

Abstract
Pancreatic cancer is an aggressive malignancy with increasing incidence. Pancreatic ductal adenocarcinoma (PDAC) accounts for > 90% of pancreatic cancer diagnoses, while other exocrine tumors are much rarer. In this review, we have focused on two rare cancers of the exocrine pancreas: adenosquamous carcinoma of the pancreas (ASCP) and pancreatic acinar cell carcinoma (PACC). The latest findings regarding their cellular and molecular pathology, clinical characteristics, prognosis, and clinical management are discussed. New genetic and transcriptomic data suggest that ASCP is related to or overlaps with the basal transcriptomic subtype of PDAC. These tumors are highly aggressive and driven by activated KRAS and MYC expression. Clinical outcomes remain poor and effective treatments are limited. PACC has no morphologic or genetic resemblance to PDAC and more favorable outcomes. Early stage PACC patients have improved survival with surgical resection and patients with advanced disease benefit most from platinum- or fluoropyrimidine-containing chemotherapy. Frequency of actionable genetic mutations is high in this disease and case reports suggest good outcomes when matched therapy is given. Dedicated clinical studies examining ASCP and PACC are limited and difficult to accrue. Further research is needed to define optimal clinical management for these rare diseases.
Keywords: Adenosquamous carcinoma of the pancreas, acinar cell carcinoma of the pancreas, rare exocrine pancreatic cancer
INTRODUCTION
Pancreatic cancer is an aggressive malignancy with a 5-year overall survival rate in the United States of just 11% despite recent advances in systemic chemotherapy that have improved outcomes for patients with both advanced and early-stage disease[1–5]. While pancreatic cancer contributes only 3.2% of new cancer cases in the US, the high mortality rate has made pancreatic cancer the third most common cause of cancer-related death in the country[5]. Since the incidence of pancreas cancer is increasing every year, it is projected to overtake colorectal cancer as the 2nd most common cause of cancer death by 2030[6]. Pancreatic cancer has a similarly grim prognosis and incidence trajectory globally[7].
Most pancreatic cancers arise from ductal and acinar cells involved in the exocrine functions of the organ. Pancreatic ductal adenocarcinoma (PDAC) is the most common histology and represents > 90% of pancreatic cancer cases. It is so common compared to other types of pancreatic cancer that mention of pancreatic cancer can be assumed synonymous with PDAC unless otherwise specified. Tumors arising from endocrine cells of the pancreas represent ~5% of all pancreas cancers[8] are mostly less aggressive than PDAC, and have entirely different standard-of-care treatment paradigms[9]. Even less common than pancreatic neuroendocrine tumors are rare tumors of the exocrine pancreas, such as adenosquamous carcinoma, acinar cell carcinoma, mucinous cystic neoplasm, colloid carcinoma, and pancreatoblastoma. These diseases are so rare that treatment paradigms for them are typically extrapolated from PDAC standard of care even though their histology and molecular underpinnings may differ markedly from PDAC.
In this review, we have described what the field presently knows about two rare exocrine cancers of the pancreas: adenosquamous carcinoma and acinar cell carcinoma. We have defined their cellular and molecular pathology, clinical characteristics, and prognosis. Basic and translational studies examining their origins and behavior have been surveyed. Case studies and epidemiologic reports which provide insights into fruitful treatment paradigms have been reviewed. It is important to note that there are no prospective clinical studies reported in the literature that examine any aspect of these diseases. Significant differences between these tumors and PDAC have been highlighted to provide insight into when clinicians should diverge from established PDAC standards of care when treating these patients. In the end, we aimed to identify the important unanswered clinical questions about these diseases, providing a guide for future research that could allow clinicians to offer the first evidence-based advice to patients.
STANDARD OF CARE TREATMENT FOR PDAC
PDAC typically presents with non-specific symptoms such as back pain, unexplained weight loss, jaundice, GI discomfort or thromboembolism[10,11]. Most patients already have distant metastasis (52%) or locoregional disease (30%) at the time of diagnosis. Primary tumors are most commonly located in the pancreatic head[12], while metastases are most often located in the liver, peritoneum and lung.
The staging for PDAC is shown in Table 1. Current standard of care for early-stage disease (Stage I and II, or Stage III that is not T4) is upfront surgical resection followed by adjuvant chemotherapy. There is no appreciable cure rate if chemotherapy is not given[13]. Choices of adjuvant chemotherapy include single-agent gemcitabine for those with poorer performance status, gemcitabine in combination with capecitabine, or modified FOLFIRINOX for those with excellent performance status[2,4,13]. More than 50% of patients who complete these potentially curative regimens will recur and die of their disease. Currently, neoadjuvant strategies are being evaluated and may prove more beneficial in patients with resectable disease[14]. Notably, complete neoadjuvant treatment is considered the standard of care for patients with borderline resectable and locally advanced diseases at many pancreatic cancer centers. The benefit of chemoradiation has not been clearly established, but it is commonly incorporated in neoadjuvant paradigms, especially in cases of borderline resectable or locally advanced disease[15]. Locally advanced (Stage III that is T4) and metastatic (Stage IV) PDAC are treated with palliative chemotherapy. Appropriate regimens for fit patients include FOLFIRINOX or gemcitabine/nanoalbumin-bound (nab-) paclitaxel (GnP), which can extend median survival to 11.5 months[1,16]. Single-agent gemcitabine can be given to patients with poorer performance status to provide clinical benefit[17]. Of note, PDAC is generally unresponsive to immunotherapy[18,19]. Markers of response to immune checkpoint inhibitors - high microsatellite instability (MSI-H) and mismatch repair deficiency (dMMR) - occur in less than 2% of PDAC patients[20,21], but even in this small group, responses to immunotherapy are lower when compared to other patients with MSI-H/dMMR solid tumors[22].
Table 1.
AJCC staging for PDAC
| Stage 0 | Tis | N0 | M0 |
|---|---|---|---|
| Stage IA | T1 | N0 | M0 |
| Stage IB | T2 | N0 | M0 |
| Stage IIA | T3 | N0 | M0 |
| Stage IIB | T1, T2, T3 | N1 | M0 |
| Stage III | T1, T2, T3, T4 | N2 | M0 |
| Any | M0 | ||
| Stage IV | Any | Any | M1 |
| T | Primary Tumor | ||
| TX | Primary tumor cannot be assessed | ||
| T0 | No evidence of primary tumor | ||
| Tis | Carcinoma in situ, including: High-grade pancreatic intraepithelial neoplasia Intraductal papillary mucinous neoplasm with high-grade dysplasia Intraductal tubulopapillary neoplasm with high-grade dysplasia Mucinous cystic neoplasm with high-grade dysplasia |
||
| T1 | Largest tumor diameter < 2 cm | ||
| T1a | Largest tumor diameter ≤ 0.5 cm | ||
| T1b | Largest tumor diameter > 0.5 cm and < 1 cm | ||
| T1c | Largest tumor diameter 1-2 cm | ||
| T2 | Largest tumor diameter > 2 cm and ≤ 4 cm | ||
| T3 | Largest tumor diameter > 4 cm | ||
| T4 | Tumor involves the celiac axis, superior mesenteric artery, and/or common hepatic artery, regardless of size | ||
| N | Regional Lymph Nodes | ||
| Nx | Regional lymph nodes cannot be assessed | ||
| N0 | No regional lymph node metastasis | ||
| N1 | Metastasis in 1-3 regional lymph nodes | ||
| N2 | Metastasis in ≥ 4 regional lymph nodes | ||
| M | Distant Metastasis | ||
| M0 | No distant metastasis | ||
| M1 | Distant Metastasis | ||
The poor response of PDAC to both chemo- and immunotherapies has most often been attributed to the tumor’s unique microenvironment. PDAC typically has a desmoplastic stroma which makes up approximately 70% of the tumor mass, leading to vascular collapse and hypoxia[23]. This results in an “immunologically cold” microenvironment that is mainly infiltrated by immunosuppressive myeloid cells such as tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), tumor-associated neutrophils (TANs), and FOXP3+ CD4+ regulatory T (Treg) cells, with exclusion of tumor-restraining effector T cells (reviewed in[24]).
The genomic landscape of PDAC is well known. The four most commonly mutated genes are KRAS, TP53, CDKN2A, and SMAD4, all of which have been considered “undruggable” until recently[25,26]. Notably, mutations in KRAS are the primary driver of PDAC oncogenesis, occurring in > 90% of patient cases[27]. No other mutations occur more frequently than 10%. Around 5%-9% of PDAC tumors exhibit germline or somatic mutations in homologous recombination (HR) repair-related genes such as BRCA1/2 or PALB2[28,29], and these tumors have significant sensitivity to platinum chemotherapy[30]. Treatment of these tumors with platinum-containing regimens doubles patient survival[31–33]. These patients may also benefit from maintenance treatment with olaparib, a PARP inhibitor[34].
Transcriptomic profiling has revealed two main PDAC subtypes - a less-aggressive, better differentiated classical subtype, and a more aggressive basal (also called quasimesenchymal or squamous) subtype[35–37]. It is important to note that some tumors fail to fall distinctly into a single transcriptomic category[38], transcriptomic subtype can be heterogeneous across individual tumors[39,40], and transcriptomic subtype may not be static as treatment or environmental factors may select for a particular subtype[41,42].
ADENOSQUAMOUS CARCINOMA OF THE PANCREAS (ASCP)
Pathologic characteristics
Histology of ASCP
ASCP is a rare exocrine cancer that includes both a glandular and a malignant squamous component. It was first reported by Herxheimer in 1907 as a “cancroide” tumor. Subsequently, other researchers have called it mucoepidermoid carcinoma, adenoacanthoma, and mixed squamous and adenocarcinoma[43]. ASCP is defined by the presence of a malignant squamous component in at least 30% of cancer cells in a background of malignant glandular epithelium[44,45]. The arbitrary cutoff of 30% has been disputed in the literature[45,46], with opponents noting that the evaluation is subjective and highly dependent on sampling[47], especially when fine-needle aspiration is used[48]. This was further substantiated by a retrospective trial showing that the percentage of cells below or above the 30% cutoff did not correlate with different clinical outcomes[49]. Conversely, a very high percentage of squamous cell component (> 60%) was associated with worse survival in resectable patients[50].
Under light microscopy, ASCP is more likely to have increased necrosis, to be poorly differentiated and to have increased vascular invasion compared to PDAC[51]. Malignant glandular components of ASCP appear similar to PDAC, while the squamous component has well-defined cell borders with intercellular bridges and keratinization of the cytoplasm[52]. Representative H&E-stained images of ASCP are shown in Figure 1. Immunohistochemistry differs between the two components. The glandular component stains positive for CK8/18, CK7, CEA, and CA19-9, similar to PDAC, while appreciable expression of CK5/6, p63, and p40 is seen in the squamous component. The squamous area also exhibits frequent loss of E-cadherin and p16, and increased EGFR and vimentin expression[52–55]. Positive nuclear p63 expression has been validated as a more sensitive marker for ASCP in tumors that are difficult to classify by routine H&E stains[52]. Others have noted that metastases from an ASCP primary may appear as a pure adenocarcinoma, or even dedifferentiate to giant cell or pleomorphic-appearing anaplastic carcinomas with multi-nucleated forms, markedly bizarre nuclei, and prominent nucleoli[45]. Interestingly, a recent study of the transitional area between glandular and squamous components identified an intermediate component that appeared glandular but stained heterogeneously for squamous and glandular markers (i.e., CK8/18 and p63 double-positive)[55]. This type of dual staining was also seen in some PDAC samples that lacked any histologic squamous component. Recently, a retrospective study identified a sarcomatous component to ASCP in 7 of 7 cases[54], but this finding has not been mentioned by others except for one case report describing a particularly large ASCP[56]. Immunologically, the adenocarcinoma component of ASCP resembles PDAC; however, ASCP has been noted to frequently express PD-L1 within the squamous component[57,58].
Figure 1. Histologic appearance of ASCP.
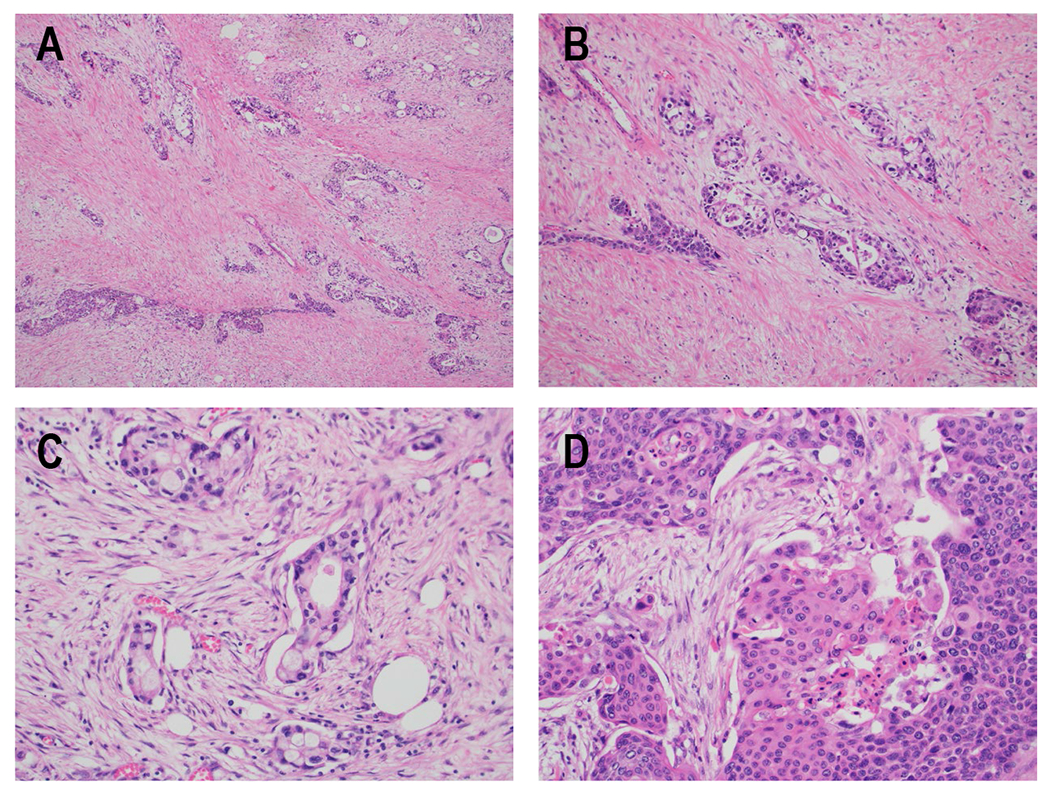
H&E specimens of ASCP. A) Areas resembling conventional ductal adenocarcinoma of the pancreas (top right) and squamous cell carcinoma (bottom left) are apparent in this sample (100x). Note the striking amount of desmoplastic stroma present between tumor nests. B) In this area of the tumor, the glandular and squamous components have merged together so that both morphologies can be seen within a single group of tumor cells (100x). C) Infiltrative glands making up the adenocarcinoma component of the tumor are shown at higher power (200x), with some cells containing possible mucin vacuoles. D) Solid sheets of tumor cells with densely eosinophilic cytoplasm and areas of keratinization, consistent with the squamous cell carcinoma component of the tumor, are shown at higher power (200x).
Molecular profiling of ASCP
Normal pancreatic tissue has no squamous cells, leading to the obvious question of how a squamous component can appear in a primary pancreatic malignancy. Three etiologies were proposed: collision of two separate primaries to form one tumor, metaplasia caused by inflammation resulting in a transition to squamous differentiation in the pre-malignant stage, or trans-differentiation of existing adenocarcinoma to a squamous component[59]. Presently, the latter theory is favored given the genomic and transcriptomic data described below, which are consistent with ASCP appearing following activation of squamous differentiation programs in a pre-existing epithelial cancer cell[35,39].
The genomics of ASCP have been assessed by several groups and are summarized in Figure 2. An initial study by Brody et al. analyzed samples from 8 ASCP patients and identified KRAS codon 12 mutations in all samples, as well as frequent DPC4 and TP53 alterations[53]. Subsequently, others also demonstrated KRAS mutation in 100% of 33 combined tumor samples, making the prevalence of this mutation even higher than that seen in PDAC[60,61]. In addition, mutation of TP53, the second most frequently mutated gene in PDAC, was found in 88% of 17 ASCP cases[61]. Frequent amplification of the MYC oncogene was also noted[62]. Through the use of laser capture microdissection to separately isolate adenomatous and squamous components of ASCP, it was shown that both histotypes have similar genomic changes, consistent with both components deriving from a common progenitor[61]. This was also seen in an ASCP arising from intraductal papillary mucinous neoplasm[63]. In summary, the genomics of both the squamous and glandular components of ASCP closely resemble what is typical for PDAC. Novel mutations unique to ASCP have been difficult to identify. Liu et al. reported frequent somatic mutations in the UPF1 RNA surveillance gene of ASCP tumors and proposed this could be a unique genetic driver of ASCP[64]; however, three succeeding studies in other ASCP cohorts were unable to replicate these findings[39,61,65]. It was later noted that 45% of UPF1 mutations purportedly present in ASCPs were identical to normal genetic variants, and functional studies of these mutations identified no pathogenic characteristics[66]. Taken together, these findings suggested that ASCP and PDAC are genetically similar and may represent the same tumor type.
Figure 2. Molecular characteristics of ASCP and PACC.
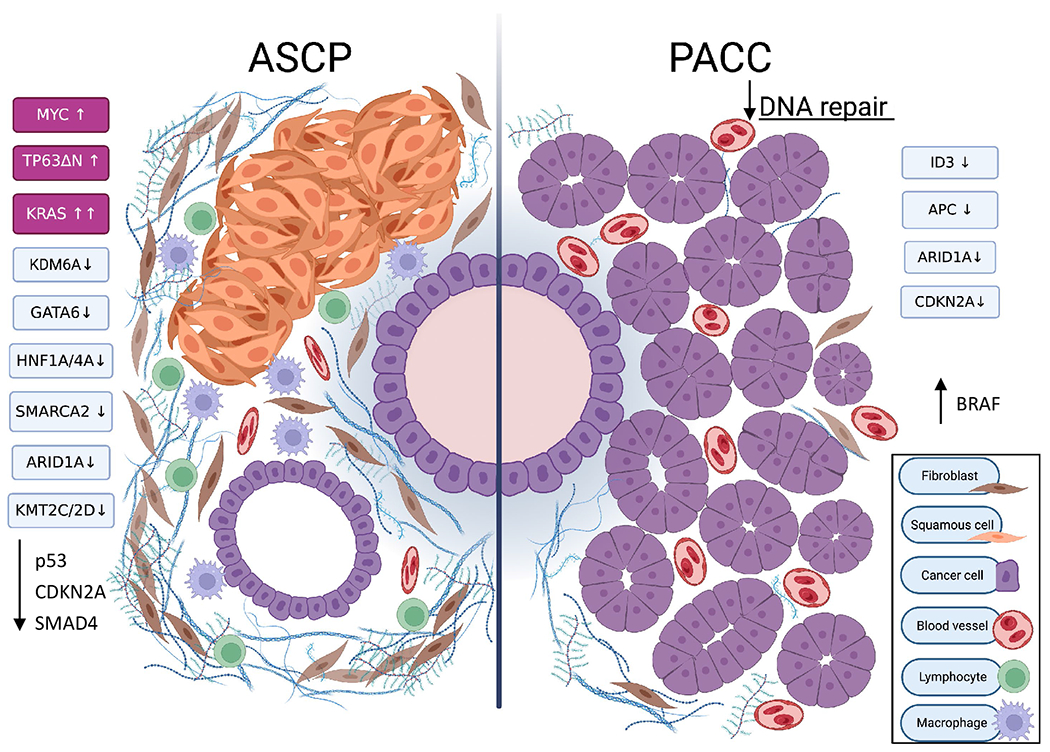
Left- Adenosquamous carcinoma of the pancreas (ASCP) is characterized by KRAS, MYC and TP63ΔN activation. Epigenetic changes cause loss of expression of chromatin modifying genes such as KDM6A and KMT2C/D as well as molecular determinants of endoderm fate leading to expression of squamous programs. Many of the usual mutations present in PDAC also occur in ASCP. Like PDAC, ASCP is known to have a dense and prolific desmoplastic stroma that is enriched in immunosuppressive immune cells. Right- Pancreatic acinar cell carcinoma (PACC) is not defined by changes in single genes. Mutational signatures suggest that impairment of the DNA damage repair system is frequent in this tumor type. Methylation and copy number changes cause downregulation of several tumor suppressors at the protein level. Activating BRAF/RAF1 mutations and fusion products have been frequently observed. The tumor is highly cellular with little stroma, extensive vasculature, and limited immune cell infiltration.
Created with BioRender.com.
ASCP has numerous similarities to the basal/squamous transcriptomic subtype of PDAC[35]. Characteristic gene programs found in basal-type PDAC include MYC pathway activation, upregulated expression of transcription factor TP63ΔN and target genes, and activated EGF and TGF-β signaling. The basal/squamous subtype was also associated with epigenetic downregulation of genes involved in pancreatic endodermal cell fate determination, such as GATA6. Hayashi et al. extended these observations in their integrated analysis of the histologic, genomic, and transcriptional characteristics of PDAC[39]. A new classification system was developed, identifying PDAC tumors with > 30% keratinization or immunohistochemical labeling with squamous markers p63 or CK5/6 as having squamous differentiation (SD), while those tumors with some, but ≤ 30%, of cancer cells having these characteristics were termed PDAC with squamoid features (SF). Overall, 15.7% of 123 exocrine pancreatic cancer cases showed SF or SD, while 5.7% of cases met the pathologic definition for ASCP. SF/SD tumor samples, whether pathologically defined as ASCP or not, overlaid with the basal transcriptomic subtype of PDAC, meaning that as many as 10% of PDAC cases contain squamous components and have a transcriptomic profile indistinguishable from ASCP. MYC amplification was much more frequent in SF/SD/ASCP tumors, and MYC copy number was highest in areas of SF/SD within tumors heterogeneous for squamous and glandular components, although it was noted that overexpression of MYC in glandular-type PDAC did not induce squamous histology. MYC is a major instigator of metastasis in PDAC. MYC expression has also been shown to drive glycolysis and to stimulate recruitment of immunosuppressive cell types to the tumor microenvironment[67–71]. Of note, SF/SD tumors had a higher likelihood of mutation in a chromatin modifying gene than tumors lacking SF/SD, and the presence of these changes in conjunction with MYC amplification or overexpression were associated with expression of squamous differentiation markers. It is important to note that mutations in chromatin modifying genes did not exceed 50% of SF/SD cases, meaning that other genetic or epigenetic changes that lead to squamous marker expression must exist[39].
Epigenetic changes cause squamous trans-differentiation and a more aggressive phenotype
The squamous program is initiated in pancreatic cancer as in other squamous tumors by upregulation of transcription factor TP63. Specifically, expression of the ΔN isoform of TP63 (TP63ΔN) is sufficient to drive the squamous differentiation program in PDAC; forced expression of Tp63ΔN in mice resulted in more aggressive tumors that grew faster, were more motile and invasive and metastasized more frequently[72,73].
The upstream genetic and epigenetic changes required to stimulate TP63ΔN expression are slowly becoming clearer. Upregulation of TP63ΔN can result from inactivating mutation or loss of the histone demethylase KDM6A, a genomic change that has been observed in ASCP and PDAC with SF/SD[74]. Recently, it was shown that loss of HNF1A, a homeodomain transcriptional regulator that recruits KDM6A to genomic binding sites, can phenocopy KDM6A loss and produce a sarcomatoid tumor morphology in conjunction with Kras mutation in mouse pancreas[75]. Trans-differentiation to a squamous morphology appears to require concomitant loss of endodermal cell fate determinants such as GATA6, although the loss of GATA6 alone is insufficient to drive the squamous program[76]. It is important to note that KDM6A loss is not ubiquitous in squamous trans-differentiated pancreatic cancers, and therefore one might hypothesize that other chromatin regulatory genes found to be mutated in ASCP and PDAC with SF/SD (such as ARID1A, KMT2C, KMT2D, SMARCA2, ARID2, ASXL2, TET1 and MSL2[39,62]) may also lead to upregulation of TP63ΔN.
Summary of ASCP histology and molecular pathology
In conclusion, ASCP is an extreme form of the basal/squamous subtype of PDAC that contains both glandular and squamous regions. Squamous differentiation is primarily driven by epigenetic reprogramming through loss of chromatin regulatory elements and endodermal cell fate determinants, with consequent activation of TP63 and upregulation of MYC.
Epidemiology and prognosis
ASCP comprises 0.38%-10% of exocrine pancreatic cancers and appears to have a slight male predominance[49,60,77]. ASCP is typically considered more aggressive than standard PDAC. Multiple registry studies that have evaluated large cohorts of patients concur that ASCP patients have similar demographic characteristics to PDAC patients, but present with larger and more poorly differentiated tumors[78–81]. Median survival in the unselected population is similar between ASCP and PDAC patients and is ~4-6 months [Table 2]. In patients who undergo resection for local or locoregional disease, those with ASCP have a lower median overall survival (OS), but similar long-term outcomes, with less than 20% of patients surviving 5 years or more [Table 3]. Adjusting for the more aggressive clinical characteristics of ASCP at diagnosis did not change the poorer outcome in two studies[79,80], but matching ASCP patients to similar PDAC patients using a nested case-control design did find that survival differences in resected patients resolved[78].
Table 2.
Comparison of ASCP to PDAC populations
| Study | Data source | No. Of patients | Poor diff (%) | Node-positive (%) | Tumor size (cm) | Median OS (mo) | OS (%) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||||
| ASCP | PDAC | ASCP | PDAC | ASCP | PDAC | ASCP | PDAC | ASCP | PDAC | ASCP | PDAC | Time | ||
| [79] | SEER | 415 (0.9%) | 45693 | 71.4 | 45 | 52.8 | 47.1 | 5.7 | 4.3 | 4 | 5 | 10.8 | 10.9 | 2y |
| [80] | NCDB | 1745 (0.8%) | 205328 | 40.6 | 17.3 | 21.9 | 14.8 | 56%* | 33.1%* | 5.7 | 6.2 | 13 | 13.8 | 2y |
| [81] | CA Cancer Reg | 95 (0.6%) | 14746 | 4 | NR | |||||||||
Digits in italics indicate the study found a statistically significant difference in this measure.
Indicates measurement of percent tumors ≥ 4 cm. Poor diff: Poorly differentiated tumor on histology; OS: overall survival; NR: not reported.
Table 3.
Comparison of resected ASCP to PDAC
| Study | Data source | No. Of patients | Poor diff (%) | Node-positive (%) | Tumor size (cm) | Median OS (mo) | OS (%) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||||
| ASCP | PDAC | ASCP | PDAC | ASCP | PDAC | ASCP | PDAC | ASCP | PDAC | ASCP | PDAC | Time | ||
| [78] | single institution | 91 (2.3%) | 3918 | 79.5 | 35.9 | 88 | 78 | 4.0 | 3.2 | 10.8 | 20.5 | 18.2 | 17.5 | 5y |
| [79] | SEER | 176 (1.0%) | 17411 | 49.4 | 27.2 | 51.4 | 48.4 | 5.3 | 3.9 | 12 | 16 | 29 | 35.8 | 2y |
| [80] | NCDB | 503 (1.4%) | 35492 | 57 | 33 | 55 | 61 | 53%* | 30%* | 14.8 | 22 | 18.2 | 19.2 | 5y |
| [81] | CA Cancer Reg | 31 (1.5%) | 2071 | 57.7 | 60.2 | 4.6 | 3.3 | 12 | NR | |||||
Digits in italics indicate the study found a statistically significant difference in this measure.
Indicates measurement of percent tumors ≥ 4 cm. Poor diff: Poorly differentiated tumor on histology; OS: overall survival; NR: not reported.
Recently, a nomogram utilizing 9 independent clinical and demographic risk factors was found to be predictive of overall survival and cancer-specific survival in ASCP patients[82]. Protective factors for increased overall survival included resection, radiotherapy, chemotherapy, negative lymph node involvement, smaller tumor size, multiple tumors, localized tumors, and being married. Female sex was also associated with prolonged cancer-specific survival. In the metastatic/recurrent disease setting, presence of lung or peritoneal metastases, anemia, high serum C-reactive protein, and CA 19-9 ≥ 1,000 conferred poorer prognosis in patients receiving chemotherapy[83]. These factors are largely consistent with those identified as pro-survival determinants in other studies.
Diagnosis and imaging
Pathological diagnosis of ASCP is most straightforward in resected patients where the large quantity of tissue available allows for more conclusive determination of the percentage of squamous component in the tumor. Unfortunately, the majority of patients with ASCP do not present with resectable disease and diagnosis must be made from fine needle aspiration (FNA) or core needle biopsy. Diagnosis using these methods is possible[84]; however, definitive ASCP diagnosis is more difficult as patients with some squamous component noted in the sample may be diagnosed with PDAC due to the pathologist’s inability to conclusively quantitate a ≤ 30% percentage of squamous component. Note in Table 3 that ASCP constitutes at least 1% of diagnoses in resected patients, while Table 2 shows that this percentage is much lower in the full populations which include non-resected patients, although the incidence of metastatic ASCP is anticipated to be higher given its more aggressive behavior. These data suggest that the percentage of patients with ASCP in the metastatic exocrine pancreatic cancer population is likely underestimated and may be due to limitations of diagnosing ASCP from non-surgical samples.
On CT imaging, ASCP is more likely to be round or lobulated than PDAC, to have extensive central necrosis and to have more frequent accompanying portal vein thrombosis[85]. Indeed, central necrosis may be a predominant feature, with at least 75% of cases exhibiting this characteristic[86], although others have found more evidence of ring enhancement than widespread necrosis[87,88]. MRI was found to be more sensitive and specific than CT for detecting ring enhancement and necrosis, and concurrence of both findings is indicative of ASCP[88]. Ren et al. have developed a novel radiomics signature that can preoperatively differentiate between ASCP and PDAC with a positive predictive value of 92% and a negative predictive value of 98%[89]. If this signature could be accurately applied in the non-operative setting, it might increase diagnostic accuracy for ASCP when used in conjunction with pathologic review of needle biopsy specimens.
Clinical manifestations
ASCP patients have been reported to present with anorexia, weight loss, abdominal pain, nausea and vomiting, fever, and occasionally jaundice[90–96]. Jaundice may be less prominent given that primary tumors in the head of the pancreas are less frequent than seen in PDAC, with a higher incidence of body and tail tumors observed. While malignant hypercalcemia is almost never seen in PDAC, multiple cases of hypercalcemia due to elevated PTHrP (Parathyroid hormone-related protein) have been reported in ASCP patients[97–99]. This is likely driven by the squamous component, given that malignant hypercalcemia is not infrequently seen in other squamous solid tumors that arise from lung, esophagus, and head and neck[100].
The PDAC serum tumor marker Carbohydrate Antigen 19-9 (CA 19-9) is frequently expressed in ASCP and can be used as surrogate marker of tumor response similar to PDAC[45]. CEA elevation can also be seen.
Clinical management of ASCP
There are currently no treatment guidelines for ASCP. All treatment is extrapolated from standards of care for PDAC as no prospective or randomized trials examining treatment paradigms have been reported. Staging of ASCP is as per PDAC staging [Table 1].
Resectable disease
Resection is recommended for patients with early-stage disease and several population-based studies have demonstrated a significant survival advantage for patients with locoregional disease who undergo surgery versus those who do not[79–81]. R1 resection was associated with worse outcomes in one smaller single-institution study that included only one patient with this resection status[50], but no contribution of R0 status to survival was found in two larger population-based studies[78,80]. A separate retrospective single-institution study found that a positive resection margin was negatively associated with survival[101]. Unfortunately, margin positive rates exceeded 20% in one study examining ASCP cases in the National Cancer Database and another large surgical case series[49]. Although better tumor removal during surgery appears advantageous to patients, the risks of surgery remain significant. The post-operative mortality rate at 90 days reached 6.5% for ASCP patients in one population-based study. Higher mortality was associated with more advanced age and increasing co-morbidities[102]. Surgery is an important part of treatment for patients with resectable disease.
Addition of chemotherapy before or after surgery appears to be beneficial. Recent retrospective population-based studies have shown improved survival outcomes in patients who receive adjuvant chemotherapy, with a > 50% decrease in mortality as compared to surgery alone. However, patients receiving multi-modal therapy were more likely to be younger and to have fewer co-morbidities than those who did not receive post-operative chemotherapy, confounding survival assessments[80,102]. Nevertheless, chemotherapy does appear active in ASCP patients: patients with resectable disease who received chemotherapy alone had similar median OS as those who received surgery alone, suggesting it has a similar magnitude of benefit[102]. In a separate, but smaller, single institution, retrospective case series where the populations receiving or not receiving adjuvant therapy were similar, receipt of adjuvant therapy was still strongly associated with improved survival. Backbones of the administered regimens were either fluoropyrimidine or gemcitabine; however, only inclusion of a platinum chemotherapy (cisplatin or oxaliplatin) improved survival in this population[101]. Notably, delivery of chemotherapy in the neoadjuvant versus adjuvant setting did not influence overall survival[102]. This is difficult to understand, given that at least 50% of pancreatic cancer patients are unable to complete their planned adjuvant therapy[102–104]. The optimal timing and absolute benefit of chemotherapy in resectable ASCP patients are still unclear.
Radiotherapy
Radiation monotherapy is associated with poor survival. Amongst all treatment modalities, using radiotherapy alone to treat ASCP patients resulted in the shortest median survival, estimated at just 2.3 months[102]. The relative insensitivity of ASCP to radiation therapy is somewhat surprising given that true squamous cell cancers are well known to have a higher sensitivity to radiation than adenocarcinoma and radiation can be used as definitive treatment in some clinical circumstances[105–108]. Although the population offered radiation alone may have been less fit than those offered other modalities confounding an accurate assessment of the absolute benefit of radiation, it is still clear that radiation will not serve as a potentially curative treatment in ASCP. Even so, neoadjuvant chemoradiation was associated with improved survival as compared to observation following resection or adjuvant chemotherapy alone and therefore should have a role in managing this stage of disease[49].
The use of intraoperative radiation therapy has been documented, but data is limited to a few case studies and small case series. One such case utilized a multidisciplinary treatment approach which included upfront resection with intraoperative radiotherapy followed by adjuvant chemotherapy and radiotherapy. The reported patient enjoyed a 40-month survival[109]. A separate case series also documented the use of intraoperative radiation in two patients with death reported 44 and 22 months post-diagnosis[110]. Intraoperative radiation remains a rare treatment in this and other pancreatic diseases.
Use of chemotherapy in advanced disease
Palliative chemotherapy is typically given to patients with locally advanced or metastatic disease. Significant questions remain regarding what treatment regimen is best for ASCP patients. There are 2 retrospective multicenter studies and 3 case reports that have documented the response of advanced ASCP to chemotherapy. Yoshida et al. examined the outcomes of 116 patients with recurrent or metastatic ASCP who received chemotherapy at 24 Japanese institutions from 2001-2017[83]. Those receiving combination chemotherapy regimens (n = 57) had a trend of increased survival compared to those receiving monotherapy (n = 59) and a significantly higher disease control rate, and clinical characteristics were well-balanced between the cohorts. This study was large enough that response to PDAC standards GnP and FOLFIRINOX could be compared. No difference in median OS, PFS or other clinical parameters of response was found in the 28 patients receiving GnP versus the 10 treated with FOLFIRINOX, and only 5 patients receiving these treatments survived 18 months or longer. Similarly, a different retrospective multicenter study reported individual outcomes of 16 ASCP patients receiving chemotherapy for advanced disease and identified no statistical or anecdotal evidence for the superiority of a specific regimen[111]. Two case reports of patients with advanced disease describe rapid disease progression on standard PDAC regimens, but a third documents a patient achieving a partial response to 5-FU given in combination with cytokines IFNα and TNFα that allowed for subsequent surgical resection[90,91,112]. There are no reports of common regimens given for pure squamous cell cancer, such as platinum with taxane being tested in this tumor type. Current data are unclear on what constitutes an optimal chemotherapy regimen for patients with advanced ASCP.
Clinical research
Three clinical trials specific for advanced, previously treated ASCP are currently enrolling; these are the first prospective clinical trials specific for this tumor type [Table 4]. Sequencing of several ASCPs identified FGFR activation as a lesion that is frequently present in ASCP, and at least one organoid model bearing FGFR fusion was found to be sensitive to FGFR inhibition with a pharmacologic agent[62]. Based upon this, a Phase 2 study of the anti-FGFR drug pemigatinib was recently initiated (NCT05216120). ASCP is now well understood to have strong overexpression/amplification of MYC. It was recently found that triptolide, the active ingredient of a Chinese herbal remedy, exhibits anti-cancer efficacy, at least in part, through inhibition of superenhancer complexes that drive expression of oncogenes like MYC. Treatment with the triptolide pro-drug minnelide can inhibit activity of MYC and other oncogenic superenhancers[113]. Based upon this data, we have initiated a Phase 2 study of Minnelide for ASCP patients, which is currently in recruitment (NCT04896073)[114]. ASCP has been noted to have expression of PD-L1 in the squamous components and a more tumor inflamed phenotype than standard PDAC[51,57,58]. Another Phase 2 study (NCT05216120) is testing the anti-PD-1 antibody retifanlimab (INCMGA00012) in ASCP patients with hopes that this tumor type will be more responsive to immunotherapy than PDAC. Results from these studies of targeted therapies are anxiously awaited.
Table 4.
Clinical trials specific to ASCP and PACC
| NCT# | Tumor | Stage | Agent | Mechanism | Phase |
|---|---|---|---|---|---|
| NCT04116073 | ASCP | advanced | retifanlimab | anti-PD-1 | 2 |
| NCT04896073 | ASCP | advanced | minnelide | superenhancer inhibitor | 2 |
| NCT05216120 | ASCP | advanced | pemigatinib | FGFR2 inhibitor | 2 |
| NCT05286827 | PACC | previously treated | olaparib | PARP inhibitor | 2 |
Several rationally designed Phase 2 clinical trials of targeted agents are currently recruiting patients with these rare exocrine tumor types.
PANCREATIC ACINAR CELL CARCINOMA (PACC)
Pathologic characteristics
Grossly, PACC appears as a well-circumscribed, lobular, tan or pink mass emerging from pancreatic acini. Hemorrhage and necrosis are frequently present as the tumor is well-vascularized. Mucin production is not seen in PACC. Microscopically, PACC is a highly cellular tumor with no desmoplasia and limited stromal elements. Well-to-moderately differentiated cases appear as nests of pyramidal-shaped cells that cluster around a lumen, the so-called acinar pattern[115] [Figure 3]. These PACC cells have abundant eosinophilic cytoplasm that contains frequent, fine apical zymogen granules, basally located and relatively uniform nuclei, and a prominent nucleolus. As the tumor progresses, the acinar differentiation can become less pronounced, and nuclei may lose their basal orientation. Sheets of cells lacking obvious acinar structures may be present in poorly-differentiated tumors and some tumors can develop a solid or trabecular pattern with large rows of cells[116]. Sometimes the acinar pattern can be confused with neuroendocrine tumor rosettes and mixed acinar-neuroendocrine tumors also exist[117]. The absence of squamoid corpuscles differentiates PACC from pancreatoblastoma[118]. Immunohistochemistry and special stains can help to distinguish PACC from other tumor types. Positive staining for lipase, trypsin, chymotrypsin, and/or BCL10 is considered definitive for PACC diagnosis[115,119]. Trypsin and chymotrypsin together are the most sensitive markers of acinar differentiation and are positive in over 95% of cases[120]. Anti-BCL10 and carboxyl ester lipase are additional, highly sensitive markers[119,121,122]. Recently, staining for CPA1 (carboxypeptidase A1) and REG1α (lithostathine-1-alpha) were found to have excellent sensitivity and specificity for PACC, essentially excluding the diagnosis if the tumor does not stain positively[123,124]. PACC is morphologically and histologically distinct from PDAC.
Figure 3. Histologic appearance of PACC.
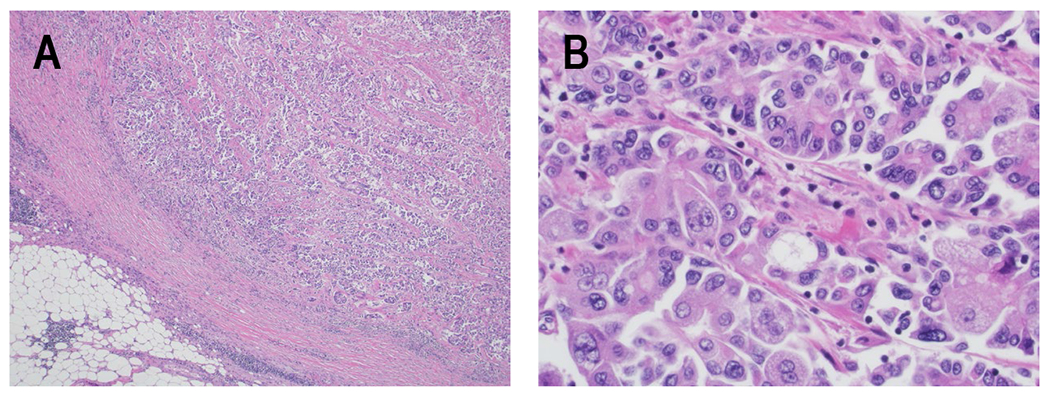
H&E specimens of PACC. A) Low power image showing how PACC recapitulates the architecture of the exocrine pancreatic parenchyma with back-to-back acinar structures containing lumina. The tumor clusters are irregular in size and shape and are approaching the peri-pancreatic adipose tissue (40x). B) Higher power image showing the classic cytologic features of PACC with basally located nuclei containing single prominent nucleoli, and abundant apical amphophilic and granular cytoplasm (400x).
Molecular profiling of PACC
Multiple studies have examined the molecular profile of PACC and identified actionable genetic changes in this tumor type[125–127]. Findings are summarized in Figure 2. From the first series examining whole exome sequencing of PACC, it was apparent that PACC had higher somatic mutation rates than PDAC and a high frequency of large chromosomal changes, mostly unaccompanied by mutations or methylation in known DNA strand break repair genes[125]. In fact, no genes were mutated at a frequency higher than 30% of cases[125,127]. Some genes which had somatic mutations or rearrangements in more than 1 case were: APC, PTEN, ARID1A, SMAD4, JAK, and BRAF. One series identified BRAF or RAF1 rearrangements in 23% of cases and determined that at least some of these are sensitive to pharmacologic MEK inhibition[126,128]. Other studies have also reported BRAF gene arrangements in PACC[129,130]; however, a subsequent study could not replicate the high incidence of this finding in a larger cohort[127]. Cases of RET gene rearrangements have also been reported and these may be susceptible to RET-directed therapy[131]. One case of NTRK fusion sensitive to larotrectinib has been described[132]. Overall, recurrent point mutations, rearrangements or fusions of a specific gene do not appear to define this disease. Interestingly, ~10% of PACC tests positive for MSI-H/dMMR (8 of 72 tested in[118,125,133]) and could potentially be treated with anti-PD-1 therapy.
Moreover, analysis of mutational signatures revealed that 14 and 15 of 22 PACC cases displayed signatures related to tobacco or associated with defective DNA repair, respectively[127]. Inactivating genetic changes in DNA repair genes that might explain the latter signature were found at high incidence in two studies[126,128], and changes in BRCA1/2 may be particularly frequent[134]. However, this finding was not replicated in two other studies[125,127]. Uniquely, Jakel et al. discovered that expression of specific tumor suppressors is frequently lost in PACC through copy number aberrations and changes in methylation[127]. These include ARID1A, APC, CDKN2A and ID3. Downregulation of ID3 at the protein level was nearly ubiquitous. Interestingly, this protein has been linked to DNA repair processes through a novel mechanism and to sensitivity to PARP inhibitors[135–137]. Actionable genetic mutations in PACC are not uncommon, such that mutational analysis of these tumors may be clinically useful in the majority of PACC patients.
It is important to note that the tumor samples used for larger -omics studies are all derived from surgical specimens, presumably representing early-stage disease exclusively. It is unclear whether similar patterns of genetic changes occur in advanced diseases. One study specifically examining TP53 mutation in PACC did include specimens from patients with metastatic disease, and matched primary tumor specimens were available for 10 cases[138]. It was noted that the rate of deleterious TP53 mutations was higher in metastatic specimens, but that the corresponding primary tumors demonstrated the same mutational profile. Moreover, in this in-depth analysis incorporating focused mutational analysis, FISH, methylation analysis and immunohistochemistry, 44% of cases contained at least one TP53 alteration that was anticipated to affect gene function. Despite this, survival only correlated with true TP53 loss (which occurred in only 5 of 43 cases) and was not impacted by TP53 mutation alone. It was concluded that TP53 loss may define a subset of more aggressive PACC tumors that have high rates of metastasis.
Summary of PACC anatomic and molecular pathology
PACC is not similar to PDAC at the anatomic or genetic level. It is highly cellular, never desmoplastic, and most often lacks KRAS mutation. The frequency of actionable gene mutation appears high, but these do not seem to cluster to a single gene.
Epidemiology and prognosis of PACC
PACC is an extremely rare tumor; it represents just 0.2%-2% of all pancreatic malignancies in adults[139]. In children, PACC accounts for up to 15% of pancreatic tumors and has some clinical and morphological overlap with pancreatoblastoma[119]. Adult patients present with PACC about a decade earlier on average than the median age for diagnosis of PDAC. Males are more commonly affected; the male to female ratio is 2:1[140–143]. The reason for the male predominance is unknown. Interestingly, clinical outcomes are superior in females with PACC[143]. Retrospective population-based studies suggest that 32%-54% of patients present with distant metastatic disease[142–145]. The most frequent site of metastasis is the liver. While this proportion is similar (in some studies) to that for PDAC, PACC patients have improved median survival compared to PDAC at every disease stage[145]. This is particularly notable in patients with PACC who undergo resection, where median OS can exceed 70 months, but rarely reaches 36 months in PDAC patients. PACC tumors are typically larger at diagnosis than those seen in PDAC. Interestingly, primary tumor size did not correlate with the time to onset of metastatic disease or with the presence of metastatic lesions[141]. Those with mixed acinar-neuroendocrine and acinar-ductal pathologies appear to have similar prognosis and tumor characteristics[146]. PACC is still an aggressive tumor, but prognosis is superior to PDAC. PACC retrospective case series examining clinical outcomes are summarized in Table 5 [142–148].
Table 5.
Clinical outcomes in PACC
| Reference | Data source | Years | n | Age at diagnosis (median) | Median tumor size (cm) | With metastasis (%) | OS (median) | Stage 4 OS (median) | Early stage OS (median) |
|---|---|---|---|---|---|---|---|---|---|
| [142] | NCDB | 1985-2005 | 865 | 67 | ]5.9 | 32.1 | N/A | 17.2%* | NR-22.6m |
| [145] | SEER | 1988-2003 | 672 | 56 | 53.1 | 47 | 22%* | 72%*^ | |
| [144] | SEER | 2004-2016 | 252 | 63.8 | N/A | 54.4 | 10 | 7 | 18,29^ |
| [143] | German Cancer Registry Group | 2000-2019 | 233 | 66 | N/A | 33.9 | 22 | 6 | 34^ |
| [141] | SI-Harvard Hospitals | 1996-2019 | 66 | 64 | 4.3 | 42 | 13 | 15 | 38 |
| [147] | Korean Tumor Registry System | 2003-2018 | 59 | 59.2 | 4.6 | 0 | N/A | N/A | 78.8^ |
| [148] | SI-West China Hospital | 2006-2016 | 52 | 50.8 | 5.0 | 30.8 | 39 | N/A | 48^ |
| Seo 2017 | SI-Asan Medical Center | 1997-2015 | 20 | 57 | 4.0 | 0 | N/A | N/A | 75^ |
| Matos 2009 | MI | 1998-2008 | 17 | 59 | 5.3 | 23.5 | 19 | N/A | 61^ |
Multiple retrospective case series examining survival in PACC patients have been conducted.
indicates that 5-year overall survival percentage is documented;
indicates value is for resected patients only;
SI: Single institution; MI: multi-institution; n: number of patient cases; OS: overall survival; N/A: not available; NR: not reached.
PACC has been seen in kindreds with familial adenomatous polyposis[149], Lynch syndrome[150], and BRCA mutation[134,151], but it is difficult to determine how influential these genetic syndromes are in this disease given the small number of cases overall. Specific clinical or environmental risk factors for PACC remain unknown.
PACC diagnosis and imaging
Diagnosis of PACC is by pathology. Tumor samples can be obtained through surgical resection or needle aspiration. Diagnosing PACC from a fine needle aspiration can present a challenge as cytologic samples can be easily confused with normal acinar cells or sampling from a neuroendocrine tumor. Labate et al. were the first to describe the cytopathology of PACC in detail and were able to correctly diagnose PACC from a cytology specimen in just 2 of 7 cases[152]. Subsequently, the diagnostic yield has not appeared to improve, as only 2 of 7 cases reported since could be definitively diagnosed as PACC based on cytology alone[153–156].
CT is the standard and first line choice for imaging studies of PACC. Visible lymph nodes were noted to be more frequent in PACC than PDAC. Tumor hypoattenuation in arterial phase compared to the uninvolved pancreas was much less frequent in PACC and PACC was much more likely to have well-defined margins than PDAC. Lack of bile duct dilatation was also much more common in PACC than PDAC[157]. MRI characteristics of PACC were investigated in a small study of 5 patients, and it was concluded that PACC presents as a round, encapsulated tumor with moderate and heterogeneous enhancement after gadolinium administration[158]. Either CT or MRI can adequately detect PACC.
Clinical manifestations of PACC
The most common symptoms in newly diagnosed PACC patients are generally non-specific: abdominal pain and weight loss occur most frequently[141]. Jaundice is an infrequent symptom even when PACC presents in the pancreatic head. Jaundice is very common in PDAC and PACC tumors are typically larger at diagnosis[141,148,159], so the rarity of jaundice in PACC has been difficult to understand. It has been suggested that the more encapsulated biology of PACC is less susceptible to causing jaundice than the more infiltrative biology of PDAC.
Lipase hypersecretion syndrome (LHS) is a paraneoplastic syndrome uniquely observed with PACC. Many PACCs produce lipase and excessive release of this enzyme into the circulation occurs in about 10%-15% of PACC patients, leading to accumulation of pancreatic enzymes in peripheral tissues and subsequent enzymatic digestion. Very elevated blood lipase levels can cause eosinophilia and panniculitis that most often manifests as painful subcutaneous nodules that resemble erythema nodosum. Panniculitis of LHS has a predilection for pressure points in the lower extremities, especially on the shins[160,161]. Another common feature of LHS is painful and inflammatory polyarthritis that resembles rheumatic fever or gout. It is thought to be secondary to periarticular fat necrosis. Joint aspirates are typically sterile but occasionally crystals are visualized[162,163]. Patients experiencing LHS usually have advanced metastatic disease. LHS in PACC has been recently well-reviewed by Taskin et al.[164].
Levels of the PDAC serum tumor marker CA 19-9 are elevated in less than 30% of patients with PACC and may be related to non-specific biliary duct irritation rather than tumor production in most cases[141]. Nevertheless, poor survival outcomes were observed in patients with tumors that do express CA 19-9[146,147]. Larger cohorts would be required to assess the prognostic value of this marker in PACC. Elevation of CEA has been observed in about one-fifth of PACC patients[141], and elevated blood Alpha-fetoprotein (AFP) levels in younger patients and should raise suspicion of PACC, especially when associated with a pancreatic mass[165]. Serum lipase might be used as a surrogate marker of tumor response in some patients, as PACC frequently produces high levels of lipase, but this has not been well characterized in the literature.
Clinical management of PACC
There are currently no treatment guidelines for PACC. All treatment is extrapolated from standards of care for PDAC as no prospective or randomized trials examining treatment paradigms have been reported. Staging of PACC is as per PDAC staging [Table 1].
Resectable disease
Surgical resection is the treatment of choice for early-stage disease. Resection has been shown to result in a dramatic improvement in cohort overall survival as compared to no resection in multiple retrospective case series and population-based studies[141–144,166]. In fact, resection was the most predictive factor for overall survival in some studies. In one study, 5-year survival for Stage I or II patients receiving resection was 42% compared to 9% for those who did not get surgery[167]. Negative margins are predictive of better long-term survival in some studies.
Recurrence following surgery is estimated to be > 50%, especially in patients with Stage II or III disease; however, it remains unclear whether addition of adjuvant therapy is of benefit to PACC patients. Patients receiving adjuvant therapy had better OS than those that did not in one large retrospective population-based study[142]. Others found that the benefit was only associated with administration of platinum-based chemotherapy[141] or presence of node-positive disease[166,168]. A fourth study found no survival benefit for patients who received adjuvant chemotherapy compared to those who did not[143]. Choice of chemotherapy regimen may prove a confounder in the latter study; single-agent gemcitabine was the chemotherapy regimen administered to 60% of patients in that study, and as discussed below, the benefit of gemcitabine in PACC appears to be limited. Another smaller study of 9 patients found that patients receiving 5-FU-based adjuvant chemotherapy had superior overall survival to those receiving a gemcitabine-based regimen[169]. A randomized study of adjuvant therapy in PACC could resolve this question but is unlikely to be feasible given the rarity of the tumor.
Neoadjuvant therapy has not been thoroughly explored in this disease. One case study reported on a 65-year-old PACC patient who had a complete pathologic response to neoadjuvant modified FOLFIRINOX and remained in remission at 33 months post-resection without adjuvant treatment. It is anticipated that more resectable PACC patients may receive neoadjuvant therapy, given the trend towards this paradigm in PDAC.
Recurrent disease following resection
Re-resection at the time of recurrence has been performed in selected cases of PACC with some success[170,171]. One case report identified a patient with recurrent disease who appeared to be cured after treatment with radiofrequency ablation (RFA) and combination chemotherapy and a second who experienced a long remission after RFA[172]. These case reports suggest that local therapy may still be curative in some cases of post-surgical recurrence.
Advanced disease
In metastatic or locally advanced PACC multiple therapeutic modalities have been used. Unlike in PDAC, metastasectomy is sometimes performed in conjunction with resection of the primary tumor. One single institution case series of 64 PACC patients found that patients with distant metastases who received surgery had no difference in overall survival compared to those who did not[141], but it is unclear how many received surgery with curative intent versus a palliative procedure. Multiple case reports describe metastasectomy being beneficial in individual patients. Sumiyoshi et al. reported ongoing recurrence-free survival at 73 months for one patient following resection of the primary tumor in combination with removal of several synchronous peritoneal nodules followed by continuous S-1 chemotherapy[173]. Another case study described a patient who had multiple metastasectomies between sequential lines of combination chemotherapy that has survived over 10 years since initial diagnosis[174]. A third case report outlined the course of a metastatic patient who received neoadjuvant capecitabine/oxaliplatin followed by resection of the primary tumor with hepatectomy resulting in over 30 months of disease-free survival before recurrence. The patient subsequently restarted chemotherapy and remained in complete response for the last 3 years[175]. These inspiring cases suggest that outcomes of metastasectomy in PACC may more closely resemble those of colon cancer patients than PDAC patients and that surgical intervention could be of benefit to patients with oligometastatic disease in selected cases. Further study of this treatment paradigm is needed.
For patients with locally advanced disease, upfront FOLFIRINOX chemotherapy can be successful enough at debulking to allow resection[176]. Descriptions of successful debulking with other regimens have not been described.
Systemic chemotherapy is the mainstay of treatment for patients with advanced PACC. Several groups of investigators have analyzed retrospective case series to identify the most beneficial regimens for advanced PACC patients[169,177–179]. Overall, the objective response rate to FOLFIRINOX appears to be higher than for any other regimen, with 16 of 21 patients receiving this regimen in the first-line setting achieving objective responses[169,178,180,181]. All studies also agree that gemcitabine monotherapy has poor efficacy in PACC, with no responses reported in 10 patients described in 2 studies. Similarly, gemcitabine-based regimens overall are inferior to fluoropyrimidine-based regimens, with significantly lower progression-free survival observed in multiple studies[169,178]. Intriguingly, objective responses have been noted to single-agent infusional 5-fluorouracil, S-1 and capecitabine; however, addition of irinotecan or platinum agents clearly increases efficacy. In a study of 58 advanced disease patients, Takahashi et al. reported a 40% response rate to platinum-containing regimens and a 29% response rate to irinotecan-containing regimens in the first-line advanced disease setting. In addition, patients who received these regimens had improved overall survival[177]. One important caveat of these findings is that almost all of these case series are from Asia, and it is unclear whether similar benefits to irinotecan and fluoropyrimidine containing regimens will be reproducible in a majority Western population who metabolize these drugs differently. However, smaller case series in Western countries suggest the relationship holds true regardless of race/ethnicity.
Targeted therapies have been demonstrated to produce clinical benefits in PACC patients with sensitizing mutations. Responses to platinum-containing regimens appear particularly strong in PACC patients with BRCA mutation, and a beneficial treatment effect of olaparib in this setting has also been reported[151,178]. A complete response to the RAF/MEK inhibitor combination of dabrafenib/trametinib has been reported in 2 patients with tumors bearing BRAF V600E mutation[182,183]. Treatment with alectinib of a metastatic PACC patient with a somatic KANK4/ALK fusion resulted in a partial response ongoing at 1 year[184]. The use of immunotherapy in a PACC patient with high tumor mutational burden and tumor PD-L1 expression resulted in a partial response that permitted subsequent re-resection with continued absence of disease[185]. It is very clear that precision medicine is of significant benefit to PACC patients with actionable tumor genetics and providers should advocate for sequencing of available tumor material to expand treatment options.
Immunotherapy has not been extensively evaluated in PACC. MSI-H/dMMR disease is not uncommon in PACC and would be expected to respond to anti-PD-1 therapies. It is unclear whether patients lacking microsatellite instability would respond to single-agent immunotherapy treatment. However, combination of lenvatinib with an anti-PD-1 therapy was reported effective in one case study, resulting in a partial response ongoing for 21 months[186]. Combination of anti-PD-(L)1 therapies with anti-angiogenic agents like lenvatinib has produced exciting results in several tumor types[187,188]. PACC is a highly vascular tumor, like renal and hepatocellular carcinomas which have been responsive to this combination. Evaluation of this combination in PACC would be an intriguing prospect.
Research on PACC
We have recently opened a Phase 2 study evaluating olaparib in patients with previously treated PACC (NCT05286827). To our knowledge, this is the first-ever PACC-specific prospective treatment study [Table 4]. This study is based upon the observation that over 90% of PACC patients have down-regulation of ID3, and that PARP inhibitor is active in ID3-deficient tumors[127,136]. Dedicated clinical studies of PACC are extremely difficult to accrue, given the extreme rarity of the disease.
SUMMARY AND CONCLUSIONS
Consensus guidelines for rare exocrine pancreatic tumors like ASCP and PACC have not been published. Consequently, these tumors are most frequently treated using standards of care established for PDAC. While ASCP may represent an extreme variant of the basal transcriptomic subtype of PDAC, PACC shares few morphologic, histologic or molecular characteristics with PDAC and application of PDAC treatment paradigms to PACC is inappropriate.
Based on our review of the existing literature, it is clear that ASCP is a more aggressive tumor than PDAC and responses to current PDAC treatments are inadequate. Solving the puzzle of how best to control ASCP is likely to benefit the larger PDAC population, given the overlap of ASCP and the most aggressive subtypes of PDAC that harbor SF or SD. Research into treatment paradigms for basal-type PDAC, which is largely synonymous with PDAC with SF/SD, has expanded significantly over the last several years, but has resulted in limited clinical advances thus far. Recent studies have suggested that basal-type PDAC is less sensitive to platinum chemotherapy than classical-type[189,190]. Intriguingly, the available ASCP data hint at platinum chemotherapy providing benefits to this patient population. It is worth considering whether the more extensive squamous differentiated component of ASCP may have sensitivities to unique chemotherapy regimens that are less effective in pure adenocarcinoma. Perhaps more importantly for this population, new drugs targeting activated KRAS appear to be imminent and could turn the tide in this KRAS-driven disease.
PACC is a tumor that shares few characteristics with PDAC other than arising in the pancreas and preferentially metastasizing to the liver. It is clear that surgical resection of this less aggressive tumor provides significant benefit to early-stage patients, and investigation into the role of metastasectomy in treating PACC is also warranted. A benefit of adjuvant chemotherapy to cure early-stage patients is likely to present for PACC patients with lymph node involvement, but unclear in patients with more limited disease. Systemic therapy with platinum agents produces the best outcomes in PACC patients. Regimens with a fluoropyrimidine backbone are preferred in treating PACC, as the use of gemcitabine in this disease has resulted in inferior outcomes. All patients with PACC should undergo molecular testing for somatic mutations as these occur at high incidence and are frequently actionable, and treatment with matched therapy has been highly beneficial in case reports. In addition, the high rate of MSI-H/dMMR occurrence in PACC should prompt universal evaluation of these markers which could demonstrate sensitivity to immunotherapy. Given the extreme rarity of this disease, a coordinated research effort across many institutions will likely be required to generate sufficient cases to inform evidence-based clinical care for PACC.
Dedicated clinical trials for ASCP and PACC are uncommon. These studies are difficult to accrue given the rarity of the diseases. It is also unclear how many ASCP and PACC patients understand that their pancreatic cancer is different from “standard” pancreatic cancer and would be interested in seeking more tailored treatment. We have found that at least one-third of pancreas cancer trials testing medical interventions permit the accrual of patients with any exocrine pancreatic tumor rather than limiting eligibility to PDAC [Figure 4]. While this offers benefits to patients by providing additional treatment options, these treatments may not be well justified in ASCP or PACC and are unlikely to advance our understanding of these rare diseases when it is likely that, at most, a single rare tumor patient will be enrolled, and their availability could further reduce the patient pool willing to seek out ASCP- or PACC-specific studies.
Figure 4. Inclusion of rare exocrine tumor patients on clinical studies of pancreatic cancer.
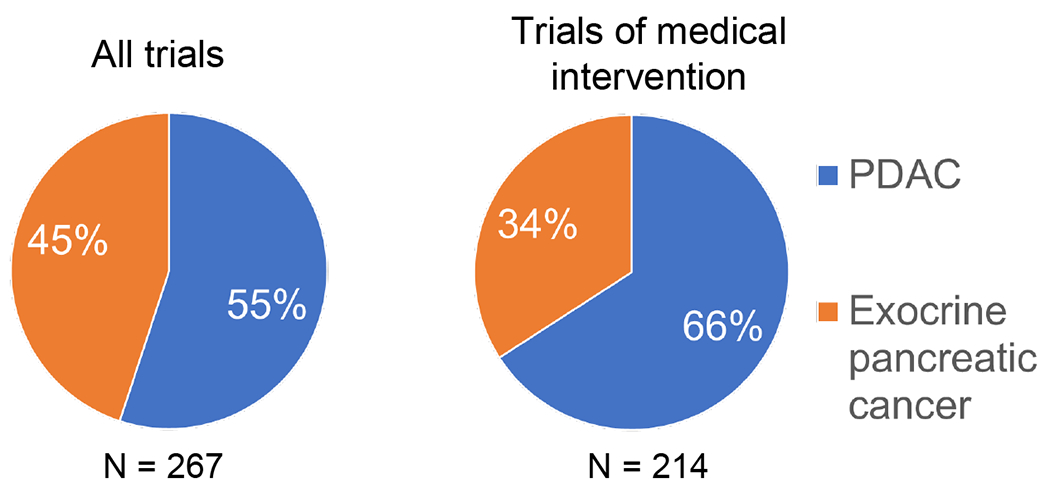
Study registrations on clinicaltrials.gov over the last two years (from 8/2020 through 8/2022) were searched for those that were interventional, enrolled adults, and included the term pancreas cancer, resulting in identification of 425 records. Eliminating those specific for PNET, carcinoid syndrome or endocrine tumors resulted in 405 entries. 267 of these were specific for pancreas cancer and did not include other tumor types. After exclusion of those evaluating screening regimens, diagnostic procedures (imaging, molecular profiling), interventional procedures (endoscopy, surgery, anesthesia), and psychological, dietary, or lifestyle modification, 214 studies testing medical interventions were identified. The percentage of clinical studies specific for pancreas cancer that allow only PDAC patients versus any exocrine pancreatic cancer patient are depicted.
In summary, rare exocrine tumors of the pancreas present a clinical challenge since treatment guidelines are lacking. However, existing data, as described above, could form the nucleus of what constitutes best practice for these diseases.
Acknowledgments
The authors would like to thank Cynthia Hurlbert for assistance with document formatting.
Financial support and sponsorship
This work was supported by the NCI Center for Cancer Research (ZIA BC 012041 and ZIE BC 011653).
Footnotes
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
REFERENCES
- 1.Conroy T, Desseigne F, Ychou M, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 2011;364:1817–25. [DOI] [PubMed] [Google Scholar]
- 2.Conroy T, Hammel P, Hebbar M, et al. FOLFIRINOX or gemcitabine as adjuvant therapy for pancreatic cancer. N Engl J Med 2018;379:2395–406. [DOI] [PubMed] [Google Scholar]
- 3.Von Hoff DD, Ramanathan RK, Borad MJ, et al. Gemcitabine plus nab-paclitaxel is an active regimen in patients with advanced pancreatic cancer: a phase I/II trial. J Clin Oncol 2011;29:4548–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neoptolemos JP, Palmer DH, Ghaneh P, et al. Comparison of adjuvant gemcitabine and capecitabine with gemcitabine monotherapy in patients with resected pancreatic cancer (ESPAC-4): a multicentre, open-label, randomised, phase 3 trial. Lancet 2017;389:1011–24. [DOI] [PubMed] [Google Scholar]
- 5.Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin 2022;72:7–33. [DOI] [PubMed] [Google Scholar]
- 6.Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res 2014;74:2913–21. [DOI] [PubMed] [Google Scholar]
- 7.Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2021;71:209–49. [DOI] [PubMed] [Google Scholar]
- 8.Gordon-Dseagu VL, Devesa SS, Goggins M, Stolzenberg-Solomon R. Pancreatic cancer incidence trends: evidence from the surveillance, epidemiology and end results (SEER) population-based data. Int J Epidemiol 2018;47:427–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Falconi M, Eriksson B, Kaltsas G, et al. ENETS consensus guidelines update for the management of patients with functional pancreatic neuroendocrine tumors and non-functional pancreatic neuroendocrine tumors. Neuroendocrinology 2016;103:153–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holly EA, Chaliha I, Bracci PM, Gautam M. Signs and symptoms of pancreatic cancer: a population-based case-control study in the San Francisco Bay area. Clin Gastroenterol Hepatol 2004;2:510–7. [DOI] [PubMed] [Google Scholar]
- 11.Khorana AA, Fine RL. Pancreatic cancer and thromboembolic disease. Lancet Oncol 2004;5:655–63. [DOI] [PubMed] [Google Scholar]
- 12.Artinyan A, Soriano PA, Prendergast C, Low T, Ellenhorn JD, Kim J. The anatomic location of pancreatic cancer is a prognostic factor for survival. HPB 2008;10:371–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oettle H, Post S, Neuhaus P, et al. Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative-intent resection of pancreatic cancer: a randomized controlled trial. JAMA 2007;297:267–77. [DOI] [PubMed] [Google Scholar]
- 14.Versteijne E, van Dam JL, Suker M, et al. Neoadjuvant chemoradiotherapy versus upfront surgery for resectable and borderline resectable pancreatic cancer: long-term results of the dutch randomized preopanc trial. J Clin Oncol 2022;40:1220–30. [DOI] [PubMed] [Google Scholar]
- 15.Versteijne E, Suker M, Groothuis K, et al. Preoperative chemoradiotherapy versus immediate surgery for resectable and borderline resectable pancreatic cancer: results of the dutch randomized phase III preopanc trial. J Clin Oncol 2020;38:1763–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 2013;369:1691–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burris HA 3rd, Moore MJ, Andersen J, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol 1997;15:2403–13. [DOI] [PubMed] [Google Scholar]
- 18.Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012;366:2455–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O’Reilly EM, Oh DY, Dhani N, et al. Durvalumab with or without tremelimumab for patients with metastatic pancreatic ductal adenocarcinoma: a phase 2 randomized clinical trial. JAMA Oncol 2019;5:1431–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu ZI, Shia J, Stadler ZK, et al. Evaluating mismatch repair deficiency in pancreatic adenocarcinoma: challenges and recommendations. Clin Cancer Res 2018;24:1326–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Humphris JL, Patch AM, Nones K, et al. Hypermutation in pancreatic cancer. Gastroenterology 2017; 152:68–74.e2. [DOI] [PubMed] [Google Scholar]
- 22.Bian J, Almhanna K. Pancreatic cancer and immune checkpoint inhibitors-still a long way to go. Transl Gastroenterol Hepatol 2021;6:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012;21:418–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Truong LH, Pauklin S. Pancreatic cancer microenvironment and cellular composition: current understandings and therapeutic approaches. Cancers 2021; 13:5028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jaffee EM, Hruban RH, Canto M, Kern SE. Focus on pancreas cancer. Cancer Cell 2002;2:25–8. [DOI] [PubMed] [Google Scholar]
- 26.Jones S, Zhang X, Parsons DW, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008;321:1801–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luo J. KRAS mutation in pancreatic cancer. Semin Oncol 2021;48:10–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Holter S, Borgida A, Dodd A, et al. Germline BRCA mutations in a large clinic-based cohort of patients with pancreatic adenocarcinoma. J Clin Oncol 2015;33:3124–9. [DOI] [PubMed] [Google Scholar]
- 29.Shindo K, Yu J, Suenaga M, et al. Deleterious germline mutations in patients with apparently sporadic pancreatic adenocarcinoma. J Clin Oncol 2017;35:3382–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O’Reilly EM, Lee JW, Zalupski M, et al. Randomized, multicenter, phase II trial of gemcitabine and cisplatin with or without veliparib in patients with pancreas adenocarcinoma and a germline BRCA/PALB2 mutation. J Clin Oncol 2020;38:1378–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fogelman D, Sugar EA, Oliver G, et al. Family history as a marker of platinum sensitivity in pancreatic adenocarcinoma. Cancer Chemother Pharmacol 2015;76:489–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reiss KA, Yu S, Judy R, Symecko H, Nathanson KL, Domchek SM. Retrospective survival analysis of patients with advanced pancreatic ductal adenocarcinoma and germline BRCA or PALB2 mutations. JCO Precis Oncol 2018;2:1–9. [DOI] [PubMed] [Google Scholar]
- 33.Yu S, Agarwal P, Mamtani R, et al. Retrospective survival analysis of patients with resected pancreatic ductal adenocarcinoma and a germline BRCA or PALB2 mutation. JCO Precis Oncol 2019;3:1–11. [DOI] [PubMed] [Google Scholar]
- 34.Golan T, Hammel P, Reni M, et al. Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. N Engl J Med 2019;381:317–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bailey P, Chang DK, Nones K, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016;531:47–52. [DOI] [PubMed] [Google Scholar]
- 36.Collisson EA, Sadanandam A, Olson P, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med 2011;17:500–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moffitt RA, Marayati R, Flate EL, et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat Genet 2015;47:1168–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Topham JT, Karasinska JM, Lee MKC, et al. Subtype-discordant pancreatic ductal adenocarcinoma tumors show intermediate clinical and molecular characteristics. Clin Cancer Res 2021;27:150–7. [DOI] [PubMed] [Google Scholar]
- 39.Hayashi A, Fan J, Chen R, et al. A unifying paradigm for transcriptional heterogeneity and squamous features in pancreatic ductal adenocarcinoma. Nat Cancer 2020;1:59–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chan-Seng-Yue M, Kim JC, Wilson GW, et al. Transcription phenotypes of pancreatic cancer are driven by genomic events during tumor evolution. Nat Genet 2020;52:231–40. [DOI] [PubMed] [Google Scholar]
- 41.Raghavan S, Winter PS, Navia AW, et al. Microenvironment drives cell state, plasticity, and drug response in pancreatic cancer. Cell 2021;184:6119–6137.e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Porter RL, Magnus NK, Thapar V, Morris R, Szabolcs A, Neyaz A, Kulkarni AS, Tai E, Chougule A, Hillis A. Epithelial to mesenchymal plasticity and differential response to therapies in pancreatic ductal adenocarcinoma. Proc Natl Acad Sci USA 2019;116:26835–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Madura JA, Jarman BT, Doherty MG, Yum MN, Howard TJ. Adenosquamous carcinoma of the pancreas. Arch Surg 1999; 134:599–603. [DOI] [PubMed] [Google Scholar]
- 44.Board WCoTE. Digestive system tumors. Lyon, France; 2019. [Google Scholar]
- 45.Kardon DE, Thompson LD, Przygodzki RM, Heffess CS. Adenosquamous carcinoma of the pancreas: a clinicopathologic series of 25 cases. Mod Pathol 2001;14:443–51. [DOI] [PubMed] [Google Scholar]
- 46.Murakami Y, Yokoyama T, Yokoyama Y, et al. Adenosquamous carcinoma of the pancreas: preoperative diagnosis and molecular alterations. J Gastroenterol 2003;38:1171–5. [DOI] [PubMed] [Google Scholar]
- 47.Collisson EA, Bailey P, Chang DK, Biankin AV. Molecular subtypes of pancreatic cancer. Nat Rev Gastroenterol Hepatol 2019;16:207–20. [DOI] [PubMed] [Google Scholar]
- 48.Olson MT, Siddiqui MT, Ali SZ. The differential diagnosis of squamous cells in pancreatic aspirates: from contamination to adenosquamous carcinoma. Acta Cytol 2013;57:139–46. [DOI] [PubMed] [Google Scholar]
- 49.Voong KR, Davison J, Pawlik TM, et al. Resected pancreatic adenosquamous carcinoma: clinicopathologic review and evaluation of adjuvant chemotherapy and radiation in 38 patients. Hum Pathol 2010;41:113–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ito T, Sugiura T, Okamura Y, et al. Long-term outcomes after an aggressive resection of adenosquamous carcinoma of the pancreas. Surg Today 2019;49:809–19. [DOI] [PubMed] [Google Scholar]
- 51.Lee SM, Sung CO. PD-L1 expression and surgical outcomes of adenosquamous carcinoma of the pancreas in a single-centre study of 56 lesions. Pancreatology 2021;21:920–7. [DOI] [PubMed] [Google Scholar]
- 52.Viswanathan K, Rao R. Pancreatic ductal adenocarcinoma and its variants. In: Pancreas and biliary tract cytohistology. Cham: Springer International Publishing; 2019. pp. 95–145. [Google Scholar]
- 53.Brody JR, Costantino CL, Potoczek M, et al. Adenosquamous carcinoma of the pancreas harbors KRAS2, DPC4 and TP53 molecular alterations similar to pancreatic ductal adenocarcinoma. Mod Pathol 2009;22:651–9. [DOI] [PubMed] [Google Scholar]
- 54.Taniwaki S, Hisaka T, Sakai H, et al. Sarcomatous component in pancreatic adenosquamous carcinoma: a clinicopathological series of 7 cases. Anticancer Res 2019;39:4575–80. [DOI] [PubMed] [Google Scholar]
- 55.Boecker W, Tiemann K, Boecker J, et al. Cellular organization and histogenesis of adenosquamous carcinoma of the pancreas: evidence supporting the squamous metaplasia concept. Histochem Cell Biol 2020;154:97–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lu BC, Wang C, Yu JH, Shen ZH, Yang JH. A huge adenosquamous carcinoma of the pancreas with sarcomatoid change: an unusual case report. World J Gastroenterol 2014;20:16381–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Silvestris N, Brunetti O, Pinto R, et al. Immunological mutational signature in adenosquamous cancer of pancreas: an exploratory study of potentially therapeutic targets. Expert Opin Ther Targets 2018;22:453–61. [DOI] [PubMed] [Google Scholar]
- 58.Tanigawa M, Naito Y, Akiba J, et al. PD-L1 expression in pancreatic adenosquamous carcinoma: PD-L1 expression is limited to the squamous component. Pathol Res Pract 2018;214:2069–74. [DOI] [PubMed] [Google Scholar]
- 59.Motojima K, Tomioka T, Kohara N, Tsunoda T, Kanematsu T. Immunohistochemical characteristics of adenosquamous carcinoma of the pancreas. J Surg Oncol 1992;49:58–62. [DOI] [PubMed] [Google Scholar]
- 60.Borazanci E, Millis SZ, Korn R, et al. Adenosquamous carcinoma of the pancreas: molecular characterization of 23 patients along with a literature review. World J Gastrointest Oncol 2015;7:132–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fang Y, Su Z, Xie J, et al. Genomic signatures of pancreatic adenosquamous carcinoma (PASC). J Pathol 2017;243:155–9. [DOI] [PubMed] [Google Scholar]
- 62.Lenkiewicz E, Malasi S, Hogenson TL, et al. Genomic and epigenomic landscaping defines new therapeutic targets for adenosquamous carcinoma of the pancreas. Cancer Res 2020;80:4324–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Matsuzaka S, Karasaki H, Ono Y, et al. Tracking the clonal evolution of adenosquamous carcinoma, a rare variant of intraductal papillary mucinous neoplasm of the pancreas. Pancreas 2016;45:915–8. [DOI] [PubMed] [Google Scholar]
- 64.Liu C, Karam R, Zhou Y, et al. The UPF1 RNA surveillance gene is commonly mutated in pancreatic adenosquamous carcinoma. Nat Med 2014;20:596–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Witkiewicz AK, McMillan EA, Balaji U, et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun 2015;6:6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Polaski JT, Udy DB, Escobar-Hoyos LF, et al. The origins and consequences of UPF1 variants in pancreatic adenosquamous carcinoma. Elife 2021;2021:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dey P, Li J, Zhang J, et al. Oncogenic KRAS-driven metabolic reprogramming in pancreatic cancer cells utilizes cytokines from the tumor microenvironment. Cancer Discov 2020;10:608–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Maddipati R, Norgard RJ, Baslan T, et al. MYC levels regulate metastatic heterogeneity in pancreatic adenocarcinoma. Cancer Discov 2022;12:542–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Muthalagu N, Monteverde T, Raffo-Iraolagoitia X, et al. Repression of the type I interferon pathway underlies MYC- and KRAS-dependent evasion of NK and B cells in pancreatic ductal adenocarcinoma. Cancer Discov 2020;10:872–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shukla SK, Gunda V, Abrego J, et al. MUC16-mediated activation of mTOR and c-Myc reprograms pancreatic cancer metabolism. Oncotarget 2015;6:19118–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Karasinska JM, Topham JT, Kalloger SE, et al. Altered gene expression along the glycolysis-cholesterol synthesis axis is associated with outcome in pancreatic cancer. Clin Cancer Res 2020;26:135–46. [DOI] [PubMed] [Google Scholar]
- 72.Somerville TDD, Xu Y, Miyabayashi K, et al. TP63-mediated enhancer reprogramming drives the squamous subtype of pancreatic ductal adenocarcinoma. Cell Rep 2018;25:1741–1755.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hamdan FH, Johnsen SA. DeltaNp63-dependent super enhancers define molecular identity in pancreatic cancer by an interconnected transcription factor network. Proc Natl Acad Sci USA 2018;115:E12343–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Andricovich J, Perkail S, Kai Y, Casasanta N, Peng W, Tzatsos A. Loss of KDM6A activates super-enhancers to induce gender-specific squamous-like pancreatic cancer and confers sensitivity to BET inhibitors. Cancer Cell 2018;33:512–526.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kalisz M, Bernardo E, Beucher A, et al. HNF1A recruits KDM6A to activate differentiated acinar cell programs that suppress pancreatic cancer. EMBO J 2020;39:e102808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kloesch B, Ionasz V, Paliwal S, et al. A GATA6-centred gene regulatory network involving HNFs and ΔNp63 controls plasticity and immune escape in pancreatic cancer. Gut 2022;71:766–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Imaoka H, Shimizu Y, Mizuno N, et al. Clinical characteristics of adenosquamous carcinoma of the pancreas: a matched case-control study. Pancreas 2014;43:287–90. [DOI] [PubMed] [Google Scholar]
- 78.Kaiser J, Hinz U, Mayer P, et al. Clinical presentation and prognosis of adenosquamous carcinoma of the pancreas - Matched-pair analysis with pancreatic ductal adenocarcinoma. Eur J Surg Oncol 2021;47:1734–41. [DOI] [PubMed] [Google Scholar]
- 79.Boyd CA, Benarroch-Gampel J, Sheffield KM, Cooksley CD, Riall TS. 415 patients with adenosquamous carcinoma of the pancreas: a population-based analysis of prognosis and survival. J Surg Res 2012; 174:12–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hester CA, Augustine MM, Choti MA, et al. Comparative outcomes of adenosquamous carcinoma of the pancreas: an analysis of the National Cancer Database. J Surg Oncol 2018; 118:21–30. [DOI] [PubMed] [Google Scholar]
- 81.Katz MH, Taylor TH, Al-Refaie WB, et al. Adenosquamous versus adenocarcinoma of the pancreas: a population-based outcomes analysis. J Gastrointest Surg 2011;15:165–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yang Z, Shi G, Zhang P. Development and validation of nomograms to predict overall survival and cancer-specific survival in patients with pancreatic adenosquamous carcinoma. Front Oncol 2022; 12:831649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yoshida Y, Kobayashi S, Ueno M, et al. Efficacy of chemotherapy for patients with metastatic or recurrent pancreatic adenosquamous carcinoma: a multicenter retrospective analysis. Pancreatology 2022;22:1159–66. [DOI] [PubMed] [Google Scholar]
- 84.Lozano MD, Panizo A, Sola IJ, Pardo-Mindán FJ. FNAC guided by computed tomography in the diagnosis of primary pancreatic adenosquamous carcinoma: a report of three cases. Acta Cytol 1998;42:1451–4. [DOI] [PubMed] [Google Scholar]
- 85.Toshima F, Inoue D, Yoshida K, et al. Adenosquamous carcinoma of pancreas: CT and MR imaging features in eight patients, with pathologic correlations and comparison with adenocarcinoma of pancreas. Ahdom Radiol 2016;41:508–20. [DOI] [PubMed] [Google Scholar]
- 86.Feng YF, Chen JY, Chen HY, et al. 110 patients with adenosquamous carcinomas of the pancreas (PASC): imaging differentiation of small (≤ 3 cm) versus large (> 3 cm) tumors. Ahdom Radiol 2019;44:2466–73. [DOI] [PubMed] [Google Scholar]
- 87.Zhao R, Jia Z, Chen X, et al. CT and MR imaging features of pancreatic adenosquamous carcinoma and their correlation with prognosis. Abdom Radiol 2019;44:2822–34. [DOI] [PubMed] [Google Scholar]
- 88.Schawkat K, Tsai LL, Jaramillo-Cardoso A, et al. Use of ring-enhancement and focal necrosis to differentiate pancreatic adenosquamous carcinoma from pancreatic ductal adenocarcinoma on CT and MRI. Clin Imaging 2021;73:134–8. [DOI] [PubMed] [Google Scholar]
- 89.Ren S, Zhao R, Cui W, et al. Computed tomography-based radiomics signature for the preoperative differentiation of pancreatic adenosquamous carcinoma from pancreatic ductal adenocarcinoma. Front Oncol 2020; 10:1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Arco H, Chakiba-Brugère C, Salabert L, Béchade D. Adenosquamous carcinoma of the pancreas. Clin Med Insights Oncol 2019; 13:1179554919886587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Inomata N, Masuda A, Itani T, et al. An autopsy case of granulocyte colony-stimulating factor-producing pancreatic adenosquamous carcinoma. Clin J Gastroenterol 2020;13:448–54. [DOI] [PubMed] [Google Scholar]
- 92.Skafida E, Grammatoglou X, Glava C, et al. Adenosquamous carcinoma of the pancreas: a case report. Cases J 2010;3:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Na YJ, Shim KN, Cho MS, et al. Primary adenosquamous cell carcinoma of the pancreas: a case report with a review of the Korean literature. Korean J Intern Med 2011;26:348–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kelly K, Moore C. Two rare cases of pancreatic adenosquamous carcinoma: a review of the literature with focus on radiologic findings. Radiol Case Rep 2019;14:809–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nabae T, Yamaguchi K, Takahata S, et al. Adenosquamous carcinoma of the pancreas: report of two cases. Am J Gastroenterol 1998;93:1167–70. [DOI] [PubMed] [Google Scholar]
- 96.Kurdi YM, Peck JR, Roth R, Conwell DL. A case of pancreatic adenosquamous carcinoma obstructing the common bile and pancreatic ducts, duodenum, and gastric outlet. Pancreas 2016;45:e9–el0. [DOI] [PubMed] [Google Scholar]
- 97.López-Tomassetti-Fernández EM, Favre-Rizzo J, Delgado-Plasencia L, Hernández-Hemández JR. Hypercalcemia associated with adenosquamous pancreatic carcinoma: a reason to initiate palliative treatment. Rev Esp Enferm Dig 2013;105:425–8. [DOI] [PubMed] [Google Scholar]
- 98.Kobayashi N, Higurashi T, Iida H, et al. Adenosquamous carcinoma of the pancreas associated with humoral hypercalcemia of malignancy (HHM). J Hepatobiliary Pancreat Surg 2008;15:531–5. [DOI] [PubMed] [Google Scholar]
- 99.Inoue T, Nagao S, Tajima H, et al. Adenosquamous pancreatic cancer producing parathyroid hormone-related protein. J Gastroenterol 2004;39:176–80. [DOI] [PubMed] [Google Scholar]
- 100.Stewart AF. Clinical practice. Hypercalcemia associated with cancer. N Engl J Med 2005;352:373–9. [DOI] [PubMed] [Google Scholar]
- 101.Wild AT, Dholakia AS, Fan KY, et al. Efficacy of platinum chemotherapy agents in the adjuvant setting for adenosquamous carcinoma of the pancreas. J Gastrointest Oncol 2015;6:115–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hue JJ, Katayama E, Sugumar K, et al. The importance of multimodal therapy in the management of nonmetastatic adenosquamous carcinoma of the pancreas: analysis of treatment sequence and strategy. Surgery 2021; 169:1102–9. [DOI] [PubMed] [Google Scholar]
- 103.Altman AM, Wirth K, Marmor S, et al. Completion of adjuvant chemotherapy after upfront surgical resection for pancreatic cancer is uncommon yet associated with improved survival. Ann Surg Oncol 2019;26:4108–16. [DOI] [PubMed] [Google Scholar]
- 104.DePeralta DK, Ogami T, Zhou JM, et al. Completion of adjuvant therapy in patients with resected pancreatic cancer. HPB 2020;22:241–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Romano E, Janati S, Monnier L, et al. Outcomes of vaginal squamous cell carcinoma of patients treated with radiation therapy: a series of 37 patients from a single expert center. Clin Transl Oncol 2020;22:1345–54. [DOI] [PubMed] [Google Scholar]
- 106.Maubec E. Update of the management of cutaneous squamous-cell carcinoma. Acta Derm Venereol 2020;100:adv00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mendenhall WM, Strojan P, Lee AWM, et al. Radiotherapy in the management of glottic squamous cell carcinoma. Head Neck 2020;42:3558–67. [DOI] [PubMed] [Google Scholar]
- 108.Lin HN, Chen LQ, Shang QX, Yuan Y, Yang YS. A meta-analysis on surgery with or without postoperative radiotherapy to treat squamous cell esophageal carcinoma. Int J Surg 2020;80:184–91. [DOI] [PubMed] [Google Scholar]
- 109.Yamaue H, Tanimura H, Onishi H, et al. Adenosquamous carcinoma of the pancreas: successful treatment with extended radical surgery, intraoperative radiation therapy, and locoregional chemotherapy. Int J Pancreatol 2001;29:53–8. [DOI] [PubMed] [Google Scholar]
- 110.Hsu JT, Yeh CN, Chen YR, et al. Adenosquamous carcinoma of the pancreas. Digestion 2005;72:104–8. [DOI] [PubMed] [Google Scholar]
- 111.Brunetti O, Aprile G, Marchetti P, et al. Systemic chemotherapy for advanced rare pancreatic histotype tumors: a retrospective multicenter analysis. Pancreas 2018;47:759–71. [DOI] [PubMed] [Google Scholar]
- 112.Tanaka N, Ohoida J, Matuno T, et al. Response of adenosquamous carcinoma of the pancreas to interferon-alpha, tumor necrosis factor-alpha and 5-fluorouracil combined treatment. Anticancer Res 1994;14:2739–42. [PubMed] [Google Scholar]
- 113.Noel P, Hussein S, Ng S, et al. Triptolide targets super-enhancer networks in pancreatic cancer cells and cancer-associated fibroblasts. Oncogenesis 2020;9:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Skorupan N, Ahmad MI, Steinberg SM, et al. A phase II trial of the super-enhancer inhibitor Minnelide™ in advanced refractory adenosquamous carcinoma of the pancreas. Future Oncol 2022;18:2475–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Klimstra DS, Heffess CS, Oertel JE, Rosai J. Acinar cell carcinoma of the pancreas. A clinicopathologic study of 28 cases. Am J Surg Pathol 1992;16:815–37. [DOI] [PubMed] [Google Scholar]
- 116.Toll AD, Hruban RH, Ali SZ. Acinar cell carcinoma of the pancreas: clinical and cytomorphologic characteristics. Korean J Pathol 2013;47:93–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Klimstra DS, Rosai J, Heffess CS. Mixed acinar-endocrine carcinomas of the pancreas. Am J Surg Pathol 1994;18: 765–78. [DOI] [PubMed] [Google Scholar]
- 118.Abraham SC, Wu TT, Hruban RH, et al. Genetic and immunohistochemical analysis of pancreatic acinar cell carcinoma: frequent allelic loss on chromosome 11p and alterations in the APC/beta-catenin pathway. Am J Pathol 2002;160:953–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Klimstra DS, Adsay V. Acinar neoplasms of the pancreas-A summary of 25 years of research. Semin Diagn Pathol 2016;33:307–18. [DOI] [PubMed] [Google Scholar]
- 120.Thompson ED, Wood LD. Pancreatic neoplasms with acinar differentiation: a review of pathologic and molecular features. Arch Pathol Lab Med 2020;144:808–15. [DOI] [PubMed] [Google Scholar]
- 121.La Rosa S, Franzi F, Marchet S, et al. The monoclonal anti-BCL10 antibody (clone 331.1) is a sensitive and specific marker of pancreatic acinar cell carcinoma and pancreatic metaplasia. Virchows Arch 2009;454:133–42. [DOI] [PubMed] [Google Scholar]
- 122.Hosoda W, Sasaki E, Murakami Y, Yamao K, Shimizu Y, Yatabe Y. BCL10 as a useful marker for pancreatic acinar cell carcinoma, especially using endoscopic ultrasound cytology specimens. Pathol Int 2013;63:176–82. [DOI] [PubMed] [Google Scholar]
- 123.Said S, Kurtin PJ, Nasr SH, et al. Carboxypeptidase A1 and regenerating islet-derived 1α as new markers for pancreatic acinar cell carcinoma. Hum Pathol 2020; 103:120–6. [DOI] [PubMed] [Google Scholar]
- 124.Uhlig R, Contreras H, Weidemann S, et al. Carboxypeptidase A1 (CPA1) immunohistochemistry is highly sensitive and specific for acinar cell carcinoma (ACC) of the pancreas. Am J Surg Pathol 2022;46:97–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Jiao Y, Yonescu R, Offerhaus GJ, et al. Whole-exome sequencing of pancreatic neoplasms with acinar differentiation. J Pathol 2014;232:428–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Chmielecki J, Hutchinson KE, Frampton GM, et al. Comprehensive genomic profiling of pancreatic acinar cell carcinomas identifies recurrent RAF fusions and frequent inactivation of DNA repair genes. Cancer Discov 2014;4:1398–405. [DOI] [PubMed] [Google Scholar]
- 127.Jäkel C, Bergmann F, Toth R, et al. Genome-wide genetic and epigenetic analyses of pancreatic acinar cell carcinomas reveal aberrations in genome stability. Nat Commun 2017;8:1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Furukawa T, Sakamoto H, Takeuchi S, et al. Whole exome sequencing reveals recurrent mutations in BRCA2 and FAT genes in acinar cell carcinomas of the pancreas. Sci Rep 2015;5:8829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Chou A, Kim Y, Samra JS, Pajic M, Gill AJ. BRAF gene rearrangements can be identified by FISH studies in pancreatic acinar cell carcinoma. Pathology 2018;50:345–8. [DOI] [PubMed] [Google Scholar]
- 130.Prall OWJ, Nastevski V, Xu H, et al. RAF1 rearrangements are common in pancreatic acinar cell carcinomas. Mod Pathol 2020;33:1811–21. [DOI] [PubMed] [Google Scholar]
- 131.Chou A, Brown IS, Kumarasinghe MP, et al. RET gene rearrangements occur in a subset of pancreatic acinar cell carcinomas. Mod Pathol 2020;33:657–64. [DOI] [PubMed] [Google Scholar]
- 132.Gupta M, Sherrow C, Krone ME, et al. Targeting the NTRK fusion gene in pancreatic acinar cell carcinoma: a case report and review of the literature. J Natl Compr Canc Netw 2021;19:10–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Liu W, Shia J, Gönen M, Lowery MA, O’Reilly EM, Klimstra DS. DNA mismatch repair abnormalities in acinar cell carcinoma of the pancreas: frequency and clinical significance. Pancreas 2014;43:1264–70. [DOI] [PubMed] [Google Scholar]
- 134.Kryklyva V, Haj Mohammad N, Morsink FHM, et al. Pancreatic acinar cell carcinoma is associated with BRCA2 germline mutations: a case report and literature review. Cancer Biol Ther 2019;20:949–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lee JH, Park SJ, Hariharasudhan G, et al. ID3 regulates the MDC1-mediated DNA damage response in order to maintain genome stability. Nat Commun 2017;8:903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Bakr A, Hey J, Sigismondo G, et al. ID3 promotes homologous recombination via non-transcriptional and transcriptional mechanisms and its loss confers sensitivity to PARP inhibition. Nucleic Acids Res 2021;49:11666–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Xu J, Palestino Dominguez M, Alewine C. Loss of ID3 in pancreatic cancer cells increases DNA damage without impairing MDC1 recruitment to the nuclear foci. Cancer Commun 2022;42:269–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.La Rosa S, Bernasconi B, Frattini M, et al. TP53 alterations in pancreatic acinar cell carcinoma: new insights into the molecular pathology of this rare cancer. Virchows Arch 2016;468:289–96. [DOI] [PubMed] [Google Scholar]
- 139.Matsuno S, Egawa S, Fukuyama S, et al. Pancreatic cancer registry in japan: 20 years of experience. Pancreas 2004;28:219–30. [DOI] [PubMed] [Google Scholar]
- 140.Mustafa S, Hruban RH, Ali SZ. Acinar cell carcinoma of the pancreas: a clinicopathologic and cytomorphologic review. J Am Soc Cytopathol 2020;9:586–95. [DOI] [PubMed] [Google Scholar]
- 141.Sridharan V, Mino-Kenudson M, Cleary JM, et al. Pancreatic acinar cell carcinoma: a multi-center series on clinical characteristics and treatment outcomes. Pancreatology 2021:1119–26. [DOI] [PubMed] [Google Scholar]
- 142.Schmidt CM, Matos JM, Bentrem DJ, Talamonti MS, Lillemoe KD, Bilimoria KY. Acinar cell carcinoma of the pancreas in the United States: prognostic factors and comparison to ductal adenocarcinoma. J Gastrointest Surg 2008;12:2078–86. [DOI] [PubMed] [Google Scholar]
- 143.Petrova E, Wellner J, Nording AK, et al. Survival outcome and prognostic factors for pancreatic acinar cell carcinoma: retrospective analysis from the german cancer registry group. Cancers 2021; 13:6121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Duorui N, Shi B, Zhang T, et al. The contemporary trend in worsening prognosis of pancreatic acinar cell carcinoma: a population-based study. PLoS One 2020;15:e0243164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Wisnoski NC, Townsend CM Jr, Nealon WH, Freeman JL, Riall TS. 672 patients with acinar cell carcinoma of the pancreas: a population-based comparison to pancreatic adenocarcinoma. Surgery 2008;144:141–8. [DOI] [PubMed] [Google Scholar]
- 146.Seo S, Yoo C, Kim KP, et al. Clinical outcomes of patients with resectable pancreatic acinar cell carcinoma. J Dig Dis 2017;18:480–6. [DOI] [PubMed] [Google Scholar]
- 147.Shin SH, Hwang HK, Jang JY, et al. Clinical characteristics of resected acinar cell carcinoma of the pancreas: a korean multi-institutional study. Careens- 2021; 13:5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Huang X, Li M, Zhang L, Xiong J, Lu H, Tian B. Clinical characteristics and treatment analysis of pancreatic acinar cell carcinoma: a single institutional comparison to pancreatic ductal adenocarcinoma. Surg Oncol 2021;37:101528. [DOI] [PubMed] [Google Scholar]
- 149.Seket B, Saurin JC, Scoazec JY, Partensky C. [Pancreatic acinar cell carcinoma in a patient with familial adenomatous polyposis]. Gastroenterol Clin Biol 2003;27:818–20. [PubMed] [Google Scholar]
- 150.Karamurzin Y, Zeng Z, Stadler ZK, et al. Unusual DNA mismatch repair-deficient tumors in Lynch syndrome: a report of new cases and review of the literature. Hum Pathol 2012;43:1677–87. [DOI] [PubMed] [Google Scholar]
- 151.Li M, Mou Y, Hou S, Cao D, Li A. Response of germline BRCA2-mutated advanced pancreatic acinar cell carcinoma to olaparib: a case report. Medicine 2018;97:e13113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Labate AM, Klimstra DL, Zakowski MF. Comparative cytologic features of pancreatic acinar cell carcinoma and islet cell tumor. Diagn Cytopathol 1997; 16:112–6. [DOI] [PubMed] [Google Scholar]
- 153.Stelow EB, Bardales RH, Shami VM, et al. Cytology of pancreatic acinar cell carcinoma. Diagn Cytopathol 2006;34:367–72. [DOI] [PubMed] [Google Scholar]
- 154.Tharian B, Canipe AL, Krall K, Hawes RH, Hébert-Magee S. Ring around the ROSE: pancreatic acinar cell carcinoma diagnosed on site by EUS-FNA. Gastrointest Endosc 2015;81:1049–50. [DOI] [PubMed] [Google Scholar]
- 155.Toyonaga Y, Yamazaki K, Yamada M, Koyasu T, Koyama Y, Ishida Y. Brush cytology of acinar cell carcinoma of the pancreas with intraductal growth: a case report. Diagn Cytopathol 2014;42:321–4. [DOI] [PubMed] [Google Scholar]
- 156.Peng HQ, Darwin P, Papadimitriou JC, Drachenberg CB. Liver metastases of pancreatic acinar cell carcinoma with marked nuclear atypia and pleomorphism diagnosed by EUS FNA cytology: a case report with emphasis on FNA cytological findings. Cytojournal 2006;3:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Barat M, Dohan A, Gaujoux S, et al. Computed tomography features of acinar cell carcinoma of the pancreas. Diagn Interv Imaging 2020;101:565–75. [DOI] [PubMed] [Google Scholar]
- 158.Jornet D, Soyer P, Terris B, et al. MR imaging features of pancreatic acinar cell carcinoma. Diagn Interv Imaging 2019;100:427–35. [DOI] [PubMed] [Google Scholar]
- 159.Matos JM, Schmidt CM, Turrini O, et al. Pancreatic acinar cell carcinoma: a multi-institutional study. J Gastrointest Surg 2009;13:1495–502. [DOI] [PubMed] [Google Scholar]
- 160.Singh S, Gorouhi F, Konia T, Burrall B. Pancreatic acinar cell carcinoma-induced panniculitis. JAAD Case Rep 2018;4:719–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Nizam W, Shah AA, Rajack F, Ramdath A, Naab T, Williams M. Lipase hypersecretion syndrome: a rare cutaneous manifestation of advanced pancreatic acinar cell carcinoma. Clin Case Rep 2020;8:905–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Good AE, Schnitzer B, Kawanishi H, Demetropoulos KC, Rapp R. Acinar pancreatic tumor with metastatic fat necrosis: report of a case and review of rheumatic manifestations. Am J Dig Dis 1976;21:978–87. [DOI] [PubMed] [Google Scholar]
- 163.Burns WA, Matthews MJ, Hamosh M, Vander Weide G, Blum R, Johnson FB. Lipase-secreting acinar cell carcinoma of the pancreas with polyarthropathy. A light and electron microscopic, histochemical, and biochemical study. Cancer 1974;33:1002–9. [DOI] [PubMed] [Google Scholar]
- 164.Taskin OC, Adsay V. Lipase hypersecretion syndrome: a distinct form of paraneoplastic syndrome specific to pancreatic acinar carcinomas. Semin Diagn Pathol 2019;36:240–5. [DOI] [PubMed] [Google Scholar]
- 165.Askan G, Deshpande V, Klimstra DS, et al. Expression of markers of hepatocellular differentiation in pancreatic acinar cell neoplasms: a potential diagnostic pitfall. Am J Clin Pathol 2016;146:163–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Patel DJ, Lutfi W, Sweigert P, et al. Clinically resectable acinar cell carcinoma of the pancreas: is there a benefit to adjuvant systemic therapy? Am J Surg 2020;219:522–6. [DOI] [PubMed] [Google Scholar]
- 167.Landa K, Freischlag K, Nussbaum DP, Youngwirth LM, Blazer DG 3rd. Underutilization of surgical resection in patients with pancreatic acinar cell carcinoma. HPB 2019;21:687–94. [DOI] [PubMed] [Google Scholar]
- 168.Burchard PR, Chacon AC, Melucci A, et al. Defining the role of systemic therapy in resectable pancreatic acinar cell carcinoma. J Surg Oncol 2022;125:856–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Yoo C, Kim BJ, Kim KP, et al. Efficacy of chemotherapy in patients with unresectable or metastatic pancreatic acinar cell carcinoma: potentially improved efficacy with oxaliplatin-containing regimen. Cancer Res Treat 2017;49:759–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Hiyoshi M, Kai K, Hamada T, Yano K, Imamura N, Nanashima A. Curative remnant total pancreatectomy for recurrent pancreatic acinar cell carcinoma: a case report. Int J Surg Case Rep 2022;94:107091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Ohara Y, Oda T, Enomoto T, et al. Surgical resection of hepatic and rectal metastases of pancreatic acinar cell carcinoma (PACC): a case report. World J Surg Oncol 2018; 16:158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Di Marco M, Carloni R, De Lorenzo S, et al. Long-term survival of two patients with recurrent pancreatic acinar cell carcinoma treated with radiofrequency ablation: a case report. World J Clin Cases 2020;8:1241–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Sumiyoshi T, Shima Y, Okabayashi T, et al. Long-term survival following pancreatectomy and s-1 chemotherapy for pancreatic acinar cell carcinoma with peritoneal dissemination: a case report and literature review. Medicine 2015;94:e378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Cananzi FC, Jayanth A, Lorenzi B, et al. “Chronic” metastatic pancreatic acinar cell carcinoma. Pancreatology 2013;13:549–52. [DOI] [PubMed] [Google Scholar]
- 175.Jauch SF, Morris VK, Jensen CT, Kaseb AO. Multimodal approach and long-term survival in a patient with recurrent metastatic acinar cell carcinoma of the pancreas: a case report. Pancreatology 2016; 16:153–6. [DOI] [PubMed] [Google Scholar]
- 176.Jimbo M, Batista PM, Baliff JP, Yeo CJ. Neoadjuvant chemotherapy and appleby procedure for pancreatic acinar cell carcinoma: a case report. Case Rep Pancreat Cancer 2016;2:46–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Takahashi H, Ikeda M, Shiba S, et al. Multicenter retrospective analysis of chemotherapy for advanced pancreatic acinar cell carcinoma: potential efficacy of platinum- and irinotecan-containing regimens. Pancreas 2021;50:77–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Xu JY, Guan WL, Lu SX, et al. Optimizing chemotherapy of pancreatic acinar cell carcinoma: our experiences and pooled analysis of literature. Clin Med Insights Oncol 2022; 16:11795549221090186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Lowery MA, Klimstra DS, Shia J, et al. Acinar cell carcinoma of the pancreas: new genetic and treatment insights into a rare malignancy. Oncologist 2011;16:1714–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Schempf U, Sipos B, König C, Malek NP, Bitzer M, Plentz RR. FOLFIRINOX as first-line treatment for unresectable acinar cell carcinoma of the pancreas: a case report. Z Gastroenterol 2014;52:200–3. [DOI] [PubMed] [Google Scholar]
- 181.Hashimoto M, Hikichi T, Suzuki T, et al. Successful chemotherapy with modified FOLFIRINOX for pancreatic acinar cell carcinoma. Clin J Gastroenterol 2017; 10:564–9. [DOI] [PubMed] [Google Scholar]
- 182.Busch E, Kreutzfeldt S, Agaimy A, et al. Successful BRAF/MEK inhibition in a patient with BRAF(V600E)-mutated extrapancreatic acinar cell carcinoma. Cold Spring Harb Mol Case Stud 2020;6:a005553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Cramer S, Marcus MA, Ramkissoon S, Szabo S, Pressey JG. Pediatric BRAF (V600E)-mutated pancreatic acinar cell carcinoma with complete and durable response to dabrafenib and trametinib. JCO Precis Oncol 2020;4:801–5. [DOI] [PubMed] [Google Scholar]
- 184.Gaule M, Pesoni C, Quinzii A, et al. Exceptional clinical response to alectinib in pancreatic acinar cell carcinoma with a novel ALK-KANK4 gene fusion. JCO Precis Oncol 2022;6:e2100400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Maehira H, Iida H, Mori H, et al. Pathological complete response in a patient with metastatic pancreatic acinar cell carcinoma who received a chemotherapy regimen containing cisplatin and irinotecan. Clin J Gastroenterol 2021; 14:1772–8. [DOI] [PubMed] [Google Scholar]
- 186.Qin L, Shen J, Yang Y, Zou Z. Rapid response to the combination of lenvatinib and sintilimab in a pancreatic acinar cell carcinoma patient with elevated alpha-fetoprotein: a case report. Front Oncol 2021; 11:692480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Finn RS, Qin S, Ikeda M, et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med 2020;382:1894–905. [DOI] [PubMed] [Google Scholar]
- 188.Makker V, Rasco D, Vogelzang NJ, et al. Lenvatinib plus pembrolizumab in patients with advanced endometrial cancer: an interim analysis of a multicentre, open-label, single-ann, phase 2 trial. Lancet Oncol 2019;20:711–8. [DOI] [PubMed] [Google Scholar]
- 189.Tiriac H, Belleau P, Engle DD, et al. Organoid profiling identifies common responders to chemotherapy in pancreatic cancer. Cancer Discov 2018;8:1112–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.O’Kane GM, Grünwald BT, Jang GH, et al. GATA6 expression distinguishes classical and basal-like subtypes in advanced pancreatic cancer. Clin Cancer Res 2020;26:4901–10. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.