

Abstract
Generating artificial protein sequences using artificial intelligence could enable breakthrough solutions for biomedical and environmental challenges. Viewing amino acid sequences as a language, we demonstrate that a deep learning-based language model can generate functional artificial protein sequences across large protein families, akin to generating grammatically and semantically correct natural language sentences on diverse topics. Our protein language model is trained by simply learning to predict the next amino acid for over 280 million protein sequences from thousands of protein families, without explicit biophysical modeling. We experimentally evaluate model-generated artificial proteins fine-tuned to five distinct antibacterial lysozyme families. Artificial proteins show similar activities and catalytic efficiencies as representative natural lysozymes, including hen egg white lysozyme, while maintaining activity with as low as 31.4% identity to any known naturally-evolved protein. The X-ray crystal structure of an enzymatically active artificial protein recapitulates the conserved fold and positioning of active site residues found in natural proteins. We show our language model’s ability to be adapted to different protein families by accurately predicting the functionality of artificial chorismate mutase and malate dehydrogenase proteins. These results indicate that neural language models successfully perform artificial protein generation across protein families and may prove to be a tool to shortcut evolution.
Protein engineering can have a transformative impact on biomedicine, environmental science, and nanotechnology, among other fields. Traditional evolutionary protein engineering methods perform iterative screening/selection to identify proteins with desired functional and structural properties. In contrast, rational or de novo protein design methods aim to create artificial proteins, with the goal of improved efficiency and precision. Structure-based de novo design methods1–5 employ biophysical principles to design proteins that will fold into a desired structure using simulation-based methods and statistical analysis of experimental data. Coevolutionary methods6–10 build statistical models from evolutionary sequence data to specify novel sequences with desired function or stability. Both structural and coevolutionary approaches are not without limitations. Structural methods rely on scarce experimental structure data and difficult or intractable biophysical simulations3,11. Coevolutionary statistical models are tailored to specific protein families, frequently rely on multiple sequence alignments, and struggle to operate in space outside of the defined multiple sequence alignment11. Recently, deep neural networks have shown promise as generative and discriminative models for protein science and engineering12–20. Their ability to learn complex representations could be essential to effectively utilize an exponentially growing source of diverse and relatively unannotated protein data—public databases containing millions of raw unaligned protein sequences21–23, enabled by the dramatic reduction in sequencing costs.
Inspired by the success of deep learning-based natural language models (LMs) trained on large text corpora that generate realistic text with varied topics and sentiments24–28, we develop ProGen, a protein language model trained on millions of raw protein sequences that generates artificial proteins across multiple families and functions. While prior work has shown that natural language-inspired statistical representations of proteins are useful for protein informatics tasks such as stability prediction, remote homology detection, and secondary structure prediction11,29–31, we show that the latest advances in deep learning-based language modeling can be adopted to generate artificial protein sequences, from scratch, that function as well as natural proteins.
ProGen is iteratively optimized by learning to predict the probability of the next amino acid given the past amino acids in a raw sequence, with no explicit structural information or pairwise coevolutionary assumptions. Trained in this unsupervised manner from a large, varied protein sequence database, ProGen learns a universal, domain-independent representation of proteins that subsumes local and global structure motifs, analogous to natural language models learning semantic and grammatical rules. After training, ProGen can be prompted to generate full-length protein sequences for any protein family from scratch, with a varying degree of similarity to natural proteins. In the common case where some sequence data from a protein family is available, we can utilize the technique of fine-tuning pre-trained language models32–35 with family-specific sequences to further improve ProGen’s ability to capture the distribution of local sequence neighborhoods corresponding to the protein family.
ProGen is a 1.2 billion-parameter neural network trained using a publicly available dataset of 280 million protein sequences. A key component of ProGen is conditional generation28,36–38, i.e., sequence generation controlled by property tags provided as input to the language model. In the case of natural language, these control tags may be style, topics, dates, and other entities (Fig. 1a). For proteins, the control tags are properties such as protein family, biological process, and molecular function, that are available for a large fraction of sequences in public protein databases (Fig. 1b, Fig. S1). By specifying one or more control tags (e.g., Protein Family: Pfam ID PF16754, Pesticin) and none or a few amino acids at the beginning of a sequence, we can significantly constrain the sequence space for generation and improve generation quality, as shown by our prior computational experiments39.
Figure 1: Artificial protein generation with conditional language modeling.
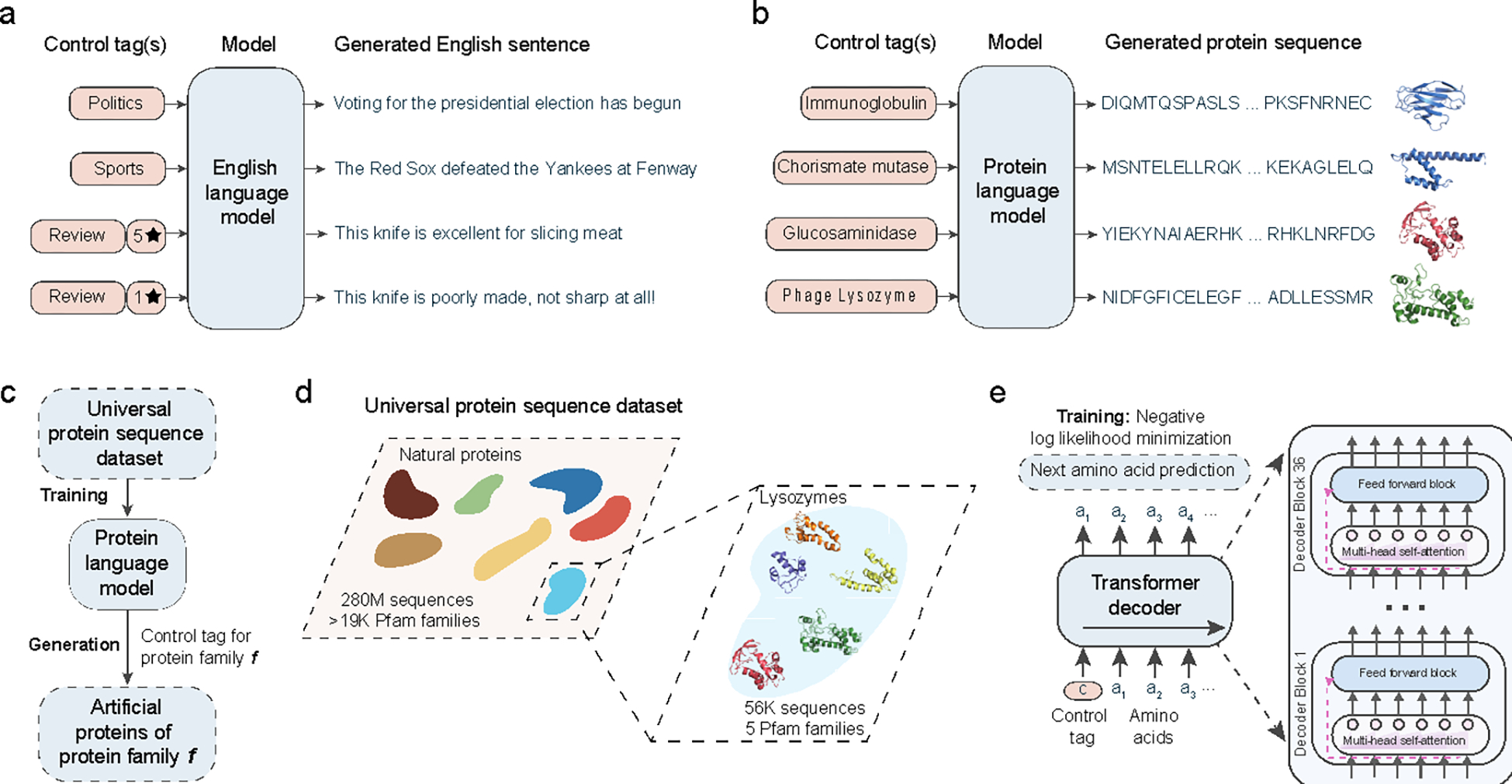
(a) Conditional language models are deep neural networks that can generate semantically and grammatically correct, yet novel and diverse natural language text, steerable using input control tags that govern style, topic, and other entities. Analogous to natural language models, we develop ProGen, a conditional protein language model (b) that generates diverse artificial protein sequences across protein families based on input control tags (c). ProGen is trained using a large, universal protein sequence dataset (d) of 280 million naturally-evolved proteins from thousands of families, of which five diverse lysozyme families are experimentally characterized in this study. ProGen is a 1.2 billion parameter neural network (e) based on the Transformer architecture which utilizes a self-attention mechanism for modeling comprehensive residue-residue interactions. ProGen is trained to generate artificial sequences by minimizing the loss over the next amino acid prediction problem on the universal protein sequence dataset.
Let be a sequence of amino acids that specifies a protein of length appended with an “end of sequence” token. Let be an associated set of descriptors such as protein family or source organism, i.e., ‘control tags’, through which we would like to control generation of amino acid sequences. Let be the sequence formed by prepending a control tag sequence to an amino acid sequence. The probability over such a combined sequence of length is then . Language modeling decomposes the problem of generating into a next-token prediction problem40, where a token can either be an amino acid or a control tag. We train a neural network with parameters to minimize the negative log-likelihood over a dataset
| (Eq. 1) |
A new protein of length with desired properties encoded by a control tag sequence of length can then be generated by sequentially sampling its constituent tokens: 39. Generation continues until the model generates an “end of sequence” token.
We utilize a state-of-the-art transformer-based24 neural network architecture for constructing ProGen. The transformer learns long-range context within sequences using a series of stacked layers, each containing a self-attention mechanism (Fig. 1e). The self-attention mechanism in each layer infers pairwise interaction relationships between all positions in its input sequence. Stacking multiple self-attention layers allows us to learn multiple-residue interactions41. The transformer-based approach has been shown to be related to co-evolutionary methods for sequence design such as MRFs42, Potts models43, and Hopfield networks44. In contrast to transformer-based language models that encode amino acid sequences for discriminative protein prediction tasks30,45,46, ProGen is a decoder transformer tailored for autoregressive generation: it generates a sequence in a left-to-right manner, token-by-token, where the next token is conditioned on all previously generated tokens.
ProGen’s transformer architecture has 36 layers, and 8 self-attention heads per layer and a total of 1.2 billion trainable neural network parameters (see Methods). To train ProGen, we collected a universal protein sequence dataset containing 280 million protein sequences (from >19,000 Pfam23 families) and associated metadata (as control tags) from UniParc21, UniprotKB22, Pfam23, and NCBI taxonomic information47 (Fig. 1d, Table S1). We trained ProGen to minimize the negative log-likelihood defined in Eq. 1 using this dataset with a batch size of 2048 for one million iterations. Training was performed across 256 Google Cloud TPU v3 cores for two weeks. Once trained, ProGen could be used to generate novel protein sequences from scratch by specifying a control tag (e.g. protein family identifier from Pfam), Fig. 1c.
We experimentally evaluated ProGen’s ability to generate functional artificial amino acid sequences by testing its generations from the lysozyme clan23,48. Lysozymes are antibacterial enzymes that disrupt the integrity of the cell wall by catalyzing the hydrolysis of 1,4-beta-linkages between N-acetylmuramic acid and N-acetyl-D-glucosamine residues. Within the lysozyme clan, we chose to work with five families that lack disulfide bonds as to be amenable to high-throughput cell-free expression: Phage lysozyme, Pesticin, Glucosaminidase, Glycoside hydrolase, and Transglycosylase. These lysozyme families contain significant protein sequence diversity (Table S3) with average sequence length varying between 84–167 across families. The sequences also show large structural diversity and multiple structural folds (Fig. S2). As a whole, this represents a challenging design space for a model that is not constrained in generation to local sequence neighborhoods near known functional wild-types and also not provided with structural priors. We collected a dataset of 55,948 sequences from these five families from Pfam and UniprotKB sources for obtaining positive controls and for fine-tuning32–35 ProGen. Fine-tuning involves making limited, computationally inexpensive, gradient updates to the parameters of the trained model (see Methods).
After fine-tuning ProGen using the curated lysozyme dataset, we generated one million artificial sequences using ProGen by providing the Pfam ID for each family as a control tag. Our artificial lysozymes span the sequence landscape of natural lysozymes, Fig. 2a, across five families which contain diverse protein folds, active site architectures, and enzymatic mechanisms49,50. As our model can generate full-length artificial sequences within milliseconds, a large database can be created to expand the plausible sequence diversity beyond natural libraries (Table S3). Although artificial sequences may diverge from natural sequences purely from a sequence identity calculation, (Fig. 2b, Fig. S3), they demonstrate similar residue position entropies when forming separate multiple sequence alignments of natural and artificial proteins within each family, Fig. 2c. This indicates that the model has captured evolutionary conservation patterns without training on explicit alignment information such as with Potts models51, as implemented in direct coupling analysis7,52–55.
Figure 2: Generated artificial antibacterial proteins are diverse and express well in our experimental system.
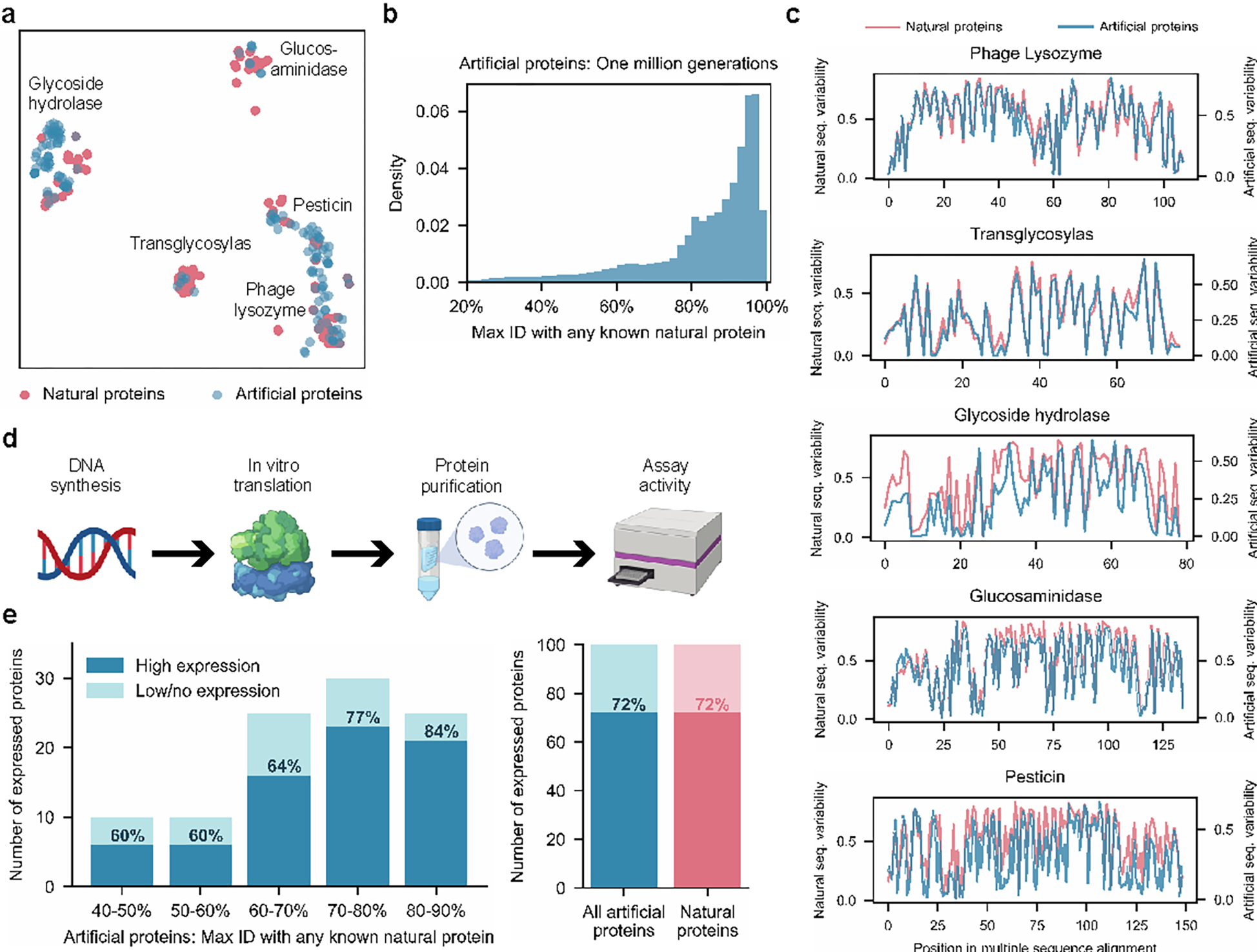
When analyzed using t-SNE as a dimensionality reduction technique for visualization purposes, artificial sequences from our model are shown to span the landscape of natural proteins from five lysozyme families (a). Each point represents a natural or generated sequence embedded in a two-dimensional t-SNE space. With sufficient sampling, ProGen can generate sequences that are highly dissimilar from natural proteins (b). Max ID measures the maximum identity of an artificial protein with any publicly available natural protein. (c) Artificial proteins maintain similar evolutionary conservation patterns as natural proteins across families. Plots demonstrate the variability at each aligned position for a library of proteins. Conserved positions are represented as curve dips. From our generated proteins, we select one hundred proteins for synthesis and characterization in our experimental setup (d). Artificial proteins express well even with increasing dissimilarity from nature (40–50% max ID) and yield comparable expression quality to one hundred representative natural proteins (e).
To experimentally evaluate ProGen performance across a range of sequence divergences from natural proteins, we selected one hundred sequences filtered based on generation quality and diversity to natural sequences, measured as top-hit identities (“max ID”) to any protein in our training dataset containing 280 million proteins, which is primarily composed of UniParc21 (Fig. S4). We first ranked sequences for generation quality using a combination of an adversarial discriminator27,56 and model log-likelihood scoring57 (see Methods) and then sampled from top-ranked sequences to span a range of max IDs between 40–90%. We also ensured that no two generated samples share over 80% identity with each other. Our final selection included 100 artificial sequences (Table S2), with a minimum of 8 proteins from each protein family. The average sequence length for artificial proteins varies between 93–179 across families, comparable to natural lysozymes in our curated dataset from Pfam. Artificial proteins included specific amino acids and pairwise interactions never before observed in aligned positions in lysozyme family-specific alignments. We also selected a positive control group from the 55,948 curated lysozyme sequences. We clustered the natural sequences with MMseqs258 and chose roughly 20 cluster-representative sequences from each of the five families (see Methods).
To evaluate function, full-length genes were synthesized and purified via cell-free protein synthesis and affinity chromatography (see Methods). In the positive control set of 100 natural proteins, 72% were well-expressed based on chromatography peaks and band visualization. The ProGen-generated proteins express equally well (72/100 total) across all bins of sequence identity to any known natural protein (maxID 40–90%), Fig 2e. In addition, we designed artificial proteins using bmDCA7, a statistical model based on direct coupling analysis, which explicitly approximates first and second-order residue dependencies. Starting from their publicly available code, we tried to make the bmDCA model converge on the same sequences as ProGen and using additional alignment information and searched over a wide range of hyperparameters (see Methods). bmDCA was unable to fit three out of the five lysozyme families, and exhibited 60% detectable expression (30/50 proteins) for the remaining two protein families. These results indicate that ProGen can generate artificial proteins that are structurally well-folded for proper expression as compared to a batch of natural proteins, even when sequence alignment size and quality limit the success of alternative approaches.
Next we examined activity based on quench release of fluorescein labeled Micrococcus lysodeikticus cell wall (Molecular Probes EnzChek Lysozyme kit) using 90 randomly chosen proteins out of each expressed set of 100. Proteins were prepared in 96-well plate format to extract fluorescence curves over time, Fig 3a. Hen egg white lysozyme (HEWL), a naturally-evolved exemplar protein, was measured as positive control, in addition to ubiquitin as negative control. Proteins that generated fluorescence one standard deviation above the maximum fluorescence of any negative control were considered functional. Among our artificial proteins, 73% (66/90) were functional and exhibited high levels of functionality across families, Fig. 3c. The representative natural proteins exhibited similar levels of functionality with 59% (53/90) of total proteins considered functional. None of the bmDCA artificial proteins exhibited a detectable level of functionality, Fig. S10, which may be due to convergence, sampling, or other specific model run issues further highlighting the versatility of ProGen providing a potentially more robust alternative. These results indicate that ProGen generates protein sequences that not only can express well but also maintain enzymatic function for diverse sequence landscapes across protein families.
Figure 3: Artificial protein sequences are functional while reaching as low as 44% identity to any known protein, exhibit comparable catalytic efficiencies to a highly-evolved natural protein, and demonstrate similar structures to known natural folds.
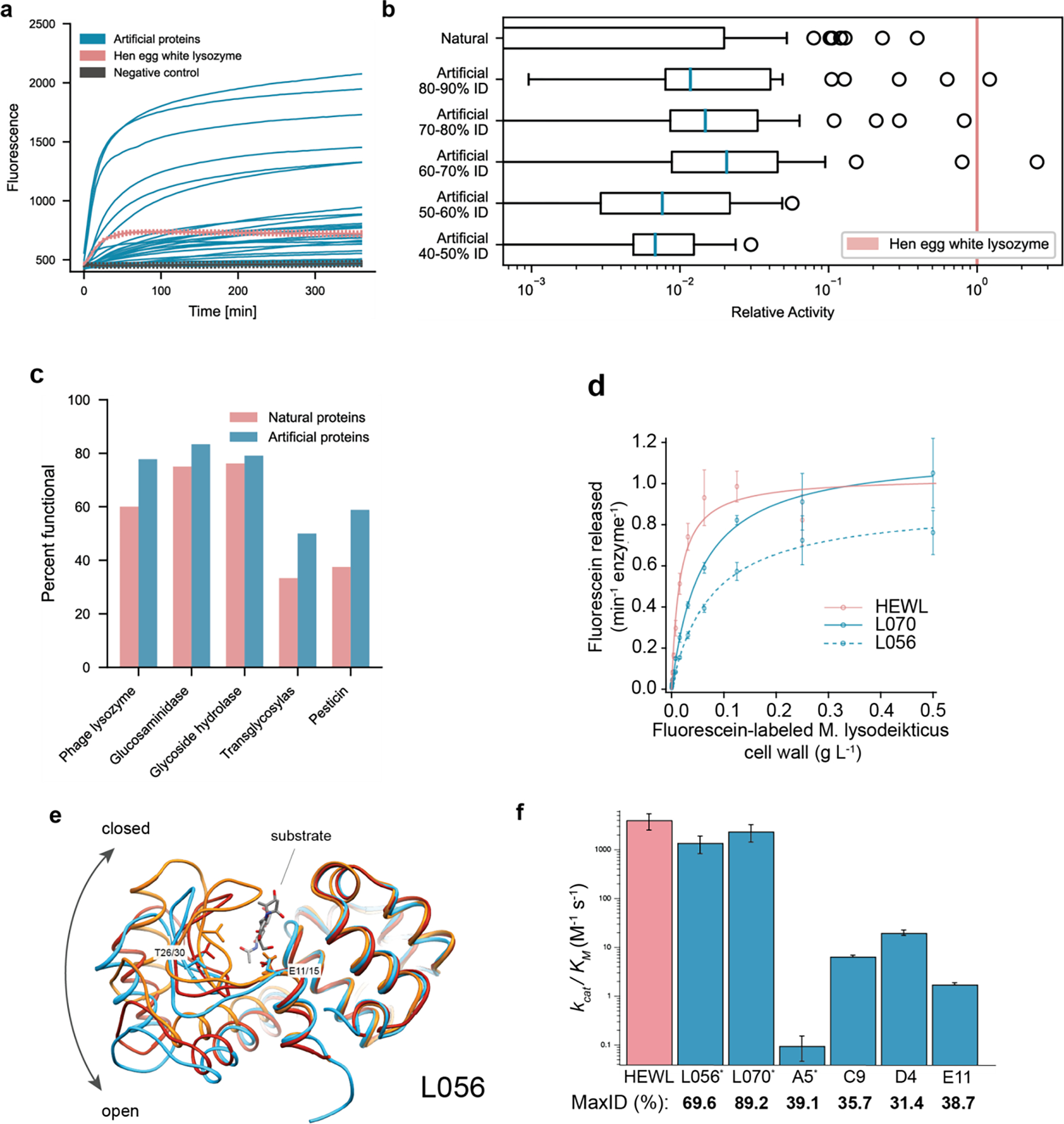
(a) Artificial proteins bind well to substrates and exhibit high fluorescence responses over time. Error bars (minimum and maximum) are shown for hen egg white lysozyme, HEWL, and negative (ubiquitin) controls. (b) Artificial proteins remain active even while being dissimilar (40–50% max ID i.e., top hit-identity) from known natural proteins. Outliers indicate high activity samples where relative activity is computed with respect to HEWL. (c) Artificial proteins are functional across protein families. Functional is defined as a fluorescence one standard deviation above the maximum value of all negative controls. (d) Michaelis-Menten kinetics of HEWL natural lysozyme (red) and two generated lysozymes (blue; L056 and L070) against cell-wall substrate show comparable performance ( technical replicates). (e) We determined a 2.5 Å resolution crystal of L056 artificial lysozyme. A global overlay of L056 crystal structure with two representative T4 lysozyme conformations is shown with L056 presented in sky blue, ‘open’ conformation of M6I T4 lysozyme (PDB:150L) in dark red, ‘closed’ conformation of wild-type T4 lysozyme (PDB:3FA0) in orange, and substrate (PDB:148L) colored by element. Catalytic threonine (T30 in L056 and T26 in T4 lysozyme) and first catalytic glutamate (E15 in L056 and E11 in T4 lysozyme) are represented as sticks. (f) Michaelis-Menten constants derived for lysozyme variants demonstrating a range of catalytic activities across variants of varied maximal sequence IDs to known natural protein. Error bars represent propagated standard deviations (). * denotes derived from initial rate analysis and unit converted (Table S4).
In addition to a binary value for functionality, we calculated a relative activity score with respect to HEWL for the in vitro assay (see Methods). Our artificial proteins match activity levels of natural proteins even at lower levels of sequence identity to any known natural protein, (Fig 3b, Fig. S5). Notably a small number of proteins, both within the natural and artificial proteins, were within an order of magnitude of HEWL, which was substantially more active than all negative controls. These highly active outliers demonstrate the potential for our model to generate sequences that may rival natural proteins that have been highly optimized through evolutionary pressures.
From the 100 artificial proteins, we selected five proteins that spanned a wide range of max IDs (48–89%) to recombinantly express in E. coli (see Methods). Of these, only one, L008, generated no detectable expression (Fig. S6). Two (L013 and L038) expressed robustly to inclusion bodies and were not pursued further. Two proteins, L056 (max ID 69.6%) and L070 (max ID 89.2%) expressed well and incurred bactericidal activities towards the E. coli BL21(DE3) strain used during overnight induction at 16C. Spent media harbored enzymatic activity (see Methods), therefore, enzymes were purified from this material.
While both enzymes purified as monomers at the expected size by size exclusion chromatography, we also observed a defined later eluting (apparent lower molecular weight, likely due to binding the dextran component of the column) species for each enzyme that corresponded to full-length enzyme by SDS-PAGE (Fig. S6). The values of both monomers were too weak to be measured using a heterogeneous, fluorescein-labeled Micrococcus lysodeikticus cell wall substrate (Molecular Probes EnzChek Lysozyme kit); however, both monomers were active using a pseudo-first order kinetic assay (Fig. S7). In contrast, we could readily measure the values for the purified apparent lower molecular weight species, where both L056 and L070 were highly active and had comparable Michaelis-Menten parameters to HEWL (Fig. 3e). Taken together, L056 and L070 harbor potent catalytic activity and bactericidal capabilities that are comparable to HEWL, while diverging from their nearest known natural sequence by 53 and 18 amino acids respectively. We also found that there is no bias to location or structural element to the mutations that diverge L056 and L070 from their respective nearest sequence homolog in nature. Differing residues are instead uniformly distributed. Some mutations are even found within the active site cleft and within regions that influence conformational state (for L056). Despite having comparable enzymatic activities, L070 and L056 only share 17.9% sequence ID. In sum, these results demonstrate that ProGen can generate artificial proteins with near native activity.
Next, we examined the structural divergence of the artificial proteins. We determined a 2.5 Å resolution crystal of L056 (see Methods, Figure 3e). The global fold was similar to predictions, with a Cα RMSD of 2.9 Å from the backbone structure predicted by trRosetta and 2.3 Å RMSD from a WT T4 Lysozyme structure59,60. The largest structural divergence occurs in the beta hairpin comprising residues 18–31. This region forms the bottom of the substrate binding cleft61 and is part of a hinge binding motion that is important for substrate binding62. The structure of the M6I mutant of T4 lysozyme (PDB 150L) is used as a model of the “open” state of this hinge and more closely resembles the structure of L056 (1.0Å Cα RMSD). Alignment with a structure featuring a covalently trapped substrate (PDB 148L) reveals that the active site cleft is well formed with the key catalytic residue Glu15 (Glu 11 in T4L) and key substrate binding residue Thr30 (Thr26 in T4L) correctly positioned. In addition, the hydrophobic core of L056 is well packed, with only two small packing voids of less than 5Å3 in volume, which is typical for structures of this size63.
To examine whether ProGen could generate functional proteins in the “twilight zone” sequence identity64 where two proteins are not assumed to share functional similarity65, we generated 95 new artificial sequences with maximum sequence identities lower than 40% to any known natural protein for two lysozyme families (PF00959 and PF05838). Of the selected sequences, 78 out of the 89 (88%) expressed well and 24 out of the 78 (31%) were soluble (Supplemental Figure 8). We purified 6 highly expressed proteins and found that they were all active, but with significantly lower Michaelis-Menten activities than HEWL or the previously generated artificial proteins L056 and L070 (Fig 3f). The protein with the lowest sequence identity to a natural protein, D4 (31.4% ID to a protein from an Arcobacter nitrofigilis organism), had a of 20.2 M−1s−1, approximately 200-fold below HEWL. While the activity is significantly lower for these distant proteins, directed evolution could be employed to improve activity. Collectively, these results demonstrate a procedure for generating soluble, active proteins that are distant enough in sequence space that they might not be considered traditional sequence homologs.
To additionally compare across structural representations, we utilized AlphaFold214 to predict the structure of functional artificial sequences. We performed structure prediction without an MSA as input so as to not heavily bias the structure prediction toward a known natural mode. As in the crystal structure of L056, the predicted artificial structures roughly match known structures found in nature, Fig S11, including for low identity (<40%) artificial sequences. Collectively, the experimental crystal structure and the AlphaFold2 calculations demonstrate that even though ProGen is not trained using any structural information or biophysical priors, it has likely learned structural invariances, presented as latent coevolutionary signatures, within large-scale evolutionary sequence databases. Furthermore, ProGen seems to capture these structural invariances in a generalizable fashion to design artificial proteins that reside within known natural protein folds while having high sequence dissimilarity, Fig S11.
Trained on a universal protein sequence dataset spanning many families, ProGen designs proteins from any family when provided with the corresponding control tag as input. To explore this capability beyond the lysozyme clan, we evaluated ProGen’s performance in generating and predicting functional full-length sequences from families where other methods have previously been applied: chorismate mutase (CM)7 and malate dehydrogenase (MDH). We fine-tuned ProGen to unaligned protein sequences from these families and generated 64,000 unique sequences for each family (see Methods). Generated proteins exhibit similar conservation patterns to natural sequence libraries (Fig. 4ad). After aligning the generations to a sequence with known structure in Fig 4be, we observed that the conserved positions in generated sequences correlate with ligand-binding and buried residues. Using previously published sequences and their experimentally-measured assay data for CM7 and MDH66 proteins, we also evaluated the concordance of ProGen’s model likelihood for these sequences to their relative activity and compared it with the generative methods utilized in the original studies—bmDCA7 and proteinGAN66. Specifically, we measured per-token log-likelihoods for artificial sequences using ProGen (see Methods) and used them to predict if artificial sequences should function. On CM function data, ProGen log-likelihoods had an area under the curve (AUC) of 0.85, significantly better (, two-tailed test, ) than bmDCA, which had an AUC of 0.78 (Fig. 4c). On MDH function data, ProGen log-likelihoods had an AUC of 0.94 (Fig. 4f), which was better than ProteinGAN discriminator scores, with an AUC of 0.87 (, two-tailed test, ). In sum, ProGen’s model likelihoods are better aligned with experimentally-measured assay data on two diverse protein datasets—chorismate mutase and malate dehydrogenase—than the sequence generation methods from original studies specifically tailored for these families.
Figure 4: Applicability of conditional language modeling to other protein systems.
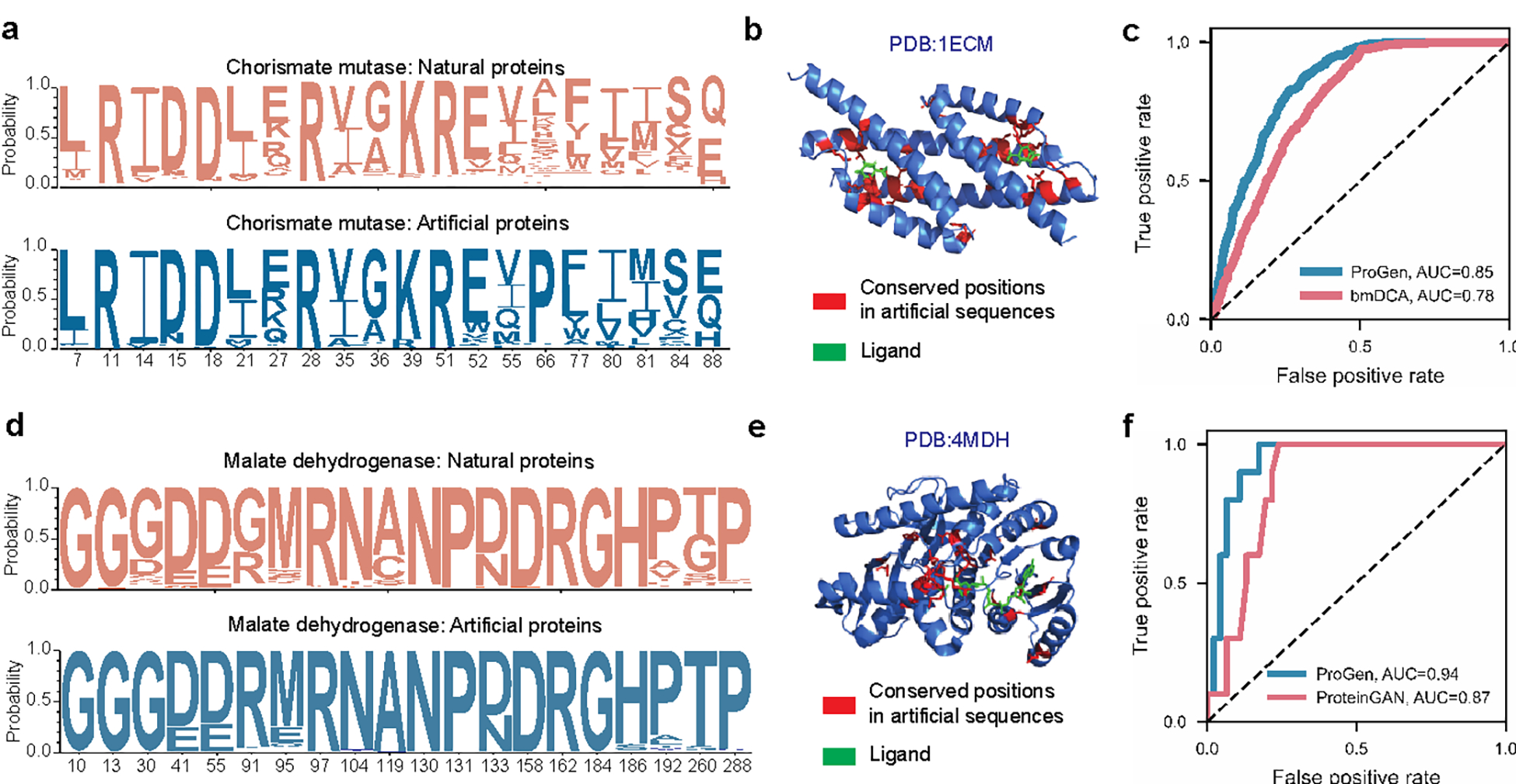
Using the appropriate control tag, our language model, ProGen, can generate sequences for distinct protein families. Here we show that ProGen can generate chorismate mutase (CM) enzymes that exhibit a similar residue distribution to nature (a) and the conserved residues among generated sequences correlate to ligand-binding sites (b). ProGen’s model likelihoods can also accurately predict the functionality of CM variants from published data, slightly better than the coevolutionary bmDCA7 algorithm from the original study (c). ProGen can also generate malate dehydrogenase (MDH) proteins that exhibit a similar residue distribution to nature (d). The conserved residues among generated sequences correlate to buried residues (e). ProGen’s model likelihoods are also accurate in predicting functionality of published variants of MDH, similar to the generative proteinGAN66 model used in the original study (f).
To understand the relative impact of the universal sequence dataset and of sequences from the protein family of interest on ProGen’s generation ability, we perform two ablation studies using the CM and MDH experimentally-measured assay data. First, we evaluated the performance of ProGen trained only with the universal sequence dataset. We measured per-token log-likelihoods for artificial sequences for this version of ProGen using control tags for CM and MDH (see Methods). These likelihoods showed a significant drop in AUC of 0.18 for CM (, two-tailed test, ) and 0.08 for MDH (, two-tailed test, ), as compared to fine-tuned ProGen when predicting if an artificial sequence should function. Conversely, the ProGen architecture trained on CM and MDH protein sequences alone without the benefit of initial training on the universal sequence dataset also showed a significant drop in performance as compared to fine-tuned ProGen—the AUC reduced by 0.11 (, two-tailed test, ) and 0.04 (, two-tailed test, ) on the CM and MDH data respectively.
These results indicate that both components of our training strategy—initial training on the universal sequence dataset and fine-tuning on the protein family of interest—contribute significantly to final model performance. Training with the universal sequence dataset containing many protein families enables ProGen to learn a generic and transferable sequence representation that encodes intrinsic biological properties. Fine-tuning on the protein family of interest steers this representation to improve generation quality in the local sequence neighborhood. Similar to the adaptability shown by neural networks trained on large datasets using transfer learning and fine-tuning in natural language processing25,34,67 and computer vision68,69, protein language models have the potential to be a versatile tool for generating tailored proteins with desired properties. In Figure S12, the distribution of available sequences for different protein families indicates there is a large portion of the protein universe where our current technique would be useful. We extrapolate that it may be possible to successfully generate artificial proteins with functional activity without fine-tuning, especially for larger protein families; however, it would likely do so at a small success rate. We did not attempt to experimentally test generated sequences without additional fine-tuning in our study.
In conclusion, our study shows that a state-of-the-art transformer-based conditional language model trained only with evolutionary sequence data generates functional artificial proteins across protein families. Additional analyses suggest that our model has learned a flexible protein sequence representation which can be applied to diverse families such as lysozymes, chorismate mutase, and malate dehydrogenase. While we do not expect our language model to generate proteins that belong to a completely different distribution or domain (for example creating a new fold that catalyzes an unnatural reaction), it can significantly expand the space of protein sequences from those sampled by evolution. Combining biophysical modeling with generative models could further help us explore data distributions that are completely distinct from those sampled by evolution17,70,71. Applications of our model could include generating synthetic libraries of highly-likely functional proteins for novel discovery or iterative optimization. In combination with ever-increasing sources of sequence data and more expressive control tags, our work points to an exciting potential for the use of deep learning-based language models for precise design of novel proteins to solve important problems in biology, medicine, and the environment.
Methods
Training Data Curation
The initial training data consisted of over 281 million non-redundant, annotated protein sequences with associated metadata, formulated as control tags. The amino acid vocabulary consisted of the standard 25 amino acids designations in IUPAC72. The control tags were divided into two categories: (1) keyword tags and (2) taxonomic tags. Following the definitions laid out in the UniprotKB controlled, hierarchical vocabulary of keywords (many of which are derived from Gene Ontology (GO) terms73), the control keyword tags included 1100 terms ranging from cellular component, biological process, and molecular function terms. The taxonomic tags include 100k terms from the NCBI taxonomy across the eight standard taxonomic ranks. The aggregated dataset was split into a training set of size 280 million and two test sets, an out-of-distribution test set (OOD-test) of size 100k from 20 protein families and a randomly sampled in-domain test set (ID-test) of size 1 million, that were held-out for training and used for evaluation. After model training on the training database, the model was further trained, i.e. fine-tuned, to the following datasets for generation and classification tasks.
For fine-tuning on lysozyme proteins, five protein families from the Pfam database were selected, Phage lysozyme (PF00959), Pesticin (PF16754), Glucosaminidase (PF01832), Glycoside hydrolase family 108 (PF05838), and Transglycosylase (PF06737), yielding a total of 55,948 sequences. Proteins were provided to the model during fine-tuning as unaligned protein sequences with one control tag prepended for the protein family designation. For fine-tuning on chorismate mutase proteins, a search with HHBlits and blastp was performed with residues 1–95 of EcCM (the CM domain of the E. coli CM-prephenate dehydratase, the P-protein) yielding 20214 sequences. For fine-tuning on malate dehydrogenase proteins, the L-lactate/malate dehydrogenase protein family from Interpro IPR001557 was selected with 17094 sequences.
Conditional Language Modeling
Our conditional language model, named ProGen, is a decoder style Transformer24-variant with 36 layers, 8 attention heads per layer, and dimensions and . To learn the conditional distributions over amino acids and control tags, a sequence containing n tokens is embedded as a sequence of corresponding vectors in . This sequence of vectors is stacked into a matrix , and is processed by a series of pre-activation layer-normalized Transformer self-attention layers28. Logits for each token in the vocabulary are computed via a learned linear transformation that is applied to each output of the last layer. During training, these logits input to a cross-entropy loss. During generation, the logits for the final token in the sequence are normalized with a softmax to get a distribution for sampling a new token.
ProGen Training
For training, we included each sequence and its reverse. We prepended each sequence with a corresponding subset of control tags. For a given sequence, there can be multiple versions across databases, each with their own associated control tags. We randomly sampled which set of control tags to utilize, but biased sampling toward SwissProt tags as they are manually verified. Additionally, we always included a sample with the sequence alone without control tags so that ProGen could be used to complete proteins using sequence data alone. We truncated all sequences to a maximum length of 512. Sequences of length less than 512 were padded, and padded tokens were excluded from the cost function used for training. The average token length of control tags during pretraining was 8. Our model was implemented in TensorFlow and trained with a global batch size of 2048 distributed across 256 cores of a Cloud TPU v3 Pod for a fixed number of 1 million iterations, with no specific stopping criterion. The perplexity on a held-out test set was monitored and did not exceed training set perplexity throughout model training. Training took approximately two weeks using Adagrad with linear warmup from 0 to 1e−2 over the initial 40k steps with a linear decay for the remainder of training. The model was initialized with pretrained weights of CTRL28, which was trained on an English language corpus.
Lysozyme Generation
We fine-tuned ProGen to the 55,948-sequence fine-tuning dataset using the conditional language modeling loss function introduced in Eq. 1, using a separate control tag for each of the five lysozyme families. The fine-tuning dataset was clustered at 80% sequence identity and 10% of the clusters were held-out as a validation set for hyper-parameter optimization and stopping criteria. The model was fit for 4 epochs using the Adam optimizer74 with a learning rate of 0.0001, batch size of 2, gradient norm clipping75 threshold of 0.25, and a dropout76 rate of 0.1. We then applied sampling using the final checkpoint of the fine-tuned model. We generated one million artificial sequences from the learned conditional probability distribution using each of the 5 lysozyme families as a control tag , and applying top-p sampling77, which zeros out the probability of the tail of the distribution during sampling, and uses a hyperparameter to determine what fraction of the original distribution to keep. Lower values result in sequences with a higher likelihood under the model, but lower diversity. We generated a batch of one million synthetic sequences (Fig. S3) using values that varied in [0.25,0.50,0.75], and applied the sequence selection criteria in the next section to determine which sequences to synthesize.
Lysozyme Sequence Selection
We selected sequences for synthesis by ranking them using the combination of an adversarial discriminator27,56 and generative model log-likelihood scoring57. First, we trained an adversarial discriminator to distinguish between natural lysozymes and ProGen-generated lysozymes. A higher discriminator score indicates a protein sequence that is “semantically” and “grammatically” closer to natural sequences, but not necessarily one of high sequence identity to natural proteins. To train the discriminator, we generated a batch of samples from fine-tuned ProGen (with nucleus sampling turned off, or ) that was the same size and distribution of families as our dataset of natural lysozymes. The discriminator architecture was a fine-tuned TAPE-BERT46. For robustness, we trained three discriminators using different random seeds. We assigned each sequence a discriminator score as the geometric mean of the probability of the sample being a natural sequence as predicted by the three discriminators. We also assigned each sequence a log-likelihood score as the average per-token log-likelihood for each sample computed using the fine-tuned ProGen model and conditioned on the control tag used to generate the sequence, given by
| (Eq. 2) |
A higher log-likelihood score indicates a sequence close to the probability distribution of sequences seen in training. Model log-likelihoods are directly correlated with perplexity as a language modeling evaluation metric. We selected artificial sequences using separate rankings based on the discriminator and log-likelihood scores. We separately ranked candidate sequences in maximum sequence identity ranges of 40–50%, 50–60%, 60–70%, 70–80%, and 80–90%. For each range, we added the top discriminator-ranked sequences, skipping any sequences that were >80% identical to any previously selected sequence, for a total of 90 sequences. Ten more sequences were added based on ranking by generative model log-likelihood scores in each range, again skipping any sequences with >80% identity to any previously selected sequence.
Evaluating ProGen on Other Protein Systems
We also evaluated ProGen on generation of Chorismate Mutase (CM) and Malate Dehydrogenase (MDH) proteins. We separately fine-tuned ProGen on datasets of CM and MDH proteins using the Adam optimizer, a learning rate of 1e-4, a gradient norm clipping threshold of 0.25, and a dropout rate of 0.1. We also preappended the CM and MDH data with control tags that corresponded to CM and MDH families in ProGen’s original training. After fine-tuning, we generated a set of 64k sequences using top-p sampling () from the CM and MDH fine-tuned models respectively. We measured concordance of our model’s log-likelihoods with protein function data on CM and MDH sequences, and compared with bmDCA7 and ProteinGAN66 baselines respectively. We computed the area under the curve (AUC) in receiver operating characteristic (ROC) curves for predicting binary function labels from model scores. We computed model scores for each sequence in both CM and MDH by using the per token model log-likelihood in Eq. 2. We used model scores for bmDCA given by negative energy of each CM sequence provided by the study’s authors7. We also applied thresholding at 0.42 norm relative enrichment (r.e.) to obtain binary labels for CM function, which roughly corresponds to the cut-off point between two modes that exist in CM function data, to be used for ROC curves, following the original study7.
Since model likelihoods for GANs are intractable, we used discriminator scores corresponding to the probability at which the ProteinGAN discriminator predicted each sample was real as a ProteinGAN model score for each MDH sequence. The MDH functional labels are binary, so no thresholding was needed to compute AUCs. For an ablation study on ProGen, we also evaluate 1. a randomly initialized LM that has the same architecture as ProGen and is finetuned to the same task-specific data as ProGen (CM or MDH), but is not pretrained on a larger dataset 2. ProGen without task specific fine-tuning, conditioning on control tags for CM or MDH from ProGen’s original pretraining data. After measuring the AUC of each model for each dataset, we used bootstrapping to compute the statistical significance of the difference in AUC of fine-tuned ProGen vs. the reference method (bmDCA and ProGen ablations for CM, ProteinGAN and ProGen ablations for MDH). At each bootstrapping iteration, we resampled a new dataset of fitness and model score pairs the same size as the original dataset by randomly selecting data points from the original dataset with replacement. For each sample dataset, we compute the difference in AUC score between fine-tuned ProGen and the reference method. We drew a total of 10,000 bootstrapping samples, and the p-value is given by the percentage of the samples where the baseline achieves an AUC greater than or equal to fine-tuned ProGen, multiplied by two to give two-tailed.
Materials
All reagents were purchased from Thermo Fisher Scientific (Waltham, MA, USA) unless otherwise noted. DNAs utilized for in vitro translation were purchased from Twist Bioscience (San Francisco, CA, USA) and DNAs utilized for E. coli expression and purification were purchased from VectorBuilder (Chicago, IL, USA).
High-throughput cell-free expression of lysozymes
Lysozymes were expressed using the Tierra Bioscience (San Leandro, CA) cell-free expression platform. Cell-free extracts for protein expression were prepared according to the methods of Sun et al. (2013)78 with the following modifications: Terrific Broth was used in lieu of 2xYT, cells were lysed in a single pass by French® press at 10,000 PSI, dithiothreitol was omitted from wash buffers, and run-off and dialysis steps were removed to streamline extract processing. Expression reactions were composed of cell-free extract, an energy buffer, and a linear DNA template containing a promoter sequence, the protein sequence of interest, the sequence of a strep purification tag and a terminator sequence; reactions were carried out at 29 °C for 6 hours. Expression reactions for screening optimal affinity purification tag terminus were performed in 10μL volumes; selected reactions with good expression were then scaled to 200μL. Lysozymes were purified from expression reactions by affinity chromatography with elution by enzymatic cleavage with 3C protease leaving a small sequence scar.
High-throughput screening of lysozyme activity
Purified cell-free synthesized lysozymes were assayed with the EnzChek® Lysozyme Assay Kit (Thermo Fisher Scientific, Waltham, MA). The assay was performed according to protocol with minimal modifications. Hen egg white (HEW) lysozyme standards and purified proteins in buffer (100mM Tris pH 7.4, 150mM NaCl, 2mM TCEP, 20% glycerol) were brought to 50μL with reaction buffer (100mM sodium phosphate pH 7.5, 100mM NaCl, 2mM NaN3) in a 96 well plate. 50μL of DQ lysozyme substrate, fluorescein conjugate (1 mg/mL) was added to each well and fluorescence (ex 485/20; em 528/20) was collected every 5 minutes with a Synergy 2 multi-mode microplate reader (BioTek, Winooski, VT) for 6 hours at 37 °C.
For each 96-well plate, three random wells were dedicated for egg white lysozyme controls and three wells were dedicated for a negative control of ubiquitin expressed and purified on the Tierra Biosciences cell-free expression platform. A purified protein was considered functional if it exhibited a higher fluorescence than one standard deviation above the maximum fluorescence value of all negative controls. The relative activity for each protein was calculated by the following equation:
| (Eq. 3) |
Where r is the linear rate of fluorescence increase in the initial twenty minutes of the fluorogenic assay and m is the mass of protein as determined by Bradford assay concentration and measured volumes.
E. coli Expression of Lysozyme Variants
We chose five generated lysozyme variants ( L008, L013, L038, L056, L070) for expression in E. coli based on strength of signal in the in vitro assay, expression level in the cell-free system, and max ID to natural proteins. Generated lysozyme variants, were codon optimized for E. coli (Integrated DNA Technologies) with an HRV3C protease site N-terminal of the open reading frame. DNA was synthesized and cloned in-frame with a 5’ His6-tag in a pET vector and transformed into BL21(DE3) (Vectorbuilder). One liter of Terrific Broth (Fisher) was pre-warmed to 37°C before being inoculated with 10mL of an overnight starter culture. Cultures were grown to 0.6 < OD600 < 1.0 before temperature was dropped to 16°C for expression. Cultures were induced with 0.5 mM IPTG (source) and protein expression allowed to continue overnight. For induced cultures of L056 and L070, significant turbidity was observed in the spent media after cells were pelleted at 3,500 r.c.f. for 30 mins at 4°C. Spent media also harbored lysozyme activity as ascertained through fluorescence increase over time of the Fluorescein-labeled M. Lysodeikticus cell wall substrate (EnzChek kit; ThermoFisher). Spent media was saved for protein purification (outlined below) and cell pellet frozen and stored at −20°C. Variant L008 did not express under multiple different conditions. L013 and L038 expressed highly to inclusion bodies.
Purification of L056 and L070 from spent media
Media was split into two 0.5 L pools each. The first pools were loaded onto a 5 mL HisTrap FF NiNTA column (GE) using a peristaltic pump at room temperature. Columns were washed with 200 mL of 30 mM HEPES pH 7.6, 150mM NaCl, 25 mM imidazole, 0.5 mM TCEP. Columns were eluted with 25 mL of 30 mM HEPES pH 7.6, 150mM NaCl, 250 mM imidazole, 0.5 mM TCEP. Eluates were concentrated to 8–10mL and dialyzed against 30 mM HEPES pH 7.6, 150mM NaCl, 0.5 mM TCEP with HRV3C protease added overnight at 4°C. Dialyzed protein was put through an ortho 5 mL HisTrap FF NiNTA column (GE) to remove HRV3C protease and uncleaved lysozyme. Though highly pure by SDS-PAGE analysis, protein was further purified by size-exclusion chromatography and loaded on an S75 10/300 gl column pre-equilibrated with 30 mM HEPES pH 7.6, 150mM NaCl, 0.5 mM TCEP. Two peaks were resolved for each variant and that harbored lysozyme activity against the fluorescein-labeled M. Lysodeikticus cell wall substrate (EnzChek kit; ThermoFisher). Individual peaks were pooled and protein concentration determined either by Bradford assay (Biorad) or by SDS-PAGE using colloidal coomassie (ThermoFisher) and HEWL in-gel standards.
The second spent media pools were batch-bound to 5 mL of HisPur NiNTA resin (ThermoFisher) at 4°C for 1 hour before protein-bound resin was pelleted through centrifugation at 3000 r.c.f. for 5 mins at 4°C. Protein bound resin was resuspended with 25 mL 30 mM HEPES pH 7.6, 150mM NaCl, 25 mM imidazole, 0.5 mM TCEP and applied to a gravity flow column (BioRad) at room temperature. Columns were washed with 200 mL of 30 mM HEPES pH 7.6, 150mM NaCl, 25 mM imidazole, 0.5 mM TCEP. Columns were eluted with 25 mL of 30 mM HEPES pH 7.6, 150mM NaCl, 250 mM imidazole, 0.5 mM TCEP. Eluates were concentrated to 8–10mL and dialyzed against 30 mM HEPES pH 7.6, 150mM NaCl, 0.5 mM TCEP with HRV3C protease added overnight at 4°C. Lysozyme was separated from HRV3C protease by size-exclusion chromatography on an S75 10/300 gl column pre-equilibrated with 30 mM HEPES pH 7.6, 150mM NaCl, 0.5 mM TCEP. Two peaks were resolved for each variant and that harbored lysozyme activity against the fluorescein-labeled M. Lysodeikticus cell wall substrate (EnzChek kit; ThermoFisher) that corresponded to peaks observed in the first pool purification. Individual peaks were pooled and protein concentration determined either by Bradford assay (Biorad) or by SDS-PAGE using colloidal coomassie (ThermoFisher) and HEWL in-gel standards.
Michaelis-Menten kinetics of lysozyme variants using fluorescein-labeled M. lysodeikticus cell wall
Fluorescein-labeled M. Lysodeikticus cell wall substrate (EnzChek kit; ThermoFisher) was reconstituted in 30mM HEPES pH 7.6, 150mM NaCl to 1 mg/mL, aliquoted and stored at −20°C until use. A serial two-fold dilution series of substrate was prepared in 30mM HEPES pH 7.6, 150mM NaCl and treated as a 2X solution for enzymatic assays. Enzyme concentration was calculated either through Bradford assay (Bio-Rad) or by SDS-PAGE, in-gel using Novex or Abcam Colloidal Coomassie stain against a hen egg white lysozyme (HEWL; Alfa Aesar) standard. Enzymes were diluted to between 10 and 100nM in 30mM HEPES pH 7.6, 150mM NaCl (HEWL) or 30mM HEPES pH 7.6, 150mM NaCl, 0.5mM TCEP (L056 and L070) and these stocks treated as a 2X solution for enzymatic assays. Kinetic assays were performed in a Tecan Spark 10M plate reader using monochrometers with a fixed 20 nm bandpass filter in a 384-well black-bottom plate (Corning) at 10 μL final volume. Reactions were initiated by pipetting 5 μL of substrate into appropriate wells followed immediately by 5 μL of enzyme, mixed by pipetting before starting data acquisition. The dead-time from reaction initiation to acquisition of first read was measured to be 24 s. For reactions carried out above ambient temperature (25°C), the plate was preincubated at temperature for at least 5 mins prior to reaction initiation. Initial velocities were calculated through linearly fitting fluorescence intensity (au) versus time for the first 2 minutes of each reaction. Finally, velocities were converted from au to fluorescein liberated through application of a fluorescein (Sigma) standard curve (Figure S6) and normalized to enzyme concentration. Averaged data ( technical replicates) were nonlinearly fit to the Michaelis Menten model (Eq. 4) in IgorPro 7 to report in units of fluorescein liberated enzyme−1 min−1 and in units of g L−1 (the average molecular weight of the fluorescein-labeled M. Lysodeikticus cell wall substrate was unknown and likely heterogeneous).
| (Eq. 4) |
For lowID lysozyme A5 the above protocol was altered slightly to accommodate lower catalytic activity of these variants: reaction volumes were increased to 20 μL, plate was covered with an optically transparent seal (Microseal ® ‘B’ seal; BioRad) to mitigate sample evaporation, fluorescent reads were taken every 5 mins for 16 hours with 5 s of linear plate shaking prior to each measurement to minimize photobleaching of substrate and ensure substrate maintained homogeneous dispersion during longer reactions. The rate of substrate photobleaching was measured using a buffer-only control and used as a background rate subtraction for initial rate determination.
Lysozyme extrapolation from pseudo first-order kinetic data
For the higher molecular weight L056 and L070 species whose ’s were beyond the concentration regime of the fluorescein-labeled M. Lysodeikticus cell wall substrate (EnzChek kit; ThermoFisher), the ratio was measured through pseudo first-order kinetics where when [Enyzme] >> Substrate the Michaelis-Menten model simplifies to Eq. 5. Fluorescein-labeled M. Lysodeikticus cell wall substrate was diluted to 0.01 g L−1 and this stock was treated as 2X for kinetic assays. Kinetic assays were performed in a Tecan Spark 10M plate reader using monochrometers with a fixed 20 nm bandpass filter in a 384-well black-bottom plate (Corning) at 10 μL final volume. The dead-time from reaction initiation to acquisition of first read was measured at 24 s and the 0 s fluorescence intensity was measured through dilution of substrate with buffer. Reactions were initiated by pipetting 5 μL of 2X enzyme into 5 μL of 2X substrate in a prewarmed 384-well black assay plate (Corning). Five technical replicates were performed across four enzyme concentrations. The resultant data were not described by a single exponential model but were described by a double exponential model (Eq. 6), likely due to the heterogeneity of the substrate, and all data were fit in IgorPro7. The reciprocal of the weighted sum of each tau component was taken to estimate a single value for subsequent analysis (Eq. 7). To estimate values were plotted against enzyme concentration where the slope of a linear fitting is equal to .
| (Eq. 5) |
| (Eq. 6) |
| (Eq. 7) |
For lowID lysozyme variants the above protocol was altered slightly to accommodate lower catalytic activity of these variants: reaction volumes were increased to 20 μL where 2 μL of 0.05 mg/mL (0.005 mg/mL final) of fluorescein-labeled M. Lysodeikticus cell wall substrate was diluted with 18 μL of lysozyme variant to initiate reactions, plate was covered with an optically transparent seal (Microseal ® ‘B’ seal; BioRad) to mitigate sample evaporation, fluorescent reads were taken every 5 mins for 16 hours with 5 s of linear plate shaking prior to each measurement to minimize photobleaching of substrate and ensure substrate maintained homogeneous dispersion during longer reactions. At least four enzyme concentrations were tested. Initial rates from these data (first two hours of reaction) were also collected to determine enzyme relative activity according to Eq. 3 (Supplemental Figure 9).
Crystallization and structure determination of L056
Purified L056 was concentrated to 18.6 mg/mL in 30 mM HEPES pH 7.6, 150 mM NaCl, 0.5 mM TCEP. Crystals were identified from sitting drop vapor diffusion experiments set at 20°C with a 1:1 ratio of 200nL protein and 200nL well solution (0.1 M CHES 9.5 pH, 30 %w/v PEG 3K). Diffraction data were collected from a single crystal at Beamline 8.3.1. at the Advanced Light Source. Data were processed using XDS79 and a molecular replacement solution was identified using phaser80 with a trRosetta model of L056 as a search model. Significant translational non-crystallographic symmetry and differences with the search model resulted in maps that were initially hard to interpret. The initial model was improved using Refmac jelly body refinement81 prior to rebuilding using phenix.autobuild82 and the CCP4 buccaneer_pipeline83. The model was finalized and iteratively improved with multiple rounds of manual modification in Coot84 and refinement using phenix.refine85. The model is deposited as PDB 7RGR.
Low max ID lysozyme sequence selection, expression, and assay
To evaluate whether ProGen can generate low max ID sequences, we generated an additional batch of sequences selected to have maximum sequence identities under 40% with respect to any natural protein. Since we could only test a limited number of proteins in vitro for this experiment, we modified our earlier generation procedure to bias the distribution of generations towards lysozyme families with higher measured functionality in previous experiments. We finetuned an ensemble of four ProGen models only to lysozymes in PF00959 and PF05838 families. During generation, we used control tags for the two families, as well as control tags to indicate proteins with at least a 30% sequence similarity to L056 and L070, two proteins that we were able to successfully measure catalytic efficiency for in the previous batch. We then used a geometric ensemble of these four models to generate 1 million samples across these control tag settings with varying top-p values. We only kept generations with maximum sequence identities between 20–40%, and ranked these generations using discriminator scores using the same methodology as before, except with a larger 5B parameter discriminator that was pretrained as the T586 model, instead of TAPE-BERT. Our final batch included 12 sequences with the PF00959 control tag, 13 with the PF05838 control tag, 20 with the “L056 similar” control tag, 20 with the “L070 similar” control tag, 13 across control tags with under 30% maximum sequence identity, and 20 sequences from the 1 million generated for the original batch (with 10 at least 30% similar to L056 or L070, and 10 not similar), ranked by both the TAPE-BERT and T5 discriminators.
High-Throughput Expression Testing of low max ID lysozyme variants
Variant sequences were appended an N-terminal His6 and HRV3C tagged on their N-termini, codon optimized (VectorBuilder), cloned into a pET vector (VectorBuilder), transformed into BL21(DE3) and shipped from VectorBuilder as a glycerol stock in 96-well block. Variants were inoculated into 1 mL of ZYM-5052 autoinduction media87 supplemented with 100 μg/mL carbenicillin in a 96-well deep block, covered with a gas-permeable seal and allowed to grow and expressed by shaking at 37°C overnight (16 hours). High density expressed cultures were lysed by addition of detergent (Promega Fast Break™ Cell Lysis Reagent) supplemented with lysis buffer (30 mM HEPES pH 7.6, 150 mM NaCl, 0.5 mM TCEP, cOmplete mini EDTA free protease inhibitor cocktail (Roche), benzonase nuclease) with incubation under gentle shaking for at least 15 mins at room temperature before whole expression SDS-PAGE gel samples were taken. Individual wells from 96-well block were transferred to microcentrifuge tubes, centrifuged at 21,000 rcf for 5 mins at room temperature, and the soluble fraction was transferred to a new 96-well block for soluble protein SDS-PAGE sample collection.
Expression and Purification of lowID lysozyme variants
Variants A5, B6, C9, D4, D10, and E11 were chosen for follow up biochemical characterization based on their high expression and solubility (Fig. S9). Variants were inoculated into 50 – 200 mL of ZYM-5052 autoinduction media87 supplemented with 100 μg/mL carbenicillin and allowed to grow and express constructs overnight (16 hours) at 37°C. High density cell culture was pelleted by centrifugation at 4000 rcf for 20 mins at 4°C and resuspended to half the total culture volume in 30 mM HEPES pH 7.6, 150 mM NaCl, 0.5 mM TCEP, cOmplete mini EDTA free protease inhibitor cocktail (Roche), benzonase nuclease. Resuspended cells were being lysed by addition of detergent (Promega Fast Break™ Cell Lysis Reagent) by rotating end-over-end at 4°C for at least 15 minutes. Lysate was clarified by centrifugation at 4000 rcf for 20 mins at 4°C. Clarified lysate was batch bound to 0.5 – 1 mL dry volume of HisPur NiNTA resin (ThermoFisher) for 45 mins at 4°C by rotating end-over-end. NiNTA bound variants were purified by either gravity or vacuum flow by washing resin with 75 – 125 mL of 30 mM HEPES pH 7.6, 150 mM NaCl, 0.1 mM TCEP, 25 mM imidazole before eluting with 4 mL of 30 mM HEPES pH 7.6, 150 mM NaCl, 0.5 mM TCEP, 250 mM imidazole. His6 tags were removed through addition of HRV3C protease and cleavage was allow to proceed either at room temperature for 2 hours followed by buffer exchange using EconoPac 10 DG desalting columns (BioRad) equilibrated with 30 mM HEPES pH 7.6, 150 mM NaCl, 0.5 mM TCEP or dialyzed overnight at 4°C against 30 mM HEPES pH 7.6, 150 mM NaCl, 0.5 mM TCEP. If total protein concentration was low, protein was concentrated in 3k MWCO Amico centrifugal filters. In-gel Coomassie quantification against HEWL standard curve was performed for all preparations and used for variant enzymology.
Structure prediction methods
To predict structure for the functional artificial sequences, we utilized AlphaFold214 in single sequence mode (without MSA information), with PDB templates, and 12 recycles. The highest ranked predicted structure among five models was used. We attempted structure prediction without templates under varying settings (1–48 recycles) using three different implementations (AlphaFold2 run locally, ColabFold88 run on Google Colab, and ColabFold run locally), however all predictions for our functional artificial sequences yielded unreliable results with pLDDT scores below 60.
Supplementary Material
Acknowledgements
We thank Bryan McCann, Christine Gee, Erik Procko, Kelly Trego, Lav Varshney, Nitish Shirish Keskar, and Silvio Savarese for their feedback at various stages of this project. We thank Audrey Cook, Denise Lo, and Vanita Nemali for operational support. Thanks also to the Salesforce Research Computing Infrastructure team and the Google Cloud TPU team for their help with computing resources, in addition to Twist Bioscience for DNA synthesis support. Beamline 8.3.1 at the Advanced Light Source is operated by the University of California Office of the President, Multicampus Research Programs and Initiatives grant MR-15–328599, the National Institutes of Health (R01 GM124149 and P30 GM124169), Plexxikon Inc., and the Integrated Diffraction Analysis Technologies program of the US Department of Energy Office of Biological and Environmental Research. The Advanced Light Source (Berkeley, CA) is a national user facility operated by Lawrence Berkeley National Laboratory on behalf of the US Department of Energy under contract number DE-AC02–05CH11231, Office of Basic Energy Sciences. JSF was supported by NIH GM123159 and a Sanghvi-Agarwal Innovation Award.
Footnotes
Code Availability
Our code and experiments can be reproduced using the details provided in the Methods section on data preparation, model architecture, and training protocol. Major components of our model architecture and training protocol can be reproduced using CTRL (https://github.com/salesforce/ctrl). A comprehensive codebase for methods described in this paper will be made publicly available at https://github.com/salesforce/progen.
Data Availability
All sequence databases used in this study are publicly available and include UniprotKB, UniParc, NCBI Taxonomy, Pfam, Uniref30, NCBI nr database, and Interpro. Please refer to Table S1 for more details. Sequences and activity data for natural and artificial lysozymes tested are in the Supplementary Material. Evaluation data for the chorismate mutase experiments can be found in Russ et al.6 Evaluation data for the malate dehydrogenase experiments can be found in Repecka et al.52 The crystal structure datasets generated during the current study are available in the Protein Data Bank repository, under accession 7RGR.
References
- 1.Koga N et al. Principles for designing ideal protein structures. Nature 491, 222–227 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lin Y-R et al. Control over overall shape and size in de novo designed proteins. Proc. Natl. Acad. Sci. U. S. A. 112, E5478–85 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang P-S, Boyken SE & Baker D The coming of age of de novo protein design. Nature 537, 320–327 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Huang P-S et al. De novo design of a four-fold symmetric TIM-barrel protein with atomic-level accuracy. Nat. Chem. Biol. 12, 29–34 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boyken SE et al. De novo design of protein homo-oligomers with modular hydrogen-bond network–mediated specificity. Science 352, 680–687 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lapedes AS, Giraud Bertrand G., Liu LonChang & Stormo GD Correlated Mutations in Models of Protein Sequences: Phylogenetic and Structural Effects. Lect. Notes Monogr. Ser. 33, 236–256 (1999). [Google Scholar]
- 7.Russ WP et al. An evolution-based model for designing chorismate mutase enzymes. Science 369, 440–445 (2020). [DOI] [PubMed] [Google Scholar]
- 8.Hopf TA et al. The EVcouplings Python framework for coevolutionary sequence analysis. Bioinformatics 35, 1582–1584 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morcos F et al. Direct-coupling analysis of residue coevolution captures native contacts across many protein families. Proc. Natl. Acad. Sci. U. S. A. 108, E1293–301 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ovchinnikov S, Kamisetty H & Baker D Robust and accurate prediction of residue-residue interactions across protein interfaces using evolutionary information. Elife 3, e02030 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alley EC, Khimulya G, Biswas S, AlQuraishi M & Church GM Unified rational protein engineering with sequence-based deep representation learning. Nat. Methods 16, 1315–1322 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu Z et al. Signal Peptides Generated by Attention-Based Neural Networks. ACS Synth. Biol. 9, 2154–2161 (2020). [DOI] [PubMed] [Google Scholar]
- 13.Shin J-E et al. Protein design and variant prediction using autoregressive generative models. Nat. Commun. 12, 2403 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jumper J et al. Highly accurate protein structure prediction with AlphaFold. Nature 1–11 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bryant DH et al. Deep diversification of an AAV capsid protein by machine learning. Nat. Biotechnol. 39, 691–696 (2021). [DOI] [PubMed] [Google Scholar]
- 16.Das P et al. Accelerated antimicrobial discovery via deep generative models and molecular dynamics simulations. Nat Biomed Eng 5, 613–623 (2021). [DOI] [PubMed] [Google Scholar]
- 17.Anishchenko I et al. De novo protein design by deep network hallucination. Nature 600, 547–552 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moffat L, Kandathil SM & Jones DT Design in the DARK: Learning Deep Generative Models for De Novo Protein Design. bioRxiv 2022.01.27.478087 (2022) doi: 10.1101/2022.01.27.478087. [DOI] [Google Scholar]
- 19.Ferruz N, Schmidt S & Höcker B A deep unsupervised language model for protein design. bioRxiv 2022.03.09.483666 (2022) doi: 10.1101/2022.03.09.483666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang B et al. A backbone-centred energy function of neural networks for protein design. Nature 602, 523–528 (2022). [DOI] [PubMed] [Google Scholar]
- 21.Leinonen R et al. UniProt archive. Bioinformatics 20, 3236–3237 (2004). [DOI] [PubMed] [Google Scholar]
- 22.Bairoch A et al. The Universal Protein Resource (UniProt). Nucleic Acids Res. 33, D154–9 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Finn RD et al. Pfam: the protein families database. Nucleic Acids Res. 42, D222–30 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vaswani A et al. Attention Is All You Need. arXiv [cs.CL] (2017). [Google Scholar]
- 25.Devlin J, Chang M-W, Lee K & Toutanova K BERT: Pre-training of Deep Bidirectional Transformers for Language Understanding. arXiv [cs.CL] (2018). [Google Scholar]
- 26.Brown TB et al. Language Models are Few-Shot Learners. arXiv [cs.CL] (2020). [Google Scholar]
- 27.Zellers R et al. Defending Against Neural Fake News. arXiv [cs.CL] (2019). [Google Scholar]
- 28.Keskar NS, McCann B, Varshney LR, Xiong C & Socher R CTRL: A Conditional Transformer Language Model for Controllable Generation. arXiv [cs.CL] (2019). [Google Scholar]
- 29.AlQuraishi M The Future of Protein Science will not be Supervised. https://moalquraishi.wordpress.com/2019/04/01/the-future-of-protein-science-will-not-be-supervised/ (2019).
- 30.Rives A et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc. Natl. Acad. Sci. U. S. A. 118, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Elnaggar A et al. ProtTrans: Towards Cracking the Language of Lifes Code Through Self-Supervised Deep Learning and High Performance Computing. IEEE Trans. Pattern Anal. Mach. Intell. PP, (2021). [Google Scholar]
- 32.Peters ME et al. Deep contextualized word representations. arXiv [cs.CL] (2018). [Google Scholar]
- 33.Howard J & Ruder S Universal Language Model Fine-tuning for Text Classification. arXiv [cs.CL] (2018). [Google Scholar]
- 34.Radford A, Narasimhan K, Salimans T & Sutskever I Improving language understanding by generative pre-training. (2018).
- 35.Biswas S, Khimulya G, Alley EC, Esvelt KM & Church GM Low-N protein engineering with data-efficient deep learning. Nat. Methods 18, 389–396 (2021). [DOI] [PubMed] [Google Scholar]
- 36.Pfaff CW Constraints on Language Mixing: Intrasentential Code-Switching and Borrowing in Spanish/English. Language 55, 291–318 (1979). [Google Scholar]
- 37.Poplack S Sometimes I’ll start a sentence in Spanish Y TERMINO EN ESPAÑOL: toward a typology of code-switching1. Linguistics vol. 18 (1980). [Google Scholar]
- 38.Dathathri S et al. Plug and Play Language Models: A Simple Approach to Controlled Text Generation. arXiv [cs.CL] (2019). [Google Scholar]
- 39.Madani A et al. ProGen: Language Modeling for Protein Generation. arXiv [q-bio.BM] (2020). [Google Scholar]
- 40.Bengio Y, Ducharme R, Vincent P & Janvin C A neural probabilistic language model. J. Mach. Learn. Res. 3, 1137–1155 (2003). [Google Scholar]
- 41.Vig J et al. BERTology Meets Biology: Interpreting Attention in Protein Language Models. in International Conference on Learning Representations (2020). [Google Scholar]
- 42.Goyal K, Dyer C & Berg-Kirkpatrick T Exposing the Implicit Energy Networks behind Masked Language Models via Metropolis--Hastings. arXiv [cs.LG] (2021). [Google Scholar]
- 43.Bhattacharya N et al. Single Layers of Attention Suffice to Predict Protein Contacts. doi: 10.1101/2020.12.21.423882. [DOI] [Google Scholar]
- 44.Ramsauer H et al. Hopfield Networks is All You Need. arXiv [cs.NE] (2020). [Google Scholar]
- 45.Alley E, Khimulya G, Biswas S, AlQuraishi M & Church GM Unified rational protein engineering with sequence-based deep representation learning. doi: 10.21203/rs.2.13774/v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rao R et al. Evaluating Protein Transfer Learning with TAPE. Adv. Neural Inf. Process. Syst. 32, 9689–9701 (2019). [PMC free article] [PubMed] [Google Scholar]
- 47.Federhen S The NCBI Taxonomy database. Nucleic Acids Res. 40, D136–43 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Matthews BW Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim. Biophys. Acta 405, 442–451 (1975). [DOI] [PubMed] [Google Scholar]
- 49.Broendum SS, Buckle AM & McGowan S Catalytic diversity and cell wall binding repeats in the phage-encoded endolysins. Mol. Microbiol. 110, 879–896 (2018). [DOI] [PubMed] [Google Scholar]
- 50.Love MJ, Abeysekera GS, Muscroft-Taylor AC, Billington C & Dobson RCJ On the catalytic mechanism of bacteriophage endolysins: Opportunities for engineering. Biochim. Biophys. Acta: Proteins Proteomics 1868, 140302 (2020). [DOI] [PubMed] [Google Scholar]
- 51.Martin PP Potts Models And Related Problems In Statistical Mechanics. (World Scientific, 1991). [Google Scholar]
- 52.Thomas J, Ramakrishnan N & Bailey-Kellogg C Graphical models of residue coupling in protein families. IEEE/ACM Trans. Comput. Biol. Bioinform. 5, 183–197 (2008). [DOI] [PubMed] [Google Scholar]
- 53.Weigt M, White RA, Szurmant H, Hoch JA & Hwa T Identification of direct residue contacts in protein–protein interaction by message passing. Proc. Natl. Acad. Sci. U. S. A. 106, 67–72 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Balakrishnan S, Kamisetty H, Carbonell JG, Lee S-I & Langmead CJ Learning generative models for protein fold families. Proteins 79, 1061–1078 (2011). [DOI] [PubMed] [Google Scholar]
- 55.Stein RR, Marks DS & Sander C Inferring Pairwise Interactions from Biological Data Using Maximum-Entropy Probability Models. PLoS Comput. Biol. 11, e1004182 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Goodfellow IJ et al. Generative Adversarial Networks. arXiv [stat.ML] (2014). [Google Scholar]
- 57.Koehn P Pharaoh: A Beam Search Decoder for Phrase-Based Statistical Machine Translation Models. in Machine Translation: From Real Users to Research 115–124 (Springer Berlin Heidelberg, 2004). [Google Scholar]
- 58.Mirdita M, Steinegger M, Breitwieser F, Söding J & Levy Karin E Fast and sensitive taxonomic assignment to metagenomic contigs. Bioinformatics (2021) doi: 10.1093/bioinformatics/btab184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mooers BHM, Tronrud DE & Matthews BW Evaluation at atomic resolution of the role of strain in destabilizing the temperature-sensitive T4 lysozyme mutant Arg 96 → His. Protein Science vol. 18 863–870 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Baase WA, Liu L, Tronrud DE & Matthews BW Lessons from the lysozyme of phage T4. Protein Sci. 19, 631–641 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kuroki R, Weaver LH & Matthews BW A covalent enzyme-substrate intermediate with saccharide distortion in a mutant T4 lysozyme. Science 262, 2030–2033 (1993). [DOI] [PubMed] [Google Scholar]
- 62.Mchaourab HS, Oh KJ, Fang CJ & Hubbell WL Conformation of T4 lysozyme in solution. Hinge-bending motion and the substrate-induced conformational transition studied by site-directed spin labeling. Biochemistry 36, 307–316 (1997). [DOI] [PubMed] [Google Scholar]
- 63.Kim J-K et al. BetaCavityWeb: a webserver for molecular voids and channels. Nucleic Acids Res. 43, W413–8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rost B Twilight zone of protein sequence alignments. Protein Eng. 12, 85–94 (1999). [DOI] [PubMed] [Google Scholar]
- 65.Pearson WR An introduction to sequence similarity (‘homology’) searching. Curr. Protoc. Bioinformatics Chapter 3, Unit 3.1 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Repecka D et al. Expanding functional protein sequence spaces using generative adversarial networks. Nature Machine Intelligence 3, 324–333 (2021). [Google Scholar]
- 67.Ruder S, Peters ME, Swayamdipta S & Wolf T Transfer Learning in Natural Language Processing. in Proceedings of the 2019 Conference of the North American Chapter of the Association for Computational Linguistics: Tutorials 15–18 (Association for Computational Linguistics, 2019). [Google Scholar]
- 68.Huh M, Agrawal P & Efros AA What makes ImageNet good for transfer learning? arXiv [cs.CV] (2016). [Google Scholar]
- 69.LeCun Y, Bengio Y & Hinton G Deep learning. Nature 521, 436–444 (2015). [DOI] [PubMed] [Google Scholar]
- 70.Norn C et al. Protein sequence design by conformational landscape optimization. Proc. Natl. Acad. Sci. U. S. A. 118, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Anand N et al. Protein sequence design with a learned potential. Nat. Commun. 13, 746 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.The IUPAC stability constants database. Chem. Int. 28, (2006). [Google Scholar]
- 73.Ashburner M et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25–29 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kingma DP & Ba J Adam: A Method for Stochastic Optimization. arXiv [cs.LG] (2014). [Google Scholar]
- 75.Pascanu R, Mikolov T & Bengio Y On the difficulty of training recurrent neural networks. in Proceedings of the 30th International Conference on Machine Learning (eds. Dasgupta S & McAllester D) vol. 28 1310–1318 (PMLR, 2013). [Google Scholar]
- 76.Srivastava N, Hinton G, Krizhevsky A, Sutskever I & Salakhutdinov R Dropout: a simple way to prevent neural networks from overfitting. J. Mach. Learn. Res. 15, 1929–1958 (2014). [Google Scholar]
- 77.Holtzman A, Buys J, Du L, Forbes M & Choi Y The Curious Case of Neural Text Degeneration. in Eighth International Conference on Learning Representations (2020). [Google Scholar]
- 78.Sun ZZ et al. Protocols for implementing an Escherichia coli based TX-TL cell-free expression system for synthetic biology. J. Vis. Exp. e50762 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kabsch W XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.McCoy AJ et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kovalevskiy O, Nicholls RA, Long F, Carlon A & Murshudov GN Overview of refinement procedures within REFMAC5: utilizing data from different sources. Acta Crystallogr D Struct Biol 74, 215–227 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Terwilliger TC et al. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr. D Biol. Crystallogr. 64, 61–69 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hoh SW, Burnley T & Cowtan K Current approaches for automated model building into cryo-EM maps using Buccaneer with CCP-EM. Acta Crystallogr D Struct Biol 76, 531–541 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Emsley P, Lohkamp B, Scott WG & Cowtan K Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Afonine PV et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Raffel C et al. Exploring the limits of transfer learning with a unified text-to-text transformer. arXiv preprint arXiv:1910. 10683 (2019). [Google Scholar]
- 87.Studier FW Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 41, 207–234 (2005). [DOI] [PubMed] [Google Scholar]
- 88.Mirdita M et al. ColabFold-Making protein folding accessible to all. (2021). [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequence databases used in this study are publicly available and include UniprotKB, UniParc, NCBI Taxonomy, Pfam, Uniref30, NCBI nr database, and Interpro. Please refer to Table S1 for more details. Sequences and activity data for natural and artificial lysozymes tested are in the Supplementary Material. Evaluation data for the chorismate mutase experiments can be found in Russ et al.6 Evaluation data for the malate dehydrogenase experiments can be found in Repecka et al.52 The crystal structure datasets generated during the current study are available in the Protein Data Bank repository, under accession 7RGR.