


Keywords: Pck1, mitoribosome, diabetic nephropathy
Abstract
Significance Statement
Renal gluconeogenesis plays an important role in the pathogenesis of diabetic nephropathy (DN). Proximal tubular phosphoenolpyruvate carboxykinase1 (PEPCK1) is the rate-limiting enzyme in gluconeogenesis. However, the functions of PEPCK1 have not been elucidated. We describe the novel role of PEPCK1 as a mitoribosomal protector using Pck1 transgenic (TG) mice and knockout mice. Pck1 blocks excessive glycolysis by suppressing the upregulation of excess HK2 (the rate-limiting enzyme of glycolysis). Notably, Pck1 overexpression retains mitoribosomal function and suppresses renal fibrosis. The renal and mitoribosomal protective roles of Pck1 may provide important clues for understanding DN pathogenesis and provide novel therapeutic targets.
Background
Phosphoenolpyruvate carboxykinase (PEPCK) is part of the gluconeogenesis pathway, which maintains fasting glucose levels and affects renal physiology. PEPCK consists of two isoforms—PEPCK1 and PEPCK2—that the Pck1 and Pck2 genes encode. Gluconeogenesis increases in diabetic nephropathy (DN), escalating fasting and postprandial glucose levels. Sodium–glucose cotransporter-2 inhibitors increase hepatic and renal gluconeogenesis. We used genetically modified mice to investigate whether renal gluconeogenesis and Pck1 activity are renoprotective in DN.
Methods
We investigated the expression of Pck1 in the proximal tubule (PTs) of streptozotocin (STZ)-treated diabetic mice. We studied the phenotypic changes in PT-specific transgenic (TG) mice and PT-specific Pck1 conditional knockout (CKO) mice.
Results
The expression of Pck1 in PTs was downregulated in STZ-treated diabetic mice when they exhibited albuminuria. TG mice overexpressing Pck1 had improved albuminuria, concomitant with the mitigation of PT cell apoptosis and deposition of peritubular type IV collagen. Moreover, CKO mice exhibited PT cell apoptosis and type IV collagen deposition, findings also observed in STZ-treated mice. Renal fibrotic changes in CKO mice were associated with increasing defects in mitochondrial ribosomes (mitoribosomes). The TG mice were protected against STZ-induced mitoribosomal defects.
Conclusion
PCK1 preserves mitoribosomal function and may play a novel protective role in DN.
Introduction
Diabetic nephropathy (DN) can exacerbate chronic kidney disease, leading to end-stage renal disease. The paucity of medications to treat kidney injury under diabetic conditions may reflect limited understanding of the details of renal glucose metabolism in DN.
We previously reported that proximal tubular metabolic changes in NAD and the NAD-dependent deacetylation enzyme Sirtuin 1 (SIRT1) affect glomerular phenotypes,1 termed the tubuloglomerular interplay,2 and the role of the nicotinomide mononucleotide (NMN)–producing enzyme, Nampt, in suppressing renal fibrosis.3 NMN supplementation maintained NAD levels and reversed diabetic podocyte injury.4 Glycolysis consumes NAD and generates ATP, whereas gluconeogenesis consumes ATP and generates NAD. Thus, NAD metabolism is significantly associated with glucose metabolism.
Claudin-1, a tight junction protein, is present in parietal epithelial cells of control, healthy mice, but not in podocytes. However, under pathological conditions, such as focal segmental glomerulosclerosis5 and DN,6 claudin-1 is ectopically expressed in podocytes. In addition, we recently reported that NMN administration suppressed diabetic podocyte damage by repressing the ectopic expression of claudin-1 in podocytes.4 Thus, in general, renal tubular metabolic changes affect diabetic glomerular function, and specifically, the Sirt1–NMN–claudin-1 axis is a promising therapeutic target for DN. The function of proximal tubular carnitine palmitoyl-transferase 1A in kidney fibrosis has also recently been reported.7
We found that sodium–glucose cotransporter-2 (SGLT2) inhibitors elevate renal SIRT1 levels in murine DN models, leading to renal protection.8 Other studies have reported that SGLT2 inhibitors activate SIRT1 and peroxisome proliferator–activated receptor-γ coactivator-1 α, increasing renal gluconeogenesis.9 It has been suggested that SGLT2 inhibitors protect against diabetic kidney damage by upregulating SIRT1 levels and gluconeogenesis in addition to optimizing tubuloglomerular feedback (TGF). However, whether SGLT2 inhibitors increase or decrease renal gluconeogenesis remains controversial.10 In addition, it is not known whether renal gluconeogenesis is protective or pathogenic in DN.10 This study investigated whether enzymes that participate in gluconeogenesis reverse renal damage.
Phosphoenolpyruvate carboxykinase (PEPCK) catalyzes the conversion of oxaloacetate to phosphoenolpyruvate in gluconeogenesis. Two genes encode isozymes of PEPCK. The Pck1 gene encodes PEPCK1, which localizes in the cytosol, whereas the PcK2 gene encodes PEPCK2, which is restricted to mitochondria.11 Studies using Pck1-knockout mice confirmed the importance of Pck1 in hepatic gluconeogenesis and glucose homeostasis because Pck1−/− mice showed severe hypoglycemia.12 However, kidney-specific Pck1 TG mice have not been reported. We describe them here.
Three key enzymes regulate glycolysis: hexokinase (HK), phosphofructokinase, and pyruvate kinase (PK). The three key enzymes that regulate gluconeogenesis are glucose-6-phosphatase, PEPCK, and fructose-1,6-bisphosphatase.13 Because renal PEPCK is significantly elevated in human DN14 and, along with glucose-6-phosphatase is rate-limiting in gluconeogenesis, it may be critical in DN pathogenesis. Further evidence is that single-cell RNA-sequencing analyses of kidneys reveal that Pck1 is elevated in DN but Pck2 is not and is more significant.15 This the rational for focusing on Pck1 in this study.
The enzymes that catalyze gluconeogenesis are localized to the proximal tubule (PT), whereas glycolytic enzymes are only in the distal tubule.11 However, recent reports identify glycolytic enzymes, such as pyruvate kinase M2 (PKM2), also in the PT, and implicate them in the aberrant glycolysis that promotes renal fibrosis.16 Thus, the PT apparently regulates glycolysis and gluconeogenesis, in addition to glucose reabsorption that SGLT2 and GLUT2 mediate. Therefore, we targeted proximal tubular cells to clarify the function of PCK1 in the kidneys. To investigate the pathogenic role of PCK1 in DN, we created TG mice that overexpress Pck1 and Pck1-knockout (Pck1 CKO) mice in the PT.
Methods
Detailed descriptions are in Supplemental Methods.
Creation of TG and CKO Mice
A TG expression vector containing a mouse sodium phosphate cotransporter IIa (Npt2) promoter and murine Pck1 cDNA tagged with an 8-amino acid FLAG epitope was developed, as previously described,1 to create renal PT-specific Pck1 TG mice on a C57BL/6J background. These mice were then propagated as heterozygous TG mice by breeding them with wild-type (WT) C57BL/6 mice. F2 mice were used as TG mice, and age-matched and sex-matched littermates were used as WT control mice. Renal PT-specific Pck1-knockout (Pck1−/−) mice (CKO mice) were created by crossing Pck1 flox/flox mice on a C57BL/6J background with γGT-Cre mice (Jackson Laboratory). The following three types of control mice were created using this breeding process: Pck1 flox/flox, Pck1 flox/+, and Pck1 flox/+/γGT-Cre tg/+. In the subsequent studies, Pck1 flox/+/γGT-Cre tg/+ mice were crossed with Pck1 flox/flox mice to create Pck1 flox/flox/γGT-Cre tg/+ (PT-specific Pck1−/−) mice (Pck1 CKO mice), and Pck1 flox/flox mice were used as controls. All animal studies were conducted according to the animal experimentation guidelines of the School of Medicine, Tokushima University. In each experiment, we used Pck1 TG and WT (N=8), Pck1 CKO, and control (N=8) mice.
Animal Experiment Protocols of Streptozotocin-Treated Mice
Eight-week-old male mice were treated with streptozotocin (STZ) (Sigma-Aldrich, St. Louis, MO) dissolved in 100 mmol/L citrate buffer (pH 4.5) or phosphate-buffered saline dissolved in citrate buffer. Multiple injections of low-dose (50 mg/kg body weight) STZ were administered intraperitoneally for 5 days in nonfasted mice.
Animal Experiment Protocols Using db/db Mice
To assess the effect of PCK1 knockdown in CKO on db/db, we developed Pck1 CKO-db/db mice (Supplemental Figures 1–4).
Terminal Deoxynucleotidyl Transferase dUTP Nick-End Labeling (TUNEL) Assay
The TUNEL assay and immunohistochemical staining for the percentage of apoptotic proximal renal tubular cells (TUNEL kit; Roche Diagnostics) were assessed semiquantitatively, as described in a previous study.3
Microarray Analysis
Mice in the control and Pck1 CKO groups were euthanized at 32 weeks and whole kidneys harvested for microarray analyses using the Affymetrix Mouse Genome 430 2.0 Microarray Chips according to the Affymetrix standard protocols (http://www.affymetrix.com, Affymetrix, Santa Clara, CA). Genes were excluded if signal strength did not significantly exceed background levels and expression did not reach a threshold for reliable detection (P < 0.05) in any of the three studies. The remaining genes were subjected to nonparametric Welch t tests and indicated by their respective fold changes and P-values. Microarray data are available in GEO database (GEO: GSE228053).
RNA Isolation, Reverse Transcription, and Quantitative PCR
The RNeasy Plus Mini Kit (QIAGEN, Hilden, Germany) was used to extract total RNA from culture cells and tissues. Real-time PCR was performed using the ABI Prism 7700 Sequence Detection System (Applied Biosystems, Foster City, CA) and SYBR GREEN System (Applied Biosystems). The relative mRNA levels for each gene were normalized to the level of mRNA expression for the housekeeping gene, GAPDH. The following primers were used: Pck1, F(5′-AAGTGCCTGCACTCTGTGG-3′) and R(5′-CAGGCCCAGTTGTTGACC-3′); HK2, F(5′-TCTACCACATGCGCCTCTCT-3′) and R(5′-GCCCATTGTCCGTTACTTTC-3′); NDUFA9, F(5′-ACTGTGTTTGGGGCTACAGG-3′) and R(5′-GATTGATGACCACGTTGCTG-3′); FP, F(5′-ACACAGACCTGGTGGAGACC-3′) and R(5′-GCACAGTCAGCCTCATTCAA-3′); UQCRC2, F(5′-ATCAAAAGGGGCAACAACAC-3′) and R(5′-CACTCAGGAAGCCCTCTGAC-3′); COX1, F(5′-ATTCGAGCAGAATTAGGTCA-3′) andR(5′-CTCCGATTATTAGTGGGACA-3′); ATP5A1, F(5′-AGGCCTATCCTGGTGATGTG-3′) and R(5′-CTTCATGGTACCTGCCACCT-3′); ND1, F(5′-ACGCAAAATCTTAGGGTACA-3′) and R (5′-GAGTGATAGGGTAGGTGCAA-3′); MRPL13, F(5′-ACATAAACCTGTGTACCATG CAC-3′) and R(5′-GGTAGCCAGTATGCGAAGAGT-3′); MRPS15, F(5′-ATCCGTTCTAGAAGCACCAAGAG) and R(5′-CTCAGCATAGCGTTGATAGTGAG); VDAC, F(5′-CTCCCACATACGCCGATCTT-3′) and R(5′-GCCGTAGCCCTTGGTGAAG-3′); LaminB, F(5′-GGGCGTCAGATTGAGTATGAG-3′) and R(5′-TTAGAGAGCTGTGAGGAGAGG-3′); CR6-interacting factor 1 (CRIF1), F(5′-TATCTCCTGCGGCTCTCTGT-3′) and R(5′-CTTCTGCTTTCGCCAGTTTT-3′); and GAPDH, F(5′-CCAGGGCTGCTTTTAACTC-3′) and R(5′-GCTCCCCCCTGCAAATGA-3′). Changes in albumin uptake-related molecules in Pck1 CKO and TG were also analyzed using real-time PCR systems (Supplemental Figure 5).
Histopathological Examination
The kidney tissue specimens were fixed in 10% neutral-buffered formaldehyde, embedded in paraffin, and sliced into 4-μm thick sections. Immunohistochemistry was performed as described previously.4 In brief, 4-μm thick paraffin sections were fixed in 3% formaldehyde and stained with primary antibodies against Pck1 (Abcam, Cambridge, MA), HK2 (Cell Signaling Technology, MA), albumin (Nordic-MUbio, Rangeerweg, The Netherlands), type IV collagen (Millipore, Bedford, MA), and TGF-β (Santa Cruz Biotechnology, Santa Cruz, CA). These sections were stained with biotin-labeled goat anti-rabbit immunoglobulin G (Vector Laboratories, UK) or biotin-labeled anti-mouse immunoglobulin G (Vector) and then treated with the Vectastain Elite ABC Kit (Vector). Each image of the stained sections was scanned using a 3CCD camera (Olympus Optical, Tokyo, Japan). For morphometric analyses of the tubulointerstitium, areas positive for type IV collagen staining were measured per high-power field in a blinded manner. Changes in tissue inhibitor metalloproteinase-1 levels were also assessed by immunohistochemistry (Supplemental Figure 6).
Mitochondrial Function
Isolated primary kidney epithelial cells from CKO or control mice and four groups of mice (i.e., WT+Sal, TG+Sal, WT+STZ, and TG+STZ groups) were used to assess mitochondrial function. Isolation was performed as described previously.7 The endogenous cellular oxygen consumption rate (OCR) was measured using an XF-24 extracellular flux analyzer (Seahorse Bioscience, Santa Clara, CA) per the provided protocol. For measuring the mitochondrial membrane potential, the JC-1 mitochondrial membrane potential detection kit (Peninsula Laboratories, Inc.) was used per the manufacturer's instructions. To measure mitochondrial reactive oxygen species, harvested cells were stained with MitoSOX red (Molecular Probes) as described previously.7 ATP was also measured as described previously.7 We also measured the activity of oxidative phosphorylation (OXPHOS) complexes (Supplemental Figure 7).
Electron Microscopy (EM)
For EM, the kidney tissue specimens were embedded in Epon epoxy resin. Electron micrographs of ten PTs per kidney were randomly obtained for each mouse to evaluate the morphometry. Glomerular histological changes in CKO and TG mice were also evaluated (Supplemental Figure 8).
Measurement of Extracellular Acidification Rate (ECAR)
Mitochondrial ECAR was measured (Supplemental Figure 9).
Immunofluorescence Staining
Five-μm thick cryostat kidney sections were incubated overnight and labeled with two primary antibodies—rabbit polyclonal anti-Pck1 (1:500, ab28455, Abcam, Cambridge, MA) and mouse polyclonal anti–aquaporin-1 (1:100, B-11, Santa Cruz Biotechnology, Santa Cruz, CA). The secondary antibodies were from Jackson ImmunoResearch Laboratories (West Grove, PA). Double immunofluorescence staining of TUNEL and AQP-1 in the kidney (Supplemental Figures 10–15) revealed localized STZ-induced or Pck1 defect-induced apoptosis.
Statistical Analyses
Prism 8 (GraphPad Software, CA) was used to perform all statistical analyses. Data were expressed as mean±SEM. Comparisons among several groups used the one-way analysis of variance and the Tukey post hoc test. A P-value of <0.05 was considered statistically significant.
Results
Downregulation of Pck1 Expression in DN
We evaluated the expression levels of PCK1 in the PTs using STZ-treated mice as models of diabetes (Figure 1A). Immunofluorescence revealed decreasing Pck1 staining 8 weeks after STZ treatment and distinctly reduced staining after 24 weeks (Figure 1B).
Figure 1.

Effects of Pck1 CKO on urinary albuminuria in DN. (A) A schematic illustration showing the STZ-induced diabetic model procedure. Eight-week-old C57BL/6 mice were stimulated with STZ (50 mg/kg per day×5 d, i.p.) for 5 consecutive days, and Pck1 levels were determined at age 8, 16, and 32 weeks. STZ, streptozotocin; i.p., intraperitoneal injection. (B) Temporary changes in the protein expression of Pck1 in DN. Representative images of kidney cryosections derived from STZ-treated mice were stained by immunofluorescence for Pck1 (green) and costained for AQP1 (red), a proximal tubule marker. Scale bar, 50 μm; N=3. (C) Schematic showing the breeding of Pck1 flox/flox γGT-cre+/− mice. Pck1 flox/flox mice were mated with γGT-cre+/− mice; subsequently, the F2 Pck1 flox/− γGT-cre+/− mice were created. Pck1 flox/flox γGT-cre+/− mice were created by crossing Pck1 flox/− γGT-cre+/− mice with Pck1 flox/flox mice. (D) Pck1 immunofluorescence intensity levels in CKO and control mice. Kidney tissue specimens derived from CKO or control mice at age 32 weeks were stained using immunofluorescence for Pck1 (green) and AQP1 (red). The right panel illustrates the quantitative analysis of the Pck1 fluorescence intensity. Scale bar, 50 µm; N=3. (E) Real-time quantitative reverse transcription analysis of renal mRNA levels of Pck1 (N=7) in control and CKO mice at age 32 weeks. (F) Temporal changes in the mean plasma glucose concentrations in the mice of each group. We examined the mice at age 8, 12, 16, 20, 24, 28, and 32 weeks, respectively. N=7 mice per group. (G) Body weight changes from age 8 to 32 weeks in control or CKO mice. N=7 mice per group. (H) Representative images of sections immune-stained with HK2 in the kidneys from control and CKO mice at age 32 weeks. Negative and positive controls for Pck1 immunohistochemistry. Kidney tissues were used as negative controls and liver tissues as positive controls. Scale bar, 50 μm. The right panel shows proportional staining areas for HK2. N=7. The right schematic shows the metabolic map indicating Pck1 was downregulated, and HK2 was augmented in CKO mice. Statistically significant differences in each group are represented by horizontal bars. *P < 0.05.
Effects of Pck1 Deficiency in the PTs on Proteinuria
To further evaluate the role of PCK1 in the PTs, we created PT-specific Pck1-deficient mice (CKO mice, Pck1 CKO mice) (Figure 1C). In immunofluorescence staining, 32-week-old Pck1 CKO mice demonstrated reduced Pck1 expression compared with control mice (Figure 1D). RT-PCR for Pck1 revealed similar results (Figure 1E). Serum glucose levels did not differ between control and CKO mice at age 8, 12, 16, 20, 24, 28, 32, and 36 weeks (Figure 1F). No significant differences in body weights were observed at 8, 16, 24, and 32 weeks (Figure 1G). Although we predicted that Pck1 deficiency may affect glucose metabolism and body weights, the serum glucose concentrations and body weights remained unchanged. To elucidate the molecular mechanisms through which CKO did not influence glucose levels, we performed DNA microarray analyses of kidney samples to examine the differences in gene expression between control and CKO mice (Tables 1 and 2). CKO mice showed a significant increase in HK2 compared with control mice. HK, the first enzyme in the glycolysis pathway, converts glucose to glucose-6-P. In CKO mice, Pck1 decreased and HK2 increased (Figure 1H). This effect seems to balance glucose metabolism, resulting in no change in blood glucose levels and body weight in these mice. To exclude antigen-independent staining, negative controls omitted the incubation step with the primary antibody. Negative controls did not stain. Pck1 immunoreactivity was initially detected in positive controls (livers) known to express Pck1 (the Human Protein Atlas) (Figure 1H).
Table 1.
Genes that were upregulated in the kidneys of CKO compared with Cont mice
| Gene Symbol | Gene Name | Fold Change | Genbank Accession |
|---|---|---|---|
| Ucp1 | Uncoupling protein 1 (mitochondrial, proton carrier) | 26.625 | NM_009463 |
| Cox8b | Cytochrome c oxidase subunit 8B | 11.979 | NM_007751 |
| Ctxn3 | Cortexin 3 | 11.540 | NM_001134697 |
| Cidea | Cell death–inducing DNA fragmentation factor, α subunit-like effector A | 10.868 | NM_007702 |
| Hp | Haptoglobin | 10.756 | NM_001329965 |
| Rgs14 | Regulator of G-protein signaling 14 | 5.484 | NM_016758 |
| Mrap | Melanocortin 2 receptor accessory protein | 4.802 | NM_029844 |
| Bcl6 | B-cell leukemia/lymphoma 6 | 4.584 | AK046709 |
| Rem1 | rad and gem–related GTP binding protein 1 | 4.571 | AK078772 |
| Nat8f5 | N-acetyltransferase 8 (GCN5-related) family member 5 | 4.514 | NM_023493 |
| Car3 | Carbonic anhydrase 3 | 4.496 | NM_007606 |
| Slc16a14 | Solute carrier family 16 (monocarboxylic acid transporters), member 14 | 4.429 | NM_027921 |
| Adig | Adipogenin | 4.065 | NM_145635 |
| Slc16a14 | Solute carrier family 16 (monocarboxylic acid transporters), member 14 | 4.011 | XM_006496551 |
| Apoh | Apolipoprotein H | 3.935 | NM_013475 |
| Ppbp | Proplatelet basic protein | 3.862 | NM_023785 |
| Cyp4a12a | Cytochrome P450, family 4, subfamily a, polypeptide 12a | 3.734 | NM_177406 |
| Cyp4a12b | Cytochrome P450, family 4, subfamily a, polypeptide 12B | 3.681 | NM_172306 |
| Plin4 | Perilipin 4 | 3.677 | NM_020568 |
| Apoc1 | Apolipoprotein C-I | 3.647 | NM_007469 |
| Clstn3 | Calsyntenin 3 | 3.616 | NM_153508 |
| Zfp458 | Zinc finger protein 458 | 3.615 | NM_001001152 |
| Sfpq | Splicing factor proline/glutamine rich (polypyrimidine tract binding protein associated) | 3.596 | XR_001784200 |
| Hk2 | Hexokinase 2 | 3.593 | NM_013820 |
| Derl3 | Der1-like domain family, member 3 | 3.535 | NM_024440 |
| Gm8016 | Predicted gene 8016 | 3.459 | NR_152173 |
| Prkar2b | Protein kinase, cAMP-dependent regulatory, type II β | 3.351 | NM_011158 |
| F13a1 | Coagulation factor XIII, A1 subunit | 3.343 | NM_028784 |
| S100a8 | S100 calcium binding protein A8 (calgranulin A) | 3.282 | NM_013650 |
| Rbp7 | Retinol binding protein 7, cellular | 3.269 | NM_022020 |
| Thrsp | Thyroid hormone responsive | 3.126 | NM_009381 |
| Gm16982 | Predicted gene, 16982 | 3.066 | NR_040337 |
| Slc36a2 | Solute carrier family 36 (proton/amino acid symporter), member 2 | 3.050 | AK079219 |
| S100a9 | S100 calcium binding protein A9 (calgranulin B) | 3.025 | NM_001281852 |
| Col19a1 | Collagen, type XIX, α 1 | 2.924 | NM_007733 |
| Adamts4 | A disintegrin-like and metallopeptidase (reprolysin type) with thrombospondin type-1 motif, 4 | 2.851 | NM_172845 |
| Atp1a2 | ATPase, Na+/K+transporting, α 2 polypeptide | 2.829 | NM_178405 |
| Kcnk3 | Potassium channel, subfamily K, member 3 | 2.804 | NM_010608 |
| Pisd-ps3 | Phosphatidylserine decarboxylase, pseudogene 3 | 2.741 | NR_003518 |
| Gp9 | Glycoprotein 9 (platelet) | 2.696 | NM_018762 |
| Oxct2b | 3-oxoacid CoA transferase 2B | 2.644 | NM_181859 |
| Sec14l4 | SEC14-like lipid binding 4 | 2.620 | NM_146013 |
| Fabp4 | Fatty acid binding protein 4, adipocyte | 2.573 | NM_024406 |
| B3galt2 | UDP-Gal:βGlcNAc β 1,3-galactosyltransferase, polypeptide 2 | 2.559 | NM_020025 |
| Meox2 | Mesenchyme homeobox 2 | 2.555 | NM_008584 |
| Apod | Apolipoprotein D | 2.545 | NM_007470 |
| Inhbb | Inhibin β-B | 2.525 | NM_008381 |
| S100b | S100 protein, β polypeptide, neural | 2.522 | NM_009115 |
| Nrg4 | Neuregulin 4 | 2.500 | AK080089 |
| Gm40812 | Predicted gene, 40812 | 2.443 | XR_872234 |
| Gm33272 | Predicted gene, 33272 | 2.421 | XR_001782921 |
| Ttr | Transthyretin | 2.413 | NM_013697 |
| Vmn1r197 | Vomeronasal 1 receptor 197 | 2.408 | NM_134244 |
| Penk | Preproenkephalin | 2.400 | NM_001002927 |
| Gm33272 | Predicted gene, 33272 | 2.389 | XR_001782921 |
| Fabp4 | Fatty acid binding protein 4, adipocyte | 2.375 | NM_024406 |
| Zfp874a | Zinc finger protein 874a | 2.364 | AK136394 |
| Serpina1a | Serine (or cysteine) peptidase inhibitor, clade A, member 1A | 2.358 | NM_001252569 |
| Gm33272 | Predicted gene, 33272 | 2.350 | XR_001782921 |
| Wnt10a | Wingless-type MMTV integration site family, member 10A | 2.345 | NM_009518 |
| Nrg4 | Neuregulin 4 | 2.336 | NM_032002 |
| Uqcc1 | Ubiquinol–cytochrome c reductase complex assembly factor 1 | 2.334 | XM_011239706 |
| Maged1 | Melanoma antigen, family D, 1 | 2.327 | LF202122 |
| Trarg1 | Trafficking regulator of GLUT4 (SLC2A4) 1 | 2.318 | NM_177709 |
| Tnfrsf25 | Tumor necrosis factor receptor superfamily, member 25 | 2.303 | NM_001291010 |
| Pgbd5 | piggyBac transposable element derived 5 | 2.303 | NM_171824 |
| Nrg4 | Neuregulin 4 | 2.268 | NM_032002 |
| 1700018B24Rik | Enhancer of rudimentary homolog pseudogene | 2.257 | NR_003617 |
| Gm8765 | Predicted gene 8765 | 2.250 | NM_001244649 |
| Arxes2 | Adipocyte-related X-chromosome expressed sequence 2 | 2.250 | NM_029823 |
| C78197 | Expressed sequence C78197 | 2.246 | AK041628 |
| Mmp3 | Matrix metallopeptidase 3 | 2.241 | NM_010809 |
| Ttr | Transthyretin | 2.239 | NM_013697 |
| 4930502E18Rik | RIKEN cDNA 4930502E18 gene | 2.238 | XM_006535359 |
| Apod | Apolipoprotein D | 2.208 | NM_007470 |
| Hesx1 | Homeobox gene expressed in ES cells | 2.205 | NM_010420 |
| Cyp4f15 | Cytochrome P450, family 4, subfamily f, polypeptide 15 | 2.188 | NM_134127 |
| Cpe | Carboxypeptidase E | 2.185 | NM_013494 |
| Nrg4 | Neuregulin 4 | 2.182 | NM_032002 |
| Nr4a1 | Nuclear receptor subfamily 4, group A, member 1 | 2.177 | NM_010444 |
| Uqcc1 | Ubiquinol–cytochrome c reductase complex assembly factor 1 | 2.175 | AK054294 |
| 4930535E02Rik | RIKEN cDNA 4930535E02 gene | 2.174 | AK015977 |
| Myom3 | Myomesin family, member 3 | 2.146 | NM_001085509 |
| Ccdc33 | Coiled-coil domain containing 33 | 2.145 | NM_001311085 |
| Igf2bp1 | Insulin-like growth factor 2 mRNA binding protein 1 | 2.119 | NM_009951 |
| Slc36a2 | Solute carrier family 36 (proton/amino acid symporter), member 2 | 2.113 | AK086373 |
| Rbm3 | RNA binding motif (RNP1, RRM) protein 3 | 2.094 | NM_016809 |
| Polr3e | Polymerase (RNA) III (DNA directed) polypeptide E | 2.087 | NM_025298 |
| Gm41572 | Predicted gene, 41572 | 2.060 | XR_876756 |
| Tfec | Transcription factor EC | 2.049 | AK157426 |
| Slc25a48 | Solute carrier family 25, member 48 | 2.047 | NM_177809 |
| C4b | Complement component 4B (Chido blood group) | 2.041 | NM_009780 |
| Pzp | PZP, α-2 macroglobulin like | 2.039 | NM_007376 |
| C4a | Complement component 4A (Rodgers blood group) | 2.036 | NM_011413 |
| Il34 | Interleukin 34 | 2.023 | NM_029646 |
| Fcamr | Fc receptor, IgA, IgM, high affinity | 2.014 | NM_144960 |
| Pzp | PZP, α-2 macroglobulin like | 2.013 | NM_007376 |
| Slco4a1 | Solute carrier organic anion transporter family, member 4a1 | 2.001 | LF193827 |
| Dgkb | Diacylglycerol kinase, β | 1.992 | NM_001361686 |
| Pcdh7 | Protocadherin 7 | 1.989 | NM_018764 |
| Dmkn | Dermokine | 1.986 | NM_001166173 |
| Pparg | Peroxisome proliferator–activated receptor γ | 1.985 | NM_011146 |
| Atp1a2 | ATPase, Na+/K+transporting, α 2 polypeptide | 1.960 | NM_178405 |
| Catsper3 | Cation channel, sperm associated 3 | 1.950 | NM_029772 |
| Aldh3a2 | Aldehyde dehydrogenase family 3, subfamily A2 | 1.950 | NR_138545 |
| Nnmt | Nicotinamide N-methyltransferase | 1.947 | NM_010924 |
| C4b | Complement component 4B (Chido blood group) | 1.937 | NM_009780 |
| Treml1 | Triggering receptor expressed on myeloid cells like 1 | 1.936 | NM_027763 |
| Terc | Telomerase RNA component | 1.925 | NR_001579 |
| Rpp40 | Ribonuclease P 40 subunit | 1.916 | AK048810 |
| Il34 | Interleukin 34 | 1.912 | NM_029646 |
| Noct | Nocturnin | 1.908 | NM_009834 |
| Gm9951 | Predicted gene 9951 | 1.905 | AK040144 |
| Necab2 | N-terminal EF-hand calcium binding protein 2 | 1.904 | NM_054095 |
| Psat1 | Phosphoserine aminotransferase 1 | 1.903 | NM_001205339 |
| Serpina1a | Serine (or cysteine) peptidase inhibitor, clade A, member 1A | 1.903 | NM_001252569 |
| Gm5105 | Predicted gene 5105 | 1.901 | NR_037975 |
| Arhgef1 | Rho guanine nucleotide exchange factor (GEF) 1 | 1.901 | XM_006539565 |
| Duox2 | Dual oxidase 2 | 1.892 | AK079781 |
| Pappa2 | Pappalysin 2 | 1.882 | NM_001085376 |
| B3gat1 | β-1,3-glucuronyltransferase 1 (glucuronosyltransferase P) | 1.881 | NM_001310766 |
| Slc1a3 | Solute carrier family 1 (glial high affinity glutamate transporter), member 3 | 1.880 | NM_148938 |
| Nnmt | Nicotinamide N-methyltransferase | 1.879 | NM_001311062 |
| Noct | Nocturnin | 1.876 | NM_009834 |
| Serpina1b | Serine (or cysteine) preptidase inhibitor, clade A, member 1B | 1.876 | NM_009244 |
| Ebf3 | Early B cell factor 3 | 1.868 | NM_010096 |
| Prr18 | Proline rich 18 | 1.867 | NM_178774 |
| Chil3 | Chitinase-like 3 | 1.865 | NM_009892 |
Table 2.
Genes that were downregulated in the kidneys of CKO mice compared with Cont mice
| Gene Symbol | Gene Name | Fold Change | Genbank Accession |
|---|---|---|---|
| Hmgcs2 | 3-Hydroxy-3-methylglutaryl-coenzyme A synthase 2 | 0.187 | NM_008256 |
| Zdhhc19 | Zinc finger, DHHC domain containing 19 | 0.206 | NM_199309 |
| Hmgcs2 | 3-Hydroxy-3-methylglutaryl-coenzyme A synthase 2 | 0.206 | NM_008256 |
| Kynu | Kynureninase | 0.222 | NM_001289593 |
| Slc7a12 | Solute carrier family 7 (cationic amino acid transporter, y+system), member 12 | 0.227 | NM_080852 |
| Kynu | Kynureninase | 0.249 | NM_027552 |
| Crabp1 | Cellular retinoic acid binding protein I | 0.267 | NM_001284507 |
| Prlr | Prolactin receptor | 0.309 | NM_001253781 |
| Creb3l3 | cAMP responsive element binding protein 3-like 3 | 0.315 | NM_145365 |
| Prlr | Prolactin receptor | 0.335 | NM_011169 |
| Prlr | Prolactin receptor | 0.342 | NM_011169 |
| Antxr1 | Anthrax toxin receptor 1 | 0.346 | NM_054041 |
| Fabp1 | Fatty acid binding protein 1, liver | 0.383 | NM_017399 |
| Mbl2 | Mannose-binding lectin (protein C) 2 | 0.390 | NM_010776 |
| Antxr1 | Anthrax toxin receptor 1 | 0.393 | AK031465 |
| Depp1 | DEPP1 autophagy regulator | 0.397 | NM_145980 |
| Rab6b | RAB6B, member RAS oncogene family | 0.405 | NM_173781 |
| S100a14 | S100 calcium binding protein A14 | 0.425 | NM_025393 |
| Ang3 | Angiogenin, ribonuclease A family, member 3 | 0.426 | NM_001123394 |
| Ang | Angiogenin, ribonuclease, RNase A family, 5 | 0.427 | NM_007447 |
| B930025P03Rik | RIKEN cDNA B930025P03 gene | 0.428 | NR_040705 |
| Lrriq1 | Leucine-rich repeats and IQ motif containing 1 | 0.429 | NM_001163559 |
| Igfbp1 | Insulin-like growth factor binding protein 1 | 0.431 | NM_008341 |
| Klra22 | Killer cell lectin-like receptor subfamily A, member 22 | 0.436 | NM_053152 |
| Otos | Otospiralin | 0.438 | NM_153114 |
| Cry2 | Cryptochrome 2 (photolyase-like) | 0.442 | AK037778 |
| Ang4 | Angiogenin, ribonuclease A family, member 4 | 0.447 | NM_177544 |
| Gzma | Granzyme A | 0.450 | NM_010370 |
| Cyp24a1 | Cytochrome P450, family 24, subfamily a, polypeptide 1 | 0.451 | NM_009996 |
| Slc13a1 | Solute carrier family 13 (sodium/sulfate symporters), member 1 | 0.454 | AK047580 |
| 2900060L22Rik | RIKEN cDNA 2900060L22 gene | 0.458 | XR_869289 |
| 5730405A17Rik | RIKEN cDNA 5730405A17 gene | 0.458 | AK017489 |
| Baiap3 | BAI1-associated protein 3 | 0.459 | NM_001163270 |
| Ang6 | Angiogenin, ribonuclease A family, member 6 | 0.460 | NM_001011876 |
| Gm40787 | Predicted gene, 40787 | 0.460 | XR_872127 |
| Lgr5 | Leucine-rich repeat containing G protein coupled receptor 5 | 0.461 | NM_010195 |
| Sema5b | Sema domain, seven thrombospondin repeats (type 1 and type 1-like), transmembrane domain (TM), and short cytoplasmic domain, (semaphorin) 5B | 0.462 | NM_013661 |
| Gm16010 | Predicted gene 16010 | 0.462 | AK085341 |
| Rnase4 | Ribonuclease, RNase A family 4 | 0.464 | NM_021472 |
| Slc13a1 | Solute carrier family 13 (sodium/sulfate symporters), member 1 | 0.466 | AK019876 |
| Gm525 | Predicted gene 525 | 0.475 | NM_001033266 |
| Gm36283 | Predicted gene, 36283 | 0.480 | XR_380771 |
| Pde6h | Phosphodiesterase 6H, cGMP-specific, cone, γ | 0.482 | BC057290 |
| 0.483 | AK051762 | ||
| Gm4340 | Predicted gene 4340 | 0.486 | NM_001177535 |
| LOC101056011 | Uncharacterized LOC101056011 | 0.492 | AK140221 |
| 8430419K02Rik | RIKEN cDNA 8430419K02 gene | 0.494 | XR_872264 |
| Scgb1c1 | Secretoglobin, family 1C, member 1 | 0.495 | NM_001099742 |
| Aqp6 | Aquaporin 6 | 0.498 | DQ826418 |
| Nmrk1 | Nicotinamide riboside kinase 1 | 0.498 | LF257405 |
| Slc26a7 | Solute carrier family 26, member 7 | 0.498 | NM_145947 |
| Procr | Protein C receptor, endothelial | 0.505 | NM_011171 |
| 4732416N19Rik | RIKEN cDNA 4732416N19 gene | 0.508 | NR_015615 |
| Sema5b | Sema domain, seven thrombospondin repeats (type 1 and type 1-like), transmembrane domain (TM), and short cytoplasmic domain, (semaphorin) 5B | 0.510 | AK078659 |
| Cry2 | Cryptochrome 2 (photolyase-like) | 0.511 | AK041696 |
| 2900052L18Rik | RIKEN cDNA 2900052L18 gene | 0.512 | NR_151480 |
| Slc51a | Solute carrier family 51, α subunit | 0.512 | NM_145932 |
| Aplnr | Apelin receptor | 0.513 | NM_011784 |
| Itga8 | Integrin α 8 | 0.513 | AK044910 |
| Slc40a1 | Solute carrier family 40 (iron-regulated transporter), member 1 | 0.514 | AK165333 |
| Gm10824 | Predicted gene 10824 | 0.514 | AK033378 |
| Gm40798 | Predicted gene, 40798 | 0.516 | AK042269 |
| Tnfsf15 | Tumor necrosis factor (ligand) superfamily, member 15 | 0.517 | NM_177371 |
| Olfr1500 | Olfactory receptor 1500 | 0.518 | NM_001011831 |
| Slc51a | Solute carrier family 51, α subunit | 0.518 | AK147155 |
| Ccdc116 | Coiled-coil domain containing 116 | 0.519 | XM_006522712 |
| Gm32133 | Predicted gene, 32133 | 0.519 | XR_379717 |
| Aqp6 | Aquaporin 6 | 0.520 | NM_175087 |
| Tekt1 | Tektin 1 | 0.520 | NM_001282006 |
| Pdia2 | Protein disulfide isomerase associated 2 | 0.521 | NM_001081070 |
| Gm15230 | Predicted gene 15230 | 0.522 | XR_387114 |
| Igf1r | Insulin-like growth factor I receptor | 0.523 | AK155079 |
| Gm26588 | Predicted gene, 26588 | 0.523 | XR_377840 |
| Ptch1 | Patched 1 | 0.523 | XM_011244497 |
| 4930539N22Rik | RIKEN cDNA 4930539N22 gene | 0.524 | NR_040598 |
| AI662168 | Expressed sequence AI662168 | 0.525 | AI662168 |
| Eif4e1b | Eukaryotic translation initiation factor 4E family member 1B | 0.527 | NM_001033269 |
| Acot3 | Acyl-CoA thioesterase 3 | 0.529 | NM_001346701 |
| Acot3 | Acyl-CoA thioesterase 3 | 0.529 | NM_134246 |
| Ypel1 | Yippee like 1 | 0.530 | AK163365 |
| Pik3ip1 | Phosphoinositide-3-kinase interacting protein 1 | 0.530 | NM_178149 |
| Actn3 | Actinin α 3 | 0.532 | NM_013456 |
| Enpp1 | Ectonucleotide pyrophosphatase/phosphodiesterase 1 | 0.533 | AK035402 |
| Cryaa | Crystallin, α A | 0.533 | NM_001278569 |
| Prnd | Prion like protein doppel | 0.534 | NM_023043 |
| Tcstv3 | 2 Cell-stage, variable group, member 3 | 0.534 | NM_153523 |
| Nek5 | NIMA (never in mitosis gene a)-related expressed kinase 5 | 0.534 | AK054168 |
| Iigp1 | Interferon inducible GTPase 1 | 0.536 | AK172047 |
| Clec7a | C-type lectin domain family 7, member a | 0.537 | NM_020008 |
| Timm8a2 | Translocase of inner mitochondrial membrane 8A2 | 0.538 | NM_001037744 |
| Bfsp1 | Beaded filament structural protein 1, in lens-CP94 | 0.539 | NM_001291061 |
| Gm17762 | Predicted gene, 17762 | 0.539 | AK033161 |
| Tnnt2 | Troponin T2, cardiac | 0.541 | AK086260 |
| Nr5a2 | Nuclear receptor subfamily 5, group A, member 2 | 0.541 | NM_030676 |
| Olfr10 | Olfactory receptor 10 | 0.542 | NM_206822 |
Although serum creatinine concentrations were the same (Figure 2A), urinary albumin excretion was significantly higher in CKO mice than in control mice (Figure 2B). To confirm this finding, SDS-PAGE analyzed urinary protein excretion, which indicated that CKO mice had excessive albuminuria (Figure 2C).
Figure 2.

Renal tubular damage and fibrosis in proximal tubule-specific Pck1 CKO mice. (A) Serum creatinine levels in control and CKO mice at age 32 weeks. N=7 per mice group. (B) Urinary albumin excretion in control and CKO mice at age 32 weeks. N=7 per mice group. (C) SDS-PAGE of mouse urine samples. Urine samples of 32-week-old CKO mice were subjected to a 15% SDS-PAGE before staining with Coomassie blue. N=2 mice per group. (D) Representative images of albumin staining in control and CKO mice. Arrows indicate albumin casts in CKO mice. The right panel depicts the relative staining intensity. N=7 mice per group. (E) Apoptotic tubular cells in control and CKO mice. Arrows indicate apoptotic cells. The bar graph illustrates the quantitative analysis of apoptotic tubular cells. N=7 mice per group. (F) Representative photomicrographs showing TGF-β immunostaining in each group. The bar graph illustrates the quantitative analysis of TGF-β staining. N=7 per mice group. (G) Representative photomicrographs showing collagen IV immunostaining in each group. The bar graph illustrates the quantitative analysis of collagen IV staining. N=7 per mice group. (D, E, F, and G) Light micrograph; scale bar=100 μm. Kidney tissue specimens for immunostaining were obtained from CKO and control mice at age 32 weeks. All data are shown as mean±SEM. Statistically significant differences in each group are represented by horizontal bars. *P < 0.05.
Effects of Pck1 Deficiency in the PTs on Renal Fibrotic Changes
Albumin staining in the intracellular tubular area demonstrated lower reabsorption in CKO mice than in control mice (Figure 2D). CKO mice had intratubular albumin casts, indicating loss of albumin reuptake. Moreover, the number of apoptotic tubular cells was higher in CKO mice than in control mice, as TUNEL staining (Figure 2E) showed. Data from double immunofluorescence for TUNEL and AQP-1 demonstrated apoptotic cells originating in the PT (Supplemental Figures 10–11). These data indicated that tubular damage in CKO mice mitigated albumin reabsorption with a concomitant increase in albuminuria. CKO mice were also stained for TGF-β and deposition of type IV collagen, resulting in renal fibrotic changes (Figure 2, F and G). These findings were consistent with changes observed in DN when PCK1 expression in the PTs decreased, which the following results from STZ-treated mice demonstrate. Our data revealed that CKO mice have peritubular fibrotic changes even in nondiabetic conditions, indicating that proximal tubular Pck1 deficiency has profibrotic effects on the kidney.
To further elucidate whether morphological or functional changes in intracellular organelles were associated with such profibrotic phenotypes and tubular injuries, we comprehensively examined the EM findings. EM images of the mitochondria clearly revealed that the number of tiny black (electron dense) spots inside the mitochondria was fewer in CKO mice than in control mice (Figure 3A). These tiny black spots appeared as praying beads, corresponding to ribosomes in the mitochondria (mitochondrial ribosomes [mitoribosomes]; Figure 3B). Mitoribosomes were fewer in CKO mice and appeared gray because of the low density of black spots. By contrast, the mitochondria seemed black (darker) in control mice. The number of dense black spots in the cytoplasm was not altered in CKO mice compared with control mice. These cytoplasmic spots corresponded to the ribosomes in the cytoplasm (cytoplasmic ribosomes; cytoribosomes).
Figure 3.
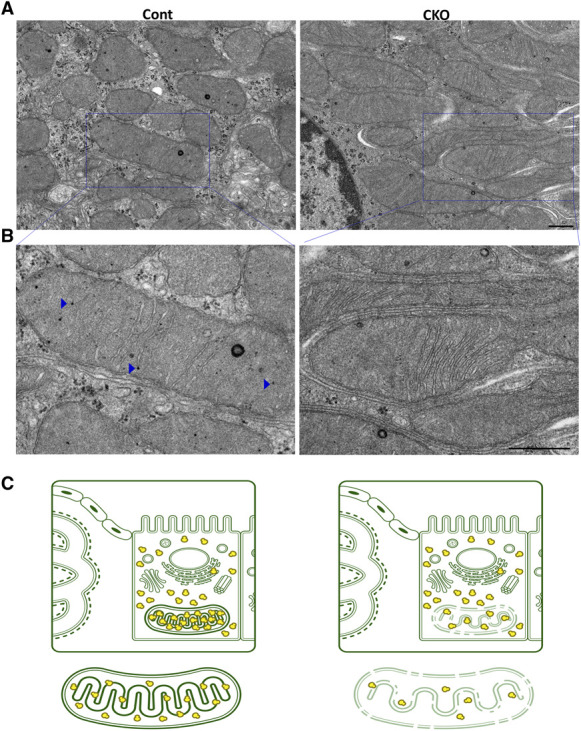
Mitoribosomal defect in Pck1 CKO. (A) Representative electron micrograph in each group. Scale bar=500 nm. Blue squares indicate the enlarged regions. (B) Expanded images of (A) are presented. Blue arrowheads indicate mitoribosomes. Scale bar=500 nm. (C) Illustration depicting the mitoribosomal defect in the two groups of mice. For EM, the kidney tissue specimens were embedded in Epon epoxy resin. Electron micrographs of ten PTs per kidney were randomly obtained for each mouse to evaluate the morphometry of the PTs.
Expanded images of Figure 3A are presented in Figure 3B. Arrowheads indicate mitoribosomes, which several articles have described.17,18 However, because the black dots were different sizes, it is uncertain whether they were all mitoribosomes or other granules. Therefore, we confirmed the results of RT-PCR analysis for correctly measuring the number of mitoribosomes by counting mitoribosomal proteins (MRPs), shown in Figure 4.
Figure 4.

The dysfunctional mitoribosomes and their concomitant mitochondrial dysfunction in CKO mice. (A) Real-time quantitative reverse transcription analysis of the renal mRNA levels of OXPHOS subunits encoded by nDNA and mtDNA. (B) Real-time quantitative reverse transcription analysis of renal mRNA levels of intracellular organelle markers. MRPL13 and MRPS15 are mitoribosomal proteins, whereas VDAC is a mitochondrial protein, and Lamin B is a nuclear protein. (C) Real-time quantitative reverse transcription analysis of the renal mRNA levels of CRIF1, a mitochondrial ribosomal synthesis regulator. Illustration depicting the dysfunctional mitoribosomes and their concomitant mitochondrial dysfunction in CKO mice. These functions are intact in control mice. (A–C) Glyceraldehyde 3-phosphate dehydrogenase was used as a control. The kidney tissue specimens for RT-PCR were obtained from CKO and control mice at age 32 weeks. N=3 mice per group. (D) OCR of TECs isolated from CKO and control mice was measured using a Seahorse XF-24 flux analyzer. N=3. (E) The ratio of red/green fluorescence of JC-1 of TECs isolated from CKO and control mice as a measure of mitochondrial membrane potential. N=7. (F) Fluorescence of MitoSox of TECs isolated from CKO and control mice as a measure of mitochondrial ROS levels. N=7. (G) ATP content of TECs isolated from CKO and control mice. N=7. All data are presented as mean±SEM. Horizontal bars indicate statistically significant differences between groups. *P < 0.05. (H) Scheme depicting the new mitoribosomes-mediated mechanism of renal profibrotic changes and tubular injury in CKO mice. The downregulation of Pck1 increased HK2 expression. Increased HK2 expression decreased the expression of CRIF1, causing mitoribosomal defects. The upregulation of mitoribosomal defects leads to the deposition of collagen IV in addition to the OXPHOS impairment and tubular injury.
Our novel findings suggest that PCK1 is essential for maintaining the integrity of mitoribosomes in the kidneys (Figure 3C). A previous report19 indicated that mitoribosome defects enhance TGF-β expression, a finding consistent with our results. Mitoribosomal defect-mediated TGF-β upregulation promoted the deposition of type IV collagen, which is the main component of renal fibrosis.
Mitoribosomal defect and CRIF1 downregulation are responsible for diabetic peritubular fibrotic changes by Pck1 deficiency.
We investigated whether mitoribosomal defects with fewer mitochondrial ribosomes were associated with mitoribosomal dysfunction to detect the expression of proteins synthesized by mitoribosomes and encoded by mit-DNA. Mitoribosomes generate several OXPHOS enzyme subunits, such as of ND1 and COX1 (Figure 4A), and the expression of these subunits is largely reduced in CKO mice. However, the nuclear DNA-encoded and cytoribosome-synthesized OXPHOS subunits, such as NDUFA9, FP, UQCRC2, and ATP5A1, were unaltered in both CKO and control mice (Figure 4A). These findings demonstrate that the mitoribosomal defect is associated with loss of mitoribosomal function. To quantify the mitoribosomal defect in CKO mice, we monitored MRPs as mitoribosomal markers and compared these levels with those of other organelle marker proteins. MRPS15 and MRPL13 are mitoribosomal markers, whereas VDAC and Lamin B are mitochondrial and nuclear markers, respectively. We found a strong reduction in the levels of MRPs, such as MRPS15 and MRPL13, in CKO mice (Figure 4B). In addition, Western blotting (WB) confirmed RT-PCR data (Supplemental Figure 16). The WB findings were consistent with the PCR data. However, VDAC and Lamin B levels were unchanged in both CKO and control mice. The synthesis of MRPs requires CRIF1, which is a key regulator of MRP synthesis. Furthermore, we confirmed that CRIF1 was greatly reduced in CKO mice (Figure 4C). Thus, CRIF1 reduction induced mitoribosomal defects in CKO mice. Altogether, mitoribosomal defects in CKO mice mitigated CRIF1 and MRPs.
Upregulation of Pck1 Expression in db/db
To further assess the effect of PCK1 knockdown in CKO on db/db, we developed Pck1 CKO-db/db mice (Supplemental Figures 1–4). First, we assessed the expression levels of PCK1 in the PTs using db/db mice (Supplemental Figure 1A). Immunofluorescence examination showed that Pck1 staining rose in the PTs at 16 weeks of db/db. At 32 weeks of db/db, Pck1 staining further increased (Supplemental Figure 1B).
Effects of Pck1 Elevation in the PTs on Proteinuria
In immunofluorescence staining, 32-week-old Pck1 CKO-db/db mice showed reduced Pck1 expression compared with Cont-db/db mice (Supplemental Figure 1D). Cont-db/db mice exhibited elevated Pck1 expression compared with Cont-ND mice. In addition, serum glucose levels of Cont-db/db or CKO-db/db were higher than those of Cont-ND or CKO-ND at age 8, 16, 24, and 32 weeks (Supplemental Figure 1E). Increase in body weights of Cont-db/db and CKO-db/db was seen at age 8, 16, 24, and 32 weeks (Supplemental Figure 1F). HK2 immunostaining levels were markedly lower in Cont-db/db mice than in Cont-ND mice. Furthermore, immunostaining demonstrated that HK2 levels in CKO-db/db mice did not vary from Cont-db/db (Supplemental Figure 1G). Despite having the same serum creatinine levels (Supplemental Figure 2A), urinary albumin excretion was significantly higher in CKO-ND mice, Cont-db/db mice, or CKO-db/db mice than in Cont-ND mice (Supplemental Figure 2B). However, the levels of albuminuria of CKO-db/db mice were similar to Cont-db/db mice. To validate this finding, the urinary protein excretion was evaluated using SDS-PAGE, which also implied that CKO-db/db did not further increase in albuminuria compared with Cont-db/db mice (Supplemental Figure 2C).
Effects of Pck1 Deficiency in the PTs on Renal Fibrotic Changes in db/db
Albumin staining showed elevated intratubular albumin casts in CKO-ND mice compared with Cont-ND mice (Supplemental Figure 2D). Cont-db/db and CKO-db/db mice manifested a slight increase in albumin casts. Furthermore, the proportion of apoptotic tubular cells was higher in CKO-ND mice than in Cont-ND mice, as TUNEL staining (Supplemental Figure 2E) showed. Cont-db/db and CKO-db/db also denote the apoptotic cells, of which levels were also lower than in CKO-ND mice. These data suggest that CKO-ND mice exhibit tubular damage that reduces albumin reabsorption with a concomitant increase in albuminuria. CKO-ND mice also revealed staining of TGF-β and deposition of type IV collagen, causing renal fibrotic changes (Supplemental Figure 2, F and G). Cont-db/db and CKO-db/db mice also showed these changes, also lower than CKO-ND.
HK2 Levels in db/db
To elucidate how Cont-db/db and CKO-db/db mice develop intratubular albumin casts, tubular apoptotic cells, and ECM production, all of which were lower than those of CKO-ND mice, we conducted HK2 immunostaining (Supplemental Figure 3A). CKO-ND mice demonstrated a significant increase in HK2 compared with Cont-ND mice. We hypothesized that Cont-db/db mice had lower HK2 expression, and CKO-db/db mice had higher HK2 expression because Pck1 levels were higher in Cont-db/db and lower in CKO-db/db (Supplemental Figure 1D). However, HK2 levels were commonly lower in Cont-db/db and CKO-db/db than in Cont-ND and CKO-ND (Supplemental Figure 3A). So we hypothesized how db/db models suppress HK2 levels, illustrating the molecular mechanisms underlying the mitoribosome-dependent and mitoribosome-independent renal injuries in type-1 diabetes mellitus (T1DM) and type-2 diabetes mellitus (T2DM), respectively (Supplemental Figure 3B). Supplemental Figure 3C illustrates the presumed mechanisms that did not worsen renal injury in CKO-db/db mice.
We evaluated the effect of db/db on mitoribosome number using EM (Supplemental Figure 4, A and B) and counting mitoribosome protein markers using RT-PCR systems (Supplemental Figure 4C). Mitoribosomes were fewer in CKO-ND mice than in Cont-ND mice but more numerous in both Cont-db/db and CKO-db/db mice. Therefore, we hypothesize that the increased number of mitoribosomes (Supplemental Figure 4D) protect against TGF-β upregulation and type IV collagen deposition, both observed in Cont-db/db and CKO-db/db mice.
Impaired Mitochondrial OXPHOS Function in Pck1 Deficiency
To investigate the influence of mitoribosomal defects in CKO mice on mitochondrial dysfunction, we analyzed OCR and JC-1 as mitochondrial membrane potentials and MitoSox as mitochondrial reactive oxygen species. We also investigated ATP production by primary tubular epithelial cells isolated and harvested from the kidneys of CKO and control mice. The OCR in CKO mice was lower than that in control mice (Figure 4D), and the OCR in CKO mice did not respond to treatment with chemical inhibitors of the mitochondrial respiratory chains, indicating that CKO lost its OXPHOS activity in the basal state. The marked decreases in mitochondrial JC-1 incorporation (Figure 4E) in CKO cells suggested that the mitochondria in these cells were depolarized and dysfunctional. CKO cells exhibited higher mitochondrial superoxide levels, as MitoSox analysis (Figure 4F) shows. However, within 4 days after selection, the ATP levels in CKO cells did not differ from those in control cells grown in a high-glucose culture medium. By contrast, CKO cells had decreased ATP levels when cultured in a glucose-free medium, suggesting that enhanced glycolysis in CKO cells is a main pathway for cellular ATP production (Figure 4G). Therefore, CKO mice exhibited elevated levels of HK2—a key enzyme in glycolysis (Figure 1H). Overall, mitoribosomal defects in CKO mice enhanced OXPHOS dysfunction because of the loss of OXPHOS subunits, which could stimulate proximal tubular apoptosis (Figure 2E). However, glycolytic pathways were increased in CKO mice, as increased expression of HK2 (Figure 1H) and the ATP assay (Figure 4G) show. In CKO mice, serum glucose levels did not differ from those in Cont mice (Figure 1F). However, the glycolytic pathway may compensate for the reduced ATP production in kidneys, as the data from the high-glucose medium culture (Figure 4G) show. Our results demonstrate that mitoribosomal deficiency led to mitochondrial OXPHOS dysfunction (Figure 4H).
Effects of Pck1 Overexpression in the PTs on Proteinuria
We examined the protective role of Pck1 overexpression in the PTs in DN, and we created PT-specific Pck1 overexpressing (Pck1 TG) mice (Figure 5, A and B). We compared kidney phenotypes of the four experimental groups: WT littermate mice treated with normal saline (WT+Sal), WT mice treated with STZ (WT+STZ), Pck1 TG+Sal mice, and Pck1 TG+STZ mice (Figure 5C). Immunofluorescence revealed lower Pck1 levels in WT+STZ mice than in WT+Sal mice and significantly higher in Pck1 TG+Sal and Pck1 TG+STZ mice than in WT+Sal mice (Figure 5D). RT-PCR for Pck1 (Figure 5E) had similar results.
Figure 5.

Generation and antialbuminuric phenotypes of TG mice. (A) Constructs used for generating TG mice. A fragment composed of the Npt2 promoter, murine Pck1 cDNA, and RBG poly(A) sequences. (B) Southern blotting shows copies of the Pck1 transgene in mice. Arrows indicate bands corresponding to transgene-derived Pck1. (C) A schematic illustration of the STZ-induced diabetic model procedure. Eight-week-old WT or TG mice were stimulated with STZ (50 mg/kg per day, i.p.) or saline for 5 consecutive days. (D) Representative immunofluorescence images from kidney cryosections derived from each experimental group: WT (wild-type mice)+Sal (saline), WT+STZ, TG+Sal, and TG+STZ mice at age 32 weeks. These sections were stained by immunofluorescence for Pck1 (green) and AQP1 (red). N=3. (E) The real-time PCR reveals the presence of Pck1 in all four groups of mice. N=3. (F) Temporal changes in mean plasma glucose concentrations in mice from each group. We examined the mice 4, 8, 12, 16, 20, 24, and 28 weeks after the treatment, corresponding to age 8, 12, 16, 20, 24, 28, and 32 weeks, respectively. N=7 mice per group. (G) Body weight changes from 0 to 24 weeks after STZ or saline treatment in WT or TG mice. N=7 mice per group. (H) Representative images of sections immune-stained with HK2 in the kidneys from each group of mice at age 32 weeks. Scale bar, 50 μm. The right panel shows the proportional staining areas for HK2. N=7. The right schematic shows the metabolic map where Pck1 was upregulated and HK2 was downregulated in TG mice. All data are shown as mean±SEM. Statistically significant differences in each group are represented by horizontal bars. *P < 0.05.
Serum glucose levels did not differ between WT+Sal and TG+Sal mice or WT+STZ and TG+STZ mice at age 8, 12, 16, 20, 24, 28, and 32 weeks (Figure 5F). No significant difference in body weight was observed between WT+Sal and TG+Sal mice or WT+STZ and TG+STZ mice at age 8, 16, 24, and 32 weeks (Figure 5G). HK2 immunostaining levels were significantly lower in Pck1 TG+Sal mice than in WT+Sal mice. In addition, immunostaining revealed that in contrast to TG+STZ mice, WT+STZ mice showed higher levels of HK2 than WT+Sal mice (Figure 5H).
Serum creatinine levels were unaltered in all four experimental groups (Figure 6A). However, TG+STZ mice showed reduced diabetic albuminuria compared with WT+STZ mice, indicating that Pck1 has antialbuminuric effects in the PTs (Figure 6B). SDS-PAGE analysis of urine protein excretion further demonstrated that TG had a protective effect on albuminuria (Figure 6C). Consistent with this, RT-PCR assays revealed that albumin reuptake molecules such as megalin, cubilin, and amnioless in the PT were retained in TG+STZ mice (Supplemental Figure 5). On the other hand, in CKO or WT+STZ mice, levels of these molecules were reduced (Supplemental Figure 5), which could result in albumin casts in tubular lumens and concomitant albuminuria.
Figure 6.

Antiapoptotic and antifibrotic phenotypes of TG mice. (A) Serum creatinine levels in each group of mice at age 32 weeks. N=7. (B) Urinary albumin excretion in the four groups of mice at age 32 weeks. N=7. (C) SDS-PAGE of mouse urine samples. Urine samples of the four groups of mice at age 32 weeks were subjected to a 15% SDS-PAGE before staining with Coomassie blue. N=2 mice per group. (D) Representative images of albumin staining. Arrows indicate albumin casts. The right panel depicts the relative staining intensities. N=7 mice per group. (E) Apoptotic tubular cells detected by TUNEL staining. Arrows indicate apoptotic cells. The bar graph illustrates the quantitative analysis of apoptotic tubular cells. N=7 mice per group. (F) Representative photomicrographs showing collagen IV immunostaining in each group. The bar graph illustrates the quantitative analysis of collagen IV staining. N=7 per mice group. (G) Representative photomicrographs showing TGF-β immunostaining in each group. The bar graph illustrates the quantitative analysis of TGF-β staining. N=7 per mice group. (D, E, F, and G) Light micrograph; scale bar=100 μm. Kidney tissue specimens for immunostaining were obtained from the four groups of mice at age 32 weeks. All data are shown as mean±SEM. Statistically significant differences in each group are represented by horizontal bars. *P < 0.05.
Protective Effects of Pck1 Overexpression in the PTs on Renal Fibrotic Changes
To identify the underlying mechanism of the protective effects of Pck1 overexpression on albuminuria, we used an anti–albumin-specific antibody to examine changes in albumin reabsorption in PT cells. In contrast to TG+STZ mice, WT+STZ mice had reduced albumin staining in the intracellular tubular area, indicating reduction in albumin reabsorption. Formation of intratubular albumin casts was elevated in WT+STZ mice, but this was blocked in TG+STZ mice (Figure 6D).
TUNEL staining revealed that the number of positively stained apoptotic cells was higher in the tubular regions in WT+STZ mice than in WT+Sal mice, and the number of these cells was reduced in TG+STZ mice. Therefore, PT-specific Pck1 overexpression prevented damage from PT apoptosis in DN (Figure 6E). Additional data of double immunofluorescence for TUNEL and AQP-1 demonstrated origin of apoptotic cells in the PT (Supplemental Figures 12–15).
The levels of TGF-β deposition were significantly higher in WT+STZ mice than in WT+Sal mice, but these changes were attenuated in TG+STZ mice (Figure 6F). Similar findings applied to deposition of type IV collagen (Figure 6G).
Immunostaining of tissue inhibitor metalloproteinase-1 revealed higher expression in WT+STZ mice than in WT+Sal mice (Supplemental Figure 6). Our results demonstrate that PCK1 in the PTs protects against STZ-induced renal peritubular fibrotic changes and proximal tubular cell apoptosis, in addition to reducing STZ-induced albuminuria.
Mitochondrial Ribosome Protection and CRIF1 Upregulation are Responsible for Protecting Peritubular Fibrotic Changes in Diabetes by Pck1 Overexpression
EM images of mitochondria indicate fewer tiny black spots corresponding to mitoribosomes in WT+STZ mice than in TG+STZ mice (Figure 7A), and the lower density corresponds to the gray appearance of the organelles. By contrast, the mitochondria seemed black in TG+STZ mice (Figure 7B). The number of dense black spots in the cytoplasm, indicating cytoribosomes, was not altered in WT+STZ mice compared with TG+STZ mice. Figure 7B shows expanded images of Figure 7A. We confirmed the results of RT-PCR analysis for correctly measuring the number of mitoribosomes by counting MRPs, as described in the following results (Figure 8).
Figure 7.
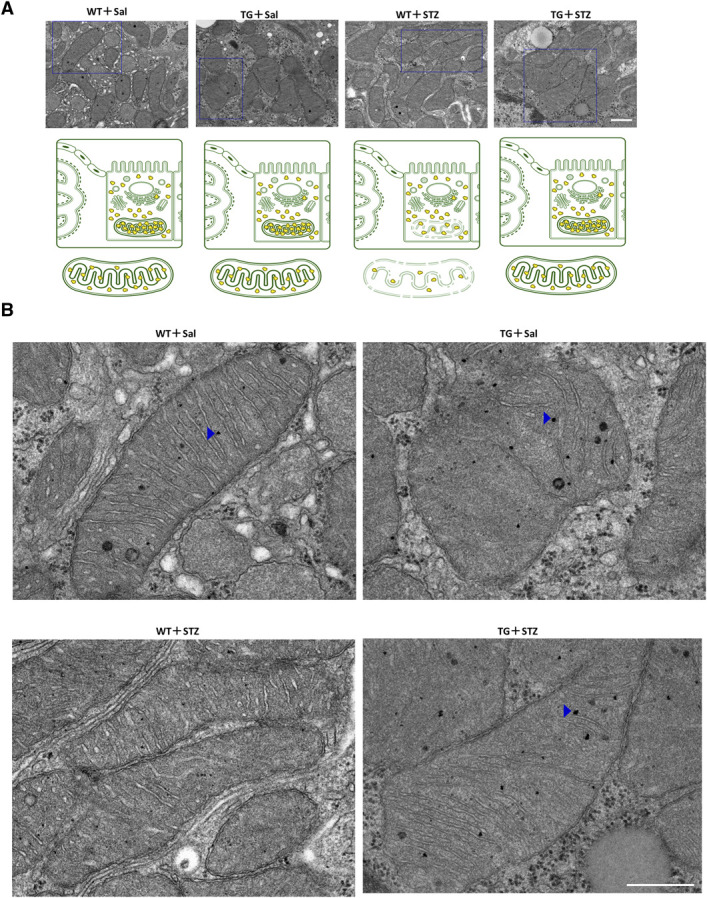
Mitoribosomal protection in TG mice. (A) Representative electron micrograph in each group. Scale bar=500 nm. Blue squares indicate the enlarged regions. Illustration depicts the mitoribosomes in the four groups of mice. (B) Expanded images of Figure 7A. Blue arrowheads indicate mitoribosomes. Scale bar=500 nm. For EM, the kidney tissue specimens were embedded in Epon epoxy resin. Electron micrographs of ten PTs per kidney were randomly obtained for each mouse to evaluate the morphometry of the PTs.
Figure 8.

Mitochondrial protection in TG mice. (A) Real-time quantitative reverse transcription analysis of renal mRNA levels of OXPHOS subunits encoded by nDNA and mtDNA. (B) Real-time quantitative reverse transcription analysis of the renal mRNA levels of intracellular organelle markers. MRPL13 and MRPS15 are mitoribosomal proteins, whereas VDAC is a mitochondrial protein, and Lamin B is a nuclear protein. (C) The real-time quantitative reverse transcription analysis of renal mRNA levels of CRIF1, a mitochondrial ribosomal synthesis regulator. (A–C) Glyceraldehyde 3-phosphate dehydrogenase was used as a control. The kidneys for RT-PCR were obtained from each group of mice at age 32 weeks. N=3 mice per group. Illustration depicting the dysfunctional mitoribosomes and their concomitant mitochondrial dysfunction in WT+STZ mice. These changes in WT+STZ mice were resisted by TG+STZ mice. (D) OCR of TECs isolated from each group of mice was measured using a Seahorse XF-24 flux analyzer. N=3. (E) The ratio of red/green fluorescence of JC-1 of TECs isolated from each group of mice as a measure of the mitochondrial membrane potential. N=6. (F) Fluorescence of MitoSox of TECs isolated from each group of mice as a measure of mitochondrial ROS levels. N=7. (G) ATP content of TECs isolated from each group of mice. N=7. All data are presented as mean±standard errors of the mean. Horizontal bars indicate statistically significant differences between groups. *P < 0.05. (H) Scheme depicting the new mitoribosomes-mediated mechanism of renal profibrotic changes and tubular injury in DN. Under STZ-induced diabetic conditions, the downregulation of Pck1 increased HK2 expression. Increased HK2 expression decreased the expression of CRIF1, causing mitoribosomal defects. The upregulation of mitoribosomal defects leads to deposition of collagen IV in addition to OXPHOS impairment and tubular injury. TG blocked these changes.
We next investigated whether TG mice could be protected from diabetes-induced mitochondrial ribosome defects. TG mice significantly retained mtDNA-encoded OXPHOS subunits synthesized in mitoribosomes, including COX1 and ND1, whereas the nDNA-encoded OXPHOS subunits synthesized in cytoribosomes, including NDUFA9, FP, UQCRC2, and ATP5A1, remained unchanged (Figure 8A). Because TG retained the mitoribosomal function of synthesizing mt-DNA–encoded OXPHOS subunits, we evaluated MRPs to confirm whether mitoribosomes were maintained. A marked reduction in expression levels of MRPL13 and MRPS15 was observed in WT+STZ mice but was significantly blocked in TG+STZ mice (Figure 8B).
WB experiments confirmed Figure 8B RT-PCR data (Supplemental Figure 17) and were consistent with the PCR data. Supplemental Figures 18–27 show uncropped blots. We also measured the activity of OXPHOS complexes (Supplemental Figure 7). WT+STZ mice exhibited reduced activities of complexes I, III, IV, and V, but not complex II. Major component proteins of complexes I, III, and IV were synthesized in mitoribosomes, and complex II–related proteins were synthesized in cytoplasmic ribosomes. Therefore, the mitoribosome defect in WT+STZ mice reduced the activity of complexes I, III, IV, and V.
To confirm whether MRP synthesis was maintained in TG+STZ mice, we tested levels of CRIF1, which were attenuated in WT+STZ mice but improved in TG+STZ mice (Figure 8C). Mitoribosomal defects were seen in WT+STZ mice and were reversed in TG+STZ mice (Figure 8C). EM analysis (Supplemental Figure 8) indicated GBM thickening and elevated foot process effacement in WT+STZ mice as well as in TG+STZ animals. Therefore, lower albuminuria in TG+STZ mice may indicate mitigation of proximal tubular damage from mitoribosomal protection.
Protected Mitochondrial OXPHOS Function in Pck1 Overexpression
To examine the effect of mitoribosomal function preserved in TG mice on mitochondrial function, we examined OCR, JC-1, MitoSox, and ATP production in primary tubular epithelial cells isolated from the kidneys of four groups of mice: WT+Sal, TG+Sal, WT+STZ, and TG+STZ. The OCR was lower in WT+STZ cells than in WT+Sal cells, and the OCR in TG+STZ cells was conserved at values similar to those of WT+Sal cells. The OCR in WT+STZ did not respond to treatment with chemical inhibitors of the mitochondrial respiratory chain. This suggests the loss of OXPHOS activity in WT+STZ mice under basal conditions (Figure 8D). In contrast to TG+STZ cells, the remarkable decrease in the rate of mitochondrial JC-1 incorporation (Figure 8E) in WT+STZ cells suggests that the mitochondria are depolarized and dysfunctional. WT+STZ cells exhibited higher mitochondrial superoxide levels (Figure 8F), which were also retained in TG+STZ cells. However, within 4 days after selection, the ATP level in WT+STZ cells and WT+Sal cells grown in a high-glucose medium were the same. By contrast, WT+STZ cells had decreased ATP levels when grown in a glucose-free medium, suggesting that enhanced glycolysis in WT+STZ cells is a key pathway for cellular ATP production (Figure 8G). Furthermore, the reduction in ATP levels in a glucose-free medium of WT+STZ cells was reversed in TG+STZ cells. These results were consistent with those indicating elevated levels of HK2 in WT+STZ mice. However, these levels were suppressed in TG+STZ mice (Figure 5H). Therefore, a mitoribosomal defect disrupted OXPHOS function in WT+STZ cells, but it was reversed in TG+STZ cells (Figure 8H).
We also assessed glycolysis levels with ECAR assays using primary tubular cells from each group of mice (Supplemental Figure 9). Cells from WT+Sal responded to blocked mitochondrial respiration (treatment with oligomycin) by raising glycolytic flux. By contrast, Pck1-lacking cells from WT+STZ had elevated glycolysis already at baseline that did not increase after blocking OXPHOS. These outcomes indicate that cells without Pck1 rely on glycolysis rather than OXPHOS for energy generation.
In summary, Pck1 expression level was downregulated in mice with STZ-induced DN. Pck1 downregulation elevated HK2 expression, leading to mitochondrial ribosome defects. Mitoribosomal dysfunction elevated TGF-β levels and impaired OXPHOS. TGF-β induced type IV collagen deposition in Pck1 CKO and WT+STZ mice (Figure 4H). To the best of our knowledge, this is the first report on the novel roles of Pck1 in mitoribosomal function.
Discussion
Nephropathy caused by STZ-induced diabetes is associated with reduced expression of Pck1 in the PT. We investigated the role of Pck1 deficiency in PT-specific Pck1-knockout (CKO) mice with mitoribosomal defects and increased albuminuria. PT-specific Pck1 TG mice retained mitoribosomal function and attenuated albuminuria.
HK2 levels increased in CKO mice. This reciprocal change was also observed in mice with hepatic Pck1 overexpression,20 which suppressed HK2. However, the mechanisms are still unknown. Future studies will need to further examine how renal Pck1 downregulation causes HK2 upregulation.
Measurement of ECAR indicated increased glycolysis in step with increased HK2 levels in WT+STZ and CKO mice. However, mitochondrial inhibitors had no effect on glycolytic capacity in the mice because mitoribosomal dysfunction was implicated.
Ribosomes are in the cytoplasm (cytoribosomes) as well as in mitochondria (mitoribosomes).19 A novel finding is that Pck1 affects the function of mitoribosomes, which typically synthesize 13 OXPHOS proteins, two rRNAs, and 22 tRNAs (Supplemental Figure 28). Deficiency of the mitoribosome-synthesized OXPHOS complex in CKO mice underlies their phenotype.
The CR6/GADD45-interacting protein CRIF1 was recently reported to regulate the synthesis and insertion of OXPHOS polypeptides.21 Some studies have reported mitoribosomal dysfunctions in Crif1 CKO mice in specific tissues, such as the brain,22 heart,23 intestine,24 adipose tissues,25 skin,26 pancreatic islets,27 renal collecting ducts,28 and podocytes.29 showed. However, to date, PT-specific CRIF1 has not been reported. Our findings reveal that PT CRIF1 plays an important role in mitoribosomal and mitochondrial functions. According to previous reports,21–29 CRIF1 induces MRP synthesis in a translational system. However, CRIF1 induced production of ND1, COX1, MRPL13, and MPPS15 in mRNA in this study. This finding indicates that renal CRIF1 upregulates mitoribosomal biogenesis in a transcriptional manner. However, further studies are needed to elucidate the detailed mechanism. CRIF1 function may be cell type–defined.
In addition to mitoribosomal defects, renal tubular apoptotic changes and renal fibrosis were detected in CKO and STZ-induced DN mice. A recent report stated that mitoribosomal defects increase TGF-β and OXPHOS defects.19,30 TGF-β promotes renal fibrosis and a diseased tubular cell phenotype. CKO and STZ-treated mice showed an increase in mitoribosome defects and TGF-β, which may cause renal fibrosis and tubular apoptotic changes.
This study had limitations. First, we did not show how Pck1 levels are reduced in diabetic STZ-treated mice. Second, although previous reports indicated that Pck1 was upregulated in DN, one recent report indicated that STZ-induced DN downregulates Pck1 expression, consistent with our results.31 The authors of this recent report emphasized that insulin is not only a regulator of renal gluconeogenesis.31
In STZ-treated mice, Pck1 levels were relatively low in the kidneys, probably because of the STZ-induced tubular toxicity.32,33 Future studies are required to determine whether other murine models with T1DM also downregulate Pck1. Possible detection of the low levels of Pck1 in these T1DM models would indicate that T1DM-specific molecular changes, besides STZ-induced toxicity, may also affect Pck1 levels. This study was limited by the use of STZ and db/db models.
Pck1 levels decreased in the CKO-induced and STZ-induced mice, leading to HK2 upregulation, mitoribosomal downregulation, and concomitant renal injury (Supplemental Figure 3B). CKO mice showed mitoribosomal defects and renal damage under basal conditions without a diabetic milieu, indicating that Pck1 deficiency and mitoribosomal defects are important factors in renal injury. The phenotypes of CKO mice were similar to those of the STZ-treated mice, suggesting the effect of Pck1 deficiency on the pathophysiology of T1DM-induced renal ECM deposition and albuminuria. Furthermore, TG mice were protected against renal damage from STZ-induced T1DM. Therefore, overexpression of Pck1 may be a potential novel therapeutic target for T1DM-induced renal injury. Further studies are needed to assess utility of Pck1 in other T1DM models.
Our data also showed that db/db-induced T2DM elevates Pck1 and mitigates HK2, increasing the number of mitoribosomes (Supplemental Figure 3B). These molecular changes in T2DM seem to mirror the changes observed in T1DM. Despite the reciprocal changes in the Pck1-HK2-mitoribosomes axis in T1DM and T2DM, Pck1 regulates HK2, which further regulates mitoribosomes.
Pck1 deficiency in CKO mice increased the expression of HK2, whereas db/db decreased expression (Supplemental Figure 3C). However, CKO-db/db did not alter levels of HK2 and renal injury. This finding has two implications. First, other regulatory mechanisms more effectively downregulate HK2 expression in T2DM than Pck1-mediated HK2 regulation. Second, other highly effective mechanisms affect renal damage in T2DM. Therefore, the Pck1-HK2-mitoribosome axis is not able to account for db/db-induced renal injury. However, the results remain inconclusive. Future studies are required to assess whether other T2DM models with insulin deficiency exhibit low Pck1 levels, high HK2 levels, and mitoribosomal defects and whether renal injury can be prevented by TG or augmented by CKO.
Two recent studies address the role of HK2 in the PTs in enhancing aberrant glycolysis in renal pathophysiology.34,35 HK2 in PTs was elevated in murine DN models. PKM2 in aberrant glycolysis is important in driving renal fibrosis in diabetes.35 Therefore, HK2 is important in aberrant or elevated glycolysis and PKM2 under diabetic profibrotic conditions.
This investigation demonstrated the association of Pck1 downregulation with mitoribosomal defects. Interventions targeting this novel pathway may provide a therapeutic strategy against DN.
Supplementary Material
Disclosures
All authors have nothing to disclose.
Funding
This work was supported by the Scientific Research Fund of the Ministry of Education, Culture, Sports, Science, and Technology of Japan (Grant no. 22K0835400).
Author Contributions
Conceptualization: Kazuhiro Hasegawa, Yusuke Sakamaki.
Data curation: Kazuhiro Hasegawa, Masanori Tamaki.
Formal analysis: Kazuhiro Hasegawa.
Funding acquisition: Shu Wakino.
Investigation: Kazuhiro Hasegawa, Yusuke Sakamaki.
Methodology: Kazuhiro Hasegawa, Yusuke Sakamaki.
Project administration: Shu Wakino.
Resources: Masanori Tamaki.
Supervision: Yusuke Sakamaki.
Validation: Yusuke Sakamaki.
Visualization: Masanori Tamaki.
Writing – original draft: Kazuhiro Hasegawa.
Writing – review & editing: Masanori Tamaki, Shu Wakino.
Data Sharing Statement
The data that support the findings of this study are available in the methods of this article. Further information and requests for resources and reagents are available from the corresponding author (kazuhiro@tokushima-u.ac.jp).
Supplemental Material
This article contains the following supplemental material online at http://links.lww.com/JSN/E433.
Supplemental Figure 1. Effects of Pck1 CKO on urinary albuminuria in db/db mice.
Supplemental Figure 2. Renal phenotypes in proximal tubule-specific Pck1 CKO-db/db mice.
Supplemental Figure 3. HK2 expression in Pck1 CKO-db/db.
Supplemental Figure 4. Mitoribosomal phenotypes in Pck1 CKO-db/db.
Supplemental Figure 5. Changes in albumin uptake-related molecules in Pck1 CKO and TG.
Supplemental Figure 6. The role of TIMP-1 in diabetic fibrotic changes in Pck1 deficiency.
Supplemental Figure 7. Measurement of mitochondrial ETC complex enzyme activities in the kidneys of CKO and TG mice.
Supplemental Figure 8. Glomerular histological changes in CKO and TG mice.
Supplemental Figure 9. Glycolysis assay in CKO and TG mice.
Supplemental Figures 10–15. Apoptotic cells in proximal tubule of CKO and TG.
Supplemental Figure 16. Reduced mitoribosomes in CKO mice.
Supplemental Figure 17. Reduced mitoribosomes in WT+STZ mice.
Supplemental Figures 18–27. Uncropped blots of Supplemental Figures 16 and 17.
Supplemental Figure 28. Gene map depicting mit-DNA–encoded genes.
References
- 1.Hasegawa K, Wakino S, Simic P, Sakamaki Y, Minakuchi H, Fujimura K. Renal tubular Sirt1 attenuates diabetic albuminuria by epigenetically suppressing Claudin-1 overexpression in podocytes. Nat Med. 2013;19(11):1496–1504. doi: 10.1038/nm.3363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hasegawa K. Novel tubular-glomerular interplay in diabetic kidney disease mediated by sirtuin 1, nicotinamide mononucleotide, and nicotinamide adenine dinucleotide Oshima Award Address 2017. Clin Exp Nephrol. 2019;23(8):987–994. doi: 10.1007/s10157-019-01719-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Muraoka H, Hasegawa K, Sakamaki Y, Minakuchi H, Kawaguchi T, Yasuda I. Role of Nampt-Sirt6 axis in renal proximal tubule in extracellular matrix deposition in diabetic nephropathy. Cell Rep. 2019;27(1):199–212.e5. doi: 10.1016/j.celrep.2019.03.024 [DOI] [PubMed] [Google Scholar]
- 4.Yasuda I, Hasegawa K, Sakamaki Y, Muraoka H, Kawaguchi T, Kusahana E. Pre-emptive short-term nicotinamide mononucleotide treatment in a mouse model of diabetic nephropathy. J Am Soc Nephrol. 2021;32(6):1355–1370. doi: 10.1681/ASN.2020081188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bukosza EN, Kratochwill K, Kornauth C, Schachner H, Aufricht C, Gebeshuber CA. Podocyte RNA sequencing reveals Wnt- and ECM-associated genes as central in FSGS. PLoS One. 2020;15(4):e0231898. doi: 10.1371/journal.pone.0231898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gong Y, Sunq A, Roth RA, Hou J. Inducible expression of claudin-1 in glomerular podocytes generates aberrant tight junctions and proteinuria through slit diaphragm destabilization. J Am Soc Nephrol. 2017;28(1):106–117. doi: 10.1681/ASN.2015121324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miguel V, Tituaña J, Herrero JI, Herrero L, Serra D, Cuevas P. Renal tubule Cpt1a overexpression protects from kidney fibrosis by restoring mitochondrial homeostasis. J Clin Invest. 2021;131(5):e140695. doi: 10.1172/JCI140695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Umino H, Hasegawa K, Minakuchi H, Muraoka H, Kawaguchi T, Kanda T. High basolateral glucose increases sodium-glucose cotransporter 2 and reduces sirtuin-1 in renal tubule through glucose transporter-2 detection. Sci Rep. 2018;8(1):6791. doi: 10.1038/s41598-018-25054-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Packer MJ. Role of ketogenic starvation sensors in mediating the renal protective effects of SGLT2 inhibitors in type 2 diabetes. J Diabetes Complications. 2020;34(9):107647. doi: 10.1016/j.jdiacomp.2020.107647 [DOI] [PubMed] [Google Scholar]
- 10.Fernandes R. The controversial role of glucose in the diabetic kidney. Porto Biomed J. 2021;6(1):e113. doi: 10.1097/j.pbj.0000000000000113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu L, Yin Q, Irwin DM, Zhang S. Phosphoenolpyruvate carboxykinase 1 gene (Pck1) displays parallel evolution between old world and new world fruit bats. PLoS One. 2015;10(3):e0118666. doi: 10.1371/journal.pone.0118666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Y, Guan Q, Liu Y, Zhang Y, Chen Y, Chen J. Regulation of hepatic gluconeogenesis by nuclear factor Y transcription factor in mice. J Biol Chem. 2018;293(20):7894–7904. doi: 10.1074/jbc.ra117.000508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beale EG, Harvey BJ, Forest C. PCK1 and PCK2 as candidate diabetes and obesity genes. Cell Biochem Biophys. 2007;48(2-3):89–95. doi: 10.1007/s12013-007-0025-6 [DOI] [PubMed] [Google Scholar]
- 14.Liu Q, Zhang L, Zhang W, Hao Q, Qiu W, Wen Y. Inhibition of NF-κB reduces renal inflammation and expression of PEPCK in Type 2 diabetic mice. Inflammation. 2018;41(6):2018–2029. doi: 10.1007/s10753-018-0845-0 [DOI] [PubMed] [Google Scholar]
- 15.Wilson PC, Wu H, Kirita Y, Uchimura K, Ledru N, Rennke HG. The single-cell transcriptomic landscape of early human diabetic nephropathy. Proc Natl Acad Sci U S A. 2019;116(39):19619–19625. doi: 10.1073/pnas.1908706116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu H, Takagaki Y, Kumagai A, Kanasaki K, Koya D. The PKM2 activator TEPP-46 suppresses kidney fibrosis via inhibition of the EMT program and aberrant glycolysis associated with suppression of HIF-1α accumulation. J Diabetes Investig. 2021;12(5):697–709. doi: 10.1111/jdi.13478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Amikura R, Kashikawa M, Nakamura A, Kobayashi S. Presence of mitochondria-type ribosomes outside mitochondria in germ plasm of Drosophila embryos. Proc Natl Acad Sci U S A. 2001;98(16):9133–9138. doi: 10.1073/pnas.171286998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Watson K. The organization of ribosomal granules within mitochondrial structures of aerobic and anaerobic cells of Saccharomyces cerevisae. J Cell Biol. 1972;55(3):721–726. doi: 10.1083/jcb.55.3.721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kwon SM, Lee YK, Min S, Woo HG, Wang HJ, Yoon G. Mitoribosome defect in hepatocellular carcinoma promotes an aggressive phenotype with suppressed immune reaction. iScience. 2020;23(6):101247. doi: 10.1016/j.isci.2020.101247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tang Y Zhang Y Wang C, et al. Overexpression of pck1 gene antagonizes hepatocellular carcinoma through the activation of gluconeogenesis and suppression of glycolysis pathways. Cell Physiol Biochem. 2018;47(1):344–355. doi: 10.1159/000489811 [DOI] [PubMed] [Google Scholar]
- 21.Kim SJ, Kwon MC, Ryu MJ, Chung HK, Tadi S, Kim YK. CRIF1 is essential for the synthesis and insertion of oxidative phosphorylation polypeptides in the mammalian mitochondrial membrane. Cell Metab. 2012;16(2):274–283. doi: 10.1016/j.cmet.2012.06.012 [DOI] [PubMed] [Google Scholar]
- 22.Byun J, Son SM, Cha MY, Shong M, Hwang YJ, Kim Y. CR6-interacting factor 1 is a key regulator in Aβ-induced mitochondrial disruption and pathogenesis of Alzheimer’s disease. Cell Death Differ. 2015;22(6):959–973. doi: 10.1038/cdd.2014.184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ryu MJ, Kim SJ, Choi MJ, Kim YK, Lee MH, Lee SE. Mitochondrial oxidative phosphorylation reserve is required for hormone- and PPARγ agonist-induced adipogenesis. Mol Cells. 2013;35(2):134–141. doi: 10.1007/s10059-012-2257-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ryu MJ, Kim SJ, Kim YK, Choi MJ, Tadi S, Lee MH. Crif1 deficiency reduces adipose OXPHOS capacity and triggers inflammation and insulin resistance in mice. PLoS Genet. 2013;9(3):e1003356. doi: 10.1371/journal.pgen.1003356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shin J, Lee SH, Kwon MC, Yang DK, Seo HR, Kim J. Cardiomyocyte specific deletion of Crif1 causes mitochondrial cardiomyopathy in mice. PLoS One. 2013;8(1):e53577. doi: 10.1371/journal.pone.0053577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shin JM, Choi DK, Sohn KC, Kim JY, Im M, Lee Y. Targeted deletion of Crif1 in mouse epidermis impairs skin homeostasis and hair morphogenesis. Sci Rep. 2017;7(1):44828. doi: 10.1038/srep44828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim YK, Joung KH, Ryu MJ, Kim SJ, Kim H, Chung HK. Disruption of CR6-interacting factor-1 (CRIF1) in mouse islet beta cells leads to mitochondrial diabetes with progressive beta cell failure. Diabetologia. 2015;58(4):771–780. doi: 10.1007/s00125-015-3506-y [DOI] [PubMed] [Google Scholar]
- 28.Jeong JY Na KR Shin JA, et al. Collecting duct-specific CR6-interacting Factor-1-deletion aggravates renal inflammation and fibrosis induced by unilateral ureteral obstruction. Int J Mol Sci. 2021;22(21):11699. doi: 10.3390/ijms222111699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Na KR, Jeong JY, Shin JA, Chang YK, Suh KS, Lee KW. Mitochondrial dysfunction in podocytes caused by CRIF1 deficiency leads to progressive albuminuria and glomerular sclerosis in mice. Int J Mol Sci. 2021;22(9):4827. doi: 10.3390/ijms22094827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee YK, Lim JJ, Jeoun UW, Min S, Lee EB, Kwon SM. Lactate-mediated mitoribosomal defects impair mitochondrial oxidative phosphorylation and promote hepatoma cell invasiveness. J Biol Chem. 2017;292(49):20208–20217. doi: 10.1074/jbc.m117.809012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sasaki M, Sasako T, Kubota N, Sakurai Y, Takamoto I, Kubota T. Dual regulation of gluconeogenesis by insulin and glucose in the proximal tubule of the kidney. Diabetes. 2017;66(9):2339–2350. doi: 10.2337/db16-1602 [DOI] [PubMed] [Google Scholar]
- 32.Tay YC, Wang Y, Kairaitis L, Rangan GK, Zhang C, Harris DC. Can murine diabetic nephropathy be separated from superimposed acute renal failure? Kidney Int. 2005;68(1):391–398. doi: 10.1111/j.1523-1755.2005.00405.x [DOI] [PubMed] [Google Scholar]
- 33.Brouwers B, Pruniau VPEG, Cauwelier EJG, Schuit F, Lerut E, Ectors N. Phlorizin pretreatment reduces acute renal toxicity in a mouse model for diabetic nephropathy. J Biol Chem. 2013;288(38):27200–27207. doi: 10.1074/jbc.m113.469486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cui X Shi E Li J, et al. GPR87 promotes renal tubulointerstitial fibrosis by accelerating glycolysis and mitochondrial injury. Free Radic Biol Med. 2022;189:58–70. doi: 10.1016/j.freeradbiomed.2022.07.004 [DOI] [PubMed] [Google Scholar]
- 35.Li J, Liu H, Takagi S, Nitta K, Kitada M, Srivastava SP. Renal protective effects of empagliflozin via inhibition of EMT and aberrant glycolysis in proximal tubule. JCI Insight. 2020;5(6):e129034. doi: 10.1172/jci.insight.129034 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available in the methods of this article. Further information and requests for resources and reagents are available from the corresponding author (kazuhiro@tokushima-u.ac.jp).