

Abstract
Infection and delayed wound healing are two major serious complications related to traumatic injuries and cause a significant burden to patients and society. Most currently available drug delivery materials typically carry a single drug, lack protection from drug loading, and face challenges in on-demand and precisely controlled drug release. Here, we report a flower (Cirsium arvense)-inspired capsule-integrated multilayer nanofilm (FICIF), synthesized using a layer-by-layer self-assembly, for programmed multiple drug co-delivery for trauma (open fracture as an example) treatments. Our approach allows polypeptide multilayer nanofilms and innovative impregnated capsules to assemble hierarchical reservoirs with specific drug binding sites, shielding protection capability, and ordered packing structures. The resultant FICIF nanocarriers enable sustained and on-demand co-delivery of a unique immune-tuning cytokine (interleukin 12p70) and a growth factor (bone morphogenetic protein 2) in clinical use, resulting in extraordinary anti-infection (3 orders of magnitude improved bacterial killing) and bone regeneration (5 times enhanced bone healing) in treating infected rat femur fractures. The successful synthesis of these biomimetic high-performance delivery nanocoatings is expected to serve as a source of inspiration for the development of biomaterials for various clinical applications.
Keywords: self-assembly, implant nanocoating, drug delivery, anti-infection, bone regeneration, fracture, trauma
Graphical Abstract:

INTRODUCTION
Wars and traffic accidents cause a significant number of open fractures. For instance, in the recent U.S. military operations, approximately 13% of the more than 52,000 combat injuries were open fractures.1–5 There are also more than 150,000 open fractures every year among U.S. civilians. Overall, open fractures worldwide are increasingly common because of increased survivability of high energy trauma. Medical and surgical advancements, including early antibiotic treatment, tetanus prophylaxis, surgical debridement, and fracture stabilization, have been utilized by orthopedic surgeons to treat open fractures.6–8 Biomedical implants have been widely used, but unfortunately, they usually suffer from complications, two major ones are implant-associated infection and delayed wound healing,9,10 which typically result in poor clinical outcome, surgical failure, and even high-risk disability or death. For instance, military personnel with open fracture infections had a significantly higher rate of amputation (34.3 vs 15.4%, p = 0.02) and significantly longer time to union (11.0 vs 8.6 mons, p = 0.001) compared to those injuries without infections.11 As a result, infected military patients showed long hospital stays (average 17.9 days) and high inpatient resource utilization ($65.2 million) along with $170 million in possible disability benefits,12 and combat-related infections have led to more deaths than direct battle-related injuries.13 Therefore, a variety of implants (e.g., for open fracture fixation) may demand to be anti-infective and/or tissue regenerative, exceeding the traditionally passive biocompatibility requirements.14–16 Drug delivery materials, which can be coated on various implants in forms of films, particles, capsules, and so forth.17–19 may achieve direct action with low toxicity to reduce postoperative complications, providing an effective strategy to functionalize implants without compromising their original structural and physicochemical properties.
The layer-by-layer (LbL) self-assembly technique enables highly versatile and facile synthesis of drug delivery vehicles from various materials (polymers, lipids, particles, proteins, etc.), and the resultant materials combine nanoscale thickness control, component diversity, and remarkable assembly flexibility (such as diverse substrates with different topographies).20–24 Most of the currently developed vehicles for drug delivery, prepared using the LbL self-assembly technique, are mainly found in the forms of multilayer nanofilms or capsules.25–29 The multilayer films typically deliver only one single drug with a low drug loading capacity. The capsules usually suffer from limited structural controllability and unsustained burst release despite their improved loading capability. Some multidrug delivery vehicles have been developed using drugs as components during the LbL self-assembly; however, the self-assembly process inevitably results in the loss of drug bioactivity and heavy storage burden. The challenge, therefore, is to develop an innovative strategy to create delivery vehicles with the capability of multiple drug loading (in particular protein drugs with stability concerns) and achieve on-demand and sustained release in programmed sequences to reduce infectious diseases and to promote wound healing.
Inspired by flowers’ self-adaptive pollen spread we see in nature, here, we demonstrate a facile approach based on LbL self-assembly to create flower-inspired capsule-integrated nanofilms (FICIF) from biocompatible polypeptides for pH-responsive delivery of protein drugs. The premise of our design is that the capsule impregnation, capsule deposition, and shielded nanofilm constitution are tailored to assemble capsule-embedded multilayer structures with specific drug binding sites and highly ordered packing structures. The resultant FICIF nanocoatings enabled the co-delivery of various protein drugs with diverse properties (e.g., positive/negative charge, molecular type, and molecular weight), achieving a unique IL-12 loading of 178 pg cm–2 with a desired 10-day release and BMP-2 loading of 286 ng cm–2 with a long 42-day release. Both in vitro cell culture and in vivo rat fracture studies indicated that drug-loaded FICIF coatings resulted in remarkably high bone regeneration and osseointegration and significantly reduced bacterial infection.
RESULTS AND DISCUSSION
Flower-Inspired Design.
Natural flowers like Cirsium arvense display a typical structure with a set of stacking petals wrapping the flower buds, which show a biological function to gradually make sepals and then petals open to expose the buds because of temperature and humidity stimuli, which facilitates buds to transfer pollen (Figure 1a). Inspired by this unique biostructure, we created capsule-integrated, pH-responsive biomimetic implant nanocoatings via a LbL self-assembly for multiple drug delivery to reduce the two commonly seen complications (i.e., infection and delayed healing) of open fractures (Figure 1b). The FICIF nanocoatings were developed based on two criteria: (i) the coatings can load multiple protein drugs with different properties any time before their clinical uses and (ii) the coatings must achieve an on-demand and programmed release of the required drugs (such as anti-infection and bone healing drugs) upon pathological stimulation. The first requirement was met by using impregnated capsules, where biocompatible polypeptides (i.e., poly-l-lysine or PL, poly-l-glutamic acid, or PG) rather than conventional polymers were chosen to prepare the porous capsule reservoirs (functioning as biomimetic flower buds), in which abundant polyelectrolytes were impregnated to create specific loading sites for diverse drugs based on the electrostatic interaction. A unique structural design originating from flower biostructures was used to satisfy the other criterion–controllable pH-triggered drug release. Diverse impregnated capsules, as main drug reservoirs, were interspersed in multilayer films in an elaborate self-assembly density and sequence and with the desirable layers of nanofilms. These multilayer nanofilms function as flower sepals and petals to separate or cover the flower bud-like capsules, in order to achieve sustained and on-demand drug release to meet the pathological responsive requirement. Different from conventional antibiotic approaches, here, an immune-tuning antimicrobial agent (i.e., interleukin 12p70 or IL-12)30–32 and a clinically approved growth factor (i.e., recombinant human bone morphogenetic protein 2 or BMP-2)33,34 were used to reduce infection and to promote healing of trauma injuries; rapid healing reduces the chances of bacterial colonization thereby contributing to infection reduction.
Figure 1.
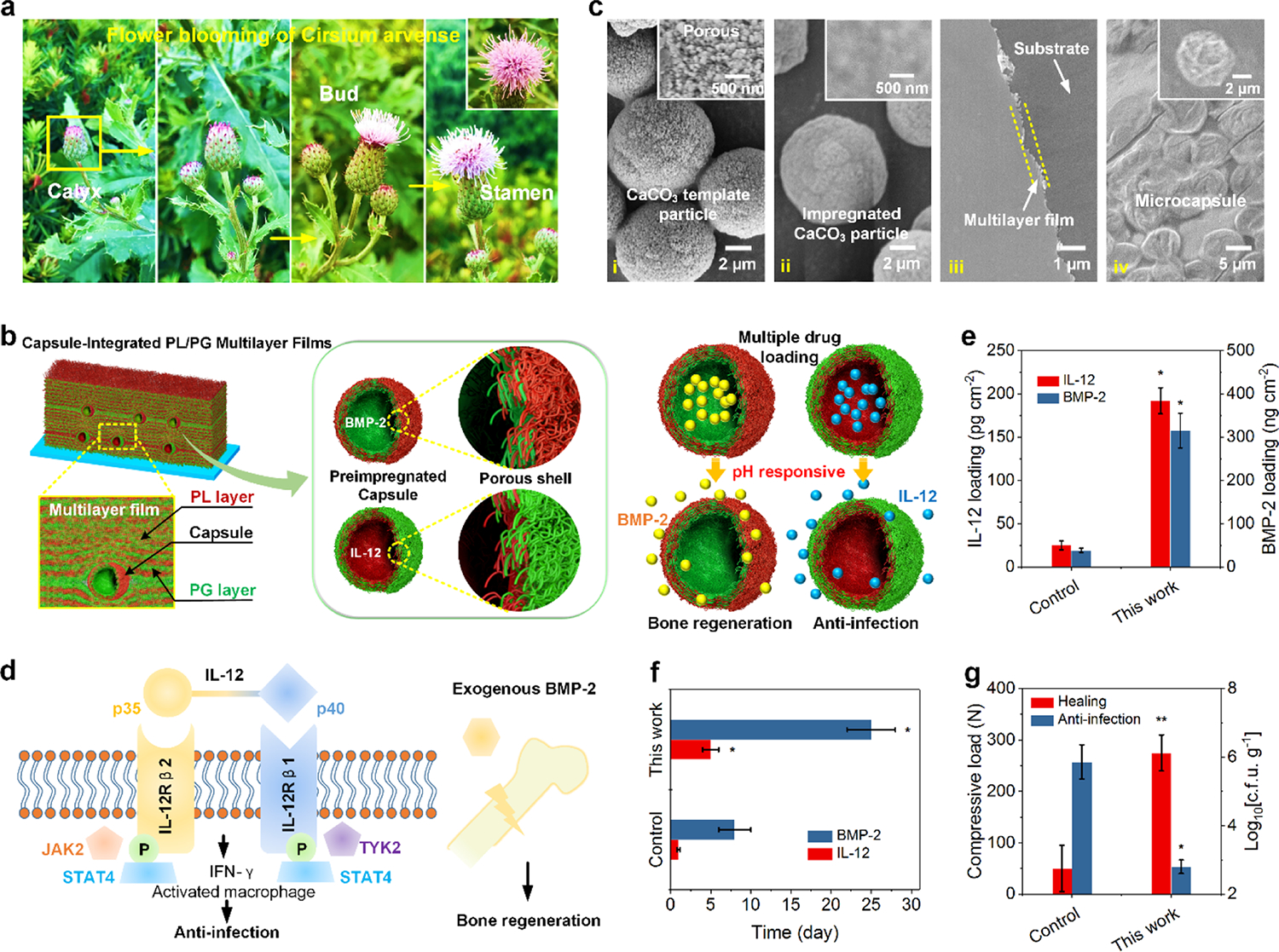
(a) Pictures showing the flower blooming of C. arvense for pollen spread in nature. (b) Schematic demonstrating the capsule-integrated PL/PG multilayer nanocoatings capable of delivering multiple protein drugs for bone regeneration and anti-infection. (c) SEM images of (i) CaCO3 template particles, (ii) polypeptide-impregnated CaCO3 particles, (iii) (PL/PG)10 multilayer nanofilms, and (iv) microcapsules assembled on PL/PG films. (d) Schematic showing the functional mechanism for anti-infection using immune-tuning antimicrobial agent of IL-12 and bone healing based on the osteogenic growth factor of BMP-2. The comparison of (e) loading capacity (IL-12 and BMP-2), (f) release period (release 80% of their total loading), and (g) bone healing and anti-infection performances (loaded with IL-12 and BMP-2) between the controls (nanofilms) and capsule-integrated nanocoatings. *p < 0.05 and **p < 0.01 compared to the control.
Scanning electron microscopy (SEM) imaging indicated that, after impregnation in a PL or PG solution (3 mg mL−1), the mesoporous pores on CaCO3 template particles (particle size of ~6 μm) were almost filled up and disappeared (Figure 1c(i,ii)). This change resulted in obviously decreased surface roughness and generated abundant drug loading sites within subsequently formed capsules. The impregnation was mainly driven by the capillary effect of the CaCO3 particle pore channels.35 Figure 1c(iii,iv) presents the typical structures of the (PL/PG)10 multilayer nanofilms, which possess a shield effect like flower sepals/petals, and the deposition of (PL/PG)5-shelled microcapsules that are utilized to simulate the function of flower buds. With increasing deposition of PL/PG bilayers, the film thickness increased linearly regardless of substrates, showing a bilayer thickness of ~5 nm (Supporting Information Figure 1). In order to reduce the use of antibiotics and related antibiotic resistance, a unique immune-tuning cytokine IL-12 was studied for anti-infection. IL-12 with subunits of IL-12p35 and IL-12p40 signals through the IL-12 receptors (IL-12Rβ1 and IL-12Rβ2) and stimulates the activities of nonreceptors like Janus kinase 2 (JAK2) and tyrosine kinase 2 (TYK2),36,37 resulting in the phosphorylation of the signal transducer and activator of transcription (Figure 1d left). IL-12 drives naive CD4 T cells to differentiate into TH1 cells and secrete interferon (IFN-γ), activating macrophages to destroy bacteria. BMP-2 was used to stimulate bone regeneration (Figure 1d right), which may also provide a seal to reduce bacterial attachment therefore reducing infections.
As shown in Figure 1e, in contrast to the multilayer films (25 pg cm−2 IL-12 and 38.5 ng cm−2 BMP-2), the unique capsule-integration design (flower bud-inspired structures as drug reservoirs) allowed our FICIF nanocoatings to achieve more than 7 times the loading capability for both IL-12 and BMP-2 (192 pg cm−2 IL-12 and 315 ng cm−2 BMP-2). Besides the high drug loadings, the FICIF nanocarriers were prepared in water-based solutions and can load multiple protein drugs after coating preparation. These features overcome the limitations of existing cutting-edge drug-delivering implant coatings, which typically load bioactive drug molecules during coating preparation, and thus inevitably suffer from the loss of their drug bioactivities during the coating process. Moreover, the embedded multilayer nanofilms (flower petal-inspired protection/shielding structures) covered on capsules reduced the typical “burst release” of conventional delivery systems, and a sustained, programmed, and controllable release (>5 days and >25 days for 80% IL-12 and BMP-2 release, respectively, compared to <1 day and <8 days for the controls, Figure 1f) was achieved. These FICIF nanocoatings loaded with IL-12 and BMP-2 significantly improved anti-infection and bone healing (Figure 1g), for example, 5-times enhanced bone strength and 3 orders of magnitude higher Staphylococcus aureus killing efficacy were achieved, demonstrating promising clinical applications.
Structural Control and Drug Loading/Release.
Unlike most current drug-loading approaches that use drugs as self-assembly components of coatings, we mainly utilized two routines—pre-impregnation capacity and capsule density—to tailor the loading capacity of the FICIF nanocarriers. As illustrated in Figure 2a left, with increasing incubation time (10–60 min) in a 3 mg mL−1 PL solution, the amount of the PL polypeptide immobilized in each CaCO3 particle increased from 1.8 to 5.5 ng and achieved saturation within 40 min. Their confocal laser scanning microscopy or CLSM images at 10 and 40 min (Figure 2a right) directly confirmed the loading of PLFITC in CaCO3 particles. The manipulation of capsule density by the control of incubation time in a 5 × 106 particle mL−1 suspension also enabled the control of drug loading capability. Obviously, the particle assembly was quite fast at the beginning, and then slowed and achieved 11,850 particle cm−2 within 90 min followed by a plateau (Figure 2b). In dramatic contrast to the conventional drug delivery systems that typically require complex and tedious structural designs while having limited applications, these two innovative tailoring strategies were facile and easily tunable based on specific application requirements. Besides the assembly structures, two properties (i.e., surface wettability and surface roughness) of FICIF nanocoatings, which play a vital role in biomedical applications, were also evaluated. Compared to nanocoatings having PL as the outermost layer [such as L5.5 and L10.5, water contact angles (WCAs): 40–50°], using PG as the outermost coating resulted in significantly decreased WCA to 20–30° because of the numerous carboxyl side groups in the PG polypeptide (Figure 2c). The coating’s surface roughness increased from 0.03 to 0.12 μm with increasing deposition of both nanofilms and capsules (Figure 2c). The resultant properties of improved hydrophilicity and surface roughness of the FICIF nanocoatings provide a desirable platform to facilitate cell adhesion and proliferation,38,39 revealing their promising tissue engineering applications.
Figure 2.
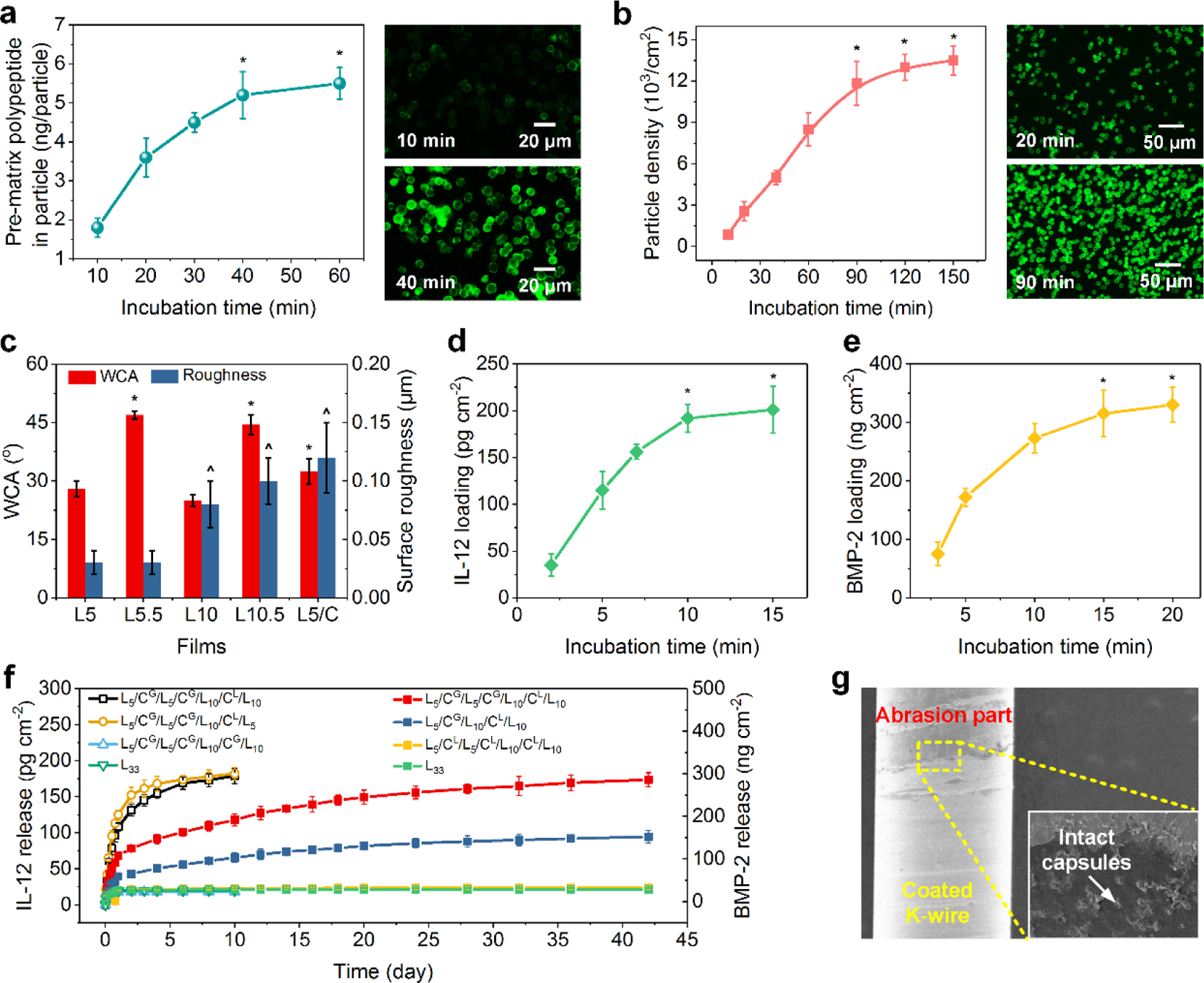
(a) Left, the impregnation of polyelectrolyte (e.g., PL). in CaCO3 particles incubated in a polypeptide solution (3 mg mL−1) for various times. Right, CLSM images of the impregnated CaCO3 particles (CaCO3PL) after different incubation times. PL was labeled using FITC. (b) Left, density of CaCO3PL particles on PL/PG nanofilms incubated in a 5 × 106 particle mL−1 suspension for different times. Right, CLSM images of CaCO3PL particles assembled on the films at different times. (c) WCA and surface roughness of various nanocoatings. L5, L5.5, L10, L10.5, and L5/C are (PL/PG)5, (PL/PG)5/PL, (PL/PG)10, (PL/PG)10/PL, and (PL/PG)5/capsule coatings, respectively. Loading profiles of (d) IL-12 and (e) BMP-2 of L5/CG/L5/CG/L10/CL/L10 FICIF coatings. L indicates the PL/PG bilayer, C represents capsules, and CG and CL are capsulePG and capsulePL, respectively. (f) Cumulative release profiles of IL-12 and BMP-2 from different PL/PG nanocoatings with and without capsules. (g) SEM images of the FICIF nanocoatings on the Kirschner wire (K-wire) after implantation. *p < 0.05 in (a,b,d,e) compared to the shorter incubation times (unmarked data points). *p < 0.05 in (c) compared to L5 and L10 films. ˆp< 0.05 in (c) compared to L5 and L5.5 films.
After structural tailoring of the FICIF nanocoatings, we further evaluated their delivery capability for two heat/solvent-sensitive protein drugs of IL-12 and BMP-2 (Figure 2d,e). The previous antibiotic delivery systems, such as poly(methyl methacrylate)-based bone cements, usually lack the delivery capability for these drugs because of the inevitable handling of drugs with heat or denaturing solvents.40–42 In contrast, our FICIF nanocarriers can deliver IL-12 and BMP-2 molecules with high loading capability and sustained release, yet without sacrificing their bioactivity. An increase in incubation time in the drug solutions led to rapid loading of both IL-12 and BMP-2 (Figure 2d,e). For instance, using L5/CG/L5/CG/L10/CL/L10 FICIF coatings, the loading of IL-12 achieved 192 pg cm−2 within 10 min (Figure 2d), and the BMP-2 reached 315 ng cm−2 within 15 min (Figure 2e). Such a rapid and high-efficiency loading manner for multiple drugs with very different properties confirmed the remarkable clinical application potential (e.g., desired drugs loaded just minutes before a surgical implantation). By mimicking the temperature/humidity stimuli of flower blooming, the drug delivery of the FICIF nanocoatings was manipulated by external pH. The drug loading was because of the large pH gap above/below the drug isoelectric point based on the electrostatic attraction caused by net charges, while the release was triggered by the disappearance of “binding sites” under a pH similar to their isoelectric point.19 Because of the shield effect of the multilayer nanofilms covered on capsules, the conventional “burst release” of the drug delivery systems was significantly reduced. More outside PL/PG layers resulted in a slower drug release. This result was confirmed obviously by comparing the release profiles of the L5/CG/L5/CG/L10/CL/L5 and L5/CG/L5/CG/L10/CL/L10 FICIF nanocoatings, as demonstrated in Figure 2f. The control L33 nanofilms and FICIF coatings with like charges as drugs (such as L5/CG/L5/CG/L10/CG/L10 coatings for IL-12 or L5/CL/L5/CL/L10/CL/L10 coatings for BMP-2) indicated an obvious burst release within 1 day along with quite low release amounts (<20 pg cm−2 IL-12 and <35 ng cm−2 BMP-2) (Figure 2f). In contrast, the L5/CG/L5/CG/L10/CL/L10 FICIF nanocoatings achieved programmed loadings of 178 pg cm−2 IL-12 within 10 days and 286 ng cm−2 BMP-2 within 42 days, indicating more than 8 times higher release amount and duration, satisfying the pathological requirements (maximally suppressed anti-infection of 10 days and physiological bone healing of 6 weeks). In addition, the SEM observation (Figure 2g) of FICIF nanocoatings after implantation indicated that effective mechanical protection to the embedded capsules was provided by the multilayer nanofilms, achieving >90% of the coatings that remained after in vivo rat femur implantation. The observed high stability was consistent with other polypeptide nanocoatings, which have also shown high stability both in vitro and in vivo.18,19,37
In Vitro Anti-infection and Bone Regeneration.
Besides outstanding loading/release performance, the capability of our FICIF nanocarriers was further confirmed by their remarkable bone-regeneration properties after BMP-2 loading (Figure 3a–c). The PL/PG polypeptide FICIF coatings offered a pH-triggered, biocompatible, and fully degradable vehicle for drug BMP-2 delivery to achieve local therapeutic delivery at the fractures and to overcome the side effects from drug burst-release and use of nonbiodegradable carriers. As shown in Figure 3a, in contrast to the control (blank quartz slides), more OB cells were found to attach after 2 h culturing on the FICIF nanocoatings (unloaded L5/CG/L10/CL/L10 or F-I and L5/CG/L5/CG/L10/CL/L10 or F-II), and BMP-2 loading (loaded F-I and F-II) resulted in further significantly enhanced cell adhesion at 4, 8, and 12 h culturing. At 8 h, the cell density on the BMP-2-loaded F-II FICIF coatings was 1.2 × 104 cell cm−2, confirming the improved cell adhesion because of the unique hierarchical coating structure (Figure 2c) and the therapeutic effect of BMP-2 (Figure 2g). As expected, their cell proliferation within 1–5 days was also significantly improved (Figure 3b). After 5 day culture, the BMP-2-loaded F-II FICIF coatings achieved a high cell density (i.e., 3.5 × 104 cell cm−2), which was more than 5 times higher than the control group (0.68 × 104 cell cm−2). Strikingly, the controlled and sustained BMP-2 release led to a significantly higher cell viability (>90% on both loaded F-I and F-II nanocoatings), reducing conventional drug toxicity because of high-concentration burst release, compared to those cultured on the control quartz slides (<75%), as shown in Figure 3c. Compared to OB cells, the human mesenchymal stem cell (hMSC) showed similar attachment and proliferation results on different culture substrates (Figure 3d), further demonstrating the benefit of the BMP-2 delivery using the FICIF nanocoatings. By examining the gene expression of secreted phosphoprotein 1 (SPP1) and Runt-related transcription factor 2 (RUNX2), we also cultured hMSCs on different surfaces and assessed their osteogenic potential; the obtained result is shown in Figure 3e. Benefiting from the BMP-2 delivery, significantly improved expression of SPP1 and RUNX2 levels was achieved because of the 5 times improved SPP1 expression and 3 times RUNX2 expression on BMP-2-loaded F-II culture surfaces in contrast to the control samples.
Figure 3.
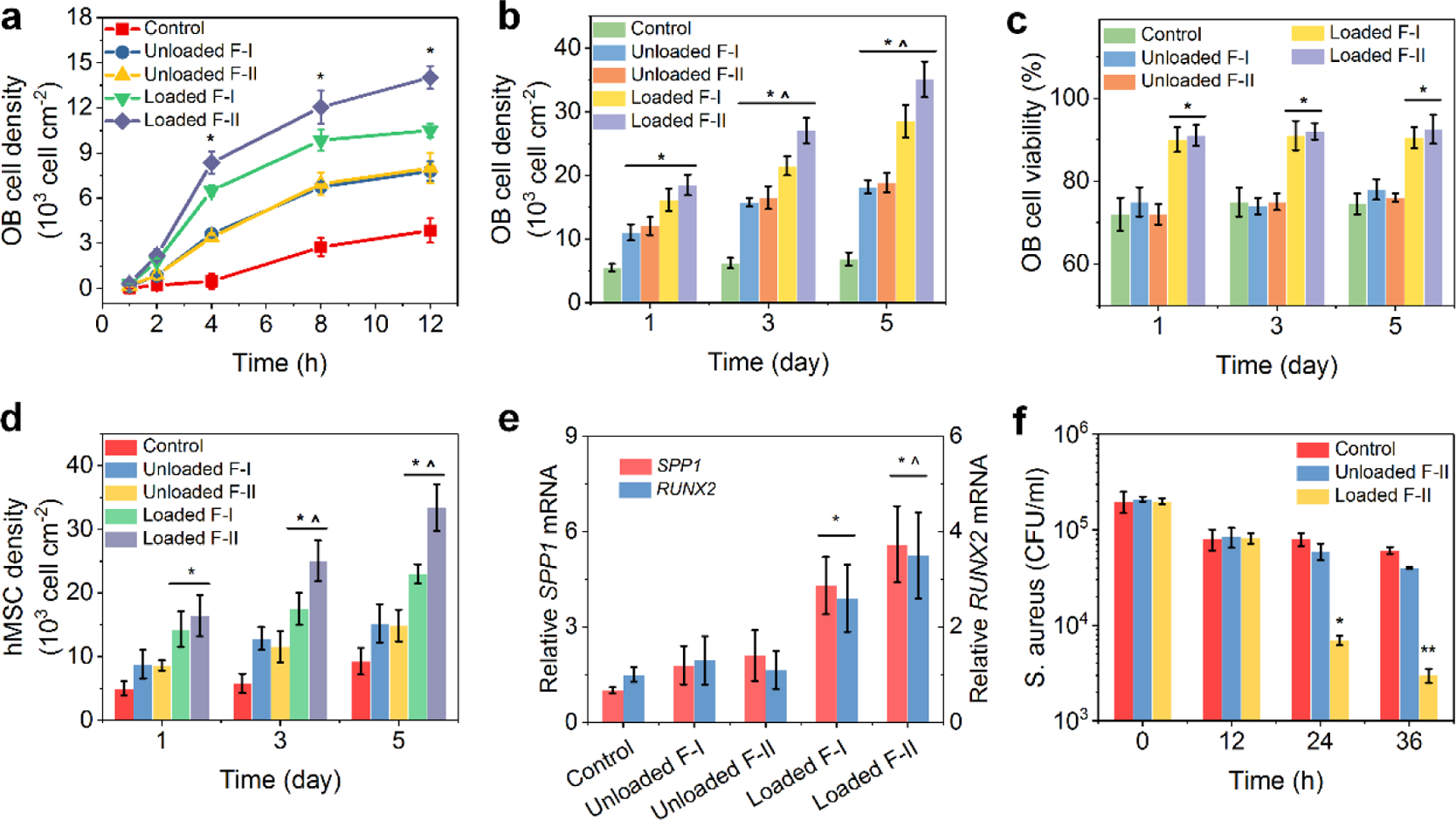
(a) Cell density of OB cells within 12 h cultured on the control (quartz slides), L5/CG/L10/CL/L10 (F-I) and L5/CG/L5/CG/L10/CL/L10 (F-II) nanocoatings loaded without and with BMP-2. (b) OB proliferation, (c) OB viability, and (d) hMSC proliferation on various culture substrates at 1, 3, and 5 days. (e) Osteogenic potential of hMSCs cultured on various culture substrates. Glyceraldehyde-3-phosphate-dehydrogenase was used as the endogenous control. (f) S. aureus survived (cultured with macrophages) at different times on the control (quartz slides), L5/CG/L5/CG/L10/CL/L10 (F-II) loaded without and with IL-12. *p < 0.05 in (a) compared to the control and unloaded F-I and F-II. *p < 0.05 in (b) compared to the control. ˆp < 0.05 in (b) compared to the corresponding samples cultured at 1 day. *p < 0.05 in (c–e) compared to the control and unloaded F-I and F-II. ˆp < 0.05 in (d) compared to the corresponding samples cultured at 1 day. ˆp < 0.05 in (e) compared to the loaded F-I. *p < 0.05 in (f) compared to the control and unloaded F-II.
Antibiotic resistance has increasingly become one of the leading issues in modern medicine and poses a huge burden to the public health.43–46 For instance, in the U.S. alone, infections from antibiotic resistant bacteria increased 359% between 1997 and 2006,47 causing more than 2 million infections, 23,000 deaths,48 as well as $55–70 billion economic cost each year in recent years.49,50 Unfortunately, open fractures, especially those commonly seen among military injuries, have shown substantial contamination with antibiotic resistant bacteria;51–53 for instance, alarmingly high percentages (e.g., 32.2%) of open tibia fracture associated infections from combat injuries have been reported to be associated with antibiotic resistant bacteria.11 Here, besides the improved bone regeneration, we innovatively applied IL-12 to tune the host immune responses against bacteria, which was distinctly different from antibiotic treatments.54 Instead of direct administration of IL-12, we delivered IL-12 using FICIF nanocarriers to reduce infection in a programmed and on-demand strategy, realizing both sustained long-term effects and minimal side effects. To assess the anti-infection capability of the IL-12-loaded FICIF nanocoatings, we performed in vitro co-culture of immune cells (like macrophages) and clinical S. aureus. The results (Figure 3f) indicated that the release of exogenous IL-12 enabled significantly improved capability in killing S. aureus at 24 and 36 h post-treatment. After 36 h, the surviving S. aureus density on IL-12-loaded F-II FICIF nanocoatings was 3 × 103 CFU mL−1, less than 5% of the control group (6.1 × 104 CFU mL−1). We attributed the enhancement of macrophages’ bacterial killing to the improved secretion level of interferon (IFN-γ) from macrophages because of IL-12 stimuli (Figure 1d).
In Vivo Anti-infection and Bone Healing.
Infection and delayed wound healing are the two commonly seen clinical challenges in treating open fractures and other traumatic injuries and have placed a significant burden on public health. Currently, many studies on antibiotic delivery have been conducted; however, the in vivo study, in particular, co-delivery of IL-12 and BMP-2 has never been reported. Here, we evaluated the in vivo bone regeneration and anti-infection using BMP-2-and IL-12-loaded FICIF implant nanocoatings in an open fracture rat model (Supporting Information Methods). X-ray and micro-CT analyses showed that the BMP-2-loaded coating groups had a significantly larger callus width and area compared to those of the control group at postoperative weeks 2 and 4 (Figure 4a,b and Supporting Information Figure. 2), for example, 25 mm2 area and 17 mm width of the F-II group compared to 18 mm2 area and 13.5 mm width of the controls. Micro-CT results (Figure 4b) revealed that, in contrast to the control group, significantly greater total bone tissue volume (TV) and high-density bone volume (BV), but lower BV/TV for the F-II groups were observed at week 4 after implantation. At week 6, the TV of the F-I and F-II groups became lower (such as, from 235 at week 4 to 195 mm2 at week 6 for F-II group). The BV of BMP-2-loaded FICIF groups remained higher than the control group, while their BV/TV were similar at week 6 after implantation. The bone mineral density of TV of the F-II groups continuously increased from 450 to 700 HA cm−3, which was lower at week 2 but higher at week 6 compared to that of the control groups. These results confirmed that, during fracture healing, in contrast to the control group, because of BMP-2 release, more calluses were formed in the early stage (before week 3), and the mineralization was enhanced in the later stage (after week 4) for the BMP-2-loaded F-I and F-II groups (Figure 4c). The biomechanical measure, using a three-point bending test, further confirmed the better bone regeneration of BMP-2-loaded FICIF groups (Figure 4d and Supporting Information Figure 3). At week 6, the F-II group showed a maximum compressive load of 275 N, a bending stiffness of 540 N mm−1, and a bending energy of 270 N mm, suggesting at least 4 times improvement compared to those of the control group.
Figure 4.
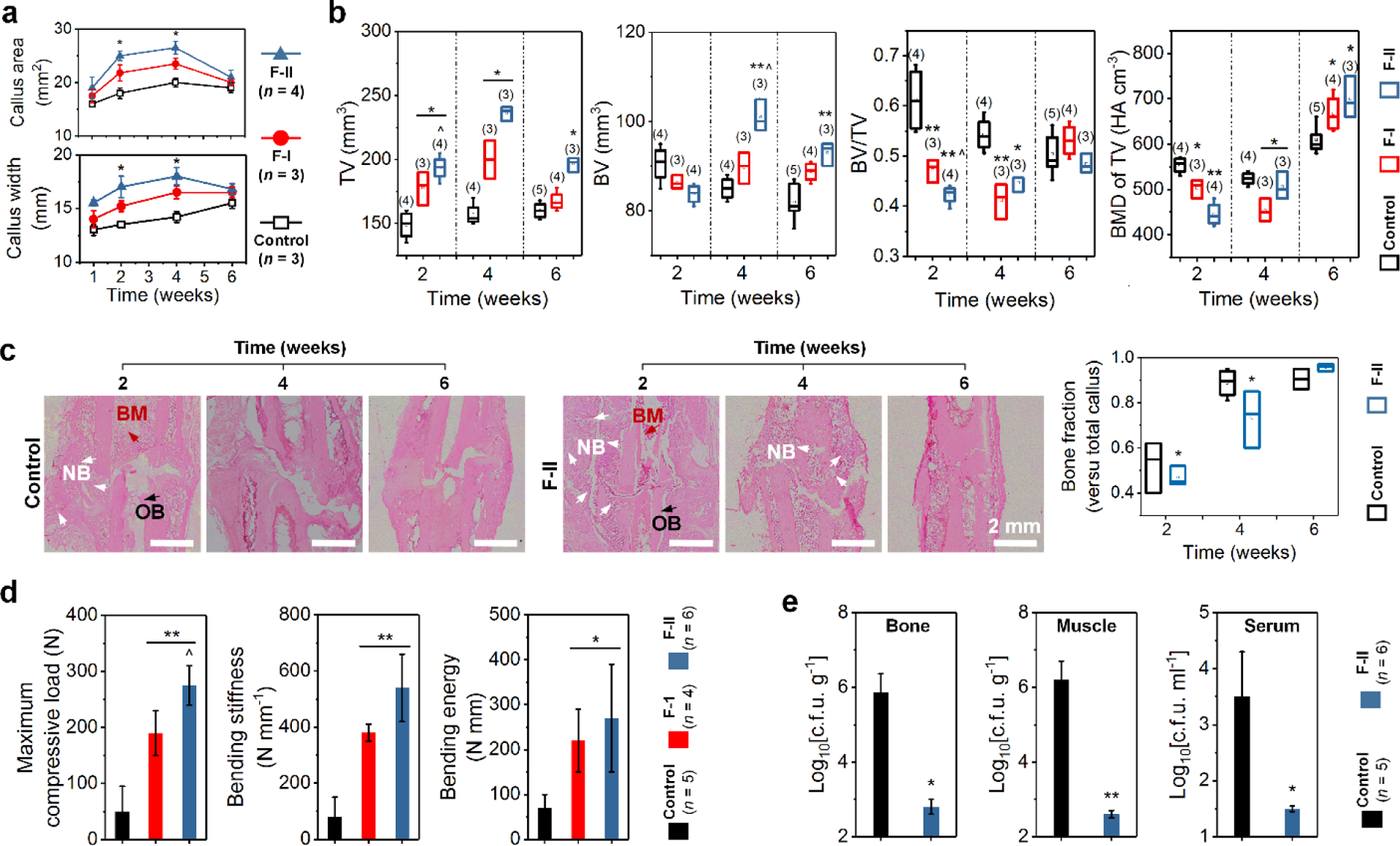
(a) Quantification of the area (top) and width (bottom) of the fracture callus (1–6 weeks) in a rat fracture model after implantation using the control (nanofilms), and drug-loaded F-I and F-II nanofilm-coated K-wires. (b) Micro-CT measurements of total bone TV, high-density BV, BV/TV, and TV density of fractured rat femur 2–6 weeks after implantation with the control and drug-loaded F-I and F-II nanocoatings. (c) Representative images of H&E staining (left) in mid-sagittal section of the fracture callus 2–6 weeks after implantation (control and F-II groups) with quantitation (right) of bone fractions. (d) Biomechanical test of maximum compressive load, bending stiffness, and bending energy of the fractured rat femurs, and (e) infection of the rats 6 weeks after implantation with the control and drug-loaded F-I and F-II nanocoatings. The bones studied in (d) were from fracture callus and the muscles in (e) were from surrounding the fracture. *p < 0.05, **p < 0.01 compared to the control. ˆp < 0.05 compared to the F-I group. n is shown in parentheses for each group.
The rat fracture was left open for 1 h, mimicking a trauma patient’s so-called golden hour, and infection was established by inoculating 100 μL of bacterial inoculum of 102 CFU/0.1 mL S. aureus clinical isolate at the fracture site (Supporting Information Figure 4). After 6 weeks, bone, muscle, and serum samples were harvested for infection evaluation (Supporting Information Methods), the obtained results are shown in Figure 4e and Supporting Information Figure 5. Significantly, the FICIF nanocoatings loaded with IL-12 resulted in a substantial reduction in infection levels; the F-II groups had significantly reduced S. aureus from the bone (2.8 log10 CFU g−1), muscle (2.6 log10 CFU g−1), and serum (1.5 log10 CFU mL−1). These data further demonstrated that the FICIF implant nanocoatings successfully delivered antimicrobial IL-12 to reduce bacteria. Our findings show that such implant coatings with the capability of multiple drug delivery can serve as a biocidal layer, indicating intriguing potential applications in bioprotective materials against increasing threats of emerging infectious diseases.
CONCLUSIONS
In summary, we have demonstrated an innovative route, based on LbL self-assembly, to develop unique FICIF nanocarriers to co-deliver multiple protein drugs for reducing infections and stimulating bone regeneration and osseointegration. Tailoring of the capsule impregnation, capsule deposition, and shielded nanofilm structures allows polypeptides to assemble capsule-embedded multilayer structures with specific drug binding sites and packing structures. Postpreparation loading and programmed release were realized for multiple protein drugs using the developed unique nanocarriers, achieving high and tunable loadings of IL-12 of 178 pg cm−2 and BMP-2 of 286 ng cm−2 and sustained and on-demand release profiles (10 days for IL-12 and 6 weeks for BMP-2). This successful delivery of unique immune-tuning antimicrobial IL-12 and clinically approved growth factor of BMP-2 enabled 5-times enhanced healing bone strength and 3 orders of magnitude higher S. aureus killing efficacy. Such bio-inspired encapsulation and delivery materials are expected to offer exciting opportunities for a variety of applications in medicine.
METHODS
Materials.
Poly-l-lysine (PL) hydrobromide (Mw 150–300 kDa), poly-l-glutamic acid (PG) sodium salt (Mw 50–100 kDa), sodium chloride, glutaraldehyde, BMP-2, and BMP-2 enzyme-linked immunosorbent assay (ELISA) kit were purchased from Sigma-Aldrich (St. Louis, MO). Disodium ethylenediaminetetraacetic acid (EDTA), IL-12, and IL-12 ELISA kit were bought from Thermo Fisher Scientific (Fair Lawn, NJ). Calcium carbonate (CaCO3) particles were obtained from PlasmaChem GmbH (Rudower Chaussee, Berlin, Germany). Stainless steel Kirschner wires (K-wires) were supplied by Smith & Nephew, Inc. (Memphis, TN). Quartz microscope slides were purchased from Electron Microscopy Sciences (Hatfield, PA).
LbL Self-Assembly of PL/PG Nanofilms and Capsules.
The preparation of PL/PG multilayer nanofilms was carried out using a dipping robot (Riegler and Kirstein GmbH, Berlin, Germany). To construct multilayer films, the substrates (e.g., K-wire, quartz slide, silica wafer, etc.) were alternately immersed in PL and PG solutions (1 mg mL−1), and the rinsing and drying processes were performed between each dipping. To prepare polypeptide capsules, CaCO3 particles were immersed in 3 mg mL−1 of PL or PG solutions (3 Torr vacuum pressure, 30 min) to impregnate polyelectrolytes for drug loading, then PL and PG were alternately deposited onto these particles to form multilayer shells. Subsequently, the shelled particles were cross-linked (incubated in 25% glutaraldehyde for 5 h) and CaCO3 removed (incubated in 0.1 M EDTA for 1 h, and then rinsed with phosphate buffered saline) to obtain PL/PG capsules.
Polypeptide multilayer films integrated with capsules were prepared based on the LbL self-assembly of multilayer nanofilms and microcapsules; the typical procedure includes: (i) LbL self-assembly of (PL/PG)a nanofilms, (ii) deposition of CaCO3 particles (density of 5 × 106 particles mL−1) impregnated with PG, (iii) repetition of the processes (i,ii,iv) LbL self-assembly of (PL/PG)b nanofilms, (v) deposition of CaCO3 particles (density of 5 × 106 particles mL−1) impregnated with PL, (vi) LbL self-assembly of (PL/PG)c nanofilms, and (vii) removal of CaCO3 templates. The obtained capsule integrated nanofilms were ((PL/PG)a/capsulePG)x/(PL/PG)b/capsulePL/(PL/PG)c, simplified as (La/CG)x/Lb/CL/Lc, in which L indicates a PL/PG bilayer, CL and CG represent PL and PG impregnated capsules, a, b, and c are the number of PL/PG bilayers, and x is the repetition cycles of La/CG structures.
Characterization.
The morphology of CaCO3 particles, microcapsules, and nanocoatings was characterized using SEM (S-4700, Hitachi Ltd. Japan), and their surface roughness was studied by NanoScope atomic force microscopy (PicoSPM II, Tempe, AZ). The thickness of nanocoatings was examined using ellipsometry (M-2000, JA Woollam Co., Lincoln, NE), and WCA (3 μL) was measured using a SL200B contact angle goniometer (Kino Industry Co., Ltd., MA). The observation of impregnation of fluorescence-labeled polypeptides was performed using confocal laser scanning microscopy (CLSM, LSM 510, Zeiss, Thornwood, NY), and their quantitative loading was measured using a hybrid multimode microplate reader (Synergy H4, Winooski, VT). The loading and release profiles of BMP-2 and IL-12 were carried out using ELISA kits, as described in Supporting Information Methods.
In Vitro and In Vivo Study.
The human osteoblast (OB) cell line CRL-11372 (American Type Cell Culture, Manassas, VA, cell density of 1 × 104 cell cm−2) was cultured in 24-well culture plates, in which the quartz slides with and without coatings were placed, for cell adhesion and proliferation studies. Clinical S. aureus (Ruby Memorial Hospital, Morgantown, WV, log-phase growth) was cultured with macrophages to evaluate the anti-infection performance of the IL-12-loaded FICIF nanocoatings. The cell adhesion, proliferation, and viability were measured after detaching cells from the substrates and counted using a hemocytometer, while the S. aureus killing data were obtained by diluting medium samples in 24-well culture plates and then agar plate counting. In vivo studies using Sprague-Dawley rats were approved by the Institutional Animal Care and Use Committee, and the detailed procedures are presented in Supporting Information Methods. The data presented are mean ± standard deviation. Statistical significance was determined using Student’s t-test, one-way ANOVA followed by Tukey’s honestly significant difference test. A p value < 0.05 was considered statistically significant.
Supplementary Material
ACKNOWLEDGMENTS
This work is supported by the Office of the Assistant Secretary of Defense for Health Affairs, through the Peer Reviewed Medical Research Program, Discovery Awards under award nos. W81XWH1710603 and W81XWH1810203. We also acknowledge the financial support from AO Foundation, Osteosynthesis & Trauma Care Foundation, the West Virginia National Aeronautics and Space Administration Experimental Program to Stimulate Competitive Research (WV NASA EPSCoR), WVU PSCoR, and WVCTSI. In addition, we acknowledge the use of the WVU Shared Research Facilities that are supported by NIH grants 2U54GM104942-02, 5P20RR016477, U57GM104942, P30GM103488, P20GM109098, and P20GM103434. Opinions, interpretations, conclusions, and recommendations are those of the authors and are not necessarily endorsed by the funding agencies. We acknowledge Suzanne Danley for proofreading.
Footnotes
The authors declare no competing financial interest.
Complete contact information is available at: https://pubs.acs.org/10.1021/acsami.0c20332
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsami.0c20332.
Supplementary methods, growth profile of multilayer nanofilms, biomechanical testing of healing rat femurs, procedures in creating open femur fracture and subsequent fixation, images of infected rat fracture and agar plate counting of S. aureus from the bone tissue (PDF)
Contributor Information
Shichao Zhang, Department of Orthopaedics, School of Medicine, West Virginia University, Morgantown, West Virginia 26506, United States.
Justin Vaida, Department of Orthopaedics, School of Medicine, West Virginia University, Morgantown, West Virginia 26506, United States.
Josh Parenti, Department of Orthopaedics, School of Medicine, West Virginia University, Morgantown, West Virginia 26506, United States.
Brock A. Lindsey, Department of Orthopaedics, School of Medicine, West Virginia University, Morgantown, West Virginia 26506, United States
Malcolm Xing, Department of Mechanical Engineering, University of Manitoba, and the Children’s Hospital Research Institute of Manitoba, Winnipeg, Manitoba R3E 3P4, Canada.
Bingyun Li, Department of Orthopaedics, School of Medicine, West Virginia University, Morgantown, West Virginia 26506, United States.
REFERENCES
- (1).Lewandowski LR; Potter BK; Murray CK; Petfield J; Stinner DJ; Krauss M; Weintrob AC; Tribble DR Osteomyelitis Risk Factors Related to Combat Trauma Open Femur Fractures: A Case-Control Analysis. J. Orthop. Traumatol. 2019, 33, e110–e119. [DOI] [PubMed] [Google Scholar]
- (2).Andersen RC; D’Alleyrand J-CG; Swiontkowski MF; Ficke JR Extremity War Injuries VIII: Sequelae of Combat Injuries. J. Am. Acad. Orthop. Surg. 2014, 22, 57–62. [DOI] [PubMed] [Google Scholar]
- (3).Belmont PJ Jr.; McCriskin BJ; Sieg RN; Burks R; Schoenfeld AJ Combat wounds in Iraq and Afghanistan from 2005 to 2009. J. Trauma Acute Care Surg. 2012, 73, 3–12. [DOI] [PubMed] [Google Scholar]
- (4).Owens BD; Kragh JF Jr.; Wenke JC; Macaitis J; Wade CE; Holcomb JB Combat Wounds in Operation Iraqi Freedom and Operation Enduring Freedom. J. Trauma 2008, 64, 295–299. [DOI] [PubMed] [Google Scholar]
- (5).Owens BD; Wenke JC; Svoboda SJ; White DW Extremity Trauma Research in the United States Army. J. Am. Acad. Orthop. Surg. 2006, 14, S37. [DOI] [PubMed] [Google Scholar]
- (6).Mathieu L; Bilichtin E; Durand M; de l’Escalopier N; Murison JC; Collombet J-M; Rigal S Masquelet Technique for Open Tibia Fractures in a Military Setting. Eur. J. Trauma Emerg. Surg. 2020, 46, 1099–1105. [DOI] [PubMed] [Google Scholar]
- (7).Brauner A; Fridman O; Gefen O; Balaban NQ Distinguishing between Resistance, Tolerance and Persistence to Antibiotic Treatment. Nat. Rev. Microbiol. 2016, 14, 320–330. [DOI] [PubMed] [Google Scholar]
- (8).Garner MR; Sethuraman SA; Schade MA; Boateng H Antibiotic Prophylaxis in Open Fractures. J. Am. Acad. Orthop. Surg. 2020, 28, 309–315. [DOI] [PubMed] [Google Scholar]
- (9).Arciola CR; Campoccia D; Montanaro L Implant Infections: Adhesion, Biofilm Formation and Immune evasion. Nat. Rev. Microbiol. 2018, 16, 397–409. [DOI] [PubMed] [Google Scholar]
- (10).Forbes SJ; Rosenthal N Preparing the Ground for Tissue Regeneration: from Mechanism to Therapy. Nat. Med. 2014, 20, 857–869. [DOI] [PubMed] [Google Scholar]
- (11).Burns TC; Stinner DJ; Mack AW; Potter BK; Beer R; Eckel TT; Possley DR; Beltran MJ; Hayda RA; Andersen RC; Keeling JJ; Frisch HM; Murray CK; Wenke JC; Ficke JR; Hsu JR Microbiology and Injury Characteristics in Severe Open Tibia Fractures from Combat. J. Trauma Acute Care Surg. 2012, 72, 1062–1067. [DOI] [PubMed] [Google Scholar]
- (12).Masini BD; Waterman SM; Wenke JC; Owens BD; Hsu JR; Ficke JR Resource Utilization and Disability Outcome Assessment of Combat Casualties from Operation Iraqi Freedom and Operation Enduring Freedom. J. Orthop. Traumatol. 2009, 23, 261–266. [DOI] [PubMed] [Google Scholar]
- (13).Murray CK; Hinkle MK; Yun HC History of Infections associated with Combat-related Injuries. J. Trauma 2008, 64, S221. [DOI] [PubMed] [Google Scholar]
- (14).Lee J; Byun H; Madhurakkat Perikamana SK; Lee S; Shin H Current Advances in Immunomodulatory Biomaterials for Bone Regeneration. Adv. Healthcare Mater. 2019, 8, 1801106. [DOI] [PubMed] [Google Scholar]
- (15).Chen R; Wang J; Liu C Biomaterials Act as Enhancers of Growth Factors in Bone Regeneration. Adv. Funct. Mater. 2016, 26, 8810–8823. [Google Scholar]
- (16).Jin X; Xiong YH; Zhang XY; Wang R; Xing Y; Duan S; Chen D; Tian W; Xu FJ Self-Adaptive Antibacterial Porous Implants with Sustainable Responses for Infected Bone Defect Therapy. Adv. Funct. Mater. 2019, 29, 1807915. [Google Scholar]
- (17).Lee JS; Murphy WL Functionalizing Calcium Phosphate Biomaterials with Antibacterial Silver Particles. Adv. Mater. 2013, 25, 1173–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Zhang S; Zhou S; Liu H; Xing M; Ding B; Li B Pinecone-Inspired Nanoarchitectured Smart Microcages Enable Nano/Microparticle Drug Delivery. Adv. Funct. Mater. 2020, 30, 2002434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Zhang S; Xing M; Li B Capsule-Integrated Polypeptide Multilayer Films for Effective pH-Responsive Multiple Drug Co-Delivery. ACS Appl. Mater. Interfaces 2018, 10, 44267–44278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Richardson JJ; Bjornmalm M; Caruso F Multilayer Assembly. Technology-driven Layer-by-layer Assembly of Nanofilms. Science 2015, 348, aaa2491. [DOI] [PubMed] [Google Scholar]
- (21).Alkekhia D; Hammond PT; Shukla A Layer-by-layer Biomaterials for Drug Delivery. Annu. Rev. Biomed. Eng. 2020, 22, 1–24. [DOI] [PubMed] [Google Scholar]
- (22).Tang Z; Wang Y; Podsiadlo P; Kotov NA Biomedical Applications of Layer-by-Layer Assembly: From Biomimetics to Tissue Engineering. Adv. Mater. 2006, 18, 3203–3224. [Google Scholar]
- (23).Zhang S; Xing M; Li B Biomimetic Layer-by-layer Self-assembly of Nanofilms, Nanocoatings, and 3D Scaffolds for Tissue Engineering. Int. J. Mol. Sci. 2018, 19, 1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Yuan W; Weng G-M; Lipton J; Li CM; Van Tassel PR; Taylor AD Weak Polyelectrolyte-based Multilayers via Layer-by-layer Assembly: Approaches, Properties, and Applications. Adv. Colloid Interface Sci. 2020, 282, 102200. [DOI] [PubMed] [Google Scholar]
- (25).De Cock LJ; De Koker S; De Geest BG; Grooten J; Vervaet C; Remon JP; Sukhorukov GB; Antipina MN Polymeric Multilayer Capsules in Drug Delivery. Angew. Chem., Int. Ed. 2010, 49, 6954–6973. [DOI] [PubMed] [Google Scholar]
- (26).Boudou T; Crouzier T; Ren K; Blin G; Picart C Multiple Functionalities of Polyelectrolyte Multilayer Films: New Biomedical Applications. Adv. Mater. 2010, 22, 441–467. [DOI] [PubMed] [Google Scholar]
- (27).Shi D; Ran M; Zhang L; Huang H; Li X; Chen M; Akashi M Fabrication of Biobased Polyelectrolyte Capsules and Their Application for Glucose-Triggered Insulin Delivery. ACS Appl. Mater. Interfaces 2016, 8, 13688–13697. [DOI] [PubMed] [Google Scholar]
- (28).Hsu BB; Hagerman SR; Hammond PT Rapid and Efficient Sprayed Multilayer Films for Controlled Drug Delivery. J. Appl. Polym. Sci. 2016, 133, 43563. [Google Scholar]
- (29).Liu XQ; Picart C Layer-by-Layer Assemblies for Cancer Treatment and Diagnosis. Adv. Mater. 2016, 28, 1295–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Vignali DAA; Kuchroo VK IL-12 Family Cytokines: Immunological Playmakers. Nat. Immunol. 2012, 13, 722–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Scott P IL-12: Initiation Cytokine for Cell-mediated Immunity. Science 1993, 260, 496–497. [DOI] [PubMed] [Google Scholar]
- (32).Hsieh C; Macatonia S; Tripp C; Wolf S; O’Garra A; Murphy K Development of TH1 CD4+ T Cells through IL-12 Produced by Listeria-induced Macrophages. Science 1993, 260, 547–549. [DOI] [PubMed] [Google Scholar]
- (33).Sebald H-J; Klenke FM; Siegrist M; Albers CE; Sebald W; Hofstetter W Inhibition of Endogenous Antagonists with an Engineered BMP-2 Variant Increases BMP-2 Efficacy in Rat Femoral Defect Healing. Acta Biomater. 2012, 8, 3816–3820. [DOI] [PubMed] [Google Scholar]
- (34).Hunziker EB; Enggist L; Küffer A.; Buser D.; Liu Y. Osseointegration: the Slow Delivery of BMP-2 Enhances Osteoinductivity. Bone 2012, 51, 98–106. [DOI] [PubMed] [Google Scholar]
- (35).Wang C; He C; Tong Z; Liu X; Ren B; Zeng F Combination of Adsorption by Porous CaCO3 Microparticles and Encapsulation by Polyelectrolyte Multilayer Films for Sustained Drug Delivery. Int. J. Pharm. 2006, 308, 160–167. [DOI] [PubMed] [Google Scholar]
- (36).Teng MWL; Bowman EP; McElwee JJ; Smyth MJ; Casanova J-L; Cooper AM; Cua DJ IL-12 and IL-23 Cytokines: from Discovery to Targeted Therapies for Immune-mediated Inflammatory Diseases. Nat. Med. 2015, 21, 719–729. [DOI] [PubMed] [Google Scholar]
- (37).Li B; Jiang B; Boyce BM; Lindsey BA Multilayer Polypeptide Nanoscale Coatings Incorporating IL-12 for the Prevention of Biomedical Device-associated Infections. Biomaterials 2009, 30, 2552–2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Zankovych S; Bossert J; Faucon M; Finger U; Jandt KD Selectively Promoting or Preventing Osteoblast Growth on Titanium Functionalized with Polyelectrolyte Multilayers. Adv. Eng. Mater. 2011, 13, B454. [Google Scholar]
- (39).Papenburg BJ; Rodrigues ED; Wessling M; Stamatialis D Insights into the Role of Material Surface Topography and Wettability on Cell-material Interactions. Soft Matter 2010, 6, 4377–4388. [Google Scholar]
- (40).Soleymani Eil Bakhtiari S; Bakhsheshi-Rad HR; Karbasi S; Tavakoli M; Razzaghi M; Ismail AF; RamaKrishna S; Berto F Polymethyl Methacrylate-based Bone Cements Containing Carbon Nanotubes and Graphene Oxide: An Overview of Physical, Mechanical, and Biological Properties. Polymers 2020, 12, 1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Shen S-C; Ng WK; Shi Z; Chia L; Neoh KG; Tan RBH Mesoporous Silica Nanoparticle-functionalized Poly(methyl methacrylate)-based Bone Cement for Effective Antibiotics Delivery. J. Mater. Sci.: Mater. Med. 2011, 22, 2283–2292. [DOI] [PubMed] [Google Scholar]
- (42).Wei W; Abdullayev E; Hollister A; Mills D; Lvov YM Clay Nanotube/poly (methyl methacrylate) Bone Cement Composites with Sustained Antibiotic Release. Macromol. Mater. Eng. 2012, 297, 645–653. [Google Scholar]
- (43).Palmer AC; Kishony R Understanding, Predicting and Manipulating the Genotypic Evolution of Antibiotic Resistance. Nat. Rev. Genet. 2013, 14, 243–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Andersson DI; Hughes D Antibiotic Resistance and its Cost: Is it Possible to Reverse Resistance? Nat. Rev. Microbiol. 2010, 8, 260–271. [DOI] [PubMed] [Google Scholar]
- (45).MacFadden DR; McGough SF; Fisman D; Santillana M; Brownstein JS Antibiotic Resistance Increases with Local Temperature. Nat. Clim. Change 2018, 8, 510–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Hernando-Amado S; Coque TM; Baquero F; Martínez JL Defining and Combating Antibiotic Resistance from One Health and Global Health Perspectives. Nat. Microbiol. 2019, 4, 1432–1442. [DOI] [PubMed] [Google Scholar]
- (47).Mainous AG III; Diaz VA; Matheson EM; Gregorie SH; Hueston WJ. Trends in Hospitalizations with Antibiotic-resistant Infections: U.S., 1997–2006. Publ. Health Rep. 2011, 126, 354–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Kaur S; Rana D; Matsuura T; Sundarrajan S; Ramakrishna S Preparation and Characterization of Surface Modified Electrospun Membranes for Higher Filtration Flux. J. Membr. Sci. 2012, 390–391, 235–242. [Google Scholar]
- (49).Jang W; Yun J; Jeon K; Byun H PVdF/graphene Oxide Hybrid Membranes via Electrospinning for Water Treatment Applications. RSC Adv. 2015, 5, 46711–46717. [Google Scholar]
- (50).Roberts RR; Hota B; Ahmad I; Scott II RD II; Foster SD; Abbasi F; Schabowski S; Kampe LM; Ciavarella GG; Supino M; Naples J; Cordell R; Levy SB; Weinstein RA Hospital and Societal Costs of Antimicrobial-resistant Infections in a Chicago Teaching Hospital: Implications for Antibiotic Stewardship. Clin. Infect. Dis. 2009, 49, 1175–1184. [DOI] [PubMed] [Google Scholar]
- (51).Murray CK; Yun HC; Griffith ME; Thompson B; Crouch HK; Monson LS; Aldous WK; Mende K; Hospenthal DR Recovery of Multidrug-resistant Bacteria from Combat Personnel Evacuated from Iraq and Afghanistan at a Single Military Treatment Facility. Mil. Med. 2009, 174, 598–604. [DOI] [PubMed] [Google Scholar]
- (52).Calhoun JH; Murray CK; Manring MM Multidrug-resistant Organisms in Military Wounds from Iraq and Afghanistan. Clin. Orthop. Relat. Res. 2008, 466, 1356–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Murray CK; Hsu JR; Solomkin JS; Keeling JJ; Andersen RC; Ficke JR; Calhoun JH Prevention and Management of Infections associated with Combat-related Extremity Injuries. J. Trauma 2008, 64, S239. [DOI] [PubMed] [Google Scholar]
- (54).Hamza T; Barnett JB; Li B Interleukin 12 a Key Immunoregulatory Cytokine in Infection Applications. Int. J. Mol. Sci. 2010, 11, 789–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.