

Abstract
Aims
To study the brain metabolic signature in Chinese amyotrophic lateral sclerosis (ALS) patients and compare the difference in brain metabolic patterns between ALS with and without genetic variants.
Methods
We included 146 patients with ALS and 128 healthy controls (HCs). All patients with ALS underwent genetic testing to screen for ALS related genetic variants and were then divided into genetic (n = 22) and nongenetic ALS (n = 93) subgroups. All participants underwent brain 18F‐FDG‐PET scans. Group comparisons were performed using the two‐sample t‐test model of SPM12.
Results
We identified a large of hypometabolic clusters in ALS patients as compared with HCs, especially in the bilateral basal ganglia, midbrain, and cerebellum. Moreover, hypometabolism in the bilateral temporal lobe, precentral gyrus and hypermetabolism in the left anterior cingulate, occipital lobe, and bilateral frontal lobe were also found in ALS patients as compared with HCs. Compared with nongenetic ALS patients, genetic ALS patients showed hypometabolism in the right postcentral gyrus, precuneus, and middle occipital gyrus. The incidence of sensory disturbance in patients with genetic ALS was higher than that in patients with nongenetic ALS (5 of 22 [22.72%] vs. 7 of 93 [7.52%], p = 0.036).
Conclusions
Our investigation provided unprecedented evidence of relative hypometabolism in the midbrain and cerebellum in ALS patients. Genetic ALS patients showed a specific signature of brain metabolism and a higher incidence of sensory disturbance, indicating that genetic factors may be an underlying cause affecting the brain metabolism and increasing the risk of sensory disturbance in ALS.
Keywords: 18F‐FDG‐PET, amyotrophic lateral sclerosis, brain metabolism, genetic, whole exome sequencing
This is the first study to measure compare the difference in brain metabolic patterns between ALS with and without genetic variants. Interestingly, we found that genetic ALS patients showed a specific signature of brain metabolism and a higher incidence of sensory disturbance, indicating that genetic factors may be an underlying cause affecting the brain metabolism and increasing the risk of sensory disturbance in ALS.
1. INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a rare neurodegenerative disorder characterized by weakness and atrophy due to loss of both upper and lower motor neurons, causing death due to respiratory paralysis within 3‐5 years from the onset. 1 Although its etiology is still poorly understood, the interaction between genetic background, environmental, and lifestyle factors is a potential cause of ALS. 2 About 10% of cases are familial ALS (fALS), while the remaining 90% of cases are sporadic ALS (sALS). Since the SOD1 gene was reported in 1993, more than 40 genes have been reported to be linked with ALS. 1 , 2 , 3 , 4 ALS patients carrying specific mutations or variants may show distinct clinical phenotypes and prognosis. 5 Uncovering the relationship between genotype and phenotype has important implications for pathogenetic explanations in ALS.
18F‐fluorodeoxyglucose positron emission tomography (18F‐FDG‐PET) is a powerful tool to display the brain metabolic signature in ALS. 6 , 7 , 8 , 9 Previous studies have shown that different ALS phenotypes displayed their specific brain metabolic changes. For example, ALS with cognitive impairment or frontotemporal dementia (FTD) demonstrated prefrontal, anterior cingulate, and insular hypometabolism when compared with ALS with normal cognition. 10 , 11 , 12 However, a large 18F‐FDG‐PET study is still lacking in Chinese mainland. Moreover, most 18F‐FDG‐PET studies focused on the metabolic features in characterize patients carrying GGGGCC repeat expansion in C9orf72, 13 , 14 , 15 While the GGGGCC repeat expansion is rarely found in ALS patients in Asia, especially in China. 16 Moreover, genome‐wide pathogenic mutation metabolic pattern in ALS has not been systematically studied yet. Herein, the aim of this study was twofold: first, to elucidate the brain metabolic pattern in ALS patients in the mainland of China; second, to explore the brain metabolic changes characterizing genetic ALS as compared with nongenetic ALS.
2. MATERIALS AND METHODS
2.1. Participants
The present study recruited a cohort of 146 patients with ALS from the Department of Neurology, Xiangya Hospital, Central South University from January 1, 2014, to January 1, 2021. All patients with ALS were diagnosed with clinically definite‐, probable‐, or probable laboratory‐supported ALS by at least two experienced neurologists according to the revised El Escorial criteria 2015. 17 Demographic and clinical data of ALS, including age at onset (AAO), age at PET, sex, family history, site of onset, disease duration, and ALS Functional Rating Scale‐Revised (ALSFRS‐R) scores were collected by specialists. ALS patients with sensory symptoms or abnormal sensory nerve conductions detected by electromyogram were regarded as ALS patients with sensory disturbances. We enrolled 128 sex‐ and age‐matched healthy controls (HCs) from the Department of Nuclear Medicine in Xiangya Hospital, of which, healthy was defined as (i) absence of no oncologic disease, (ii) with brain 18F‐FDG‐PET scan reported as normal by at least two nuclear medicine doctors, (iii) with normal neurological examination, and (iv) lack of a history of neurological diseases.
2.2. Genetic analysis
In this study, 115 patients with ALS in this study underwent genetic testing by whole exome sequencing (WES), standard polymerase chain reaction (PCR) and repeat primed polymerase chain reaction (RP‐PCR) assay. The genomic data was stored in our inhouse data base (National Geriatric Clinical Medical Research Center, Xiangya Hospital, Bioinformatics Center). According to previous studies (Table S1), 2 , 18 we focused on 52 known ALS related pathogenic genes, of which we used WES examined 50 genes. WES was completed by the Illumina HiSeq4000 using the Agilent SureSelect Human All Exon V6. Variants were annotated by software (UCSC hg19). 19 The criteria used to define rare disease variants (RDVs) referred to the previous research. 18 Variant frequencies were initially determined in 1000 Genomes, esp6500s, gnomAD, ExAC, and our inhouse WES data of 1258 controls without any nervous neurological disease to draft rare single nucleotide polymorphisms (SNP). Heterozygous variants in dominant ALS‐causative genes with a minor allele frequency (MAF) less than 0.1% across all population databases. Homozygous or compound heterozygous variants in recessive ALS‐causative genes with a MAF less than 1% across all population databases. All stop gain/loss, frameshift, and splice‐site variants (falling within 2 bp of exon‐intron junctions) were selected for further analysis. Rare missense variants are functionally predicted to be deleterious with a ReVe value more than 0.7. 20 Polynucleotide repeat expansions in the ATXN2 and C9orf72 genes were detected by standard PCR and RP‐PCR assay. The CAG repeat expansion more than 27 in ATXN2 was consider ALS susceptibility, and the GGGGCC repeat expansion more than 30 in C9orf72 was considered pathological. We define genetic variant ALS group as ALS patients who carry RDVs in the known ALS genes, nongenetic variant ALS group as ALS patients who do not carry RDVs in the known ALS genes.
2.3. 18F‐FDG‐PET imaging acquisition
Brain 18F‐FDG‐PET was performed according to the published criteria. 21 Participants fasted at least 6 h before the examination. The fasting blood glucose was lower than 7.2 mmoL/L in all subjects before the test. A dose of 3.7 MBq/kg of 18F‐FDG was injected intravenously through the cubital vein over 1 min. The PET images were acquired in three dimensions for 5 min, starting at 60 min after intravenous 18F‐FDG injection. The full width of the scan at half‐maximum was 5.4 mm. PET/computed tomography (CT) images were acquired by a Discovery Elite PET/CT scanner (GE Healthcare, Waukesha, USA). Participants were placed in the PET scanner so that slices were parallel to the canthomeatal line. All images were reconstructed as a 256 × 256 trans‐axial matrix using the 3D VUE Point (GE Healthcare, Waukesha, USA) ordered‐subset expectation–maximization algorithm with 6 iterations and 6 subsets, which produced 47 trans‐axial images at 3.25‐mm intervals. A low‐dose CT scan was obtained simultaneously for photon attenuation correction. Participants were also monitored on‐site for other signs of adverse effects for 90 min after injection of 18F‐FDG and asked to report any ensuing adverse effects.
2.4. Statistical analysis
Data are shown as the mean (standard deviation, SD) or percentage. Statistical analysis was performed using SPSS v25.0 (IBM, New York, USA). Differences with p < 0.05 were considered statistically significant. Kolmogorov–Smirnov tests were utilized to check the normality of the clinical data. All data should be subject to tests for normality. The demographic and clinical characteristics were compared as follows. The χ 2 test was used to analyze the differences among categorical variables. The differences among quantitative, continuous variables were analyzed with Student's t‐test or the Mann–Whitney U test.
Image processing was performed using the SPM12 toolkit (https://www.fil.ion.ucl.ac.uk/spm/) implemented in MATLAB R2013b (MathWorks). Individual 18F‐FDG‐PET image volumes were spatially normalized into standard stereotactic Montreal Neurological Institute (MNI) space, resliced to 2 × 2 × 2 mm. An 8‐mm full‐width half‐maximum Gaussian kernel was used to improve between‐participant spatial alignment and smooth data for statistical analysis. 22 Once the images were spatially normalized and smoothed, a general linear model was used to carry out the appropriate voxel‐by‐voxel univariate statistical tests. Image intensity was normalized between participants to prevent interparticipant variability in cerebral tracer uptake from masking regional changes. 22 This was done using proportional scaling, which scales each image proportionally to the mean global brain activity.
Comparisons between different groups were performed using the two‐sample t‐test model of SPM12. When each patient's group was compared with HCs, sex, age at PET, and years of education were used as covariates. In the comparison of subgroups of patients with ALS, sex, age at PET, disease duration, years of education, and ALSFRS‐R score were used as covariates. The height threshold of metabolic changes was set at p ˂ 0.001 (p ˂ 0.05 Family‐wise error [FEW]‐corrected at cluster). If no cluster of significant difference was identified, a height threshold of p < 0.005 (p < 0.05 FWE‐corrected at cluster) was set to perform further exploratory analyses. After data was preprocessed using SPM12, significant clusters were visualized, reported, and anatomically labeled using the xjView (http://www.alivelearn.net/xjview) and BrainNet Viewer. 23
2.5. Ethics approval
Written informed consent was obtained from all participants, and the study protocol was approved by the Ethics Committee and the Expert Committee of Xiangya Hospital, Central South University.
3. RESULTS
3.1. Demographic and clinical features of subjects
The demographic and clinical features of all subjects are listed in Table 1, Table S5 and Table S6. In the ALS cohort, the mean AAO was 54.84 ± 10.22 years, the mean age at PET is 55.90 ± 10.10 years (97 male, 49 female). The mean disease duration was 14.44 ± 15.11 months. There were 34 ALS patients with bulbar onset and 112 ALS patients with spinal onset. The mean ALSFRS‐R score was 38.86 ± 6.60. Patients with ALS and HCs were well matched for age and sex as shown in Table 1. There was no significant difference between the genetic group and nongenetic group in terms of age at PET, AAO, sex, site of onset, disease duration, years of education, diagnostic categories, and ALSFRS‐R score (Table 1). The incidence of sensory disturbance in patients with genetic ALS was higher than that in patients with nongenetic ALS (χ 2 test, 5 of 22 [22.72%] vs. 7 of 93 [7.52%], p = 0.036).
TABLE 1.
Demographic and clinical features of genetic ALS, nongenetic ALS, and HCs.
| Total ALS patients | HCs | p | Genetic ALS | Nongenetic ALS | p | |
|---|---|---|---|---|---|---|
| Number | 146 | 128 | 22 | 93 | ||
| Age at PET (years) | 55.90 ± 10.10 | 55.24 ± 7.75 | 0.545 a | 52.18 ± 10.93 | 56.31 ± 9.23 | 0.072 a |
| Sex | ||||||
| Male (%) | 97 (66.4) | 88 (68.8) | 0.684 | 18 (81.8) | 60 (64.5) | 0.118 c |
| Female (%) | 49 (33.6) | 40 (31.2) | 4 (18.2) | 33 (35.5) | ||
| Years of education | 8.75 ± 3.45 | 8.82 ± 2.77 | 0.781 a | 8.12 ± 2.49 | 8.85 ± 2.62 | 0.314 a |
| Age at onset (years) | 54.84 ± 10.22 | 51.50 ± 10.97 | 55.29 ± 9.55 | 0.107 b | ||
| Diagnostic category (Revised El Escorial criteria 2015) | ||||||
| Definite ALS (%) | 32 (21.9) | 4 (18.1) | 22 (23.7) | 0.160 c | ||
| Probable ALS (%) | 31 (21.2) | 2 (9.1) | 23 (24.7) | |||
| Laboratory support probable ALS (%) | 83 (56.9) | 16 (72.8) | 48 (51.6) | |||
| Site of onset | ||||||
| Bulbar onset (%) | 34 (23.29) | 4 (18.2) | 26 (28.0) | 0.348 c | ||
| Spinal onset (%) | 112 (76.71) | 18 (81.8) | 67 (72.0) | |||
| Disease duration (months) | 14.44 ± 15.11 | 9.91 ± 12.50 | 14.01 ± 11.62 | 0.145 b | ||
| ALSFRS‐R | 38.86 ± 6.60 | 40.00 ± 5.07 | 38.72 ± 6.07 | 0.362 b | ||
| Sensory symptoms (%) | ||||||
| Positive | 12 (8.2) | 5 (22.7) | 7 (7.5) | 0.036 c | ||
| Negative | 134 (97.8) | 17 (77.3) | 86 (92.5) | |||
| Genetic status | ||||||
| SOD1 | 5 (5/115) | 5 (5/22) | ||||
| OPTN | 3 (3/115) | 3 (3/22) | ||||
| Other genes | 14 (14/115) | 14 (14/22) | ||||
Abbreviations: ALS, Amyotrophic lateral sclerosis; ALSFRS‐R, Amyotrophic Lateral Sclerosis Functional Rating Scale‐Revised; HCs, healthy controls.
Student's t‐test, p < 0.05 was considered significant.
Mann–Whitney U test, p < 0.05 was considered significant.
χ 2 test, p < 0.05 was considered significant.
Bold value indicate statistically significant differences with P<0.05.
3.2. Genetic features of the ALS patients
One hundred and fifteen patients with ALS completed genetic testing, and 22 of them were detected to carry causative genetic variants, contributing to 19.13% (22/115) of all ALS patients. The most common mutant gene was SOD1, followed by OPTN and CACNA1H. Mutations that we identified are listed in Table S2.
3.3. Group comparison of 18F‐FDG‐PET data
3.3.1. Patients with ALS versus HCs (height threshold at p < 0.001, p < 0.05 FWE‐corrected at cluster level)
Compared with HCs, patients with ALS showed relative hypometabolism in the bilateral temporal lobe, precentral gyrus, basal ganglia, midbrain, and cerebellum as compared with HCs (Figure 1; Table 2). Some regions with relatively increased metabolism in ALS were found in the left anterior cingulate, occipital lobe, and bilateral prefrontal lobe as compared with HCs (Figure 1; Table 2).
FIGURE 1.
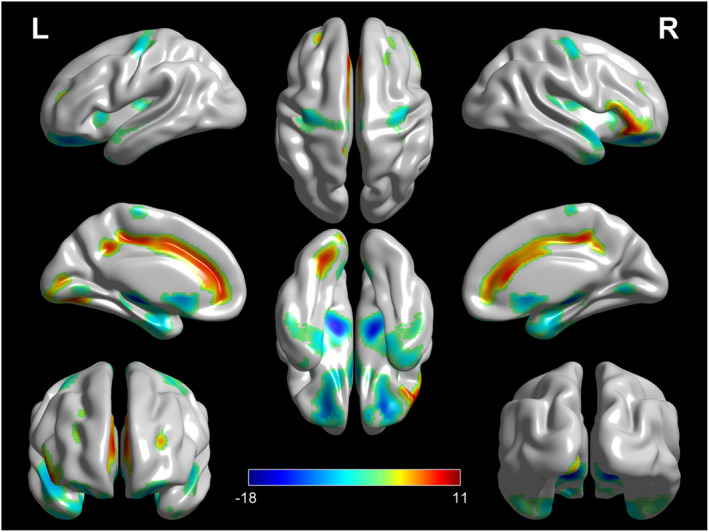
Patients with ALS versus HCs (height threshold at p < 0.001, p < 0.05 FWE‐corrected at cluster level). The regions showing statistically significant relative hypometabolism or hypermetabolism in patients with ALS as compared with HCs are reported on the brain surface.
TABLE 2.
Clusters showing a statistically significant relative hypermetabolism or hypometabolism in ALS patients as compared to HCs.
| P (FWE‐corrected) | Cluster extent | T‐score | Peak coordinates (x, y, z) (mm) | Anatomical region | Cortical region | BA | ||
|---|---|---|---|---|---|---|---|---|
| 0.000 | 6692 | 10.45 | −14 | 44 | 0 | Left limbic lobe | Anterior cingulate | |
| 10.09 | −30 | −36 | 36 | Left frontal lobe | Subgyral | |||
| 9.99 | 32 | −32 | 36 | Right frontal lobe | Subgyral | |||
| 0.002 | 722 | 10.21 | 48 | 32 | −8 | Right frontal lobe | Inferior frontal gyrus | |
| 9.17 | 52 | 34 | 0 | Right frontal lobe | Inferior frontal gyrus | |||
| 7.43 | 56 | 26 | 8 | Right frontal lobe | Inferior frontal gyrus | |||
| 0.006 | 587 | 7.62 | −24 | −74 | −6 | Left occipital lobe | Subgyral | |
| 5.27 | −10 | −82 | 8 | Left occipital lobe | Cuneus | 17 | ||
| 4.11 | −10 | −92 | −4 | Left occipital lobe | Lingual gyrus | |||
| 0.024 | 407 | 8.57 | −50 | 30 | −2 | Left frontal lobe | Inferior frontal gyrus | |
| 7.25 | −44 | 28 | −10 | Left frontal lobe | Inferior frontal gyrus | 47 | ||
| 7.03 | −54 | 20 | 8 | Left frontal lobe | Inferior frontal gyrus | 45 | ||
| 0.000 | 26,535 | −17.59 | −42 | −28 | −28 | Left temporal lobe | Subgyral | |
| −17.25 | −24 | −2 | 12 | Left sublobar | Lentiform nucleus | |||
| −16.66 | 42 | −32 | −28 | Right temporal lobe | Fusiform gyrus | 36 | ||
| 0.000 | 2012 | −5.86 | −36 | −20 | 64 | Left frontal lobe | Precentral gyrus | |
| −5.67 | −32 | −14 | 52 | Left frontal lobe | Precentral gyrus | |||
| −5.61 | 32 | −14 | 52 | Right frontal lobe | Precentral gyrus | |||
Abbreviations: ALS, amyotrophic lateral sclerosis; BA, Brodmann area; FWE, Family‐wise error; HCs, healthy controls.
3.3.2. Patients with genetic ALS versus patients with nongenetic ALS (height threshold at p < 0.005, p < 0.05 FWE‐corrected at cluster level)
Since we did not identify any significant difference when setting the threshold at p < 0.001, we performed an exploratory analysis with the height threshold at p < 0.005. The genetic ALS group showed a cluster of relative hypometabolism, including the right precuneus, postcentral gyrus, and middle occipital gyrus as compared with the nongenetic ALS group (Figure 2; Table 3). No cluster of relative hypermetabolism was found in the genetic ALS group as compared to the nongenetic ALS group.
FIGURE 2.
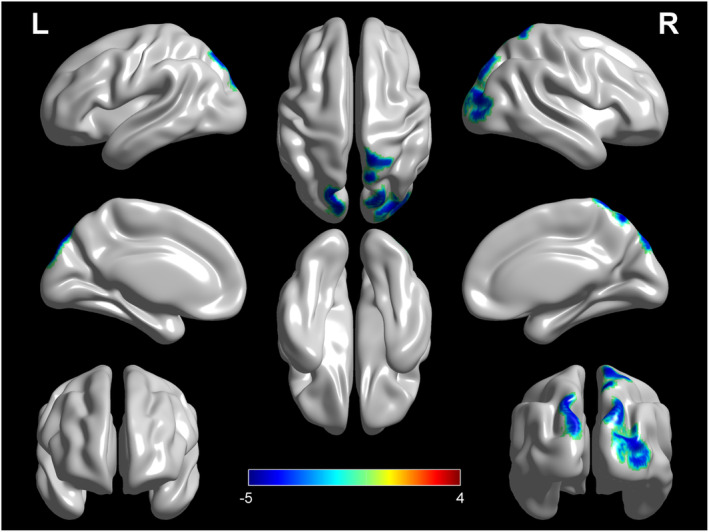
Patients with genetic ALS versus patients with nongenetic ALS (height threshold at p < 0.005, p < 0.05 FWE‐corrected at cluster level). The regions showing statistically significant relative hypometabolism in patients with genetic ALS as compared with patients with nongenetic ALS are reported on the brain surface. ALS, amyotrophic lateral sclerosis.
TABLE 3.
Clusters showing a statistically significant relative hypometabolism in genetic ALS patients as compared to nongenetic ALS patients.
| P (FWE‐corrected) | Cluster extent | T‐score | Peak coordinates (x, y, z) (mm) | Anatomical region | Cortical region | BA | ||
|---|---|---|---|---|---|---|---|---|
| 0.000 | 2347 | −4.57 | 8 | −64 | 60 | Right parietal lobe | Precuneus | 7 |
| −4.23 | 14 | −50 | 72 | Right parietal lobe | Postcentral gyrus | 7 | ||
| −4.13 | 32 | −88 | 18 | Right occipital lobe | Middle occipital gyrus | |||
Abbreviations: ALS, amyotrophic lateral sclerosis; BA, Brodmann area; FWE, Family‐wise error.
3.3.3. Patients with genetic ALS versus HCs (height threshold at p < 0.001, p < 0.05 FWE‐corrected at cluster level)
Patients with genetic ALS showed relative hypometabolism in the bilateral sublobar, parahippocampal gyrus, occipital lobe, and cerebellum, and right temporal lobe as compared with HCs (Figure 3A; Table S3). A cluster of hypermetabolism was found in the bilateral frontal lobe and left sublobar in patients with genetic ALS as compared with HCs (Figure 3A; Table S3).
FIGURE 3.
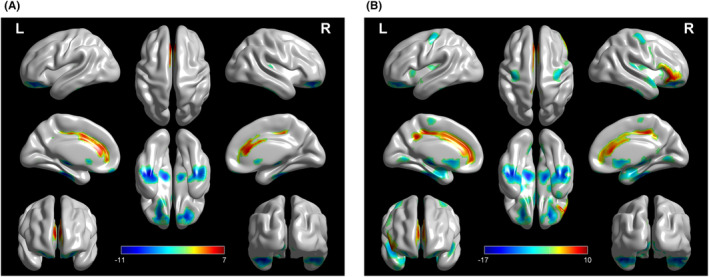
Patients with genetic ALS versus HCs and patients with nongenetic versus HCs (height threshold at p < 0.005, p < 0.05 FWE‐corrected at cluster level). (A) The regions showing statistically significant relative hypometabolism or hypermetabolism in patients with genetic ALS as compared with HCs are reported on the brain surface. (B) The regions showing statistically significant relative hypometabolism or hypermetabolism in patients with nongenetic ALS as compared with HCs are reported on the brain surface. ALS, amyotrophic lateral sclerosis; HCs, healthy controls.
3.3.4. Patients with nongenetic ALS versus HCs (height threshold at p < 0.001, p < 0.05 FWE‐corrected at cluster level)
Patients with nongenetic ALS showed relative hypometabolism in the left temporal lobe and precentral gyrus and bilateral sublobar (Figure 3B; Table S4). We identified a cluster of relative hypermetabolism in patients with nongenetic ALS as compared to HCs, including the left cingulate gyrus and occipital lobe and bilateral inferior frontal gyrus (Figure 3B; Table S4).
3.3.5. ALS patients with sensory disturbance versus ALS patients with sensory normal (height threshold at p < 0.001, p < 0.05 FWE‐corrected at cluster level)
ALS patients with sensory disturbance showed hypometabolism in postcentral gyrus, precentral gyrus and other frontal–parietal lobe regions. (Figure S2; Table S7).
3.3.6. ALS patients with sensory disturbance versus HCs (height threshold at p < 0.001, p < 0.05 FWE‐corrected at cluster level)
ALS patients with sensory abnormal showed hypometabolism in postcentral gyrus too. (Figure S3a; Table S8).
3.3.7. ALS patients with sensory normal versus HCs (height threshold at p < 0.001, p < 0.05 FWE‐corrected at cluster level)
ALS patients showed relative hypometabolism in sublobar and temporal lobe and relative hypermetabolism in frontal lobe and limbic system as compared with HCs. There were no differences of metabolism between ALS patients with sensory normal and HCs in postcentral gyrus. (Figure S3b; Table S9).
4. DISCUSSION
In the present study, we specifically investigated brain metabolic disturbances in ALS patients and explored the relationship between brain metabolism and genotypes in a Chinese ALS cohort. As compared to HCs, we found hypometabolism in the primary motor cortex, frontal lobe, and temporal lobe in ALS patients in agreement with the results of previous studies. 9 , 24 Interestingly, as compared with HCs, we found that ALS patients also showed hypometabolism in the midbrain and cerebellum, which was inconsistent with previous studies. 9 , 14 Moreover, we also found that patients with genetic ALS showed hypometabolism in the right parietal and occipital lobe as compared to patients with nongenetic ALS.
Although previous studies have investigated the pattern of brain metabolism in ALS, an 18F‐FDG‐PET study in a large Chinese ALS cohort is still lacking. We performed a brain metabolic signature in ALS patients in the mainland of China in this study. And we found relative hypometabolism in the precentral gyrus of the frontal lobe and temporal lobe as described in previous studies. 9 , 24 Our findings further strengthen the evidence that 18F‐FDG‐PET could be used as a biomarker to evaluate the degeneration of upper motor neurons and cognitive dysfunction in ALS.
Interestingly, we found brain hypometabolism in the region of midbrain and cerebellum in patients with ALS as compared with HCs (Figure S1), which was inconsistent with previous 18F‐FDG‐PET studies. 9 , 14 Most of previous studies revealed the relative hypermetabolism in midbrain and cerebellum in ALS patients. 8 , 9 , 25 , 26 Some researchers suggest that hypermetabolism in the midbrain and cerebellum may be resulted by the astrocytosis and activated microglia. 9 , 27 Nevertheless, theoretically, ALS is a neurodegenerative disease characterized by the progressive loss of motor neurons in the brain, brainstem, and spinal cord, 1 , 3 thus the expected effect of ALS is hypometabolism in the midbrain caused by neuronal loss. Recently, an MRI study disclosed that patients with ALS exhibited focal cerebellar degeneration and cerebro‐cerebellar connectivity alterations. 28 As with hypometabolism in the frontal and temporal in ALS, 10 , 12 , 27 the degeneration of the cerebellum and midbrain inevitably leads to a reduction in tissue metabolic rate. Hence, we proposed a hypothesis that the metabolic states of the midbrain and cerebellum in patients with ALS are determined by which of the two pathological states of inflammation and degeneration is dominant. Further postmortem or specific PET tracer studies are required to validate the hypothesis.
In this study, 115 patients with ALS underwent genetic test. The most common mutant gene was SOD1, followed by OPTN and CACNA1H. This result was in line with previous studies in China. 16 , 18 As we all know, SPG11 is a common AR‐inherited ALS causative gene. 29 However, no mutations were detected in SPG11. There are two possible reasons listed below. First, in our cohort, none of the patients with ALS have autosomal recessive family history. Second, the AR‐inherited mutations in SPG11 in Chinese ALS patients were rare. 16 , 18
As compared with HCs, the cluster of relative hypometabolism of patients with nongenetic ALS was major located in the temporal and frontal cortex, in line with the previous studies. 9 , 24 As compared with HCs, patients with genetic ALS showed relative hypometabolism in the occipital lobe, sublobar, and limbic lobe, which indicated that genetic ALS patients showed a specific brain metabolism signature.
Another interesting finding was hypermetabolism in the postcentral gyrus in patients with genetic ALS. The primary somatosensory cortex is located in the postcentral gyrus and widely interconnected with other brain regions, including the primary motor cortex. A previous study found that the number of neurons in the motor cortex and the somatosensory cortex were a positively correlated in ALS, suggesting that the somatosensory cortex is affected, once the degeneration of the motor cortex is initiated. 30 These findings were also supported by other clinical researches on ALS. An MRI study found that patients with ALS showed parietal lobe atrophy during disease progression. 31 Functional evaluation of the sensory cortex in patients with ALS using the high‐frequency somatosensory evoked potentials (HF‐SEP) disclosed significant somatosensory cortex dysfunction in patients with a disease duration of more than 2 years. 32 In familial ALS patients, a more frequent occurrence of sensory features at presentation was reported. 33 A previous study found that sensory disturbance was a more frequent feature in C9orf72‐associated ALS patients than nonC9orf72‐associated ALS patients. 34 Numerous studies reported that patients with ALS carrying SOD1 causative mutations were more likely to have sensory abnormalities during the course of the disease. 35 , 36 , 37 Consistent with previous studies, we also found the incidence of sensory disturbance in genetic ALS patients was higher than nongenetic ALS patients. Moreover, we further found that ALS patients with sensory disturbance displayed hypometabolism in postcentral gyrus and other regions of frontal–parietal lobe. ALS patients with sensory normal have no significant changes of metabolism in postcentral gyrus as compared with HCs. We provide direct and indirect evidence on the anatomo‐clinical correlations between hypometabolism in sensory brain regions and sensory disturbances. Recently, a neuroimaging study confirmed the degeneration of somatosensory, in ALS, which is more marked in C9orf72 positive patients. 38 A study about clusters of anatomical disease‐burden patterns in ALS confirmed the imaging signatures of sensory cortex could be distinct disease subtypes. 39 Thus, these studies suggest that patients with ALS have somatosensory cortex involvement, although the molecular mechanism is unclear. Combining with our results, we hypothesize that genetic factors may be an underlying cause of sensory disturbances in ALS.
Similar to the previous PET studies, 9 , 40 we also found hypometabolism in occipital lobes in genetic ALS patients as compared with nongenetic ALS patients. Several MRI studies reported reductions of cortical thickness, gray matter volume, and functional connectivity in occipital lobes in patients with ALS. 41 , 42 , 43 The above studies indicated the abnormal alterations in the occipital lobes of ALS patients, while the molecular mechanism is unknown. C9orf72‐linked FTD‐ALS patients were found to present parietal and occipital lobe atrophy using structural MRI scans. 44 A postmortem study found that two aberrant SOD1 mRNAs were detected from occipital cortex of ALS patients. 45 These findings suggested that mutations in known causative ALS genes may affect the metabolism of the occipital lobes of ALS patients. Neuroaxonal retinal abnormalities were detected in neurodegenerative diseases like Parkinson's Disease, progressive supranuclear palsy, multiple system atrophy by optical coherence tomography. 46 , 47 Moreover, a previous study reported that retinal nerve fiber layer thinning was associated with the atrophy of occipital lobe in Alzheimer's disease. 48 These studies indicate that pathological changes of visual pathway were solid evidence in neurodegenerative diseases. Recently, reported alteration of the retinal nerve in ALS was reported implied that the ALS visual pathway may be damaged. 49 The effects of genetic factors on pathological changes in the visual pathway in ALS require more investigation.
There are several limitations in our study. First, the relatively small sample size of the genetic group might influence the results of our study. Second, ALS patients were recruited from a single center in China and our findings regarding the correlations between genotype and brain metabolism in Chinese ALS patients cannot be generalized to European or American ALS populations. Third, we could not correct the effects of cortical atrophy, since not all patients underwent brain MRI scans in this study. Nevertheless, previous studies showed that the result of brain metabolism is relatively independent of cortical atrophy. 50 Fourth, since the specific functional scale of the occipital lobe in our study is lacking, we were unable to assess the difference in the function of the occipital lobe between patients with genetic ALS and nongenetic ALS. The correlation between clinical phenotype and hypometabolism in the occipital lobe in ALS needs further studies to discover. Moreover, cognitive screening is not available in part of ALS patients in our study. It does make some bias of the results in our study. Some patients overlapping ALS and other dementia diseases were not excluded in this study due to the lack of cognitive screening.
In conclusion, our observations strengthen the evidence that 18F‐FDG‐PET is a reliable tool to assess the motor in ALS, and our investigation provided unprecedented evidence of relative hypometabolism in midbrain and cerebellum in ALS patients as compared with HCs. Genetic ALS patients showed a specific signature of brain metabolism and a higher incidence of sensory disturbance, indicating that genetic factors may be an underlying cause affecting the brain metabolism and increasing the risk of sensory disturbance in ALS. Further studies may be required to confirm our preliminary findings.
CONFLICT OF INTEREST STATEMENT
The authors declare that there is no conflict of interest associated with the contents of this article.
Supporting information
Appendix S1
ACKNOWLEDGMENTS
This work was supported by the Science and Technology Innovation 2030 (STI2030‐Major Projects:2021ZD0201803); the National Key Research and Development Program of China (2021YFA0805202 and 2018YFC1312003), the Program of the National Natural Science Foundation of China (82171431, 81671120, 81300981, 91859207, and 81801740), the Natural Science Fund for Distinguished Young Scholars of Hunan Province, China (2020JJ2057), the National Science Foundation of Hunan Province, China (2020JJ5922), the Project Program of National Clinical Research Center for Geriatric Disorders at Xiangya Hospital (2020LNJJ13), The Degree and Postgraduate Education Reform Project of Central South University (2020JGB136). We are grateful to the participating patients for their involvement. This work was supported in part by the Bioinformatics Center, Xiangya Hospital, Central South University.
Liu P, Tang Y, Li W, et al. Brain metabolic signatures in patients with genetic and nongenetic amyotrophic lateral sclerosis. CNS Neurosci Ther. 2023;29:2530‐2539. doi: 10.1111/cns.14193
The first two authors contributed equally to this work as cofirst authors. The last two authors contributed equally to this work as cosenior authors.
Contributor Information
Shuo Hu, Email: hushuo2018@163.com.
Junling Wang, Email: junling.wang@csu.edu.cn.
DATA AVAILABILITY STATEMENT
Datasets analyzed in this study are not publicly available. Further information about the datasets is available from the senior author (J.W.) and (S.H.) on reasonable request.
REFERENCES
- 1. Brown RH, Al‐Chalabi A. Amyotrophic lateral sclerosis. N Engl J Med. 2017;377(2):162‐172. [DOI] [PubMed] [Google Scholar]
- 2. Chia R, Chio A, Traynor BJ. Novel genes associated with amyotrophic lateral sclerosis: diagnostic and clinical implications. Lancet Neurol. 2018;17(1):94‐102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hardiman O, Al‐Chalabi A, Chio A, et al. Amyotrophic lateral sclerosis. Nat Rev Dis Primers. 2017;3:17071. [DOI] [PubMed] [Google Scholar]
- 4. Mathis S, Goizet C, Soulages A, Vallat JM, Masson GL. Genetics of amyotrophic lateral sclerosis: a review. J Neurol Sci. 2019;399:217‐226. [DOI] [PubMed] [Google Scholar]
- 5. Li HF, Wu ZY. Genotype‐phenotype correlations of amyotrophic lateral sclerosis. Transl Neurodegener. 2016;5:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ludolph AC, Langen KJ, Regard M, et al. Frontal lobe function in amyotrophic lateral sclerosis: a neuropsychologic and positron emission tomography study. Acta Neurol Scand. 1992;85(2):81‐89. [DOI] [PubMed] [Google Scholar]
- 7. Hoffman JM, Mazziotta JC, Hawk TC, Sumida R. Cerebral glucose utilization in motor neuron disease. Arch Neurol. 1992;49(8):849‐854. [DOI] [PubMed] [Google Scholar]
- 8. Cistaro A, Valentini MC, Chio A, et al. Brain hypermetabolism in amyotrophic lateral sclerosis: a FDG PET study in ALS of spinal and bulbar onset. Eur J Nucl Med Mol Imaging. 2012;39(2):251‐259. [DOI] [PubMed] [Google Scholar]
- 9. Pagani M, Chio A, Valentini MC, et al. Functional pattern of brain FDG‐PET in amyotrophic lateral sclerosis. Neurology. 2014;83(12):1067‐1074. [DOI] [PubMed] [Google Scholar]
- 10. Canosa A, Pagani M, Cistaro A, et al. 18F‐FDG‐PET correlates of cognitive impairment in ALS. Neurology. 2016;86(1):44‐49. [DOI] [PubMed] [Google Scholar]
- 11. Renard D, Collombier L, Castelnovo G, Fourcade G, Kotzki PO, LaBauge P. Brain FDG‐PET changes in ALS and ALS‐FTD. Acta Neurol Belg. 2011;111(4):306‐309. [PubMed] [Google Scholar]
- 12. Cistaro A, Pagani M, Montuschi A, et al. The metabolic signature of C9ORF72‐related ALS: FDG PET comparison with nonmutated patients. Eur J Nucl Med Mol Imaging. 2014;41(5):844‐852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. De Vocht J, Blommaert J, Devrome M, et al. Use of multimodal imaging and clinical biomarkers in Presymptomatic carriers of C9orf72 repeat expansion. JAMA Neurol. 2020;77(8):1008‐1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Van Laere K, Vanhee A, Verschueren J, et al. Value of 18fluorodeoxyglucose‐positron‐emission tomography in amyotrophic lateral sclerosis: a prospective study. JAMA Neurol. 2014;71(5):553‐561. [DOI] [PubMed] [Google Scholar]
- 15. Diehl‐Schmid J, Licata A, Goldhardt O, et al. FDG‐PET underscores the key role of the thalamus in frontotemporal lobar degeneration caused by C9ORF72 mutations. Transl Psychiatry. 2019;9(1):54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zou ZY, Zhou ZR, Che CH, Liu CY, He RL, Huang HP. Genetic epidemiology of amyotrophic lateral sclerosis: a systematic review and meta‐analysis. J Neurol Neurosurg Psychiatry. 2017;88(7):540‐549. [DOI] [PubMed] [Google Scholar]
- 17. Ludolph A, Drory V, Hardiman O, et al. A revision of the El Escorial criteria—2015. Amyotroph Lateral Scler Frontotemporal Degener. 2015;16(5–6):291‐292. [DOI] [PubMed] [Google Scholar]
- 18. Liu Z, Yuan Y, Wang M, et al. Mutation spectrum of amyotrophic lateral sclerosis in central South China. Neurobiol Aging. 2021;107:181‐188. [DOI] [PubMed] [Google Scholar]
- 19. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li J, Zhao T, Zhang Y, et al. Performance evaluation of pathogenicity‐computation methods for missense variants. Nucleic Acids Res. 2018;46(15):7793‐7804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Varrone A, Asenbaum S, Vander Borght T, et al. EANM procedure guidelines for PET brain imaging using [18F]FDG, version 2. Eur J Nucl Med Mol Imaging. 2009;36(12):2103‐2110. [DOI] [PubMed] [Google Scholar]
- 22. Canosa A, Moglia C, Manera U, et al. Metabolic brain changes across different levels of cognitive impairment in ALS: a 18F‐FDG‐PET study. J Neurol Neurosurg Psychiatry. 2020;92:357‐363. [DOI] [PubMed] [Google Scholar]
- 23. Xia M, Wang J, He Y. BrainNet viewer: a network visualization tool for human brain connectomics. PLoS One. 2013;8(7):e68910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jeong Y, Park KC, Cho SS, et al. Pattern of glucose hypometabolism in frontotemporal dementia with motor neuron disease. Neurology. 2005;64(4):734‐736. [DOI] [PubMed] [Google Scholar]
- 25. Matias‐Guiu JA, Pytel V, Cabrera‐Martin MN, et al. Amyloid‐ and FDG‐PET imaging in amyotrophic lateral sclerosis. Eur J Nucl Med Mol Imaging. 2016;43(11):2050‐2060. [DOI] [PubMed] [Google Scholar]
- 26. Zanovello M, Soraru G, Campi C, et al. Brain stem glucose hypermetabolism in amyotrophic lateral sclerosis/frontotemporal dementia and shortened survival: an 18F‐FDG PET/MRI study. J Nucl Med. 2022;63(5):777‐784. [DOI] [PubMed] [Google Scholar]
- 27. Chio A, Pagani M, Agosta F, Calvo A, Cistaro A, Filippi M. Neuroimaging in amyotrophic lateral sclerosis: insights into structural and functional changes. Lancet Neurol. 2014;13(12):1228‐1240. [DOI] [PubMed] [Google Scholar]
- 28. Bede P, Chipika RH, Christidi F, et al. Genotype‐associated cerebellar profiles in ALS: focal cerebellar pathology and cerebro‐cerebellar connectivity alterations. J Neurol Neurosurg Psychiatry. 2021;92(11):1197‐1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Orlacchio A, Babalini C, Borreca A, et al. SPATACSIN mutations cause autosomal recessive juvenile amyotrophic lateral sclerosis. Brain. 2010;133(Pt 2):591‐598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mochizuki Y, Mizutani T, Shimizu T, Kawata A. Proportional neuronal loss between the primary motor and sensory cortex in amyotrophic lateral sclerosis. Neurosci Lett. 2011;503(1):73‐75. [DOI] [PubMed] [Google Scholar]
- 31. Shimizu T, Nakayama Y, Funai A, et al. Progressive deterioration of sensory cortex excitability in advanced amyotrophic lateral sclerosis with invasive ventilation. Amyotroph Lateral Scler Frontotemporal Degener. 2020;21(1–2):147‐149. [DOI] [PubMed] [Google Scholar]
- 32. Nardone R, Golaszewski S, Thomschewski A, et al. Disinhibition of sensory cortex in patients with amyotrophic lateral sclerosis. Neurosci Lett. 2020;722:134860. [DOI] [PubMed] [Google Scholar]
- 33. Li TM, Alberman E, Swash M. Comparison of sporadic and familial disease amongst 580 cases of motor neuron disease. J Neurol Neurosurg Psychiatry. 1988;51(6):778‐784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pegat A, Bouhour F, Mouzat K, et al. Electrophysiological characterization of C9ORF72‐associated amyotrophic lateral sclerosis: a retrospective study. Eur Neurol. 2019;82(4–6):106‐112. [DOI] [PubMed] [Google Scholar]
- 35. Sakamoto H, Akamatsu M, Hirano M, et al. Multiple system involvement in a Japanese patient with a V31A mutation in the SOD1 gene. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15(3–4):312‐314. [DOI] [PubMed] [Google Scholar]
- 36. Nishiyama A, Warita H, Takahashi T, et al. Prominent sensory involvement in a case of familial amyotrophic lateral sclerosis carrying the L8V SOD1 mutation. Clin Neurol Neurosurg. 2016;150:194‐196. [DOI] [PubMed] [Google Scholar]
- 37. Dalla Bella E, Rigamonti A, Mantero V, et al. Heterozygous D90A‐SOD1 mutation in a patient with facial onset sensory motor neuronopathy (FOSMN) syndrome: a bridge to amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2014;85(9):1009‐1011. [DOI] [PubMed] [Google Scholar]
- 38. Chipika RH, Mulkerrin G, Murad A, Lope J, Hardiman O, Bede P. Alterations in somatosensory, visual and auditory pathways in amyotrophic lateral sclerosis: an under‐recognised facet of ALS. J Integr Neurosci. 2022;21(3):88. [DOI] [PubMed] [Google Scholar]
- 39. Bede P, Murad A, Lope J, Hardiman O, Chang KM. Clusters of anatomical disease‐burden patterns in ALS: a data‐driven approach confirms radiological subtypes. J Neurol. 2022;269(8):4404‐4413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dalakas MC, Hatazawa J, Brooks RA, Di Chiro G. Lowered cerebral glucose utilization in amyotrophic lateral sclerosis. Ann Neurol. 1987;22(5):580‐586. [DOI] [PubMed] [Google Scholar]
- 41. Agosta F, Valsasina P, Riva N, et al. The cortical signature of amyotrophic lateral sclerosis. PLoS One. 2012;7(8):e42816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhang Y, Fang T, Wang Y, et al. Occipital cortical gyrification reductions associate with decreased functional connectivity in amyotrophic lateral sclerosis. Brain Imaging Behav. 2017;11(1):1‐7. [DOI] [PubMed] [Google Scholar]
- 43. Bede P, Bokde A, Elamin M, et al. Grey matter correlates of clinical variables in amyotrophic lateral sclerosis (ALS): a neuroimaging study of ALS motor phenotype heterogeneity and cortical focality. J Neurol Neurosurg Psychiatry. 2013;84(7):766‐773. [DOI] [PubMed] [Google Scholar]
- 44. Boxer AL, Mackenzie IR, Boeve BF, et al. Clinical, neuroimaging and neuropathological features of a new chromosome 9p‐linked FTD‐ALS family. J Neurol Neurosurg Psychiatry. 2011;82(2):196‐203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kawata A, Kato S, Shimizu T, et al. Aberrant splicing of human Cu/Zn superoxide dismutase (SOD1) RNA transcripts. Neuroreport. 2000;11(12):2649‐2653. [DOI] [PubMed] [Google Scholar]
- 46. Huang L, Zhang D, Ji J, Wang Y, Zhang R. Central retina changes in Parkinson's disease: a systematic review and meta‐analysis. J Neurol. 2021;268(12):4646‐4654. [DOI] [PubMed] [Google Scholar]
- 47. Schneider M, Müller HP, Lauda F, et al. Retinal single‐layer analysis in parkinsonian syndromes: an optical coherence tomography study. J Neural Transm (Vienna). 2014;121(1):41‐47. [DOI] [PubMed] [Google Scholar]
- 48. Ong YT, Hilal S, Cheung CY, et al. Retinal neurodegeneration on optical coherence tomography and cerebral atrophy. Neurosci Lett. 2015;584:12‐16. [DOI] [PubMed] [Google Scholar]
- 49. Zhang Y, Liu X, Fu J, et al. Selective and inverse U‐shaped curve alteration of the retinal nerve in amyotrophic lateral sclerosis: a potential Mirror of the disease. Front Aging Neurosci. 2021;13:783431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ibanez L, Dube U, Davis AA, et al. Pleiotropic effects of variants in dementia genes in Parkinson disease. Front Neurosci. 2018;12:230. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1
Data Availability Statement
Datasets analyzed in this study are not publicly available. Further information about the datasets is available from the senior author (J.W.) and (S.H.) on reasonable request.