 90 °C until the staring material compound 7 disappeared as indicated by TLC. The mixture was partitioned between ethyl acetate and saturated solution of sodium bicarbonate, the organic layer was washed with brine, dried over Na2SO4, and concentrated in vacuum. The residue was purified by column chromatography on silica to provide 7I (133 mg, yield 43%) as white solid.
General procedure for preparation of 6J, 7J and 8J
A 35 mL pressure tube was charge with magnetic stirring bar, compound 6A (440 mg, 2.0 mmol). Next 4-(Chloromethyl)benzaldehyde (310 mg, 2.0 mmol), K2CO3 (608 mg, 4.4 mmol) and acetonitrile (10 mL) was added. Then the reaction mixture was stirred at 80 °C until the staring material compound 7 disappeared as indicated by TLC. The mixture was partitioned between ethyl acetate and water, the organic layer was washed with brine, dried over Na2SO4, and concentrated in vacuum. The residue was purified by column chromatography on silica to provide 6J (162 mg, yield 24%) as white solid.
General procedure for preparation of 4K, 5K, 6K, 7K and 8K
A 35 mL pressure tube was charge with magnetic stirring bar, compound 4A (620 mg, 3.0 mmol). Next tert-Butyl bromoacetate (0.465 mL, 3.15 mmol), K2CO3 (910 mg, 6.6 mmol) and acetonitrile (15 mL) was added. Then the reaction mixture was stirred at 80 °C until the staring material compound 7 disappeared as indicated by TLC. The mixture was partitioned between ethyl acetate and water, the organic layer was washed with brine, dried over Na2SO4, and concentrated in vacuum. The residue was purified by column chromatography on silica to provide 4K (662 mg, yield 69%) as white solid.
General procedure for preparation of 6M and 7M by Heck Reaction
A 25 mL flask was charge with magnetic stirring bar, compound 7 (165 mg, 0.5 mmol), Pd(OAc)2 (11 mg, 0.05 mmol) and P(o-tol)3 (38 mg, 0.12 mmol), evacuated and backfilled with argon. Dimethylformamide (DMF) (3.0 mL), Triethylamine (NEt3) (0.21 mL, 1.5 mmol) and tert-Butyl acrylate (0.22 mL, 1.5 mmol) were successively added by syringe. The reaction mixture stirred at 100 °C until the staring material compound 7 disappeared as indicated by TLC. The mixture was partitioned between ethyl acetate and saturated solution of sodium bicarbonate, the organic layer was washed with brine, dried over Na2SO4, and concentrated in vacuum. The residue was purified by column chromatography on silica to provide 7M (101 mg, yield 61%) as white solid.
General procedure for preparation of 6G, 7G, 6L and 7L
A 20 mL vial was charge with magnetic stirring bar and compound 6F (50 mg, 0.14 mmol). Next methanol (3.0 mL) and palladium on carbon (10 wt. %) (20 mg) was added. A hydrogen balloon was connected with the reaction mixture through a needle. The reaction mixture stirred at room temperature until the staring material compound 6F disappeared as indicated by TLC. The mixture was filtrated by celite and wash by methanol (3×5 mL). The methanol solution was concentrated in vacuum to provide 6G (48 mg, yield 95%) as white solid.
General procedure for preparation of 9 and 10.
A 20 mL vial was charge with magnetic stirring bar and compound 6K (67 mg, 0.2 mmol). Next dichloromethane (DCM) (0.5 mL) and trifluoroacetic acid (TFA) (0.75 mL) was added. The reaction mixture stirred at room temperature until the staring material compound 6K disappeared as indicated by TLC. The mixture was concentrated in vacuum to provide white solid. In the same vial, DMF (1.0 mL), piperidine (0.02 mL, 0.2 mmol), triethylamine (0.112 mL, 0.8 mmol) and 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU) (91 mg, 0.24 mmol) were successively added. The reaction mixture stirred at room temperature until the staring material disappeared as indicated by TLC. The mixture was partitioned between ethyl acetate and saturated solution of sodium bicarbonate, the organic layer was washed with brine, dried over Na2SO4, and concentrated in vacuum. The residue was purified by column chromatography on silica to provide 9 (42 mg, yield 61%) as white solid.
Procedure for preparation of 11.
A 20 mL vial was charge with magnetic stirring bar and tert-butyl (4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-2-methylphenyl)but-3-yn-1-yl)carbamate (74 mg, 0.2 mmol). Next, HCl/dioxane (1.0 mL, 4M) was added by syringe. The mixture was stirred at room temperature for half-hour, TLC indicated that the reaction was completed. The reaction mixture was concentrated under high vacuum at room temperature to provide solid. The solid was dissolved by DMF (1.0 mL) in the same vial, triethylamine (0.136 mL, 0.98 mmol) and acetic anhydride (0.021 mL, 0.22 mmol) were successively added. The reaction mixture stirred at room temperature until the staring material disappeared as indicated by TLC. The mixture was partitioned between ethyl acetate and saturated solution of sodium bicarbonate, the organic layer was washed with brine, dried over Na2SO4, and concentrated in vacuum. The residue was purified by column chromatography on silica to provide 11 (30 mg, yield 48%) as white solid.
General procedure for preparation of 12A, 12B and 12C.
A 20 mL vial was charge with magnetic stirring bar and 6F (54 mg, 0.15 mmol). Next, HCl/dioxane (1.0 mL, 4M) was added by syringe. The mixture was stirred at room temperature for half-hour, TLC indicated that the reaction was completed. The reaction mixture was concentrated under high vacuum at room temperature to provide solid. The solid was dissolved by DMF (1.0 mL) in the same vial, N,N-diisopropylethylamine (DIPEA) (0.107 mL, 0.6 mmol), JQ-1 (carboxylic acid) (Purchase from MedChemExpress Part NO.: HY-78695) (60 mg, 0.15 mmol) and HATU (63 mg, 0.165 mmol) were successively added. The mixture was stirred at room temperature for half-hour, TLC indicated that the reaction was completed. The mixture was partitioned between ethyl acetate and saturated solution of sodium bicarbonate, the organic layer was washed with brine, dried over Na2SO4, and concentrated in vacuum. The residue was purified by column chromatography on silica to provide 12A (83 mg, yield 86%) as white solid.
Procedure for preparation of 6K-Me tert-butyl 2-(2-methyl-3-(3-methyl-2,4-dioxotetrahydropyrimidin-1(2H)-yl)phenoxy)acetate
A 20 mL vial was charge with magnetic stirring bar, compound 6K (100 mg, 0.3 mmol) and Cs2CO3 (108 mg, 0.33 mmol). Next, DMF (1.0 mL) and iodomethane (MeI) (0.023 mL, 0.36 mmol) was added by syringe. The mixture was stirred at room temperature until the staring material 6K disappeared as indicated by TLC. The mixture was partitioned between ethyl acetate and saturated solution of sodium bicarbonate, the organic layer was washed with brine, dried over Na2SO4, and concentrated in vacuum, the residue was directly used for next step reaction without further purification.
General procedure for preparation of 13, 13NT and 14
A 20 mL vial was charge with magnetic stirring bar and 6K (51 mg, 0.15 mmol). Next DCM (0.5 mL) and TFA (0.75 mL) was added. The reaction mixture stirred at room temperature until the staring material compound 6K disappeared as indicated by TLC. The mixture was concentrated in vacuum to provide white solid. In the same vial, DMF (0.5 mL), 4-(N-Boc-amino)piperidine (30 mg, 0.15 mmol), DIPEA (0.107 mL, 0.6 mmol) and HATU (63 mg, 0.165 mmol) were successively added. The reaction mixture stirred at room temperature until the staring material disappeared as indicated by TLC. The mixture was partitioned between ethyl acetate and saturated solution of sodium carbonate, the organic layer was washed with brine, dried over Na2SO4, and concentrated in vacuum. The residue was treated by DCM (0.75 mL) and TFA (0.75 mL) at room temperature for half-hour, TLC indicated that the reaction was completed. The mixture was concentrated in high vacuum, in the same vial, DIPEA (0.107 mL, 0.6 mmol), JQ-1 (carboxylic acid) (60 mg, 0.15 mmol) and HATU (63 mg, 0.165 mmol) were successively added. The mixture was stirred at room temperature for half-hour, TLC indicated that the reaction was completed. The mixture was partitioned between ethyl acetate and saturated solution of sodium bicarbonate, the organic layer was washed with brine, dried over Na2SO4, and concentrated in vacuum. The residue was purified by column chromatography on silica to provide 13 (85 mg, yield 76%) as white solid.
Compound Characterization Data
1-phenyldihydropyrimidine-2,4(1H,3H)-dione (2)
1H NMR (400 MHz, DMSO-d6) δ 10.36 (s, 1H), 7.43 – 7.35 (m, 2H), 7.39 – 7.29 (m, 2H), 7.27 – 7.19 (m, 1H), 3.79 (t, J = 6.7 Hz, 2H), 2.70 (t, J = 6.7 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 170.6, 152.1, 142.1, 128.6, 125.8, 125.3, 44.5, 31.1. HPLC purity: 97.8%.
1-(2-hydroxyphenyl)dihydropyrimidine-2,4(1H,3H)-dione (3A)
1H NMR (400 MHz, DMSO-d6) δ 7.45 (s, 1H), 7.34 – 7.26 (m, 2H), 7.24 – 7.17 (m, 1H), 7.15 – 7.07 (m, 1H), 6.91 (s, 1H), 4.00 (t, J = 6.9 Hz, 2H), 2.53 (t, J = 6.9 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ = 171.6, 153.6, 141.9, 131.0, 123.8, 122.1, 109.6, 109.5, 38.5, 33.1. HRMS (ESI/[M+H]+) Calcd for [C10H10N2O3+H]+: 207.0764, found: 207.0765. HPLC purity: 98.6%.
1-(o-tolyl)dihydropyrimidine-2,4(1H,3H)-dione (3B)
1H NMR (400 MHz, DMSO-d6) δ 10.33 (s, 1H), 7.33 – 7.18 (m, 4H), 3.82 – 3.71 (m, 1H), 3.55 – 3.45 (m, 1H), 2.86 – 2.60 (m, 2H), 2.19 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ = 170.8, 151.7, 140.9, 135.5, 130.6, 127.4, 127.2, 126.8, 44.6, 31.1, 17.5. HRMS (ESI/[M+H]+) Calcd for [C11H12N2O2+H]+: 205.0972, found: 205.0973. HPLC purity: 97.5%.
1-(2-methoxyphenyl)dihydropyrimidine-2,4(1H,3H)-dione (3C)
1H NMR (400 MHz, DMSO-d6) δ 10.29 (s, 1H), 7.36 – 7.28 (m, 1H), 7.27 – 7.21 (m, 1H), 7.14 – 7.07 (m, 1H), 7.01 – 6.92 (m, 1H), 3.80 (s, 3H), 3.57 (t, J = 6.7 Hz, 2H), 2.67 (t, J = 6.6 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ = 170.8, 154.8, 152.1, 130.1, 129.2, 128.8, 120.5, 112.4, 55.7, 44.5, 31.1. HRMS (ESI/[M+H]+) Calcd for [C11H12N2O3+H]+: 221.0921, found: 221.0924. HPLC purity: 98.2%.
1-(2-ethylphenyl)dihydropyrimidine-2,4(1H,3H)-dione (3D)
1H NMR (400 MHz, DMSO-d6) δ 10.34 (s, 1H), 7.36 – 7.23 (m, 4H), 3.84 – 3.72 (m, 1H), 3.52 – 3.41 (m, 1H), 2.83 – 2.62 (m, 2H), 2.54 (tt, J = 7.6, 3.8 Hz, 2H), 1.15 (t, J = 7.6 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 170.7, 152.2, 141.2, 140.4, 128.8, 127.8, 127.6, 126.8, 45.0, 31.1, 23.5, 14.4. HRMS (ESI/[M+H]+) Calcd for [C12H14N2O2+H]+: 219.1128, found: 219.1131. HPLC purity: 97.6%.
1-(2-chlorophenyl)dihydropyrimidine-2,4(1H,3H)-dione (3E)
1H NMR (400 MHz, DMSO-d6) δ 10.47 (s, 1H), 7.61 – 7.54 (m, 1H), 7.53 – 7.46 (m, 1H), 7.45 – 7.34 (m, 2H), 3.77 – 3.66 (m, 1H), 3.65 – 3.54 (m, 1H), 2.77 – 2.69 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 170.6, 151.8, 139.0, 131.8, 130.1, 129.9, 129.3, 128.2, 44.3, 31.0. HRMS (ESI/[M+H]+) Calcd for [C10H9ClN2O2+H]+: 225.0425, found: 225.0425. HPLC purity: 95.8%.
1-(3-iodophenyl)dihydropyrimidine-2,4(1H,3H)-dione (4)
1H NMR (400 MHz, DMSO-d6) δ 10.42 (s, 1H), 7.76 – 7.70 (m, 1H), 7.63 – 7.56 (m, 1H), 7.39 – 7.32 (m, 1H), 7.23 – 7.15 (m, 1H), 3.78 (t, J = 6.6 Hz, 2H), 2.69 (t, J = 6.6 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 170.6, 152.1, 143.3, 134.4, 133.9, 130.5, 124.7, 94.0, 44.4, 31.0.
1-(3-hydroxyphenyl)dihydropyrimidine-2,4(1H,3H)-dione (4A)
1H NMR (400 MHz, DMSO-d6) δ 10.31 (s, 1H), 9.54 (s, 1H), 7.20 – 7.12 (m, 1H), 6.76 – 6.69 (m, 2H), 6.67 – 6.60 (m, 1H), 3.73 (t, J = 6.6 Hz, 2H), 2.68 (t, J = 6.6 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 170.6, 157.5, 152.0, 143.1, 129.3, 115.6, 112.9, 112.6, 44.6, 31.1. HRMS (ESI/[M+H]+) Calcd for [C10H10N2O3+H]+: 207.0764, found: 207.0764. HPLC purity: 95.4%.
1-(3-methoxyphenyl)dihydropyrimidine-2,4(1H,3H)-dione (4C)
1H NMR (400 MHz, DMSO-d6) δ 10.35 (s, 1H), 7.33 – 7.24 (m, 1H), 6.94 – 6.86 (m, 2H), 6.86 – 6.78 (m, 1H), 3.77 (t, J = 6.7 Hz, 2H), 3.75 (s, 3H), 2.69 (t, J = 6.7 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 170.6, 159.4, 152.1, 143.2, 129.3, 117.5, 111.4, 111.4, 55.2, 44.6, 31.1. HRMS (ESI/[M+H]+) Calcd for [C11H12N2O3+H]+: 221.0921, found: 221.0922. HPLC purity: 95.3%.
tert-butyl (3-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)phenyl)prop-2-yn-1-yl)carbamate (4F)
1H NMR (400 MHz, DMSO-d6) δ 10.41 (s, 1H), 7.49 – 7.32 (m, 4H), 7.29 – 7.22 (m, 1H), 3.98 (d, J = 5.8 Hz, 2H), 3.79 (t, J = 6.6 Hz, 2H), 2.70 (t, J = 6.6 Hz, 2H), 1.40 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ 170.6, 155.3, 152.1, 142.2, 129.0, 128.5, 128.0, 125.4, 122.6, 88.0, 80.9, 78.3, 44.3, 31.0, 30.1, 28.2. HRMS (ESI/[M+Na]+) Calcd for [C18H21N3O4+Na]+: 366.1424, found: 366.1420. HPLC purity: 98.9%.
tert-butyl 4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)phenoxy)piperidine-1-carboxylate (4H)
1H NMR (400 MHz, DMSO-d6) δ 10.35 (s, 1H), 7.32 – 7.23 (m, 1H), 6.98 – 6.93 (m, 1H), 6.92 – 6.82 (m, 2H), 4.60 – 4.49 (m, 1H), 3.77 (t, J = 6.7 Hz, 2H), 3.70 – 3.60 (m, 2H), 3.24 – 3.14 (m, 2H), 2.69 (t, J = 6.7 Hz, 2H), 1.98 – 1.85 (m, 2H), 1.57 – 1.47 (m, 2H), 1.40 (s, 8H). 13C NMR (101 MHz, DMSO-d6) δ 170.6, 157.0, 153.9, 152.1, 143.3, 129.4, 117.7, 113.3, 113.2, 78.7, 71.9, 48.6, 44.6, 31.0, 30.3, 28.1. HRMS (ESI/[M+H]+) Calcd for [C20H27N3O5+H]+: 390.2023, found: 390.2022. HPLC purity: 95.6%.
tert-butyl 2-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)phenoxy)acetate (4K)
1H NMR (400 MHz, DMSO-d6) δ 10.36 (s, 1H), 7.34 – 7.24 (m, 1H), 6.97 – 6.88 (m, 2H), 6.81 – 6.73 (m, 1H), 4.65 (s, 2H), 3.76 (t, J = 6.6 Hz, 2H), 2.69 (t, J = 6.6 Hz, 2H), 1.43 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ 170.6, 167.7, 157.8, 152.0, 143.1, 129.3, 118.1, 112.1, 111.6, 81.4, 65.1, 44.6, 31.0, 27.7. HRMS (ESI/[M+H]+) Calcd for [C16H20N2O5+H]+: 321.1445, found: 321.1444. HPLC purity: 97.4%.
1-(4-hydroxyphenyl)dihydropyrimidine-2,4(1H,3H)-dione (5A)
1H NMR (400 MHz, DMSO-d6) δ 10.25 (s, 1H), 9.46 (s, 1H), 7.14 – 7.06 (m, 2H), 6.79 – 6.71 (m, 2H), 3.67 (t, J = 6.7 Hz, 2H), 2.67 (t, J = 6.7 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 170.7, 155.6, 152.3, 133.5, 127.0, 115.2, 45.1, 31.1. HRMS (ESI/[M+H]+) Calcd for [C10H10N2O3+H]+: 207.0764, found: 207.0763. HPLC purity: 99.0%.
tert-butyl 2-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)phenoxy)acetate (5K)
1H NMR (400 MHz, DMSO-d6) δ 10.30 (s, 1H), 7.28 – 7.19 (m, 2H), 6.95 – 6.86 (m, 2H), 4.65 (s, 2H), 3.72 (t, J = 6.7 Hz, 2H), 2.69 (t, J = 6.7 Hz, 2H), 1.44 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ 170.6, 167.8, 155.6, 152.3, 135.4, 126.8, 114.5, 81.4, 65.1, 44.9, 31.1, 27.7. HRMS (ESI/[M+H]+) Calcd for [C16H20N2O5+H]+: 321.1445, found: 321.1448. HPLC purity: 98.8%.
1-(3-iodo-2-methylphenyl)dihydropyrimidine-2,4(1H,3H)-dione (6)
1H NMR (400 MHz, DMSO-d6) δ 10.40 (s, 1H), 7.82 (dd, J = 7.9, 1.2 Hz, 1H), 7.33 (dd, J = 7.9, 1.2 Hz, 1H), 7.07 – 6.98 (m, 1H), 3.83 – 3.72 (m, 1H), 3.57 – 3.46 (m, 1H), 2.86 – 2.73 (m, 1H), 2.73 – 2.62 (m, 1H), 2.28 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ = 170.7, 151.8, 141.0, 138.8, 138.1, 128.6, 127.7, 102.2, 44.6, 31.0, 23.5.
1-(3-hydroxy-2-methylphenyl)dihydropyrimidine-2,4(1H,3H)-dione (6A)
1H NMR (400 MHz, DMSO-d6) δ 10.28 (s, 1H), 9.49 (s, 1H), 7.06 – 6.98 (m, 1H), 6.81 – 6.74 (m, 1H), 6.73 – 6.67 (m, 1H), 3.77 – 3.66 (m, 1H), 3.52 – 3.42 (m, 1H), 2.81 – 2.70 (m, 1H), 2.70 – 2.59 (m, 1H), 1.97 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 170.8, 156.0, 151.7, 141.9, 126.3, 122.3, 117.6, 113.7, 44.7, 31.1, 10.7. HRMS (ESI/[M+H]+) Calcd for [C11H12N2O3+H]+: 221.0921, found: 221.0922. HPLC purity: 98.0%.
tert-butyl (3-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-2-methylphenyl)prop-2-yn-1-yl)carbamate (6F)
1H NMR (400 MHz, DMSO-d6) δ 10.37 (s, 1H), 7.46 – 7.36 (m, 1H), 7.36 – 7.18 (m, 3H), 4.00 (d, J = 5.8 Hz, 2H), 3.85 – 3.69 (m, 1H), 3.58 – 3.44 (m, 1H), 2.87 – 2.58 (m, 2H), 2.24 (s, 3H), 1.40 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ 170.7, 155.4, 151.8, 141.3, 137.7, 130.7, 127.6, 126.7, 123.6, 91.9, 79.9, 78.2, 44.5, 31.1, 30.3, 28.2, 15.6. HRMS (ESI/[M+Na]+) Calcd for [C19H23N3O4+Na]+: 380.1581, found: 380.1575. HPLC purity: 98.6%.
tert-butyl (3-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-2-methylphenyl)propyl)carbamate (6G)
1H NMR (400 MHz, DMSO-d6) δ 10.30 (s, 1H), 7.19 – 7.08 (m, 3H), 6.95 – 6.87 (m, 1H), 3.79 – 3.67 (m, 1H), 3.53 – 3.42 (m, 1H), 3.03 – 2.94 (m, 2H), 2.84 – 2.72 (m, 1H), 2.72 – 2.62 (m, 1H), 2.62 – 2.54 (m, 2H), 2.09 (s, 3H), 1.62 (p, J = 7.0 Hz, 2H), 1.38 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ 170.8, 155.7, 151.9, 141.4, 141.2, 133.6, 128.0, 126.2, 124.9, 77.4, 44.8, 31.1, 30.5, 30.1, 28.3, 13.2. HRMS (ESI/[M+H]+) Calcd for [C19H27N3O4+H]+: 362.2074, found: 362.2080. HPLC purity: 98.3%.
tert-butyl 4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-2-methylphenoxy)piperidine-1-carboxylate (6H)
1H NMR (400 MHz, DMSO-d6) δ 1H NMR (400 MHz, DMSO) δ 10.32 (s, 1H), 7.23 – 7.14 (m, 1H), 7.04 – 6.97 (m, 1H), 6.90 – 6.84 (m, 1H), 4.59 (tt, J = 7.3, 3.5 Hz, 1H), 3.80 – 3.69 (m, 1H), 3.66 – 3.54 (m, 2H), 3.54 – 3.44 (m, 1H), 3.33 – 3.22 (m, 2H), 2.83 – 2.61 (m, 2H), 2.02 (s, 3H), 1.96 – 1.84 (m, 2H), 1.66 – 1.52 (m, 2H), 1.42 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ 170.7, 155.4, 153.9, 151.7, 142.0, 126.6, 125.1, 119.3, 112.1, 78.7, 71.9, 44.7, 31.1, 30.4, 28.1, 10.8. HRMS (ESI/[M+H]+) Calcd for [C21H29N3O5+H]+: 404.2180, found: 404.2182. HPLC purity: 96.3%.
3’-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-2’-methyl-[1,1’-biphenyl]-4-carbaldehyde (6I)
1H NMR (400 MHz, DMSO-d6) δ 10.38 (s, 1H), 10.08 (s, 1H), 8.04 – 7.97 (m, 2H), 7.62 – 7.55 (m, 2H), 7.41 – 7.35 (m, 2H), 7.29 – 7.20 (m, 1H), 3.89 – 3.77 (m, 1H), 3.62 – 3.52 (m, 1H), 2.87 – 2.75 (m, 1H), 2.75 – 2.65 (m, 1H), 2.09 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 192.8, 170.7, 151.9, 147.0, 141.7, 141.5, 134.9, 133.1, 130.0, 129.5, 128.6, 127.2, 126.7, 44.7, 31.1, 15.3. HRMS (ESI/[M+H]+) Calcd for [C18H16N2O3+H]+: 309.1234, found: 309.1239. HPLC purity: 95.5%.
4-((3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-2-methylphenoxy)methyl)benzaldehyde (6J)
1H NMR (400 MHz, DMSO-d6) δ 10.34 (s, 1H), 10.02 (s, 1H), 7.99 – 7.92 (m, 2H), 7.73 – 7.67 (m, 2H), 7.25 – 7.16 (m, 1H), 7.04 – 6.97 (m, 1H), 6.94 – 6.88 (m, 1H), 5.26 (s, 2H), 3.81 – 3.70 (m, 1H), 3.55 – 3.45 (m, 1H), 2.85 – 2.73 (m, 1H), 2.73 – 2.62 (m, 1H), 2.09 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 192.8, 170.7, 156.5, 151.8, 144.2, 141.8, 135.6, 129.8, 127.6, 126.7, 124.3, 119.6, 110.8, 69.0, 44.7, 31.1, 10.8. HRMS (ESI/[M+H]+) Calcd for [C19H18N2O4+H]+: 339.1339, found: 339.1336. HPLC purity: 97.8%.
tert-butyl 2-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-2-methylphenoxy)acetate (6K)
1H NMR (400 MHz, DMSO-d6) δ 10.33 (s, 1H), 7.21 – 7.13 (m, 1H), 6.93 – 6.87 (m, 1H), 6.83 – 6.76 (m, 1H), 4.70 (s, 2H), 3.80 – 3.69 (m, 1H), 3.54 – 3.43 (m, 1H), 2.84 – 2.72 (m, 1H), 2.72 – 2.61 (m, 1H), 2.05 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 170.7, 167.8, 156.3, 151.7, 141.8, 126.5, 124.4, 119.8, 110.5, 81.4, 65.6, 44.7, 31.1, 27.7, 10.7. HRMS (ESI/[M+H]+) Calcd for [C17H22N2O5+H]+: 335.1601, found: 335.1604. HPLC purity: 96.4%.
tert-butyl 3-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-2-methylphenyl)propanoate (6L)
1H NMR (400 MHz, Acetone-d6) δ 7.22 – 7.11 (m, 3H), 3.92 – 3.81 (m, 1H), 3.70 – 3.59 (m, 1H), 2.98 – 2.91 (m, 2H), 2.90 – 2.73 (m, 2H), 2.57 – 2.46 (m, 2H), 2.22 (s, 3H), 1.42 (s, 9H). 13C NMR (101 MHz, Acetone-d6) δ = 172.4, 170.9, 152.6, 142.4, 141.4, 135.0, 128.9, 127.2, 126.2, 80.4, 46.1, 36.2, 32.1, 29.5, 28.2, 13.8. HRMS (ESI/[M+H]+) Calcd for [C18H24N2O4+H]+: 333.1809, found: 333.1813. HPLC purity: 97.9%.
tert-butyl (E)-3-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-2-methylphenyl)acrylate (6M)
1H NMR (400 MHz, DMSO-d6) δ 10.37 (s, 1H), 7.83 (d, J = 15.8 Hz, 1H), 7.71 – 7.65 (m, 1H), 7.38 – 7.33 (m, 1H), 7.33 – 7.24 (m, 1H), 6.42 (d, J = 15.8 Hz, 1H), 3.83 – 3.71 (m, 1H), 3.55 – 3.44 (m, 1H), 2.87 – 2.76 (m, 1H), 2.72 – 2.63 (m, 1H), 2.22 (s, 3H), 1.49 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ 170.7, 165.4, 152.0, 141.6, 140.9, 135.1, 134.3, 129.0, 126.8, 125.9, 121.8, 80.2, 44.6, 31.0, 27.8, 13.7. HRMS (ESI/[M+H]+) Calcd for [C18H22N2O4+H]+: 331.1652, found: 331.1665. HPLC purity: 99.6%.
1-(5-iodo-2-methylphenyl)dihydropyrimidine-2,4(1H,3H)-dione (7)
1H NMR (400 MHz, DMSO-d6) δ 10.42 (s, 1H), 7.67 (d, J = 1.9 Hz, 1H), 7.58 (dd, J = 8.0, 1.9 Hz, 1H), 7.09 (d, J = 8.1 Hz, 1H), 3.82 – 3.71 (m, 1H), 3.55 – 3.44 (m, 1H), 2.84 – 2.60 (m, 2H), 2.13 (s, 3H).13C NMR (101 MHz, DMSO-d6) δ 170.8, 151.7, 142.4, 136.1, 135.8, 135.7, 132.6, 90.7, 44.4, 31.1, 17.1.
1-(5-hydroxy-2-methylphenyl)dihydropyrimidine-2,4(1H,3H)-dione (7A)
1H NMR (400 MHz, DMSO-d6) δ 10.28 (s, 1H), 9.35 (s, 1H), 7.07 – 7.01 (m, 1H), 6.68 – 6.60 (m, 2H), 3.77 – 3.66 (m, 1H), 3.52 – 3.41 (m, 1H), 2.87 – 2.55 (m, 2H), 2.05 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 170.8, 156.0, 151.6, 141.4, 131.0, 125.2, 114.5, 113.9, 44.5, 31.1, 16.6. HRMS (ESI/[M+H]+) Calcd for [C11H12N2O3+H]+: 221.0921, found: 221.0916. HPLC purity: 99.2%.
tert-butyl (3-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-4-methylphenyl)prop-2-yn-1-yl)carbamate (7F)
1H NMR (400 MHz, DMSO-d6) δ 10.30 (s, 1H), 7.51 – 7.10 (m, 4H), 3.97 (d, J = 5.6 Hz, 2H), 3.82 – 3.71 (m, 1H), 3.54 – 3.43 (m, 1H), 2.80 – 2.60 (m, 2H), 2.18 (s, 3H), 1.39 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ 171.1, 155.3, 152.1, 141.3, 136.5, 130.9, 130.2, 130.1, 120.7, 87.6, 80.7, 78.3, 44.5, 31.2, 30.1, 28.2, 17.4. HRMS (ESI/[M+Na]+) Calcd for [C19H23N3O4+Na]+: 380.1581, found: 380.1598. HPLC purity: 99.1%.
tert-butyl (3-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-4-methylphenyl)propyl)carbamate (7G)
1H NMR (400 MHz, DMSO-d6) δ 7.17 (d, J = 7.9 Hz, 1H), 7.05 (dd, J = 7.8, 1.8 Hz, 1H), 6.98 (d, J = 1.8 Hz, 1H), 5.97 (s, 1H), 4.68 (s, 1H), 3.82 – 3.71 (m, 1H), 3.63 – 3.53 (m, 1H), 3.17 – 3.07 (m, 2H), 2.77 (t, J = 6.8 Hz, 2H), 2.59 (t, J = 7.7 Hz, 2H), 2.20 (s, 3H), 1.77 (p, J = 7.3 Hz, 2H), 1.43 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ 171.3, 156.1, 152.9, 140.9, 140.2, 133.0, 131.3, 128.3, 126.9, 79.3, 77.4, 45.4, 40.0, 32.4, 31.6, 28.6, 17.6. HRMS (ESI/[M+H]+) Calcd for [C19H27N3O4+H]+: 362.2074, found: 362.2065. HPLC purity: 96.1%.
tert-butyl 4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-4-methylphenoxy)piperidine-1-carboxylate (7H)
1H NMR (400 MHz, DMSO-d6) δ 10.32 (s, 1H), 7.16 (d, J = 8.4 Hz, 1H), 6.91 (d, J = 2.6 Hz, 1H), 6.86 (dd, J = 8.4, 2.6 Hz, 1H), 4.50 (tt, J = 7.8, 3.6 Hz, 1H), 3.81 – 3.70 (m, 1H), 3.70 – 3.60 (m, 2H), 3.54 – 3.43 (m, 1H), 3.22 – 3.12 (m, 2H), 2.81 – 2.63 (m, 2H), 2.09 (s, 3H), 1.93 – 1.84 (m, 2H), 1.58 – 1.45 (m, 2H), 1.40 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ 170.8, 155.5, 153.9, 151.6, 141.7, 131.1, 127.4, 114.9, 114.8, 78.7, 72.0, 44.5, 31.1, 30.3, 28.1, 16.6. HRMS (ESI/[M+H]+) Calcd for [C21H29N3O5+H]+: 404.2180, found: 404.2196. HPLC purity: 96.4%.
3’-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-4’-methyl-[1,1’-biphenyl]-4-carbaldehyde (7I)
1H NMR (400 MHz, DMSO-d6) δ 10.40 (s, 1H), 10.05 (s, 1H), 8.03 – 7.96 (m, 2H), 7.96 – 7.89 (m, 2H), 7.75 (d, J = 2.0 Hz, 1H), 7.67 (dd, J = 7.9, 2.1 Hz, 1H), 7.42 (d, J = 8.1 Hz, 1H), 3.93 – 3.82 (m, 1H), 3.67 – 3.54 (m, 1H), 2.87 – 2.67 (m, 2H), 2.24 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 192.7, 170.8, 151.8, 144.9, 141.7, 137.4, 136.2, 135.1, 131.4, 130.2, 127.1, 126.0, 125.9, 44.6, 31.2, 17.3. HRMS (ESI/[M+H]+) Calcd for [C18H16N2O3+H]+: 309.1234, found: 309.1239. HPLC purity: 95.2%.
4-((3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-4-methylphenoxy)methyl)benzaldehyde (7J)
1H NMR (400 MHz, DMSO-d6) δ 10.34 (s, 1H), 10.01 (s, 1H), 7.97 – 7.90 (m, 2H), 7.70 – 7.63 (m, 2H), 7.18 (d, J = 8.5 Hz, 1H), 7.01 (d, J = 2.7 Hz, 1H), 6.92 (dd, J = 8.4, 2.7 Hz, 1H), 5.20 (s, 2H), 3.82 – 3.71 (m, 1H), 3.54 – 3.44 (m, 1H), 2.82 – 2.62 (m, 2H), 2.10 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 192.8, 170.8, 156.8, 151.6, 143.9, 141.6, 135.6, 131.1, 129.7, 127.8, 127.8, 113.8, 113.8, 68.7, 44.5, 31.1, 16.6. HRMS (ESI/[M+H]+) Calcd for [C19H18N2O4+H]+: 339.1339, found: 339.1334. HPLC purity: 98.4%.
tert-butyl 2-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-4-methylphenoxy)acetate (7K)
1H NMR (400 MHz, DMSO-d6) δ 10.32 (s, 1H), 7.17 (d, J = 8.4 Hz, 1H), 6.87 (d, J = 2.7 Hz, 1H), 6.78 (dd, J = 8.4, 2.7 Hz, 1H), 4.62 (s, 2H), 3.81 – 3.70 (m, 1H), 3.53 – 3.43 (m, 1H), 2.86 – 2.57 (m, 2H), 2.10 (s, 3H), 1.43 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ 170.7, 167.8, 156.3, 151.6, 141.5, 131.0, 127.9, 113.5, 113.4, 81.4, 65.2, 44.5, 31.1, 27.7, 16.6. HRMS (ESI/[M+H]+) Calcd for [C17H22N2O5+H]+: 335.1601, found: 335.1592. HPLC purity: 97.5%.
tert-butyl 3-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-4-methylphenyl)propanoate (7L)
1H NMR (400 MHz, CDCl3) δ 7.43 (s, 1H), 7.21 (d, J = 7.8 Hz, 1H), 7.11 (dd, J = 7.8, 1.8 Hz, 1H), 7.02 (d, J = 1.8 Hz, 1H), 3.86 – 3.74 (m, 1H), 3.68 – 3.57 (m, 1H), 2.93 – 2.79 (m, 4H), 2.53 (t, J = 7.1 Hz, 2H), 2.23 (s, 3H), 1.42 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ 171.5, 170.8, 151.6, 140.7, 139.3, 133.0, 130.4, 127.3, 126.8, 79.7, 44.6, 36.1, 31.2, 29.8, 27.7, 17.1. HRMS (ESI/[M+H]+) Calcd for [C18H24N2O4+H]+: 333.1809, found: 333.1802. HPLC purity: 97.9%.
tert-butyl (E)-3-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-4-methylphenyl)acrylate (7M)
1H NMR (400 MHz, DMSO-d6) δ 10.38 (s, 1H), 7.69 (d, J = 1.8 Hz, 1H), 7.57 – 7.47 (m, 2H), 7.31 (d, J = 8.0 Hz, 1H), 6.52 (d, J = 15.9 Hz, 1H), 3.86 – 3.75 (m, 1H), 3.58 – 3.48 (m, 1H), 2.84 – 2.66 (m, 2H), 2.20 (s, 3H), 1.48 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ 170.8, 165.5, 151.7, 142.7, 141.5, 138.1, 133.1, 131.1, 127.5, 126.7, 119.7, 79.9, 44.6, 31.1, 27.8, 17.5. HRMS (ESI/[M+H]+) Calcd for [C18H22N2O4+H]+: 331.1652, found: 331.1647. HPLC purity: 98.5%.
1-(4-hydroxy-2-methylphenyl)dihydropyrimidine-2,4(1H,3H)-dione (8A)
1H NMR (400 MHz, DMSO-d6) δ 1H NMR (400 MHz, DMSO) δ 10.23 (s, 1H), 9.40 (s, 1H), 7.01 (d, J = 8.5 Hz, 1H), 6.64 (d, J = 2.8 Hz, 1H), 6.60 (dd, J = 8.4, 2.8 Hz, 1H), 3.71 – 3.60 (m, 1H), 3.50 – 3.39 (m, 1H), 2.79 – 2.59 (m, 2H), 2.08 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 170.8, 156.4, 152.0, 136.5, 132.3, 128.2, 116.8, 113.3, 44.9, 31.2, 17.5. HRMS (ESI/[M+H]+) Calcd for [C11H12N2O3+H]+: 221.0921, found: 221.0916. HPLC purity: 97.9%.
tert-butyl 4-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-methylphenoxy)piperidine-1-carboxylate (8H)
1H NMR (400 MHz, DMSO-d6) δ 10.28 (s, 1H), 7.14 (d, J = 8.6 Hz, 1H), 6.88 (d, J = 2.9 Hz, 1H), 6.82 (dd, J = 8.6, 2.9 Hz, 1H), 4.54 (tt, J = 7.8, 3.6 Hz, 1H), 3.73 – 3.60 (m, 3H), 3.52 – 3.42 (m, 1H), 3.23 – 3.14 (m, 2H), 2.80 – 2.60 (m, 2H), 2.14 (s, 3H), 1.94 – 1.84 (m, 2H), 1.58 – 1.45 (m, 2H), 1.41 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ 170.8, 155.8, 153.9, 151.9, 136.9, 133.9, 128.3, 117.5, 113.7, 78.7, 71.9, 44.8, 31.1, 30.4, 28.1, 17.6. HRMS (ESI/[M+H]+) Calcd for [C21H29N3O5+H]+: 404.2180, found: 404.2169. HPLC purity: 96.3%.
4-((4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-methylphenoxy)methyl)benzaldehyde (8J)
1H NMR (400 MHz, DMSO-d6) δ 10.29 (s, 1H), 10.01 (s, 1H), 7.97 – 7.91 (m, 2H), 7.70 – 7.64 (m, 2H), 7.17 (d, J = 8.6 Hz, 1H), 6.96 (d, J = 2.9 Hz, 1H), 6.88 (dd, J = 8.6, 3.0 Hz, 1H), 5.23 (s, 2H), 3.75 – 3.64 (m, 1H), 3.52 – 3.41 (m, 1H), 2.81 – 2.60 (m, 2H), 2.15 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 192.8, 170.8, 156.9, 151.9, 144.1, 136.9, 135.6, 134.2, 129.7, 128.4, 127.7, 116.5, 112.8, 68.6, 44.8, 31.1, 17.6. HRMS (ESI/[M+H]+) Calcd for [C19H18N2O4+H]+: 339.1339, found: 339.1335. HPLC purity: 98.5%.
tert-butyl 2-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-methylphenoxy)acetate (8K)
1H NMR (400 MHz, DMSO-d6) δ 10.28 (s, 1H), 7.16 (d, J = 8.6 Hz, 1H), 6.81 (d, J = 3.0 Hz, 1H), 6.75 (dd, J = 8.6, 3.0 Hz, 1H), 4.64 (s, 2H), 3.75 – 3.64 (m, 1H), 3.52 – 3.42 (m, 1H), 2.81 – 2.60 (m, 2H), 2.14 (s, 3H), 1.44 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ170.8, 167.8, 156.5, 151.9, 136.8, 134.3, 128.2, 116.2, 112.4, 81.4, 65.0, 44.8, 31.1, 27.7, 17.6. HRMS (ESI/[M+H]+) Calcd for [C17H22N2O5+H]+: 335.1601, found: 335.1593. HPLC purity: 95.8%.
1-(2-methyl-3-(2-oxo-2-(piperidin-1-yl)ethoxy)phenyl)dihydropyrimidine-2,4(1H,3H)-dione (9)
1H NMR (400 MHz, DMSO-d6) δ 10.32 (s, 1H), 7.20 – 7.12 (m, 1H), 6.91 – 6.80 (m, 2H), 3.80 – 3.69 (m, 1H), 3.53 – 3.35 (m, 5H), 2.84 – 2.61 (m, 2H), 2.04 (s, 3H), 1.64–1.39 (m, 6H). 13C NMR (101 MHz, DMSO-d6) 170.7, 165.4, 156.6, 151.7, 141.7, 126.5, 124.1, 119.4, 110.6, 66.7, 45.2, 44.7, 42.2, 31.1, 26.0, 25.3, 24.0, 10.8. HRMS (ESI/[M+H]+) Calcd for [C18H23N3O4+H]+: 346.1761, found: 346.1760. HPLC purity: 98.9%.
2-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-2-methylphenoxy)-N-ethylacetamide (10)
1H NMR (400 MHz, DMSO-d6) δ 10.33 (s, 1H), 8.08 – 7.91 (m, 1H), 7.23 – 7.14 (m, 1H), 6.95 – 6.88 (m, 1H), 6.86 – 6.79 (m, 1H), 4.48 (s, 2H), 3.80 – 3.69 (m, 1H), 3.54 – 3.43 (m, 1H), 3.23 – 3.12 (m, 2H), 2.84 – 2.62 (m, 2H), 2.09 (s, 3H), 1.05 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 170.7, 167.3, 156.5, 151.8, 141.8, 126.6, 124.6, 119.9, 110.8, 67.7, 44.7, 33.3, 31.1, 14.8, 10.9. HRMS (ESI/[M+H]+) Calcd for [C15H19N3O4+H]+: 306.1448, found: 306.1444. HPLC purity: 99.1%.
N-(4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-2-methylphenyl)but-3-yn-1-yl)acetamide (11)
1H NMR (400 MHz, DMSO-d6) δ 10.36 (s, 1H), 8.11 – 8.04 (m, 1H), 7.38 – 7.32 (m, 1H), 7.30 – 7.18 (m, 2H), 3.82 – 3.71 (m, 1H), 3.55 – 3.45 (m, 1H), 3.25 (t, J = 6.9 Hz, 2H), 2.85 – 2.72 (m, 1H), 2.75 – 2.62 (m, 1H), 2.59 (t, J = 6.9 Hz, 2H), 2.24 (s, 3H), 1.82 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 170.7, 169.3, 151.8, 141.2, 137.5, 130.8, 127.3, 126.6, 124.2, 92.7, 79.7, 44.5, 37.9, 31.1, 22.6, 19.9, 15.7. HRMS (ESI/[M+H]+) Calcd for [C17H19N3O3+H]+: 314.1499, found: 314.1491. HPLC purity: 95.4%.
(S)-2-(4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)-N-(3-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-2-methylphenyl)prop-2-yn-1-yl)acetamide (12A)
1H NMR (400 MHz, CDCl3) δ 8.07 (d, J = 5.3 Hz, 1H), 7.42 – 7.36 (m, 3H), 7.35 – 7.30 (m, 1H), 7.25 – 7.13 (m, 4H), 4.68 – 4.60 (m, 1H), 4.50 – 4.38 (m, 1H), 4.29 – 4.17 (m, 1H), 3.81 – 3.68 (m, 1H), 3.63 – 3.40 (m, 3H), 2.84 – 2.75 (m, 2H), 2.65 (d, J = 1.5 Hz, 3H), 2.39 (s, 3H), 2.28 (d, J = 3.7 Hz, 3H), 1.68 – 1.62 (m, 3H). 13C NMR (101 MHz, CDCl3) δ 13C NMR (101 MHz, CDCl3) δ 170.4, 169.7, 164.2, 155.6, 151.6, 150.1, 140.1, 138.2, 136.8, 136.7, 132.4, 132.3, 131.0, 131.0, 130.5, 130.0, 128.8, 127.4, 126.9, 124.9, 90.1, 81.4, 54.5, 45.3, 39.1, 31.5, 30.2, 16.2, 14.5, 13.2, 11.9. HRMS (ESI/[M+H]+) Calcd for [C33H30ClN7O3S+H]+: 640.1892, found: 640.1883. HPLC purity: 96.0%.
(S)-2-(4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)-N-(4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-2-methylphenyl)but-3-yn-1-yl)acetamide (12B)
1H NMR (400 MHz, MeOD-d4) δ 7.50 – 7.41 (m, 2H), 7.39 – 7.27 (m, 3H), 7.26 – 7.13 (m, 2H), 4.69 (dd, J = 8.8, 5.2 Hz, 1H), 3.86 – 3.69 (m, 1H), 3.65 – 3.42 (m, 4H), 3.38 – 3.32 (m, 2H), 2.91 – 2.73 (m, 3H), 2.71 (s, 3H), 2.43 (s, 3H), 2.31 (s, 3H), 1.73 – 1.63 (m, 3H). 13C NMR (101 MHz, MeOD-d4) δ 172.8, 172.8, 166.5, 156.9, 154.1, 152.4, 141.9, 139.2, 138.2, 137.8, 133.7, 133.3, 132.9, 132.2, 132.0, 131.4, 129.8, 128.2, 127.8, 126.6, 93.2, 81.0, 55.0, 46.3, 39.9, 38.5, 32.1, 21.0, 16.3, 14.4, 13.0, 11.6. HRMS (ESI/[M+H]+) Calcd for [C34H32ClN7O3S+H]+: 654.2049, found: 654.2031. HPLC purity: 95.6%.
(S)-2-(4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)-N-(5-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-2-methylphenyl)pent-4-yn-1-yl)acetamide (12C)
1H NMR (101 MHz, MeOD-d4) δ 7.51 – 7.44 (m, 2H), 7.44 – 7.29 (m, 3H), 7.25 – 7.15 (m, 2H), 4.69 (dd, J = 9.0, 5.3 Hz, 1H), 3.87 – 3.76 (m, 1H), 3.67 – 3.57 (m, 1H), 3.54 – 3.37 (m, 3H), 3.36 – 3.26 (m, 1H), 2.93 – 2.70 (m, 2H), 2.73 (s, 3H), 2.59 (t, J = 7.1 Hz, 2H), 2.45 (s, 3H), 2.32 (d, J = 11.6 Hz, 3H), 1.90 (p, J = 6.9 Hz, 2H), 1.73 – 1.67 (m, 3H). 13C NMR (101 MHz, MeOD-d4) δ 172.9, 172.7, 166.6, 156.9, 154.2, 152.5, 141.9, 139.0, 138.3, 137.7, 133.8, 133.4, 132.8, 132.2, 132.0, 131.5, 129.9, 128.0, 127.8, 126.8, 95.1, 80.5, 55.1, 46.3, 39.6, 38.5, 32.1, 29.7, 17.7, 16.3, 14.4, 12.9, 11.5. HRMS (ESI/[M+H]+) Calcd for [C35H34ClN7O3S+H]+: 668.2205, found: 668.2195. HPLC purity: 96.7%.
(S)-2-(4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)-N-(1-(2-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-2-methylphenoxy)acetyl)piperidin-4-yl)acetamide (13)
1H NMR (400 MHz, Acetone-d6) δ 7.53 – 7.38 (m, 4H), 7.22 – 7.10 (m, 1H), 7.04 – 6.85 (m, 2H), 4.98 – 4.76 (m, 2H), 4.68 – 4.57 (m, 1H), 4.41 – 4.20 (m, 1H), 4.12 – 3.79 (m, 3H), 3.77 – 3.59 (m, 1H), 3.43 – 3.16 (m, 3H), 2.89 – 2.72 (m, 3H), 2.61 (s, 3H), 2.45 (s, 3H), 2.15 (s, 3H), 2.01 – 1.78 (m, 2H), 1.70 (s, 3H), 1.62 – 1.34 (m, 2H). 13C NMR (101 MHz, Acetone-d6) δ 171.0, 170.1, 166.6, 164.2, 157.9, 156.5, 152.5, 150.6, 142.9, 138.3, 136.7, 133.6, 131.7, 131.2, 131.1, 131.0, 129.3, 127.5, 125.8, 120.5, 111.6, 68.4, 55.2, 47.1, 46.0, 44.5, 41.4, 39.1, 33.1, 32.6, 32.1, 14.5, 13.0, 11.8, 11.3. HRMS (ESI/[M+H]+) Calcd for [C37H39ClN8O5S+H]+: 743.2525, found: 743.2504. HPLC purity: 96.0%.
(S)-2-(4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)-N-(1-(2-(2-methyl-3-(3-methyl-2,4-dioxotetrahydropyrimidin-1(2H)-yl)phenoxy)acetyl)piperidin-4-yl)acetamide (13NT)
1H NMR (400 MHz, Acetone-d6) δ 7.52 – 7.45 (m, 2H), 7.44 – 7.38 (m, 2H), 7.21 – 7.13 (m, 1H), 6.98 – 6.88 (m, 2H), 5.02 – 4.73 (m, 2H), 4.69–4.57 (m, 1H), 4.31 (d, J = 11.3 Hz, 1H), 4.10 – 3.92 (m, 2H), 3.90 – 3.76 (m, 1H), 3.67 – 3.56 (m, 1H), 3.45 – 3.18 (m, 3H), 3.10 (s, 3H), 2.98 – 2.75 (m, 3H), 2.61 (s, 3H), 2.44 (s, 3H), 2.15 (s, 3H), 2.02 – 1.78 (m, 2H), 1.70 (s, 3H), 1.64 – 1.38 (m, 2H). 1H NMR (400 MHz, Acetone-d6) δ 170.3, 170.1, 166.6, 164.2, 157.9, 156.5, 153.6, 150.6, 143.6, 138.2, 136.7, 133.6, 131.7, 131.2, 131.1, 131.0, 129.3, 127.5, 125.7, 120.5, 111.6, 68.5, 55.2, 47.0, 44.8, 44.6, 41.5, 39.2, 32.9, 32.5, 32.2, 27.6, 14.5, 13.0, 11.8, 11.3. HRMS (ESI/[M+H]+) Calcd for [C38H41ClN8O5S+H]+: 757.2682, found: 757.2662. HPLC purity: 97.2%.
(S)-N-(2-(1-(2-(4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)acetyl)piperidin-4-yl)ethyl)-2-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-2-methylphenoxy)acetamide (14)
1H NMR (400 MHz, Acetone-d6) δ 9.19 (t, J = 4.3 Hz, 1H), 7.53 – 7.45 (m, 3H), 7.45 – 7.38 (m, 2H), 7.27 – 7.18 (m, 1H), 7.01 – 6.95 (m, 1H), 6.92 – 6.86 (m, 1H), 4.71 (t, J = 6.6 Hz, 1H), 4.56 – 4.45 (m, 3H), 4.23 – 4.15 (m, 1H), 3.95 – 3.83 (m, 1H), 3.70 – 3.55 (m, 2H), 3.48 – 3.37 (m, 1H), 3.40 – 3.31 (m, 2H), 3.16 – 3.03 (m, 1H), 2.89 – 2.69 (m, 2H), 2.61 (s, 3H), 2.58 – 2.47 (m, 1H), 2.44 (s, 3H), 2.16 (s, 3H), 1.92 – 1.41 (m, 8H), 1.23 (s, 1H), 1.13 – 0.93 (m, 1H). 13C NMR (101 MHz, Acetone-d6) δ 170.9, 168.9, 168.5, 163.9, 157.5, 156.7, 152.5, 150.4, 143.1, 138.4, 136.6, 133.6, 131.6, 131.2, 131.1, 131.0, 129.3, 127.7, 126.0, 121.0, 111.6, 68.9, 55.6, 46.5, 46.0, 42.6, 36.9, 36.9, 36.0, 34.5, 33.5, 32.7, 32.1, 14.5, 13.0, 11.7, 11.3. HRMS (ESI/[M+H]+) Calcd for [C39H43ClN8O5S+H]+: 771.2838, found: 771.2822. HPLC purity: 95.4%.
Supplementary Material
ACKNOWLEDGMENT
W.T. thanks the financial support from the University of Wisconsin– Madison Office of the Vice Chancellor for Research and Graduate Education with funding from the Wisconsin Alumni Research Foundation (WARF) through a UW2020 award before June 30, 2022 and National Institute of General Medical Sciences of the National Institutes of Health under award number R35GM148266. I.T. thanks NIH T32 GM141013 for the pre-doctoral fellowship. This study made use of the Medicinal Chemistry Center at UW-Madison instrumentation funded by the UW Lachman Institute for Pharmaceutical Development at the School of Pharmacy and WARF, and UW Carbone Cancer Center Flow Cytometry Core. We thank Eric Fischer’s lab from Dana-Farber Cancer Institute, Harvard University, for DNA plasmid: BRD4(BD1) subcloned into mammalian pcDNA5/FRT Vector (Ampicillin and Hygromycin B resistant) modified to contain MCS-eGFP-P2A-mCherry.
Funding Sources
NIH R35GM148266
NIH T32 GM141013
ABBREVIATIONS
- CRBN
cereblon
- TLC
thin layer chromatography
- PROTAC
proteolysis targeting chimeras
- PDHU
phenyl dihydrouracil
- MD
molecular dynamics
- BRD4
Bromodomain-containing protein 4
- BD1
bromodomain 1
- GFP
green fluorescent protein
- POI
protein of interest
- SAR
structure-activity relationship
- DDB1
DNA damage-binding protein 1
- DIPEA
N,N-diisopropylethylamine
- HATU
O-(7-Azabenzotriazol-1-yl)-N,N,N’,N’-tetramethyluronium hexafluorophosphate
Footnotes
Supporting Information. This material is available free of charge via the Internet.
Compound 6F binding poses modeling, human plasma, human liver microsome and hydrolysis stability, fluorescence polarization binding assays, HPLC traces, NMR spectra (PDF).
Molecular formula strings (CSV).
A patent application for some compounds in this manuscript was filed.
REFERENCES
- (1).Schapira M; Calabrese MF; Bullock AN; Crews CM Targeted Protein Degradation: Expanding the Toolbox. Nat. Rev. Drug Discov 2019, 18 (12), 949–963. 10.1038/s41573-019-0047-y. [DOI] [PubMed] [Google Scholar]
- (2).Luh LM; Scheib U; Juenemann K; Wortmann L; Brands M; Cromm PM Prey for the Proteasome: Targeted Protein Degradation—A Medicinal Chemist’s Perspective. Angew. Chem. Int. Ed 2020, 59 (36), 15448–15466. 10.1002/anie.202004310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Dale B; Cheng M; Park K-S; Kaniskan HÜ; Xiong Y; Jin J Advancing Targeted Protein Degradation for Cancer Therapy. Nat. Rev. Cancer 2021, 21 (10), 638–654. 10.1038/s41568-021-00365-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Mullard A Targeted Protein Degraders Crowd into the Clinic. Nat. Rev. Drug Discov 2021, 20 (4), 247–250. 10.1038/d41573-021-00052-4. [DOI] [PubMed] [Google Scholar]
- (5).Békés M; Langley DR; Crews CM PROTAC Targeted Protein Degraders: The Past Is Prologue. Nat. Rev. Drug Discov 2022, 21 (3), 181–200. 10.1038/s41573-021-00371-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Lai AC; Crews CM Induced Protein Degradation: An Emerging Drug Discovery Paradigm. Nat. Rev. Drug Discov 2017, 16 (2), 101–114. 10.1038/nrd.2016.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Sakamoto KM; Kim KB; Kumagai A; Mercurio F; Crews CM; Deshaies RJ Protacs: Chimeric Molecules That Target Proteins to the Skp1–Cullin–F Box Complex for Ubiquitination and Degradation. Proc. Natl. Acad. Sci 2001, 98 (15), 8554–8559. 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Lipinski CA Drug-like Properties and the Causes of Poor Solubility and Poor Permeability. J. Pharmacol. Toxicol. Methods 2000, 44 (1), 235–249. 10.1016/S1056-8719(00)00107-6. [DOI] [PubMed] [Google Scholar]
- (9).Lipinski CA Lead- and Drug-like Compounds: The Rule-of-Five Revolution. Drug Discov. Today Technol 2004, 1 (4), 337–341. 10.1016/j.ddtec.2004.11.007. [DOI] [PubMed] [Google Scholar]
- (10).Leeson PD; Springthorpe B The Influence of Drug-like Concepts on Decision-Making in Medicinal Chemistry. Nat. Rev. Drug Discov 2007, 6 (11), 881–890. 10.1038/nrd2445. [DOI] [PubMed] [Google Scholar]
- (11).Doak BC; Over B; Giordanetto F; Kihlberg J Oral Druggable Space beyond the Rule of 5: Insights from Drugs and Clinical Candidates. Chem. Biol 2014, 21 (9), 1115–1142. 10.1016/j.chembiol.2014.08.013. [DOI] [PubMed] [Google Scholar]
- (12).Wang C; Zhang Y; Wu Y; Xing D Developments of CRBN-Based PROTACs as Potential Therapeutic Agents. Eur. J. Med. Chem 2021, 225, 113749. 10.1016/j.ejmech.2021.113749. [DOI] [PubMed] [Google Scholar]
- (13).Winter GE; Buckley DL; Paulk J; Roberts JM; Souza A; Dhe-Paganon S; Bradner JE Phthalimide Conjugation as a Strategy for in Vivo Target Protein Degradation. Science 2015, 348 (6241), 1376–1381. 10.1126/science.aab1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Lu J; Qian Y; Altieri M; Dong H; Wang J; Raina K; Hines J; Winkler JD; Crew AP; Coleman K; Crews CM Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem. Biol 2015, 22 (6), 755–763. 10.1016/j.chembiol.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Eriksson T; Bjöurkman S; Roth B; Fyge Å; Höuglund P Stereospecific Determination, Chiral Inversion in Vitro and Pharmacokinetics in Humans of the Enantiomers of Thalidomide. Chirality 1995, 7 (1), 44–52. 10.1002/chir.530070109. [DOI] [PubMed] [Google Scholar]
- (16).Chamberlain PP; Lopez-Girona A; Miller K; Carmel G; Pagarigan B; Chie-Leon B; Rychak E; Corral LG; Ren YJ; Wang M; Riley M; Delker SL; Ito T; Ando H; Mori T; Hirano Y; Handa H; Hakoshima T; Daniel TO; Cathers BE Structure of the Human Cereblon–DDB1–Lenalidomide Complex Reveals Basis for Responsiveness to Thalidomide Analogs. Nat. Struct. Mol. Biol 2014, 21 (9), 803–809. 10.1038/nsmb.2874. [DOI] [PubMed] [Google Scholar]
- (17).Mori T; Ito T; Liu S; Ando H; Sakamoto S; Yamaguchi Y; Tokunaga E; Shibata N; Handa H; Hakoshima T Structural Basis of Thalidomide Enantiomer Binding to Cereblon. Sci. Rep 2018, 8 (1), 1294. 10.1038/s41598-018-19202-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Fischer ES; Böhm K; Lydeard JR; Yang H; Stadler MB; Cavadini S; Nagel J; Serluca F; Acker V; Lingaraju GM; Tichkule RB; Schebesta M; Forrester WC; Schirle M; Hassiepen U; Ottl J; Hild M; Beckwith REJ; Harper JW; Jenkins JL; Thomä NH Structure of the DDB1–CRBN E3 Ubiquitin Ligase in Complex with Thalidomide. Nature 2014, 512 (7512), 49. 10.1038/nature13527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).FDA’S Policy Statement for the Development of New Stereoisomeric Drugs. Chirality 1992, 4 (5), 338–340. 10.1002/chir.530040513. [DOI] [PubMed] [Google Scholar]
- (20).Kazantsev A; Krasavin M Ligands for Cereblon: 2017–2021 Patent Overview. Expert Opin. Ther. Pat 2022, 32 (2), 171–190. 10.1080/13543776.2022.1999415. [DOI] [PubMed] [Google Scholar]
- (21).Jacques V; Czarnik AW; Judge TM; Van der Ploeg LHT; DeWitt SH Differentiation of Antiinflammatory and Antitumorigenic Properties of Stabilized Enantiomers of Thalidomide Analogs. Proc. Natl. Acad. Sci 2015, 112 (12), E1471–E1479. 10.1073/pnas.1417832112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Hartmann MD; Boichenko I; Coles M; Zanini F; Lupas AN; Hernandez Alvarez B Thalidomide Mimics Uridine Binding to an Aromatic Cage in Cereblon. J. Struct. Biol 2014, 188 (3), 225–232. 10.1016/j.jsb.2014.10.010. [DOI] [PubMed] [Google Scholar]
- (23).Boichenko I; Bär K; Deiss S; Heim C; Albrecht R; Lupas AN; Hernandez Alvarez B; Hartmann MD Chemical Ligand Space of Cereblon. ACS Omega 2018, 3 (9), 11163–11171. 10.1021/acsomega.8b00959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Sosič I; Bricelj A; Steinebach C E3 Ligase Ligand Chemistries: From Building Blocks to Protein Degraders. Chem. Soc. Rev 2022. 10.1039/D2CS00148A. [DOI] [PubMed] [Google Scholar]
- (25).Yamamoto T; Tokunaga E; Nakamura S; Shibata N; Toru T Synthesis and Configurational Stability of (S)- and (R)-Deuteriothalidomides. Chem. Pharm. Bull. (Tokyo) 2010, 58 (1), 110–112. 10.1248/cpb.58.110. [DOI] [PubMed] [Google Scholar]
- (26).Hansen JD; Correa M; Nagy MA; Alexander M; Plantevin V; Grant V; Whitefield B; Huang D; Kercher T; Harris R; Narla RK; Leisten J; Tang Y; Moghaddam M; Ebinger K; Piccotti J; Havens CG; Cathers B; Carmichael J; Daniel T; Vessey R; Hamann LG; Leftheris K; Mendy D; Baculi F; LeBrun LA; Khambatta G; Lopez-Girona A Discovery of CRBN E3 Ligase Modulator CC-92480 for the Treatment of Relapsed and Refractory Multiple Myeloma. J. Med. Chem 2020, 63 (13), 6648–6676. 10.1021/acs.jmedchem.9b01928. [DOI] [PubMed] [Google Scholar]
- (27).Nishimura K; Hashimoto Y; Iwasaki S (S)-Form of α-Methyl-N(α)-Phthalimidoglutarimide, But not Its (R)-Form, Enhanced Phorbol Ester-Induced Tumor Necrosis Factor-α Production by Human Leukemia Cell HL-60 : Important of Optical Resolution of Thalidomidal Effects. Chem. Pharm. Bull. (Tokyo) 1994, 42 (5), 1157–1159. 10.1248/cpb.42.1157. [DOI] [PubMed] [Google Scholar]
- (28).Min J; Mayasundari A; Keramatnia F; Jonchere B; Yang SW; Jarusiewicz J; Actis M; Das S; Young B; Slavish J; Yang L; Li Y; Fu X; Garrett SH; Yun M-K; Li Z; Nithianantham S; Chai S; Chen T; Shelat A; Lee RE; Nishiguchi G; White SW; Roussel MF; Potts PR; Fischer M; Rankovic Z Phenyl-Glutarimides: Alternative Cereblon Binders for the Design of PROTACs. Angew. Chem. Int. Ed 2021, 60 (51), 26663–26670. 10.1002/anie.202108848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Fromme JC; Verdine GL DNA Lesion Recognition by the Bacterial Repair Enzyme MutM*. J. Biol. Chem 2003, 278 (51), 51543–51548. 10.1074/jbc.M307768200. [DOI] [PubMed] [Google Scholar]
- (30).Barreiro EJ; Kümmerle AE; Fraga CAM The Methylation Effect in Medicinal Chemistry. Chem. Rev 2011, 111 (9), 5215–5246. 10.1021/cr200060g. [DOI] [PubMed] [Google Scholar]
- (31).Leung CS; Leung SSF; Tirado-Rives J; Jorgensen WL Methyl Effects on Protein–Ligand Binding. J. Med. Chem 2012, 55 (9), 4489–4500. 10.1021/jm3003697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Liao J; Nie X; Unarta IC; Ericksen SS; Tang W In Silico Modeling and Scoring of PROTAC-Mediated Ternary Complex Poses. J. Med. Chem 2022, 65 (8), 6116–6132. 10.1021/acs.jmedchem.1c02155. [DOI] [PubMed] [Google Scholar]
- (33).Schumacher H; Smith RL; Williams RT The Metabolism of Thalidomide: The Spontaneous Hydrolysis of Thalidomide in Solution. Br. J. Pharmacol. Chemother 1965, 25 (2), 324–337. 10.1111/j.1476-5381.1965.tb02053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Franks ME; Macpherson GR; Figg WD Thalidomide. The Lancet 2004, 363 (9423), 1802–1811. 10.1016/S0140-6736(04)16308-3. [DOI] [PubMed] [Google Scholar]
- (35).Nowak RP; DeAngelo SL; Buckley D; He Z; Donovan KA; An J; Safaee N; Jedrychowski MP; Ponthier CM; Ishoey M; Zhang T; Mancias JD; Gray NS; Bradner JE; Fischer ES Plasticity in Binding Confers Selectivity in Ligand-Induced Protein Degradation. Nat. Chem. Biol 2018, 14 (7), 706–714. 10.1038/s41589-018-0055-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Douglass EF; Miller CJ; Sparer G; Shapiro H; Spiegel DA A Comprehensive Mathematical Model for Three-Body Binding Equilibria. J. Am. Chem. Soc 2013, 135 (16), 6092–6099. 10.1021/ja311795d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Nawrocki ST; Griffin P; Kelly KR; Carew JS MLN4924: A Novel First-in-Class Inhibitor of NEDD8-Activating Enzyme for Cancer Therapy. Expert Opin. Investig. Drugs 2012, 21 (10), 1563–1573. 10.1517/13543784.2012.707192. [DOI] [PubMed] [Google Scholar]
- (38).Baltrushis RS; Beresnevichyus Z-IG; Vizgaitis IM 1-Phenyl-5(6)-Methyldihydrouracils and Their Transformations. Chem. Heterocycl. Compd 1981, 17 (8), 816–820. 10.1007/BF00503666. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.