

Abstract
Ena/VASP proteins are processive actin polymerases that are required throughout animal phylogeny for many morphogenetic processes, including axon growth and guidance. Here we use in vivo live imaging of morphology and actin distribution to determine the role of Ena in promoting the growth of the TSM1 axon of the Drosophila wing. Altering Ena activity causes stalling and misrouting of TSM1. Our data show that Ena has a substantial impact on filopodial morphology in this growth cone but exerts only modest effects on actin distribution. This is in contrast to the main regulator of Ena, Abl tyrosine kinase, which was shown previously to have profound effects on actin and only mild effects on TSM1 growth cone morphology. We interpret these data as suggesting that the primary role of Ena in this axon may be to link actin to the morphogenetic processes of the plasma membrane, rather than to regulate actin organization itself. These data also suggest that a key role of Ena, acting downstream of Abl, may be to maintain consistent organization and reliable evolution of growth cone structure, even as Abl activity varies in response to guidance cues in the environment.
INTRODUCTION
As a nervous system develops, each neuron extends an axon to connect with downstream elements of its neural circuit. In this process, the path followed by each growing axon is directed by chemical and physical cues in its extracellular environment (Tessier-Lavigne and Goodman, 1996; Dickson, 2002). These cues are detected and processed, in large part, in a sensory and motile structure at the tip of the growing axon, called the growth cone (Lowery and Van Vactor, 2009; Stoeckli, 2018; Dumoulin et al., 2021). One of the key cytoplasmic signaling mechanisms that integrate signals in the growth cone is the Abl protein tyrosine kinase and its associated signaling network (Lanier and Gertler, 2000; Moresco and Koleske, 2003; Bradley and Koleske, 2009). Abl is a key element downstream of many of the common, conserved cell surface receptors that direct axon growth and guidance in organisms across the animal kingdom, including DCC, Robo, Eph family receptors, Plexins, and integrins (Yu et al., 2001; Hsouna et al., 2003; Forsthoefel et al., 2005; Garbe et al., 2007; Deinhardt et al., 2011; Grossman et al., 2013). Abl is also an upstream regulator of many aspects of cytoskeletal organization, including polymerization, branching, bundling, severing, and contractility of actin networks, through processes that are mediated by Ena/VASP proteins, Arp2/3, cofilin, Myosin II, and other actin regulatory factors (Lanier and Gertler, 2000; Bradley and Koleske, 2009). This makes Abl a uniquely informative tool for dissecting the molecular mechanisms by which external signals generate neuronal morphology and connectivity.
One excellent system to connect Abl signaling to cytoskeletal dynamics and growth cone motility in vivo is provided by simultaneous live imaging of fluorescent markers for actin and plasma membrane in the extending TSM1 sensory axon of the developing Drosophila wing (Clarke et al., 2020a, 2020b). Previous studies have revealed, first, that the TSM1 growth cone is a protrusive filopodial domain near the leading tip of the axon. The position of this domain is determined by the presence of a localized mass of actin in this portion of the axon shaft, which provides the raw materials that make up and maintain the growth cone filopodia. That actin mass undergoes constant stochastic fluctuations in length, but with a spatial bias that progressively advances the position of the actin mass over time. Forward motion of the actin, however, necessarily also advances the region that is capable of supporting a high density of filopodia; that is, it advances the morphological structure that we recognize as the growth cone. Those experiments further revealed that the role of Abl kinase is to coordinate the stochastically fluctuating expansion and compaction of the actin network, maintaining the actin as a coherent mass and promoting its orderly net advance down the axon toward the axon tip. Specifically, increased Abl activity causes net expansion of the actin mass, while decreased Abl causes net compaction. Thus, a pattern of guidance cues that generates a gradient of Abl activity across the length of a growth cone could, in principle, cause preferential expansion of actin at the leading edge of the growth cone and compaction at the trailing edge. This would produce a net advance of the actin mass over time, and therefore a net advance of the morphological growth cone and growth of the axon.
While experiments to date have characterized the effect of Abl on the spatial distribution of actin in the TSM1 growth cone, it is now essential to dissect the molecular mechanism downstream of Abl by which the effects of the kinase control the distribution of actin. The best characterized, and most direct, effector linking Abl to actin dynamics is the actin polymerase Enabled (Ena; Gertler et al., 1995; Krause et al., 2003). Ena promotes growth of actin filaments, both as a processive polymerase that juxtaposes G-actin monomers to the barbed ends of F-actin filaments (Winkelman et al., 2014), and also by antagonizing the binding of actin filament–capping proteins (Bear and Gertler, 2009; Gates et al., 2009). Ena also bundles the tips of actin filaments, in part by forming tetramers that link adjacent filaments (Blanchoin et al., 2014; Bruhmann et al., 2017). Ena has profound effects on cell morphology. Overexpression of Ena stimulates formation of filopodia in many systems, though the mechanism by which it does so is complex and context-dependent (Krause et al., 2002; Lebrand et al., 2004; Gates et al., 2007; Trichet et al., 2008). For example, the properties of filopodia induced by Ena alone can be rather different from those produced in conjunction with formins, such as Drosophila Diaphanous (Homem and Peifer, 2009; Bilancia et al., 2014). Moreover, in some contexts Ena can promote or stabilize lamellipodia, in part by extending the actin filaments that line the leading edge of such structures (Rottner et al., 1999; Lacayo et al., 2007). The effects of Ena on morphogenesis are not determined solely by its direct effects on actin, however. Ena proteins have a conserved EVH1 domain that binds the peptide motif FPPPP (FP4), which is commonly found in adhesive structures, such as focal adhesions (Bear et al., 2000). Ena can therefore link the actin cytoskeleton to the plasma membrane. Ena is present in axons and has demonstrated effects on axon growth and guidance in vitro and in vivo (Wills et al., 1999; Lebrand et al., 2004; Kuzina et al., 2011). The molecular mechanisms underlying those effects have so far been enigmatic, however (Krause et al., 2002). First described in Drosophila, Ena has close orthologs in Caenorhabditis elegans (UNC-34; Sheffield et al., 2007; Fleming et al., 2010) and in mammals (MENA, VASP [vasodilator-stimulated phosphoprotein], and EVL [Ena- and VASP-like]) that have similar properties (Gertler et al., 1996). Ena was first identified as a genetic antagonist of Abl, in that the phenotypes of Drosophila Abl mutants can be suppressed by reducing the gene dosage of ena (Gertler et al., 1995), and at least some phenotypes of Abl mutant Drosophila seem to be produced by mislocalization and/or hyperactivity of Ena protein (Grevengoed et al., 2001, 2003; Kannan et al., 2014). Genetic tests show that ena acts downstream of Abl, and consistent with this, Abl regulates Ena activity, in part, by phosphorylation of conserved tyrosine residues of Ena protein (Comer et al., 1998). This cannot be the entire mechanism by which Abl regulates Ena, however, as an Ena derivative lacking these tyrosines still retains significant activity to perform its Abl-dependent functions in Drosophila (Comer et al., 1998).
Here we perform live imaging of axon morphology and actin distribution in TSM1 axons growing in their native environment of the developing Drosophila wing, both in wild type (WT), and upon increase or suppression of Ena activity in the neuron. We find that altering Ena activity has a substantial effect on filopodial number and length, but that the TSM1 growth cone is far more sensitive to reduction of Ena from its WT level than it is to increase of Ena. The effects of Ena on the distribution of actin are quantitatively much more modest than those on filopodial morphology, though a sensitive analytical method reveals that reducing Ena activity tends to broaden the actin distribution relative to higher levels of Ena. This is consistent with our previously published analysis of the effects of Abl in TSM1, but it is striking that Abl had a far more pronounced effect on actin than on morphology, opposite to our observations here of the consequences of altering Ena. Moreover, as for Abl, we find that either increasing or decreasing Ena activity causes the temporal evolution of the actin distribution to be less orderly and predictable in individual axons than that we observe in WT. Together, these data suggest that the main role of Ena in the TSM1 growth cone may not be to regulate the actin distribution itself, but rather to modify the linkage of that actin to morphogenetic processes of the plasma membrane. It also suggests that a key function of Ena may be to buffer the downstream consequences of altering Abl activity, thus maintaining the growth cone in optimal organization for orderly movement while still allowing the cue-directed modulation of Abl activity, and thus actin dynamics (Chandrasekaran et al., 2022a, 2022b), that is necessary to produce guided axon growth.
RESULTS
We performed live imaging of the TSM 1 axon as it grows through the Drosophila wing, much as we have described previously (Clarke et al., 2020a, 2020b; Figure 1A; Supplemental Figure 1). Wing imaginal discs were dissected ∼9 h after the onset of metamorphosis (APF: after puparium formation), mounted in culture media, and imaged for 90 min by collecting z-stacks using spinning disk confocal microscopy (interframe interval = 3 min). Membrane and actin distribution, respectively, were visualized by coexpression of CD4-td-Tomato and LifeAct-eGFP, both under control of neuralized-GAL4 (neur-GAL4). Axon morphology was traced in three dimensions and growth cone parameters were quantified as described previously (Clarke et al., 2020a, 2020b), including both morphological features, and actin distribution along the axon shaft (measured by radial integration of LifeAct intensity as a function of position along the axon; Figure 1, B–D; measured parameters are listed in Figure 1C, shown schematically in Figure 1D, and described in detail in Figure 1 legend and in Materials and Methods). Note that, for consistency, all projections from the axon will be referred to below as “filopodia” irrespective of their caliber, lifetime, or potential invasion by microtubules.
FIGURE 1.

Anatomy and measured parameters of TSM1. (A) Anatomy of TSM1. z-projections of wing discs fixed and dissected at the indicated times. Neuronal membranes were labeled with anti-HRP (8 h APF image) or by expression of CD4-tdTomato (20hr APF image). Position of TSM1 cell body is indicated by white arrow; scale bar is at the bottom of each image. (B) Time course of development of a typical WT TSM1 axon, with membrane and actin, labeled, respectively, by expression of td-Tomato and LifeAct-eGFP. z-projections are shown of image stacks collected at the indicated times by spinning disk confocal microscopy. Yellow arrows in the membrane images indicate a fixed position in the disk for comparison of axon length. Scale bar is indicated in the 18′ image. (C) List of parameters measured at all time points of all trajectories imaged. See Materials and Methods for details. (D) For illustrative purposes, example of some of the quantified parameters from a single time point, taken from the axon whose membrane and LifeAct channels are shown beneath. Blue line shows the distribution of integrated LifeAct fluorescence intensity along the axon, expressed in arbitrary units (a.u.); orange bars show the number of filopodial branches arising from the axon shaft in 5-μm windows along the axon. Note that alignment with the image below is not exact, as length is measured in three-dimensional space, not in projection, and as some filopodia are not visible at this projection angle. Positions of the peaks of actin and of filopodial projections were identified using a 5-μm sliding window and are indicated with arrows. The “position” of the peak is defined as the midpoint of the window that has the highest integrated actin intensity or filopodial density, respectively. The “lengths” of the peak zones of actin and of filopodia were calculated as the square root of the second moment of each distribution about its peak position, a measure corresponding essentially to ±1 SD (brackets). See Materials and Methods and Clarke et al. (2020a) for detailed explanation of the utility of this definition.
Properties and dynamics of the wild-type TSM1 growth cone
Live imaging of TSM1 revealed the growth cone to be a domain of three-dimensional filopodial protrusiveness with only very rare indications of lamellipodial structures. As reported previously for TSM1 (Clarke et al., 2020a), visual inspection of the movies showed no obvious signs of significant substratum adhesion. For example, filopodia were splayed as broadly on the z-axis as they were on the x- and y-axes, both the axon shaft and individual filopodia were commonly observed to shift laterally in position relative to the substratum between time points, and the tips of individual filopodia, including the highly dynamic leading filopodia, showed substantial fluctuation in position between time points, suggesting that they are not strongly anchored to the substratum. In addition, we could not detect any consistent relationship between localization of actin in the growth cone and localization of focal adhesion proteins, such as zyxin (A. Clarke and EG, unpublished data). We did find, however, that the protrusive region of the axon, the morphological “growth cone,” contained a high local concentration of actin intensity in the axon shaft, such that the position of the window containing the peak density of filopodial protrusions (here called “the filopodial peak”) correlated roughly with the position of the window containing the maximum integrated amount of total actin intensity (here called “the actin peak”; Figure 2A; R = 0.7; p < 10-4; Spearman correlation). When examined in detail, however, we found that the positions of the filopodial peak and the actin peak, while correlated, are not coincident. Rather, the position of the peak of filopodial density tended to lag significantly behind the peak of actin intensity (by 5.2 ± 0.7 μm; mean ± SEM; median = 1.1 μm, p < 0.0001; Wilcoxon signed rank; Figure 2B). Moreover, the magnitude of this offset between the positions of the actin and filopodial peaks in any given image correlated with the amount by which each then advanced in the next frame. Thus, for example, when the actin peak led the filopodial peak by a large amount in one image, we tended to observe greater advance of the filopodial peak in the subsequent image (Figure 2C) but less advance of the actin distribution or even its regression in that following image (Figure 2D). As discussed elsewhere, this is consistent with data published previously suggesting that progressive advance of the protrusive filopodial domain over time occurs as a response to the change in the local availability of actin as the actin mass moves forward down the axon (Goncalves-Pimentel et al., 2011; Clarke et al., 2020a).
FIGURE 2:

Dynamics of the growth of WT axons. (A) Scatterplot of the positions of the peak of filopodial density (“filopodial peak”) vs. the actin peak for the entire WT dataset. Note that many datapoints are concentrated along the line x = y (dashed black line); that is, positions of the actin and filopodial peaks correspond closely for many time points. However, it is clear by inspection that the filopodial peak lags significantly behind the actin peak at a substantial number of time points; that is, off-line datapoints have a pronounced tendency to lie well below the line, rather than above it. (B) Tabulated offset between the positions of the peaks of actin and filopodia (in micrometers). Positive values are time points where the actin peak leads the filopodial peak. Median and interquartile range are shown; note that median > 0, and that the distribution is skewed heavily to larger positive than negative values. (C) Scatterplot of the motion of the filopodial peak (in micrometers) in the time interval t → t+1 vs. the offset between actin and filopodial peaks at time t. Spearman r = 0.34; p < 0.0001. (D) Scatterplot of the motion of the actin peak (in micrometers) in the time interval t → t+1 vs. the offset between actin and filopodial peaks at time t. Spearman r = –0.26; p < 0.0001. (E) Graph of the positions of the actin peak (blue) and of the trailing (purple) and leading (green) “edges” of the actin peak as a function of time in a typical WT trajectory. Trailing and leading edge positions were calculated as, respectively, the square roots of the trailing and leading partial second moments of the actin distribution. These are indicated as dashed lines to emphasize that they are statistical measures, not discrete axonal features. Positions are given in micrometers; times in minutes. Time axis increases downward. Note the inconsistent nature of actin advance, with the peak position wriggling back and forth within the window that defines the actin mass at any given time. (F) Length of the actin peak (square root of the second moment of the distribution) as a function of time in the same trajectory shown in E. Note the fluctuating nature of the length of the actin peak. The peak length graphed here, and used in all statistical analyses below, is based on the global second moment, whereas the positions graphed in E are calculated from the separate trailing and leading partial second moments. In general, the global second moment does not equal the sum of the partial second moments because of how these properties are calculated. The global moment is more appropriate for further statistical analyses, while the partial moments are more informative for aligning to visible features of the axon. (G) Scatterplot of the offset between the positions of the actin and filopodial peaks vs. the length of the actin peak. Spearman r = 0.25; p < 0.0001.
Detailed examination of actin distribution in the TSM1 growth cone revealed that the mass of actin undergoes constant, seemingly stochastic fluctuations in position, but with a small, yet persistent forward bias that produced a net advance of the actin distribution over time (Figure 2E). Thus, while the position of peak actin intensity took a significant number of steps both forward and backward in any given trajectory, and these could be of roughly comparable magnitude, the net effect over the course of imaging was that the peak position of actin intensity preferentially moved forward along its trajectory. These fluctuations in peak position were also associated with fluctuations in the spatial extent of the actin peak along the axon (the “length” of the actin peak; Figure 2F [compare Figure 1D]), suggestive of the actin “inchworming” forward over time as it moved forward in the axon shaft. In essence, a combination of preferential forward expansion of the leading edge of the actin mass, together with preferential compaction from the rear, causes the length of the actin mass to fluctuate around a mean, but with net forward motion of the mass as a whole. Together with other experiments (Clarke et al., 2020a, 2020b), these observations demonstrate that TSM1 growth cone advance is correlated with forward-biased fluctuations of the actin distribution. This interpretation is also consistent with the observation that the magnitude of the offset between the peaks of actin vs filopodial density in any given image correlated with the length of the actin peak (Figure 2G; see also Clarke et al., 2020a, 2020b for further discussion of this hypothesis).
Overall morphological phenotypes from enabled gain and loss of function
We next determined the effect produced on TSM1 morphology and motility when we altered the activity of Ena in the neuron by taking advantage of the yeast transcriptional activator GAL4, expressed under control of regulatory sequences of the gene neuralized (neur-GAL4). Ena activity was either increased, by expressing a UASG-ena(WT) transgene, or suppressed, by sequestering Ena protein to mitochondria using expression of a UASG-FP4-mito transgene. FP4-mito has an Ena binding motif, including the sequence FPPPP, linked to a mitochondrial targeting sequence. It has been validated extensively in multiple organisms and developmental contexts and shown to provide an effective (albeit not perfect) mimic of the ena genetic loss-of-function condition (Bear et al., 2000; Lebrand et al., 2004; Gates et al., 2007; Kuzina et al., 2011). Its use here allows us to inactivate Ena selectively in neural tissue and bypass the lethality of a genetic null mutant of ena.
We first verified the efficacy of our genetic reagents in manipulation of ena activity in TSM1. To validate the effectiveness of the UAS-ena transgene for producing a gain-of-function phenotype when expressed with neur-GAL4, we needed a well characterized ena overexpression phenotype that could be assayed in neurons in vivo. In Drosophila photoreceptors, Ena protein is associated with the cis-Golgi and overexpression of Ena leads to clustering of the protein and of the Golgi and their relocalization to the basal part of the cell body (Kannan et al., 2014). Consistent with this, expression of UASG-ena(WT) under control of neur-GAL4 produced a similar pattern of clustering and relocalization of Ena in photoreceptors, demonstrating that expression of the transgene is effective for enhancing Ena activity (Figure 3B; compare control in Figure 3A; see also Supplemental Figure 2, A and B). Similarly, we wished to verify the effectiveness of the FP4-mito-eGFP transgene for producing an ena loss-of-function phenotype. Reducing ena activity has relatively mild effects on overall axon patterning in the Drosophila embryo (Gates et al., 2007), but it produces a specific and well characterized misrouting of motor axons of the “b” branch of the intersegmental nerve (ISNb) in the embryo, whereby these axons fail to form a separate fascicle and instead remain part of the main ISN (called a “bypass” phenotype; Wills et al., 1999). We therefore examined ISNb targeting in embryonic neurons expressing UASG-FP4-mito under the control of neur-GAL4 and observed ISNb axons following the aberrant “bypass” trajectory that is typical of ena mutants, validating the activity of this reagent in Drosophila neurons (Figure 3D; compare control in Figure 3C). To verify that these transgenes are expressed effectively specifically in TSM1 when placed under control of neur-GAL, we performed anti-Ena antibody staining of early prepupal wing discs expressing UASG-ena(WT) or UASG-FP4-mito-eGFP under the control of neur-GAL4. As expected, in wings expressing UAS-ena under control of neur-GAL4, we observed enhanced Ena protein immunoreactivity in wing margin sense organs in general, and TSM1 in particular (Figure 3F; compare control in Figure 3E). In contrast, upon expression of UAS-FP4-mito-eGFP with neur-GAL4, the endogenous Ena protein (visualized by anti-Ena immunoreactivity) became concentrated in small, dense puncta that colocalize with mitochondria in TSM1, as identified by the localization of the FP4-mito-eGFP protein itself. Such puncta of Ena immunoreactivity were not observed in control wings (Figure 3I; compare control in Figure 3H). Finally, the eponymous mutant phenotype of enabled is its suppression of mutant phenotypes observed in Abl loss of function. We therefore compared the frequency of defects in the mature trajectory of TSM1 in wings of flies expressing RNAi against Abl (under control of neur-GAL4) with that in Abl RNAi flies that coexpress UASG-FP4-mito. Expression of Abl RNAi in TSM1 causes a variety of axonal defects, primarily axon stalling (Clarke et al., 2020a). We found that suppression of Ena activity with FP4-mito reduced the frequency of Abl RNAi-induced TSM1 defects by nearly 50% (19% vs. 37%; p = 0.009; χ2; Figure 3M). Typical examples of the TSM1 axonal phenotype under these conditions are shown in Figure 3, J–L, and the phenotype is quantified in Figure 3M. These data verify that expressing FP4-mito suppresses the Abl loss-of-function axonal phenotype in TSM1.
FIGURE 3:

Validation of transgenes for manipulating Ena activity. (A, B) Anti-Ena immunolocalization of Ena protein in photoreceptor neurons of fixed third instar larva. A, control (neur-GAL4). Ena protein immunoreactivity (green) can be seen in small, widely distributed puncta (cis-Golgi compartment; Kannan et al., 2014) and along cell boundaries. B, neur-GAL4; UAS-ena. Upon overexpression of Ena, the Golgi compartment becomes concentrated in the basal portion of the photoreceptor cell body and recruits much of the endogenous Ena protein from the rest of the cell (Kannan et al., 2014). Coexpressed CD4-td-Tomato channel is shown (red) to facilitate visualization of neuronal membrane. Scale bar in panel B. (C, D) Anti-Fasciclin 2 immunostaining of fixed stage 17 embryos to visualize peripheral nerves. C, control (neur-GAL4). ISNb nerve is visible (brown DAB reactivity; arrow), associated with a layer of internal muscles. ISN is in a deeper focal plane (out of focus; dashed arrow). D, neur-GAL4; UAS-FP4-mito-eGFP-expressing motoneurons display the ISNb axon misrouting typical of ena loss-of-function. ISNb projections are not observed in the ventrolateral target zone (outlined with dashed oval), but rather, the axons remain associated with ISN in a deep plane of focus (arrow) and fail to arborize on muscles. Scale bar in panel C. (E–I) Anti-Enabled antibody staining of fixed wing imaginal disk at 8 h APF. E and F are single optical slices; G–I are z-projected image stacks (scale bars in panels G, H). E, control (neur-GAL4). Ena immunoreactivity in green. F, neur-GAL4; UAS-ena. Signal from overexpressed Ena protein is clearly visible above the endogenous background of Ena immunoreactivity in sense organ cells along the wing margin, and specifically in the TSM1 cell body (arrow). G, neur-GAL4, td-Tomato; UAS-ena, showing a projected z-stack of TSM1 cell body, axon, and growth cone. Green channel: anti-Ena; red channel: CD4-td-Tomato (membrane marker). In a fraction of filopodia, Enabled immunoreactivity becomes concentrated at the filopodial tip (yellow arrow), but more commonly, Ena signal is the same or weaker at the tip than in the rest of the filopodium (white arrows). Arrows are in the same position relative to the cell in both panels. Scale bar at lower left. H, control (neur-GAL4). Ena protein signal in magenta. TSM1 cell body is circled (identified by coexpression of CD4-td-Tomato; not shown). I, in neur-GAL4; UAS-FP4-mito-eGFP-expressing cells, endogenous Enabled protein colocalizes with mitochondria (green puncta, visualized with FP4-mito-eGFP). TSM1 cell body is circled. Compare anti-Enabled signal (magenta, I) with mitochondrial signal (green, I’) and overlay (I”). Scale bar in panel E. (J–L) Suppression of the Abl loss-of-function axonal phenotype upon coexpression of FP4-mito-eGFP. Anti-HRP immunostaining of fixed pupal wing (20 h APF) to visualize the mature axonal projection of TSM1 and of the L1 and L3 peripheral nerves. J, control (neur-GAL4). TSM1 and L3 nerve are indicated; scale bar is shown. K, neur-GAL4, UAS-Abl RNAi. Note stalled TSM1 growth cone (yellow arrow). L, neur-GAL4, UAS-Abl RNAi, UAS-FP4-mito-eGFP. L1 nerve is restored (white arrow) and grows to fasciculate with L3 nerve. Note that neur-GAL4, UAS-Abl RNAi flies were bred to contain an additional UAS-transgene (UAS-CD4-td-Tomato, not shown) to ensure that affected and rescued genotypes carried equal numbers of UAS transgenes in their genome. (M) Quantification of L1 nerve defects in Abl RNAi with and without expression of FP4-mito-eGFP. Frequency of defects in L1 nerve at 20 h APF is indicated by bar graph. Black numbers above bar give the frequency of defects (including stalls, misrouting, splitting, or absence of the nerve). White numbers at the base of the bar are the number of wings examined. p-values of comparisons are indicated (χ2 test). (N–P) Typical terminal phenotypes of TSM1/L1 nerve upon gain and loss of function of ena. Anti-HRP immunostaining of 20 h APF wings of the indicated genotypes. N, control (neur-GAL4). TSM1 is indicated with white arrow. O, neur-GAL4; UAS-FP4-mito. Yellow arrow indicates stalled and branched TSM1 growth cone. P, neur-GAL4; UAS-ena. Yellow arrow indicates stalled and misrouted TSM1 axon. Note that, in WT, TSM1 fasciculates with its target zone on the L3 nerve by about 12 h APF. The first few wing margin follower axons have begun to traverse this trajectory by ∼19–20 h APF, though followers seem generally to be delayed somewhat in ena gain and loss of function. (Q) Quantification of TSM1 defects upon alteration of ena activity. Frequency of defects in L1 nerve at 20 h APF is indicated by bar graph. Black numbers above bar give the frequency of defects (including stalls, misrouting, or splitting of the nerve). White numbers at the base of the bar are the number of wings examined. p-values of comparisons are indicated (χ2 test). The frequency of TSM1 defects in UAS-FP4-mito is not significantly different from that in Abl RNAi; UAS-FP4-mito, above (p = 0.2; χ2 test).
It was also of interest to investigate the localization of Ena protein in the TSM1 growth cone. Unfortunately, anti-Ena immunostaining of fixed WT wing discs was not informative on this point, since the Ena immunoreactivity of the axon and growth cone was obscured by the high levels of Ena immunoreactivity in the closely associated epithelia of the wing (Supplemental Figure 2C). However, we were able to detect reliably the enhanced Ena immunoreactivity in TSM1 growth cones expressing UAS-ena (Figure 3G). Here we saw Ena signal throughout the growth cone above the level in the associated epithelia, both in the core of the growth cone and in filopodia. In contrast to what has been reported in other contexts, however (Lebrand et al., 2004; Gates et al., 2007), we usually did not observe evidence for accumulation of Ena at the tips of filopodia. Ena signal did not seem to be excluded from the distal parts of filopodia, but neither did we generally observe it concentrated there (Figure 3G). This may perhaps be related to the failure to see adhesive interactions of filopodial tips with the substratum (see above). We emphasize, however, that this analysis of Ena localization reports the distribution of the protein when it is overexpressed. We cannot comment on Ena subcellar distribution in wild-type TSM1 growth cones. Ena protein was also not discernable above the epithelial background in TSM1 growth cones expressing FP4-mito (Supplemental Figure 2D).
Visualizing the terminal phenotype of wings expressing Ena-modifying transgenes revealed that altering Ena activity disrupted the overall patterning of the TSM1 axon at only modest expressivity, just as has been observed for many embryonic axons (Gates et al., 2007). Expression of UAS-ena with neur-GAL4 caused terminal defects in 19% of wings examined, while expression of UAS-FP4-mito-eGFP caused defects in 28% (Figure 3, N–P; quantified in Figure 3Q; n ≥ 55 wings for each genotype). Most of these defects were failures of the axon to reach its target zone on the L3 nerve (termed “stalls”), often with neurites projecting from the tip of the stalled axon. In a small number of cases, other kinds of defects were observed, such as the axon projecting along an aberrant path, leading to fasciculation with L3 at a position distant from its normal target zone. The average rate of axon growth was not significantly different in the three genetic backgrounds (average growth rate = 0.21 ± 0.15 μm/min in WT vs. 0.13 ± 0.35 μm/min in UAS-FP4-mito and 0.18 ± 0.11 μm/min in UAS-ena; differences not significant: p = 0.42; this and all parameter values are presented in the text as mean ± SEM; see Statistical Methods for details of how statistical significance was calculated; Figure 4A). Examining the pattern of motility in greater detail revealed that the mode of growth cone movement under altered-Ena conditions resembled that of WT, displaying a stuttering, inconsistent pattern of advance, overlaid by stochastic fluctuation of the length of the actin mass in the growth cone, and with the actin peak tending to lead the peak of projection density (Figure 4, B, E–H; typical examples of growth cone morphology for neur-GAL4; UAS-FP4-mito-eGFP and neuro-GAL4; UAS-ena are shown in Figure 4, C and D, respectively).
FIGURE 4:

TSM1 axons with Ena loss- or gain-of-function grow similarly to WT axons. (A) Tabulated values of the motion of the actin peak between successive time points in all genotypes. Distances in micrometers; 3′ time steps for all trajectories. Median and interquartile ranges are shown. (B) Tabulated values of the offset between the positions of the actin and filopodial peaks at all time points of ena loss of function (UAS-FP4-mito) and gain of function (UAS-ena). Median and interquartile range are shown. Median offset is significantly positive for UAS-FP4-mito (p < 0.0001; Wilcoxon signed rank); for UAS-ena the median is not different from 0 by a statistically significant amount (p > 0.05), but interquartile range shows that the distribution trends toward positive values (actin peak leading branching peak). Compare with Figure 2B for the offset values in WT. (C) Morphology of a typical TSM1 growth cone in neur-GAL4; UAS-FP4-mito-eGFP. CD4-td-Tomato signal is shown, with scale bar. (D) Morphology of a typical TSM1 growth cone in neur-GAL4; UAS-ena. CD4-td-Tomato signal is shown, with scale bar. (E, F) Graph of the positions of the actin peak (blue) and of the trailing (purple) and leading (green) “edges” of the actin peak, as a function of time, in typical trajectories of actin loss- and gain-of-function, as described in Figure 2E. (G, H) Length of the actin peak as a function of time in the same trajectories shown in C, D. Compare with wt (Figure 2F).
Effects of Ena on actin distribution and filopodial pattern in TSM1
We next examined the effects of Ena on the detailed pattern of actin distribution and filopodial morphology in TSM1. Whereas we had found previously (Clarke et al., 2020a) that altering Abl kinase activity primarily modulates the distribution of actin, with only modest effects on growth cone morphology, we now found that the effects of manipulating Ena were opposite, primarily modifying morphology, with only modest effects on actin distribution (Figure 5). First, we found that the amount of active Ena correlated directly with the number and total length of filopodia but not with their average length or branching complexity, as we now describe. Neurons overexpressing Ena (UAS-ena) had 35.6 ± 2.1 filopodia, vs. 16.9 ± 0.7 filopodia for UAS-FP4-mito (mean ± SEM; p < 10-4; ANOVA; Figure 5A). Similarly, increasing Ena also enhanced the total length of filopodial projections per axon (254.5 ± 18.1 μm for UAS-ena vs. 145.9 ± 7.6 μm in UAS-FP4-mito; p < 10-4; ANOVA; Figure 5B). For each of these parameters, comparison to the WT reveals that the neuron was far more sensitive to reduction of Ena activity than to its increase, with reduction accounting for 85% of the difference in mean filopodial number between Ena overexpression and FP4-mito mediated suppression, and essentially 100% of the difference in total filopodial length (filopodial number in WT = 30.7 ± 2.9; total filopodial length = 256.2 ± 26.9). Perhaps surprisingly, average filopodial length was not reduced upon expression of UAS-FP4-mito, as the decrease in total filopodial length was in proportion to the decrease in filopodial number (average filopodial length 8.9 ± 0.4 μm in UAS-FP4-mito vs. 8.2 ± 0.1 in WT; difference not significant: p = 0.3; ANOVA; Figure 5C). In contrast, the combination of increased filopodial number without a corresponding increase in total filopodial length in the Ena-overexpressing condition manifested as a significant decrease in average filopodial length (7.2 ± 0.1 μm; p < 10-4 compared with WT; ANOVA). This observation is different from findings in other systems, a point we will come back to in the Discussion. Finally, in contrast to filopodial number and length, altering Ena activity did not change the complexity of filopodial branching, as the average projection order (primary, secondary, tertiary, etc.) was not altered by changes in ena activity (1.44, 1.42, and 1.46, respectively, for WT, UAS-FP4-mito, and UAS-ena; p = 0.62 across genotypes; ANOVA; Figure 5D).
FIGURE 5.

Comparison of single growth cone parameters in WT vs. altered-Ena conditions. Values of indicated growth cone parameters were tabulated for all three genotypes. Statistical significance of differences is as indicated. Error bars indicate mean and SD. Statistical significance is indicated as follows for this and subsequent figures : *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. Comparisons that are not marked were not formally significant. See Materials and Methods for details of how significance was assessed. (A) Total filopodial number. (B) Total filopodial length. (C) Average filopodial length. (D) Average filopodial order. (E) Length of actin peak (square root of the global second moment of the actin distribution). (F) Plot of the ratio of the average coefficient for each spatial order of the wavelet transform for UAS-FP4-mito/UAS-ena. Wavelet analysis quantifies the spatial structure of the actin distribution, with higher-order wavelets reflecting the frequency of local actin concentrations at short length scales, and lower-order wavelets reflecting spreading of actin density at longer length scales (see text and Materials and Methods, and also Clarke et al., 2020b for additional explanation). The maximum at wavelet order 5 indicates that expression of FP4-mito causes the actin distribution to be spread out significantly on a length scale peaking at 6.5–25.6 μm, relative to that in UAS-ena, consistent with broadening of the actin peak by reduction of Ena activity. Asterisks indicate the significance of the difference in coefficient values for the indicated order between these two genotypes. For comparison of each altered-Ena genotype with WT, and table listing the spatial range corresponding to each wavelet order, see Supplemental Figure 3. (G–I) For each trajectory of each genotype, the JSD of the shape of the actin distribution was calculated between the first time point vs each subsequent time point. Divergence can vary from 0 (identical distributions) to 1 (unrelated distributions). For clarity, plots are shown here for only three illustrative trajectories of each genotype. For plots of JSD vs. time for all trajectories, see Supplemental Figure 4. (J) Tabulation of the correlation of JSD vs. time for all trajectories of each genotype (Kendall τ correlation value). Black bar indicates median value; asterisks indicate significance of genotype differences as specified above.
We next quantified the effect of Ena on parameters of actin organization. One of the key actin features of the growth cone shown previously to be regulated by Abl is the length of the actin peak (Clarke et al., 2020a). Here, for Ena, we saw a trend toward a small shift in the expected direction, with suppression of Ena activity perhaps causing expansion of the actin peak relative to Ena overexpression (14.1 μm in UAS-FP4-mito vs. 11.9 μm in UAS-Ena; 95% confidence intervals 11.8–16.5 vs. 10.7–13.2; Figure 5E). This is consistent with the expansion of the actin peak seen upon overexpression of Abl, the Ena antagonist. As we found for the filopodial parameters, the majority of the difference between the two altered Ena conditions derived from the effect of Ena suppression (WT = 12.2 μm; 95% CI 8.8–16.0). Quantitatively, however, the apparent effect of Ena on the length of the actin peak was rather small in magnitude and did not achieve formal statistical significance. Therefore, to test this apparent trend, we performed a more sensitive analysis of the spatial distribution of actin along the axon, using wavelet analysis. The wavelet transform quantifies how LifeAct fluorescence intensity is distributed along the axon: for example, is it distributed homogeneously or is it clumped, and what is the spacing between concentrations of actin along the length of the axon? Thus, the presence of small, dense clumps of actin would give rise to increased values for the coefficients of high-order wavelets, while spreading of actin across large, multi-micrometer spatial scales would be reflected in enhanced values of lower-order wavelets (for a more detailed explanation of wavelets see Supplemental Methods and also Clarke et al., 2020b). It is important to note that the amplitude values of the wavelet transform derive from the entire actin distribution, across the whole length of the axon, for any given time point. It is not calculated from selected individual features of a distribution. In addition, wavelets are calculated separately for each of the time points for a given cell, and these can then be averaged together to yield a picture of the properties of that axon across the entire imaging session. Similarly, wavelet amplitudes can be averaged together from multiple cells to give a quantitative picture of the properties of a given genotype. Here we find that plotting the ratio of (wavelet amplitude)2 for (UAS-FP4-mito/UAS-ena) vs wavelet order reveals that reducing Ena activity leads to a significant enhancement of the contribution of a narrow range of wavelets, peaking at fifth order (p < 10-4; ANOVA), corresponding to separation of actin density at a spatial scale corresponding to ∼6.5–25.6 μm (Figure 5F; Supplemental Figure 3). Stated otherwise, the wavelet analysis shows that reducing Ena activity causes actin to spread out at this multi-micrometer scale of separation, consistent with the increased length of the actin peak observed by direct measurement as Ena activity is decreased, and also consistent with our earlier wavelet analysis of the Abl gain-of-function condition (Clarke et al., 2020b).
The other major feature of actin shown previously to be regulated by Abl in TSM1 is its degree of consistency, that is, the extent to which the distribution of actin observed at one time point predicts what the distribution will be at a subsequent time point. We quantify this feature with a property termed the Jensen–Shannon divergence. To calculate the divergence of the actin distribution between two time points, one compares the magnitude of the actin signal at each position along the axon at one time with the magnitude at the corresponding position at another time. If two distributions are identical, the divergence will be 0. If two distributions have no overlap, the divergence will be 1. This metric therefore gives a quantitative measure of how much the distribution of actin has changed between any two selected times for a given cell. Note that this is simply an unbiased comparison of the spatial distribution of actin intensity measured along the length of the axon. It does not incorporate any derived quantities or assumptions about growth cone features. For each of the WT trajectories we analyzed, we found that the divergence between the actin distribution at the start of imaging (t = 0) and the distribution at the next time point (t = 1) is relatively small, but that the divergence increases systematically as the starting actin distribution is compared with later and later time points of the same trajectory (Figure 5G). In contrast, if we alter Ena activity, either by suppression with FP4-mito or by overexpression of Ena, that predictability is degraded significantly (Figure 5, H–J; Supplemental Figure 4). There are still some trajectories under the altered Ena conditions that show consistent increase in divergence with time, but there are also trajectories where divergence is uniformly high, or changes with time in unpredictable ways. Thus, we see that the dynamic reliability of the evolution of the actin pattern is disrupted when we perturb the activity of Ena.
Analysis of the global effects of Ena on the TSM1 growth cone by principal components analysis
We know from our previous analysis of Abl that single growth-cone parameters with small individual responses to perturbation can nonetheless contribute to robust consequences because of consistent correlations among some growth cone features. We therefore expanded our analysis of Ena by querying the pairwise correlations of growth cone parameters, as well as examining the global effects of the whole set of growth cone features in an unbiased principal components analysis (PCA).
Correlations between individual pairs of growth cone parameters that were found to be significant across all three genetic conditions identified core features of a well-formed growth cone and of effective growth cone advance (Figure 6A; Supplemental Figure 5). Thus, for example, filopodial number, total filopodial length, and the filopodial branching complexity (average projection order) showed significant three-way correlation in all three genotypes, suggesting that this nexus reflects a consistent feature of growth cone cell biology (Figure 6B). Consistent correlation of these three features was also observed in our previous study of TSM1 (Clarke et al., 2020a). Moreover, as discussed above for WT, the offset between the actin and filopodial peaks also showed a significant correlation with the length of the actin mass in both altered-Ena genotypes, as well as a negative correlation between the magnitude of that offset in a given time point and the degree of advance of the actin in the following interval, both suggesting a stepwise, inchworming mode of axon growth. Also consistent with this, both altered-Ena conditions recapitulate the positive correlation of the offset between the positions of the filopodial and actin peaks in any given image vs advance of the filopodial distribution in the time step that followed (Figure 6, C and D), which was shown above to be a consistent feature of WT TSM1 axon growth (Figure 2C).
FIGURE 6:

Pairwise correlations of parameters among time points of each ena genotype. (A) Table of pairwise correlation of the seven parameters reported for each time point (Kendall τ correlation). Correlation significance was calculated by the Benjamini–Hochberg method. τ value is shown for all correlations with a false discovery rate (FDR) < 5%. τ values in blue—WT; red—UAS-FP4-mito (Ena loss-of-function); green—UAS-ena (Ena gain of function). For table of all τ values and associated p-values, see Supplemental Figure 5, A and B, respectively. (B) Filopodial number, length, and order were significantly correlated in all pairwise combinations in all three genotypes. τ values are shown. (C) Scatterplot of motion of the peak of filopodial density in a given time step vs. offset between the actin and filopodial peaks at the beginning of that time step for UAS-FP4-mito. Compare with WT (Figure 2C). Spearman r = 0.24; p < 0.0001. (D) Scatterplot of motion of the peak of filopodial density in a given time step vs. offset between the actin and filopodial peaks at the beginning of that time step for UAS-ena. Compare with WT (Figure 2C). Spearman r = 0.41; p < 0.0001.
Unbiased global analysis of the interactions among growth cone parameters by PCA yielded additional insight into axon structure and dynamics and how they are modulated by Ena (Figure 7, A–F). Our previous study of TSM1 revealed that WT growth cones could be classified into two related, but distinct, morphological classes, one with a simpler filopodial branching structure and the other more complex (Clarke et al., 2020a). The current WT dataset reproduces that effect. Thus, in the current WT dataset, morphological features of filopodia dominate PC1: filopodial number, total filopodial length, and filopodial branching complexity. Examination of the PCA reveals that the WT cells fall into a bimodal distribution of PC1 values, with the time points from three of the WT trajectories having PC1 values nearly exclusively less than -0.5 and the time points from the other seven trajectories almost entirely above that value (Figure 7, G and H), reproducing the categorical distinction into two classes that was observed previously. Suppression of Ena activity by expression of FP4-mito shifted the distribution completely to the simpler morph (higher values of PC1; Figure 7, D and I; Supplemental Figure 6A). In contrast, however, upon overexpression of Ena, while the mean value of PC1 did not change by a statistically significant amount, the distribution essentially collapsed to a single peak, erasing any evidence for a categorical distinction between two different morphological classes (Figure 7, E and J; Supplemental Figure 6A). The total range of possible PC1 values was nearly as broad as in the WT, but the most common morphology in UAS-ena was essentially intermediate between the two forms observed in the WT data. We also examined PC2, which was dominated by the length of the actin distribution, and to a lesser degree by the closely correlated length of the filopodial distribution. The three genotypes showed no significant difference in the means of the distributions of PC2 values (Supplemental Figure 6B), reinforcing the interpretation that Ena has limited effects on the actin distribution itself, in contrast to its strong effects on morphological features of TSM1.
FIGURE 7:

Principal component analysis (PCA) of all parameters for each ena genotype. (A, B) Fractional contribution to each of the first two principal components is shown for each measured parameter. (Contribution)2 is shown to facilitate comparison. (C–F) PCA was performed on WT using all seven parameters, and data from UAS-FP4-mito and UAS-ena datasets were then projected into the WT PCA space. The plane corresponding to PC 1 and 2 is shown. C–E show the three genotypes individually; F shows the overlay of all three genotypes. (G) PCA of WT is shown with each imaged cell colored separately. Note separation of trajectories into a cluster of three cells with PC1 less than approximately -0.5, and a second cluster with PC1 greater than that value. (H–J) Histogram of PC1 values for the three ena genotypes, as indicated. Note the bimodal distribution of PC1 in WT, coincidence of PC1 values of FP4-mito with the higher-value WT cluster, and PC1 values of UAS-ena concentrated roughly around the value of the minimum in PC1 values of WT.
DISCUSSION
Here we have used live imaging of the TSM1 axon in the developing Drosophila wing to investigate the role of the processive actin polymerase Ena in the molecular mechanism of axon growth and guidance, and to compare it with our previous analysis of the effects of the Ena regulator, Abl tyrosine kinase (Clarke et al., 2020a, 2020b). We find that Ena has a significant effect on the number of filopodial projections in the WT growth cone, but much less effect on their length, or on where they form. We also observe an asymmetry in the effects of Ena, with suppression of Ena activity producing a large effect on growth cone morphology but increase of Ena having little effect. This may suggest that the WT level of Ena in this growth cone is already close to saturation for its morphological functions. In contrast to its substantial effects on growth cone morphology, we find that the effect of Ena on the distribution of actin in this growth cone is quite modest. The actin mass at the heart of the growth cone undergoes a slight expansion with decrease of Ena activity, but the effect size is not large. Alteration of the Ena level also impairs the orderly evolution of the growth cone actin distribution over time. The limited sensitivity of the growth cone actin distribution to the level of Ena, as opposed to the strong effect of Ena on morphological features, is in striking contrast to the effect of Abl, which has a profound impact on the distribution of actin but only modest effects on overall growth cone morphology. These observations suggest that Ena may be more important for the linkage of actin to the membrane and downstream morphogenetic processes of this axon than it is for the distribution of the actin itself. Taken together, our data also suggest that the balance of Ena with other factors regulated by Abl may serve to buffer the effects of Abl on filopodial patterning, thus maintaining an optimal growth cone organization for orderly axon growth while still allowing Abl to act as a rheostat to vary actin dynamics.
Live imaging of morphology and actin organization in a developing pioneer axon of the Drosophila wing, TSM1, recently led us to propose a novel and unexpected model for axon growth and guidance in vivo (Clarke et al., 2020a, 2020b). We found that the distal part of the axon shaft contains a region with a high local concentration of actin. This actin mass undergoes constant stochastic fluctuations in size, but with a small forward bias that produces a net advance of the actin mass over time. Because actin and associated factors are essential for building and maintaining filopodia, advance of the actin mass leads, in turn, to advance of an emergent domain of high filopodial density that is the morphological feature we recognize as “the growth cone.” We also showed that Abl tyrosine kinase is a key regulator of actin distribution and dynamics in the growth cone, and thus, indirectly, of growth cone morphology and motility. In the current work, to investigate how Abl produces these changes in actin organization, we have quantified the effects of a core Abl effector, the processive actin polymerase Ena.
The effects of Ena on growth cone morphology and actin organization in TSM1
Consistent with data from other systems(Krause et al., 2003), we found that Ena has a significant impact on the morphology of the TSM1 growth cone in vivo. Varying Ena had a significant effect on filopodial number, with fewer filopodia present under conditions of low Ena activity than with high Ena. Surprisingly, however, nearly all of that effect (85%) was due to the consequences observed upon suppressing Ena; the effect of increasing Ena (relative to WT) was far more muted. Similarly, varying Ena activity revealed a shift to less total filopodial length with lower levels of active Ena, again with the overall effect dominated by the consequences of Ena suppression. The limited effect of Ena overexpression was unexpected and may suggest that Ena is present in the WT TSM1 at a level that is already nearly maximal for Ena function. A significant effect of Ena overexpression was observed, however, upon measuring average filopodial length: the combination of increased filopodial number in UAS-Ena, relative to WT, without a corresponding increase in total filopodial length, manifested as a significant decrease in average filopodial length (though, curiously, suppression of Ena in this axon did not cause a decrease in average filopodial length, unlike many other systems where Ena has been investigated [Vasioukhin et al., 2000; Lebrand et al., 2004; Gates et al., 2007]). It is not clear why increasing Ena failed to produce a simple dose-dependent increase in average filopodial length in TSM1. It may be that Ena interacts in some complex way with other actin polymerases, such as formins, in the filopodia of this axon (Homem and Peifer, 2009; Bilancia et al., 2014), or that formin levels are regulated in a way that compensates for changes in Ena activity. Alternatively, because we typically did not observe strong accumulation of Ena protein at TSM1 filopodial tips, it may be that Ena functions differently in these nonadherent filopodia in vivo than it does in the highly adhesive contexts where most analyses of Ena/VASP filopodial function have been performed previously (Rottner et al., 1999; Bear et al., 2000; Vasioukhin et al., 2000; Krause et al., 2002; Grevengoed et al., 2003; Lebrand et al., 2004; Gates et al., 2007; Lacayo et al., 2007). In this context, it is interesting to note that in vivo analysis of the C. elegans ena ortholog, UNC-34, in growth cones of VD motoneurons revealed results very similar to those we observe here, in that mutation of UNC-34 reduced filopodial number but had no effect on filopodial length (Norris et al., 2009). Despite these Ena-dependent changes in growth cone morphology, however, altering Ena activity produced overt defects in the terminal phenotype of TSM1 in only a modest fraction of wings. This is reminiscent of results from analysis of axon patterning in Drosophila embryonic development, where ena gain or loss of function also produces axonal mutant phenotypes only in a limited set of developmental contexts (Wills et al., 1999; Gates et al., 2007), and also of development of retinal axons in Xenopus, where Ena is important for arborization of axons, but not for their targeting (Dwivedy et al., 2007).
In contrast to these strong effects of Ena on filopodial organization, it was surprising to find that Ena has only rather subtle effects on actin distribution in TSM1. Decreasing the level of available Ena is associated with an increasing overall length of the actin mass in the growth cone, as well as with spreading of the actin within that mass, as assayed by wavelet analysis, consistent with the expected complementarity of Ena to the effect of altering Abl. In the case of Ena, however, the effects on actin are quantitatively rather small. This is different from the findings in our earlier analysis of Abl, whose major effect on TSM1 is in regulation of actin organization, with effects of Abl on morphology being much more limited. Consistent with the modest effects of Ena on actin distribution in the current study, altering Ena activity did not significantly alter the average rate of advance of the growth cone actin mass. Moreover, the overall pattern of growth cone dynamics remained the same regardless of the level of Ena activity, with longitudinal expansion of the actin mass being associated with the instantaneous advance of the actin peak, and the resulting offset between actin mass and filopodial density correlating with subsequent advance of the filopodial peak.
Comparison of TSM1 observations to other experimental and theoretical analyses of Ena
The combination of effects we observe for Ena in TSM1 in some ways match those seen in published analyses of Ena in other systems, but in other ways they were rather unexpected. In contrast to our data here for TSM1, in other systems, Ena has been shown to have substantial effects on actin organization, and Ena overexpression has been shown to induce robust extension of cellular projections. We note, however, that those studies have in general investigated Ena action in cellular contexts dominated by substratum adhesion: epithelia (Vasioukhin et al., 2000; Gates et al., 2007), adherent cells (Bear et al., 2000; Lacayo et al., 2007), and axons growing in an adherent manner on rigid supports (Lebrand et al., 2004; Gupton and Gertler, 2010). In all of these cases, it may be that interaction of Ena with adhesive structures plays a critical role, feeding back on actin organization in response to morphological inputs. In TSM1, in contrast, our observations do not provide obvious suggestion of a significant adhesive contribution, similarly to the limited role of adhesion in studies from other labs investigating motion in compliant, three-dimensional media, both for growth cones and for motile cells (Lammermann et al., 2008; Santos et al., 2020). Indeed, the notion suggested above that a key role of Ena in TSM1 may lie in the linkage of actin to the plasma membrane, rather than in direct effects on actin structure, would agree well with an observation that the protein has generally stronger effects in adhesion-limited cellular contexts (Vasioukhin et al., 2000; Sheffield et al., 2007) than it does in TSM1.
The experimental observations made here showing that Ena has relatively modest effects on actin organization agree well with our recent results from computational simulations of actin networks (Chandrasekaran et al., 2022a, 2022b). There, we found that the effects on actin from changing Ena activity were manifested most strongly on fine details of network organization at very short range (sub-micrometer) length scales, beyond the resolution of our microscopy of TSM1. In the simulations, modulation of more robust actin nucleators, such as Arp2/3, and contractile elements, such as Myosin II, were needed to produce large, mesoscale (multi-micrometer-level) effects on actin distribution such as those that we observe here in the living wing disk, and that we have shown to underlie the mechanism of axon growth and guidance. This suggests that aspects of signaling downstream of Abl that are distinct from its regulation of Ena are likely to play the key role in regulating large-scale actin organization in the growth cone. A strong candidate is Abl-dependent activation of the Rac GEF, Trio, with consequent stimulation of a Rac-WAVE pathway, which we have shown to occur in parallel to the Abl–Ena interaction (Kannan and Giniger, 2017; Kannan et al., 2017), and which would be predicted to stimulate the branching actin nucleator, Arp2/3. Moreover, it has been shown that Abl regulates the activity of Myosin II (Dudek et al., 2010), another key regulator of the mesoscopic organization of nonpolarized actin assemblies in our simulations (Chandrasekaran et al., 2022b). Our computational analysis also suggested a simple mechanistic explanation for how nanometer-scale changes to actin filament length produce multi-micrometer-scale changes in the overall distribution of actin density by modifying the connectivity (percolation) of the actomyosin network (Chandrasekaran et al., 2022a, 2022b).
How and why does altering Ena activity produce terminal mutant phenotypes in TSM1?
While the overt effects of Ena on TSM1 growth cone properties are relatively subtle, they are evidently significant physiologically, since we observe defects in the final trajectory of the axon in 20–30% of wings (depending on the manipulation). One good candidate for an explanation of those terminal defects is our observation that experimental manipulations increasing or decreasing Ena activity disrupt the reliable evolution of the actin pattern in the growth cone over time. In the WT axon, the actin peak advances in an orderly way, where the distribution of actin at any given time allows one to predict the global features the distribution will have at subsequent times. In the altered Ena conditions this predictability is lost, stochastically, in a fraction of cells. We speculate that the axons that eventually stall or misroute may be the ones where the orderly progression of actin states fails to occur. The same kind of dynamic instability of the actin pattern was observed in our previous study upon increase or decrease of Abl activity, and in that case it was also correlated with more generalized disorganization of the actin distribution (Clarke et al., 2020a, 2020b). These data are therefore consistent with the hypothesis that modulation of Ena activity may be important to the mechanism by which Abl ensures that transformations of the actin distribution occur in an orderly manner in the advancing growth cone, and that this consistency is important for reliable axon growth.
There is a second possible reason that WT Ena activity may be essential for consistent growth of the TSM1 axon. The data reported here show that altering Ena activity, particularly reducing Ena activity, produces strong effects on filopodial pattern in TSM1, in contrast to Abl, which has at most a mild effect on TSM1 filopodial morphology. This is curious, however. If Abl regulates Ena, and Ena strongly modifies morphology, why does altering Abl not have a stronger effect on TSM1 filopodial morphology? Our earlier studies of Abl signaling may hint at an explanation. We have shown previously that there are at least two opposing signals downstream of Abl, suppression of Ena, and also activation of a Trio-Rac-WAVE-Arp2/3 pathway, and we have speculated that this pattern of antagonistic regulation of its two key effectors may be critical to Abl function (Kannan and Giniger, 2017; Kannan et al., 2017). Activation of Ena promotes filopodial development, as discussed above, but so does activation of Arp2/3 (Norris et al., 2009; Goncalves-Pimentel et al., 2011). It is thought that Arp2/3 promotes formation of submembranous branched actin networks that nucleate the parallel actin filaments that extrude filopodia (Biyasheva et al., 2004; Korobova and Svitkina, 2008), and indeed, experimental manipulation of Arp2/3 activity has verified that activation of this protein complex enhances filopodial number in Drosophila growth cones (Sanchez-Soriano et al., 2010). Therefore, it seems plausible that the antagonistic regulation of Ena versus Arp2/3 by Abl may have the net effect of keeping the local propensity for filopodial extension in the growth cone roughly constant, even as Abl activity changes. Stated otherwise, by this model, a key function behind the complementary regulation of Ena versus Rac-WAVE-Arp2/3 may be to buffer the effects of Abl on filopodial morphogenesis, maintaining the growth cone in an optimal morphological state for continued growth, while leaving Abl activity free to be an adjustable rheostat that can be altered to modulate actin organization in response to external cues, tuned to produce the directed expansion and compaction of the mesoscale actin network that is the engine for axon growth and guidance.
MATERIALS AND METHODS
Drosophila stocks
Drosophila stocks neur-GAL4[A101] (BDSC 6393), UAS-LifeAct-eGFP (BDSC 35544), and UAS-CD4-td-Tomato (BDSC 35837) were obtained from the Bloomington Drosophila Stock Center. UAS-ena (untagged) and UAS-FP4-mito-GFP were obtained from Julie Gates (Bucknell University) and Mark Peifer (UNC—Chapel Hill). Note that under our conditions of imaging (low intensity of the 488-nm laser to limit photodamage and low GFP detector gain to prevent saturation of axonal LifeAct-eGFP signal; see below), GFP fluorescence was not detectable in the axon upon expression of FP4-mito-eGFP. Flies were raised on standard cornmeal/molasses food (KD Medical, Columbia, MD).
Microscopy and antibody staining
Fixed samples were used only to generate the anatomical reference images of Figure 1A and controls for reagent activity in Figure 3. To prepare the early-prepupal image, white prepupae (WPP) were collected, aged 8 h at 25°C, and then dissected and fixed for 25′ in PBS containing 4% formaldehyde and 0.1% glutaraldehyde. Wings were then washed in PBS, transferred to PBS + 0.3% Triton X-100 (EM Sciences, Hatfield, PA), blocked, incubated for 90′ with TRITC-anti-HRP (Jackson ImmunoResearch, West Grove, PA; cat# 323-025-021; dilution 1:100), washed, and mounted in Prolong Gold. To visualize Ena protein in prepupal wing discs, essentially the same fixation and staining protocol was used, except that glutaraldehyde was omitted from the fix and the primary antibody was anti-Enabled antibody (mAb 5G2, 1:50 dilution, Developmental Studies Hybridoma Bank, Iowa City, IA), followed by donkey anti-mouse secondary (AlexaFluor, 1:500 dilution, Jackson ImmunoResearch)) To prepare the late-stage (pupal) image and samples for scoring the terminal phenotype of TSM1, WPP expressing CD4-td-Tomato under control of neur-GAL4 were collected and aged 20 h at 25°C. Pupae were removed from the pupal case and fixed in PBS containing 4% formaldehyde for 25′, RT. After washing, fixed pupae were dehydrated in 100% ethanol and stored in ethanol for at least 24 h at 4°C. Pupae were then rehydrated in PBS + 0.3% Triton, and wing discs were dissected and mounted in Vectashield (ThermoFisher). Wide field microscopy was performed with an AxioImager Z1 microscope, and image stacks were deconvoluted and processed in Zen.
Live imaging
Live imaging was performed by a modification of the method described in Clarke et al. (2020a). WPP of the appropriate genotype were collected and aged 8 h at 25°C. Wing discs were dissected in fresh culture medium (Schneider’s Drosophila medium [Life Technologies] containing 10% fetal bovine serum [Life Technologies]). Wing discs were transferred to a drop of culture medium (∼15 μl) in the middle of an 18 × 18–mm #1.5 coverslip and mounted by the method of Rusan and coworkers (Lerit et al., 2014; see also Supplemental Figure 1). In brief, discs were transferred in a minimum volume of culture medium using a pipet tip that had been treated with Sigmacote (Sigma-Aldrich, St. Louis, MO) and preblocked by triturating contents of the pupal abdomen. Small (∼10–15 μl) drops of #700 halocarbon oil (Sigma) were placed at the corners of the coverslip and it was stuck to the underside of a gas-permeable Lumox 35 culture dish (Sarstedt), which was then inverted. Medium and oil were allowed to spread, and a Kimwipe was used to wick away medium and oil until wings were physically restrained but not crushed. Additional oil was used as needed to seal the edges of the coverslip. Up to five discs were mounted per imaging chamber. Imaging was performed on an inverted microscope, with imaging chamber right side up and filled with ∼3 ml culture medium (to avoid reflection at the surface of the dish).
Imaging was performed with a Zeiss AxioObserver Z1 spinning-disk confocal microscope with a 25°C temperature-controlled stage. Z-stacks were taken at 0.8 μm spacing with a 63×/1.2 NA water immersion lens. Typically, two discs were imaged at once, using the multipoint feature of the software. Imaging runs were 90 min with 3′ between initiation of successive frames. Images were not deconvoluted, as previous experiments showed that deconvolution corrupts the information content of the images (Clarke et al., 2020a).
Ten control trajectories (neur-GAL4), 10 trajectories of neur-GAL4; UAS-FP4-mito-eGFP, and 13 trajectories of neur-GAL4; UAS-ena were determined. Thirty-one time steps were collected for each trajectory with a 3′ interval between the initiation of successive z-stacks.
Segmentation of images and quantification of growth cone parameters
Tracing of axons and quantification of LifeAct intensity were performed precisely as described in Clarke et al. (2020a). In brief, three-dimensional tracing in Imaris (Bitplane, version 8 or 9) was first performed of just the axon shaft and converted to Nikon ICS format. This was imported into MIPAV, which generated an SWC format description of the axon (plug-in: Drosophila creates SWC) and then calculated the LifeAct intensity as a function of position along the axon by summing signal intensity in sequential frustums centered on the axon shaft (plug-in: 3D SWC stats). Complete tracing of all projections was then performed in Imaris, and again converted to ICS format and imported to MIPAV for preparation of an SWC file. During tracing, care was taken to begin the trace at a specific position of the proximal axon that could be identified consistently in all frames of the trajectory.
Parameters describing features of morphology and actin distribution were calculated as described previously (Clarke et al., 2020a). Custom Python scripts were written to calculate the desired parameters for each image from the SWC file of projections and from the spreadsheet of actin intensity as a function of position in the axon shaft. Parameters are listed in Figure 1C. These include the total number of filopodial projections from the axon shaft, total length and average length of filopodia, and average order of filopodial projections. To calculate filopodial density along the axon, higher-order projections were assigned to the positions of their parent primary projections. The positions of maximum filopodial density and of maximum actin intensity were identified separately using 5-μm sliding windows (advanced in 1-μm steps). It was shown previously that results are robust to the choice of window length (1–10 μm; Clarke et al., 2020a). As previously, the “length” of the protrusive filopodial zone of the axon was calculated as the square root of the second moment of the distribution of filopodial density about the position of the window with the maximum value, and the “length” of the region of elevated actin concentration was similarly calculated as the square root of the second moment of the actin intensity about the position of the window with the maximum integrated signal intensity. The square root of the second moment is essentially analogous to one SD and was previously found empirically to a be a useful measure of growth cone length (Clarke et al., 2020a). The global sqrt(second moment) was used for all subsequent quantitative analyses. For purposes of representation of positions on the axon (Figures 2E and 4, E and F), it was found useful to indicate the partial sqrt(second moment) in the leading and trailing directions, but note that the global sqrt(second moment) does not, in general, equal the sum of the leading and trailing partial moments. Motion of the actin peak position was calculated between successive time points. For assessing correlation of actin peak motion during an interframe interval to static features of the axon, comparison was made to the static value at the start of that interval.
Statistical methods
Repeated measures ANOVA was used to assess the statistical significance of genotype comparisons. A linear model was generated, with first-order autoregression used as the covariance structure to account for repeated measures from each single cell. Box–Cox transformation was applied to outcome variables with nonnormal distribution, using the Shapiro–Wilk test to assess normality of model residuals. Tukey’s method was used to correct for multiple comparisons between the three genotypes. Where other statistical tests were applied, they are specified in the text and figure legends (GraphPad Prism, Version 9).
Pairwise analysis of parameter correlations was quantified by the Kendall rank correlation (τ), with significance assessed by Benjamini–Hochberg FDR. Correlations were considered significant at FDR < 5% after correction for multiple testing
PCA was performed using standard methods of principal component regression. The PCA axes were defined by the eigenvectors of the correlation matrix of the seven parameters measured for each time point in the WT dataset (specified in the text and listed in Figure 7). The first PC axis is along the eigenvector associated with the largest eigenvalue, the second PC axis is along the eigenvector with the second largest eigenvalue, etc. Data from the UAS-FP4-mito and UAS-ena datasets were then visualized by projecting them into the PCA space determined from the WT data. Determination of the PCA axes and projection of parameter data in the PCA space was coded in Python using the Numpy library routines.
Wavelet analysis.
Wavelet analysis was performed using the Daubechies type 4 (D4) wavelet transform to quantify spatial frequency components of the actin distribution, with the transform modified to account for non-periodic boundaries. As described previously (Clarke et al., 2020b), given NB bins (equal to a power of 2) along the axon length, we define S0,n as the measured actin intensity in bin n. Let w be the bin width. We define the transform from spatial resolution 2Lw to spatial resolution 2L+1w to be
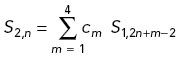 |
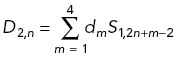 |
Here L = 0 corresponds to the original binned intensities above, and for L, n takes values 1, 2, …, NB /2L. The coefficients { c 1 , c 2, c 3 , c 4 } = { 1 + √3, 3 + √3, 3 – √3, 1 – √3}/4 × √2 specify an averaging filter, and { d1, d2, d3, d4 } = { c3, -c2, c1, -c0 } is a differentiating filter. The bin width w = 0.06 μm, NB = 2048, and the intensity of any bin outside of the axon is set to zero to avoid edge artifacts. See Supplemental Methods for a more conceptual explanation of wavelet analysis.
Jensen–Shannon Divergence.
Jensen–Shannon divergence (JSD) was calculated for each cell between the starting actin distribution (p) and the distribution at each subsequent time point (r) using the formula
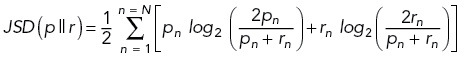 |
where p and r are the two actin distributions and n is the normalized actin intensity of the nth bin of the distribution (n = 1, 2, …, N) . For three cells expressing UAS-FP4-mito (FP4-mito cell #1, #6, and #12) the absolute intensity of the LifeAct signal was quite low (nominal integrated actin intensity per time point < 5 × 104 arbitrary units), causing the resulting actin distributions to be discontinuous. Such distributions were not appropriate for JSD analysis and were excluded from this calculation.
Reproducibility and data exclusion.
Movies were not collected or analyzed from cells that failed to show dynamics upon mounting, axons that grew out of the field of focus, or those for which image intensity was too low to detect. One cell (FP4-mito#7) had a robust CD4-td-Tomato signal, but insufficient LifeAct-GFP intensity to segment in Imaris and MIPAV. Therefore, this cell was included in analysis of morphological features but not actin parameters. Sample randomization and blinding were not relevant to the experimental design. Sample size was selected based on leave-out analyses of datasets from previous experiments using this study design.
Data and code availability.
Numerical data for all figures are included in Supplemental Datasheet S1. MIPAV code, including plug-ins, is freely available on the NIH website. Python scripts and all other primary data will be deposited in Mendeley Data upon publication.
Supplementary Material
Acknowledgments
We are grateful to all the members of our labs for their extensive contributions to the conception, execution, and interpretation of these experiments, particularly Ginger Hunter, Ram Kannan, and Arvind Shukla. We are also deeply grateful to Lenny Campanello for insightful critiques and suggestions concerning our analytical methods, Tianxia Wu for expert assistance with statistical analysis of our time course data, Camille Hanes for assisting in collection and analysis of end stage wing samples for determining terminal phenotypes of ena, and Joy Gu and Irina Kuzina for outstanding technical assistance throughout the course of these studies. We thank Holly Cline and David Miller III for their thoughtful comments on the manuscript. We are also indebted to Julie Gates for providing the flies bearing UAS-ena and UAS-FP4-mito-eGFP transgenes, without which these experiments could not have been performed. Spinning disk microscopy was performed in the Cytogenetics and Microscopy Core Facility of the National Human Genome Research Institute, with the invaluable assistance of Stephen Wincovitch. Drosophila stocks obtained from the Bloomington Drosophila Stock Center (NIH P4OOD018537) and antibodies from the Developmental Studies Hybridoma Bank were essential to these studies. This work was supported by an MURI grant to W.G. (AFOSR Grant FA9550-16-1-0052), National Science Foundation Grants CHE-1800418 and CHE-2102684 to G.A.P., and funds from the Basic Neuroscience Program of the Intramural Research Program of the National Institute of Neurological Disorders and Stroke of the National Institutes of Health (Z01-NS003013, to E.G.).
Abbreviations used:
- Arp2/3
complex of Actin-related protein 2 and Actin-related protein 3
- Ena
enabled
- EVL
Ena- and VASP-like
- MENA
Mouse Ena
- Rac GEF
guanine exchange protein for Rac GTPase
- SD
standard deviation
- TSM1
twin sensilla of the margin 1
- VASP
vasodilator-stimulated phosphoprotein
- WAVE
WASP-family verprolin-homologous protein
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E23-01-0003) on May 24, 2023.
REFERENCES
- Bear JE, Gertler FB (2009). Ena/VASP: towards resolving a pointed controversy at the barbed end. J Cell Sci 122, 1947–1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear JE, Loureiro JJ, Libova I, Fassler R, Wehland J, Gertler FB (2000). Negative regulation of fibroblast motility by Ena/VASP proteins. Cell 101, 717–728. [DOI] [PubMed] [Google Scholar]
- Bilancia CG, Winkelman JD, Tsygankov D, Nowotarski SH, Sees JA, Comber K, Evans I, Lakhani V, Wood W, Elston TC, et al. (2014). Enabled negatively regulates diaphanous-driven actin dynamics in vitro and in vivo. Dev Cell 28, 394–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biyasheva A, Svitkina T, Kunda P, Baum B, Borisy G (2004). Cascade pathway of filopodia formation downstream of SCAR. J Cell Sci 117, 837–848. [DOI] [PubMed] [Google Scholar]
- Blanchoin L, Boujemaa-Paterski R, Sykes C, Plastino J (2014). Actin dynamics, architecture, and mechanics in cell motility. Physiol Rev 94, 235–263. [DOI] [PubMed] [Google Scholar]
- Bradley WD, Koleske AJ (2009). Regulation of cell migration and morphogenesis by Abl-family kinases: emerging mechanisms and physiological contexts. J Cell Sci 122, 3441–3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruhmann S, Ushakov DS, Winterhoff M, Dickinson RB, Curth U, Faix J (2017). Distinct VASP tetramers synergize in the processive elongation of individual actin filaments from clustered arrays. Proc Natl Acad Sci USA 114, E5815–E5824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrasekaran A, Clarke A, McQueen P, Fang HY, Papoian GA, Giniger E (2022a). Computational simulations reveal that Abl activity controls cohesiveness of actin networks in growth cones. Mol Biol Cell 33, ar92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrasekaran A, Giniger E, Papoian GA (2022b). Nucleation causes an actin network to fragment into multiple high-density domains. Biophys J 121, 3200–3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke A, McQueen PG, Fang HY, Kannan R, Wang V, McCreedy E, Buckley T, Johannessen E, Wincovitch S, Giniger E (2020a). Dynamic morphogenesis of a pioneer axon in Drosophila and its regulation by Abl tyrosine kinase. Mol Biol Cell 31, 452–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke A, McQueen PG, Fang HY, Kannan R, Wang V, McCreedy E, Wincovitch S, Giniger E (2020b). Abl signaling directs growth of a pioneer axon in Drosophila by shaping the intrinsic fluctuations of actin. Mol Biol Cell 31, 466–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comer AR, Ahern-Djamali SM, Juang JL, Jackson PD, Hoffmann FM (1998). Phosphorylation of Enabled by the Drosophila Abelson tyrosine kinase regulates the in vivo function and protein-protein interactions of Enabled. Molecular and cellular biology 18, 152–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deinhardt K, Kim T, Spellman DS, Mains RE, Eipper BA, Neubert TA, Chao MV, Hempstead BL (2011). Neuronal growth cone retraction relies on proneurotrophin receptor signaling through Rac. Sci Signal 4, ra82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson BJ (2002). Molecular mechanisms of axon guidance. Science 298, 1959–1964. [DOI] [PubMed] [Google Scholar]
- Dudek SM, Chiang ET, Camp SM, Guo Y, Zhao J, Brown ME, Singleton PA, Wang L, Desai A, Arce FT, et al. (2010). Abl tyrosine kinase phosphorylates nonmuscle myosin light chain kinase to regulate endothelial barrier function. Mol Biol Cell 21, 4042–4056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumoulin A, Zuniga NR, Stoeckli ET (2021). Axon guidance at the spinal cord midline-A live imaging perspective. J Comp Neurol 529, 2517–2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwivedy A, Gertler FB, Miller J, Holt CE, Lebrand C (2007). Ena/VASP function in retinal axons is required for terminal arborization but not pathway navigation. Development 134, 2137–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming T, Chien SC, Vanderzalm PJ, Dell M, Gavin MK, Forrester WC, Garriga G (2010). The role of C. elegans Ena/VASP homolog UNC-34 in neuronal polarity and motility. Dev Biol 344, 94–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsthoefel DJ, Liebl EC, Kolodziej PA, Seeger MA (2005). The Abelson tyrosine kinase, the Trio GEF and Enabled interact with the Netrin receptor Frazzled in Drosophila. Development 132, 1983–1994. [DOI] [PubMed] [Google Scholar]
- Garbe DS, O’Donnell M, Bashaw GJ (2007). Cytoplasmic domain requirements for Frazzled-mediated attractive axon turning at the Drosophila midline. Development 134, 4325–4334. [DOI] [PubMed] [Google Scholar]
- Gates J, Mahaffey JP, Rogers SL, Emerson M, Rogers EM, Sottile SL, Van Vactor D, Gertler FB, Peifer M (2007). Enabled plays key roles in embryonic epithelial morphogenesis in Drosophila. Development 134, 2027–2039. [DOI] [PubMed] [Google Scholar]
- Gates J, Nowotarski SH, Yin H, Mahaffey JP, Bridges T, Herrera C, Homem CC, Janody F, Montell DJ, Peifer M (2009). Enabled and Capping protein play important roles in shaping cell behavior during Drosophila oogenesis. Dev Biol 333, 90–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertler FB, Comer AR, Juang JL, Ahern SM, Clark MJ, Liebl EC, Hoffmann FM (1995). enabled, a dosage-sensitive suppressor of mutations in the Drosophila Abl tyrosine kinase, encodes an Abl substrate with SH3 domain-binding properties. Genes Dev 9, 521–533. [DOI] [PubMed] [Google Scholar]
- Gertler FB, Niebuhr K, Reinhard M, Wehland J, Soriano P (1996). Mena, a relative of VASP and Drosophila Enabled, is implicated in the control of microfilament dynamics. Cell 87, 227–239. [DOI] [PubMed] [Google Scholar]
- Goncalves-Pimentel C, Gombos R, Mihaly J, Sanchez-Soriano N, Prokop A (2011). Dissecting regulatory networks of filopodia formation in a Drosophila growth cone model. PLoS One 6, e18340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grevengoed EE, Fox DT, Gates J, Peifer M (2003). Balancing different types of actin polymerization at distinct sites: roles for Abelson kinase and Enabled. J Cell Biol 163, 1267–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grevengoed EE, Loureiro JJ, Jesse TL, Peifer M (2001). Abelson kinase regulates epithelial morphogenesis in Drosophila. J Cell Biol 155, 1185–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman EN, Giurumescu CA, Chisholm AD (2013). Mechanisms of ephrin receptor protein kinase-independent signaling in amphid axon guidance in Caenorhabditis elegans. Genetics 195, 899–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupton SL, Gertler FB (2010). Integrin signaling switches the cytoskeletal and exocytic machinery that drives neuritogenesis. Developmental cell 18, 725–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homem CC, Peifer M (2009). Exploring the roles of diaphanous and enabled activity in shaping the balance between filopodia and lamellipodia. Mol Biol Cell 20, 5138–5155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsouna A, Kim YS, VanBerkum MF (2003). Abelson tyrosine kinase is required to transduce midline repulsive cues. Journal of neurobiology 57, 15–30. [DOI] [PubMed] [Google Scholar]
- Kannan R, Giniger E (2017). New perspectives on the roles of Abl tyrosine kinase in axon patterning. Fly (Austin) 11, 260–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan R, Kuzina I, Wincovitch S, Nowotarski SH, Giniger E (2014). The Abl/enabled signaling pathway regulates Golgi architecture in Drosophila photoreceptor neurons. Mol Biol Cell 25, 2993–3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan R, Song JK, Karpova T, Clarke A, Shivalkar M, Wang B, Kotlyanskaya L, Kuzina I, Gu Q, Giniger E (2017). The Abl pathway bifurcates to balance Enabled and Rac signaling in axon patterning in Drosophila. Development 144, 487–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korobova F, Svitkina T (2008). Arp2/3 complex is important for filopodia formation, growth cone motility, and neuritogenesis in neuronal cells. Mol Biol Cell 19, 1561–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause M, Bear JE, Loureiro JJ, Gertler FB (2002). The Ena/VASP enigma. J Cell Sci 115, 4721–4726. [DOI] [PubMed] [Google Scholar]
- Krause M, Dent EW, Bear JE, Loureiro JJ, Gertler FB (2003). Ena/VASP proteins: regulators of the actin cytoskeleton and cell migration. Annu Rev Cell Dev Biol 19, 541–564. [DOI] [PubMed] [Google Scholar]
- Kuzina I, Song JK, Giniger E (2011). How Notch establishes longitudinal axon connections between successive segments of the Drosophila CNS. Development 138, 1839–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacayo CI, Pincus Z, VanDuijn MM, Wilson CA, Fletcher DA, Gertler FB, Mogilner A, Theriot JA (2007). Emergence of large-scale cell morphology and movement from local actin filament growth dynamics. PLoS Biol 5, e233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammermann T, Bader BL, Monkley SJ, Worbs T, Wedlich-Soldner R, Hirsch K, Keller M, Forster R, Critchley DR, Fassler R, Sixt M (2008). Rapid leukocyte migration by integrin-independent flowing and squeezing. Nature 453, 51–55. [DOI] [PubMed] [Google Scholar]
- Lanier LM, Gertler FB (2000). From Abl to actin: Abl tyrosine kinase and associated protein in growth cone motility. Curr Opin Neurobiol 10, 80–87. [DOI] [PubMed] [Google Scholar]
- Lebrand C, Dent EW, Strasser GA, Lanier LM, Krause M, Svitkina TM, Borisy GG, Gertler FB (2004). Critical role of Ena/VASP proteins for filopodia formation in neurons and in function downstream of netrin-1. Neuron 42, 37–49. [DOI] [PubMed] [Google Scholar]
- Lerit DA, Plevock KM, Rusan NM (2014). Live imaging of Drosophila larval neuroblasts. J Vis Exp [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowery LA, Van Vactor D (2009). The trip of the tip: understanding the growth cone machinery. Nat Rev Mol Cell Biol 10, 332–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moresco EM, Koleske AJ (2003). Regulation of neuronal morphogenesis and synaptic function by Abl family kinases. Curr Opin Neurobiol 13, 535–544. [DOI] [PubMed] [Google Scholar]
- Norris AD, Dyer JO, Lundquist EA (2009). The Arp2/3 complex, UNC-115/abLIM, and UNC-34/Enabled regulate axon guidance and growth cone filopodia formation in Caenorhabditis elegans. Neural Dev 4, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottner K, Behrendt B, Small JV, Wehland J (1999). VASP dynamics during lamellipodia protrusion. Nat Cell Biol 1, 321–322. [DOI] [PubMed] [Google Scholar]
- Sanchez-Soriano N, Goncalves-Pimentel C, Beaven R, Haessler U, Ofner-Ziegenfuss L, Ballestrem C, Prokop A (2010). Drosophila growth cones: a genetically tractable platform for the analysis of axonal growth dynamics. Dev Neurobiol 70, 58–71. [DOI] [PubMed] [Google Scholar]
- Santos TE, Schaffran B, Broguiere N, Meyn L, Zenobi-Wong M, Bradke F (2020). Axon growth of CNS neurons in three dimensions is amoeboid and independent of adhesions. Cell Rep 32, 107907. [DOI] [PubMed] [Google Scholar]
- Sheffield M, Loveless T, Hardin J, Pettitt J (2007). C. elegans Enabled exhibits novel interactions with N-WASP, Abl, and cell–cell junctions. Curr Biol 17, 1791–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoeckli ET (2018). Understanding axon guidance: are we nearly there yet? Development 145. [DOI] [PubMed] [Google Scholar]
- Tessier-Lavigne M, Goodman CS (1996). The molecular biology of axon guidance. Science 274, 1123–1133. [DOI] [PubMed] [Google Scholar]
- Trichet L, Sykes C, Plastino J (2008). Relaxing the actin cytoskeleton for adhesion and movement with Ena/VASP. J Cell Biol 181, 19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasioukhin V, Bauer C, Yin M, Fuchs E (2000). Directed actin polymerization is the driving force for epithelial cell–cell adhesion. Cell 100, 209–219. [DOI] [PubMed] [Google Scholar]
- Wills Z, Bateman J, Korey CA, Comer A, Van Vactor D (1999). The tyrosine kinase Abl and its substrate enabled collaborate with the receptor phosphatase Dlar to control motor axon guidance. Neuron 22, 301–312. [DOI] [PubMed] [Google Scholar]
- Winkelman JD, Bilancia CG, Peifer M, Kovar DR (2014). Ena/VASP Enabled is a highly processive actin polymerase tailored to self-assemble parallel-bundled F-actin networks with Fascin. Proc Natl Acad Sci USA 111, 4121–4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu HH, Zisch AH, Dodelet VC, Pasquale EB (2001). Multiple signaling interactions of Abl and Arg kinases with the EphB2 receptor. Oncogene 20, 3995–4006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Numerical data for all figures are included in Supplemental Datasheet S1. MIPAV code, including plug-ins, is freely available on the NIH website. Python scripts and all other primary data will be deposited in Mendeley Data upon publication.