

Significance
The majority of previous autism spectrum disorder (ASD) genetic studies have focused on rare de novo variants, which are newly found in children but not parents, while few have studied rare inherited variation and its interaction with common genetic risk of ASD. Using the largest whole-genome sequencing cohort of families with multiple autistic children to amplify inherited risk signal, we found seven previously unrecognized risk genes. Common ASD genetic risk is overtransmitted from nonautistic parents to autistic children with rare inherited variants and is associated with social dysfunction and language delay. These findings suggest that language is a core biological feature of ASD, although it is not currently a core clinical criterion.
Keywords: autism spectrum disorder (ASD), genetics, polygenic score (PGS), inherited, multiplex families
Abstract
Autism spectrum disorder (ASD) has a complex genetic architecture involving contributions from both de novo and inherited variation. Few studies have been designed to address the role of rare inherited variation or its interaction with common polygenic risk in ASD. Here, we performed whole-genome sequencing of the largest cohort of multiplex families to date, consisting of 4,551 individuals in 1,004 families having two or more autistic children. Using this study design, we identify seven previously unrecognized ASD risk genes supported by a majority of rare inherited variants, finding support for a total of 74 genes in our cohort and a total of 152 genes after combined analysis with other studies. Autistic children from multiplex families demonstrate an increased burden of rare inherited protein-truncating variants in known ASD risk genes. We also find that ASD polygenic score (PGS) is overtransmitted from nonautistic parents to autistic children who also harbor rare inherited variants, consistent with combinatorial effects in the offspring, which may explain the reduced penetrance of these rare variants in parents. We also observe that in addition to social dysfunction, language delay is associated with ASD PGS overtransmission. These results are consistent with an additive complex genetic risk architecture of ASD involving rare and common variation and further suggest that language delay is a core biological feature of ASD.
The last decade has seen enormous progress in identifying genes imparting risk for autism spectrum disorder (ASD) (1–10), determining over 100 genes whose risk is primarily due to rare de novo copy number variants or protein-truncating variants (PTVs) (1, 2, 6, 10–19). The majority of these risk genes appear to act pleiotropically, contributing to a broad range of neurodevelopmental disorders in addition to ASD (8, 10, 20–23). These findings have delivered on the original promise of genetic investigations by galvanizing functional genomic and neurobiological studies that have begun to yield mechanistic insight into ASD pathophysiology (24–26).
However, ASD is also highly heritable (27–30) and therefore is expected to have a substantial contribution from common (27, 31) and rare variation transmitted from parents to their autistic offspring. Indeed, at least 50% of genetic risk is predicted to be due to common variation, 15 to 20% is due to de novo variation and other Mendelian forms, and the remaining genetic risk is yet to be determined (2, 4, 10, 27, 32). Even with substantial sized cohorts (greater than 25,000), genome-wide association studies (GWAS) in ASD have to date only identified five loci reaching genome-wide significance (32). This is consistent with high genetic heterogeneity, including the contribution of many low-effect common variants through polygenic risk (33, 34) and rare inherited variation.
Another potential contribution to the preponderance of genes supported by de novo variation, and less success in detecting inherited variation, may be study designs with a heavy focus on families with simplex ASD, having only a single proband and no family history (13, 35–37). Indeed, our recent study of families having two or more autistic children demonstrated a different genetic architecture in these multiplex ASD families (9, 38) with a significant signal from rare, inherited variants (9).
In this current study, we performed whole-genome sequencing (WGS) on the largest collection of ASD multiplex families to date, including 1,004 families, to further explore the contribution of rare and common inherited variation to ASD genetic risk architecture. We identified seven previously unrecognized ASD risk genes at FDR < 0.1 and assess the contribution of polygenic score (PGS) in those carrying rare variants. We show that ASD PGS is significantly overtransmitted to those harboring rare inherited protein-coding variants and those with a more severe ASD phenotype. These data provide compelling evidence supporting a complex ASD genetic architecture that includes a mix of common and rare genetic variation.
Results
We performed WGS at a mean >30× coverage in 4,551 individuals from 1,004 multiplex ASD families (Dataset S1) (SI Appendix, Fig. S1). Study individuals were selected from the Autism Genetic Resource Exchange (AGRE) (38) and some were part of an initial smaller published subset (9). This analysis of multiplex ASD families included 1,836 autistic children and 418 nonautistic children for which both biological parents were sequenced and passed QC (fully-phaseable) (Fig. 1) and is more than double in size compared to the previous study (9). Autistic individuals were diagnosed according to widely used protocols that have been well validated (39, 40), and details of clinical ascertainment in AGRE have been previously published (36, 41) (Materials and Methods).
Fig. 1.
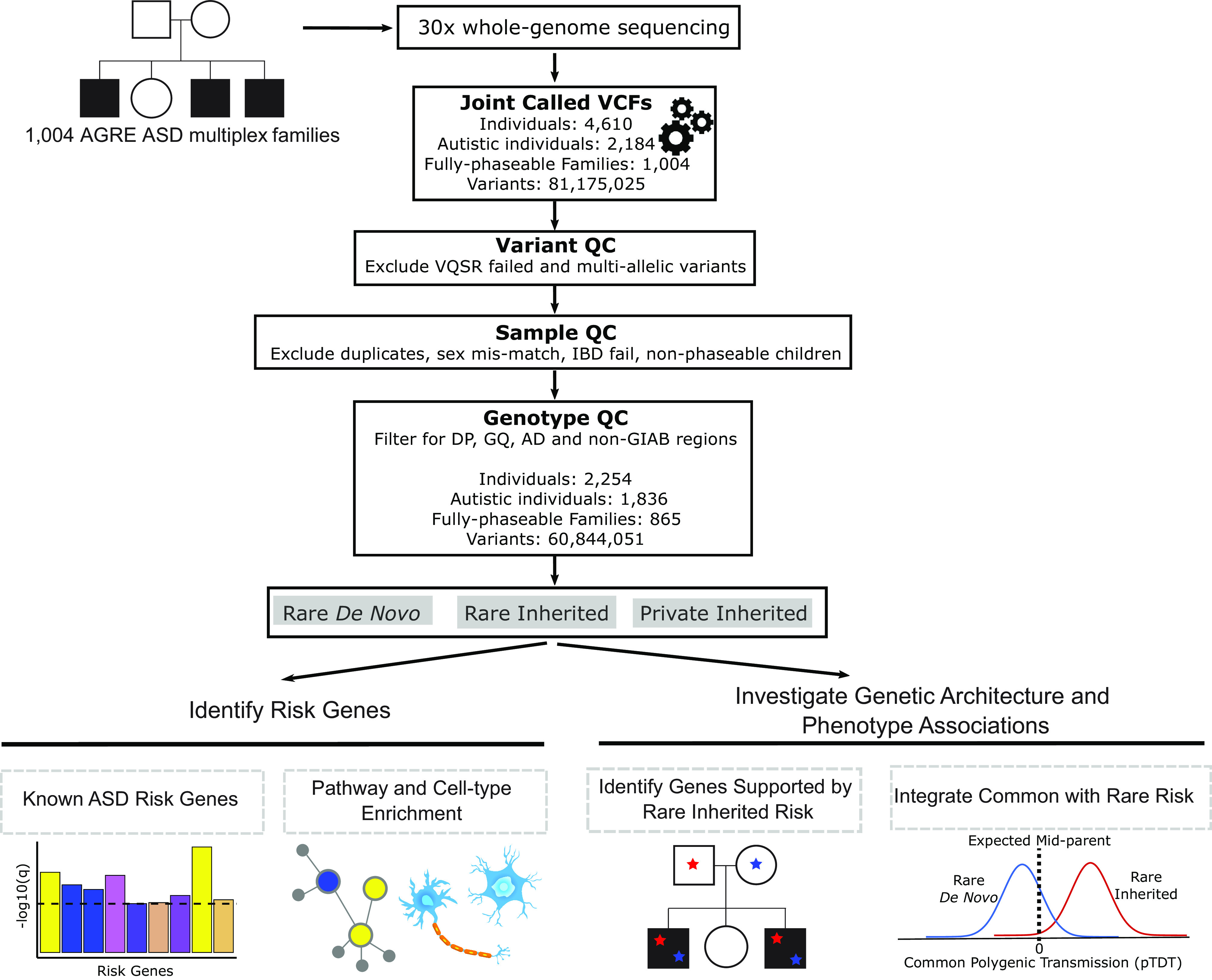
Overview diagram of study analyses. WGS was performed on 1,004 AGRE ASD multiplex families. Single-nucleotide variants and small insertions/deletions were called using the Genome Analysis Toolkit. Variant, sample, and genotype quality control was performed. Rare de novo, rare inherited, and private inherited variants were included for downstream analyses. We identified ASD risk genes at FDR < 0.1 using the Transmission and De Novo Association (TADA) analysis. Risk genes were characterized using integrative genomics. We investigated known genes supported by rare inherited risk and evaluated their variants’ coinheritance and effect on phenotypic measures. We tested if ASD polygenic score was overtransmitted to autistic and nonautistic children stratified by carrier status for rare de novo and inherited variants. VCF = Variant Call Format, QC = Quality Control, VQSR = Variant Quality Score Recalibration, IBD = Identity By Descent, DP = Total Depth, GQ = Genotype Quality, AD = Allele Depth, GIAB = Genome In A Bottle.
Overall Genetic Architecture and Transmission Patterns in Multiplex Families.
WGS yielded an average coverage of 37.5× with 80.6% of bases covered at ≥30× and 99.2% of bases covered at ≥10× (SI Appendix, Fig. S1 A–E). Quality metrics were within the expected range for WGS (42) (SI Appendix). Our WGS processing pipeline (SI Appendix, Fig. S2) retained high-quality variants and further applied a validated Artifact Removal by Classifier (ARC) (9), to distinguish true rare de novo variants (RDNVs) from likely sequencing and mapping errors or artifacts from lymphoblastoid culture (43, 44) (SI Appendix).
After ARC, we find an overall mean rate of 53 RDNVs per subject, with no differences between autistic and nonautistic children (SI Appendix, Wilcoxon rank sum test P = 0.20), consistent with published literature (7, 11, 43, 45–47), and a significant association of paternal age and de novo association rate (SI Appendix, Fig. S3, 1.02 RDNVs per year of paternal age, P < 2.2e−16) (47–50). We observed no enrichment of rare de novo protein-truncating variants (PTVs) (SI Appendix, logistic regression, P = 0.79) and missense variants [Polyphen-2 mis3 (51) P = 0.46; MPC ≥ 1 (52) P = 0.45] in autistic children compared to nonautistic children (SI Appendix, Fig. S4A). The results did not change when defining missense variants through different scoring approaches, Polyphen-2 mis3, MPC ≥ 1, and MPC ≥ 2 (MPC ≥ 2 P = 0.18) (SI Appendix, Fig. S5A). We obtained similar results when comparing the rates of these variant classes in highly constrained genes [pLI (53) ≥ 0.9, pLI ≥ 0.995] between autistic and nonautistic children (pLI ≥ 0.9 mis3 P = 0.63; pLI ≥ 0.9 MPC ≥ 1 P = 0.85; pLI ≥ 0.9 PTV P = 0.69; pLI ≥ 0.995 mis3 P = 0.07; pLI ≥ 0.995 PTV P = 0.99) (SI Appendix, Fig. S4 B and C). Although this is consistent with the hypothesized and previously confirmed depletion of de novo variation in multiplex families (9, 36), these observations could be explained by reduced statistical power due to the small sample size of the nonautistic group (54, 55). We only observed an excess of rare de novo missense variants with MPC score ≥ 1 in constrained genes defined by pLI ≥ 0.995 in autistic children compared to nonautistic subjects [pLI ≥ 0.995 MPC ≥ 1 P = 0.04, variant count odds ratio (OR) = 2.64] (SI Appendix, Fig. S4C).
To perform a more highly powered analysis to quantify the potential depletion of de novo ASD risk in AGRE multiplex families versus large published cohorts, we compared rates of rare de novo missense variants (MPC ≥ 1 and MPC ≥ 2) and PTVs in i) all genes, ii) pLI ≥ 0.9 genes, and iii) pLI ≥ 0.995 genes in AGRE versus two other large ASD simplex family-based collections, SSC (56) and ASC+SSC (57) (SI Appendix). We observed a significant depletion of RDNVs in AGRE multiplex autistic probands compared to simplex probands (SI Appendix, Fig. S6), consistent with other recent similar findings (54–56).
For rare inherited variants, we found no excess of rare inherited PTVs (P = 0.60) and missense variants (mis3 P = 0.62; MPC ≥ 1 P = 0.77, MPC ≥ 2 P = 0.56) in autistic children (SI Appendix, Figs. S4D and S5B). The same held true when limiting the analysis to rare inherited PTV and missense variants in highly constrained genes (pLI ≥ 0.9 mis3 P = 0.78; pLI ≥ 0.9 MPC ≥ 1 P = 0.40; pLI ≥ 0.9 PTV P = 0.12; pLI ≥ 0.995 mis3 P = 0.96; pLI ≥ 0.995 MPC ≥ 1 P = 0.22; pLI ≥ 0.995 PTV P = 0.89) (SI Appendix, Fig. S4 E and F). Similarly, we observed no difference in the rates of private inherited variants (SI Appendix) between autistic and nonautistic children (SI Appendix, Figs. S4 G–I and S5C). We also defined constrained genes using the LOEUF score < 0.35 (58) (SI Appendix) and obtained variant burden comparison results consistent with those based on pLI (SI Appendix, Fig. S5 D–F).
Noncoding Variation in Multiplex Families.
We extended our investigation of the distinct genetic architecture of ASD multiplex families to noncoding regions of the genome. Previous category-wide association studies carried out in simplex families have suggested a biologically plausible association of promoter regions with ASD supported by de novo variation (12, 59). Therefore, we first compared noncoding variant burden between autistic and nonautistic children from the AGRE multiplex families (SI Appendix) for variants mapping to a 3 kb window (2 kb upstream and 1 kb downstream) around the transcription start site (TSS) of all genes. We found no significant enrichment of promoter rare inherited (SI Appendix, logistic regression, P = 0.81), private inherited (P = 0.14), and rare de novo (P = 0.5) variants in autistic children (SI Appendix, Fig. S7 A–C). These results held true when focusing the rare and private inherited noncoding variant testing to promoters of ASD risk genes (SI Appendix, Fig. S7 D and E). The extension of promoter regions to 11 kb (including 1 kb downstream of TSS) did not alter the previous results (rare inherited P = 0.87; private inherited P = 0.11; and rare de novo P = 0.55) (SI Appendix, Fig. S7 A–C).
The strongest enrichment of de novo conserved promoter variants in ASD simplex individuals has been found at transcription factor binding sites (TFBSs) (59). Therefore, we filtered variants in the 11 kb promoters of all genes by retaining those in regions defined by three different genome-wide maps of TFBSs: 1) cis-regulatory modules (CRMs), which are clusters of predicted TFBSs (60), as well as 2) cistromes and 3) cismotifs, which are experimentally and technically reproducible TF binding regions (61). Again, we found no significant excess of TFBS rare inherited (CRMs P = 0.99; cistromes P = 0.91; cismotifs P = 0.9), private inherited (CRMs P = 0.21; cistromes P = 0.13; cismotifs P = 0.15), and rare de novo (CRMs P = 0.98; cistromes P = 0.58; cismotifs P = 0.76) variants in autistic children compared to nonautistic children (SI Appendix, Fig. S7 F–H).
Finally, we mapped noncoding variants within the promoters of all genes and noncoding variants across the whole genome to the brain-specific regulatory regions under sequence constraint within the human lineage, identified through the colocalization approach (62). Xu, et al previously showed these brain-specific regulatory regions were enriched for noncoding de novo variants in autistic children compared to nonautistic siblings from ASD simplex families (62). In our analysis, we found no significant burden of brain-specific constrained functional noncoding rare inherited (11 kb promoter intersected set P = 0.93; genome intersected set P = 0.82), private inherited (11 kb promoter intersected set P = 0.27; genome intersected set P = 0.15), and rare de novo (11 kb promoter intersected set P = 0.54; genome intersected set P = 0.18) variants in autistic individuals from AGRE multiplex families (SI Appendix, Fig. S7 I–K).
Across all tested sets, we found very small effect sizes for noncoding variation (SI Appendix, Table S1), which results in low power at our current sample size, consistent with previous noncoding investigation focusing on de novo variants (12, 59) (SI Appendix, Table S2). Taking advantage of the multiplex family study design and range of effect sizes for noncoding rare inherited variants, we performed power calculations, which predicted that a sample size of at least 8,000 fully phaseable children would provide sufficient power for the investigation of private inherited variants in the promoters of all genes, whereas 9,000 children would be needed to test the association of noncoding private inherited variants in functional regions such as cistromes, cismotifs, and brain-specific colocalized regions (SI Appendix, Fig. S7 L–N).
Seven Previously Unrecognized ASD Risk Genes.
We used the Transmitted And De novo Association (TADA) test (63) to identify ASD risk genes in this cohort. We estimated the distribution of the null TADA statistic for multiplex families (9) through 100,000 simulations of Mendelian transmission and de novo mutation across AGRE multiplex family structures using the observed variant counts per family (SI Appendix). This permutation approach allowed us to account for the nonindependence of autistic siblings within the same families by taking the family structure into consideration (9). Qualifying variants included rare de novo or transmitted PTVs and de novo mis3 variants predicted to damage the encoded protein (51) (Materials and Methods, Fig. 2A, and Dataset S2). We combined this extended multiplex cohort of 1,660 autistic children and their fully-phaseable parents with qualifying variants from previous ASD genetic analyses (2, 4, 6, 64) (Materials and Methods, Fig. 2A and SI Appendix, Fig. S8A). We identified 74 genes significantly associated with ASD at FDR < 0.1 (Fig. 2B). Genes with the smallest observed TADA q-values and the largest observed Bayes factors also showed the smallest simulation FDRs (SI Appendix) (SI Appendix, Fig. S8 B and C). We found a strong overlap between genes with TADA FDR < 0.1 and genes with simulation FDR < 0.05 [Fisher’s exact test, log2(OR) = 10.23; P = 2e−126]; only eight TADA genes (primarily consisting of known high-confidence ASD risk genes) showed a simulation FDR > 0.05 (SI Appendix, Fig. S8 B and C). Of the identified 74 TADA genes, 46% were supported only by de novo variants. The remaining genes (54%) were supported by inherited variants, half of which had more than 50% of genetic variants contributing to an inherited component (Fig. 2B). There were nine genes identified in this study, but not in a previous analysis on a subset of this cohort (9). Two of these nine genes (MED13L and KMT2A), supported by de novo variants, were identified as ASD risk genes in recent analyses (10, 55, 65). The remaining seven genes are previously unrecognized risk genes (PLEKHA8, PRR25, FBXL13, VPS54, SLFN5, SNCAIP, and TGM1) (Fig. 2C). We note that the potential new genes supported by de novo variants arising from LCL DNA within our cohort are also supported by inherited PTVs and/or de novo variants within other cohorts. For example, PLEKHA8 is supported by de novo mis3 variants identified within SSC and ASC cohorts (2, 4), and VPS54 and SNCAIP are also supported by inherited PTVs. Consistent with rare inherited variation having a greater contribution to ASD risk in multiplex versus simplex families, five of these genes were supported by a majority of inherited PTVs (PRR25, FBXL13, VPS54, SLFN5, and TGM1). These observations highlight how gene discovery for ASD benefits from study designs that facilitate well-powered identification of rare inherited variation, as opposed to genes only supported by de novo variants (56, 57, 66).
Fig. 2.
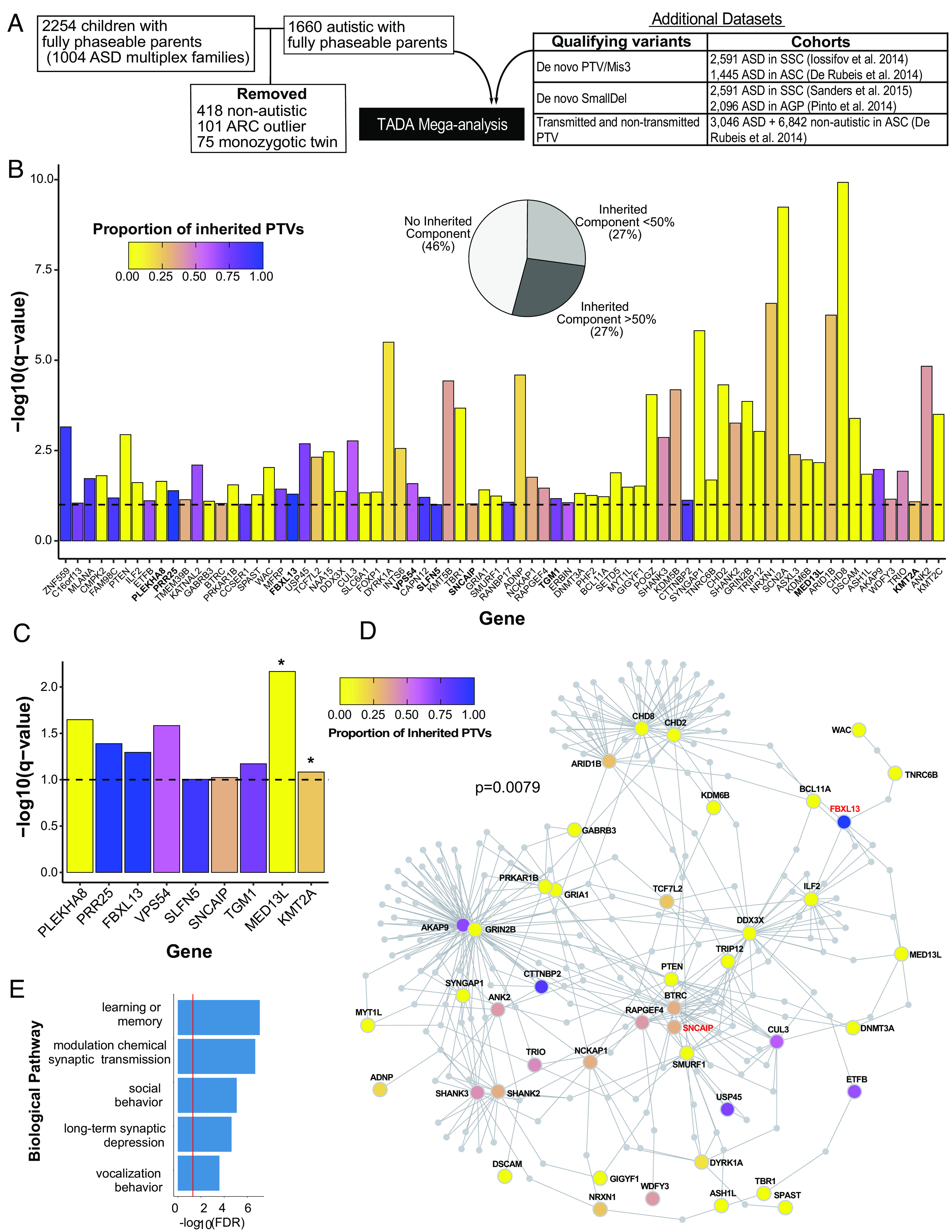
Seventy-four ASD risk genes identified by Transmission and De novo Association (TADA) analysis at FDR < 0.1. (A) Overview of cohort subjects and additional datasets included in the TADA analysis. (B) Seventy-four ASD risk genes identified in the TADA mega-analysis (FDR < 0.1) are displayed with corresponding −log10(q-value). The nine newly identified ASD risk genes from our expanded AGRE multiplex cohort in comparison to Ruzzo, et al (9) are in bold. The dashed horizontal line marks the FDR = 0.1 threshold. Bars are colored by the proportion of inherited protein-truncating variants (PTVs) for each gene (inherited PTVs/(inherited PTVs + de novo PTVs + de novo mis3 + de novo small deletions). The pie chart indicates the percentages of TADA genes with 0%, <50%, and >50% proportion of inherited PTVs. (C) The nine newly identified ASD risk genes from our expanded AGRE multiplex cohort in comparison to Ruzzo, et al (9). Bars are colored according to (B). Asterisks represent genes already identified in other recent cohort studies. Note how these genes have a very low proportion of inherited PTVs (mainly de novo support). The other seven genes are previously unrecognized risk genes for ASD. (D) Protein–protein interaction network formed by the 74 ASD risk genes using Disease Association Protein-Protein Link Evaluator (DAPPLE). Genes are colored by the proportion of inherited PTVs. The gene name for two of the seven previously unrecognized ASD risk genes are in red. (E) Gene Ontology enrichment terms for 39 significant seed genes from the protein–protein interaction network.
We next assessed the shared biological functions among the 74 ASD risk genes identified at FDR < 0.1. Notably, these genes formed a significant indirect protein–protein interaction (PPI) network (SI Appendix, P = 0.0079) involving 39 genes as significant seed genes, meaning that they were more connected to other genes in the network than expected by chance (Fig. 2D). These genes were enriched for gene ontology terms including synaptic transmission, learning, social behavior, and long-term synaptic depression (Fig. 2E).
Previous work has demonstrated that ASD risk genes are most highly expressed and converge as a group during fetal brain development (2, 4–6, 9, 10). We subsequently used human brain fetal single-cell transcriptomics (67) to determine in which cell types the 74 ASD risk genes were expressed. We observed enriched expression of ASD risk genes in newly born excitatory neurons [Expression Weighted Cell Type Enrichment (EWCE), migrating excitatory neurons, FDR = 0.0015, maturing excitatory neurons, FDR = 0.00016, Materials and Methods, Fig. 3 A and B], and interneurons (EWCE, interneurons CGE, FDR = 0.027, interneurons MGE, FDR = 0.04, Materials and Methods, Fig. 3 A and B), which is consistent with previous analyses (9, 21).
Fig. 3.
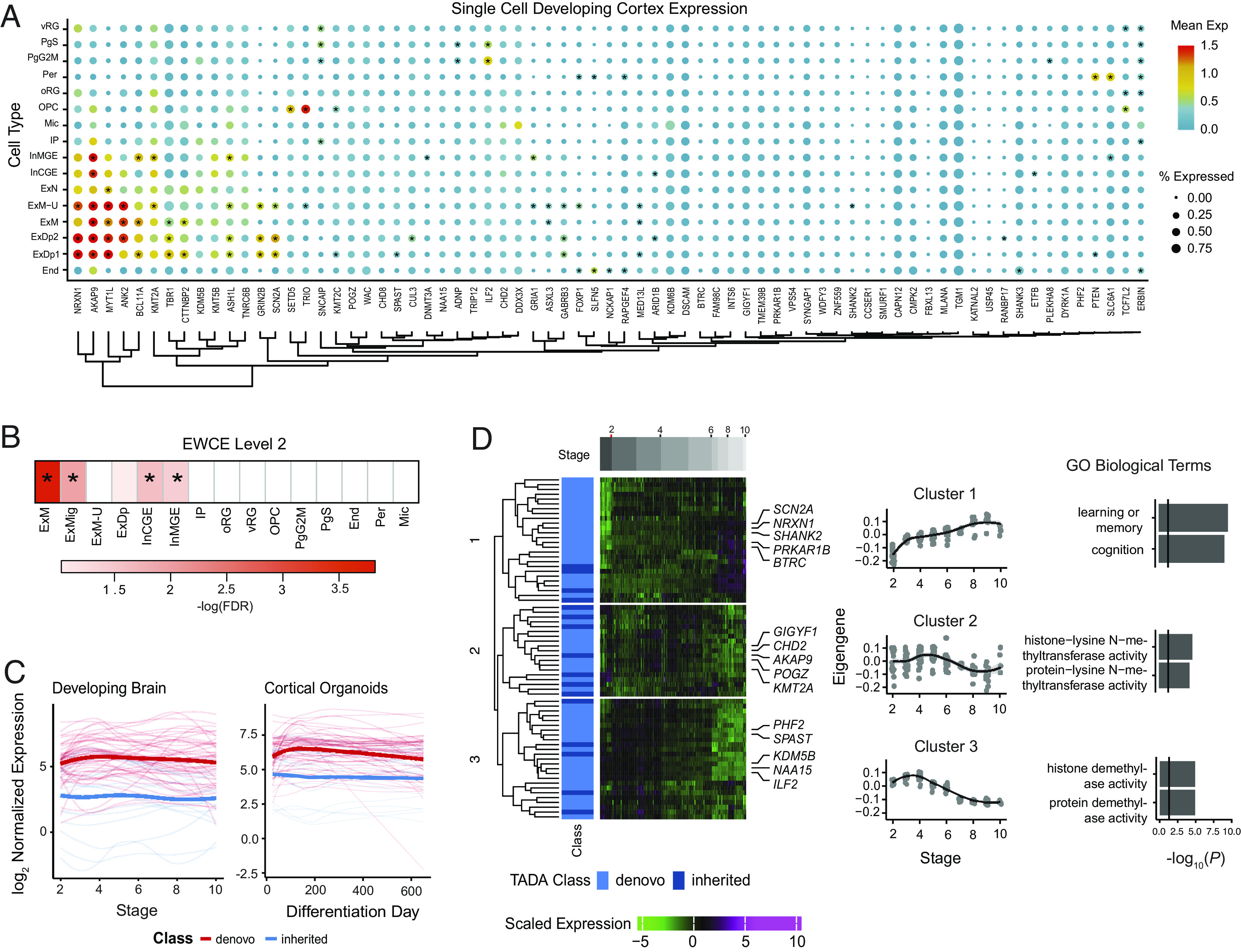
Characterization of TADA ASD risk genes at FDR < 0.1 during development. (A) Mean expression and percentage of cells expressing TADA genes from single-cell data of the human fetal brain cortex. Asterisks indicate cell type–specific TADA genes (Materials and Methods). (B) Cell types enriched in TADA genes tested using Expression Weighted Cell Type Enrichment (EWCE). Asterisks indicate FDR < 0.05. (C) TADA genes’ variation during development using long-term maturation human cortical spheroids (hCSs) and BrainSpan. Genes are stratified by those with a majority of inherited or de novo support. (D) Clustering of the TADA genes from hCS trajectory data identified three clusters. Days postconception, TADA class (majority of inherited or de novo), and scaled expression are shown. The average expression of each cluster and significant gene ontology biological terms are shown. vRG = ventral radial glia, PgS = cycling progenitor (S phase), PgG2M = cycling progenitor (G2M phase), Per = pericyte, oRG = outer radial glia, OPC = oligodendrocyte precursor, Mic = microglia, IP = intermediate progenitor, InMGE = interneuron medial ganglionic eminence, InCGE = interneuron caudal ganglionic eminence, ExN = excitatory neuron, ExM−U = maturing excitatory neuron upper enriched, ExM = maturing excitatory neuron, ExDp2 = excitatory neuron deep layer 2, ExDp1 = excitatory neuron deep layer 1, End = endothelial, ExMig = migrating excitatory neuron, ExDp = excitatory neuron deep.
We next evaluated how the expression of these 74 ASD risk genes identified at FDR < 0.1 varied during development using data from two relevant systems: i) long-term maturation in three-dimensional induced pluripotent stem cell (IPSC)-derived human forebrain organoid models in vitro, specifically human cortical spheroids (hCS), which have been shown to largely recapitulate human fetal brain development up to 600 d postnatally in vitro (68); and ii) the BrainSpan dataset, which represents an in vivo reference for cortical development (69–71). Given the developmental function of ASD risk genes identified through de novo variants (2, 10, 35), genes supported by a majority of de novo variants (<50% inherited component support) were most highly expressed early in development in both the hCS model and BrainSpan (Fig. 3C), consistent with earlier findings (72). Interestingly, in the early developmental period, the average expression of these genes was higher than that of genes whose association with ASD was supported primarily by rare inherited variants.
We also clustered the 74 ASD risk genes identified at FDR < 0.1 using the BrainSpan data, which identified three clusters. ASD risk genes with a >50% inherited component were represented in all clusters. Cluster 1 included genes that gradually rose during development and were enriched for genes involved in learning and memory (Fig. 3D). Clusters 2 and 3 included genes that were elevated early in development and were enriched for genes with histone transferase activity (Fig. 3D). Overall, these results show ASD risk genes with a majority of de novo qualifying variants have the highest expression early in fetal cortical development and decline over time, whereas a subset of ASD risk genes with both de novo or inherited qualifying variants (Cluster 1) rise more gradually across fetal neuronal development and are involved in cognition and learning.
Rare Inherited PTV Burden of Known ASD Risk Genes in Multiplex Families.
We next collated a list of all known ASD risk genes to determine their contribution to risk in multiplex families by identifying unique genes from this study and recent large ASD genetic studies (6, 10) with an FDR < 0.1, yielding a total of 152 known ASD risk genes (KARGs) (Dataset S3). Not surprisingly, autistic children had significantly more rare inherited PTVs in KARGs compared to nonautistic children (logistic regression, P = 0.020, OR = 1.73, SI Appendix, Fig. S9A). Specifically, the rate of rare inherited PTVs in KARGs for autistic children was significantly higher in AGRE compared to ASC+SSC (57) (SI Appendix, Fig. S9C). In contrast to observations in simplex families (1–8, 10) and likely due to the lack of statistical power (54), rare de novo PTVs in KARGs were not increased in autistic versus nonautistic children from AGRE (P = 0.37, SI Appendix, Fig. S9B). We confirmed a significant depletion of rare de novo PTVs in KARGs for autistic children in AGRE compared to SSC (56) and ASC+SSC (57) (SI Appendix, Fig. S9C).
We next examined whether there were autistic children who inherited more than one PTV in KARGs, referred to as oligogenic transmission (14). We identified six autistic children carrying two rare inherited PTVs in KARGs, all of whom were sibling pairs from three different families (SI Appendix, Fig. S10). Notably, we found no significant evidence of an oligogenic or two-hit inheritance model, which indicates that this is not a major mode of risk transmission or that our power to detect such an inheritance pattern is limited (SI Appendix).
Finally, we investigated the effect of rare inherited and rare de novo variants in these 152 KARGs on critical phenotypic measures describing autistic children’s cognitive, social, and motor development. We observed significantly lower verbal IQ scores as compared to standard median values in all groups of autistic children, including carriers of rare inherited variants in KARGs (one-sample sign test, FDR = 0.001), de novo carriers (FDR = 0.02), and noncarriers (FDR = 2e−20, Fig. 4A). Interestingly, we found significantly higher than population average nonverbal IQ scores (Raven population average = 100) in the autistic noncarriers (FDR = 1e−7, Fig. 4B), consistent with IQ lowering effects of rare variants observed in other studies (10, 31). Both autistic noncarriers and rare inherited carriers showed significantly delayed first steps taken (autistic noncarriers FDR = 1e−9, rare inherited carrier FDR = 0.03, Fig. 4C), first words spoken (autistic noncarriers FDR = 8e−156, rare inherited carrier FDR = 2e−23, Fig. 4D), and first phrase spoken (autistic noncarriers FDR = 8e−226, rare inherited carrier FDR = 1e−25, Fig. 4E). We observed the largest effect of rare inherited KARG variants on language development (Fig. 4 D and E), specifically on the “first phrase” milestone achievement, with a median delay of 30 mo compared to nonautistic children from the same cohort (Fig. 4E). As expected, all groups of autistic children showed significantly higher Social Responsiveness Scale (SRS) total raw scores compared to nonautistic children from the same cohort (Fig. 4F).
Fig. 4.

Phenotypic effect of rare and common risk for ASD in autistic children from AGRE multiplex families. (A–F) Phenotypic distributions in autistic children, stratified by those carrying rare inherited (RI) or rare de novo (RDN) variants in known ASD risk genes (KARGs) and those not carrying such variants. (A) Verbal IQ distribution for autistic children. Standard scores from PPVT-3 (Peabody Picture Vocabulary Test, 3rd edition) were used and compared to a standard hypothesized median value of 100. Autistic noncarrier: n = 767, mu = 89; autistic RI carrier: n = 70, mu = 88; autistic RDN carrier: n = 10, mu = 84.5. (B) Nonverbal IQ distribution for autistic children. Standard scores from the Raven test were used and compared to a standard hypothesized median value of 100. Autistic noncarrier: n = 987, mu = 104; autistic RI carrier: n = 98, mu = 100; autistic RDN carrier: n = 12, mu = 108.5. (C) Age of walking distribution for autistic children whose ADI-R (Autism Diagnostic Interview-Revised) data were available. A standard hypothesized median value of 12 mo was used for comparison (SI Appendix). Autistic noncarrier: n = 1180, mu = 12; autistic RI carrier: n = 120, mu = 13; autistic RDN carrier: n = 21, mu = 13. (D) Age of first word for autistic children whose ADI-R data were available. Autistic noncarrier: n = 1223, mu = 26; autistic RI carrier: n = 126, mu = 30; autistic RDN carrier: n = 21, mu = 18; nonautistic noncarrier: n = 295; mu = 12. (E) Age of first phrase for autistic children whose ADI-R data were available. Autistic noncarrier: n = 1220, mu = 42; autistic RI carrier: n = 126, mu = 48; autistic RDN carrier: n = 21, mu = 40; nonautistic noncarrier: n = 283; mu = 18. (F) SRS (Social Responsiveness Scale) total raw score distribution for autistic and nonautistic children. Autistic noncarrier: n = 709, mu = 104; autistic RI carrier: n = 73, mu = 98; autistic RDN carrier: n = 8, mu = 113.5; nonautistic noncarrier: n = 153; mu = 19. Noncarrier distributions are depicted in black, RI carrier ones in blue, and RDN ones in red. Standard hypothesized median values used for each phenotypic comparison are reported as green vertical lines. For D–F, these median values were derived from the corresponding phenotypic distributions for nonautistic noncarriers (shown in green) for whom phenotypic data were available. Each group of autistic children was tested for differences between its phenotypic median value and the standard hypothesized one by two-sided one-sample sign test. The three tests performed for each phenotypic measure were corrected for multiple hypothesis testing using the Benjamini and Hochberg (BH) procedure as an FDR-controlling method. An asterisk is used to denote an FDR < 0.05. Median values together with their 95% CI are represented by horizontal point ranges over the distributions. (G–I) ASD polygenic score transmission stratified by TADA gene carrier type and association with language and social impairment. (G) pTDT in autistic and nonautistic children, tested for all subjects (All - autistic: n = 1231; nonautistic: n = 288) and stratified by those carrying rare inherited (TADA RI carrier - autistic: n = 70; nonautistic: n = 8) or rare de novo (TADA RDN carrier - autistic: n = 12) variants in the 74 TADA genes and those without such variants (noncarriers - autistic: n = 1149; nonautistic: n = 279). (H) pTDT in autistic noncarriers of variants in the 74 TADA genes stratified by those with delayed (age of first word: n = 407; phrase: n = 564) language development (i.e., age of first word and phrase greater than 24 and 33 mo, respectively; SI Appendix) and those with nondelayed (age of first word: n = 432; phrase: n = 275) language development. (I) pTDT in autistic noncarriers of variants in the 74 TADA genes stratified by the degree of social impairment based on SRS (Social Responsiveness Scale) T-score cut-off ranges. T-scores (based on child’s sex) equal to 59 or less were considered non-ASD (n = 122), those 60 to 75 were considered mild ASD (n = 70), and those equal to or greater than 76 were considered severe ASD (n = 275). Polygenic transmission disequilibrium is shown on the y-axis of each panel as point ranges of the SD on the mid-parent distribution mean values together with their 95% CI. The probability of each pTDT deviation distribution mean being equal to zero was tested by a two-sided one-sample t-test. An asterisk is used to denote a P-value < 0.05.
Overall, these findings show that contributory rare protein-truncating variants in known ASD risk genes of autistic children from multiplex families are inherited from nonautistic parents. Furthermore, some autistic children may carry more than one of these variants in ASD risk genes, although this is relatively rare. Lastly, ASD phenotypes, particularly nonverbal IQ and language development, vary by the form of known ASD risk gene inheritance.
ASD Polygenic Score Is Overtransmitted in Autistic Children with Inherited Variants and Associated with Relevant Phenotypes.
We next assessed the degree to which common variation contributes to ASD susceptibility in those harboring either de novo or rare inherited variants in the 74 TADA genes identified at FDR < 0.1 in this cohort. We used the polygenic transmission disequilibrium test (pTDT) (31, 73) and confirmed overall that autistic children have an overtransmission of ASD PGS (one-sample t-test, P = 0.005, pTDT deviation mean = 0.12, Fig. 4G, Materials and Methods), while nonautistic siblings do not (P = 0.08, Fig. 4G).
To compare the contributions of common variation to subjects harboring different variant classes, we stratified both autistic and nonautistic children into those with rare de novo PTV and missense variants in the 74 TADA genes and those with rare inherited PTVs (Fig. 4G). Among autistic children with an inherited PTV, we observed a significant overtransmission of ASD PGS (P = 0.05, pTDT deviation mean = 0.34). In contrast and likely due to the small group sample sizes, autistic children harboring a rare de novo PTV and/or missense variant (P = 1, Fig. 4G) and nonautistic children with an inherited PTV do not exhibit an overtransmission of ASD PGS (P = 0.2, Fig. 4G). We observed a small, but significant, overtransmission of ASD PGS in both autistic (P = 0.02, pTDT deviation mean = 0.1) and nonautistic (P = 0.03, pTDT deviation mean = 0.17) rare variant noncarriers (Fig. 4G).
To investigate the effect of ASD common variation on phenotypes related to cognitive and social impairment, as well as motor and language development, we performed pTDT analysis for autistic noncarriers divided into groups based on critical cut-offs for each measure of interest (SI Appendix). Interestingly, we observed the significant overtransmission of ASD PGS in children showing delayed language development (age of first word and phrase greater than 24 and 33 mo, respectively) (first word: P = 0.01, pTDT deviation mean = 0.19; first phrase: P = 0.03, pTDT deviation mean = 0.14; Fig. 4H) and not in those with a nondelayed language development (age of first word and phrase before or at 24 and 33 mo, respectively) (first word P = 0.4, first phrase p = 0.4, Fig. 4H). The same relationship of PGS with language delay is not observed for other related trait PGS, such as those for educational attainment, schizophrenia, or bipolar disorder (SI Appendix, Fig. S11B), consistent with the relative specificity of this relationship for ASD PGS.
In addition, we stratified the autistic noncarriers by their social behavior and social interaction skills using T-score cut-off ranges from the SRS. As expected, the social impairment of these autistic children falls over a spectrum despite their diagnosis (Fig. 4I). Interestingly, we found a significant overtransmission of ASD PGS only in children with severe social impairment (p = 0.05, pTDT deviation mean = 0.17, Fig. 4I) and not in those with mild social impairment (P = 0.4, Fig. 4I). These results confirm social behavior impairment as a core diagnostic domain for ASD and show its association with ASD PGS. Finally, we did not note any suggestive link between ASD PGS overtransmission and cognitive impairment (SI Appendix, Fig. S11 D and E), or motor development (SI Appendix, Fig. S11F).
Taken together, these findings indicate that ASD PGS is overtransmitted to autistic children, especially those with inherited protein-truncating variants in ASD risk genes. Furthermore, overtransmission of ASD PGS is observed in autistic children with more severe language and social impairment, connecting the presence of language impairment to common genetic risk for ASD.
Discussion
Most genetic studies on ASD focus on simplex families, in which only one child is autistic, and that optimally isolate strongly acting, rare de novo variants (1–8, 10). These large sequencing efforts on simplex cohorts have elucidated ASD genetic architecture and facilitated the identification of risk genes. However, our previous study on a subset of the AGRE multiplex families detected for the first time a strong additional risk contribution from rare inherited variants which was not detected in simplex families (9). The study suggested a different genetic architecture for multiplex families which subsequently led to the discovery of new ASD risk genes whose association signal was derived from rare inherited variation and that impacted distinct biological processes from those hit by rare de novo variation (9). WGS analysis here reported on the extended AGRE multiplex family cohort, which is two times the size, provided the opportunity to 1) validate and extend the list of ASD risk genes at FDR < 0.1 supported by rare inherited variation, 2) confirm the distinct ASD genetic architecture of multiplex families in a sufficiently large cohort, and 3) more thoroughly assess the additive effects of both common and rare variation for ASD and their influence on the phenotypic spectrum.
We identified seven previously unrecognized ASD risk genes, five of which are impacted by a significantly higher proportion of rare inherited PTVs versus rare de novo variants. We view these as candidates needing additional confirmation. Our extended cohort analysis also validated two recently identified ASD risk genes, KMT2A and MED13L, and confirmed that their association signal is supported primarily by RDNVs (10). The overall yield of de novo rare variants in this study likely reflects diminishing returns of novel ASD risk genes harboring de novo variation with increasing sample sizes. This is consistent with recent studies relying primarily on de novo gene discovery since even large increases in sample size led to the discovery of only five new exome-wide significant risk genes (56). It is also consistent with the recent observation that rare inherited variation mostly impacts previously unknown risk genes (56, 57, 66), impacting similar pathways as known genes. We also did not identify an increased number of noncoding rare inherited, private inherited, or rare de novo variants in autistic children, despite focusing on putatively functional elements of the noncoding genome (60–62). Noncoding ASD genetic risk in simplex families has been identified from large DNase I hypersensitivity regions (7), TFBSs within the distal promoters of conserved loci (59), and noncoding variants predicted to be deleterious from deep-learning (74). Our analysis agrees with previous studies suggesting that sample sizes of roughly 8,000 fully-phaseable children would be necessary to be able to identify noncoding ASD genetic risk with 80% power (12).
The contribution of rare inherited variants to ASD risk in multiplex families is confirmed by our focused analysis of known ASD risk genes (10, 14). Autistic probands in AGRE multiplex families have a significant excess of rare inherited PTVs in known risk genes compared to their nonautistic siblings, an observation recently confirmed by analyses using a larger nonautistic sibling group from SSC (55) and consistent with findings on private inherited likely gene-disrupting variants (66). In addition to contributions from rare inherited variation, inherited risk in multiplex cases may in some instances involve the transmission of more than one rare PTV to autistic children. We identified six autistic siblings from three families inheriting the same pair of PTVs in two different known ASD risk genes from their nonautistic parents. However, this mode of transmission was not statistically significant overall, and this finding should be considered anecdotal at this point. A similar oligogenic burden in autistic children was posited for de novo variants in simplex families previously (14). Much larger multiplex family cohorts will be needed to assess the significance of rare inherited oligogenic burden as a contributor to ASD risk.
Previous analyses investigating additivity between common polygenic and rare variation in simplex families showed that ASD PGS is overtransmitted to autistic individuals carrying a major de novo variant (31). In AGRE multiplex families, ASD PGS is significantly overtransmitted to autistic children harboring rare inherited protein-coding variants in ASD risk genes. In addition, we observed a small, but significant, overtransmission of ASD PGS in both autistic and nonautistic children who did not carry rare variants in ASD risk genes. Our findings can be considered consistent with the liability threshold model (75) explaining ASD complex genetic architecture. Rare and common variants contribute incrementally to a continuum of behavioral and developmental traits, of which the severe tail results in ASD diagnosis (76, 77), and the total genetic load sufficient for diagnosis, or threshold, can be reached through differing combinations of rare and common variation (31, 78–80). The liability threshold model likely explains why nonautistic siblings and parents of autistic individuals, despite sharing ASD genetic risk factors and showing subclinical traits (81–83), do not attain an ASD diagnosis.
Another important observation is that ASD PGS is not only overtransmitted to autistic children with severe social impairment but also to those with delayed early language development. A recent study found a positive association between ASD PGS and earlier age of first word in autistic children from SSC and SPARK (84). However, this signal was no longer significant after accounting for the presence of high-impact de novo variants and full-scale IQ (84).
The link between overinheritance of ASD PGS and language delay is clinically relevant, as our data support its role as a core feature related to genetic risk for ASD, providing genetic evidence to inform and refine current ASD diagnostic criteria (85). Should this result be replicated in future studies on larger cohorts recruited more recently than AGRE, we contend that language delay should not represent a secondary consideration with regard to diagnosis, as it is in the current version of DSM-5 (86). Delayed language development has been described as a highly visible symptom associated with earlier ASD diagnosis (87). Since an association between ASD PGS and language delay has been observed now, it will be important to replicate it in additional cohorts in which ASD is diagnosed using DSM-5 criteria. Nevertheless, this work highlights the value of multiplex family cohorts in ASD research: in these cohorts, the effects of rare and common inherited variation are more easily observed and can be associated with neurobiologically relevant phenotypes, such as language or social function.
Materials and Methods
ASD Multiplex Family Samples.
Throughout this manuscript, the term ‘nonautistic’ refers to individuals who do not have an autism diagnosis at recruitment into the study, while ‘autistic’ refers to individuals who have an autism diagnosis at recruitment into the study and is a preferred identity-first term for many but not all autistic people (88, 89).
Subjects originated from the Autism Genetic Resource Exchange (AGRE) (38). AGRE's human subject research was reviewed and approved by the Western Institutional Review Board (now WCG IRB). AGRE representatives discussed, face-to-face or by phone, consent and assent documents and study participation with families. All adults capable of giving consent signed the informed consent form. All minor children and all adults unable to give consent gave their assent to the best of their ability to participate in AGRE. As a result of this study being limited to previously existing coded data and specimens, the UCLA and Stanford IRBs considered it “Not human subjects research” and thus exempt from review.
Subjects were carefully selected from families including at least two members with ASD. Sequencing was not performed on patients with known genetic causes of ASD or syndromes overlapping with ASD features. Dataset S1 contains a complete list of sequenced samples.
Of subjects selected from AGRE, 4,551 individuals from 1,004 ASD families passed quality control (Dataset S1). Our variant analysis included 4,531 individuals (2,136 autistic and 2,395 nonautistic); 20 children for whom both biological parents were not sequenced were excluded. In this manuscript, “HNPs” refers to 1,890 healthy cohort samples for which no biological parents have been sequenced (non-phaseable) (SI Appendix).
TADA Mega-Analysis.
Samples and qualifying variants.
To combine evidence found in ASD subjects from rare de novo (DN) or transmitted (inherited) PTVs as well as DN mis3 variants predicted to damage the encoded protein, we used the Transmitted and De novo Association (TADA) test (63). Our TADA analysis was conducted on 1,660 non-ARC outlier, genetically nonidentical (one MZ twin retained) ASD cases from 863 families with at least one autistic individual and both biological parents sequenced. We treated these 1,660 autistic children and their biological parents as independent trios in the TADA analysis. We confirmed the validity of this approach through 100,000 TADA simulations (SI Appendix) (9). We merged qualifying variants found in ASD individuals from our cohort with a previous TADA mega-analysis (6), which included variants discovered from the Autism Sequencing Consortium (ASC) and Simons Simplex Collection (SSC), as well as small de novo CNV deletions (SmallDel) identified from SSC and Autism Genome Project (AGP) probands (Dataset S2).
Qualifying variants in our cohort included rare DN and transmitted PTVs as well DN mis3 variants not flagged as low-confidence by the GIAB consortium (90). As in the previous TADA mega-analysis (6), transmitted PTVs were required to have an AF ≤ 0.1% among public databases (1,000g, ESP6500, ExACv3.0, cg46, gnomAD), internal controls, and HNPs (SI Appendix). DN variants were required to be absent (AF = 0) across all public databases, internal controls, and HNPs. ARC was run on all non-MZ twins to obtain high-confidence DN variants. DN variants shared in MZ twins were also considered qualifying variants and not filtered on their ARC score. For the TADA samples identified as ARC outliers (n = 421), their DN variants were excluded but their transmitted PTVs were retained as qualifying variants.
For the TADA analysis, we then aggregated the counts for all the qualifying variants from the different cohorts into a gene-by-variant-type matrix, which included variant counts for a total of 18,472 gencodeV19 genes with HGNC-approved gene names (Dataset S2).
A gene was considered previously unrecognized if it had an FDR < 0.1 in our TADA mega-analysis and lacked genome-wide statistical support across all previous studies (4, 6, 21) with statistical rigor.
Fetal Single-Cell Expression of TADA Genes.
Expression of TADA genes was examined in previously published single-cell human developing neocortex fetal dataset (67). See SI Appendix for additional details.
Developmental Trajectory Analyses.
Development trajectory RNAseq data were used from previous studies including the long-term maturation human cortical spheroid (hCS) model (68) and the BrainSpan dataset (69–71). See SI Appendix for additional details.
pTDT Analysis.
Polygenic scoring.
A PGS was calculated to provide a quantitative estimate for offspring’s genetic predisposition for ASD, SCZ, BD, and EA in the AGRE multiplex families. To generate PGS, 3,255 samples of European ancestry, identified by multidimensional scaling (MDS)-derived hard race calls, were used (Naut = 1,492, Nnonaut = 1,763). For each phenotype tested, we obtained summary statistics from a GWAS with similar ancestry: for ASD, we used the ASD iPSYCH GWAS (this cohort overlaps with the PGC (Psychiatric Genomics Consortium) ASD GWAS; Naut = 8,605, Nnonaut = 19,526) (91) (SI Appendix). Candidate variant weights were then generated by running LDpred (92). For all phenotypes tested, PGS models assuming that all SNPs were causal were selected for downstream analysis (SI Appendix). We observe a significant difference in ASD PGS between the tested autistic and nonautistic subjects (p = 0.01, beta = 0.01, difference (versus PGS null model) in Nagelkerke’s R2 = 0.003).
pTDT variant analysis.
Carriers for variants in the 74 genes identified at FDR < 0.1 in the TADA mega-analysis were included in the pTDT analysis (31). The same rare qualifying variant classes as TADA were considered. Transmitted PTVs in ASD risk genes were identified in the 1,519 fully phaseable children of European who do not identify as a parent in the cohort (Naut = 1,231, Nnonaut = 288), while DN PTV or mis3 qualifying variants in ASD risk genes were restricted to those identified in the 1,066 non-ARC outlier samples (Naut = 867, Nnonaut = 199) (SI Appendix).
Phenotypic Comparisons among Children of AGRE Multiplex Families.
We tested phenotypic measures descriptive of general intelligence, motor development, language development, and social behavior, which were collected through behavioral assessments performed by AGRE (38). The selected measures were verbal and nonverbal IQ scores, age of walking (AOW), ages of first word and phrase, and scores representing social behavior and social interaction skills. We used the standard scores from PPVT-3 (Peabody Picture Vocabulary Test, 3rd edition) (93) and the Raven test (94) as measures of verbal and nonverbal IQ, respectively. AOW and ages of first word and phrase in months were retrieved from ADI-R (Autism Diagnostic Interview-Revised, 2003 version with 93 items) (95). We made use of total raw scores and T-scores from SRS (Social Responsiveness Scale) (96) as measures of impairment in social behavior (SI Appendix).
Supplementary Material
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Dataset S04 (XLSX)
Dataset S05 (XLSX)
Acknowledgments
We thank the subjects and families from the Autism Genetic Resource Exchange (AGRE) who participated in this study, in addition to the Hartwell Foundation, and the New York Genome Center. We thank Christopher L. Hartl for his contribution to code for TADA simulations. We thank Stephanie N. Kravitz, Cheyenne L. Schloffman, David Keller, Min Sun, Tor Solli-Nowlan, Sasha Sharma, Marlena Duda, Greg Madden McInnes, Ravina Jain, Valentı´Moncunill, Josep M. Mercader, Montserrat Puiggròs, Anika Gupta, and David Torrents for technical support. We are grateful to all of the families at the participating SSC sites as well as the principal investigators (A. Beaudet, R. Bernier, J. Constantino, E. Cook, E. Fombonne, D. Geschwind, R. Goin-Kochel, E. Hanson, D. Grice, A. Klin, D. Ledbetter, C. Lord, C. Martin, D. Martin, R. Maxim, J. Miles, O. Ousley, K. Pelphrey, B. Peterson, J. Piggot, C. Saulnier, M. State, W. Stone, J. Sutcliffe, C. Walsh, Z. Warren, and E. Wijsman). This work was supported by grants from The Hartwell Foundation and the NIH (R01MH100027, R01MH064547, S10OD011939, P50HD055784, UM1HG008901, P50DC018006, K08AG065519 R01MH064547, and S10OD011939) and from the Stanford Precision Health and Integrated Diagnostics Center and from the Stanford Bio-X Center. We are grateful to The Hartwell Foundation for supporting the genetic sequencing and analyses in this study and for the opportunity to contribute to their mission of benefitting children. We are grateful to the Simons Foundation for additional support for genome sequencing. We thank the New York Genome Center for conducting sequencing and initial quality control. We thank Amazon Web Services for their grant support for the computational infrastructure and storage for the cohort database. AGRE is a program of Autism Speaks and was supported by grant NIMH U24 MH081810.
Author contributions
M.C., T.S.C., L.P.-C., E.K.R., J.K.L., D.P.W., and D.H.G. designed research; M.C., T.S.C., S.A.A., L.P.-C., E.K.R., A.G., and L.K.B. performed research; J.-Y.J. and D.P.W. contributed new reagents/analytic tools; M.C., T.S.C., S.A.A., L.P.-C., E.K.R., A.G., L.K.B., J.-Y.J., and J.K.L. analyzed data; D.P.W. supplied funding; D.H.G. supervised experimental design and analysis, interpreted results, and provided funding; and M.C., T.S.C., S.A.A., and D.H.G. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Contributor Information
Timothy S. Chang, Email: timothychang@mednet.ucla.edu.
Daniel H. Geschwind, Email: dhg@mednet.ucla.edu.
Data, Materials, and Software Availability
The whole-genome sequencing data generated during this study is available upon request and can be freely downloaded from the Autism Research and Technology Initiative (iHART) of the Hartwell Foundation, after request and approval of the data use agreement found at http://www.ihart.org (responsible PI: Dennis P. Wall; dbGaP Study Accession: phs001766.v1.p1) (9, 97). Autism Speaks and AGRE must approve the release of whole-genome sequencing data generated during this study (https://www.autismspeaks.org/applying-access-agre-data-and-biomaterials). We utilized the ARC (Artifact Removal by Classifier) code, as developed prior, available here: https://github.com/walllab/iHART-ARC (9, 98). The Simons Simplex Cohort data described in this study can be obtained through an application to SFARI Base (https://base.sfari.org).
Supporting Information
References
- 1.O’Roak B. J., et al. , Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485, 246–250 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Iossifov I., et al. , The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O’Roak B. J., et al. , Recurrent de novo mutations implicate novel genes underlying simplex autism risk. Nat. Commun. 5, 5595 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De Rubeis S., et al. , Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krumm N., et al. , Excess of rare, inherited truncating mutations in autism. Nat. Genet. 47, 582–588 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sanders S. J., et al. , Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron 87, 1215–1233 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Turner T. N., et al. , Genome sequencing of autism-affected families reveals disruption of putative noncoding regulatory DNA. Am. J. Hum. Genet. 98, 58–74 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stessman H. A. F., et al. , Targeted sequencing identifies 91 neurodevelopmental-disorder risk genes with autism and developmental-disability biases. Nat. Genet. 49, 515–526 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ruzzo E. K., et al. , Inherited and de novo genetic risk for autism impacts shared networks. Cell 178, 850–866.e26 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Satterstrom F. K., et al. , Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell 180, 568–584.e23 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yuen R. K., et al. , Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat. Neurosci. 20, 602–611 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Werling D. M., et al. , An analytical framework for whole-genome sequence association studies and its implications for autism spectrum disorder. Nat. Genet. 50, 727–736 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feliciano P., et al. , Exome sequencing of 457 autism families recruited online provides evidence for autism risk genes. NPJ Genomic Med. 4, 19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Turner T. N., et al. , Genomic patterns of de novo mutation in simplex autism. Cell 171, 710–722.e12 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dong S., et al. , De novo insertions and deletions of predominantly paternal origin are associated with autism spectrum disorder. Cell Rep. 9, 16–23 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Neale B. M., et al. , Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485, 242–245 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sebat J., et al. , Strong association of de novo copy number mutations with autism. Science 316, 445–449 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marshall C. R., et al. , Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet. 82, 477–488 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bucan M., et al. , Genome-wide analyses of exonic copy number variants in a family-based study point to novel autism susceptibility genes. PLoS Genet. 5, e1000536 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Epi4K Consortium et al. , De novo mutations in epileptic encephalopathies. Nature 501, 217–221 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Satterstrom F. K., et al. , Autism spectrum disorder and attention deficit hyperactivity disorder have a similar burden of rare protein-truncating variants. Nat. Neurosci. 22, 1961–1965 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Singh T., et al. , The contribution of rare variants to risk of schizophrenia in individuals with and without intellectual disability. Nat. Genet. 49, 1167–1173 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cross-Disorder Group of the Psychiatric Genomics Consortium, Genomic relationships, novel loci, and pleiotropic mechanisms across eight psychiatric disorders. Cell 179, 1469–1482.e11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de la Torre-Ubieta L., Won H., Stein J. L., Geschwind D. H., Advancing the understanding of autism disease mechanisms through genetics. Nat. Med. 22, 345–361 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Geschwind D. H., State M. W., Gene hunting in autism spectrum disorder: On the path to precision medicine. Lancet Neurol. 14, 1109–1120 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abrahams B. S., Geschwind D. H., Advances in autism genetics: On the threshold of a new neurobiology. Nat. Rev. Genet. 9, 341–355 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gaugler T., et al. , Most genetic risk for autism resides with common variation. Nat. Genet. 46, 881–885 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Colvert E., et al. , Heritability of autism spectrum disorder in a UK population-based twin sample. JAMA Psychiatry 72, 415–423 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sandin S., et al. , The heritability of autism spectrum disorder. JAMA 318, 1182–1184 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bai D., et al. , Association of genetic and environmental factors with autism in a 5-country cohort. JAMA Psychiatry 76, 1035–1043 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weiner D. J., et al. , Polygenic transmission disequilibrium confirms that common and rare variation act additively to create risk for autism spectrum disorders. Nat. Genet. 49, 978–985 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grove J., et al. , Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet. 51, 431–444 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barton N. H., Etheridge A. M., Véber A., The infinitesimal model: Definition, derivation, and implications. Theor. Popul. Biol. 118, 50–73 (2017). [DOI] [PubMed] [Google Scholar]
- 34.Fisher R. A., XV.—The correlation between relatives on the supposition of mendelian inheritance. Earth Environ. Sci. Trans. R. Soc. Edinb. 52, 399–433 (1919). [Google Scholar]
- 35.Ronemus M., Iossifov I., Levy D., Wigler M., The role of de novo mutations in the genetics of autism spectrum disorders. Nat. Rev. Genet. 15, 133–141 (2014). [DOI] [PubMed] [Google Scholar]
- 36.Leppa V. M., et al. , Rare inherited and de novo cnvs reveal complex contributions to ASD risk in multiplex families. Am. J. Hum. Genet. 99, 540–554 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yuen R. K., et al. , Whole-genome sequencing of quartet families with autism spectrum disorder. Nat. Med. 21, 185–191 (2015). [DOI] [PubMed] [Google Scholar]
- 38.Lajonchere C. M., AGRE Consortium, Changing the landscape of autism research: The autism genetic resource exchange. Neuron 68, 187–191 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lord C., Rutter M., Le Couteur A., Autism diagnostic interview-revised: A revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J. Autism Dev. Disord. 24, 659–685 (1994). [DOI] [PubMed] [Google Scholar]
- 40.Lord C., et al. , The autism diagnostic observation schedule-generic: A standard measure of social and communication deficits associated with the spectrum of autism. J. Autism Dev. Disord. 30, 205–223 (2000). [PubMed] [Google Scholar]
- 41.Geschwind D. H., et al. , The autism genetic resource exchange: A resource for the study of autism and related neuropsychiatric conditions. Am. J. Hum. Genet. 69, 463–466 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang J., Raskin L., Samuels D. C., Shyr Y., Guo Y., Genome measures used for quality control are dependent on gene function and ancestry. Bioinformatics 31, 318–323 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Conrad D. F., et al. , Variation in genome-wide mutation rates within and between human families. Nat. Genet. 43, 712–714 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.1000 Genomes Project Consortium et al. , A global reference for human genetic variation. Nature 526, 68–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Besenbacher S., et al. , Multi-nucleotide de novo mutations in humans. PLoS Genet. 12, e1006315 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kong A., et al. , Rate of de novo mutations and the importance of father’s age to disease risk. Nature 488, 471–475 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Michaelson J. J., et al. , Whole-genome sequencing in autism identifies hot spots for de novo germline mutation. Cell 151, 1431–1442 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Deciphering Developmental Disorders Study, Prevalence and architecture of de novo mutations in developmental disorders. Nature 542, 433–438 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Francioli L. C., et al. , Genome-wide patterns and properties of de novo mutations in humans. Nat. Genet. 47, 822–826 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Goldmann J. M., et al. , Parent-of-origin-specific signatures of de novo mutations. Nat. Genet. 48, 935–939 (2016). [DOI] [PubMed] [Google Scholar]
- 51.Adzhubei I. A., et al. , A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Samocha K. E., et al. , Regional missense constraint improves variant deleteriousness prediction. bioRxiv [Preprint] (2017). 10.1101/148353 (Accessed 24 April 2021). [DOI]
- 53.Lek M., et al. , Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yoon S., et al. , Rates of contributory de novo mutation in high and low-risk autism families. Commun. Biol. 4, 1026 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Trost B., et al. , Genomic architecture of autism from comprehensive whole-genome sequence annotation. Cell 185, 4409–4427.e18 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhou X., et al. , Integrating de novo and inherited variants in 42,607 autism cases identifies mutations in new moderate-risk genes. Nat. Genet. 54, 1305–1319 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fu J. M., et al. , Rare coding variation provides insight into the genetic architecture and phenotypic context of autism. Nat. Genet. 54, 1320–1331 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Karczewski K. J., et al. , The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.An J.-Y., et al. , Genome-wide de novo risk score implicates promoter variation in autism spectrum disorder. Science 362 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gheorghe M., et al. , A map of direct TF-DNA interactions in the human genome. Nucleic Acids Res. 47, e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vorontsov I. E., et al. , Genome-wide map of human and mouse transcription factor binding sites aggregated from ChIP-Seq data. BMC Res. Notes 11, 756 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xu D., Wang C., Kiryluk K., Buxbaum J. D., Ionita-Laza I., Co-localization between sequence constraint and epigenomic information improves interpretation of whole-genome sequencing data. Am. J. Hum. Genet. 106, 513–524 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.He X., et al. , Integrated model of de novo and inherited genetic variants yields greater power to identify risk genes. PLoS Genet. 9, e1003671 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pinto D., et al. , Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am. J. Hum. Genet. 94, 677–694 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Simons Foundation Autism Research Initiative, SFARI Gene 3.0 (2020), https://gene.sfari.org/ (Accessed 3 October 2020).
- 66.Wilfert A. B., et al. , Recent ultra-rare inherited variants implicate new autism candidate risk genes. Nat. Genet. 53, 1125–1134 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Polioudakis D., et al. , A single cell transcriptomic atlas of human neocortical development during mid-gestation. Neuron 103, 785–801.e8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gordon A., et al. , Long-term maturation of human cortical organoids matches key early postnatal transitions. Nat. Neurosci. 24, 331–342 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stein J. L., et al. , A quantitative framework to evaluate modeling of cortical development by neural stem cells. Neuron 83, 69–86 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kang H. J., et al. , Spatio-temporal transcriptome of the human brain. Nature 478, 483–489 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li M., et al. , Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science 362, eaat7615 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Parikshak N. N., Gandal M. J., Geschwind D. H., Systems biology and gene networks in neurodevelopmental and neurodegenerative disorders. Nat. Rev. Genet. 16, 441–458 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Spielman R. S., McGinnis R. E., Ewens W. J., Transmission test for linkage disequilibrium: The insulin gene region and insulin-dependent diabetes mellitus (IDDM). Am. J. Hum. Genet. 52, 506–516 (1993). [PMC free article] [PubMed] [Google Scholar]
- 74.Zhou J., et al. , Whole-genome deep-learning analysis identifies contribution of noncoding mutations to autism risk. Nat. Genet. 51, 973–980 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Falconer D. S., The inheritance of liability to certain diseases, estimated from the incidence among relatives. Ann. Hum. Genet. 29, 51–76 (1965). [Google Scholar]
- 76.Bulik-Sullivan B., et al. , An atlas of genetic correlations across human diseases and traits. Nat. Genet. 47, 1236–1241 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Robinson E. B., et al. , Genetic risk for autism spectrum disorders and neuropsychiatric variation in the general population. Nat. Genet. 48, 552–555 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Antaki D., et al. , A phenotypic spectrum of autism is attributable to the combined effects of rare variants, polygenic risk and sex. Nat. Genet. 54, 1284–1292 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Takahashi N., et al. , Association of genetic risks with autism spectrum disorder and early neurodevelopmental delays among children without intellectual disability. JAMA Netw. Open 3, e1921644 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Serdarevic F., et al. , Polygenic risk scores for developmental disorders, neuromotor functioning during infancy, and autistic traits in childhood. Biol. Psychiatry 87, 132–138 (2020). [DOI] [PubMed] [Google Scholar]
- 81.Piven J., Palmer P., Jacobi D., Childress D., Arndt S., Broader autism phenotype: Evidence from a family history study of multiple-incidence autism families. Am. J. Psychiatry 154, 185–190 (1997). [DOI] [PubMed] [Google Scholar]
- 82.Constantino J. N., Zhang Y., Frazier T., Abbacchi A. M., Law P., Sibling recurrence and the genetic epidemiology of autism. Am. J. Psychiatry 167, 1349–1356 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nayar K., Shic F., Winston M., Losh M., A constellation of eye-tracking measures reveals social attention differences in ASD and the broad autism phenotype. Mol. Autism 13, 18 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Warrier V., et al. , Genetic correlates of phenotypic heterogeneity in autism. Nat. Genet. 54, 1293–1304 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Geschwind D. H., Flint J., Genetics and genomics of psychiatric disease. Science 349, 1489–1494 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.American Psychiatric Association, Diagnostic and Statistical Manual of Mental Disorders: DSM-5 (American Psychiatric Association, Arlington, VA, 2013).
- 87.Nitzan T., et al. , The importance of language delays as an early indicator of subsequent ASD diagnosis in public healthcare settings. J. Autism Dev. Disord., 10.1007/s10803-022-05757-y (2022). [DOI] [PubMed]
- 88.Kenny L., et al. , Which terms should be used to describe autism? Perspectives from the UK autism community. Autism Int. J. Res. Pract. 20, 442–462 (2016). [DOI] [PubMed] [Google Scholar]
- 89.Roman-Urrestarazu A., Dumas G., Warrier V., Naming autism in the right context. JAMA Pediatr. 176, 633–634 (2022). [DOI] [PubMed] [Google Scholar]
- 90.Zook J. M., et al. , Integrating human sequence data sets provides a resource of benchmark SNP and indel genotype calls. Nat. Biotechnol. 32, 246–251 (2014). [DOI] [PubMed] [Google Scholar]
- 91.Gandal M. J., et al. , Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science 359, 693–697 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Vilhjálmsson B. J., et al. , Modeling linkage disequilibrium increases accuracy of polygenic risk scores. Am. J. Hum. Genet. 97, 576–592 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dunn L. M., Dunn L. M., Peabody Picture Vocabulary Test-III (American Guidance Service (AGS), 1997). [Google Scholar]
- 94.Raven J., Raven J., "Raven progressive matrices" in Handbook of Nonverbal Assessment, McCallum R. S., Ed. (Springer, US, 2003), pp. 223–237. [Google Scholar]
- 95.Rutter M., Le Couteur A., Lord C., ADI-R: Autism Diagnostic Interview Revised (Western Psychological Services, 2013). [Google Scholar]
- 96.Constantino J. N., Gruber C., The Social Responsiveness Scale (SRS) (Western Psychological Services, 2005). [Google Scholar]
- 97.Cirnigliaro M., et al. , The contributions of rare inherited and polygenic risk to ASD in multiplex families. AGRE multiplex families. http://www.ihart.org/data. Accessed 19 August 2014. [DOI] [PMC free article] [PubMed]
- 98.Ruzzo E. K., et al. , Inherited and de novo genetic risk for autism impacts shared networks. iHART-ARC. https://github.com/walllab/iHART-ARC. Accessed 10 July 2019. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Dataset S04 (XLSX)
Dataset S05 (XLSX)
Data Availability Statement
The whole-genome sequencing data generated during this study is available upon request and can be freely downloaded from the Autism Research and Technology Initiative (iHART) of the Hartwell Foundation, after request and approval of the data use agreement found at http://www.ihart.org (responsible PI: Dennis P. Wall; dbGaP Study Accession: phs001766.v1.p1) (9, 97). Autism Speaks and AGRE must approve the release of whole-genome sequencing data generated during this study (https://www.autismspeaks.org/applying-access-agre-data-and-biomaterials). We utilized the ARC (Artifact Removal by Classifier) code, as developed prior, available here: https://github.com/walllab/iHART-ARC (9, 98). The Simons Simplex Cohort data described in this study can be obtained through an application to SFARI Base (https://base.sfari.org).