

Abstract
Estrogen-related receptors (ERR) are an orphan nuclear receptor sub-family that play a critical role in regulating gene transcription for several physiological processes including mitochondrial function, cellular energy utilization and homeostasis. They have also been implicated to play a role in several pathological conditions. Herein, we report the identification, synthesis, structure-activity relationships and pharmacological evaluation of a new chemical series of potent pan-ERR agonists. This template was designed for ERR starting from the known acyl hydrazide template and compounds such as agonist GSK-4716 employing a structure-based drug design approach. This led to the preparation of a series of 2,5-disubstituted thiophenes from which several were found to be potent agonists of ERRγ in cell-based co-transfection assays. Additionally, direct binding to ERRγ was established through 1H-NMR protein-ligand binding experiments. Compound optimization revealed that the phenolic or aniline groups could be replaced with a boronic acid moiety, which was able to maintain activity and demonstrated improved metabolic stability in microsomal in vitro assays. Further pharmacological evaluation of these compounds showed that they had roughly equivalent agonist activity on ERR isoforms α and β representing an ERR pan-agonist profile. One potent agonist, SLU-PP-915 (10s), which contained a boronic acid moiety was profiled in gene expression assays and found to significantly upregulate the expression of ERR target genes such as peroxisome-proliferator activated receptor γ co-activators-1α, lactate dehydrogenase A, DNA damage inducible transcript 4 and pyruvate dehydrogenase kinase 4 both in vitro and in vivo.
Keywords: Nuclear receptor; Estrogen-related receptor; Pan-agonist; Boronic acid; 2,5-disubstituted thiophenes
Graphical Abstract

1. Introduction
Nuclear receptors (NR) are a super family of transcription factors that regulate expression of genes involved in a wide array of physiological functions, including reproduction, development and metabolism.1–2 While the transcriptional activity of most members of the NR family are mediated through binding of endogenous ligands the estrogen-related receptors (ERR) make up a subgroup within the NR family that have no endogenous ligands and thus are true orphan receptors.3–4 Instead the ERRs are constitutively active which is driven primarily by co-activator proteins such as peroxisome-proliferator activated receptor gamma co-activators-1α and 1β (PGC-1α & PGG-1β) as well other environmental factors.4–6 As their name implies, the ERRs share significant sequence homology with the estrogen receptors (ER), especially in the DNA-binding domains (DBD) and are able to bind to estrogen response elements (ERE).1, 7 However, they share less homology within their ligand-binding domains (LBD) nor do the bind endogenous ER ligands or affect ER signaling pathways.1, 4, 8
There are three known ERR isoforms (α, β & γ) with differing tissue expression patterns.6, 9 ERRα is the most abundant isoform, being found in nearly all tissues. It is most highly expressed in tissues with a high-energy demand such as heart, kidney’s, intestinal tract, skeletal muscle and brown adipose tissue. ERRβ has a more restricted expression pattern and typically in lower levels than ERRα. ERRγ, like ERRα is also expressed in tissues requiring high energy demands such as the heart, kidneys and tissues with basal metabolic functions. All three isoforms are highly expressed in the CNS. The pharmacology associated with the ERRs has become an area of increasing interest because of the critical role they play in various physiological processes as well as their potential roles in different pathological states. Potential applications across a wide range of therapeutic areas have been evaluated such as metabolic disorders,9–12 cardiovascular,13–15 various cancers16–19 and Alzheimer’s disease.20
The ERRs have received considerable attention for their potential therapeutic value in treating metabolic diseases. Both ERRα and ERRγ have been shown to play critical roles in maintaining cellular energy and homeostasis through regulation of gene transcription in a number of metabolic processes such as fatty acid oxidation, mitochondrial biogenesis, oxidative phosphorylation and the TCA cycle.9 Improvement in mitochondrial function in skeletal muscle has been demonstrated to have potential for treating type 2 diabetes since increased skeletal muscle oxidative capacity has been associated with improved insulin sensitivity as well as reduction in obesity. 12 A gain of function study in mice where ERRγ was overexpressed in skeletal muscle showed an increase in oxidative metabolism, mitochondrial biogenesis, lipid oxidation and exercise tolerance.21–22 ERRγ was also shown to be a positive regulator of oxidative myofibers and promote muscle repair in an ischemia model of muscle injury and demonstrated potential as a potential treatment for Duchenne Muscular dystrophy.23 Additionally, muscle from ERRα knockout mice displayed a reduced mitochondrial biogenesis and repair function.11, 24 All of the above suggests ERR agonists have a number of potential therapeutic applications with metabolic diseases.
While the ERRs are unable to bind endogenous ER ligands, they do bind a number of synthetic ligands, including synthetic ER ligands.25–26 For example, both 4-hydroxytamoxifen (4-OHT) (1) and diethylstilbesterol (DES) (2) are able to bind ERRγ strongly as inverse agonists,27 (See Figure 1) which has led to the development of several related ERRγ modulators—both agonists and inverse agonists—from these templates.28–31 Researchers at GlaxoSmithKline identified the acyl hydrazide compound GSK-4716, 3, which was a novel ERRβ/γ agonist.32 In biochemical and cell-based assays, it activated both ERRβ and ERRγ but had no effect on ERRα or either of the ERs (α or β). A series of related compounds with the hydrazide moiety replaced with amides were reported and found to be potent ERRγ agonists.33 Xu and coworkers described a structure-based drug design (SBDD) approach where they modified the acyl hydrazide of compounds such as 3 to generate a series of di-aryl substituted triazoles, such as 4, which were potent agonists of ERRγ and stimulated mitochondrial biogenesis both in vitro and in vivo.34
Figure 1.
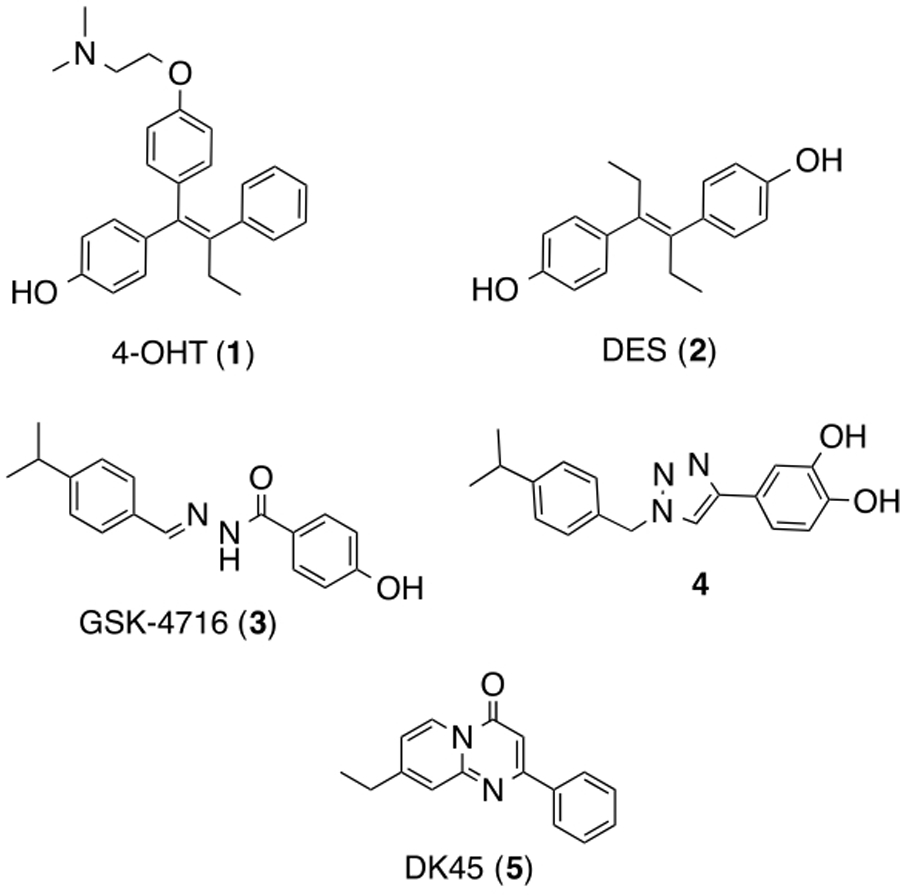
Representative ERR synthetic ligands: ERRγ inverse agonists 4-OHT(1) and DES(2), ERRβ/γ agonist GSK-4716 (3), ERRγ agonist 4 and ERRα agonist 5
A few ERRα modulators have also been developed. Novartis identified a small molecule inverse agonist of ERRα and obtained an x-ray of it bound to the LBD.8 Ligand docking studies carried out by GSK suggested that the binding pocket of the LBD of ERRα might be too small to accommodate activating ligands.35 However, Suetsugi et al, identified a group of isoflavones, including genistein, that were agonists on ERRα.36 Ding et al subsequently designed a novel small molecule ERRα agonist 5, based on genistein.37 Shinozuka and coworkers very recently disclosed a new class of diaryl ethers as potent ERRα agonists which were optimized from hits obtained via a high-throughput screen.38 There has been less attention focused on ERRβ specific modulators, though a number of ERRγ ligands, (e.g. 1-3) show activity on ERRβ. This is likely a consequence of the close homology between the LBDs of ERRβ and ERRγ.4, 26
We recently reported on the identification and characterization of SLUPP-332 (6), a structural analog of GSK compound, 3, which was found to be a potent agonist (See Figure 2) on all three ERR isoforms.39 Subsequent reexamination of the acyl hydrazide scaffold generated several analogs which profiled as pan ERR agonists with activity on ERRα in addition to ERRβ and ERRγ.40 Compound 6 was very potent for ERRα (EC50 = 98 nM), with roughly a 2-fold drop in potency on ERRβ (EC50 = 230 nM), and with a 4-fold drop on ERRγ (EC50 = 428 nM). In cell-based assays, it was shown to activate several ERR target genes involved in metabolic processes. It was found to be stable in both human and mouse liver microsomes (T1/2 = ≥ 60 min) and when dosed in mice we observed a similar gene upregulation profile in harvested muscle tissue as was observed in the cell-based experiments. Additionally, in mice, 6 induced an exercise phenotype showing increased oxidative muscle fibers, increased fatty acid oxidation and enhanced exercise endurance.39
Figure 2.
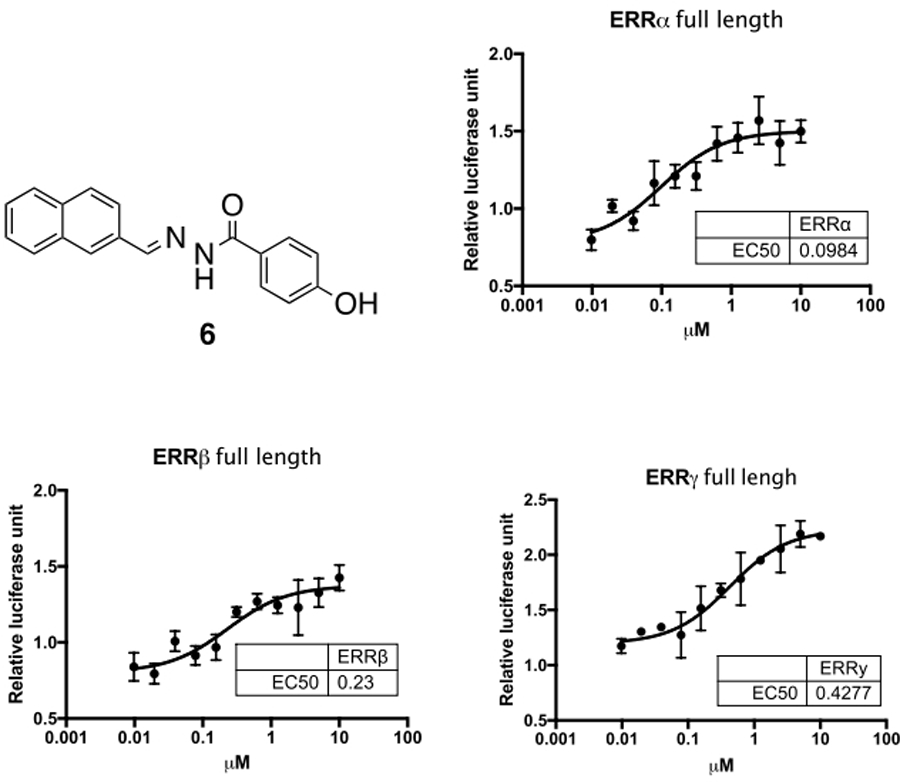
Structure and ERR co-transfection data for Pan-ERR agonist SLU-PP-332 (6)
Given the important role ERRs play in regulating the gene transcription involved in several metabolic functions as well as other pathological processes there is significant therapeutic potential for modulators of ERR, we have been engaged in efforts to generate novel ERR modulators. Herein, we describe our efforts and some of the corresponding pharmacology for a new series of ERRγ agonists utilizing a SBDD approach starting from acyl hydrazides such as 3 and 6, which ultimately led to the identification of a novel series of potent pan-ERR agonists.
2. Results and discussion
2.1. Compound design
We were interested in moving away from the acyl hydrazide scaffold due to potential concerns around the overall physicochemical properties of this scaffold. To identify new agonists of ERR we employed a structure-based approach. There is extensive X-ray crystallography data available with both APO and ligand bound structures for all 3 ERR isoforms.27–28, 41 The most studied of the 3 isoforms is ERRγ, which is the only one crystallized with an agonist ligand bound to the LBD, acyl hydrazide 3.42 Based on this structural information and the potential therapeutic importance of ERRγ, the focus of the SBDD efforts were around the ERRγ isoform. The X-ray of 3 (PDB:2GPP) bound to the LBD of ERRγ in the presence of a co-factor peptide from RIP-140 revealed several important binding features (Figure 3). The phenol acts as a hydrogen bond donor (HBD) making a strong hydrogen bond contact with ASP-328. The 4-isopropyl phenyl group sits in a lipophilic pocket and appears to be engaged in a π-stacking interaction with TYR-326. The acyl hydrazide moiety is engaged in several water-mediated contacts as well.
Figure 3.

Two-dimensional representation of GSK-4716 (3) bound in the LBD of ERRγ, PDB:2GPP
Our strategy is broadly outlined in Figure 4. The acyl hydrazide moiety was considered a potential liability from an in vivo standpoint, and replacements were sought. A ‘cyclization’ strategy where the hydrazide was replaced with a 5-membered ring leading to an acyl pyrazole structure appeared promising. Simplification leads to a generic heterocyclic central ring, i, where the acyl group is bonded directly to a carbon. To avoid a ketone moiety, further elaboration led to putting an amide substituent in the 2-position with a substituted phenyl group in the 5-position. It was speculated that the HBD group could be in either the amide moiety or the directly attached phenyl ring, giving a generic structure ii. Docking experiments were performed on representative compounds based on structure ii and gave reasonable docking scores with several of them maintaining the favorable interactions seen with 3 (Figure 3).
Figure 4.
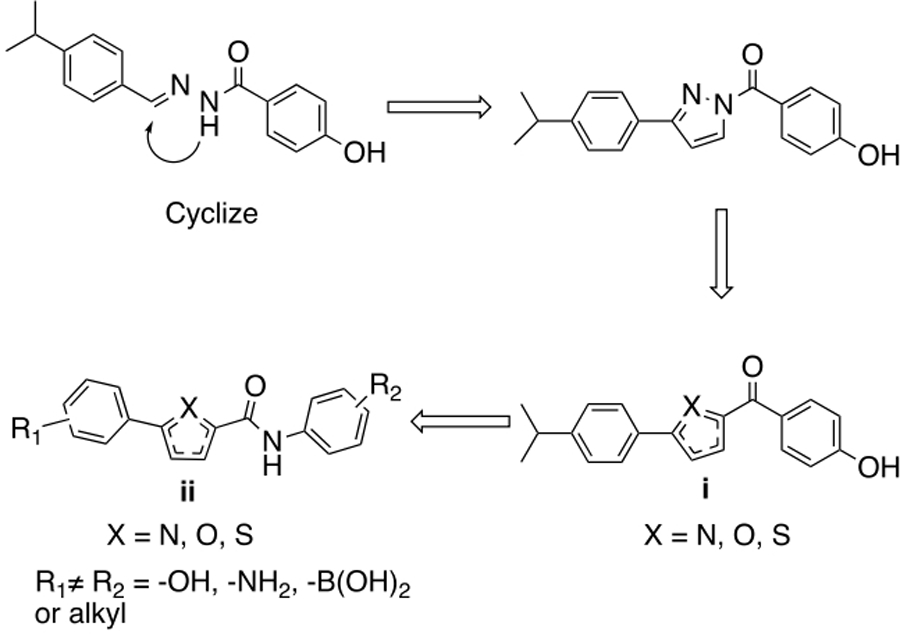
General structure-based design strategy to develop new ERR agonists
2.2. Compound synthesis.
Encouraged by the docking experiments, we set out to prepare analogs according to generic structure ii, focusing on the thiophene (X = S) as the central 5-membered ring. Thiophene was chosen because it modeled favorably in the docking experiments and the starting materials were readily available giving rapid entry into new analogs to test the design. The synthesis of the initial set of analogs with the 4-hydroxyphenyl group fixed in the C(5)-position is shown in Scheme 1. The follow up analogs where the C(5)-substituent was varied (Scheme 2) were prepared in a similar manner. These compounds were typically prepared in a two-step process starting from 5-bromothiophene-2-carboxylic acid (7). The group containing the HBD was introduced last which prevented the need for protecting groups. In general, the amide substituent was prepared via a TBTU-mediated coupling between the acid and the desired amine. In some cases, the acid was instead first converted to an acid chloride followed by direct addition of the amine. The C(5)-aromatic group was introduced via a standard Suzuki coupling reaction with aryl boronic acid and the bromothiophene. Analogs containing a boronic acid were prepared through a Suzuki coupling of the di-boronic acids. When the HBD group was in the amide moiety the Suzuki reaction was carried out first on bromide 7 followed by the amide coupling (Scheme 3).
Scheme 1.
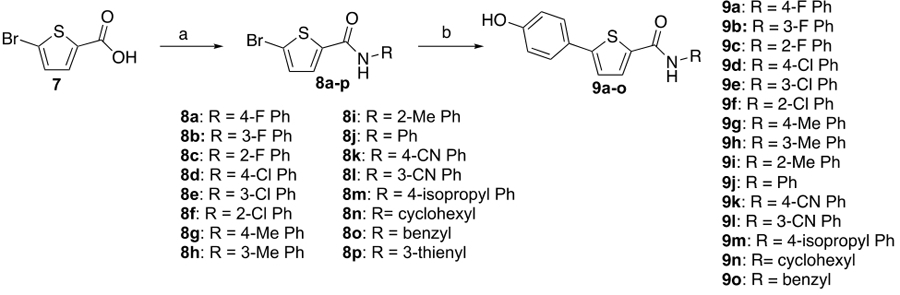
Synthesis of C(5)-hydroxyphenyl thiophene analogsa
Reaction conditions: (a) RNH2, DIPEA, TBTU, DMF (b) 4-hydroxyphenyl boronic acid, Pd(PPh3)4; K2CO3, DMF, 90 °C
Scheme 2.
Synthesis of 2,5-disubstituted thiophene analogsa
Reaction conditions: (a) Boronic acid, Pd(PPh3)4; K2CO3, EtOH or DMF, heat or Boronic acid, Pd(dppf)CI2-CH2CI2; Na2CO3, dioxane, 80°C
Scheme 3.
Synthesis of 2,5-disubstituted thiophene analogs with the HBD group in the amide substituent a
a Reaction conditions: (a) RNH2, DIPEA, TBTU, DMF (b) Boronic acid, Pd(PPh3)4; K2CO3, DMF, heat
2.3. Structure activity relationships and biological evaluation
2.3.1. Cell-based ERRγ co-transfection assays
All test compounds were evaluated for ERRγ activity in a cell-based co-transfection assay. HEK293T cells were transfected with full-length ERRγ along with a luciferase reporter driven by a synthetic ERR response element upstream of a minimal promoter vector. Compounds 1 and 6 were included in each assay to act as controls. For agonist activity, all compounds were compared against 6, which served as the agonist standard, while 4-OHT (1) was used as the inverse agonist standard. Compounds with an EC50 (≤ 1 µM) and profiling as full agonists, defined as those having a maximal efficacy ≥ 75% of that shown by 6, were considered for additional evaluation. Data for test compounds in the co-transfection assays are shown in Tables 1–3.
Table 1.
ERRγ co-transfection data for select analogs with the 2-carboxamide group modified and the C(5)hydroxyphenyl fixed

| |||||
| Cmpd # | R | EC50 (μM) (%)a | Cmpd # | R | EC50 (μM) (%)a |
|---|---|---|---|---|---|
| 9a |

|
0.343 (67) | 9i |

|
> 10b |
| 9b |

|
0.244 (122) | 9j |

|
0.220 (98) |
| 9c |

|
0.120 (50) | 9k |

|
> 10b |
| 9d |

|
1.51 (85) | 9l |

|
1.94 (90) |
| 9e |

|
0.301 (129) | 9m |

|
> 10b |
| 9f |

|
2.40 (109) | 9n |

|
0.66 (116) |
| 9g |

|
0.345 (72) | 9o |

|
3.17 (95) |
| 9h |

|
0.293 (64) | |||
% represents the % efficacy as compared to the standard, 6 (EC50 = 0.428 μM, Max = 2.24 RFUs, Min = 1.195 RFUs);
no efficacy calculated due to the lack of a saturation in the dose response curve
Table 3.
ERRγ data for representative analogs with the HBD group in the amide.

| |||||||
| Cmpd # | R1 | R2 | EC50 (μM) (%)a | Cmpd # | R1 | R2 | EC50 (μM) (%)a |
|---|---|---|---|---|---|---|---|
| 12a |

|
4-OH | >10b | 12g |

|
3-OH | > 10b |
| 12b |

|
4-OH | 0.687(60) | 12h |

|
3-OH | 0.84(83) |
| 12c |

|
4-OH | 2.87(150) | 12i |

|
3-B(OH)2 | 0.461(186) |
| 12d |

|
4-OH | 1.72(110) | 12j |

|
3-B(OH)2 | 0.526(65) |
| 12e |

|
3-OH | 1.38(67) | 12k |

|
3-B(OH)2 | 1.89(95) |
| 12f |

|
3-OH | 2.40(109) | 12l |

|
3-B(OH)2 | 9.57(60) |
% represents the % efficacy as compared to the standard, 6, (EC50 = 0.428 μM, Max = 2.24 RFUs, Min = 1.195 RFUs);
no efficacy calculated due to the lack of a saturation in the dose response curve
2.3.2. Modification of the 2-carboxyamide groups
The initial set of analogs that were prepared focused on varying substitution on the C(2)-carboxyamide group while keeping the C(5)-position on the thiophene ring fixed with the 4-hydroxyphenyl group (See Table 1). The fluorophenyl-substituted amides (9a-c) all gave good EC50 values but only the 3-fluoro analog (9b) gave efficacy (122%) comparable to that of 6. The chloro-substituted versions (9d-f) were less potent relative to their fluoro versions with 3-substitution again optimal with 9e being a potent (EC50 = 0.301 μM) agonist. Both the 2- and 4-substituted analogs had potencies > 1 μM. The methyl-substituted analogs (9g-i) were also examined. The para- (9g) and meta- (9h) compounds gave a very similar profile with potencies in the ~ 0.3 μM range and efficacies ~ 70% of 6. The ortho-substituted compound (9i) proved to be inactive, however. The unsubstituted phenyl amide 9j was a potent full agonist with an EC50 = 0.220 μM. The stronger electron-withdrawing (EW) cyano group was explored (9k-l). The 4-cyano derivative, 9k, was found to be inactive (EC50 >10 μM) while the 3-cyano analog gave diminished activity (EC50 = 1.94 μM) relative to 6. Larger alkyl substitution in the para- position also did not appear to be tolerated as the 4-isopropyl phenyl analog (9m) was found to be inactive. The saturated cyclohexyl analog, 9n, was tolerated, however, as it was a full agonist with an EC50 = 0.661 μM (116%). The more flexible benzyl derivative, 9o, gave only weak activity with an EC50 = 3.17 μM but was a full agonist (95%).
2.3.3. Modification of the C(5)-phenyl group
Next, we evaluated changes to the C(5)-phenolic group while maintaining some of the better carboxamide substituents from above (Table 2). Several analogs with the phenol moved around the phenyl or replaced by different moieties that could act either as a HBD or hydrogen bond acceptor (HBA) were prepared. Somewhat surprisingly, the 4-methyl sulfone compound, 10a, displayed comparable activity with an EC50 = 0.242 μM and efficacy 86% of 6 while the 3-sulfone analog, 10b, was found to be inactive (EC50 > 10 μM). The carboxylate derivatives were also examined. The 4-carboxy compound, 10c, did not give accurate readings due to solubility issues but the 3-carboxy analog, 10d, was found to be a weak agonist (66%) with modest activity, EC50 = 2.06 μM. The hydroxymethyl analogs, 10e-f, were both found to be inactive.
Table 2.
ERRγ data for select analogs with modifications to the C(5)-hydroxy phenyl group

| |||||||
| Cmpd # | R | Ar | EC50 (μM) (%)a | Cmpd # | R | Ar | EC50 (μM) (%)a |
|---|---|---|---|---|---|---|---|
| 10a | 4-SO2Me | 3-FPh | 0.242(86) | 10k | 3-OH | 4-MePh | 0.704(120 |
| 10b | 3-SO2Me | 3-FPh | > 10b | 10l | 3-OH | 4-ClPh | 1.74(156) |
| 10c | 4-CO2H | 3-FPh | ND | 10m | 3-OH | 3-ClPh | 0.390(78) |
| 10d | 3-CO2H | 3-FPh | 2.06 (66) | 10n | 3-OH | 3-thienyl | 1.21(60) |
| 10e | 4-CH2OH | 3-FPh | > 10b | 10o | 3-NH2 | 3-FPh | 1.11(90) |
| 10f | 3-CH2OH | 3-FPh | > 10b | 10p | 3-NH2 | 3-ClPh | 0.185(130 |
| 10g | 4-OH, 3-CF3 | 3-FPh | > 10b | 10q | 3-B(OH)2 | 3-FPh | 0.238(214 |
| 10h | 4-OH, 3-Cl | 3-FPh | 1.48(72) | 10r | 4-B(OH)2 | 3-FPh | 0.182(53) |
| 10i | 3-OH | 4-FPh | 0.740(76) | 10s | 3-B(OH)2 | 2-FPh | 0.378(167 |
| 10j | 4-OH | 3-FPh | 0.156(70) | ||||
% represents the % efficacy as compared to the standard, 6, (EC50 = 0.428 μM, Max = 2.24 RFUs, Min = 1.195 RFUs);
no efficacy calculated due to the lack of a saturation in the dose response curve
A few analogs (10g-h) were prepared with simple substituents flanking the phenol group. A representative analog (10g) with the –CF3 group in the phenyl ring was found to be inactive while the corresponding –Cl analog (10h) was active but with reduced potency ~ 5-fold relative to its corresponding des-chloro version (9b). Next, a series of analogs were prepared with the phenol moved to the meta-position in combination with different substituted amides (10i-10n). The 4-fluorophenyl amide, 10i, was an agonist (76%) with an EC50 = 0.74 μM. When the fluorine was moved to the 3-position, 10j, potency improved with an EC50 = 0.156 μM and efficacy 70% of that observed for 6. The 4-methyl analog (10k) was shown to be a full agonist with a modest EC50 (0.704 μM, 120%) while the 4-chloro analog (10l) was also a full agonist but was less potent than the methyl version with an EC50 = 1.74 μM (156%). Moving the chlorine to the 3-position (10m) yielded a more potent agonist (EC50 = 0.390 μM, 78%) albeit less potent than the corresponding 3-fluoro compound, 10j. The heterocyclic derived 3-thienyl amide, 10n, was less potent (EC50 = 1.21 μM, 60%) than the phenyl amides.
Next a series of analogs were prepared with the phenol being replaced with other groups that should be capable of acting as HBD’s, such as anilines (10o-p) and boronic acids (10q-s). Aniline compound 10o, with the 3-fluorophenyl amide was a full agonist but with ~ 10-fold weaker potency (EC50 = 1.11 μM) than the corresponding phenol, 10j. When the fluorine on the amide was changed to chlorine 10p, the potency was substantially improved (EC50 = 0.185 μM) and it was a full agonist (130%). Boronic acid moieties (10q-10s) were also examined. The pKa of phenyl boronic acids typically are in the 8.0 – 9.0 range suggesting they should be protonated at physiological pH and capable of acting as a HBD. When the boronic acid was in the meta-position (10q & 10s) the compounds were both potent full agonists (EC50 = 0.238 and 0.378 μM, respectively) while the 4-boronic acid compound, 10r, had affinity (EC50 = 0.182 μM) but profiled more as a partial agonist (53%).
2.3.4. Analogs where the HBD group is located in the amide substituent
A small series of analogs with the HBD group in the amide moiety and the lipophilic group attached directly to the thiophene as the C(5) substituent where also prepared and evaluated. (See Table 3). A set of C(5)-fluorophenyl analogs with either the 4-hydroxyphenyl amide (12a-d) or 3-hydroxyphenylamides (12e-h) were explored. In general, the potency of these compounds was lower than their corresponding analogs where HBD group was in the C(5) phenyl ring. The best of this group was 12h, which had an EC50 = 0.84 μM and profiled as a full agonist (83%). Analog 12b was a little more potent (EC50 = 0.687 μM) but had an efficacy only 60% that of 6. A few analogs where the C(5) phenyl ring was replaced by 3-thienyl group were also evaluated (12d-e). The activity for 12d and 12e was in the low micromolar range. A few analogs with the boronic acid in place of the phenol were also explored (12i-l). The most active in this group was 12i, with the boronic acid in the 3-position and the 4-F phenyl. It had an EC50 = 0.46 μM and was a full agonist (186%). The 3-F phenyl (12j) version was comparable in EC50 but a smaller Emax while the 2-F phenyl (12k) and thiophene analog (12l) were less potent overall.
2.3.5. NMR binding studies
To further establish that these compounds were directly binding to ERRγ, a series of protein-ligand NMR binding studies were performed and the 1H-NMR spectra of ERRγ protein in the presence and absence of test compounds were analyzed. Analysis of the solution NMR peak line shape can be used to indicate dynamic processes including conformational changes and binding events.43 We focused on a small group of active compounds (9b, 10o, 10s and 12e) and monitored the perturbation of the side-chain methyl 1H-NMR profiles of ERRγ LBD in the presence of 1.05:1 molar equivalence of the potential ligands as compared to that of unbound ERRγ protein, ERRγ bound to agonist standard, 6, and ERRγ with inverse agonist 1. Spectral data for a known liver X receptor (LXR) inverse agonist SR-9238, which does not bind to ERRγ to act as a negative control and should have no effect on the NMR spectrum of ERRγ was also obtained.44 Protein methyl side-chain peak perturbation analysis is a convenient approach to monitor ligand binding as these resonances are in shielded, less chaotic regions of the 1H NMR spectra that circumvents overlap with buffer and ligand peaks. As expected, the ERRγ side-chain methyl region peaks (i.e., chemical shift and/or peak intensity changes), which consists of shielded Leu, Ile and Val methyl resonances at 0.46 ppm, 0.33 ppm, 0.31 ppm, -0.06 ppm, and -0.15 ppm, are unchanged in the presence of SR-9238 (See Figure 5 and Supporting Figure S2) whereas there is significant perturbation of all peaks in this region when agonist 6 is present (Figure 5). In addition, the shoulder at 0.25 ppm, which is likely another methyl peak overlapping with the peak at 0.31 ppm, disappeared after adding 6. The 1H-NMR experiments for 10s and 12e show similar perturbation to that of agonist 6 suggesting a similar binding epitope. In contrast, ERRγ inverse agonist 1 demonstrated a more significant peak perturbation pattern distinct from the agonist ligands (Supplemental Figures S1 and S2). The substantial methyl peak perturbations induced by 1 binding suggest a large-scale conformational change in ERRγ when bound to inverse agonists. Agonist binding, on the other hand, could potentially induce a different conformational change that affect the side chain methyl groups to a less extent, but resulting in statistically significant peak shifts and peak intensity changes of the corresponding 1H-NMR resonances (See Supplemental Figure S2). Peak shifts are calculated as changes in peak position (in ppm or Hz) of the resonances in the presence of 1.05:1 molar equivalence of ligand minus the peak position in the presence of the DMSO-d6 vehicle. Meanwhile, peak intensity change is assessed by calculating the ratio of peak intensity in the presence of ligand with respect to that of vehicle alone. Based on three independent replicates, a peak shift > 0.002 ppm (1.3 Hz) and peak intensity ratio less than 0.99 or greater than 1.01 are determined to be statistically significant at 95% confidence level (i.e., within two standard deviations from the mean). We studied additional ligands, 9b and 10o, which also induced subtle but statistically significant perturbations to side chain methyl peaks consistent with 6, albeit less than what was observed with 10s and 12e (Supplemental Figure S1).
Figure 5.
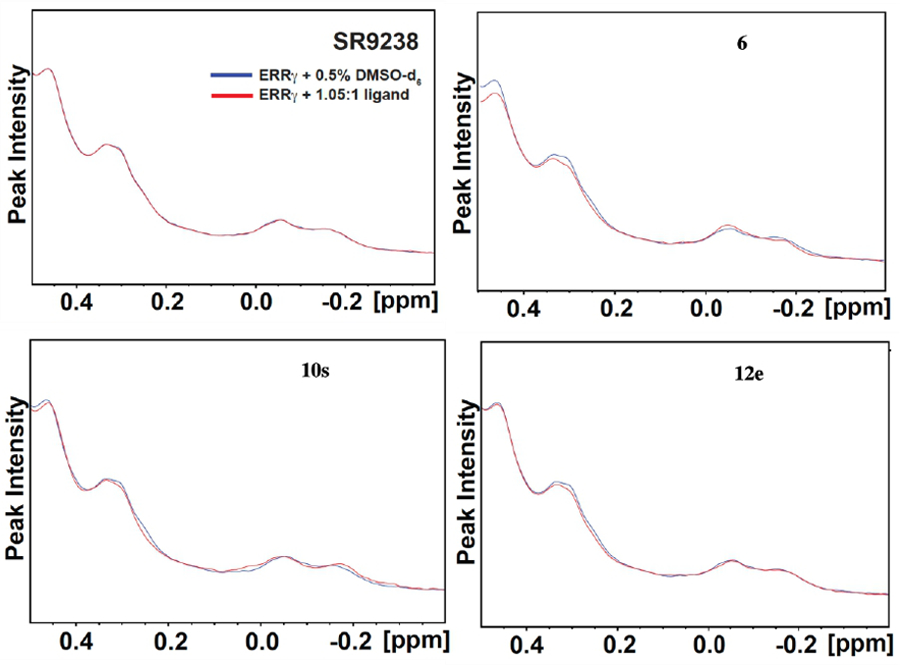
1H-NMR methyl region spectra of ERRγ with 0.5% DMSO-d6. where the blue line represents vehicle alone and the red line is protein + 1.05:1 molar equivalence of ligand. A) protein + SR-9238 B) protein + 6 C) Protein + 10s D) Protein + 12e
2.3.6. Docking Studies
Docking experiments were performed using Schrödinger Maestro.45 Several of the more potent analogs including 9b, 10o, 10s, 12e and 12i were docked and a comparison was made between their optimal poses and Glide scores to compound 6 (Glide score = -11.348).46 It was anticipated that the preferred docking pose for these compounds would be similar to that observed with acyl hydrazides 3 and 6. We expected that the HBD group would be engaged with ASP-328 while the phenyl amide moiety on the opposite side of the thiophene ring would sit in the lipophilic pocket and make the π-stacking interaction with TYR-326. For compounds 9b, 10o and 10s, however, two distinctly different low energy docking poses were seen. The anticipated pose seen with 6 described above and a second low energy conformation with the compounds flipped ~180 °C putting the HBD moiety away from the ASP-328 was also observed. Somewhat surprisingly this flipped conformation was the lower energy pose for these 3 analogs. Figure 6 shows these poses for 9b and 10s. The docking pose of 9b (Fig. 6A) in the anticipated conformation exhibited the expected interactions between the phenol and ASP-328 and the π-stacking interaction between the 3-fluorophenyl amide and TYR-326 and gave a Glide score = -10.149. One interaction not observed in these docking poses, that is seen with the acyl hydrazide, is the water mediated hydrogen-bonding between the TYR-326 and amide bond. Alternatively, the ‘flipped conformation (Fig. 6B) gave a lower Glide score = -10.627, suggesting this pose may be more favorable. In this conformation, the phenol group is flipped away from ASP-328 and sits deep in the lipophilic pocket. Here the phenolic group makes a new π-stacking interaction with PHE-435 while the thiophene makes a second π-stacking interaction with TYR-326. Additionally, there is a H-bonding interaction between TYR-326 and the amide -NH bond. The boronic acid analog 10s gave a similar result. The anticipated docking conformation mimicking 6 was found to have a good Glide score = -9.115 (Fig. 6c). In this conformation, one hydroxyl group of the boronic acid moiety engages ASP-328, as expected, and the 2-fluorophenyl makes the expected π-stacking interaction with TYR-326 along with a second π-stacking with PHE-435. The flipped conformation, however, gave a much better Glide score = -11.253, the best observed for any analog, also suggesting this pose might be more favorable. As we observed with 9b, the HBD group (boronic acid) is flipped and sits in the lipophilic pocket, but in this pose a boronic acid hydroxyl group can engage in a H-bond with a backbone LEU-268. The TYR-326 makes π-stacking interactions with both the phenyl boronic acid moiety and the thiophene ring. Additionally, H-bonding between the amide and TYR-326 is again observed in this ‘flipped conformation. The aniline analog, 10o, gave similar results to 9b and 10s (see Supplemental Figs. 3a and 3b). Interestingly though, both 12e and 12i (Supplemental Figs. 3c and 3D) only give the anticipated docking poses with Glide scores = -10.751 and 10.563, respectively. In our docking experiments, analogs with the HBD in the C(5)-phenyl rig (i.e. 9b, 10o and 10s) gave two potential low energy docking poses while analogs with the HBD group fixed in the amide phenyl group gave only the expected docking pose.
Figure 6.
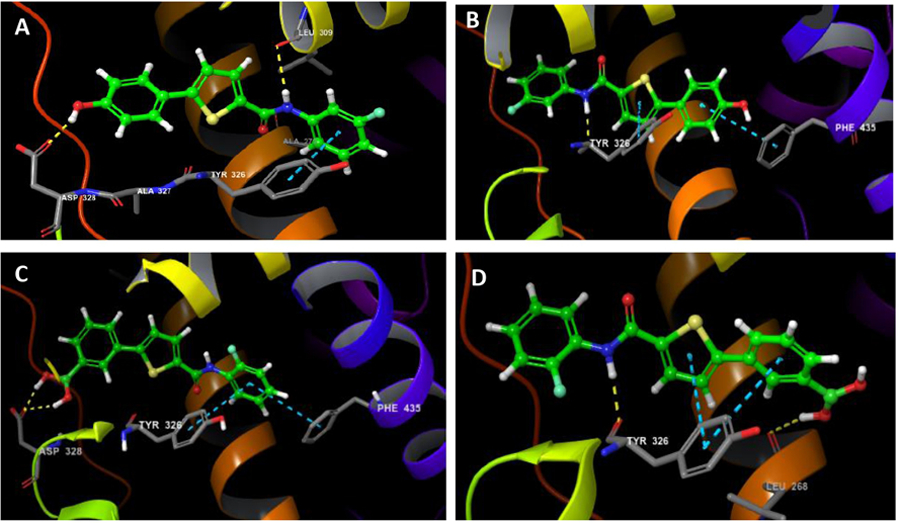
Molecular modeling of key compounds docked in the ligand binding domain of ERRγ obtained from the X-ray structure of 3 (PGB:2GPP). Compounds are shown in green. A) 9b docked in the anticipated compound 6 like orientation B) 9b docked in the ‘flipped’ conformation C) Compound 10s in the compound 6 like orientation. D) 10s in the ‘flipped’ conformation
2.3.7. Receptor Selectivity
A few analogs were evaluated for activity against both estrogen receptors, ERα and ERβ, as well as the other two ERR isoforms (α and β). A representative set of analogs (9b, 10o, 10r and 10s) was examined against both ERα or ERβ (data not shown) and found to have no activity against either. Several representative compounds were also evaluated against both ERRα and ERRβ (See Table 4). Since the current analogs were designed from the acyl hydrazide scaffold, it was not clear what to expect since 3 does not have significant activity on ERRα while 6 is a very potent agonist for it. Activity for all test compounds were again compared to 6. The analogs containing the 4-phenoxy group in the C(5)-position (9c, 9d, 9f, 9e & 9n) displayed either less potency for ERRα or gave a lower Emax’s relative to 6. The most active compound on ERRα of this set was 9e with an EC50 = 0.498 and an Emax 87% of that seen with 6. Several analogs shown in Table 2 were also evaluated and found to give a range of activities on ERRα. Compounds such as 10o, 10p and boronic acid 10s were the most active overall in terms of efficacy and Emax’s. These analogs were roughly 4, 5-fold less potent on ERRα than hydrazide 6. Interestingly, the other boronic acid analogs 10q and 10r were roughly equivalent in potency but gave lower Emax values on ERRα. Amides 12e and 12i, bearing the HBD in the amide moiety were also screened against ERRα and both analogs were found to be less potent against ERRα relative to 6 but with comparable or better Emax’s. In general, the compounds tested against ERRβ were all found to be potent agonists very comparable to 6. One exception was 9n which gave an Emax of only 56% that of 6. Also, both 12e and 12i were found to be less potent on ERRβ relative to the other analogs from this series. Overall, the disubstituted thiophene series does not appear to have activity against the ER’s but has pan-ERR activity. In general, activity against ERRα is lower than that observed for 6 but roughly equivalent on ERRβ and ERRγ
Table 4.
Co-transfection assay data for ERR isoforms for select compounds
| CMPD # | ERRα EC50(μM) (% eff)a,b | ERRα/ERRγ | ERRβ EC50(μM) (% eff)a,b | ERRβ/ERRγ | ERRγ EC50(μM) (% eff)a,b |
|---|---|---|---|---|---|
| 6 | 0.098 | 0.23 | 0.23 | 0.54 | 0.428 |
| 9c | 0.118(20) | 0.98 | ND | - | 0.120(50) |
| 9d | 0.976(51) | 4 | ND | - | 0.244(122) |
| 9e | 0.498(87) | 1.65 | 0.233(105) | 0.77 | 0.301(129) |
| 9f | 2.02(66) | 0.84 | ND | - | 2.40(109) |
| 9n | 0.577(28) | 0.87 | 0.371(56) | 0.56 | 0.661(116) |
| 10a | 0.118(69) | 0.49 | ND | - | 0.242(86) |
| 10d | 1.605(52) | 0.78 | ND | - | 2.06(66) |
| 10k | 1.06(76) | 1.5 | 0.287(110) | 0.41 | 0.704(120) |
| 10o | 0.460(259) | 0.41 | 0.258(154) | 0.23 | 1.11(90) |
| 10m | 0.816(80) | 2.1 | ND | - | 0.390(78) |
| 10p | 0.498(87) | 2.7 | 0.082(136) | 0.44 | 0.185(130) |
| 10q | 0.108(59) | 0.45 | 0.117(107) | 0.49 | 0.238(214) |
| 10r | 0.030(30) | 0.16 | 0.089(120) | 0.49 | 0.182(53) |
| 10s | 0.414(90) | 1.1 | 0.435(95) | 1.2 | 0.378(167) |
| 12e | 1.01(82) | 0.72 | 0.950(105) | 0.66 | 1.38(67) |
| 12i | 0.659(149) | 1.4 | 0.759(121) | 1.65 | 0.461(186) |
values represent the arithmetic mean from two separate runs;
% eff is defined as the Emax ratio for test compound vs. compound 6 against both ERRα (EC50 = 0.098 μM, Max = 1.50 RFUs, Min = 0.78 RFUs), ERRβ (EC50 = 0.23 μM, Max = 1.37, Min = 0.80) and ERRγ (EC50 = 0.428 μM, Max = 2.24 RFUs, Min = 1.195 RFUs); ND = not determined
2.3.8. In Vitro microsomal stability
To prioritize compounds for potential advancement into in vivo models, several analogs were evaluated for in vitro stability in both mouse and human microsomal stability assays, which were performed at Eurofins Pharma Discovery Services.47 Compounds were incubated in mouse or human liver microsomes for up to 60 minutes and analyzed for the amount of parent compound remaining. The data for a representative set in both human and mouse is shown in Table 5. In the mouse liver microsomes (MLM), ranges of stabilities were observed within the compound set. The standard aniline and the phenol analogs showed a range of half-lives with a low of ~ 7 min (12e) to 38 min (10p). The one exception was 9n, with the saturated cyclohexyl group for the amide substituent in place of the phenyl group. This compound gave a T1/2 > 60 min. The compounds containing the boronic acid (10q, 10s & 12i) moiety all displayed stabilities in the microsome assay with half-lives > 60 min. In general, the compounds all displayed greater stability in the corresponding human liver microsomes (HLM) versus the mouse liver microsomes with the boronic acid analogs again showing excellent stability (T1/2 > 60 min).
Table 5.
Human and Mouse microsomal stability measured for select compounds
| Comp # | Mouse T1/2 (min)a,c | Human T1/2 (min)b,c |
|---|---|---|
| 9b | 24.2 | 41.1 |
| 9n | > 60 | > 60 |
| 10h | 55 | ND |
| 10o | 30 | 55 |
| 10p | 38.3 | > 60 |
| 10q | > 60 | > 60 |
| 10s | > 60 | > 60 |
| 12e | 7.4 | 12.7 |
| 12i | > 60 | > 60 |
Measured in mouse liver microsomes;
measured in human liver microsomes;
Compounds were tested at 0.1 μM and half-life was measured as the average of 3 replicates. ND = not determined
2.3.9. In vitro and in vivo characterization of SLU-PP-915 (10s)
Seven of the more active compounds were evaluated for the ability to upregulate known ERR targets genes in a mouse myoblast (C2C12) cell culture at a single 5 μM dose after 24hrs of treatment. Three target genes were looked analyzed for: PGC1α, PDK4, which codes for the enzyme pyruvate dehydrogenase lipoamide kinase isozyme 4 and LDHA which codes for the enzyme lactate dehydrogenase A. PGC1α has been characterized as one potential mediator of ERR activity.48 Moreover, both PGC1α and ERR have similar expression patterns and they are induced in skeletal muscle after exercise.49 PDK4 is a well characterized ERR target gene involved in glucose metabolism while LDHA catalyzes the interconversion between pyruvate and L-lactate.50 The data is shown in Figure 7. For PGC1α, compounds 9n, 10o, 10s and 12i gave a significant increase in relative gene expression. For PDK4, however, compounds 9b, 9n, 10o, 10s and 12e demonstrated increased gene expression levels relative to control. Finally, with LDHA all seven of the compounds gave increased levels of gene expression with 9b giving the largest increase in relative gene expression.
Figure 7.
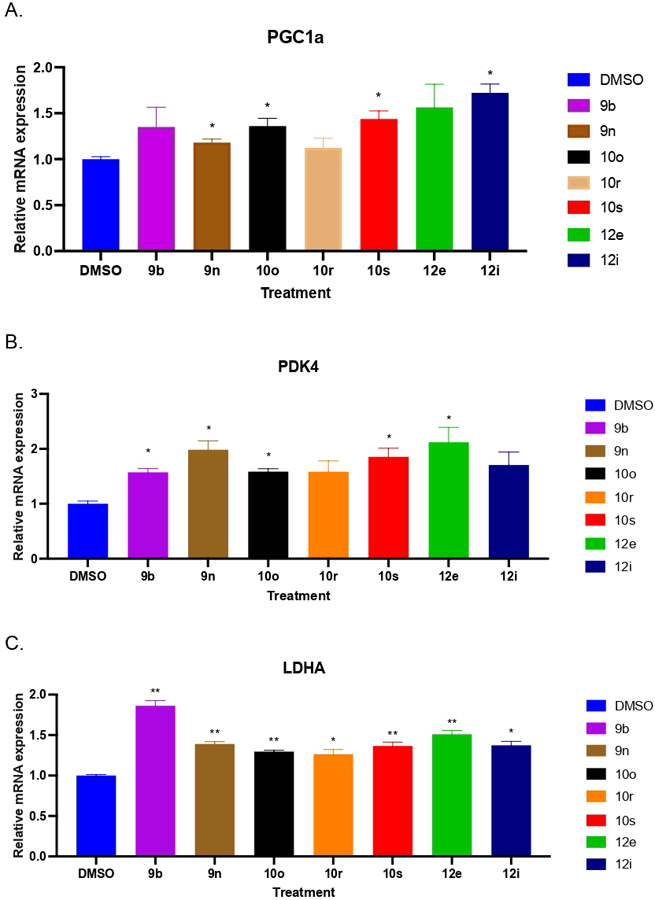
Gene expression in C2C12 myoblast cells treated with several compounds at 5uM for 24hrs. A. Relative gene expression levels for PGC1α vs control B. Relative gene expression levels for PDK4 vs control C. Relative gene expression levels for LDHA vs control. n=3 per condition, * p<0.05, ** p<0.01.
We next chose to take one of the compounds forward for preliminary evaluation in vivo. Compounds 9n, 10o and 10s gave increased gene expression against all 3 target genes and while 9n and 10s had good metabolic stability in the both the microsome assays. Compound 10s (internal designation SLU-PP-915) was selected for further evaluation in vivo because of its greater potency in the ERR co-transfection assays, especially ERRγ.
The effect on gene expression in mice when given a single dose of SLU-PP-915 was evaluated. On hour after injection with SLU-PP-915 (20mg/kg, i.p) or vehicle control, we assessed the level of expression of known ERR target genes in quadricep muscles. Quadriceps are mixed muscles composed of both glycolytic and oxidative fibers, display high mitochondrial content, and ERR expression. We evaluated both the PDK4 and PGC1α were genes. In this experiment, the DDIT4 gene, which codes for DNA-damage-inducible transcript 4 was also included in the analysis. DDIT4 is a regulator of an acute exercise genetic program in both human and rodents. Recently DDIT4 has been characterized as an ERRα specific target gene regulated by the synthetic ERR agonist SLU-PP-332 (6) after 2hrs both in vivo and in vitro.39
As expected, we observed an up regulation of DDIT4, PDK4 and PGC1α expression in muscles one hour after I.P injection of SLU-PP-915 compared to vehicle control (Figure 8A). Moreover, based on our recently reported studies with ERR pan-agonist 6 we assessed running capacity in mice treated with SLU-PP-915 compared to vehicle.39 As expected, SLU-PP-915 increased running distance and time in treated animals compare to vehicle-treated animals (Fig. 8B, see method for protocol). These data confirm the promising role of SLU-PP-915 as a specific ERR-modulator and its potential application in disease model, in particular muscle dysfunction.
Figure 8:
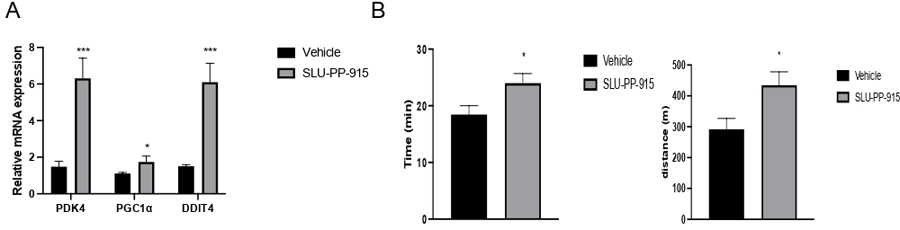
ERR agonist SLU-PP-915 (10s) induces expression of ERR target genes in vivo: A) Gene expression from muscle from mice treated with SLU-PP-915 (20mg/kg, I.P) for 1h (n=3 per group). B. Running time and distance from mice 1h after being treated with SLU-PP-915 (20mg/kg, I.P). * p<0.05, ** p<0.01.
3. Conclusion
Utilizing a SBDD strategy, a series of 2,5-disubstitued thiophene amides were prepared and their activity for ERRγ was measured. Several analogs were found to be potent agonists for ERRγ in co-transfection assays. NMR ligand-protein binding experiments were conducted with a few of the more potent agonists and confirmed binding to the LBD of ERRγ. Several of the ERRγ agonists were also found to be potent agonists on both the ERRα and ERRβ isoforms but had no activity on either of the estrogen receptors. In general, compounds displayed roughly equal activity on all three ERR isoforms (α, β and γ) in the cell-based co-transfection assays, which differed from compound SLU-PP-332 (6) which showed a preference for ERRα. NMR ligand-protein binding studies established binding to the LBD of ERRγ. Several compounds subsequently were profiled for in vitro metabolic stability. Replacement of the putative HBD phenol with a boronic acid moiety was sufficient for retaining activity while also consistently demonstrating improved metabolic stability in both human and mouse microsomes. One of the compounds was the boronic acid containing analog, SLU-PP-915 (10s), which was a potent ERR agonist able to up regulate ERR target genes such as PGC1α, LDHA and PDK4 cell-based experiments and PDK4 and DDIT4 in vivo. Recent studies with ERR agonist 6 demonstrate that activation of ERR’s lead to increase in several metabolic parameters that mimic an exercise phenotype with potential therapeutic utility in treating type-II diabetes and other metabolic diseases. This new series of ERR agonists such as SLU-PP-915, profile like 6 in terms of their ability to upregulate ERR target genes and mimic an exercise phenotype in mouse. In our hands, SLU-PP-915 was also easier to formulate for in vivo studies compared to 6. These compounds should prove extremely useful in further defining the therapeutic potential for agonists of this nuclear receptor family.
4. Experimental Section
4.1. Chemistry
General synthetic methods: Unless otherwise noted, all materials were obtained from commercial vendors and used as is without further purification. All reactions were monitored by using thin-layer chromatography (TLC) on silica gel coated glass plates or by high-pressure liquid chromatography (HPLC: Ascentis express peptide ES C-18 column, OD 3cm X 4.6cm, 6min flow rate 1 mL/min; gradient = 95/5 → 5/95 CH3CN-H2O). Flash column chromatography was performed on a Combi-Flash ISCO with prepacked silica gel columns. 1H NMR spectra were recorded on a Bruker Avance II nuclear magnetic resonance spectrometer (400 MHz) with chemical shifts reported in δ ppm relative to DMSO-d6 or CDCl3 (s = singlet, d = doublet, t = triplet, m = multiplet, dd = doublet of doublets, etc.). 13C NMR and 19F NMR spectra were obtained on the same instrument at 100 MHz and 376 MHz, respectively. Final compound purities were determined by HPLC using the conditions described above. High-resolution mass spectrometry was performed at the College of Science Major Instrumentation Center, Old Dominion University, on a Bruker 12 Tesla APEX -Qe FTICR-MS with an Apollo II ion source.
4.1.1. Synthetic procedures
4.1.1.1. 5-bromo-N-(4-fluorophenyl)thiophene-2-carboxamide (8a). To a solution of 5-bromo-2-thiophenecarboxylic acid (0.100 g, 0.4830 mmol) and TBTU (0.1551 g, 0.4830 mmol) in dry DMF (4.8 mL) was added DIPEA (0.210 mL, 1.207 mmol). The reaction was stirred at room temperature for 20 min and then 4-fluoroaniline (0.0644 g, 0.5795 mmol) was added. The reaction was stirred overnight at room temperature. The reaction was quenched with water to precipitate the product. The mixture was filtered through a fritted funnel by vacuum suction. The solid was triturated with cold methanol, the solvent was removed by vacuum suction, and the product dried. The product was isolated as a tan solid in 77% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.33 (s, 1H), 7.85 (d, J = 4.0 Hz, 1H), 7.71 (dd, J = 5.0 Hz, 9.1 Hz, 2H), 7.38 (d, J = 4.0 Hz, 1H), 7.21 (dd, J = 8.9 Hz, 8.9 Hz, 2H); LCMS: m/z calcd. for C11H17BrFNOS [M+H]+: 299.9 & 301.9; found [M+H]+: 300.0 & 302.0.
4.1.1.2. N-(4-fluorophenyl)-5-(4-hydroxyphenyl)thiophene-2-carboxamide (9a) Potassium carbonate (0.0368 g, 0.2666 mmol) and 4-(hydroxyphenyl)boronic acid (0.0184 g, 0.1333 mmol) were added to a 2-dram scintillation vial with deionized water (0.25 mL) and absolute EtOH (2 mL). The vial was purged with argon. Next 5-bromo-N-(4-fluorophenyl)thiophene-2-carboxamide (0.040 g, 0.1333 mmol) was added, followed by Pd(PPh3)4 (0.0055 g, 0.0048 mmol). The vial was purged with argon again then heated at reflux for 10 h. The reaction was cooled, acidified with 10% HCl, and extracted with ethyl acetate. The organic layer was washed with saturated aqueous NaHCO3, dried over anhydrous Na2SO4, and concentrated by rotary evaporation. The crude material was purified by flash chromatography (47% EtOAc:Hex). The desired product was isolated in ≥ 95% purity as a pale-yellow solid in 63% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.24 (s, 1H), 9.85 (s, 1H), 7.96 (d, J = 4.0 Hz, 1H), 7.75 (dd, J = 5.0 Hz, 9.2 Hz, 2H), 7.57 (d, J = 8.7 Hz, 2H), 7.42 (d, J = 4.0 Hz, 1H), 7.20 (t, J = 8.9 Hz, 2H), 6.85 (d, J = 8.7 Hz, 2H); LC-MS: 2.530 min HRMS m/z calcd for (C17H12FNO2S)2Na+ dimer 649.1038, found of spray dimer 649.1029.
4.1.1.3. 5-bromo-N-(3-fluorophenyl)thiophene-2-carboxamide (8b) was prepared from 7 and 3-fluoroaniline according to the procedure of 8a and obtained as a light brown solid in 36% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.44 (s, 1H), 7.87 (d, J = 4.1 Hz, 1H), 7.67 (dt, J = 11.7 Hz, 2.2 Hz, 1H), 7.49 (d, J = 8.2 Hz, 1H), 7.44–7.37 (m, 2H), 6.96 (td, J = 8.3 Hz, 2.5 Hz, 1H); LCMS: 2.706 min, m/z calcd. for C11H17BrFNOS [M+H]+: 299.9 & 301.9; found [M+H]+: 299.9 & 302.0.
4.1.1.4. N-(3-fluorophenyl)-5-(4-hydroxyphenyl)thiophene-2-carboxamide (9b) was prepared from 8b and 4-(hydroxyphenyl)boronic acid according to the procedure described for 9a.The desired product was isolated in ≥ 95% purity as a tan solid in 67% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.34 (s, 1H), 9.84 (s, 1H), 7.98 (d, J = 4.0 Hz, 1H), 7.71 (dt, J = 11.7 Hz, 2.2 Hz, 1H), 7.58 (d, J = 8.6 Hz, 2H), 7.52 (d, J = 8.2 Hz, 1H), 7.44 (d, J = 4.0 Hz, 1H), 7.40 (dt, J = 6.9 Hz, 8.2 Hz, 1H), 6.93 (dt, J = 2.0 Hz, 8.3 Hz, 1H), 6.85 (d, J = 8.7 Hz, 2H); 13C NMR (100 MHz, DMSO-d6) δ 160.5, 158.7, 150.2, 141.0, 137.0, 131.2, 130.8, 130.7, 127.8, 124.5, 123.0, 116.5, 116.3, 107.5, 107.2; 19F NMR (376 MHz, DMSO-d6) δ -112.13; LC-MS: 2.586 min; HRMS m/z calcd for (C17H12FN2O2S)Na+ dimer 649.1038, found of spray dimer 649.1039.
4.1.1.5. 5-bromo-N-(2-fluorophenyl)thiophene-2-carboxamide (8c) was prepared from 7 and 2-fluoroaniline according to the procedure for 8a. The product was purified by flash chromatography (15% EtOAc:Hex) and isolated as a dark oil in 55%. 1H NMR (400 MHz, DMSO-d6) δ 10.30 (s, 1H), 7.91 (d, J = 4.0 Hz, 1H), 7.61 (t, J = 7.1 Hz, 1H), 7.43 (d, J = 4.0 Hz, 1H), 7.38–7.32 (m, 2H), 7.30–7.25 (m, 1H), 2.74 (s, 1H). LCMS: m/z calcd. for C11H17BrFNOS [M+H]+: 299.9 & 301.9; found [M+H]+: 299.9 & 302.0.
4.1.1.6. N-(2-fluorophenyl)-5-(4-hydroxyphenyl)thiophene-2-carboxamide (9c) was prepared from 8c and 4-(hydroxyphenyl)boronic acid according to the procedure for 9a. The product was isolated in 94% purity as a light brown solid in 83% yield. 1H NMR (400 MHz, DMSO-d6) δ 11.00–10.20 (broad s, 1H), 10.11 (s, 1H), 7.99 (d, J = 4.0 Hz, 1H), 7.60 (d, J = 8.6 Hz, 2H), 7.56 (d, J = 1.7 Hz, 1H), 7.44 (d, J = 3.9 Hz, 1H), 7.28 (dd, J = 1.8 Hz, 8.4 Hz, 1H), 6.95 (d, J = 8.4 Hz, 1H), 6.88 (d, J = 8.6 Hz, 2H). LC-MS: 2.471 min; HRMS: m/z calcd. for (C17H12FNO2S)H+ 314.0646, found 314.0642.
4.1.1.7. 5-bromo-N-(4-chlorophenyl)thiophene-2-carboxamide (8d) was prepared from 7 and 4-chloroaniline according to the procedure for 8a. The product was isolated as a tan solid in 77% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.39 (s, 1H), 7.86 (d, J = 4.0 Hz, 1H), 7.74 (d, J = 8.9 Hz, 2H), 7.42 (d, J = 8.9 Hz, 2H), 7.38 (d, J = 4.0 Hz, 1H); LCMS: m/z calcd. for C11H7BrClNOS [M+H]+: 315.9 & 317.9; found [M+H]+: 315.9 & 317.9.
4.1.1.8. N-(4-chlorophenyl)-5-(4-hydroxyphenyl)thiophene-2-carboxamide (9d) was prepared from 8d and 4-(hydroxyphenyl)boronic acid according to the procedure for 9a. The product was purified by flash chromatography (37% EtOAc:Hex) and isolated in ≥ 95% purity as a tan solid in 26% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.28 (s, 1H), 9.83 (s, 1H), 7.97 (d, J = 4.0 Hz, 1H), 7.77 (d, J = 9.0 Hz, 2H), 7.57 (d, J = 8.6 Hz, 2H), 7.44–7.39 (m, 3H), 6.84 (d, J = 8.7 Hz, 2H); LC-MS: 2.701 min; HRMS: m/z calcd. for (C11H12ClNO2S)H+: 330.0350, found 330.0348.
4.1.1.9. 5-bromo-N-(3-chlorophenyl)thiophene-2-carboxamide (8e) was prepared from 7 and 3-chloroaniline according to the procedure for 8a. The product was isolated as an orange solid in 58% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.39 (s, 1H), 7.88–7.82 (m, 2H), 7.64–7.60 (m, 1H), 7.42–7.36 (m, 2H), 7.20–7.17 (m, 1H); LCMS: m/z calcd. for C11H7BrClNOS [M+H]+: 315.9 & 317.9; found [M+H]+: 315.9 & 317.9.
4.1.1.10. N-(3-chlorophenyl)-5-(4-hydroxyphenyl)thiophene-2-carboxamide (9e) was prepared from 8e and 4-(hydroxyphenyl)boronic acid according to the procedure for 9a. The product was isolated in ≥ 95% purity as a tan solid in 53% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.28 (s, 1H), 9.83 (s, 1H), 7.97 (d, J = 4.0 Hz, 1H), 7.90–7.88 (m, 1H), 7.67 (d, J = 8.0 Hz, 1H), 7.60–7.54 (m, 2H), 7.44–7.35 (m, 2H), 7.16–7.10 (m, 1H), 6.86–6.82 (m, 2H); LC-MS: 2.718 min; HRMS: m/z calcd. for (C11H12ClNO2S)H+: 330.0350, found 330.0349.
4.1.1.11. 5-bromo-N-(2-chlorophenyl)thiophene-2-carboxamide (8f). To a 2-dram vial containing 5-bromothiophene-2-carboxylic acid (0.207g, 1.0 mmol) was slowly added a solution of cyanuric chloride (0.074 g, 0.40 mmol) in anhydrous CH3CN (3.0 mL). To this mixture was added DIPEA (0.18 mL, 1.05 mmol) and the resulting solution was allowed to stir at rt for 1 hour. To this mixture was then added 2-chloroaniline (0.124 mg, 1.05 mmol). The resulting dark mixture was allowed to stir overnight at rt. The reaction was judged complete by HPLC, and the resulting reaction mixture was loaded on a C18 reverse phase column and purified. The pure fractions were combined and concentrated overnight with passing of air over the top of the flask. The product was isolated as a yellow solid in 27% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.21 (s, 1H), 7.83 (d, J = 8.0 Hz, 1H), 7.58–7.50 (m, 2H), 7.42–7.35 (m, 2H), 7.34–7.30 (m, 1H); LCMS: m/z calcd. for C11H7BrClNOS [M+H]+: 315.9 & 317.9; found [M+H]+: 315.9 & 317.9.
4.1.1.12. N-(2-chlorophenyl)-5-(4-hydroxyphenyl)thiophene-2-carboxamide (9f) was prepared from 8f and 4-(hydroxyphenyl)boronic acid according to the procedure for 9a. The product was isolated in ≥ 95% purity as a tan solid in 17% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.03 (s, 1H), 9.83 (s, 1H), 7.95 (d, J = 4.8 Hz, 1H), 7.57 (m, 4H), 7.40 (m, 2H), 7.30 (td, J = 8.0, 1.6 Hz, 1H), 6.84 (d, J = 8.4 Hz, 2H); LC-MS: 2.280 min; HRMS: m/z calcd. for (C17H12ClNO2S)H+: 330.0350, found 330.0349.
4.1.1.13. 5-bromo-N-(p-tolyl)thiophene-2-carboxamide (8g) was prepared from 7 and p-toluidine according to the procedure for 8f. The product was isolated as a tan solid in 78% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.20 (s, 1H), 7.84 (d, J = 4.0 Hz, 1H), 7.57 (d, J = 8.4 Hz, 2H), 7.36 (d, J = 4.0 Hz, 1H), 7.16 (d, J = 8.3 Hz, 2H), 2.28 (s, 3H): LCMS: m/z calcd. for C12H10BrFNOS [M+H]+: 296.0 & 298.0; found [M+H]+: 296.0 & 298.0.
4.1.1.14. 5-(4-hydroxyphenyl)-N-(p-tolyl)thiophene-2-carboxamide (9g) was prepared from 8g and 4-(hydroxyphenyl)boronic acid according to the procedure outlined of 8a. The product was isolated in 95% purity as a yellow solid in 62% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.09 (s, 1H), 9.81 (s, 1H), 7.94 (d, J = 4.0 Hz, 1H), 7.60 (d, J = 8.4 Hz, 2H), 7.56 (d, J = 8.7 Hz, 2H), 7.40 (d, J = 4.0 Hz, 1H), 7.15 (d, J = 8.3 Hz, 2H), 6.84 (d, J = 8.7 Hz, 2H), 2.28 (s, 3H); LC-MS: 2.569 min; HRMS: m/z calcd. for (C18H15NO2S)H+: 310.0896, found 310.0895.
4.1.1.15. 5-bromo-N-(m-tolyl)thiophene-2-carboxamide (8h) was prepared from 7 and m-toluidine according to the procedure for 8f. The product was isolated as a while solid in 74% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.20 (s, 1H), 7.86 (d, J = 4.1 Hz, 1H), 7.53 (s, 1H), 7.50 (d, J = 8.1 Hz, 1H), 7.36 (d, J = 4.1 Hz, 1H), 7.24 (t, J = 7.8 Hz, 2H), 6.94 (d, J = 7.5 Hz, 1H) 2.31 (s, 3H); LCMS: m/z calcd. for C12H10BrNOS [M+H]+: 296.0; found [M+H]+: 296.0.
4.1.1.16. 5-(4-hydroxyphenyl)-N-(m-tolyl)thiophene-2-carboxamide (9h) was prepared from 8h and 4-(hydroxyphenyl)boronic acid according to the procedure outlined for 9a. The product was in 95% purity isolated as a tan solid in 55% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.08 (s, 1H), 9.82 (s, 1H), 7.97 (d, J = 4.0 Hz, 1H), 7.55 (m, 4H), 7.41 (d, J = 4.0 Hz, 1H), 7.23 (t, J = 8.0 Hz, 1H), 6.92 (d, J = 7.6 Hz, 1H), 6.84 (d, J = 8.8 Hz, 2H), 2.31 (s, 3H); LC-MS: 2.220 min; HRMS: m/z calcd. for (C18H15NO2S)H+: 310.0896, found 310.0895.
4.1.1.17. 5-bromo-N-(o-tolyl)thiophene-2-carboxamide (8i) was prepared from 7 and o-toluidine according to the procedure for 8f. The product was isolated as white solid in 48% yield. 1H NMR (400 MHz, DMSO-d6) δ 9.99 (s, 1H), 7.82 (d, J = 4.0 Hz, 1H), 7.37 (d, J = 4.0 Hz, 1H), 7.31–7.27 (m, 2H), 7.25–7.17 (m, 2H), 2.22 (s, 3H). LCMS: m/z calcd. for C12H10BrNOS [M+H]+: 296.0; found [M+H]+: 296.0.
4.1.1.18. 5-(4-hydroxyphenyl)-N-(o-tolyl)thiophene-2-carboxamide (9i) was prepared from 8i and 4-(hydroxyphenyl)boronic acid according to the procedure outlined for 9a. The product was obtained in 94% purity as a white solid in 48% yield. 1H NMR (400 MHz, DMSO-d6) δ 9.85 (s, 1H), 9.81 (s, 1H), 7.92 (d, J = 4.0 Hz, 1H), 7.55 (d, J = 9.2 Hz, 2H), 7.40 (d, J = 4.4 Hz, 1H), 7.30 (m, 2H), 7.20 (m, 2H), 6.84 (d, J = 8.4 Hz, 2H), 2.24 (s, 3H); LC-MS: 2.174 min; HRMS: m/z calcd. for (C18H15NO2S)H+: 310.0896, found 310.0896.
4.1.1.19. 5-bromo-N-phenylthiophene-2-carboxamide (8j) was prepared from 7 and aniline according to the procedure for 8a. The product was isolated as a white solid in 83% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.27 (s, 1H), 7.87 (d, J = 4.1 Hz, 1H), 7.69 (d, J = 7.8 Hz, 2H), 7.38–7.34 (m, 3H), 7.12 (t, J = 7.4 Hz, 1H). LCMS: m/z calcd. for C11H8BrNOS [M+H]+: 282.0; found [M+H]+: 281.9.
4.1.1.20. N-phenyl-5-(4-hydroxyphenyl)thiophene-2-carboxamide (9j) was prepared from 8j and 4-(hydroxyphenyl)boronic acid according to the procedure outlined for 9a. The product was obtained in ≥ 95 purity as a yellow solid in 44% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.15 (s, 1H), 9.82 (s, 1H), 7.97 (d, J = 3.6 Hz, 1H), 7.72 (m, 2H), 7.56 (d, J = 8.8 Hz, 2H), 7.41 (d, J = 4.4 Hz, 1H), 7.35 (m, 2H), 7.10 (m, 1H), 6.84 (d, J = 8.8 Hz, 2H); LC-MS: 2.491 min; HRMS: m/z calcd. for (C17H13NO2S)H+: 296.0740, found 296.0738.
4.1.1.21. 5-bromo-N-(4-cyanophenyl)thiophene-2-carboxamide (8k) was prepared from 7 and 4-cyanoaniline according to the procedure for 8f. The product was obtained as a yellow solid in 34% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.63 (s, 1H), 7.93–7.90 (m, 3H), 7.84–7.81 (m, 2H), 7.41 (d, J = 4.0 Hz, 1H). LCMS: m/z calcd. for C12H7BrN2OS [M+H]+: 307.94; found [M+H]+: 308.9.
4.1.1.22 N-(4-cyanophenyl)-5-(4-hydroxyphenyl)thiophene-2-carboxamide (9k) was prepared from 8k and 4-(hydroxyphenyl)boronic acid according to the procedure outlined for 9a. The product was isolated in ≥ 95% purity as a brown solid in 17% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.61 (s, 1H), 9.72 (s, 1H), 8.06 (d, J = 4.8 Hz, 1H), 7.96 (d, J = 8.8 Hz, 2H), 7.83 (d, J = 9.6 Hz, 2H), 7.57 (d, J = 4.4 Hz, 1H), 7.26 (t, J = 8.0 Hz, 1H), 7.18 (m, 1H), 7.11 (t, J = 2.0 Hz, 1H), 6.81 (m, 1H); LC-MS: 2.169 min; HRMS: m/z calcd. for (C18H12N2O2S)H+: 321.0690, found 321.0692.
4.1.1.23. 5-bromo-N-(3-cyanophenyl)thiophene-2-carboxamide (8l) was prepared from 7 and 3-cyanoaniline according to the procedure for 8f. The product was isolated as a white solid in 24% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.58 (s, 1H), 8.0–7.95 (m, 1H), 7.87 (d, J = 4.1 Hz, 1H), 7.61–7.56 (m, 2H), 7.40 (d, J = 4.1 Hz, 1H). LCMS: m/z calcd. for C12H7BrN2OS [M+H]+: 307.94; found [M+H]+: 308.9.
4.1.1.24. N-(3-cyanophenyl)-5-(4-hydroxyphenyl)thiophene-2-carboxamide (9l) was prepared from 8l and 4-(hydroxyphenyl)boronic acid as outlined according to the procedure for 9a. The product was isolated in ≥ 95% purity as a tan solid in quantitative yield. 1H NMR (400 MHz, DMSO-d6) δ 10.48 (s, 1H), 9.86 (s, 1H), 8.21–8.20 (m, 1H), 8.04–7.99 (m, 2H), 7.64–7.55 (m, 4H), 7.45 (d, J = 4.0 Hz, 1H), 6.86–6.81 (m, 1H); LC-MS: 2.521 min; HRMS: m/z calcd. for (C18H12N2O2S)H+: 321.0692, found 321.0691.
4.1.1.25. 5-bromo-N-(4-isopropylphenyl)thiophene-2-carboxamide (8m) was prepared from 7 and 4-isopropylaniline according to the procedure outlined for 9a. The product was isolated as a yellow solid in 34% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.21 (s, 1H), 7.85 (d, J = 4.0 Hz, 1H), 7.62–7.58 (m, 2H), 7.36 (d, J = 4.0 Hz, 1H), 7.25–7.20 (m, 2H), 2.92–2.82 (m, 1H), 1.20 (d, J = 6.9 Hz, 6H). LCMS: m/z calcd. for C14H14BrNOS [M+H]+: 324.0 & 326.0; found [M+H]+: 324.2 & 326.2.
4.1.1.26. 5-(4-hydroxyphenyl)-N-(4-isopropylphenyl)thiophene-2-carboxamide (9m) was prepared from 8m and 4-(hydroxyphenyl)boronic acid as outlined in the procedure for 9a. The product was obtained in ≥ 95% purity as a tan solid in 29% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.10 (s, 1H), 9.81 (s, 1H), 7.95 (d, J = 4.4 Hz, 1H), 7.63 (d, J = 8.8 Hz, 2H), 7.56 (d, J = 8.8 Hz, 2H), 7.40 (d, J = 4.0 Hz, 1H), 7.22 (d, J = 8.0 Hz, 2H), 6.84 (d, J = 9.2 Hz, 2H), 2.86 (m, 1H), 1.20 (d, J = 7.2 Hz, 6H); LCMS: 2.395 min, m/z calcd. for C20H19NO2S [M+H]+: 338.1; found [M+H]+: 338.1.
4.1.1.27. 5-bromo-N-cyclohexylthiophene-2-carboxamide (8n) was prepared from 7 and cyclohexylamine as outlined in the procedure for 8a. The product was obtained as a brown solid in 89% yield. 1H NMR (400 MHz, DMSO-d6) δ 8.24 (d, J = 7.9 Hz, 1H), 7.62 (d, J = 4.0 Hz, 1H), 7.26 (d, J = 4.0 Hz, 1H), 3.75–3.60 (m, 1H), 1.85–1.77 (m, 2H), 1.77–1.68 (m, 2H), 1.65–1.56 (m, 1H), 1.32–1.24 (m, 4H), 1.18–1.07 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 142.8, 131.8, 129.0, 116.9, 48.9, 32.9, 25.7, 25.3; HRMS m/z calcd for (C11H14BrNOS)Cl-321.9673, found 321.9673.
4.1.1.28. N-cyclohexyl-5-(4-hydroxyphenyl)thiophene-2-carboxamide (9n) was obtained from 8p and 4-(hydroxyphenyl)boronic acid as outlined in the procedure for 9a. The product was isolated in 94% purity as a tan solid in 45% yield. 1H NMR (400 MHz, DMSO-d6) δ 9.77 (s, 1H), 8.15 (d, J = 8.0 Hz, 1H), 7.72 (d, J = 4.0 Hz, 1H), 7.51 (d, J = 8.7 Hz, 2H), 7.31 (d, J = 4.0 Hz, 1H), 6.82 (d, J = 8.7 Hz, 2H), 3.77–3.64 (m, 1H), 1.86–1.78 (m, 2H), 1.78–1.68 (m, 2H), 1.66–1.57 (m, 1H), 1.35–1.26 (m, 4H), 1.21–1.07 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 160.6, 158.4, 148.4, 138.1, 129.3, 127.5, 124.8, 122.7, 116.4, 48.8, 33.0, 25.7, 25.4; LC-MS: 2.493 min; HRMS m/z calcd. for (C17H19NO2S)Cl-336.0831, found 336.0831.
4.1.1.29. N-benzyl-5-bromothiophene-2-carboxamide (8o) was prepared from 7 and benzylamine according to the procedure for 8a. The product was obtained as a white solid in 66% yield. 1H NMR (400 MHz, DMSO-d6) δ 9.11 (t, J = 5.9 Hz, 1H), 7.64 (d, J = 4.1 Hz, 1H), 7.36–7.23 (m, 6H), 4.43 (d, J = 6.0 Hz, 2H). LCMS: m/z calcd. for C12H10BrNOS [M+H]+: 296.0 & 298.0; found [M+H]+: 296.0 & 298.0.
4.1.1.30 N-benzyl-5-(4-hydroxyphenyl)thiophene -2-carboxamide (9o) was prepared from 8o and 4-(hydroxyphenyl)boronic acid as outlined for 9a. The product was obtained in 95% purity as a white solid in 26% yield. White solid, yield 19%; 1H NMR (400 MHz, DMSO-d6) δ 9.77 (s, 1H), 8.99 (t, J = 5.6 Hz, 1H), 7.74 (d, J = 4.0 Hz, 1H), 7.52 (d, J = 8.8 Hz, 2H), 7.33 (m, 5H), 7.25 (m, 1H), 6.82 (d, J = 8.8 Hz, 1H), 4.45 (d, J = 6.4 Hz, 2H); LC-MS: 2.405 min; HRMS: m/z calcd. for (C18H15NO2S)H+: 310.0896, found 310.0896.
4.1.1.31. 5-bromo-N-(thiophen-3-yl)thiophene-2-carboxamide (8p) was prepared from 5 and 3-aminothiophene according to the procedure outlined for 6a. The product was isolated as a white solid in 83% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.74 (s, 1H), 7.80 (d, J = 4.0 Hz, 1H), 7.64 (dd, J =0.9 Hz, 3.1 Hz, 1H), 7.51 (dd, J = 3.2 Hz, 5.2 Hz, 1H), 7.37 (d, J = 4.0 Hz, 1H), 7.27 (dd, J = 0.9 Hz, 5.1 Hz, 1H); ). LCMS: m/z calcd. for C9H6BrNOS2 [M+H]+: 287.9 & 289.9; found [M+H]+: 287.9 & 289.9.
4.1.1.32. N-(3-fluorophenyl)-5-(4-(methylsulfonyl)phenyl)thiophene-2-carboxamide (10a) was prepared from 8b and 4-(methylsulfonyl)phenylboronic acid as outlined in the procedure for 9a. The product was isolated in ≥ 95% purity as a white solid in 42% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.50 (s, 1H), 8.11–7.98 (m, 5H), 7.84 (d, J = 3.6 Hz, 1H), 7.71 (d, J = 6.0 Hz, 1H), 7.54 (d, J = 6.0 Hz, 1H), 7.41 (quar, J = 8.0 Hz, 1H), 6.96 (t, J = 8.0 Hz, 1H), 3.33 (s, 3H); LC-MS: 2.197 min; HRMS: m/z calcd. for (C18H14FNO3S2)H+: 376.0472, found 376.0472.
4.1.1.33. N-(3-fluorophenyl)-5-(3-(methylsulfonyl)phenyl)thiophene-2-carboxamide (10b) was prepared from 8b and 3-(methylsulfonyl)phenylboronic acid as outlined in the procedure for 9a. The product was isolated in ≥ 95% purity as a white solid in 55% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.47 (s, 1H), 8.24 (s, 1H), 8.11–8.06 (m, 2H), 7.92 (d, J = 8.0 Hz, 1H), 7.83 (d, J = 3.6 Hz, 1H), 7.73 (m, 2H), 7.53 (d, J = 8.0 Hz, 1H), 7.41 (quar, J = 8.0 Hz, 1H), 6.95 (t, J = 8.4 Hz, 1H), 3.32 (s, 3H); LC-MS: 2.268 min; HRMS: m/z calcd. for (C18H14FNO3S2)H+: 376.0472, found 376.0472.
4.1.1.34. 4-(5-((3-fluorophenyl)carbamoyl)thiophen-2-yl)benzoic acid (10c) was prepared from 8b and 4-carboxyphenylboronic acid in ≥ 95% purity as outlined in the procedure for 9a. The product was isolated as a gray solid in quantitative yield. 1H NMR (400 MHz, DMSO-d6) δ 10.63 (s, 1H), 8.09 (d, J = 4.0 Hz, 1H), 7.90 (d, J = 8.3 Hz, 2H), 7.74 (dt, J = 11.8 Hz, 2.2 Hz, 1H), 7.64 (d, J = 8.3 Hz, 2H), 7.62 (d, J = 4.0 Hz, 1H), 7.59–7.55 (m, 1H), 7.44–7.36 (m, 1H), 6.93 (td, J = 8.4 Hz, 2.2 Hz, 1H); LC-MS: 2.618 min HRMS: m/z calcd. for (C18H12FNO3S)H+: 342.0595, found 342.0593.
4.1.1.35. 3-(5-((3-fluorophenyl)carbamoyl)thiophen-2-yl)benzoic acid (10d) was prepared from 8b and 3-carboxyphenylboronic acid as outlined in the procedure for 9a. The product was isolated in ≥ 95% purity as a beige solid in 55%. 1H NMR (400 MHz, DMSO-d6) δ 13.21 (s, 1H), 10.45 (s, 1H), 8.25 (t, J = 1.6 Hz, 1H), 8.07 (d, J = 4.0 Hz, 1H), 8.04–7.99 (m, 1H), 7.96 (dt, J = 7.8 Hz, 1.2 Hz, 1H), 7.76–7.69 (m, 2H), 7.61 (t, J = 7.8 Hz, 1H), 7.56–7.51 (m, 1H), 7.41 (td, J = 8.2 Hz, 6.9 Hz, 1H), 6.95 (td, J = 8.4 Hz, 2.2 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 167.3, 162.5 (d, 1JCF = 239.8 Hz), 160.2, 148.0, 140.9 (d, 3JCF = 11.1 Hz), 139.4, 133.7, 132.3, 131.2, 130.8 (d, 3JCF = 9.5 Hz), 130.4, 130.2, 129.8, 126.6, 125.7, 116.4 (d, 4JCF = 2.6 Hz), 110.7 (d, 2JCF = 20.8 Hz), 107.4 (d, 2JCF = 26.0 Hz); LC-MS: 2.601 min; HRMS m/z calcd for (C18H12FNO3S)-H 340.0449, found 340.0447.
4.1.1.36. N-(3-fluorophenyl)-5-(4-(hydroxymethyl)phenyl)thiophene-2-carboxamide (10e) was prepared from 8b and 4-(hydroxymethyl)phenylboronic acid according to the procedure outlined for 9a. The product was obtained in 94% purity as a white solid in 44% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.40 (s, 1H), 8.03 (d, J = 4.4 Hz, 1H), 7.70 (m, 3H), 7.61 (d, J = 3.6 Hz, 1H), 7.53 (m, 1H), 7.40 (m, 3H), 6.94 (m, 1H), 5.28 (t, J = 6.0 Hz, 1H), 4.53 (d, J = 5.6 Hz, 2H); LC-MS: 2.545 min; HRMS: m/z calcd. for (C18H14FNO2S)H+: 328.0802, found 328.0800.
4.1.1.37. N-(3-fluorophenyl)-5-(3-(hydroxymethyl)phenyl)thiophene-2-carboxamide (10f) was prepared from 8b and 3-(hydroxymethyl)phenyl)boronic acid according to the procedure outlined for 9a. The product was obtained in 94% purity as a white solid in 47% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.41 (s, 1H), 8.04 (d, J = 4.4 Hz, 1H), 7.71 (m, 2H), 7.63 (m, 2H), 7.53 (m, 1H), 7.38 (m, 3H), 6.94 (m, 1H), 5.30 (t, J = 6.0 Hz, 1H), 4.56 (d, J = 5.6 Hz, 2H); LC-MS: 2.561 min; HRMS: m/z calcd. for (C18H14FNO2S)H+: 328.0802, found 328.0801.
4.1.1.38. N-(3-fluorophenyl)-5-(4-hydroxy-3-(trifluoromethyl)phenyl)thiophene-2-carboxamide (10g) was prepared from 8b and (4-hydroxy-3-trifluoromethylphenyl)boronic acid as outlined in the procedure for 9a. The product was isolated in ≥ 95% as a beige solid in 34% yield. 1H NMR (400 MHz, DMSO-d6) δ 11.00 (s, 1H), 10.38 (s, 1H), 8.02 (d, J = 4.0 Hz, 1H), 7.87–7.81 (m, 2H), 7.71 (dt, J = 11.8 Hz, 2.1 Hz, 1H), 7.59 (d, J = 4.0 Hz, 1H), 7.53 (d, J = 8.2 Hz, 1H), 7.44–7.36 (m, 1H), 7.12 (d, J = 9.2 Hz, 1H), 6.94 (td, J = 8.5 Hz, 2.5 Hz, 1H); LC-MS: 2.561 min; HRMS: m/z calcd. for (C18H11F4NO2S)H+: 382.0519, found 328.0515.
4.1.1.39. 5-(3-chloro-4-hydroxyphenyl)-N-(3-fluorophenyl)thiophene-2-carboxamide (10h) was prepared from 8b and (4-hydroxy-3-chloromethylphenyl)boronic acid as outlined in the procedure for 9b. The product was isolated in 95% purity as a yellow solid in 91% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.61 (s, 1H), 10.36 (s, 1H), 7.99 (d, J = 4.0 Hz, 1H), 7.75 (d, J = 2.3 Hz, 1H), 7.71 (dt, J = 11.8 Hz, 2.2 Hz, 1H), 7.56–7.50 (m, 3H), 7.40 (dt, J = 6.9 Hz, 8.2 Hz, 1H), 7.04 (d, J = 8.5 Hz, 1H), 6.94 (td, J = 8.2 Hz, 2.3 Hz, 1H); LC-MS: 2.507 min; HRMS: m/z calcd. for (C17H11ClFNO2S)H+: 348.0256, found 348.0253.
4.1.1.40. N-(4-fluorophenyl)-5-(3-hydroxyphenyl)thiophene-2-carboxamide (10i) was prepared from 8a and 3-hydroxyphenylboronic acid as outlined in the procedure for 9a. The product was isolated in ≥ 95% as a tan solid in 60% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.29 (s, 1H), 9.70 (s, 1H), 7.99 (d, J = 4.0 Hz, 1H), 7.78–7.72 (m, 2H), 7.55 (d, J = 4.0 Hz, 1H), 7.27 (t, J = 7.8 Hz, 1H), 7.24–7.15 (m, 3H), 7.10 (t, J = 2.0 Hz, 1H), 6.80 (ddd, J = 8.0 Hz, 2.3 Hz, 0.8 Hz, 1H); LC-MS: 2.184 min HRMS m/z calcd for (C17H12FNO2S)H+: 314.0645, found 314.0644.
4.1.1.41. N-(3-fluorophenyl)-5-(3-hydroxyphenyl)thiophene-2-carboxamide (10j) was prepared from 8b and 3-hydroxyphenylboronic acid as outlined in the procedure for 9a. The product was isolated in ≥ 95% purity as a beige solid in quantitative yield. 1H NMR (400 MHz, DMSO-d6) δ 10.41 (s, 1H), 9.72 (s, 1H), 8.02 (d, J = 4.0 Hz, 1H), 7.71 (dt, J = 11.7 Hz, 2.2 Hz, 1H), 7.57 (d, J = 4.0 Hz, 1H), 7.53 (d, J = 8.2 Hz, 1H), 7.41 (dt, J = 6.9 Hz, 8.2 Hz, 1H), 7.27 (t, J = 7.8 Hz, 1H), 7.18 (d, J = 8.1 Hz, 1H), 7.11 (t, J = 2.0 Hz, 1H), 6.95 (td, J = 8.3 Hz, 2.0 Hz, 1H), 6.80 (dd, J = 2.3 Hz, 8.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 162.5 (d, 1JCF = 239.9 Hz), 160.4, 158.4, 149.5, 141.0 (d, 3JCF = 10.9 Hz), 138.5, 134.5, 131.1, 130.9, 130.8 (d, 3JCF = 10.5 Hz), 124.8, 117.1, 116.4, 112.9, 110.6 (d, 2JCF = 20.8 Hz), 107.4 (d, 2JCF = 26.3 Hz); LC-MS: 2.613 min; HRMS m/z calcd for (C17H12FNO2S)2Na+ dimer 649.1038, found of spray dimer 649.1039.
4.1.1.42. 5-(3-hydroxyphenyl)-N-(p-tolyl)thiophene-2-carboxamide (10k) was prepared from 8g and 3-hydroxyphenylboronic acid as outlined in the procedure for 9a. The product was isolated in ≥ 95% purity as a yellow solid in 84% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.16 (s, 1H), 9.69 (s, 1H), 7.99 (d, J = 4.0 Hz, 1H), 7.61 (d, J = 8.4 Hz, 2H), 7.53 (d, J = 4.0 Hz, 1H), 7.26 (t, J = 7.9 Hz, 1H), 7.19–7.14 (m, 3H), 7.10 (t, J = 2.0 Hz, 1H), 6.79 (dd, J = 5.8 Hz, 2.2 Hz, 1H), 2.28 (s, 3H); LC-MS: 2.242 min; HRMS: m/z calcd. for (C18H15NO2S)H+: 310.0896, found 310.0896.
4.1.1.43. N-(4-chlorophenyl)-5-(3-hydroxyphenyl)thiophene-2-carboxamide (10l) was prepared from 8d and 4-hydroxyphenylboronic acid as outlined in the procedure for 9a. The product was isolated in ≥ 95 purity as a yellow solid in 64% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.35 (s, 1H), 9.71 (s, 1H), 8.01 (d, J = 4.0 Hz, 1H), 7.78 (d, J = 9.0 Hz, 2H), 7.55 (d, J = 4.0 Hz, 1H), 7.42 (d, J = 8.9 Hz, 2H), 7.26 (t, J = 7.8 Hz, 1H), 7.18 (dt, J = 8.2 Hz, 1.0 Hz, 1H), 7.10 (t, J = 2.0 Hz, 1H), 6.80 (ddd, J = 8.0 Hz, 2.3 Hz, 0.8 Hz, 1H); LC-MS: 2.072 min; HRMS: m/z calcd. for (C17H12ClNO2S)H+: 330.0350, found 330.0348.
4.1.1.44. N-(3-chlorophenyl)-5-(3-hydroxyphenyl)thiophene-2-carboxamide (10m) was prepared from 8e and 3-hydroxyphenylboronic acid as outlined in the procedure for 9a. The product was isolated as a yellow solid in 50% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.38 (s, 1H), 9.71 (s, 1H), 8.02 (d, J = 4.4 Hz, 1H), 7.92 (t, J = 2.4 Hz, 1H), 7.68 (m, 1H), 7.55 (d, J = 4.0, 1H), 7.39 (t, J = 8.0 Hz, 1H), 7.26 (t, J = 8.0 Hz, 1H), 7.17 (m, 2H), 7.10 (t, J = 2.4Hz, 1H) 6.80 (dd, J = 2.4, 2.4, 1H); LC-MS: 2.368 min; HRMS m/z calcd for (C17H12ClNO2S)H+: 330.0350, found 330.0350.
4.11.45. 5-(3-hydroxyphenyl)-N-(thiophen-3-yl)thiophene-2-carboxamide (10n) was prepared from 8p and 3-hydroxyphenylboronic acid according to the procedure outlined for 9a. The product was isolated as a tan solid in quantitative yield. 1H NMR (400 MHz, DMSO-d6) δ 10.05 (s, 1H), 9.41 (br s, 1H), 7.97 (d, J = 4.0 Hz, 1H), 7.90 (dd, J = 1.1 Hz, 3.1 Hz, 1H), 7.53 (m, 1H), 7.50–7.46 (m,, 2H), 7.29 (, , s, 1H), 7.11( m, 2H), 6.79 ( m, 1H); 13C NMR (100 MHz, CDCl3) δ 159.6, 157.6, 143.5, 139.7, 134.4, 129.9, 127.8, 126.0, 124.4, 122.0; LC-MS: 2.495 min; HRMS m/z calcd for (C15H11NO2S2)-H 300.0158, found 300.0146.
4.11.46. 5-(3-aminophenyl)-N-(3-fluorophenyl)thiophene-2-carboxamide (10o). Potassium carbonate (0.0460 g, 0.3332 mmol) and (3-aminophenyl)boronic acid (0.0228 g, 0.1666 mmol) were added to a 2 dram scintillation vial with deionized water (0.25 mL) and absolute EtOH (2 mL). The vial was purged with argon. Next 5-bromo-N-(3-fluorophenyl)thiophene-2-carboxamide (8b) (0.050 g, 0.1666 mmol) was added, followed by Pd(PPh3)4 (0.0069 g, 0.0060 mmol). The vial was purged with argon again then heated at reflux for 10 h. The reaction was cooled and diluted with water. The precipitate was filtered in a sintered funnel and the solid was washed with saturated aqueous NaHCO3. The solvent was removed by vacuum aspiration. The crude solid was purified by flash chromatography (53% EtOAc:Hex). Product was isolated as a red semi-solid in ≥ 95% purity in quantitative yield. 1H NMR (400 MHz, DMSO-d6) δ 10.38 (s, 1H), 8.00 (d, J = 4.0 Hz, 1H), 7.71 (dt, J = 11.8 Hz, 2.2 Hz, 1H), 7.52 (d, J = 8.2 Hz, 1H), 7.47 (d, J = 4.0 Hz, 1H), 7.40 (dt, J = 6.9 Hz, 1H), 7.10 (t, J = 7.8 Hz, 1H), 6.98–6.86 (m, 3H), 6.59 (dd, J = 1.3 Hz, 8.0 Hz, 1H), 5.29 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 160.0, 150.0, 149.4, 140.6, 137.5, 133.4, 130.5, 130.4, 130.0, 123.6, 115.9, 114.5, 113.3, 110.8, 110.2, 107.3; LC-MS: 2.010 min; HRMS m/z calcd for (C17H13FN2OS)H+ 313.0805, found 313.0804.
4.1.1.47. 5-(3-aminophenyl)-N-(3-chlorophenyl)thiophene-2-carboxamide (10p) The product was prepared from 8e and (3-aminophenyl)boronic acid as outlined above for 10o. The product was obtained in ≥ 95% purity as a yellow solid in 36% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.35 (s, 1H), 7.99 (d, J = 4.0 Hz, 1H), 7.91 (t, J = 2.2 Hz, 1H), 7.68–7.65 (m, 1H), 7.47 (d, J = 4.0 Hz, 1H), 7.39 (t, J = 8.0 Hz, 1H), 7.18–7.10 (m, 2H), 6.93–6.91 (m, 2H), 6.61 (d, J = 8.0 Hz, 1H), 5.60 (br s, 1H); LCMS: 2.350 min, m/z calcd. for C17H13ClN2OS [M+H]+: 329.0; found [M+H]+: 329.0.
4.1.1.48. (3-(5-((3-fluorophenyl)carbamoyl)thiophen-2-yl)phenyl)boronic acid (10q) was prepared from 8b and 1,3-phenylenediboronic acid according to the procedure outlined for 10r. The product was isolated in 94% purity as a tan solid in 28% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.46 (s, 1H), 8.15 (s, 2H), 8.08 (d, J = 4.0 Hz, 1H), 7.88 (d, J = 8.1 Hz, 2H), 7.76–7.70 (m, 3H), 7.69 (d, J = 4.0 Hz, 1H), 7.55 (d, J = 8.2 Hz, 1H), 7.40 (dt, J = 7.0 Hz, 8.2 Hz, 1H), 6.95 (td, J = 8.4 Hz, 2.4 Hz, 1H); LC-MS: 2.519 min HRMS m/z calcd for (C17H13BFNO3S)-H 320.0620, found 320.0621.
4.1.1.49. (4-(5-((3-fluorophenyl)carbamoyl)thiophen-2-yl)phenyl)boronic acid (10r). A mixture of 5-bromo-N-(3-fluorophenyl)thiophene-2-carboxamide (8b) (0.040 g, 0.1333 mmol) potassium carbonate (0.1842 g, 1.333 mmol), 1,4-phenylenediboronic acid (0.0663 g, 0.3998 mmol), and Pd(PPh3)4 (0.0031 g, 0.0027 mmol) were added to a 2 dram scintillation vial with deionized water (0.66 mL) and dioxane (2.6 mL). The vial was purged with argon then heated at 80 °C for 3 h. The reaction was cooled, diluted with water, and filtered in a sintered funnel. The solid was washed with saturated aqueous NaHCO3 and the solvent removed by vacuum aspiration. The crude solid was purified by flash chromatography (2% MeOH:CH2Cl2). Product was isolated in 94% purity as a tan solid in 28% yield (0.0128 g). 1H NMR (400 MHz, DMSO-d6) δ 10.46 (s, 1H), 8.15 (s, 2H), 8.08 (d, J = 4.0 Hz, 1H), 7.88 (d, J = 8.1 Hz, 2H), 7.76–7.70 (m, 3H), 7.69 (d, J = 4.0 Hz, 1H), 7.55 (d, J = 8.2 Hz, 1H), 7.40 (dt, J = 7.0 Hz, 8.2 Hz, 1H), 6.95 (td, J = 8.4 Hz, 2.4 Hz, 1H); LC-MS: 2.500 min; HRMS m/z calcd for (C17H13BFNO3S)-H 320.0620, found 320.0621.
4.1.1.50. (3-(5-((2-fluorophenyl)carbamoyl)thiophen-2-yl)phenyl)boronic acid (10s) Tetrakis(triphenylphosphine)palladium(0) (0.077 g, 0.66 mmol), Cs2CO3 (0.470 g, 1.33 mmol), 1,3-benzenediboronic acid (0.1143 mg, 0.86 mmol), and 8c (0.200 g, 0.66 mmol) were dissolved in 4:1 mixture of dioxane:H2O (10 mL) and sealed in a microwave tube.The reaction was heated to 120°C for 40 min in the microwave and cooled to RT. The reaction mixture was washed with EtOAc through pad of celite and evaporated to afford a black solid. The crude product was purified by high pressure reverse phase column. The combined fractions were dried, sonicated with hexane and then washed with DCM several times and then filtered under gravity to afford 10s (0.061 mg, 53.8%) in ≥ 95% purity as a white solid.1H NMR (400 MHz, DMSO-d6) δ 10.17 (s, 1H), 8.23 (s, 2H), 8.17 (s, 1H), 8.03 (d, J = 4.0 Hz, 1H), 7.84–7.77 (m, 2H), 7.62–7.59 (m, 2H), 7.44 (t, J = 7.6 Hz, 1H), 7.34–7.27 (m, 2H), 7.27–7.21 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 159.9, 154.0, 149.1, 137.6, 134.5, 133.6, 132.01, 131.8, 131.49, 130.69, 128.35, 127.3, 125.3, 124.4, 124.2, 116.0, 115.8; LC-MS: 2.399 min HRMS m/z calcd for (C17H13BFNO3S)-H 340.0620, found 340.0620.
4.1.1.51. 5-(4-fluorophenyl)thiophene-2-carboxylic acid (11a). Potassium carbonate (0.667 g, 4.830 mmol) and 4-fluorophenylboronic acid (0.338 g, 2.415 mmol) were added to a 20 mL scintillation vial with deionized water (1 mL) and absolute EtOH (8 mL). The vial was purged with argon. Next 5-bromo-2-thiophenecarboxylic acid (0.500 g, 2.415 mmol) was added, followed by Pd(PPh3)4 (0.100 g, 0.0865 mmol). The vial was purged with argon again then heated at reflux for 10 h. The reaction was cooled, acidified with 10% HCl, and extracted with EtOAc three times. The combined organic layers were washed with sat. aq. NaHCO3 three times. The basic aqueous layer was then acidified with 10% HCl, resulting in precipitation of the product. The precipitate was removed by gravity filtration and dried overnight open to the atmosphere. The resulting gray precipitate was isolated in quantitative yield and used as is without further purification. 1H NMR (400 MHz, DMSO-d6) δ 7.83–7.77 (m, 2H), 7.72 (d, J = 3.9 Hz, 1H), 7.55 (d, J = 3.9 Hz, 1H), 7.30 (dd, J = 8.8 Hz, 8.8 Hz, 2H). LCMS: m/z calcd. for C11H7FO2S [M+H]+: 223.0; found [M+H]+: 223.0.
4.1.1.52. 5-(2-fluorophenyl)-N-(4-hydroxyphenyl)thiophene-2-carboxamide (12a). To a solution of 5-(4-fluorophenyl)thiophene-2-carboxylic acid (11a) (0.1000 g, 0.4499 mmol) and TBTU (0.1444 g, 0.4499 mmol) in dry DMF (4.5 mL) was added DIPEA (0.196 mL, 1.1247 mmol). The reaction was stirred at room temperature for 20 min and then 4-aminophenol (0.0589 g, 0.5399 mmol) was added. The reaction was stirred overnight at room temperature. The reaction was quenched with water to precipitate the product. The mixture was filtered in a sintered funnel by vacuum suction. The solid was triturated with cold methanol, the solvent removed by vacuum suction, and the product dried. The product was isolated in ≥ 95% purity as a tan solid in 26% yield (0.037 g). 1H NMR (400 MHz, DMSO-d6) δ 10.4 (s, 1H), 9.29 (s, 1H), 7.96 (s, 1H), 7.79 (s, 2H), 7.57 (s, 1H), 7.31 (s, 2H), 6.76 (d, J = 8.9 Hz, 2H); 19F NMR (376 MHz, CDCl3) δ -113.10; LC-MS: 2.524 min; HRMS m/z calcd for (C17H12FNO2S)2Na+ dimer 649.1038, found of spray dimer 649.1038.
4.1.1.53. 5-(3-fluorophenyl)thiophene-2-carboxylic acid (11b) was prepared from 7 and 3-fluorophenylboronic acid according to the procedure of 11a and obtained in ≥ 95% purity as a tan solid in 86% yield. 1H NMR (400 MHz, DMSO-d6) δ 7.74 (d, J = 3.9 Hz, 1H), 7.67 (d, J = 3.9 Hz, 1H), 7.64 (td, J = 2.2 Hz, 10.3 Hz, 1H), 7.58 (td, J = 1.5 Hz, 7.9 Hz, 1H), 7.51 (dt, J = 6.1 Hz, 8.1 Hz, 1H)7.24 (dt, J = 2.5 Hz, 8.5 Hz, 1H); LCMS: m/z calcd. for C11H7FO2S [M+H]+: 223.0; found [M+H]+: 223.0.
4.1.1.54. 5-(3-fluorophenyl)-N-(4-hydroxyphenyl)thiophene-2-carboxamide (12b) was prepared from the acid 11b and 4-aminophenol according to the procedure of 12a and obtained in 95% purity as a purple solid in 69% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.00 (s, 1H), 9.23 (s, 1H), 7.90 (d, J = 3.8 Hz, 1H), 7.62 (d, J = 3.8 Hz, 1H), 7.55 (d, J = 10.2 Hz, 1H), 7.51 (d, J = 7.9 Hz, 1H), 7.47–7.38 (m, 3H), 7.15 (t, J = 7.5 Hz, 1H), 6.69 (d, J = 8.8 Hz, 2H); 19F NMR (376 MHz, CDCl3) δ -112.30; LC-MS: 2.542 min; HRMS m/z calcd for (C17H12FNO2S)Na+: 336.0465, found 336.0462.
4.1.1.54. 5-(2-fluorophenyl)thiophene-2-carboxylic acid (11c) was prepared from 7 and 4-fluorophenylboronic acid according to the procedure for 11a and obtained in 94% yield. 1H NMR (400 MHz, DMSO-d6) δ 7.83–8.77 (m, 2H), 7.72 (d, J = 3.9 Hz, 1H), 7.55 (d, J = 3.9 Hz, 1H), 7.30 (dd, J = 8.8 Hz, 8.8 Hz, 2H); LCMS: m/z calcd. for C11H7FO2S [M+H]+: 223.0; found [M+H]+: 223.0.
4.1.1.55. 5-(2-fluorophenyl)-N-(4-hydroxyphenyl)thiophene-2-carboxamide (12c) was prepared from acid 11c and 2-aminophenol according to the procedure for 12a and obtained as a purple solid in 76% yield. 1H NMR (400 MHz, DMSO-d6) δ 7.88 (dt, J = 1.6 Hz, 7.9 Hz, 1H), 7.76 (dd, J = 1.3 Hz, 4.0 Hz, 1H), 7.64 (d, J = 3.8 Hz, 1H), 7.49–7.42 (m, 1H), 7.38 (ddd, J = 1.2 Hz, 8.3 Hz, 11.7 Hz, 1H), 7.32 (dt, J = 1.3 Hz, 7.7 Hz, 1H). LC-MS: 2.524 min; HRMS m/z calcd for (C17H12FNO2S)H+: 314.0645, found 314.0643.
4.1.1.56. [2,3'-bithiophene]-5-carboxylic acid (11d) was prepared from 7 and thiophene-3-ylboronic acid according to the procedure of 11a and obtained as a tan solid in 84% yield (0.4290 g). 1H NMR (400 MHz, DMSO-d6) δ 13.09 (broad s, 1H), 7.95 (dd, J = 1.3 Hz, 2.9 Hz, 1H), 7.69 (d, J = 5.0 Hz, 1H), 7.68 (d, J = 4.0 Hz, 1H), 7.51 (dd, J = 1.4 Hz, 5.0 Hz, 1H), 7.46 (d, J = 3.9 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 163.3, 145.3, 134.7, 134.6, 132.6, 128.4, 126.5, 125.0, 123.0. LCMS: m/z calcd. for C9H6O2S2 [M+H]+: 211.0; found [M+H]+: 211.0.
4.1.1.57. 5-(4-fluorophenyl)-N-(4-hydroxyphenyl)thiophene-2-carboxamide (12d) was prepared from 11d and 4-aminophenol according to the procedure of 12a and obtained in ≥ 95% purity as a brown solid in 71% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.02 (s, 1H), 9.29 (s, 1H), 7.91 (dd, J = 3.9 Hz, 9.7 Hz, 2H), 7.68 (dd, J = 2.9 Hz, 4.9 Hz, 1H), 7.52–7.45 (m, 4H), 6.75 (d, J = 8.8 Hz, 2H); LC-MS: 2.418 min, HRMS m/z calcd for (C15H11NO2S2)H+: 324.0123, found 324.0120.
4.1.1.58. N-(3-hydroxyphenyl)-[2,3'-bithiophene]-5-carboxamide (12e) was prepared from 11d and 3-aminophenol according to procedure of 12a and was obtained as a white solid in 49% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.08 (s, 1H), 9.43 (s, 1H), 7.98 (d, J = 4.0 Hz, 1H), 7.92 (dd, J = 1.3 Hz, 2.9 Hz, 1H), 7.69 (dd, J = 2.9 Hz, 5.0 Hz, 1H), 7.52 (dd, J = 1.3 Hz, 5.0 Hz, 1H), 7.49 (d, J = 3.9 Hz, 1H), 7.30 (s, 1H), 7.13 (d, J = 5.2 Hz, 2H), 6.54–6.48 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ 160.1, 158.0, 143.9, 140.2, 138.4, 134.9, 130.4, 129.8, 128.4, 126.4, 124.8, 122.5, 111.5, 111.3, 107.9; LC-MS: 2.475 min; HRMS m/z calcd for (C15H11NO2S2)Na+ 324.0123, found 324.0125.
4.1.1.59. 5-(4-fluorophenyl)-N-(3-hydroxyphenyl)thiophene-2-carboxamide (12f) was prepared from 11a and 3-aminophenol according to the procedure described for 12a and obtained in ≥ 95% purity as a yellow solid in 33% yield (0.0238 g). 1H NMR (400 MHz, DMSO-d6) δ 10.10 (s, 1H), 9.43 (s, 1H), 8.01 (d, J = 4.0 Hz, 1H), 7.84–7.76 (m, 2H), 7.58 (d, J = 4.0 Hz, 1H), 7.34–7.27 (m, 3H), 7.12 (d, J = 5.1 Hz, 2H), 6.54–6.48 (m, 1H); LC-MS: 2.572 min; HRMS m/z calcd for (C17H12FNO2S)Na+: 336.0465, found 336.0462.
4.1.1.60. 5-(3-fluorophenyl)-N-(3-hydroxyphenyl)thiophene-2-carboxamide (12g) was prepared from 11b and 3-aminophenol according to the procedure described for 12a and obtained in ≥ 95% purity as a yellow solid in 79% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.14 (s, 1H), 9.45 (s, 1H), 8.04 (d, J = 4.0 Hz, 1H), 7.70 (d, J = 4.0 Hz, 1H), 7.64 (dt, J = 10.3 Hz, 2.1 Hz, 1H), 7.59 (dt, J = 7.7 Hz, 1.5 Hz, 1H), 7.50 (td, J = 8.1 Hz, 6.1 Hz, 1H), 7.31 (t, J = 1.2 Hz, 1H), 7.23 (td, J = 8.7 Hz, 2.4 Hz, 1H), 7.16–7.12 (m, 2H), 6.56–6.50 (m, 1H): LC-MS: 2.268 min; HRMS m/z calcd for (C17H12FNO2S)Na+: 336.0465, found 336.0463.
4.1.1.61. 5-(2-fluorophenyl)-N-(3-hydroxyphenyl)thiophene-2-carboxamide (12h) was prepared from 11c and 3-aminophenol according to the procedure outlined for 12a and was obtained in ≥ 95% purity as a yellow solid in 25% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.15 (s, 1H), 9.44 (s, 1H), 8.06 (dd, J = 4.1 Hz, 1.3 Hz, 1H), 7.88 (td, J = 7.9 Hz, 1.6 Hz, 1H), 7.68 (d, J = 3.7 Hz, 1H), 7.49–7.28 (m, 4H), 7.15–7.11 (m, 2H), 6.54–6.49 (m, 1H); LC-MS: 2.268 min; HRMS m/z calcd for (C17H12FNO2S)H+: 314.0645, found 314.0644.
4.1.1.62. (3-(5-(4-fluorophenyl)thiophene-2-carboxamido)phenyl)boronic acid (12i) was prepared from 11a and (3-aminophenyl)boronic acid according to the procedure described for 12a and obtained in 95% purity as a tan solid in 58% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.22 (s, 1H), 8.07–8.04 (m, 3H), 8.00 (s, 1H), 7.86–7.77 (m, 3H), 7.59 (d, J = 4.0 Hz, 1H), 7.55 (d, J = 7.3 Hz, 1H), 7.36–7.27 (m, 3H); LC-MS: 2.495 min; HRMS m/z calcd for (C17H13BFNO3S)Na+: 364.0585, found 364.0583.
4.1.1.63. (3-(5-(3-fluorophenyl)thiophene-2-carboxamido)phenyl)boronic acid (12j) was prepared from 11b and (3-aminophenyl)boronic acid according to the procedure described for 12a and obtained in 95% purity as a tan solid in 75% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.27 (s, 1H), 8.07–8.03 (m, 3H), 8.01 (s, 1H), 7.84 (d, J = 8.0 Hz, 1H), 7.71 (J = 4.0 Hz, 1H), 7.67–7.47 (m, 4H), 7.34 (t, J = 8.0 Hz, 1H), 7.23 (dt, J = 2.0 Hz, 8.0 Hz, 1H); LC-MS: 2.514 min; HRMS m/z calcd for (C17H13BFNO3S)-H+: 340.0620, found 340.0621.
4.1.1.64. (3-(5-(2-fluorophenyl)thiophene-2-carboxamido)phenyl)boronic acid (12k) (3-(5-(3-fluorophenyl)thiophene-2-carboxamido)phenyl)boronic acid (12j) was prepared from 11c and (3-aminophenyl)boronic acid according to the procedure described for 12a and obtained in 95% purity as a tan solid in 30% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.28 (s, 1H), 8.10 (dd, J = 1.2 Hz, 4.0 Hz, 1H), 8.06 (s, 2H), 8.01 (s, 1H), 7.92–7.82 (m, 2H), 7.69 (d, J = 3.6 Hz, 1H), 7.56 (d, J = 7.2 Hz, 1H), 7.48–7.30 (m, 4H); LC-MS: 2.491 min; HRMS m/z calcd for (C17H13BFNO3S)Na+: 364.0585, found 364.0584.
4.1.1.65. (3-([2,3'-bithiophene]-5-carboxamido)phenyl)boronic acid (12l) was prepared from 11d and (3-aminophenyl)boronic acid according to the procedure described for 12a and was obtained in 95% purity as a beige solid in 19% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.19 (s, 1H), 8.05 (s, 2H), 8.02 (d, J = 4.0 Hz, 1H), 8.00 (s, 1H), 7.92 (dd, J = 1.2 Hz, 2.8 Hz, 1H), 7.83 (d, J = 8.0 Hz, 1H), 7.69 (dd, J = 2.9 Hz, 5.0 Hz, 1H), 7.55 (d, J = 7.3 Hz, 1H), 7.52 (dd, J = 1.2 Hz, 5.0 Hz, 1H), 7.49 (d, J = 4.0 Hz, 1H), 7.33 (t, J = 7.8 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 160.1, 143.9, 138.4, 134.9, 130.3, 130.1, 128.4, 128.1, 127.0, 126.4, 124.9, 122.9, 122.5; LC-MS: 2.398 min; HRMS m/z calcd for (C15H12BNO3S2)-H 328.0279, found 328.0279.
4.2. PAINS.
All of the biologically tested compounds were evaluated for structural attributes consistent with classification as pan-assay interference compounds (PAINS).51 The results for each compound were negative.
4.3. Molecular Modeling.
LigPrep in Schrödinger Maestro45 was used to produce low energy 3D structures of compounds. The ionization/tautomeric states were generated using Epik. Chirality of the compounds was retained from the original state. All the conformations were minimized using OPLS3e force field and at the most 32 conformations per ligands were generated. Protein crystallographic structure 2GPP42 was loaded from the RCSB Protein Data Bank (PDB) and prepared by using Protein Preparation Wizard. For the dimer, the ligand bound fragment was selected for processing. It was then pre-processed by assigning the bond orders, added hydrogen, filled in the missing loops and the side chains using Prime. Waters were deleted beyond 10 Å from the ligand and ionization/tautomeric states were generated at pH 7.0 ± 4.0 using Epik. Subsequently, we refined the protein by optimizing the hydrogen bonds (H-bonds) and the sample water orientations. Finally, the Imprefminimization was carried out using OPLS3e force field. Some of the structures were represented using PyMOL software.52
4.4. Biological assays
4.4.1. Luciferase assay
HEK293 cells were maintained in Dulbecco’s modified Eagles medium (DMEM) supplemented with 10% fetal bovine serum at 37 °C under 5% CO2. Cells were plated in 96-well plates at a density of 2.5x104 cells/well and transiently transfected using Lipofectamine™ 2000 (Invitrogen) according to manufacturer’s instructions. Cells were transfected with ERRE reporter construct and pcDNA3.1 ERRγ. 24-hour post-transfection, the cells were treated with vehicle or compound for 24 hours. Luciferase activity was measured using the One-Glo luciferase reporter assay system (Promega). The values indicated represent the means ± S.E. from four independently transfected wells.
4.4.2. Protein purification
DNA encoding the human ERRγ LBD (residues 229–458), which includes an N-terminal human rhinovirus (HRV) 3C protease recognition site was purchased from Integrated DNA technologies Inc. (San Diego, CA) and cloned into pET45 b(+) vector at KpnI and BamHI sites to incorporate an N-terminal His6 epitope. His6-ERRγ LBD protein was expressed using Escherichia coli BL21 (DE3) cells in autoinduction media containing 1 mM ampicillin. Cells diluted 500-fold from LB pre-culture were grown at 37 °C with vigorous shaking. The temperature was subsequently lowered to 22 °C when the optical density at 600 nm (Od600) reaches 1.2 and protein expression was allowed to proceed for 14 h. Protein purification was performed based on a modified version of a previously published protocol.42 Briefly, cell pellets were resuspended in lysis buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 50 mM imidazole, 5 mM TCEP, 1 mM PMSF and EDTA-free mini protease inhibitor cocktail tablets from Roche) and subsequently lysed via an Evanston cell disruptor. The lysate was clarified by centrifugation at 22,000 x g, filtered, and loaded onto a HisTrap FF Crude column (GE Life Sciences). Proteins in wash buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 5 mM TCEP, 50 mM imidazole) were eluted against a 500 mM imidazole gradient. His6-ERRγ was subsequently loaded onto a size exclusion column (HiLoad Superdex 75 pg, GE Life Sciences) and eluted using NMR buffer (20 mM KPO4, pH 7.4, 50 mM KCl, 0.5 mM EDTA, 5 mM TCEP).
4.4.3. NMR Spectroscopy
1H-NMR spectra were acquired in a Bruker Avance HDTM 700 MHz spectrometer at 298 K for 116 μM ERRγ supplemented with 10% D2O in the presence of 0.5% DMSO-d6 (vehicle control) or 1.05:1 molar equivalence of ligands (6, 9b, 10o, 10s, 12e, SR-9238, and 4-OH-Tamoxifen) from 20 mM DMSO-d6 stock solutions. SR9238, a known LXR inverse agonist, was used as a negative control.
4.4.4. Gene Expression
C2C12 cells (ATCC CRL-1722) were maintained in Gibco Dulbecco’s Modified Eagle Medium (DMEM) supplemented with GlutaMAX and 10% FBS. Cells were incubated at 37 0C in a humidified atmosphere at 5% CO2. Cells were seeded in 12 well plates at a density of 1x105 cells/well and treated with 5 μM compounds or control for 24 hours (3 replicates per condition). RNA extraction was performed using manufacturer’s protocol (RNeasy kit Qiagen). 1 ug of RNA was reverse transcribed into cDNA using qScript cDNA Synthesis Kit (Quanta Biosciences). Quantitative PCR (qPCR) was performed with SYBR Select Master Mix (Applied Biosystems). Gene expression were normalized to 36b4.
Primer sequence:
36b4 Forward – ACCTCCTTCTTCCAGGCTT
36b4 Reverse – CCCACCTTGTCTCCAGTCTTT
PDK4 Forward- ATCTAACATCGCCAGAATTAAACC
PDK4 Reverse- GGAACGTACACAATGTGGATTG
PGC1α Forward- CCCTGCCATTGTTAAGACC
PGC1α Reverse- TGCTGCTGTTCCTGTTTTC
DDIT4 Forward- CCTGCGCGTTTGCTCATGCC
DDIT4 Reverse- GGCCGCACGGCTCACTGTAT
4.4.4 : Animal study:
General mouse study
Male mice were obtained from Jackson Laboratories (Bar Harbor, ME). All procedures were approved and conducted in accordance to Washington University Institutional Animal Care and Use Committee.
For all experiment males C57BL6/J mice (n=4, 12 weeks of age, chow diet) were administered a single dose of SLU-PP-915 20 mg/kg or vehicle control (12%DMSO, 15%Tween20 and 75% PBS, i.p). One hour after injection, mice were euthanized, and muscles were collected for gene expression analysis by real time QPCR using methods previously described. Gene expression was normalized to 36B4 in all experiments.
Exercise endurance in WT mice
For exhaustion protocol, 6 males C57BL6/J, 12 weeks old, were with vehicle control (10% tween, 10% DMSO, 80% PBS) or SLU-PP-915 (20mg/kg, IP) for 6 days before testing. Mice were allowed to acclimate to the treadmill for 10 minutes / day, every day at 2m/min. The day of the test, mice were run 1h after the last dose of vehicle or drug. Mice were running for 2 minutes at 10m/min, then 6 minutes at 12m/min then ran until exhaustion by increasing the speed of the belt for 2m/min every 2 minutes 53. Exhaustion was assessed by mice allowing 10 consecutive 3ms electrical shock without moving. Mice were sacrificed by CO2 asphyxiation just after exhaustion. Tissues were collected, snap freeze in liquid nitrogen and stored at -80C until analysis.
4.4.5. Statistical Analysis
Data are expressed as mean +/- SEM. Student’s test or Two-way ANOVA were used to calculate statistical significance. P<0.05 was considered significant.
Supplementary Material
Highlights:
Identifies a novel series of 2,5-disubstiuted thiophenes that act as potent pan-ERR nuclear receptor agonists
The best compound, 10s (SLU-PP-915), contains a boronic acid moiety that is acting as a hydrogen-bond donor and an isostere for the phenol group.
Compounds containing the phenyl boronic acid moiety were found to have improved in vitro microsomal stability as compared to the corresponding aniline or phenol
Performed ligand-protein NMR experiments to highlight binding of key compounds to the Ligand-binding domain of ERRγ
Compound 10s was able to significantly up-regulate known ERR target genes both in vitro and in vivo and improved exercise capacity in an in vivo mouse experiment
Acknowledgements
We thank the National Institutes of Health (NIH) for their financial support for this work, R01 AR069280 (T.P.B and J. K. W).
Abbreviations:
- ER
estrogen receptor
- ERR
estrogen receptor-related receptor
- HBD
Hydrogen bond donor
- LBD
ligand binding domain
- NR
nuclear receptor
- PK
pharmacokinetics
Footnotes
Declaration of competing interest
T.B.P and J.K.W. are co-founders and stockholders in Myonid Therapeutics, Inc., which focuses on ERR based therapeutics.
Appendix A. Supplementary Data
Supporting Information. Supplemental Figure 1; Additional 1H NMR methyl region spectra of ERRγ in the presence of ligands 9b, 9n, 10o, and 4-OHT. Supplemental Figure 2; NMR peak shift and intensities for protein-ligand binding experiments. Supplemental Figure 3; Additional docking poses for compounds 10o, 12i and 12m. 1H-NMR, 13C-NMR, and Hi-Res MS data for representative compounds 9b, 9n, 10j, 10n, 10o, 10s. 1H-NMR spectra for all other compounds.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests:
John K Walker reports financial support was provided by National Institutes of Health. Thomas P. Burris reports financial support was provided by National Institutes of Health. John K. Walker has patent #WO2019036562; Heteroaromatics as ERR inverse agonists and their preparation licensed to Myonid Therapeutics. Thomas P. Burris has patent #WO2019036562; Heteroaromatics as ERR inverse agonists and their preparation licensed to Myonid Therapeutics. Carissa Hampton has patent #WO2019036562; Heteroaromatics as ERR inverse agonists and their preparation licensed to Myonid Therapeutics. Keith Haynes has patent #WO2019036562; Heteroaromatics as ERR inverse agonists and their preparation licensed to Myonid Therapeutics. Kristine Griffett has patent #WO2019036562; Heteroaromatics as ERR inverse agonists and their preparation licensed to Myonid Therapeutics. Cyrielle Billon has patent #WO2019036562; Heteroaromatics as ERR inverse agonists and their preparation licensed to Myonid Therapeutics. Sadichha Situala has patent #WO2019036562; Heteroaromatics as ERR inverse agonists and their preparation licensed to Myonid Therapeutics. Founders in Myonid Therapeutics which is trying to develop ERR modulators as novel therapeutics (J.K.W. & T.P.B)
References
- 1.Gallastegui N; Mackinnon JA; Fletterick RJ; Estebanez-Perpina E, Advances in our structural understanding of orphan nuclear receptors. Trends Biochem Sci 2015, 40 (1), 25–35. [DOI] [PubMed] [Google Scholar]
- 2.Mazaira GI; Zgajnar NR; Lotufo CM; Daneri-Becerra C; Sivils JC; Soto OB; Cox MB; Galigniana MD, The nuclear receptor field: a historical overview and future challenges. Nucl. Recept. Res 2018, 5, 101320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Horard B; Vanacker JM, Estrogen receptor-related receptors: orphan receptors desperately seeking a ligand. Journal of Mol. Endocrinol 2003, 31 (3), 349–357. [DOI] [PubMed] [Google Scholar]
- 4.Greschik H; Wurtz J-M; Sanglier S; Bourguet W; van Dorsselaer A; Moras D; Renaud J-P, Structural and Functional Evidence for Ligand-Independent Transcriptional Activation by the Estrogen-Related Receptor 3. Mol. Cell 2002, 9 (2), 303–313. [DOI] [PubMed] [Google Scholar]
- 5.Hong H; Yang L; Stallcup MR, Hormone-independent Transcriptional Activation and Coactivator Binding by Novel Orphan Nuclear Receptor ERR3. J. Biol. Chem 1999, 274 (32), 22618–22626. [DOI] [PubMed] [Google Scholar]
- 6.Crevet L; Vanacker J-M, Regulation of the expression of the estrogen related receptors (ERRs). Cell. Mol. Life Sci 2020, 77 (22), 4573–4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ranhotra HS, The estrogen-related receptors: orphans orchestrating myriad functions. J. Recept. Signal Transduct 2012, 32 (2), 47–56. [DOI] [PubMed] [Google Scholar]
- 8.Kallen J; Lattmann R; Beerli R; Blechschmidt A; Blommers MJJ; Geiser M; Ottl J; Schlaeppi J-M; Strauss A; Fournier B, Crystal Structure of Human Estrogen-related Receptor α in Complex with a Synthetic Inverse Agonist Reveals Its Novel Molecular Mechanism. J. Biol. Chem 2007, 282 (32), 23231–23239. [DOI] [PubMed] [Google Scholar]
- 9.Audet-walsh É; Giguére V, The multiple universes of estrogen-related receptor α and γ in metabolic control and related diseases. Acta Pharmacol. Sin 2014, 36, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fan W; Evans R, PPARs and ERRs: molecular mediators of mitochondrial metabolism. Curr. Opin. Cell Biol 2015, 33, 49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luo J; Sladek R; Carrier J; Bader J-A; Richard D; Giguère V, Reduced Fat Mass in Mice Lacking Orphan Nuclear Receptor Estrogen-Related Receptor α. Mol. Cell. Biol 2003, 23 (22), 7947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hesselink MKC; Schrauwen-Hinderling V; Schrauwen P, Skeletal muscle mitochondria as a target to prevent or treat type 2 diabetes mellitus. Nat. Rev. Endocrinol 2016, 12, 633. [DOI] [PubMed] [Google Scholar]
- 13.Misra J; Kim D-K; Choi H-S, ERRγ: a Junior Orphan with a Senior Role in Metabolism. Trends Endocrinol. Metab 2017, 28 (4), 261–272. [DOI] [PubMed] [Google Scholar]
- 14.Sakamoto T; Matsuura TR; Wan S; Ryba DM; Kim JU; Won KJ; Lai L; Petucci C; Petrenko N; Musunuru K; Vega RB; Kelly DP, A Critical Role for Estrogen-Related Receptor Signaling in Cardiac Maturation. Circ. Res 2020, 126 (12), 1685–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alaynick WA; Kondo RP; Xie W; He W; Dufour CR; Downes M; Jonker Johan W.; Giles W; Naviaux RK; Giguère V; Evans RM, ERRg Directs and Maintains the Transition to Oxidative Metabolism in the Postnatal Heart. Cell Metab 2007, 6 (1), 13–24. [DOI] [PubMed] [Google Scholar]
- 16.Torrano V; Valcarcel-Jimenez L; Cortazar AR; Liu X; Urosevic J; Castillo-Martin M; Fernández-Ruiz S; Morciano G; Caro-Maldonado A; Guiu M; Zúñiga-García P; Graupera M; Bellmunt A; Pandya P; Lorente M; Martín-Martín N; Sutherland JD; Sanchez-Mosquera P; Bozal-Basterra L; Zabala-Letona A; Arruabarrena-Aristorena A; Berenguer A; Embade N; Ugalde-Olano A; Lacasa-Viscasillas I; Loizaga-Iriarte A; Unda-Urzaiz M; Schultz N; Aransay AM; Sanz-Moreno V; Barrio R; Velasco G; Pinton P; Cordon-Cardo C; Locasale JW; Gomis RR; Carracedo A, The metabolic co-regulator PGC1α suppresses prostate cancer metastasis. Nat. Cell Biol 2016, 18 (6), 645–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schoepke E; Billon C; Haynes KM; Avdagic A; Sitaula S; Sanders R; Adeyemi CM; Walker JK; Burris TP, A Selective ERRα/γ Inverse Agonist, SLU-PP-1072, Inhibits the Warburg Effect and Induces Apoptosis in Prostate Cancer Cells. ACS Chem. Biol 2020, 15 (9), 2338–2345. [DOI] [PubMed] [Google Scholar]
- 18.Zhang L; Liu P; Chen H; Li Q; Chen L; Qi H; Shi X; Du Y, Characterization of a selective inverse agonist for estrogen related receptor α as a potential agent for breast cancer. Eur. J. Pharmacol 2016, 789, 439–448. [DOI] [PubMed] [Google Scholar]
- 19.Deblois G; St-Pierre J; Giguere V, The PGC-1/ERR signaling axis in cancer. Oncogene 2013, 32 (30), 3483–3490. [DOI] [PubMed] [Google Scholar]
- 20.Tang Y; Min Z; Xiang X-J; Liu L; Ma Y-L; Zhu B-L; Song L; Tang J; Deng X-J; Yan Z; Chen G-J, Estrogen-related receptor alpha is involved in Alzheimer's disease-like pathology. Exp. Neurol 2018, 305, 89–96. [DOI] [PubMed] [Google Scholar]
- 21.Narkar Vihang A.; Fan W; Downes M; Yu Ruth T.; Jonker Johan W.; Alaynick William A.; Banayo E; Karunasiri Malith S.; Lorca S; Evans Ronald M., Exercise and PGC-1α-Independent Synchronization of Type I Muscle Metabolism and Vasculature by ERRγ. Cell Metab 2011, 13 (3), 283–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fan W; He N; Lin CS; Wei Z; Hah N; Waizenegger W; He M-X; Liddle C; Yu RT; Atkins AR; Downes M; Evans RM, ERRγ Promotes Angiogenesis, Mitochondrial Biogenesis, and Oxidative Remodeling in PGC1α/β-Deficient Muscle. Cell Rep 2018, 22 (10), 2521–2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matsakas A; Yadav V; Lorca S; Narkar V, Muscle ERRγ mitigates Duchenne muscular dystrophy via metabolic and angiogenic reprogramming. FASEB J 2013, 27 (10), 4004–4016. [DOI] [PubMed] [Google Scholar]
- 24.Perry M-C; Dufour CR; Tam IS; B'Chir W; Giguère V, Estrogen-Related Receptor-α Coordinates Transcriptional Programs Essential for Exercise Tolerance and Muscle Fitness. Mol. Endocrinol 2014, 28 (12), 2060–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu X; Peng L; Lv M; ding K, Recent Advance in the Design of Small Molecular Modulators of Estrogen-Related Receptors. Curr. Pharm. Des 2012, 18 (23), 3421–3431. [DOI] [PubMed] [Google Scholar]
- 26.Yao B; Zhang S; Wei Y; Tian S; Lu Z; Jin L; He Y; Xie W; Li Y, Structural Insights into the Specificity of Ligand Binding and Coactivator Assembly by Estrogen-Related Receptor β. J. Mol. Biol 2020, 432 (19), 5460–5472. [DOI] [PubMed] [Google Scholar]
- 27.Greschik H; Flaig R; Renaud J-P; Moras D, Structural Basis for the Deactivation of the Estrogen-related Receptor γ by Diethylstilbestrol or 4-Hydroxytamoxifen and Determinants of Selectivity. J. Biol. Chem 2004, 279 (32), 33639–33646. [DOI] [PubMed] [Google Scholar]
- 28.Chao EYH; Collins JL; Gaillard S; Miller AB; Wang L; Orband-Miller LA; Nolte RT; McDonnell DP; Willson TM; Zuercher WJ, Structure-guided synthesis of tamoxifen analogs with improved selectivity for the orphan ERRγ. Bioorg. Med. Chem. Lett 2006, 16 (4), 821–824. [DOI] [PubMed] [Google Scholar]
- 29.Kim J; Woo SY; Im CY; Yoo EK; Lee S; Kim H-J; Hwang H-J; Cho J.-h.; Lee WS; Yoon H; Kim S; Kwon O.-b.; Hwang H; Kim K-H; Jeon J-H; Singh TD; Kim SW; Hwang SY; Choi H-S; Lee I-K; Kim SH; Jeon YH; Chin J; Cho SJ, Insights of a Lead Optimization Study and Biological Evaluation of Novel 4-Hydroxytamoxifen Analogs as Estrogen-Related Receptor γ (ERRγ) Inverse Agonists. J. Med. Chem 2016, 59 (22), 10209–10227. [DOI] [PubMed] [Google Scholar]
- 30.Yu DD; Huss JM; Li H; Forman BM, Identification of novel inverse agonists of estrogen-related receptors ERRγ and ERRβ. Bioorg. Med. Chem 2017, 25 (5), 1585–1599. [DOI] [PubMed] [Google Scholar]
- 31.Kim Y; Koh M; Kim D-K; Choi H-S; Park SB, Efficient Discovery of Selective Small Molecule Agonists of Estrogen-Related Receptor γ using Combinatorial Approach. ACS Comb Sci 2009, 11 (5), 928–937. [DOI] [PubMed] [Google Scholar]
- 32.Zuercher WJ; Gaillard S; Orband-Miller LA; Chao EYH; Shearer BG; Jones DG; Miller AB; Collins JL; McDonnell DP; Willson TM, Identification and Structure−Activity Relationship of Phenolic Acyl Hydrazones as Selective Agonists for the Estrogen-Related Orphan Nuclear Receptors ERRβ and ERRγ. J. Med. Chem 2005, 48 (9), 3107–3109. [DOI] [PubMed] [Google Scholar]
- 33.Lin H; Doebelin C; Patouret R; Garcia-Ordonez RD; Chang MR; Dharmarajan V; Bayona CR; Cameron MD; Griffin PR; Kamenecka TM, Design, synthesis, and evaluation of simple phenol amides as ERRγ agonists. Bioorg. Med. Chem. Lett 2018, 28 (8), 1313–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu S; Mao L; Ding P; Zhuang X; Zhou Y; Yu L; Liu Y; Nie T; Xu T; Xu Y; Liu J; Smaill J; Ren X; Wu D; Ding K, 1-Benzyl-4-phenyl-1H-1,2,3-triazoles improve the transcriptional functions of estrogen-related receptor gamma and promote the browning of white adipose. Bioorg. Med. Chem 2015, 23 (13), 3751–60. [DOI] [PubMed] [Google Scholar]
- 35.Hyatt SM; Lockamy EL; Stein RA; McDonnell DP; Miller AB; Orband-Miller LA; Willson TM; Zuercher WJ, On the Intractability of Estrogen-Related Receptor α as a Target for Activation by Small Molecules. J. Med. Chem 2007, 50 (26), 6722–6724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suetsugi M; Su L; Karlsberg K; Yuan Y-C; Chen S, Flavone and Isoflavone Phytoestrogens Are Agonists of Estrogen-Related Receptors. Mol. Cancer Res 2003, 1 (13), 981. [PubMed] [Google Scholar]
- 37.Peng L; Gao X; Duan L; Ren X; Wu D; Ding K, Identification of Pyrido[1,2-α]pyrimidine-4-ones as New Molecules Improving the Transcriptional Functions of Estrogen-Related Receptor α. J. Med. Chem 2011, 54 (21), 7729–7733. [DOI] [PubMed] [Google Scholar]
- 38.Shinozuka T; Ito S; Kimura T; Izumi M; Wakabayashi K, Discovery of a Novel Class of ERRα Agonists. ACS Med. Chem. Lett 2021, 12 (5), 817–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Billon C; Sitaula S; Banerjee S; Welch R; Elgendy B; Hegazy L; Oh TG; Kazantzis M; Chatterjee A; Chrivia J; Hayes ME; Xu W; Hamilton A; Huss JM; Zhang L; Walker JK; Downes M; Evans RM; Burris TP, Synthetic ERRα/β/γ Agonist Induces an ERRα-Dependent Acute Aerobic Exercise Response and Enhances Exercise Capacity. ACS Chem. Biol 2023, 18 (4), 756–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shahien M; Elagawany M; Sitaula S; Goher SS; Burris SL; Sanders R; Avdagic A; Billon C; Hegazy L; Burris TP; Elgendy B, Modulation of estrogen-related receptors subtype selectivity: Conversion of an ERRβ/γ selective agonist to ERRα/β/γ pan agonists. Bioorg. Chem 2020, 102, 104079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abad MC; Askari H; O’Neill J; Klinger AL; Milligan C; Lewandowski F; Springer B; Spurlino J; Rentzeperis D, Structural determination of estrogen-related receptor γ in the presence of phenol derivative compounds. J. Steroid Biochem. Mol. Biol 2008, 108 (1), 44–54. [DOI] [PubMed] [Google Scholar]
- 42.Wang L; Zuercher WJ; Consler TG; Lambert MH; Miller AB; Orband-Miller LA; McKee DD; Willson TM; Nolte RT, X-ray Crystal Structures of the Estrogen-related Receptor-γ Ligand Binding Domain in Three Functional States Reveal the Molecular Basis of Small Molecule Regulation. J. Biol. Chem 2006, 281 (49), 37773–37781. [DOI] [PubMed] [Google Scholar]
- 43.Kleckner IR; Foster MP, An introduction to NMR-based approaches for measuring protein dynamics. Biochimica et Biophysica Acta 1864 2011, 1814 (8), 942–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Griffett K; Solt LA; El-Gendy Bel D; Kamenecka TM; Burris TP, A liver-selective LXR inverse agonist that suppresses hepatic steatosis. ACS Chem. Biol 2013, 8 (3), 559–67. [DOI] [PubMed] [Google Scholar]
- 45.Schrödinger Maestro, Schrödinger LLC, New York, NY: 2021. [Google Scholar]
- 46.Friesner RA; Murphy RB; Repasky MP; Frye LL; Greenwood JR; Halgren TA; Sanschagrin PC; Mainz DT, Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein−Ligand Complexes. J. Med. Chem 2006, 49 (21), 6177–6196. [DOI] [PubMed] [Google Scholar]
- 47.Eurofins Discovery; 6 Research Park Dr, St. Charles MO.; www.eurofinsdiscoveryservices.com. [Google Scholar]
- 48.Huss JM; Kopp RP; Kelly DP, Peroxisome Proliferator-activated Receptor Coactivator-1α (PGC-1α) Coactivates the Cardiac-enriched Nuclear Receptors Estrogen-related Receptor-α and -γ. J. Biol. Chem 2002, 277 (43), 40265–40274. [DOI] [PubMed] [Google Scholar]
- 49.Schreiber SN; Knutti D; Brogli K; Uhlmann T; Kralli A, The transcriptional coactivator PGC-1 regulates the expression and activity of the orphan nuclear receptor estrogen-related receptor alpha (ERRalpha). J Biol. Chem 2003, 278 (11), 9013–8. [DOI] [PubMed] [Google Scholar]
- 50.Wende AR; Huss JM; Schaeffer PJ; Giguere V; Kelly DP, PGC-1α coactivates PDK4 gene expression via the orphan nuclear receptor ERRα: A mechanism for transcriptional control of muscle glucose metabolism. Mol. Cell. Biol 2005, 25 (24), 10684–10694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Baell JB; Holloway GA, New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem 2010, 53 (7), 2719–2740. [DOI] [PubMed] [Google Scholar]
- 52.Schrödinger L, The PyMOL Molecular Graphics System, Version 2.0 [Google Scholar]
- 53.Castro B; Kuang S, Evaluation of Muscle Performance in Mice by Treadmill Exhaustion Test and Whole-limb Grip Strength Assay. Bio. Protoc 2017, 7 (8). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.