

Abstract
The fan mussel Pinna nobilis is currently on the brink of extinction due to a multifactorial disease mainly caused to the highly pathogenic parasite Haplosporidium pinnae, meaning that the selection pressure outweighs the adaptive potential of the species. Hopefully, rare individuals have been observed somehow resistant to the parasite, stretching the need to identify the traits underlying this better fitness. Among the candidate to explore at first intention are fast‐evolving immune genes, of which toll‐like receptor (TLR). In this study, we examined the genetic diversity at 14 TLR loci across P. nobilis, Pinna rudis and P. nobilis × P. rudis hybrid genomes, collected at four physically distant regions, that were found to be either resistant or sensitive to the parasite H. pinnae. We report a high genetic diversity, mainly observed at cell surface TLRs compared with that of endosomal TLRs. However, the endosomal TLR‐7 exhibited unexpected level of diversity and haplotype phylogeny. The lack of population structure, associated with a high genetic diversity and elevated dN/dS ratio, was interpreted as balancing selection, though both directional and purifying selection were detected. Interestingly, roughly 40% of the P. nobilis identified as resistant to H. pinnae were introgressed with P. rudis TLR. Specifically, they all carried a TLR‐7 of P. rudis origin, whereas sensitive P. nobilis were not introgressed, at least at TLR loci. Small contributions of TLR‐6 and TLR‐4 single‐nucleotide polymorphisms to the clustering of resistant and susceptible individuals could be detected, but their specific role in resistance remains highly speculative. This study provides new information on the diversity of TLR genes within the P. nobilis species after MME and additional insights into adaptation to H. pinnae that should contribute to the conservation of this Mediterranean endemic species.
Keywords: adaptation, critically endangered, fan mussel, genetic diversity, Haplosporidium pinnae, hybrids, introgression, pathogens, toll‐like receptors
The endemic Mediterranean mussel Pinna nobilis struggles to cope with mass mortality events that are likely caused by the protozoan parasite Haplosporidium pinna. As the congeneric species Pinna rudis and P. nobilis × P. rudis hybris are naturally resistant to the pathogen, we performed a comparative analysis on 14 toll‐like receptor loci, between groups of resistant and susceptible individuals. Our research highlights adaptive introgression and suggests that TLR‐7 might be involved in the resistance to H. pinnae.
1. INTRODUCTION
Filtering‐feeder marine invertebrates, such as bivalves, are exposed to a wide range of potentially pathogenic microbes including bacteria, viruses, and parasites. In most cases, host and pathogens have co‐evolved over long evolutionary periods, allowing populations to cope effectively with the usual periodic epizootics (Altizer et al., 2003; Bean et al., 2013). This balance between pathogens and adapted host is currently challenged by ongoing anthropogenic changes, of which the introduction of new pathogens, global warming and pollution (Fey et al., 2015; Natalotto et al., 2015).
Newly introduced pathogens representing obvious threats for naive populations can cause dramatic demographic and genetic changes, eventually reducing biodiversity and altering ecosystem functioning and services (Elmqvist et al., 2003; Tompkins & Begon, 1999). From a biological conservation perspective, it is therefore necessary to quickly assess whether and to what extent a species is able to rapidly adapt to new infections and naturally recover (Mable, 2019). This knowledge is indeed crucial in determining the risk of global spread of pathogens, the magnitude of potential mass mortality events (MME) as well as the development of conservation strategies.
To this end, rapidly evolving immune genes are among the most relevant candidates genetic loci to study at first intention, in particular those encoding toll‐like receptors (TLR) (Grueber et al., 2015; McCallum & Dobson, 2002; Turner et al., 2011). TLR indeed have a pivotal role in the recognition of a wide diversity of pathogens, through the binding of conserved pathogen‐associated molecular patterns (PAMP) (Uematsu & Akira, 2008), as well as in the stimulation and regulation of both innate and adaptive immunity (Akira & Takeda, 2004; Barreiro et al., 2009). Previous research performed on different species has also shown that TLR genes have undergone pervasive or episodic positive selection (Fornarino et al., 2011; Grueber et al., 2015; Jiang et al., 2017; Tschirren et al., 2013), in that variations in the sequences of TLR genes could be associated with both pathogen resistance and disease tolerance (Chevillard et al., 2018; Destoumieux‐Garzón et al., 2020; Imai et al., 2008; Jiang et al., 2017; Madan‐Lala et al., 2017; Nahrendorf et al., 2021; Pila et al., 2016; Rakoff‐Nahoum et al., 2004; Tschirren et al., 2013). Moreover, determining the diversity of adaptive loci may likely help to understand the evolutionary processes underlying adaptation to pathogens and provide biomarkers that could be used to identify eventually most adapted individuals (Mable, 2019; McCallum & Dobson, 2002).
Different patterns of diversity could actually be expected according to the relative influence of selection, driven by one or multiple infectious agents for instance (Connallon & Clark, 2012; Hedrick, 1999), drift and demographic fluctuations. Selection can have antagonist effects by either maintaining or increasing diversity or reducing diversity by promoting the fixation of adaptive alleles in certain groups or local populations (Holderegger et al., 2006; Spurgin & Richardson, 2010). On the contrary, distinct scenarios of population size fluctuations can as well lead to contrasted measure of diversity (Sutton et al., 2015; Taylor & Jamieson, 2008). For instance, excess of low‐frequency variants would sign a strong long‐lasting bottleneck, while an excess of intermediate‐frequency variants would be related to a recent bottleneck of weak intensity, or to a balancing selection.
The diversity of TLRs has been mainly studied in vertebrates, globally highlighting potentially large number of haplotypes as well as high number of segregating sites (Liu et al., 2020). Noteworthy, in vertebrates, dN/dS ratios are generally less than unity, indicating that selection rather suppresses changes in protein sequences (i.e. negative or purifying selection) (Kryazhimskiy & Plotkin, 2008). Regarding mollusc taxa, less knowledge is available, but a high level of polymorphism has been evidenced overall the transcriptome of the bivalve species Macoma balthica (Pante et al., 2012).
The possibility of obtaining a fairly complete repertoire of full‐length coding DNA sequences is now possible thanks to next‐generation sequencing, making it possible to understand how these genes are eventually shaped by selective pressures, of which pathogens.
The Mediterranean endemic pen shell Pinna nobilis (Linnaeus, 1758) is an iconic species living in Posidonia oceanica meadows, in the coastal fringes from 0 m to 60 m depth. Pinna nobilis plays a central ecological role, reducing the turbidity of the water as a filtering organism, sheltering a number of symbiotic species and providing a hard substrate for the development of many epibiotic species.
In 1992, P. nobilis was declared a protected species with the category of vulnerable (European Council Directive 92/43/EEC), mainly to counteract intensive captures and habitat loss. These conservation measures proved efficient in several regions, for instance in France, Croatia, Spain and Italy, where populations regained vitality (Marrocco et al., 2019; Siletic & Peharda, 2003; Trigos‐Santos & Vicente, 2018). The most recent research on the dynamics and structure of the P. nobilis population indeed demonstrated high effective population sizes, overall genetic homogeneity between populations (i.e. based on F ST calculations) and a potentially high level of connectivity (Nebot‐Colomer et al., 2022; Peyran et al., 2021; Wesselmann et al., 2018).
Unfortunately, the species has been experiencing mass mortality events (MME) since 2016, attributed to a multifactorial disease mainly conveyed not only to the protozoan parasite Haplosporidium pinnae but also to the bacteria Vibrio mediterranei and Mycobacterium sp., among potentially yet uncharacterized other pathogens, such as viruses and micro‐eukaryotes (Hamelin et al., 2019; Künili et al., 2021; Lattos et al., 2020; Scarpa et al., 2020). These MME led to dramatic demographic bottlenecks, throughout the species distribution area (Andree et al., 2020; Catanese et al., 2018; Lattos et al., 2020; Šarić et al., 2020; Vázquez‐Luis et al., 2017), that are expected to critically reduce the genetic diversity, increase population fragmentation and probably negatively influence the natural rescue effect through larval connectivity, due to reduced reproduction success (Cowen et al., 2007). Therefore, although few populations have recently been reported to be safe in some places (Cinar et al., 2021; Peyran et al., 2022), the vast majority of populations have been affected by those pathogens. As a consequence, the species has been included in the IUCN Red List as Critically Endangered (Kersting et al., 2019), and concerns have internationally resurfaced about the abilities of P. nobilis to cope with those new pathogenic threats and conservation actions are being set up.
Noteworthy, few observations of P. nobilis individuals surviving MMEs, primarily caused by H. pinnae, have been reported, and the phylogenetically related Pinna rudis, as well as the P. nobilis × P. rudis hybrids, seem to be naturally resistant to the parasite (Vázquez‐Luis et al., 2021), suggesting that their innate immunity could be efficient enough to prevent from infection development and spreading. Moreover, there is growing evidence that invertebrates, of which bivalves, are able to develop some kind of innate immune memory, that might be transmitted across generations (Arala‐Chaves & Sequeira, 2000; Green & Speck, 2018; Melillo et al., 2018; Wang et al., 2020) which would be a glimmer of hope for the conservation of the species.
In this study, our main objective was to bring new insights on the molecular mechanisms involved in the adaptation to H. pinnae epizootic (Salis et al., 2022). We chose a candidate approach focussing on TLR genes to test the hypothesis that TLRs are involved in such adaptation. Towards this aim, we obtained individuals (P. nobilis, P. rudis and their hybrids) from four distant geographical regions covering the largest part of all three taxa geographic ranges, sampled after epizootic diseases, in which H. pinnae was mainly involved. Therefore, we here described the genetic polymorphism and diversity at 14 predicted TLRs identified in the shotgun genome of P. nobilis (Bunet et al., 2021), which displayed classical extracellular leucine‐rich repeats (LRRs) and intracellular Toll/Interleukin‐1 receptor (TIR) domains (Gay & Gangloff, 2007). Then, we assessed whether and to what extent the observed genetic polymorphism could be associated with better MME survival.
2. MATERIALS AND METHODS
2.1. Pinna spp. samples
No experimentation involving removal, dislocation or killing of Pinna spp. individuals was performed. A nonlethal sampling method was specifically developed to avoid the manipulation of animals. A small piece (14 mm2, ~20 mg) of the mantle tissue was quickly excised using biopsy forceps before the fan mussel closes its valves. The piece of tissue was transferred in a tube filled with 95% ethanol and stored at −80°C until use.
Overall DNA or tissue samples of 43 adult individuals were obtained from Greece, Italy, Spain and France (Table 1, Table S1). Among those, 38 were identified as Pinna nobilis, 1 as Pinna rudis and 4 as P. nobilis × P. rudis hybrids. These individuals were sampled during or after epizootic disease mainly due to Haplosporidium pinnae. Dead or moribund individuals at the time of sampling are termed ‘sensitive’ (S). For convenience, the others that were alive without obvious symptom of infection are termed ‘resistant’ (R) (see Section 4). The Pinna rudis and hybrids individuals did not present any signs of disease and are termed ‘naturally resistant’ (NR). Dead specimen presented heavy lesions in the digestive gland connective tissue and signs of degenerative process in epithelial tissue. Moribund specimen presented half‐open valves and a retracted body (Figure S1).
TABLE 1.
Summary of Pinna spp. samples.
| Phenotype | Code | Species | Exposed to H. pinnae | Infected | Greece (N = 13) | Italy (N = 14) | Spain (N = 10) | France (N = 6) | Total |
|---|---|---|---|---|---|---|---|---|---|
| Alive, without signs of disease | R | P. nobilis | Yes | Yes | 6 | 2 | 8 | ||
| No | 3 | 3 | 6 | ||||||
| NR | P. rudis | Yes | No | 1 | 1 | ||||
| hybrids | No | 4 | 4 | ||||||
| Dead or with signs of disease | S | P. nobilis | Yes | Yes | 7 | 8 | 2 | 17 | |
| No | 1 | 2 a | 3 | ||||||
| Alive | U | P. nobilis | nd | No | 4 | 4 |
Abbreviations: nd, not determined; NR, naturally resistant; R, resistant; S, sensitive; U, undetermined.
These individuals were sampled before the parasite reached the population, explaining why no parasite DNA could be detected (for further details see Nebot‐Colomer et al., 2022). However, these individuals died after the H. pinnae epizootic.
The presence of several pathogens has been investigated as described below, and using a diagnostic quantitative PCR as well for the detection of H. pinnae, as previously described (López‐Sanmartín et al., 2019) (Table 1). Pinna nobilis and Pinna rudis species status was all checked using mitogenome sequences (data not shown) (Catanese et al., 2022). Hybrid status, as well as potentially low introgressed individuals, was assessed by quantifying the proportion of P. rudis alleles, at toll‐like receptors and microsatellites, using neighbour‐joining phylogenies and visual inspection of multiple sequence alignments.
2.2. Illumina sequencing and reads filtering
Genomic DNAs were purified using the DNeasy Blood and Tissue Kit (Qiagen), following the manufacturer's instructions. Concentrations were determined by measuring the absorbance at 260 nm using Quantus (Promega) with quantifluor reactant. DNA integrity was assessed on an agarose gel at 1%.
DNAs were sequenced by Genewiz using Illumina sequencing generating paired‐end (PE) reads (2× 150 bp). The PE reads were processed as follows: PE reads were first filtered using Trimmomatic v0.36 in order to remove adapters and low‐quality leading and trailing bases (parameters: PE ILLUMINACLIP:TrueSeq‐PE.fa:2:30:10:4 LEADING:3 TRAILING:3). Mate‐pairs were then filtered using Fastp which resulted in PE reads flagged as MP and UNMERGED, which were concatenated (parameters: ‐t 4 ‐f 4 ‐T 4 ‐F 4 ‐c ‐g ‐x ‐m ‐‐include_unmerged). Finally, mate‐paired reads were filtered again using Trimmomatic v0.36 as described above including a filtering if the average base quality dropped below 20. Finally, reads of <60 nucleotides in length were discarded (parameters: PE ILLUMINACLIP:TrueSeq‐PE.fa:2:30:10:4 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 AVGQUAL:20 MINLEN:60). FastQC was run at each step.
2.3. Read mapping and phasing
High‐quality paired reads (Q > 40) with a read length higher than 150 nucleotides were aligned against the reference shotgun genome, as well as the partial sequences of Haplosporidium pinnae coding for the small subunit ribosomal RNA (LC338065), Mycobacterium sp. coding for hsp65 (AY379077) and ITS (MN637877), Vibrio sp. coding for ATPaseA (MN201557), Perkinsus mediterraneus, P. atlanticus and P. marinus coding for the small subunit ribosomal RNA (AY486139, AF509333 and AF126013, respectively), using bwa‐mem (Li & Durbin, 2009). Sam files were processed using samtools (Li et al., 2009) in order to obtain bam files of aligned reads. The observed sequencing depth at coding DNA sequence was at least 10×. Reads that aligned the sequences of 19 neutral microsatellites, 14 TLRs and the cytoplasmic dynein (i.e. used as a nonimmune control gene), were phased using the ‘phase’ module of samtools with default parameters, and haplotypes were then inferred using SPADES (parameters: ‐k 21,33,55,77,99,127 ‐‐careful ‐‐only‐assembler) (Bankevich et al., 2012). All reconstructed sequences were visually checked using IGV (Thorvaldsdóttir et al., 2013). SNP present at less than 20% of the reads was not considered. Finally, each TLR haplotype was phased a second time using the phase module of DNAsp v6.0 allowing events of recombination.
2.4. TLR structure, phylogenetic analyses and ligand‐binding sites analyses
Once the haplotypes of predicted TLR gene‐coding sequences were obtained, the coding DNA sequences and predicted protein sequences were deduced using Augustus (Stanke & Morgenstern, 2005) using default parameters. Then, the protein structures were cross‐checked using SMART (Letunic et al., 2021), PROSITE, TMHMM‐2.0 (Krogh et al., 2001) and BLASTP in order to provide information about the presence of leucine‐rich repeat motifs (LRR) and Toll/IL‐1 resistance (TIR) domains which are functionally important and transmembrane domains.
Then, sequence consensus for each TLR protein was obtained from multisequence alignment using EMBOSS Cons available online (https://www.ebi.ac.uk/Tools/msa/emboss_cons/), for both P. nobilis and P. rudis. Consensus sequences were used to model the 3D structure, using I‐TASSER (Yang et al., 2015) and PyMOL (http://www.pymol.org/pymol), and to draw a phylogenetic tree depicting the relationships between TLR proteins based on multiple sequence alignments, using MEGA 11 and visualized using iTOLv6 (Letunic & Bork, 2021).
2.5. Genetic diversity and structure
Neutral microsatellites and TLR haplotypes were aligned with MEGA11, using CLUSTAL and MUSCLE (codon), respectively. Singletons of TLR sequences were removed. These alignments were used in DNAsp (Rozas et al., 2017) to determine haplotype diversity, which were translated into genotype data. Regarding TLR haplotypes specifically, the following diversity statistics were assessed using DNAsp (Rozas et al., 2017): the number of polymorphic sites (S), the average number of nucleotide differences among all alleles (k), the nucleotide diversity (π), the number of nonsynonymous (dN) and synonymous (dS) changes. For each Pinna nobilis TLR, median‐joining haplotype networks were drawn using PopART (http://popart.otaga.ac.nz).
For both TLR and microsatellite loci, FSTAT v2.9.3.2 (Goudet, 2001) software was used to determine allele diversity (N A), allelic richness based on a sample of eight individuals (A R), observed and expected heterozygosity (H O and H E, respectively) and calculate estimates of F IS, with 10,000 genotype randomizations performed among samples to test for significance. Then, significant deviation from the Hardy–Weinberg equilibrium (HWE) was tested using the randomization procedure (100,000 steps in Markov Chain and 1000,000 dememorization steps) implemented in ARLEQUIN v3.5.1.2 (Excoffier & Lischer, 2010).
Prior to the genetic structure analysis, ARLEQUIN v3.5.1.2 (Excoffier & Lischer, 2010) was also used to check that the retained loci were not in linkage disequilibrium, to assess the distribution of genetic diversity by an analysis of molecular variance (AMOVA) and to calculate the pairwise genetic differentiations (F ST) among each population. The corrected significance threshold for multiple tests was set using the Benjamini–Hochberg correction procedure (Benjamini & Hochberg, 1995). Subsequently, genetic structures were depicted using principal component analysis (PCA) using R.
2.6. Assessment of selection acting at TLR loci
First, the evolutionary pressures acting on each TLR locus were assessed by a McDonald and Kreitman test and by estimating the Tajima's and Fu and Li′s D parameters using DNAsp (Rozas et al., 2017), either globally or regionally with a window length of 80 nucleotides and a step size of 20 nucleotides. Then, candidate SNPs under diversifying (i.e. positively selected sites – PSS) or purifying (i.e. negatively selected sites—NSS) selection were detected based on an outlier‐F ST approach using BAYESCAN and a phylogenetically controlled test using a fast unconstrained Bayesian approximation model (FUBAR), accessible online from the DataMonkey server (https://www.datamonkey.org/) (Weaver et al., 2018). The analyses with BAYESCAN were implemented with a total of 150,000 iterations, a burn‐in period of 50,000 and 20 pilot runs of 10,000 iterations each and did not yield any significant results. The analysis using FUBAR (Murrell et al., 2013) was performed once potential recombination events were assessed for each TLR locus using GARD algorithm (Kosakovsky Pond et al., 2006), also accessible from the DataMonkey server. This is a recommended preprocessing step for selection inference, so that any recombination effects could be considered in the FUBAR model. Only sites under putative selection with a posterior probability higher than 0.9 were retained for further analysis. Finally, DAPC analysis was performed using R, in order to depict the relative contributions of SNP retaining the three to four clusters of individuals (i.e. according to their phenotypes), and with n.pca = 6; n.da = 4.
3. RESULTS
3.1. Pinna spp. samples and infectious status
The 43 individuals were sampled and sorted according to their phenotype at the sampling period after a mass mortality event. Pinna rudis and hybrids (i.e. termed naturally resistant ‘NR’) were always alive, without signs of disease, and no trace of H. pinnae could be detected in the genomic data. Sixteen P. nobilis did not present any obvious sign of disease (i.e. termed resistant ‘R’) of which eight were infected by H. pinnae, while 20 P. nobilis were dead or presented obvious signs of disease (i.e. termed sensitive ‘S’), of which 17 were infected by H. pinnae. Four French P. nobilis could not be defined as resistant (i.e. termed undefined ‘U’) since we only have the suspicion that H. pinnae affected the whole population (Table 1, Table S1).
Regarding the other sought microorganisms, genomic DNA of Vibrio mediterranei was detected in three P. nobilis from Greece and four P. nobilis from Italy, while Mycobacterium sp. was detected in nine P. nobilis from Greece, one P. nobilis of Italy and two P. nobilis from France. Both Vibrio and Mycobacterium were detected in equal proportions within the resistant and sensitive phenotypes. Perkinsus sp. was not detected in any specimen.
3.2. Candidate TLR structure and description of genetic polymorphism
Among the 14 contigs carrying TLR gene candidates, all encoded proteins exhibited the expected typical leucine‐rich regions, transmembrane domains, one or two TIR motifs, and horseshoe 3D structures. The detected open reading frames ranged from 1659 nt to 3624 nt in length.
In both P. nobilis and P. rudis species, we found one or several members of TLR‐1, ‐2, ‐3, ‐4, ‐6, ‐7, ‐13 and toll‐like families (Table S2). The phylogenetic relationships between TLR coding sequences of both species, as well as the protein domains of each TLR, are depicted in Figure S2.
Across the 14 P. nobilis TLR loci, a total of 228 haplotypes were observed with a mean haplotype per loci of 16.4 ± SD 8.1 and a mean haplotype diversity (H d) of 0.771 ± SD 0.166. Overall, one or a few haplotypes predominate at each TLR locus (Figure 3, Figure S3). The density of segregating sites is rather homogenous across loci, thus between both the extra and intracellular domains of the protein, with a mean of roughly 8.2 ± SD 5.8 substitutions per 1000 nt, except for the TLR‐7 in which 27 substitutions per 1000 nt was observed (Table 2).
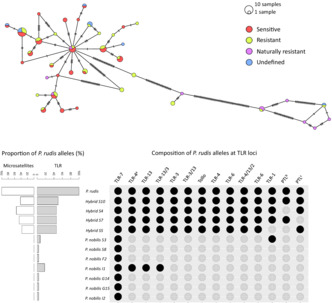
FIGURE 3.
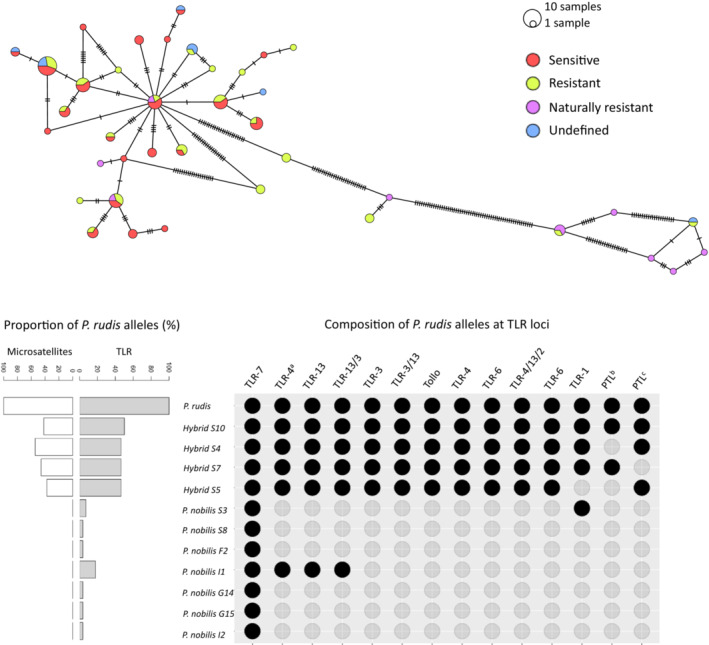
Median‐joining networks of TLR‐7 coding DNA sequence haplotypes and composition of P. rudis alleles at microsatellites and TLR loci within introgressed individuals. Above: Eighty‐six sequences are considered: 38, 30 and 8 haplotypes of sensitive, resistant and undefined P. nobilis phenotypes, respectively, and 10 haplotypes of either P. rudis or hybrids, naturally resistant phenotypes. The number of mutations between haplotypes is mentioned by dashes on connecting lines. Size of pie charts reflects the number of individuals with the observed haplotype. Below: Twelve and 14 loci were considered for microsatellites and TLR loci, respectively. Left: Proportion of P. rudis alleles within individuals. Right: Composition of P. rudis alleles at TLR loci. PTL, protein toll‐like, (a) contig 38,093, (b) contig 84,580, (c) contig 39,158.
TABLE 2.
Genetic polymorphism of TLR loci.
| TLR locus | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TLR‐4 | TLR‐4 | TLR‐4/13/2 | TLR‐3 | TLR‐3/13 | TLR‐13/3 | TLR‐13 | Tollo | TLR‐7 | TLR‐6 | TLR‐6 | TLR‐1 | Protein toll‐like | Protein toll‐like | ||
| Contig | 38,093 | 473 | 17,440 | 21,890 | 67,982 | 7594:g1 | 7594:g2 | 50,674 | 12,778 | 48,600 | 39,119 | 21,812 | 84,580 | 39,158 | |
| CDS length (nt) | 2485 | 2535 | 2353 | 2571 | 2091 | 1941 | 1659 | 3606 | 3624 | 3240 | 3372 | 2013 | 1941 | 1971 | |
| P. nobilis sequences | S | 22 | 14 | 21 | 8 | 7 | 12 | 11 | 38 | 99 | 27 | 24 | 15 | 12 | 11 |
| S/1000 nt | 8.85 | 5.52 | 8.92 | 3.11 | 3.35 | 6.18 | 6.63 | 10.54 | 27.32 | 8.33 | 7.12 | 7.45 | 6.18 | 5.58 | |
| dN/dS | 1.20 | 0.75 | 3.75 | 0.60 | 1.33 | 3.00 | 0.57 | 1.24 | 0.90 | 0.50 | 0.71 | 1.50 | 1.00 | 2.67 | |
| h | 20 | 13 | 14 | 9 | 8 | 12 | 10 | 21 | 37 | 20 | 22 | 20 | 12 | 10 | |
| AA | 15 | 8 | 13 | 4 | 5 | 11 | 5 | 18 | 27 | 9 | 10 | 15 | 7 | 8 | |
| H d | 0.905 | 0.7 | 0.799 | 0.52 | 0.41 | 0.782 | 0.583 | 0.802 | 0.971 | 0.932 | 0.942 | 0.856 | 0.802 | 0.787 | |
| k | 3.385 | 1.121 | 6.423 | 0.604 | 0.565 | 1.839 | 1.834 | 6.887 | 22.781 | 5.197 | 3.467 | 2.32 | 2.903 | 2.311 | |
| π | 0.00136 | 0.00044 | 0.00273 | 0.00023 | 0.00027 | 0.00095 | 0.00111 | 0.00191 | 0.00629 | 0.00161 | 0.00103 | 0.00115 | 0.0015 | 0.00117 | |
| All sequences | S | 118 | 154 | 97 | 97 | 95 | 99 | 60 | 138 | 151 | 119 | 130 | 26 | 128 | 69 |
| S/1000 nt | 47.48 | 60.75 | 41.22 | 37.73 | 45.43 | 51.00 | 36.17 | 38.27 | 41.67 | 36.73 | 38.55 | 12.92 | 65.95 | 35.01 | |
| dN/dS | 0.72 | 1.50 | 1.87 | 0.62 | 1.46 | 0.60 | 0.88 | 1.06 | 0.86 | 0.41 | 0.85 | 2.25 | 0.56 | 1.88 | |
| h | 26 | 19 | 16 | 13 | 12 | 14 | 11 | 27 | 39 | 25 | 25 | 24 | 14 | 14 | |
| AA | 20 | 11 | 15 | 8 | 9 | 12 | 6 | 24 | 32 | 11 | 12 | 17 | 8 | 11 | |
| Hd | 0.921 | 0.741 | 0.828 | 0.584 | 0.477 | 0.813 | 0.642 | 0.833 | 0.971 | 0.941 | 0.949 | 0.872 | 0.819 | 0.819 | |
| k | 17.799 | 16.687 | 17.319 | 9.187 | 9.984 | 12.695 | 9.134 | 20.613 | 22.781 | 16.833 | 16.119 | 3.09 | 9.957 | 10.532 | |
| π | 0.00716 | 0.00658 | 0.00736 | 0.00357 | 0.00477 | 0.00654 | 0.00552 | 0.00572 | 0.00629 | 0.0052 | 0.00478 | 0.00153 | 0.00513 | 0.00534 | |
Abbreviations: AA, number of translated proteins; dN/dS, ratio of nonsynonymous over synonymous substitution; h, number of haplotypes; H d, haplotype diversity; k, average number of nucleotide differences between sequences; S, number of segregating sites; π, nucleotide diversity per site.
The dN/dS ratio observed at the full‐length sequences ranged between 0.5 and 3.8 (mean = 1.4 ± SD 1.0) and was mostly higher at the extracellular domain (mean = 2.1 ± SD 2.1) compared with that of the intracellular domain (mean = 0.6 ± SD 0.6), which reveals a greater polymorphism in the protein region harbouring binding sites. The observed mean number of translated proteins was 11 ± SD 6.2 (Table 2 and Figure 1).
FIGURE 1.
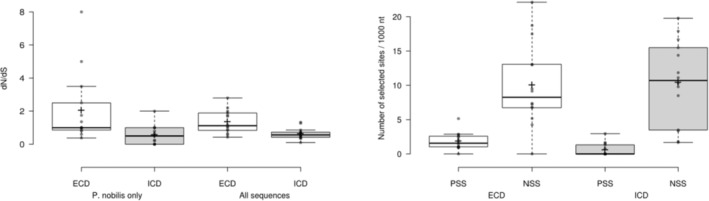
Density distribution of dN/dS ratio and positively and negatively selected sites across TLR. ECD, extracellular domain; NSS, negatively selected sites; ICD, intracellular domain; PSS, positively selected sites.
The polymorphism observed at TLR loci was found like that observed for the nonimmune comparative control gene (i.e. 15 haplotypes, H d = 0.714 and S/1000 nt = 9.4). However, for dynein, the dN/dS ratio was 0.2, that is three and seven times lower than the dN/dS ratios observed at intracellular domain and at the full‐length sequence of TLR, respectively.
As expected for TLR genes, the genetic polymorphism observed between orthologue sequences was rather high, with a mean density of segregating sites of 42.1 ± SD 12.6 sites per 1000 nt (Table 2).
3.3. Basic genetic diversity and distribution
Similar levels of genetic and gene diversities have been detected among populations as well as among phenotype groups, for both microsatellites and TLR loci. Overall, no sign of departure from Hardy–Weinberg equilibrium was observed since FIS were not significant, and specifically no excess of heterozygotes could be observed (Table 3, Table S3a,b).
TABLE 3.
Basic overall descriptive statistics.
| Species | Locality | Parameters | Microsatellites | TLR | Species | Phenotype | Parameters | Microsatellites | TLR |
|---|---|---|---|---|---|---|---|---|---|
| P. nobilis | Greece (N = 13) | N A | 160 | 131 | P. nobilis | Resistant (N = 15) | N A | 157 | 150 |
| R A | 4.8 | 4.8 | R A | 4.7 | 4.87 | ||||
| H E | 0.748 | 0.743 | H E | 0.748 | 0.731 | ||||
| F IS | 0.074 | 0.047 | F IS | 0.070 | 0.034 | ||||
| Italy (N = 14) | N A | 139 | 132 | Sensitive (N = 21) | N A | 182 | 176 | ||
| R A | 4.5 | 4.8 | R A | 4.7 | 4.98 | ||||
| H E | 0.741 | 0.747 | H E | 0.761 | 0.758 | ||||
| F IS | 0.020 | −0.085 | F IS | 0.025 | 0.013 | ||||
| France (N = 6) | N A | 115 | 80 | P. rudis & hybrids | Naturally resistant (N = 5) | N A | 98 | 92 | |
| R A | 4.8 | 4.2 | R A | 5.7 | 5.60 | ||||
| H E | 0.748 | 0.743 | H E | 0.706 | 0.78 | ||||
| F IS | −0.041 | 0.047 | F IS | 0.215 | 0.134 | ||||
| Spain (N = 5) | N A | 91 | 83 | ||||||
| R A | 4.4 | 2.6 | |||||||
| H E | 0.693 | 0.678 | |||||||
| F IS | 0.034 | −0.137 | |||||||
| All (N = 38) | N A | 228 | 228 | All (N = 41) | N A | 253 | 277 | ||
| H E | 0.777 | 0.766 | H E | 0.791 | 0.790 | ||||
| F IS | 0.031 (−0.039; 0.108) | 0.001 (−0.047; 0.062) | F IS | 0.058 (−0.010;0.132) | 0.021 (−0.047; 0.058) |
Abbreviations: N A, number of alleles; R A, allelic richness based on eight individuals; H E, expected heterozygosity; F IS: inbreeding coefficient.
The analysis of molecular variance performed considering either both species or P. nobilis only, all evidenced that the genetic diversities were mostly distributed within population or phenotype groups, regardless of the molecular marker considered. At the population level, the AMOVA's F ST were significant only when both species were considered (Table S4). Consistent with the AMOVAs, pairwise‐F ST evidenced a rather homogenous P. nobilis population. Using either molecular marker, none pairwise differentiation could be detected. Regarding phenotype groups, only the naturally resistant group was significantly differentiated from the resistant and sensitive groups, using both microsatellites and TLRs, which is obvious considering that the naturally resistant group is composed of P. rudis and hybrids individuals only (Table S5). Complementary to the F ST analysis, PCA highlights the homogeneity of P. nobilis populations, and how P. rudis and hybrid individuals separate from P. nobilis individuals (Figure 2, Figure S3).
FIGURE 2.
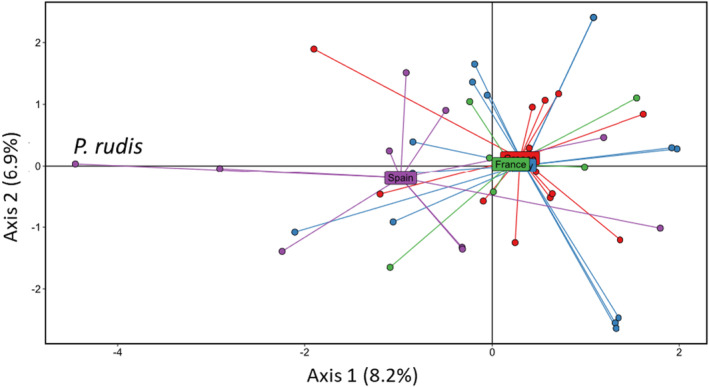
PCA performed using TLR genotypes. The analysis has been performed considering all the individuals irrespective of the species. All Pinna nobilis x Pinna rudis hybrids originated from Spain, explaining why the Spain population is remote from the three others. Individuals from Spain, Greece, Italy and France are represented in purple, red, blue and green, respectively.
3.4. Assessment of selection at TLR loci
3.4.1. Tests of departure from neutrality
As no genetic structure could be obviously evidenced, and to prevent from bias resulting from low population size, we considered the whole sequences as coming from one panmictic population. First, the departure from neutrality was tested using a McDonald–Kreitman test, which compares the amounts of polymorphism within P. nobilis and P. rudis species, to the divergence between them, at neutral and non‐neutral sites. Our results suggest that the TLR genes likely evolved under neutral genetic drift. At the intraspecific level, the Tajima's D test performed on P. nobilis haplotypes globally also meets the assumption of neutrality (Table 4), although there are signs (p < .1) of positive selection acting on TLR‐7, TLR‐4 (i.e. Contig 473) and TLR‐3. The Fu and Li′s D values were significantly positive for most of the TLR, evidencing overrepresented haplotype variants in the population, that may have been selected in the past (Table 4).
TABLE 4.
Tests of departure from neutrality.
| TLR | Contig | McDonald and Kreitman | Tajima's D | Fu and Li's D | dN/dS (ECD) | dN/dS (ICD) |
|---|---|---|---|---|---|---|
| TLR‐4 | 38,093 | 1.878 | −0.73238 | 1.76889** | 1.00 | nd |
| TLR‐4 | 473 | 0.776 | −1.70200$ | 1.55038* | 0.86 | 0.00 |
| TLR‐4/13/2 | 17,440 | 2.512 | 1.55599 | 1.76997** | 5.00 | 0.00 |
| TLR‐3 | 21,890 | 0.721 | −1.57272$ | 1.27359 | 0.60 | 0.00 |
| TLR‐3/13 | 67,982 | 1.275 | −1.45349 | 1.20882 | 1.00 | 2.00 |
| TLR‐13/3 | 7594:g1 | 0.81 | −0.66732 | 1.47375$ | 8.00 | 0.50 |
| TLR‐13/3 | 7594:g2 | 0.595 | −0.47643 | 1.43053$ | 0.80 | 0.00 |
| Tollo | 50,674 | 1.21 | −0.33431 | 2.00864** | 1.36 | 1.00 |
| TLR‐7 | 12,778 | 0.701 | −1.58454$ | 2.08624** | 0.98 | 0.64 |
| TLR‐6 | 48,600 | 1.333 | −0.14388 | 1.86558** | 0.38 | 0.80 |
| TLR‐6 | 39,119 | 0.839 | −0.86734 | 1.81176** | 1.00 | 0.20 |
| TLR‐1 | 21,812 | 0.5 | −0.66055 | 1.58483* | 1.75 | 1.00 |
| Protein toll‐like | 84,580 | 0.99 | 0.55802 | 1.47296$ | 2.50 | 0.25 |
| Protein toll‐like | 39,158 | 2.258 | 0.10234 | 1.43053$ | 3.50 | 1.00 |
Note: McDonald and Kreitman test has been performed considering P. nobilis versus P. rudis alleles. Tajima's D and Fu and Li′s D have been performed considering P. nobilis alleles. *p‐value < .05; **p‐value < .01; $.1 < p‐value < .05.
Abbreviations: ECD, extracellular domain; ICD, intracellular domain.
3.4.2. Pattern of nonsynonymous versus synonymous substitutions
Considering full‐length sequences, 7 TLR presented a dN/dS ratio higher than 1, of which three comprised between 1.5 and 3. Figure 1 highlights that contrasted pattern of selection are likely to occur at both extra and intracellular domains. Indeed, dN/dS ratios higher than 1 are mostly encountered at the extracellular domains, suggesting that positive selection is predominant over this region of the protein and that amino acid substitutions were favoured. On the contrary, with one exception, dN/dS ratios are either equal or lower than 1 at the intracellular domains, suggesting that synonymous substitutions are favoured (i.e. neutral or purifying selection).
3.4.3. Median‐joining networks of haplotypes
Overall, we observed clear star‐like phylogenies, with few high‐frequency nodes from which derive eventually rare haplotypes, which could be consistent with a population expansion model and/or a recent positive selection (Figure S3). This pattern of population expansion may reflect the increase of individuals, which is not necessarily accompanied by recovery in the loss of genetic variation.
Moreover, the networks depicted distinct phylogenies, evidencing that some TLR loci are likely under different evolutionary constraints. In line with the high level of divergence between P. nobilis and P. rudis TLR loci, the networks demonstrate a clear differentiation of naturally resistant haplotypes, separated by numerous mutational steps from sensitive, resistant or undefined ones. On the contrary, resistant and sensitive P. nobilis were evenly distributed across the nodes and branches, highlighting that there was no specific lineage associated with a particular phenotype, hence, no network evidenced strong purifying selection (Figure S3).
However, the networks obtained for TLR‐7, ‐4, ‐13, ‐13/3 and ‐1 highlighted that 7 P. nobilis were introgressed with P. rudis, which represents about 18% of the P. nobilis studied here. These individuals were identified as resistant, and all carried at least a TLR‐7 allele of P. rudis. Strikingly, P. nobilis S3, S8 and F2 carried a full‐length allele of P. rudis whereas P. nobilis I1, I2, G14 and G15 carried a partial, recombined, allele of P. rudis (Figure 3, Figure S4). Although introgressed with P. rudis, G14, G15, I1 and I2 individuals were nevertheless infected with the parasite, while S3, S8 and F2 were not infected.
3.4.4. Phylogeny‐based test of selection and discriminant analysis of principal component (DAPC)
Before the analysis using FUBAR, we assessed the putative recombination sites within TLR sequences, using GARD. A total of eight recombination sites were detected across six TLR sequences, mostly consisting in one recombination site detected in four TLR sequences and two recombination sites in TLR‐7 and a protein toll‐like (data not shown).
Among the 1481 polymorph sites observed across TLR loci (considering both P. nobilis and P. rudis), FUBAR identified 395 sites (26.6%) putatively under selection, with a posterior probability of .9. Most of these (95%) were negatively selected sites, and most of the positively selected sites (47 out of 53 sites) were found at the extracellular domain of the proteins (Figure 1).
Complementary to FUBAR analysis, DAPC performed using either all or the putatively selected SNPs evidenced that the group of naturally resistant phenotype was obviously separated from the two others, with a probability of assignment of individuals to this cluster of 100% (data not shown). On the contrary, the groups of resistant and sensitive phenotypes overlapped, though few resistant individuals were remotely distributed from the others (Figure 4).
FIGURE 4.
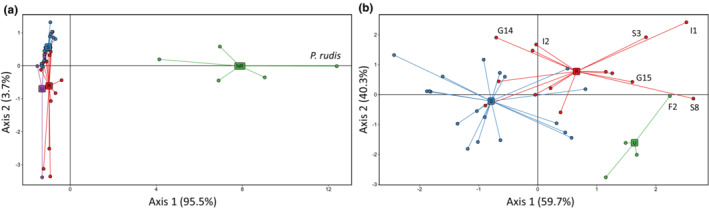
Discriminant analysis of principal component (DAPC) of the snp genotype data observed at TLR coding sequences. (a) DAPC performed according to the 1481 SNPs observed in Pinna spp. Naturally resistant (NR), resistant (R), sensitive (S) and undefined (U) phenotypes are represented in green, red, blue and purple, respectively. (b) DAPC performed according to the 395 SNPs detected by FUBAR as being under diversifying or pervasive selection in P. nobilis only. Resistant, sensitive and undefined phenotypes are represented in red, blue and green, respectively.
When the entire set of SNPs was used in the DAPC, almost all SNP of TLR‐4 (i.e. Contig 17,440), one SNP of TLR‐6 (Contig 48,600, snp771) and one SNP of TLR‐1 (Contig 21,812, snp1030) mainly contributed to the cluster according to axis 1, which separate the group of naturally resistant individuals from other groups. The distribution of individuals according to axis 2, which separated P. nobilis according to their phenotype, was driven by all SNP of TLR‐7 (i.e. Contig 12,778), eight SNPs of TLR‐4 (i.e. five SNPs for contig 17,440 and three SNPs for contig 38,093) and the snp1030 of TLR‐1, presented above.
Afterwards, the selected SNPs were used to maximize the clustering of P. nobilis groups only. Both axes explained a large part of the genetic variance, and the clustering was mainly driven by 13 SNPs belonging to the two TLR‐4 and the two TLR‐6. Among these SNP, only two were positively selected (TLR‐4, contig 473 snp96, and TLR4, contig 38,093 snp1108). Noteworthy, whatever the SNP contributing to the clustering, their overall contributions were weak, and no SNP could clearly segregate the resistant and sensitive groups.
3.5. Comparison of TLR‐7 predicted 3D structures between P. nobilis and P. rudis
I‐TASSER was used for predicting the 3D model structure of P. nobilis and P. rudis consensus TLR‐7 proteins. The software evidenced, overall, similarly shaped proteins and few 3D changes, such as the structure around the amino acids at position 733 (Figure 5). Two binding sites for guanosine analogue ligands were identified. The first one, roughly situated between amino acids 167 and 268, binds O6S ligand (PubChem reference 27,791,261). The second binding site of P. rudis TLR‐7, situated between amino acids 319 and 393, interacts with RX8 ligand (PubChem 159,603), while that of P. nobilis, situated between amino acids 369 and 443, binds the XG1 ligand (PubChem 66,958,698). Figure 5 highlights that, whatever the ligand, be it the same, the protein–ligand interactions appear specific, which are likely to induce different conformational changes and eventually protein activations.
FIGURE 5.
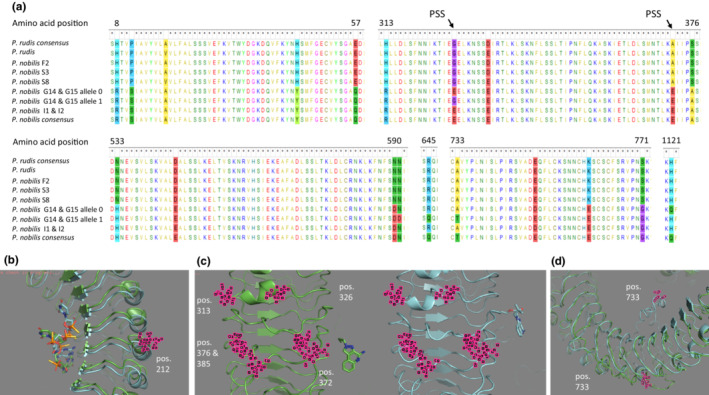
TLR‐7 polymorphism and 3D structure. (a) Sequence alignments of several regions of TLR‐7 of P. rudis origin within introgressed P. nobilis. Consensus sequences of P. nobilis and P. rudis have been included for comparison. PSS, amino acids under positive selection as detected by FUBAR. (b–d) the 3D models have been obtained using I‐TASSER software from P. nobilis and P. rudis TLR‐7 consensus sequences, represented in green and blue, respectively. (b) superimposed TLR‐7 protein at site 1 with 06S ligand; (c) P. nobilis and P. rudis TLR‐7 at site 2 with XG1 and RX8 best‐fit ligands, respectively; (d) superimposed TLR‐7 protein around site 733. Divergent amino acid substitutions are represented in pink.
The protein alignment of P. rudis and P. nobilis TLR‐7 consensus sequences reveals 30 amino acids differences of which 12 are associated with obvious physicochemical changes. Four of those are situated in or near the second binding sites of P. rudis and P. nobilis TLR‐7, respectively, and noteworthy two were also found under positive selection. Significant physicochemical changes due to amino acid substitutions could also be observed outside binding sites and even at the intracellular domain (Figure 5).
4. DISCUSSION
Determination of the evolutive capacity of threatened species is among the major keystone actions towards their conservation. Among the genes to be monitored at first intention are immune genes which act as sentinels against infectious agents. In the context of high P. nobilis mortality, our aim was to describe the current genetic polymorphism of the rapidly evolving toll‐like receptors, within P. nobilis and the phylogenetically related congeneric P. rudis. Such knowledge was lacking albeit the known association between TLR and fitness, resistance to diseases or survival.
The observation that P. rudis and hybrids were all naturally resistant to H. pinnae allowed us to set the hypothesis that their innate immune system was efficient to resist the infection, hence that putative‐resistant P. nobilis and P. rudis would share some genetic features associated with disease resistance, assuming that MMEs were mainly caused by H. pinnae.
In fact, individuals can be tolerant or resistant to a pathogen. While resistance is the ability of a host to limit parasite load or prevent infection, tolerance involves infection and is the ability to limit the negative influence on fitness for a given parasite load (Martins et al., 2019; McCarville & Ayres, 2018). Resistance and tolerance have contrasting impacts on the population dynamics of a pathogen in that resistance tends to reduce the prevalence in a population while tolerance has no influence or increases it (Kutzer & Armitage, 2016). Given interindividual variations in susceptibility to pathogens, it is conceivable that tolerance and resistance could be observed at the same time in a population.
Here, the P. nobilis that were interpreted as resistant individuals exhibited a longer postinfection survival time compared with those interpreted as sensitive (i.e. from few months for individuals from Greece, up to a year for those of Italy), but finally died, which may be attributed to other pathogens or factors as well. During MMEs, no regular monitoring of disease progression at the individual level could be done, so it was not possible to clearly compare the level of disease at the specimen level, nor the interindividual stress response over the course of infection. Given our samples and the lack of standardized sampling protocol, such an assessment could not be made neither. As a consequence, strictly speaking, these P. nobilis cannot be obviously defined as either resistant or tolerant, precisely to H. pinnae infection, despite the eventual inability to survive. In our analysis, for convenience, these P. nobilis were considered resistant.
Although we cannot exclude some assignment errors in the resistant group, we assumed that field observations were nevertheless good indicators of signs of resistance. At the molecular level, we were able to demonstrate an obvious difference between the sensitive and resistant groups since introgression at the TLR‐7 locus was observed only in the group of resistant P. nobilis. Additionally, three individuals of P. nobilis carrying the original full‐length TLR‐7 of P. rudis appeared as naturally resistant as the hybrids and P. rudis (i.e. no infection detected), suggesting that TLR‐7 was indeed probably involved in disease resistance.
This result was obtained considering a rather moderate sample size from a field study. Actually, the lack of surviving populations is a critical issue throughout the species distribution, discommoding the collection of larger number sizes of individuals, a situation that has recently been discussed (Salis et al., 2022). Nonetheless, despite the small number of TLR‐7‐introgressed naturally resistant P. nobilis of our dataset, this result appears highly consistent with the recent research evidencing that TLR‐7 can efficiently hamper the development of intracellular protozoan parasites (Baccarella et al., 2013; Fouzder et al., 2019; Heni et al., 2020; Regli et al., 2020). Therefore, we argue that it could rapidly help to develop conservation strategies for the species survival.
To clarify the relative importance of TLR‐7 introgression in the resistance to H. pinnae, further laboratory‐controlled studies should be performed. On the contrary, introgression should rapidly be more investigated in future monitoring studies as well, focussing on the TLR‐7 genetic composition of Pinna individuals collected either from mortality events or surviving populations. This knowledge will inform about the evolutionary potential of introgressed P. nobilis and the contribution of these individuals to future generations. Probably, most of the individuals identified as resistant had the same null contribution to the susceptible ones (Hasik & Siepielski, 2022). Whether the introgressed specimen contributes more to the next generation than the susceptible and the other resistant individuals, in the context of MME, needs to be assessed in future monitoring studies.
4.1. TLR validation and subtypes identification
Except for TLR‐4 (i.e. contig 473) and TLR‐4/2/13 (i.e. contig 17,440), all coding DNA sequences of our contigs consisted in one large exon, a feature that has been reported in other molluscs (Juhász & Lawton, 2022). All TLR coding sequences likely encoded functional receptors, as we did not observe any stop codon, and since the expected LRR and TIR motifs were always detected. The protein annotation performed using protein BLAST evidenced that the TLR repertoire we studied was composed of cell surface TLRs (i.e. TLR‐1/toll‐like protein, TLR‐2, TLR‐4 and TLR‐6) and endosomal TLRs (i.e. TLR‐3, TLR‐7/Tollo and TLR‐13) and some levels of gene redundancy. This is consistent with recent evidence that immune genes are often tandemly duplicated in molluscs and specially in bivalves (Batista et al., 2019; Peng et al., 2020; Zhang et al., 2013). Such an observation was, as well, observed on one contig (i.e. contig 7594) carrying the related TLR‐13 and TLR‐13/3.
Finally, the 14 TLR loci presented here are part, yet probably nearly complete, of the TLR repertoire of the two species. Indeed, 21 contigs were annotated as TLR in the shotgun genome of which five encoding partial TLR sequences (i.e. either LRR or TIR for instance) and two that aligned a high proportion of the same reads, which would have strongly biased the analysis.
4.2. Variation of TLR genes
Haplotype networks and the negative values of Tajima's D overall indicated that the diversity, at most TLR loci, was made of few high‐frequency haplotypes and a few to high number of rare alleles. The number of haplotypes and the level of polymorphism were similar to that reported in several vertebrate and invertebrate species (Babik et al., 2015). Densities of segregating sites observed across TLR genes were similar to that of the nonimmune control gene and are consistent with previous results obtained from the transcriptome sequencing in the bivalve Macoma balthica (Pante et al., 2012), which would indicate that we likely did not overestimate the level of polymorphism.
Noteworthy, TLR loci were not equally polymorphic. The observed differences in polymorphism were overall consistent with expectations in that cell surface TLRs, which bind an expanded set of ligands of different nature, exhibit high allele diversity, compared with endosomal TLRs which sense rather conserved nucleic acid structures (Georgel et al., 2009). In our study, we observed a rather unexpected high number of haplotypes at the vesicular TLR‐7 which usually presents a low diversity, at least in vertebrates. This result could be consistent with a relaxed selection acting at this locus, reflecting the high diversity of intracellular pathogens that could possibly be encountered in the environment (Künili et al., 2021; Lattos et al., 2020). Another nonexclusive explanation would be that introgression brings high levels of diversity at that locus (Fraïsse et al., 2016), which could be transmitted all in one, or partially to successive generations due to intragenic recombination. Recombination would then be a driver of diversity leading to the production of a subset of haplotypes, which then might evolve differentially than the main haplotypes. Such a contribution of recombination has already been evidenced in immune genes, including TLR (Gösser et al., 2019; Oren et al., 2019; Schaschl et al., 2006). To our knowledge, the diversity of TLR‐7 has not been described in bivalves, and a high level of TLR‐7 has just been observed in chicken (Bulumulla et al., 2011; Świderská et al., 2018). Conversely, the two other types of vesicular TLRs (i.e. TLR‐13 and TLR‐3) sense more structurally conserved double‐stranded bacterial RNAs.
4.3. Assessment of selection towards TLR loci
No population structure of P. nobilis could be detected, neither using neutral microsatellites nor TLR loci, and high and similar levels of genetic diversity were also revealed for both markers, consistent with previously published research (Peyran et al., 2021; Wesselmann et al., 2018). Moreover, considering the overall haplotypes topologies, as well as the negative Tajima's D, our results are in agreement with the hypothesis of a panmictic population that has been experiencing a recent postbottleneck expansion (Sanna et al., 2014) and suggest that demographic processes overweigh, or hamper the detection of a small effect of selection associated with the resistance to H. pinnae.
The excess of nonsynonymous mutations and high heterozygosity rates detected is, as well, congruent with at least a transient balancing selection, acting at some of the TLR loci studied (Babik et al., 2015; Fijarczyk & Babik, 2015; Minias & Vinkler, 2022). In fact, observing a clear correlation between genetic traits and a specific pathogen resistance is difficult when the populations being tested likely have contrasting life histories with respect to the microbial communities whose pathogens they have met. Accordingly, pathogens are expected to maintain high genetic polymorphisms in hosts' populations, again interpreted as balancing selection (Llaurens et al., 2017; Quéméré et al., 2018). The proposed mechanism for maintaining such a high diversity is based on either the advantage of heterozygotes (i.e. overdominance), the advantage of rare alleles (Browne & Karubian, 2018; Stefan et al., 2019), fluctuating selection (Bell, 2010; Quéméré et al., 2018) or a combination thereof. In our study, the high haplotype diversity observed mostly at cell surface TLRs and at the vesicular TLR‐7 might likely be induced and maintained by these three mechanisms, most probably by the fluctuating selection (Andree et al., 2020; Künili et al., 2021; Lattos et al., 2020; Scarpa et al., 2020) since we did not find any sign of heterozygous advantage associated to the resistance to H. pinnae. The other TLRs, of less polymorphism, are rather under directional selection.
The clustering analysis highlighted overall small contributions from each SNP, even though putatively selected, to the cluster, indicating that whether they had a role in the resistance or tolerance to H. pinnae, their effects were rather weak and more likely cumulative or synergistic. However, SNP of TLR‐7, TLR‐4 and TLR‐6 contributed the most to the clusters. At first glance, these results are in line with the scenario that Pinna nobilis mortalities are probably a multifactorial incidence, where the combination of various pathogens with abiotic factors may be responsible for high mortalities (Hamelin et al., 2019; Künili et al., 2021; Lattos et al., 2020; Scarpa et al., 2020), whereas specific TLRs may be triggered and favoured under combinations of factors.
In this study, we also report that hybrids are indeed significantly introgressed and that more than 18% of the P. nobilis of the dataset originated from ancient hybrids. Noteworthy, all P. nobilis we found introgressed at TLR loci were identified as resistant and carried a TLR‐7 allele of P. rudis origin. As discussed above, TLR‐7 is increasingly recognized as a potent modulator of protozoan infection, capable of impeding parasite growth (for an extensive review see Fouzder et al., 2019). Therefore, the fact that TLR‐7 is conserved in MME‐surviving P. nobilis could be interpreted as an obvious a sign of adaptive introgression (Edelman & Mallet, 2021; Gouy & Excoffier, 2020; Hedrick, 2013), but once again, there is a strong indication that the full‐length TLR‐7 of P. rudis is required for the effective resistance to H. pinnae.
4.4. TLR pathogens detection and immune response
TLR‐7 is an endosomal sensor of single‐stranded nucleic acids, involved in the protection against viruses, intracellular parasites and some bacteria (Baccarella et al., 2013; Barbalat et al., 2011; Fouzder et al., 2019; Heni et al., 2020; Mifsud et al., 2014; Wu & Chen, 2014). Once activated, TLR‐7 homodimers activate the interferon regulatory factors (IRF) signalling pathway and trigger both autophagy and the production of type I interferon and other inflammatory cytokines (Petes et al., 2017).
Structurally, TLR‐7 possesses two binding sites for a guanosine at site 1 and a single‐stranded RNA (ssRNA) at site 2. Both binding sites are situated at either side of a Z‐loop region which is also involved in the ssRNA recognition. Recent research evidenced that the first site was essential for TLR‐7 activation, while the second one only contributes to ssRNA‐induced activation. The TLR‐7 is thus synergistically activated in response to guanosine and ssRNA (Zhang et al., 2016, 2018). This feature of TLR‐7 likely explains why the 4 P. nobilis introgressed with a partial TLR‐7 of P. rudis could not effectively resist the infection with H. pinnae, whereas those with a full‐length P. rudis TLR‐7 were not affected.
Regarding TLR‐6 and TLR‐4, whether these TLR are indeed more or less involved in some adaptation to H. pinnae remains highly speculative and should require specific studies. Nonetheless, these TLR, once bound to their ligands, activate MyD88 and NFkB signalling pathways, hence triggering a pro‐inflammatory response (Krishnegowda et al., 2005; Molteni et al., 2016) which may potentialize the specific TLR‐7 response.
TLR‐6 typically binds diacylated lipopeptides (Drage et al., 2009) and glycoproteins glycophosphatidylinositol (Takeuchi et al., 2001) and has been implicated in the response and potential resistance to several pathogens, from viruses (Chen et al., 2015) to bacteria (Marinho et al., 2013) and protozoan (Leoratti et al., 2008; Netea et al., 2008). Functionally, TLR‐6 dimerizes with TLR‐2, directing specific interaction with a pathogen molecule, and hence triggers essentially an IL‐6 and TNF‐α mediated response (Krishnegowda et al., 2005). Interestingly, several research highlighted the role of TLR‐6 in mitigating Mycobacterium infection (Marinho et al., 2013) to which P. nobilis is regularly exposed to. Certain adaptive changes towards the bacteria could confer a better ability to cope with the H. pinnae infection as well. TLR‐4 is mostly a sensor of the bacterial lipopolysaccharide (LPS) (Qureshi et al., 1999), although a wide range of bacterial, viral and protozoan ligands have also been identified (Molteni et al., 2016; Uematsu & Akira, 2008), indicating that TLR‐4 participates in the response to a wide range of pathogens.
5. CONCLUSION
This preliminary research supports the role of TLR‐7 in the resistance of P. nobilis to H. pinnae and the possible influence of TLR‐6 and TLR‐4, although of much smaller effect. Further field and laboratory studies are needed to complete and clarify the role of these TLRs in disease resistance and tolerance. In this aim, a systematic and extensive characterization of health and infectious status will be required. From field research, it would be necessary to sample more P. nobilis that survived at least one H. pinnae‐driven MME and assess whether they are indeed introgressed by full‐length P. rudis TLR‐7. Similarly, systematically determining young recruit's haplotypes at TLR‐7 locus could, as well, help predict how resilient a population would be in the face of an H. pinnae epizootic. At the laboratory, controlled experimental infections, using genetically well‐characterized individuals would bring the evidence that TLR‐7 is indeed involved in the adaptation to H. pinnae and could also allow to assess the level of associated stress response (Box et al., 2020; Lattos et al., 2023; Natalotto et al., 2015). Moreover, prophylactic strategies to prevent infection or enhance appropriate physiological responses should be tested. Current research on this topic seems promising, by challenging the innate immune system of the host with TLR‐7 agonists which could modulate the innate immune system, thus either reducing tissue inflammation, help eliminate parasites, or prevent entry of the parasite in the host (El Hajj et al., 2018; Hamie et al., 2021). However, such experiment would probably be difficult to be performed and expensive. Finally, the fact remains that to date, no parasitic DNA has been demonstrated in P. rudis, hybrids or the P. nobilis introgressed with P. rudis full‐length TLR‐7, evidencing the likely pivotal role of this protein. Whether other pattern recognition receptors, eventually of P. rudis origin, are also involved in the protection against H. pinnae infection remains to be determined. The ongoing molecular research will probably bring additional insights in the mechanism of resistance.
AUTHOR CONTRIBUTIONS
Stéphane Coupé: Conceptualization (equal); data curation (lead); formal analysis (lead); funding acquisition (equal); investigation (lead); methodology (lead); project administration (lead); resources (equal); software (lead); supervision (lead); validation (equal); visualization (equal); writing – original draft (lead); writing – review and editing (equal). Ioannis A. Giantsis: Formal analysis (equal); resources (equal); validation (equal); writing – review and editing (equal). Maite Vázquez Luis: Formal analysis (equal); resources (equal); validation (equal); writing – review and editing (equal). Fabio Scarpa: Formal analysis (equal); resources (equal); validation (equal); writing – review and editing (equal). Mathieu Foulquié: Formal analysis (equal); resources (equal); validation (equal); writing – review and editing (equal). Jean‐Marc Prévot: Software (equal); writing – review and editing (equal). Marco Casu: Resources (equal); validation (equal); writing – review and editing (equal). Athanasios Lattos: Resources (equal); validation (equal); writing – review and editing (equal). Basile Michaelidis: Resources (equal); validation (equal); writing – review and editing (equal). Daria Sanna: Resources (equal); validation (equal); writing – review and editing (equal). José Rafa García‐March: Resources (equal); validation (equal); writing – review and editing (equal). José Tena‐Medialdea: Resources (equal); validation (equal); writing – review and editing (equal). Nardo Vicente: Resources (equal); validation (equal); writing – review and editing (equal). Robert Bunet: Conceptualization (equal); formal analysis (equal); funding acquisition (equal); investigation (equal); methodology (equal); project administration (equal); resources (equal); validation (equal); writing – review and editing (equal).
FUNDING INFORMATION
This work was supported by the University of Toulon and Toulon Provence Méditerranée (TPM) related to the PINORES project, the University Institute of Technology of the University of Toulon under the grant ‘CARTT’ and by the European Union's LIFE programme through the project LIFE PINNARCA (NAT/ES/001265). Fabio Scarpa, Marco Casu and Daria Sanna acknowledge the support of NBFC to the University of Sassari, funded by the Italian Ministry of University and Research, PNRR, Missione 4, Componente 2, ‘Dalla ricerca all'impresa’, Investimento 1.4 Project CN00000033.
CONFLICT OF INTEREST STATEMENT
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supporting information
Appendix S1
ACKNOWLEDGMENTS
We thank Dr. Santiago V. Jiménez‐Gutiérrez for providing us with some tissue samples of hybrids from Spain. We also thank an anonymous reviewer for helpfull comments.
Coupé, S. , Giantsis, I. A. , Vázquez Luis, M. , Scarpa, F. , Foulquié, M. , Prévot, J.‐M. , Casu, M. , Lattos, A. , Michaelidis, B. , Sanna, D. , García‐March, J. R. , Tena‐Medialdea, J. , Vicente, N. , & Bunet, R. (2023). The characterization of toll‐like receptor repertoire in Pinna nobilis after mass mortality events suggests adaptive introgression. Ecology and Evolution, 13, e10383. 10.1002/ece3.10383
DATA AVAILABILITY STATEMENT
Data referring to predicted coding DNA sequences of toll‐like receptors and genotypes at microsatellite loci are accessible through the Dryad https://doi.org/10.5061/dryad.z8w9ghxgn.
REFERENCES
- Akira, S. , & Takeda, K. (2004). Toll‐like receptor signalling. Nature Reviews. Immunology, 4(7), 499–511. [DOI] [PubMed] [Google Scholar]
- Altizer, S. , Harvell, D. , & Friedle, E. (2003). Rapid evolutionary dynamics and disease threats to biodiversity. Trends in Ecology & Evolution, 18(11), 589–596. [Google Scholar]
- Andree, K. B. , Carrasco, N. , Carella, F. , Furones, D. , & Prado, P. (2020). Vibrio mediterranei, a potential emerging pathogen of marine Fauna: Investigation of pathogenicity using a bacterial challenge in Pinna nobilis and development of a species‐specific PCR. Journal of Applied Microbiology, 130, 617–631. [DOI] [PubMed] [Google Scholar]
- Arala‐Chaves, M. , & Sequeira, T. (2000). Is there any kind of adaptive immunity in invertebrates? Aquaculture, 191(1–3), 247–258. [Google Scholar]
- Babik, W. , Dudek, K. , Fijarczyk, A. , Pabijan, M. , Stuglik, M. , Szkotak, R. , & Zieliński, P. (2015). Constraint and adaptation in newt toll‐like receptor genes. Genome Biology and Evolution, 7(1), 81–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baccarella, A. , Fontana, M. F. , Chen, E. C. , & Kim, C. C. (2013). Toll‐like receptor 7 mediates early innate immune responses to malaria. Infection and Immunity, 81(12), 4431–4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankevich, A. , Nurk, S. , Antipov, D. , Gurevich, A. A. , Dvorkin, M. , Kulikov, A. S. , Lesin, V. M. , Nikolenko, S. I. , Pham, S. , Prjibelski, A. D. , Pyshkin, A. V. , Sirotkin, A. V. , Vyahhi, N. , Tesler, G. , Alekseyev, M. A. , & Pevzner, P. A. (2012). SPAdes: A new genome assembly algorithm and its applications to single‐cell sequencing. Journal of Computational Biology, 19(5), 455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbalat, R. , Ewald, S. E. , Mouchess, M. L. , & Barton, G. M. (2011). Nucleic acid recognition by the innate immune system. Annual Review of Immunology, 29, 185–214. [DOI] [PubMed] [Google Scholar]
- Barreiro, L. B. , Ben‐Ali, M. , Quach, H. , Laval, G. , Patin, E. , Pickrell, J. K. , Bouchier, C. , Tichit, M. , Neyrolles, O. , Gicquel, B. , Kidd, J. R. , Kidd, K. K. , Alcaïs, A. , Ragimbeau, J. , Pellegrini, S. , Abel, L. , Casanova, J. L. , & Quintana‐Murci, L. (2009). Evolutionary dynamics of human toll‐like receptors and their different contributions to host defense. PLoS Genetics, 5(7), e1000562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista, F. M. , Churcher, A. M. , Manchado, M. , Leitão, A. , & Power, D. M. (2019). Uncovering the immunological repertoire of the carpet shell clam Ruditapes decussatus through a transcriptomic‐based approach. Aquaculture and Fisheries Studies, 4(1), 37–42. [Google Scholar]
- Bean, A. G. D. , Baker, M. L. , Stewart, C. R. , Cowled, C. , Deffrasnes, C. , Wang, L. F. , & Lowenthal, J. W. (2013). Studying immunity to zoonotic diseases in the natural host — Keeping it real. Nature Reviews Immunology, 13(12), 851–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell, G. (2010). Fluctuating selection: The perpetual renewal of adaptation in variable environments. Philosophical Transactions of the Royal Society B: Biological Sciences, 365(1537), 87–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini, Y. , & Hochberg, Y. (1995). Controlling the false discovery rate: A practical and powerful approach to multiple testing. Journal of the Royal Statistical Society: Series B: Methodological, 57(1), 289–300. [Google Scholar]
- Box, A. , Capó, X. , Tejada, S. , Catanese, G. , Grau, A. , Deudero, S. , Sureda, A. , & Valencia, J. M. (2020). Reduced antioxidant response of the fan mussel Pinna nobilis related to the presence of Haplosporidium pinnae . Pathogens, 9(11), 932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browne, L. , & Karubian, J. (2018). Rare genotype advantage promotes survival and genetic diversity of a tropical palm. The New Phytologist, 218(4), 1658–1667. [DOI] [PubMed] [Google Scholar]
- Bulumulla, P. , Silva, P. , & Jianlin, H. (2011). Genetic diversity at toll like receptor 7 (TLR7) gene of Sri Lankan indigenous chicken and Ceylon jungle fowl (Gallus lafayetti). Tropical Agricultural Research, 22(4), 367–373. [Google Scholar]
- Bunet, R. , Prévot, J. M. , Vicente, N. , García‐March, J. R. , Martinović, R. , Tena‐Medialdea, J. , Joksimovic, D. , Bonnefont, J. L. , & Coupé, S. (2021). First insight into the whole genome shotgun sequence of the endangered noble pen shell Pinna nobilis: A giant bivalve undergoing a mass mortality event. Journal of Molluscan Studies, 87, eyaa041. [Google Scholar]
- Catanese, G. , Coupé, S. , & Bunet, R. (2022). Mitogenome sequence comparison in the endangered congeneric Pinna nobilis and Pinna rudis bivalves. Molecular Biology Reports, 1‐9, 3627–3635. [DOI] [PubMed] [Google Scholar]
- Catanese, G. , Grau, A. , Valencia, J. M. , Garcia‐March, J. R. , Vázquez‐Luis, M. , Alvarez, E. , Deudero, S. , Darriba, S. , Carballal, M. J. , & Villalba, A. (2018). Haplosporidium pinnae sp. nov., a haplosporidan parasite associated with mass mortalities of the fan mussel, Pinna nobilis, in the Western Mediterranean Sea. Journal of Invertebrate Pathology, 157, 9–24. [DOI] [PubMed] [Google Scholar]
- Chen, J. , Ng, M. M. L. , & Chu, J. J. H. (2015). Activation of TLR2 and TLR6 by dengue NS1 protein and its implications in the immunopathogenesis of dengue virus infection. PLoS Pathogens, 11(7), e1005053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevillard, C. , Nunes, J. P. S. , Frade, A. F. , Almeida, R. R. , Pandey, R. P. , Nascimento, M. S. , Kalil, J. , & Cunha‐Neto, E. (2018). Disease tolerance and pathogen resistance genes may underlie Trypanosoma cruzi persistence and differential progression to Chagas disease cardiomyopathy. Frontiers in Immunology, 9, 2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cinar, M. E. , Bilecenoglu, M. , Mb, Y. , & Güçlüsoy, H. (2021). The last fortress fell: Mass mortality of Pinna nobilis in the sea of Marmara. Mediterranean Marine Science, 22(3), 669–676. [Google Scholar]
- Connallon, T. , & Clark, A. G. (2012). A general population genetic framework for antagonistic selection that accounts for demography and recurrent mutation. Genetics, 190(4), 1477–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowen, R. K. , Gawarkiewicz, G. , Pineda, J. , Thorrold, S. R. , & Werner, F. E. (2007). Population connectivity in marine systems an overview. Oceanography, 20(3), 14–21. [Google Scholar]
- Destoumieux‐Garzón, D. , Canesi, L. , Oyanedel, D. , Travers, M. , Charrière, G. M. , Pruzzo, C. , & Vezzulli, L. (2020). Vibrio–bivalve interactions in health and disease. Environmental Microbiology, 22(10), 4323–4341. [DOI] [PubMed] [Google Scholar]
- Drage, M. G. , Pecora, N. D. , Hise, A. G. , Febbraio, M. , Silverstein, R. L. , Golenbock, D. T. , Boom, W. H. , & Harding, C. V. (2009). TLR2 and its co‐receptors determine responses of macrophages and dendritic cells to lipoproteins of Mycobacterium tuberculosis . Cellular Immunology, 258(1), 29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelman, N. B. , & Mallet, J. (2021). Prevalence and adaptive impact of introgression. Annual Review of Genetics, 55, 265–283. [DOI] [PubMed] [Google Scholar]
- el Hajj, R. , Bou Youness, H. , Lachaud, L. , Bastien, P. , Masquefa, C. , Bonnet, P. A. , el Hajj, H. , & Khalifeh, I. (2018). EAPB0503: An Imiquimod analog with potent in vitro activity against cutaneous leishmaniasis caused by Leishmania major and Leishmania tropica. PLoS Neglected Tropical Diseases, 12(11), e0006854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmqvist, T. , Folke, C. , Nyström, M. , Peterson, G. , Bengtsson, J. , Walker, B. , & Norberg, J. (2003). Response diversity, ecosystem change, and resilience. Frontiers in Ecology and the Environment, 1(9), 488–494. [Google Scholar]
- Excoffier, L. , & Lischer, H. E. (2010). Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and windows. Molecular Ecology Resources, 10(3), 564–567. [DOI] [PubMed] [Google Scholar]
- Fey, S. B. , Siepielski, A. M. , Nusslé, S. , Cervantes‐Yoshida, K. , Hwan, J. L. , Huber, E. R. , Fey, M. J. , Catenazzi, A. , & Carlson, S. M. (2015). Recent shifts in the occurrence, cause, and magnitude of animal mass mortality events. Proceedings of the National Academy of Sciences, 112(4), 1083–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fijarczyk, A. , & Babik, W. (2015). Detecting balancing selection in genomes: Limits and prospects. Molecular Ecology, 24(14), 3529–3545. [DOI] [PubMed] [Google Scholar]
- Fornarino, S. , Laval, G. , Barreiro, L. B. , Manry, J. , Vasseur, E. , & Quintana‐Murci, L. (2011). Evolution of the TIR domain‐containing adaptors in humans: Swinging between constraint and adaptation. Molecular Biology and Evolution, 28(11), 3087–3097. [DOI] [PubMed] [Google Scholar]
- Fouzder, C. , Mukhuty, A. , Das, S. , & Chattopadhyay, D. (2019). TLR signaling on protozoan and helminthic parasite infection. In Rezaei, N. (Ed.), Toll‐like receptors. IntechOpen. [Google Scholar]
- Fraïsse, C. , Belkhir, K. , Welch, J. J. , & Bierne, N. (2016). Local interspecies introgression is the main cause of extreme levels of intraspecific differentiation in mussels. Molecular Ecology, 25(1), 269–286. [DOI] [PubMed] [Google Scholar]
- Gay, N. J. , & Gangloff, M. (2007). Structure and function of toll receptors and their ligands. Annual Review of Biochemistry, 76, 141–165. [DOI] [PubMed] [Google Scholar]
- Georgel, P. , Macquin, C. , & Bahram, S. (2009). The heterogeneous allelic repertoire of human toll‐like receptor (TLR) genes. PLoS One, 4(11), e7803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gösser, F. , Schartl, M. , Garcia‐De Leon, F. J. , Tollrian, R. , & Lampert, K. P. (2019). Red queen revisited: Immune gene diversity and parasite load in the asexual Poecilia formosa versus its sexual host species P. mexicana . PLoS One, 14(7), e0219000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goudet, J. (2001). FSTAT, a program to estimate and test gene diversities and fixation indices, version 2.9.3. Httpwww2 Unil Chpopgensoftwaresfstat Htm. [Google Scholar]
- Gouy, A. , & Excoffier, L. (2020). Polygenic patterns of adaptive introgression in modern humans are mainly shaped by response to pathogens. Molecular Biology and Evolution, 37(5), 1420–1433. [DOI] [PubMed] [Google Scholar]
- Green, T. J. , & Speck, P. (2018). Antiviral defense and innate immune memory in the oyster. Viruses, 10(3), 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grueber, C. E. , Knafler, G. J. , King, T. M. , Senior, A. M. , Grosser, S. , Robertson, B. , Weston, K. A. , Brekke, P. , Harris, C. L. W. , & Jamieson, I. G. (2015). Toll‐like receptor diversity in 10 threatened bird species: Relationship with microsatellite heterozygosity. Conservation Genetics, 16(3), 595–611. [Google Scholar]
- Hamelin, F. M. , Allen, L. J. , Bokil, V. A. , Gross, L. J. , Hilker, F. M. , Jeger, M. J. , Manore, C. A. , Power, A. G. , Rúa, M. A. , & Cunniffe, N. J. (2019). Coinfections by noninteracting pathogens are not independent and require new tests of interaction. PLoS Biology, 17(12), e3000551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamie, M. , Najm, R. , Deleuze‐Masquefa, C. , Bonnet, P. A. , Dubremetz, J. F. , & El Sabban, M. (2021). Imiquimod targets toxoplasmosis through modulating host toll‐like receptor‐MyD88 signaling. Frontiers in Immunology, 12, 629917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasik, A. Z. , & Siepielski, A. M. (2022). Parasitism shapes selection by drastically reducing host fitness and increasing host fitness variation. Biology Letters, 18(11), 20220323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrick, P. W. (1999). Antagonistic pleiotropy and genetic polymorphism: A perspective. Heredity, 82(2), 126–133. [Google Scholar]
- Hedrick, P. W. (2013). Adaptive introgression in animals: Examples and comparison to new mutation and standing variation as sources of adaptive variation. Molecular Ecology, 22(18), 4606–4618. [DOI] [PubMed] [Google Scholar]
- Heni, A. C. , Schmid, J. , Rasche, A. , Corman, V. M. , Drosten, C. , & Sommer, S. (2020). Pathogen‐associated selection on innate immunity genes (TLR4, TLR7) in a neotropical rodent in landscapes differing in anthropogenic disturbance. Heredity, 125(4), 184–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holderegger, R. , Kamm, U. , & Gugerli, F. (2006). Adaptive vs. neutral genetic diversity: Implications for landscape genetics. Landscape Ecology, 21(6), 797–807. [Google Scholar]
- Imai, Y. , Kuba, K. , Neely, G. G. , Yaghubian‐Malhami, R. , Perkmann, T. , van Loo, G. , Ermolaeva, M. , Veldhuizen, R. , Leung, Y. H. C. , Wang, H. , Liu, H. , Sun, Y. , Pasparakis, M. , Kopf, M. , Mech, C. , Bavari, S. , Peiris, J. S. M. , Slutsky, A. S. , Akira, S. , … Penninger, J. M. (2008). Identification of oxidative stress and toll‐like receptor 4 signaling as a key pathway of acute lung injury. Cell, 133(2), 235–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, H. , Li, J. , Li, L. , Zhang, X. , Yuan, L. , & Chen, J. (2017). Selective evolution of toll‐like receptors 3, 7, 8, and 9 in bats. Immunogenetics, 69(4), 271–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juhász, A. , & Lawton, S. P. (2022). Toll like receptors and their evolution in the lymnaeid freshwater snail species Radix auricularia and Lymnaea stagnalis, key intermediate hosts for zoonotic trematodes. Developmental and Comparative Immunology, 127, 104297. [DOI] [PubMed] [Google Scholar]
- Kersting, D. , Benabdi, M. , Čižmek, H. , Grau, A. , Jimenez, C. , & Katsanevakis, S. (2019). Pinna nobilis . IUCN Red List Threat Species, 10, 2019–2013. [Google Scholar]
- Kosakovsky Pond, S. L. , Posada, D. , Gravenor, M. B. , Woelk, C. H. , & Frost, S. D. (2006). Automated phylogenetic detection of recombination using a genetic algorithm. Molecular Biology and Evolution, 23(10), 1891–1901. [DOI] [PubMed] [Google Scholar]
- Krishnegowda, G. , Hajjar, A. M. , Zhu, J. , Douglass, E. J. , Uematsu, S. , Akira, S. , Woods, A. S. , & Gowda, D. C. (2005). Induction of proinflammatory responses in macrophages by the glycosylphosphatidylinositols of plasmodium falciparum: Cell signaling receptors, glycosylphosphatidylinositol (GPI) structural requirement, and regulation of GPI activity. The Journal of Biological Chemistry, 280(9), 8606–8616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh, A. , Larsson, B. , Von Heijne, G. , & Sonnhammer, E. L. (2001). Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. Journal of Molecular Biology, 305(3), 567–580. [DOI] [PubMed] [Google Scholar]
- Kryazhimskiy, S. , & Plotkin, J. B. (2008). The population genetics of dN/dS. PLoS Genetics, 4(12), e1000304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Künili, İ. E. , Ertürk Gürkan, S. , Aksu, A. , Turgay, E. , Çakir, F. , Gürkan, M. , & Altinağaç, U. (2021). Mass mortality in endangered fan mussels Pinna nobilis (Linnaeus 1758) caused by co‐infection of Haplosporidium pinnae and multiple vibrio infection in Çanakkale Strait, Turkey. Biomarkers, 26, 1–12. [DOI] [PubMed] [Google Scholar]
- Kutzer, M. A. , & Armitage, S. A. (2016). Maximising fitness in the face of parasites: A review of host tolerance. Zoology, 119(4), 281–289. [DOI] [PubMed] [Google Scholar]
- Lattos, A. , Giantsis, I. A. , Karagiannis, D. , & Michaelidis, B. (2020). First detection of the invasive Haplosporidian and mycobacteria parasites hosting the endangered bivalve Pinna nobilis in Thermaikos gulf, North Greece. Marine Environmental Research, 155, 104889. [DOI] [PubMed] [Google Scholar]
- Lattos, A. , Papadopoulos, D. K. , Giantsis, I. A. , Feidantsis, K. , Georgoulis, I. , Karagiannis, D. , Carella, F. , & Michaelidis, B. (2023). Investigation of the highly endangered Pinna nobilis' mass mortalities: Seasonal and temperature patterns of health status, antioxidant and heat stress responses. Marine Environmental Research, 188, 105977. [DOI] [PubMed] [Google Scholar]
- Leoratti, F. M. , Farias, L. , Alves, F. P. , Suarez‐Mútis, M. C. , Coura, J. R. , Kalil, J. , Camargo, E. P. , Moraes, S. L. , & Ramasawmy, R. (2008). Variants in the toll‐like receptor signaling pathway and clinical outcomes of malaria. The Journal of Infectious Diseases, 198(5), 772–780. [DOI] [PubMed] [Google Scholar]
- Letunic, I. , & Bork, P. (2021). Interactive tree of life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Research, 49(W1), W293–W296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic, I. , Khedkar, S. , & Bork, P. (2021). SMART: Recent updates, new developments and status in 2020. Nucleic Acids Research, 49(D1), D458–D460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , & Durbin, R. (2009). Fast and accurate short read alignment with burrows–wheeler transform. Bioinformatics, 25(14), 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Handsaker, B. , Wysoker, A. , Fennell, T. , Ruan, J. , Homer, N. , Marth, G. , Abecasis, G. , Durbin, R. , & 1000 Genome Project Data Processing Subgroup . (2009). The sequence alignment/map format and SAMtools. Bioinformatics, 25(16), 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, G. , Zhang, H. , Zhao, C. , & Zhang, H. (2020). Evolutionary history of the toll‐like receptor gene family across vertebrates. Genome Biology and Evolution, 12(1), 3615–3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llaurens, V. , Whibley, A. , & Joron, M. (2017). Genetic architecture and balancing selection: The life and death of differentiated variants. Molecular Ecology, 26(9), 2430–2448. [DOI] [PubMed] [Google Scholar]
- López‐Sanmartín, M. , Catanese, G. , Grau, A. , Valencia, J. M. , García‐March, J. R. , & Navas, J. I. (2019). Real‐time PCR based test for the early diagnosis of Haplosporidium pinnae affecting fan mussel Pinna nobilis . PLoS One, 14(2), e0212028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mable, B. K. (2019). Conservation of adaptive potential and functional diversity: Integrating old and new approaches. Conservation Genetics, 20(1), 89–100. [Google Scholar]
- Madan‐Lala, R. , Pradhan, P. , & Roy, K. (2017). Combinatorial delivery of dual and triple TLR agonists via polymeric pathogen‐like particles synergistically enhances innate and adaptive immune responses. Scientific Reports, 7(1), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinho, F. A. , de Paula, R. R. , Mendes, A. C. , de Almeida, L. A. , Gomes, M. T. , Carvalho, N. B. , Oliveira, F. S. , Caliari, M. V. , & Oliveira, S. C. (2013). Toll‐like receptor 6 senses Mycobacterium avium and is required for efficient control of mycobacterial infection. European Journal of Immunology, 43(9), 2373–2385. [DOI] [PubMed] [Google Scholar]
- Marrocco, V. , Zangaro, F. , Sicuro, A. , & Pinna, M. (2019). A scaling down mapping of Pinna nobilis (Linnaeus, 1758) through the combination of scientific literature, NATURA 2000, grey literature and citizen science data. Nature Conservation, 33, 21–31. [Google Scholar]
- Martins, R. , Carlos, A. R. , Braza, F. , Thompson, J. A. , Bastos‐Amador, P. , Ramos, S. , & Soares, M. P. (2019). Disease tolerance as an inherent component of immunity. Annual Review of Immunology, 37, 405–437. [DOI] [PubMed] [Google Scholar]
- McCallum, H. , & Dobson, A. (2002). Disease, habitat fragmentation and conservation. Proceedings of the Royal Society of London – Series B: Biological Sciences, 269(1504), 2041–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarville, J. , & Ayres, J. (2018). Disease tolerance: Concept and mechanisms. Current Opinion in Immunology, 50, 88–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melillo, D. , Marino, R. , Italiani, P. , & Boraschi, D. (2018). Innate immune memory in invertebrate metazoans: A critical appraisal. Frontiers in Immunology, 9, 1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mifsud, E. J. , Tan, A. C. , & Jackson, D. C. (2014). TLR agonists as modulators of the innate immune response and their potential as agents against infectious disease. Frontiers in Immunology, 5, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minias, P. , & Vinkler, M. (2022). Selection balancing at innate immune genes: Adaptive polymorphism maintenance in toll‐like receptors. Molecular Biology and Evolution, 39, msac102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molteni, M. , Gemma, S. , & Rossetti, C. (2016). The role of toll‐like receptor 4 in infectious and noninfectious inflammation. Mediators of Inflammation, 2016, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrell, B. , Moola, S. , Mabona, A. , Weighill, T. , Sheward, D. , Kosakovsky Pond, S. L. , & Scheffler, K. (2013). FUBAR: A fast, unconstrained bayesian approximation for inferring selection. Molecular Biology and Evolution, 30(5), 1196–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahrendorf, W. , Ivens, A. , & Spence, P. J. (2021). Inducible mechanisms of disease tolerance provide an alternative strategy of acquired immunity to malaria. eLife, 10, e63838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natalotto, A. , Sureda, A. , Maisano, M. , Spanò, N. , Mauceri, A. , & Deudero, S. (2015). Biomarkers of environmental stress in gills of Pinna nobilis (Linnaeus 1758) from Balearic Island. Ecotoxicology and Environmental Safety, 122, 9–16. [DOI] [PubMed] [Google Scholar]
- Nebot‐Colomer, E. , Vázquez‐Luis, M. , Boissin, E. , Peyran, C. , Deudero, S. , & Planes, S. (2022). Inferred family structure and dispersal patterns of a critically endangered species, Pinna nobilis, using molecular analyses: Implications for conservation. Endangered Species Research, 49, 87–102. [Google Scholar]
- Netea, M. G. , Van De Veerdonk, F. , Verschueren, I. , Van Der Meer, J. W. , & Kullberg, B. J. (2008). Role of TLR1 and TLR6 in the host defense against disseminated candidiasis. FEMS Immunology and Medical Microbiology, 52(1), 118–123. [DOI] [PubMed] [Google Scholar]
- Oren, M. , Rosental, B. , Hawley, T. S. , Kim, G. Y. , Agronin, J. , Reynolds, C. R. , Grayfer, L. , & Smith, L. C. (2019). Individual Sea urchin coelomocytes undergo somatic immune gene diversification. Frontiers in Immunology, 10, 1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pante, E. , Rohfritsch, A. , Becquet, V. , Belkhir, K. , Bierne, N. , & Garcia, P. (2012). SNP detection from de novo transcriptome sequencing in the bivalve Macoma balthica: Marker development for evolutionary studies. PLoS One, 7(12), e52302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng, J. , Li, Q. , Xu, L. , Wei, P. , He, P. , Zhang, X. , Zhang, L. , Guan, J. , Zhang, X. , Lin, Y. , Gui, J. , & Chen, X. (2020). Chromosome‐level analysis of the Crassostrea hongkongensis genome reveals extensive duplication of immune‐related genes in bivalves. Molecular Ecology Resources, 20(4), 980–994. [DOI] [PubMed] [Google Scholar]
- Petes, C. , Odoardi, N. , & Gee, K. (2017). The toll for trafficking: Toll‐like receptor 7 delivery to the endosome. Frontiers in Immunology, 8, 1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyran, C. , Boissin, E. , Morage, T. , Nebot‐Colomer, E. , Iwankow, G. , & Planes, S. (2021). Genetic homogeneity of the critically endangered fan mussel, Pinna nobilis, throughout lagoons of the Gulf of lion (North‐Western Mediterranean Sea). Scientific Reports, 11(1), 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyran, C. , Morage, T. , Nebot‐Colomer, E. , Iwankow, G. , & Planes, S. (2022). Unexpected residual habitats raise hope for the survival of the fan mussel Pinna nobilis along the Occitan coast (Northwest Mediterranean Sea). Endangered Species Research, 48, 123–137. [Google Scholar]
- Pila, E. A. , Tarrabain, M. , Kabore, A. L. , & Hanington, P. C. (2016). A novel toll‐like receptor (TLR) influences compatibility between the gastropod Biomphalaria glabrata, and the digenean trematode Schistosoma mansoni . PLoS Pathogens, 12(3), e1005513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quéméré, E. , Hessenauer, P. , Galan, M. , Fernandez, M. , Merlet, J. , Chaval, Y. , Morellet, N. , Verheyden, H. , Gilot‐Fromont, E. , & Charbonnel, N. (2021). Pathogen‐mediated selection favours the maintenance of innate immunity gene polymorphism in a widespread wild ungulate. Journal of Evolutionary Biology, 34(7), 1156–1166. [DOI] [PubMed] [Google Scholar]
- Qureshi, S. T. , Larivière, L. , Leveque, G. , Clermont, S. , Moore, K. J. , Gros, P. , & Malo, D. (1999). Endotoxin‐tolerant mice have mutations in toll‐like receptor 4 (Tlr4). The Journal of Experimental Medicine, 189(4), 615–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakoff‐Nahoum, S. , Paglino, J. , Eslami‐Varzaneh, F. , Edberg, S. , & Medzhitov, R. (2004). Recognition of commensal microflora by toll‐like receptors is required for intestinal homeostasis. Cell, 118(2), 229–241. [DOI] [PubMed] [Google Scholar]
- Regli, I. B. , Passelli, K. , Martínez‐Salazar, B. , Amore, J. , Hurrell, B. P. , Müller, A. J. , & Tacchini‐Cottier, F. (2020). TLR7 sensing by neutrophils is critical for the control of cutaneous leishmaniasis. Cell Reports, 31(10), 107746. [DOI] [PubMed] [Google Scholar]
- Rozas, J. , Ferrer‐Mata, A. , Sánchez‐DelBarrio, J. C. , Guirao‐Rico, S. , Librado, P. , Ramos‐Onsins, S. E. , & Sánchez‐Gracia, A. (2017). DnaSP 6: DNA sequence polymorphism analysis of large data sets. Molecular Biology and Evolution, 34(12), 3299–3302. [DOI] [PubMed] [Google Scholar]
- Salis, P. , Peyran, C. , Morage, T. , de Bernard, S. , Nourikyan, J. , Coupé, S. , Bunet, R. , & Planes, S. (2022). RNA‐Seq comparative study reveals molecular effectors linked to the resistance of Pinna nobilis to Haplosporidium pinnae parasite. Scientific Reports, 12(1), 21229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanna, D. , Dedola, G. L. , Scarpa, F. , Lai, T. , Cossu, P. , & Curini‐Galletti, M. (2014). New mitochondrial and nuclear primers for the Mediterranean marine bivalve Pinna nobilis . Mediterranean Marine Science, 15(2), 416–422. [Google Scholar]
- Šarić, T. , Župan, I. , Aceto, S. , Villari, G. , Palić, D. , & De Vico, G. (2020). Epidemiology of Noble pen Shell (Pinna nobilis L. 1758) mass mortality events in Adriatic Sea is characterised with rapid spreading and acute disease progression. Pathogens, 9(10), 776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarpa, F. , Sanna, D. , Azzena, I. , Mugetti, D. , Cerruti, F. , Hosseini, S. , Cossu, P. , Pinna, S. , Grech, D. , Cabana, D. , Pasquini, V. , Esposito, G. , Cadoni, N. , Atzori, F. , Antuofermo, E. , Addis, P. , Sechi, L. A. , Prearo, M. , Peletto, S. , … Casu, M. (2020). Multiple non‐species‐specific pathogens possibly triggered the mass mortality in Pinna nobilis . Life, 10(10), 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaschl, H. , Wandeler, P. , Suchentrunk, F. , Obexer‐Ruff, G. , & Goodman, S. (2006). Selection and recombination drive the evolution of MHC class II DRB diversity in ungulates. Heredity, 97(6), 427–437. [DOI] [PubMed] [Google Scholar]
- Siletic, T. , & Peharda, M. (2003). Population study of the fan shell Pinna nobilis L. in Malo and Veliko Jezero of the Mljet National Park (Adriatic Sea). Scientia Marina, 67(1), 91–98. [Google Scholar]
- Spurgin, L. G. , & Richardson, D. S. (2010). How pathogens drive genetic diversity: MHC, mechanisms and misunderstandings. Proceedings of the Royal Society B: Biological Sciences, 277(1684), 979–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke, M. , & Morgenstern, B. (2005). AUGUSTUS: A web server for gene prediction in eukaryotes that allows user‐defined constraints. Nucleic Acids Research, 33(Suppl_2), W465–W467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefan, T. , Matthews, L. , Prada, J. M. , Mair, C. , Reeve, R. , & Stear, M. J. (2019). Divergent allele advantage provides a quantitative model for maintaining alleles with a wide range of intrinsic merits. Genetics, 212(2), 553–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton, J. T. , Robertson, B. C. , & Jamieson, I. G. (2015). MHC variation reflects the bottleneck histories of New Zealand passerines. Molecular Ecology, 24(2), 362–373. [DOI] [PubMed] [Google Scholar]
- Świderská, Z. , Šmídová, A. , Buchtová, L. , Bryjová, A. , Fabiánová, A. , Munclinger, P. , & Vinkler, M. (2018). Avian toll‐like receptor allelic diversity far exceeds human polymorphism: An insight from domestic chicken breeds. Scientific Reports, 8(1), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi, O. , Kawai, T. , Mühlradt, P. F. , Morr, M. , Radolf, J. D. , Zychlinsky, A. , Takeda, K. , & Akira, S. (2001). Discrimination of bacterial lipoproteins by toll‐like receptor 6. International Immunology, 13(7), 933–940. [DOI] [PubMed] [Google Scholar]
- Taylor, S. S. , & Jamieson, I. G. (2008). No evidence for loss of genetic variation following sequential translocations in extant populations of a genetically depauperate species. Molecular Ecology, 17(2), 545–556. [DOI] [PubMed] [Google Scholar]
- Thorvaldsdóttir, H. , Robinson, J. T. , & Mesirov, J. P. (2013). Integrative genomics viewer (IGV): High‐performance genomics data visualization and exploration. Briefings in Bioinformatics, 14(2), 178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompkins, D. M. , & Begon, M. (1999). Parasites can regulate wildlife populations. Parasitology Today, 15(8), 311–313. [DOI] [PubMed] [Google Scholar]
- Trigos‐Santos, S. , & Vicente, N. (2018). Population status of Pinna nobilis in four protected areas of France and Monaco. Video MilLie life Environment, 68(2–3), 145–149. [Google Scholar]
- Tschirren, B. , Andersson, M. , Scherman, K. , Westerdahl, H. , Mittl, P. R. , & Råberg, L. (2013). Polymorphisms at the innate immune receptor TLR2 are associated with Borrelia infection in a wild rodent population. Proceedings of the Royal Society B: Biological Sciences, 280(1759), 20130364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner, A. K. , Begon, M. , Jackson, J. A. , Bradley, J. E. , & Paterson, S. (2011). Genetic diversity in cytokines associated with immune variation and resistance to multiple pathogens in a natural rodent population. PLoS Genetics, 7(10), e1002343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uematsu, S. , & Akira, S. (2008). Toll‐like receptors (TLRs) and their ligands. In Toll‐Like Receptors (TLRs) and innate immunity (pp. 1–20). Springer. [DOI] [PubMed] [Google Scholar]
- Vázquez‐Luis, M. , Álvarez, E. , Barrajón, A. , García‐March, J. R. , Grau, A. , Hendriks, I. E. , Jiménez, S. , Kersting, D. , Moreno, D. , Pérez, M. , Ruiz, J. M. , Sánchez, J. , Villalba, A. , & Deudero, S. (2017). SOS Pinna nobilis: A mass mortality event in western Mediterranean Sea. Frontiers in Marine Science, 4, 220. [Google Scholar]
- Vázquez‐Luis, M. , Nebot‐Colomer, E. , Deudero, S. , Planes, S. , & Boissin, E. (2021). Natural hybridization between pen shell species: Pinna rudis and the critically endangered Pinna nobilis may explain parasite resistance in P. nobilis . Molecular Biology Reports, 48(1), 997–1004. [DOI] [PubMed] [Google Scholar]
- Wang, W. , Wang, L. , Liu, Z. , Song, X. , Yi, Q. , Yang, C. , & Song, L. (2020). The involvement of TLR signaling and anti‐bacterial effectors in enhanced immune protection of oysters after Vibrio splendidus pre‐exposure. Developmental and Comparative Immunology, 103, 103498. [DOI] [PubMed] [Google Scholar]
- Weaver, S. , Shank, S. D. , Spielman, S. J. , Li, M. , Muse, S. V. , & Kosakovsky Pond, S. L. (2018). Datamonkey 2.0: A modern web application for characterizing selective and other evolutionary processes. Molecular Biology and Evolution, 35(3), 773–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesselmann, M. , González‐Wangüemert, M. , Serrão, E. A. , Engelen, A. H. , Renault, L. , García‐March, J. R. , Duarte, C. M. , & Hendriks, I. E. (2018). Genetic and oceanographic tools reveal high population connectivity and diversity in the endangered pen shell Pinna nobilis . Scientific Reports, 8(1), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, J. , & Chen, Z. J. (2014). Innate immune sensing and signaling of cytosolic nucleic acids. Annual Review of Immunology, 32(1), 461–488. [DOI] [PubMed] [Google Scholar]
- Yang, J. , Yan, R. , Roy, A. , Xu, D. , Poisson, J. , & Zhang, Y. (2015). The I‐TASSER suite: Protein structure and function prediction. Nature Methods, 12(1), 7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , He, X. , Yu, F. , Xiang, Z. , Li, J. , Thorpe, K. L. , & Yu, Z. (2013). Characteristic and functional analysis of toll‐like receptors (TLRs) in the lophotrocozoan, Crassostrea gigas, reveals ancient origin of TLR‐mediated innate immunity. PLoS One, 8(10), e76464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Z. , Ohto, U. , Shibata, T. , Krayukhina, E. , Taoka, M. , Yamauchi, Y. , Tanji, H. , Isobe, T. , Uchiyama, S. , Miyake, K. , & Shimizu, T. (2016). Structural analysis reveals that toll‐like receptor 7 is a dual receptor for guanosine and single‐stranded RNA. Immunity, 45(4), 737–748. [DOI] [PubMed] [Google Scholar]
- Zhang, Z. , Ohto, U. , Shibata, T. , Taoka, M. , Yamauchi, Y. , Sato, R. , Shukla, N. M. , David, S. A. , Isobe, T. , Miyake, K. , & Shimizu, T. (2018). Structural analyses of toll‐like receptor 7 reveal detailed RNA sequence specificity and recognition mechanism of agonistic ligands. Cell Reports, 25(12), 3371–3381. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1
Data Availability Statement
Data referring to predicted coding DNA sequences of toll‐like receptors and genotypes at microsatellite loci are accessible through the Dryad https://doi.org/10.5061/dryad.z8w9ghxgn.