

Abstract
Background
Fingolimod was approved in 2010 for the treatment of patients with the relapsing‐remitting (RR) form of multiple sclerosis (MS). It was designed to reduce the frequency of exacerbations and to delay disability worsening. Issues on its safety and efficacy, mainly as compared to other disease modifying drugs (DMDs), have been raised.
Objectives
To assess the safety and benefit of fingolimod versus placebo, or other disease‐modifying drugs (DMDs), in reducing disease activity in people with relapsing‐remitting multiple sclerosis (RRMS).
Search methods
We searched the Cochrane Multiple Sclerosis and Rare Diseases of the Central Nervous System (CNS) Group's Specialised Trials Register and US Food and Drug Administration reports (15 February 2016).
Selection criteria
Randomised controlled trials (RCTs) assessing the beneficial and harmful effects of fingolimod versus placebo or other approved DMDs in people with RRMS.
Data collection and analysis
We used standard methodological procedures as expected by Cochrane.
Main results
Six RCTs met our selection criteria. The overall population included 5152 participants; 1621 controls and 3531 treated with fingolimod at different doses; 2061 with 0.5 mg, 1376 with 1.25 mg, and 94 with 5.0 mg daily. Among the controls, 923 participants were treated with placebo and 698 with others DMDs. The treatment duration was six months in three, 12 months in one, and 24 months in two trials. One study was at high risk of bias for blinding, three studies were at high risk of bias for incomplete outcome reporting, and four studies were at high risk of bias for other reasons (co‐authors were affiliated with the pharmaceutical company). We retrieved 10 ongoing trials; four of them have been completed.
Comparing fingolimod administered at the approved dose of 0.5 mg to placebo, we found that the drug at 24 months increased the probability of being relapse‐free (risk ratio (RR) 1.44, 95% confidence interval (CI) (1.28 to 1.63); moderate quality of evidence), but it might lead to little or no difference in preventing disability progression (RR 1.07, 95% CI 1.02 to 1.11; primary clinical endpoints; low quality evidence). Benefit was observed for other measures of inflammatory disease activity including clinical (annualised relapse rate): rate ratio 0.50, 95% CI 0.40 to 0.62; moderate quality evidence; and magnetic resonance imaging (MRI) activity (gadolinium‐enhancing lesions): RR of being free from (MRI) gadolinium‐enhancing lesions: 1.36, 95% CI 1.27 to 1.45; low quality evidence.The mean change of MRI T2‐weighted lesion load favoured fingolimod at 12 and 24 months.
No significant increased risk of discontinuation due to adverse events was observed for fingolimod 0.5 mg compared to placebo at six and 24 months. The risk of fingolimod discontinuation was significantly higher compared to placebo for the dose 1.25 mg at 24 months (RR 1.93, 95% CI 1.48 to 2.52).
No significant increased risk of discontinuation due to serious adverse events was observed for fingolimod 0.5 mg compared to placebo at six and 24 months. A significant increased risk of discontinuation due to serious adverse events was found for fingolimod 5.0 mg (RR 2.77, 95% CI 1.04 to 7.38) compared to placebo at six months.
Comparing fingolimod 0.5 mg to intramuscular interferon beta‐1a, we found moderate quality evidence that the drug at one year slightly increased the number of participants free from relapse (RR 1.18, 95% CI 1.09 to 1.27) or from gadolinium‐enhancing lesions (RR 1.12, 95% CI 1.05 to 1.19), and decreased the relapse rate (rate ratio 0.48, 95% CI 0.34 to 0.70). We did not detect any advantage for preventing disability progression (RR 1.02, 95% CI 0.99 to 1.06; low quality evidence). We did not detect any significant difference for MRI T2‐weighted lesion load change.
We found a greater likelihood of participants discontinuing fingolimod, as compared to other DMDs, due to adverse events in the short‐term (six months) (RR 3.21, 95% CI 1.16 to 8.86), but there was no significant difference versus interferon beta‐1a at 12 months (RR 1.51, 95% CI 0.81 to 2.80; moderate quality evidence). A higher incidence of adverse events was suggestive of the lower tolerability rate of fingolimod compared to interferon‐beta 1a.
Quality of life was improved in participants after switching from a different DMD to fingolimod at six months, but this effect was not found compared to placebo at 24 months.
All studies were sponsored by Novartis Pharma.
Authors' conclusions
Treatment with fingolimod compared to placebo in RRMS patients is effective in reducing inflammatory disease activity, but it may lead to little or no difference in preventing disability worsening. The risk of withdrawals due to adverse events requires careful monitoring of patients over time. The evidence on the risk/benefit profile of fingolimod compared with intramuscular interferon beta‐1a was uncertain, based on a low number of head‐to‐head RCTs with short follow‐up duration. The ongoing trial results will possibly satisfy these issues.
Plain language summary
Fingolimod for relapsing‐remitting multiple sclerosis
Background
Considering the autoimmune pathogenesis of multiple sclerosis (MS), most of the treatments have been based on the immunomodulatory and immunosuppressive properties of drugs such as interferons, glatiramer, azathioprine, cyclophosphamide and mitoxantrone.
Fingolimod, was the first agent to gain approval as an oral treatment in 2010. It is efficiently absorbed, its absorption is unaffected by dietary intake and, as an oral therapy, it has aroused great interest in patients, having a more acceptable route of administration than injections.
Aim of the review
To assess the safety and the benefits of fingolimod in reducing disease activity in people with relapsing‐remitting MS (RRMS). A number of safety concerns have already emerged, including serious infections and adverse cardiac effects.
Study characteristics
Six studies, published between 2006 and 2014, were included in this review, comprising a total of 5152 participants suffering from RRMS. The treatment duration was six months in three studies, 12 months in one study, and 24 months in two studies.
Key results and quality of evidence
The main conclusion of this review was that fingolimod, administered as monotherapy at the approved dose of 0.5 mg once‐daily increases the probability of being relapse‐free at 24 months compared to placebo. The benefit was confirmed with disease activity measures defined by magnetic resonance imaging (MRI) scans. However, there was no effect on preventing disability worsening; treatment was not associated with an increased risk of patient withdrawals due to adverse events.
Comparing the same dose of fingolimod to intramuscular interferon beta‐1a, the drug at one year slightly increased the number of participants free from relapse or from inflammatory enhancing lesions and decreased the relapse rate. Again, we did not detect any advantage for preventing disability progression. We found a greater likelihood of discontinuation due to adverse events in the short‐term (six months) for fingolimod as compared to immunomodulating drugs, and no significant difference compared to interferon beta at 12 months.
The duration of all studies was equal or inferior to 24 months, so that the efficacy (but mostly the safety) of fingolimod over 24 months remains uncertain. This is a key point for a lifetime disease with the probability of chronic treatments as in MS.
The risk of adverse events requires careful monitoring of patients over time and suggests the need for studies with longer follow‐up, particularly considering the recent warning on the development of progressive multifocal leukoencephalopathy.
The six studies included in this review were sponsored by Novartis Pharma, and most co‐authors of the published papers were affiliated to the pharmaceutical company; this is recognised as a potential source of bias.
Summary of findings
Summary of findings for the main comparison. Fingolimod 0.5 mg versus placebo for relapsing‐remitting multiple sclerosis.
| Fingolimod 0.5 mg versus placebo for relapsing‐remitting multiple sclerosis | |||||
| Participants or population: people with relapsing‐remitting multiple sclerosis Settings: outpatients in multiple sclerosis centres Intervention: fingolimod 0.5 mg versus placebo | |||||
| Outcomes at 24 months | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of Participants (studies) | Quality of the evidence (GRADE) | |
| Assumed risk | Corresponding risk | ||||
| Control (placebo) | Fingolimod 0.5 mg | ||||
| Participants free from relapse | 49 per 100 | 70 per 100 (63 to 80) | RR 1.44 (1.28 to 1.63) | 1556 (2 studies) | ⊕⊕⊕⊝ moderatea |
| Participants free from disability worsening | 82 per 100 | 87 per 100 (83 to 91) | RR 1.07 (1.02 to 1.11) | 1556 (2 studies) | ⊕⊕⊝⊝ lowa,b |
| Withdrawals due to adverse events | 9 per 100 | 13 per 100 (8 to 21) | RR 1.42 (0.89 to 2.25) | 1556 (2 studies) | ⊕⊝⊝⊝ very lowa,b,c |
| Annualised relapse rate | ‐ | ‐ | Rate ratio 0.50 (0.40 to 0.62) | 1556 (2 studies) | ⊕⊕⊕⊝ moderatea |
| Participants free from MRI gadolinium‐enhancing lesions | 65 per 100 | 89 per 100 (83 to 94) | RR 1.36 (1.27 to 1.45) | 1226 (2 studies) | ⊕⊕⊝⊝ lowa,b |
| *For dichotomous outcomes, the corresponding risk with fingolimod 0.5 mg (and its 95% CI) is based on the assumed risk with the control group (i.e. the mean proportion of events in the control group across the two studies) and the relative effect of fingolimod (and its 95% CI). For the annualised relapse rate, only the relative effect (i.e., the rate ratio) is given, because the assumed risk with the control group is not estimable. CI: Confidence interval; RR: Risk ratio. | |||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | |||||
a Study limitations: significant differences in reasons for incomplete outcome data between treatment and control groups. b Imprecision: total number of events (i.e. the number of participants with disability worsening/gadolinium‐enhancing lesions) was less than 300 (the threshold rule‐of‐thumb value), and thus the available evidence did not meet the optimal information size criteria. Wide confidence intervals. c Inconsistency: unexplained heterogeneity.
Summary of findings 2. Fingolimod 0.5 mg versus interferon beta‐1a for relapsing‐remitting multiple sclerosis.
| Fingolimod 0.5 mg versus intramuscular interferon beta‐1a for relapsing‐remitting multiple sclerosis | |||||
| Participants or population: people with relapsing‐remitting multiple sclerosis Settings: outpatients in multiple sclerosis centres Intervention: fingolimod 0.5 mg versus intramuscular interferon beta‐1a | |||||
| Outcomes at 12 months | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of Participants (studies) | Quality of the evidence (GRADE) | |
| Assumed risk | Corresponding risk | ||||
| Control (interferon beta‐1a) | Fingolimod 0.5 mg | ||||
| Participants free from relapse | 70 per 100 | 83 per 100 (76 to 89) | RR 1.18 (1.09 to 1.27) | 860 (1 study) | ⊕⊕⊕⊝ moderatea |
| Participants free from disability worsening | 92 per 100 | 94 per 100 (91 to 98) | RR 1.02 (0.99 to 1.06) | 860 (1 study) | ⊕⊕⊝⊝ lowa,b |
| Withdrawals due to adverse events | 4 per 100 | 6 per 100 (3 to 10) | RR 1.51 (0.81 to 2.80) | 860 (1 study) | ⊕⊕⊕⊝ moderatea |
| Annualised relapse rate | ‐ | ‐ | Rate ratio 0.48 (0.34 to 0.70) | 860 (1 study) | ⊕⊕⊕⊝ moderatea |
| Participants free from MRI gadolinium‐enhancing lesions | 81 per 100 | 90 per 100 (85 to 96) | RR 1.12 (1.05 to 1.19) | 728 (1 study) | ⊕⊕⊕⊝ moderatea |
| *For dichotomous outcomes, the corresponding risk with the intervention (and its 95% CI) is based on the assumed risk with the control (i.e. the mean proportion of events in the control group across studies) and the relative effect of the intervention (and its 95% CI). For the annualised relapse rate, only the relative effect (i.e., the rate ratio) is given. CI: Confidence interval; RR: Risk ratio. | |||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | |||||
a Imprecision: total number of events (i.e. the number of participants with disability worsening/gadolinium‐enhancing lesions) was less than 300 (the threshold rule‐of‐thumb value), and thus the available evidence did not meet the optimal information size criteria.
b Indirectness: surrogate outcome (progression confirmed at three months of follow‐up).
Background
Description of the condition
Multiple sclerosis (MS) is the most common nontraumatic cause of neurologic disability in young adults (Compston 2002; Noseworthy 2000). The overall incidence of MS is between 3.5 to 6.6 people per 100,000, while the prevalence rate is between 100 to 120 people per 100,000 (Alonso 2008; Richards 2002). A recent review showed an increase in the prevalence and incidence rate over time around the Mediterranean Basin, particularly in women ( Elhami 2011). Western Europe and North America are high prevalence areas (Koch‐Henriksen 2010). The social cost associated with MS is high because of its long duration, the early loss of productivity, the need for assistance in activities of daily living and the use of highly expensive immunotherapy and multidisciplinary health care (Koutsouraki 2010). It is widely believed that MS is an immune‐mediated disease whose clinical manifestations and course, as well as response to therapy, appear to be heterogeneous, as may be the underlying pathogenic mechanisms (Compston 2008).
Different clinical subtypes of MS are distinguishable. Approximately 80% of patients have an initial disease course characterised by relapses and remissions (relapsing‐remitting MS (RRMS)). The remaining have primary or transitional progressive MS and experience progressive decline in neurological function from onset. In patients with RRMS, disability can result from one or more of the following: incomplete recovery from relapses, development of secondary progressive MS (SPMS), or cognitive impairment. However, as many as 17% of patients with benign MS never develop a clinically important physical disability (Pittock 2004).
Considering the autoimmune pathogenesis of the disease, the mainstay of treatment has been based on immunomodulatory drugs, including interferon beta and glatiramer acetate, which are generally perceived as very safe drugs. However, they allow only a partial control of the disease. The available armamentarium has been expanding in recent years, with injectable and oral agents with more selective mechanisms of action and more efficacy (Oh J 2013).
Description of the intervention
Fingolimod is a sphingosine‐1‐phosphate (S1P)–receptor modulator, 2‐amino‐2‐(2‐[4‐octylphenyl]ethyl)‐1, 3‐propanediol (Brinkmann 2002).
It is a prodrug, phosphorylated by sphingosine kinases to active phosphofingolimod. There are at least five S1P receptor subtypes, known as S1P subtypes 1‐5 (S1P1‐5), four of which bind fingolimod‐phosphate (Chun 2010). S1P1 is highly expressed on T and B lymphocytes. Fingolimod induces lymphocytes S1P1 down‐regulation, preventing the egress of cells from secondary lymphoid tissues (Chun 2010; Pinschewer 2000). Therefore, lymphocytes are retained away from the Central Nervous System (CNS) (Matloubian 2004; Pinschewer 2000). After oral administration, fingolimod is efficiently absorbed and its absorption is unaffected by dietary intake. After administration of one dose, blood concentration of fingolimod increases steadily over the first 12 hours and remains elevated during the 24‐hour period until the next dose. It has a half‐life of six to nine days. Steady‐state blood concentrations are reached within one to two months following once‐daily administration and steady‐state levels are approximately 10‐fold greater than with the initial dose (David 2012).
Other effects of fingolimod are the transient activation of S1P receptors in atrial myocytes (associated with transient reduction of heart rate), the increase of lung hyperreactivity (associated with bronchospasm and airway constriction effects), mediated by S1P1 and S1P3 and a role in the regulation of endothelial permeability and vascular tone. Fingolimod has also been reported to be a competitive antagonist of cannabinoid receptors, and cannabinoid receptor activation has been shown to stimulate sphingomyelin catabolism (Paugh 2006).
Fingolimod can prevent renal graft rejections and suppress a variety of autoimmune disorders (Liu 2013).
Fingolimod marketed as Gilenya ® (Novartis Pharma) is provided as 0.5 mg hard gelatin capsules for oral use, once‐daily. Each capsule contains 0.56 mg of fingolimod hydrochloride, equivalent to 0.5 mg of fingolimod.
How the intervention might work
Fingolimod has been shown to reduce disease activity and established neurologic deficits in animal models (Brinkmann 2002). Prophylactic administration of fingolimod to animals with experimental autoimmune encephalitis (EAE), a model of MS, completely prevents development of EAE, whereas therapeutic administration significantly reduces the clinical severity of EAE (Chun 2010). Phase II and phase III trials in RRMS patients showed significant reduction of the relapse rate and of the number of gadolinium‐enhancing, and new and/or enlarging T2 lesions on magnetic resonance imaging (MRI), as compared with placebo and with interferon beta‐1a (Oh J 2013). These effects have been related to sequestration of lymphocytes within lymph nodes, and a significant decrease in peripherally circulating lymphocytes, preventing autoaggressive lymphocytes from crossing the blood‐brain barrier. Some studies have shown that fingolimod may promote neuroprotective and reparative processes within the CNS through modulation of S1P receptors on glial and neural cells (Miron 2008; Paugh 2006; Pinschewer 2000).
Fingolimod was the first drug to gain approval as an oral treatment in the United States on 21 September, 2010, by the US Food and Drug Administration (FDA), "for the treatment of patients with relapsing forms of MS to reduce the frequency of clinical exacerbations and to delay the accumulation of physical disability." Marketing authorisation was approved only for the 0.5 mg dose, due to a more favourable safety profile of this dosage compared to the 1.25 mg dose: "the higher dose, while exposing patients to more risk, did not expose patients to significantly increased efficacy," and specific recommendations for monitoring patients and contraindications for use has been provided (FDA 2010).
The approval by the European Medicines Agency Committee for Medicinal Products for Human Use on 27 January, 2011 (EMA 2011), has been recently updated (EMA 2015). Gilenya® is indicated as single disease modifying therapy in highly active RRMS for the following adult patient groups.
Patients with high disease activity despite treatment with at least one disease modifying drug (DMD). These patients may be defined as those who have failed to respond to a full and adequate course (normally at least one year of treatment) of at least one DMD. Patients should have had at least one relapse in the previous year while on therapy, and have at least nine T2‐hyperintense lesions in cranial MRI or at least one gadolinium‐enhancing lesion. A “non‐responder” could also be defined as a patient with an unchanged or increased relapse rate or ongoing severe relapses, as compared to the previous year.
Patients with rapidly evolving severe RRMS defined by two or more disabling relapses in one year, and with one or more gadolinium‐enhancing lesions on brain MRI or a significant increase in T2 lesion load as compared to a previous recent MRI.
Why it is important to do this review
Oral therapies have aroused lively interest amongst stakeholders, suggesting a new era of MS therapies with improved efficacy and more acceptable routes of administration. After approval of the use of fingolimod for the treatment of RRMS, different descriptive reviews have been published confirming the effectiveness of fingolimod in people with MS (Del Santo 2011; Freedman 2013; Hillert 2012; Hutchinson 2014; Oh J 2013), but also raising concerns about serious adverse events (Lu 2013; Parfenov 2013). A number of safety concerns emerged with post‐marketing surveillance, including serious infections and adverse cardiac effects (AIFA 2015; EMA 2015; Oh J 2013). The advantage of this therapy (as compared to other DMDs) has been questioned, suggesting need to reserve the use of fingolimod to patients who can be closely monitored.
No systematic review of trials evaluating the benefit and safety of fingolimod has until now, been provided. By assessing and updating its benefit‐risk profile, the results of this review might clarify the use of fingolimod in clinical practice.
Objectives
To assess the safety and benefits of fingolimod versus placebo, or other disease‐modifying drugs (DMDs), in reducing disease activity in people with relapsing‐remitting multiple sclerosis (RRMS).
Methods
Criteria for considering studies for this review
Types of studies
We considered randomised controlled trials (RCTs) that studied fingolimod versus placebo or other approved disease‐modifying drugs (DMDs) in relapsing‐remitting multiple sclerosis (RRMS), irrespective of publication status and language. We excluded cross‐over studies.
Types of participants
We included participants of any age, gender and race affected by RRMS according to McDonald's diagnostic criteria (Mc Donald 2001; Polman 2005; Polman 2011).
Types of interventions
Fingolimod, any dose and route of administration, versus placebo without restriction of treatment duration.
Fingolimod, any dose and route of administration, versus any other approved DMDs without restriction of treatment duration.
Types of outcome measures
Primary outcomes
1. Number of participants relapse‐free at six,12 and 24 months after randomisation and at the end of follow‐up.
2. Number of participants free from disability worsening at 12, 24 and 36 months after randomisation and at the end of follow‐up. Disability worsening is defined as at least one point Expanded Disability Status Scale (EDSS) (Kurtzke 1983) increase, or a 0.5 point increase if the baseline EDSS was > 5.5, confirmed during two subsequent neurological examinations separated by at least 6 months' interval free of relapses. We considered separately studies that reported disability worsening defined using different criteria.
3. Number of participants who withdrew from the study due to
a) adverse events;
b) serious adverse events, i.e. death, life‐threatening, hospitalisation, disability or permanent damage, congenital anomaly/birth defect (FDA 2013).
Secondary outcomes
4. Annualised relapse rate at six, 12 and 24 months after randomisation and at the end of follow‐up.
5. Number of participants free from MRI gadolinium‐enhancing lesions at six, 12 and 24 months after randomisation and at the end of follow‐up.
6. Mean change of total MRI T2 weighted lesion load at 12 and 24 months after randomisation and at the end of follow‐up.
7. Quality of life measured by validated questionnaires such as MSQOL‐54 (Vickrey 1995).
Search methods for identification of studies
We did not apply any language restrictions.
Electronic searches
The Trials Search Co‐ordinator searched the Cochrane Multiple Sclerosis and Rare Diseases of the CNS Group Specialised Trials Register, which contained trials identified from:
Cochrane Central Register of Controlled Trials (CENTRAL; 2016, Issue 2).
MEDLINE (PubMed) (1966 to 15 February 2016).
EMBASE (Embase.com) (1974 to 15 February 2016).
Cumulative Index to Nursing and Allied Health Literature (CINAHL) (EBSCOhost) (1981 to 15 February 2016).
Latin American and Caribbean Health Science Information Database (LILACS) (Bireme) (1982 to 15 February 2016).
Clinical trial registries: clinicaltrials.gov.
World Health Organization (WHO) International Clinical Trials Registry Portal (apps.who.int/trialsearch/).
The keywords for this review are listed in (Appendix 1).
Information on the Trial Register of the Review Group and details of search strategies used to identify trials can be found in the 'Specialised Register' section within the Cochrane Multiple Sclerosis and Rare Diseases of the CNS group module.
Searching other resources
Reference list of included studies and related reviews.
Abstract books of the main MS meetings (European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS), European Neurological Society (ENS), American Academy of Neurology (AAN)) for the last eight years.
Contact with authors of primary studies, or drug manufacturers, or both.
FDA reports on fingolimod (www.fda.gov).
European Medicines Agency Committee for Medicinal Products for Human Use reports on fingolimod (EMA).
Data collection and analysis
We performed the review and meta‐analyses following the recommendations of Cochrane (Higgins 2011a). We used Review Manager 5 to perform the analyses (RevMan 2015).
Selection of studies
Three review authors (LLM, IP, RP) independently identified trials and assessed titles and abstracts of the records retrieved by the search. We excluded irrelevant studies. We obtained the full text of the remaining studies to confirm inclusion. We resolved disagreements by discussion.
Data extraction and management
Two review authors (LLM, IP) developed a data extraction form. Three review authors (LLM, IT, IP) extracted data on trial design, participants, interventions, and outcomes independently from each other. We resolved disagreements by discussion.
Assessment of risk of bias in included studies
Three review authors (LLM, IT, IP) independently assessed risk of bias for each study using the criteria outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a). We resolved any disagreement by discussion .
We followed Cochrane's recommendations for assessing risk of bias for each included study (Higgins 2011a). We graded each potential source of bias as high, low or unclear (either lack of information or uncertainty over the potential for bias) and provided a quote for the study report together with a justification for our judgment in the "Risk of Bias" table. We assessed the risk of bias according the following domains:
Random sequence generation.
Allocation concealment.
Blinding of participants and personnel.
Blinding of outcome assessment.
Incomplete outcome data.
Selective outcome reporting.
Other bias.
Outcome data were judged as low risk of bias when numbers and causes of losses to follow up were balanced between arms and when the percentage of participants lost to follow‐up was low (arbitrarily set at values lower than 15%).
In addition, we defined the following two specific criteria to assess adverse events.
Did the study provide a definition for severe adverse events?
Did the researchers actively monitor for adverse events (low risk of bias) or did they simply provide spontaneous reporting of adverse events that arose (high risk of bias)?
Measures of treatment effect
The preset outcomes involved dichotomous, continuous and count data. We used the risk ratio (RR) with 95% confidence intervals (CIs) for dichotomous data, the weighted mean difference with 95% CIs for continuous data, and the rate ratio with 95% CI for count data. The rate ratio compares the rate of events in two groups by dividing one by the other. In order to avoid the assumption that the variability between the studies was exclusively because of a random sampling variation around a fixed‐effect, we used the random‐effects model.
Unit of analysis issues
We performed a comparison with the parallel group maintaining the original randomisation of the study. In case of repeated observations, we tried to analyse each predefined outcome to reflect short‐term (six months), medium‐term (one year) and long‐term (> 2 years) follow‐up.
Dealing with missing data
In order to assess the effect of missing outcome data, we analysed data according to a likely scenario, i.e. we assumed that the treatment and control group participants who contributed to the missing data, both had experienced the outcome (relapse or disability).
Assessment of heterogeneity
We assessed between‐study heterogeneity both by inspection of graphical presentations using forest plots (Egger 1997), and by calculating the I2 statistic (significant if more than 50%) and the Chi2 test (Higgins 2011a).
Assessment of reporting biases
We did not perform a funnel plot because less than ten trials were included (Egger 1997).
Data synthesis
We performed meta‐analyses of primary and secondary outcomes using RevMan 5 (RevMan 2015).
Subgroup analysis and investigation of heterogeneity
Subgroup analyses were planned to answer specific questions, such as the effects about types of interventions (different dosages) or to investigate heterogeneous results (Deeks 2011).
Sensitivity analysis
In the case where there was evidence of trials results heterogeneity, we planned to perform a sensitivity analysis to determine the effect of excluding trials with a high risk of bias.
'Summary of findings' table
We created two 'Summary of findings' tables comparing fingolimod at the approved dose of 0.5 mg daily; one versus placebo at 24 months after randomisation (Table 1), and one versus intramuscular interferon‐beta 1a at 12 months (Table 2). In both 'Summary of findings' tables, we included five outcomes: number of participants relapse‐free; number of participants free from disability worsening ; number of withdrawals due to adverse events; annualised relapse rate; and number of participants free from MRI gadolinium‐enhancing lesions. We used the five GRADE considerations (study limitations, consistency of effect, imprecision, indirectness and publication bias) to assess the quality of the body of evidence. We followed methods and recommendations described in Section 11.5 and Chapter 12 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a).
Results
Description of studies
Results of the search
Results of the search are described in a PRISMA flow chart (Figure 1; Moher 2009). We assessed for eligibility 81 out of 481 records; 19 studies did not satisfy the inclusion criteria (Characteristics of excluded studies). We included a total of six RCTs (Calabresi 2014; Cohen 2010; Fox 2014; Kappos 2006, Kappos 2010; Saida 2012). Fifty‐six reports were ancillary to these primary studies. We found 10 ongoing trials; six are still ongoing (Characteristics of ongoing studies) and four have been completed and are awaiting classification (NCT01317004; NCT01333501; NCT01534182; NCT01623596) (Characteristics of studies awaiting classification).
1.

Study flow diagram.
Included studies
The six trials were as follows.
Calabresi 2014 (FREEDOMS II study) (phase III).
Cohen 2010 (TRANSFORMS study) (phase III).
Fox 2014 (EPOC study) (phase IV).
Kappos 2006 (phase II).
Kappos 2010 (FREEDOMS study) (phase III).
Saida 2012 (phase II).
These trials were published between 2006 and 2014. Four studies compared fingolimod to placebo (Calabresi 2014; Kappos 2006; Kappos 2010; Saida 2012), one to intramuscular interferon beta‐1a (Cohen 2010), and one to other DMDs (interferon beta‐1a, interferon beta‐1b, glatiramer acetate) (Fox 2014). Four studies used fingolimod at doses of 0.5 mg and 1.25 mg (Calabresi 2014; Cohen 2010; Kappos 2010; Saida 2012). One study used doses of 1.25 mg and 5.0 mg (Kappos 2006). One study evaluated only the dose of 0.5 mg (Fox 2014). Fingolimod was administered orally in all studies.
The primary outcome was annualised relapse rate in three studies (Calabresi 2014; Cohen 2010; Kappos 2010); MRI measures of activity in two studies: number of gadolinium‐enhancing lesions (Kappos 2006) and percentage of participants free from gadolinium‐enhancing lesions (Saida 2012); and treatment satisfaction in the other study (Fox 2014). Outcome measures and time points of assessment considered by each of the included trials are listed in Table 3.
1. Outcome measures and time points.
| Study name | Clinical outcomes | Time point assessment | MRI outcomes | Time point assessment |
| Calabresi 2014 | 1.Annualised relapse rate 2. Time to disability progression confirmed at 3 months 3. Time to disability progression confirmed at 6 months 4. Safety 5. Time to first relapse 6. Proportion of relapse‐free participants 7. Change from baseline to the end of study on the MSFC score 8. Quality of life using the Euro quality of life scale (EQ‐5D) 9. Patient Reported Indices in Multiple Sclerosis 10. Fatigue using the Modified Fatigue Impact Scale |
24 months | 1.Percent brain‐volume change from baseline at 24 months 2.Number and volume of gadolinium‐enhancing T1 lesions 3. Number of new or newly enlarged T2 lesions 4. Proportion of participants free of gadolinium‐enhancing T1 lesions 5. Proportion of participants free of new or newly enlarged T2 lesions 6. Proportion of participants free of new inflammatory activity (no gadolinium‐enhancing T1 lesions and no new or newly enlarged T2 lesions) 7. Percentage change from baseline in volume of gadolinium‐enhanced T1 lesions 8. Percentage change from baseline in volume of new or newly enlarged T2 lesions 9. Brain volume |
24 months |
| Cohen 2010 | 1. Annualised relapse rate 2. Progression of disability (confirmed at 3 months) |
12 months | Number of new or enlarged lesions on T2‐weighted scans | 12 months |
| Fox 2014 | 1. Treatment satisfaction 2. Fatigue 3. Depression 4. Activities of daily living 5. Health‐related Quality Of Life 6, Side effects |
6 months | Not included | |
| Kappos 2006 | 1. Number of participants remaining free of relapse 2. Annualised relapse rate 3, Time to the first relapse |
6 months | 1.Number of gadolinium‐enhanced lesions per participant recorded on T1‐weighted MRI at monthly intervals for 6 months 2.Total volume of gadolinium‐enhanced lesions per participants 3. Proportion of participants with gadolinium‐enhanced lesions 4. Total number of new lesion per participant on T‐weighted images 5. Changes in lesion volume on T2‐weighted images 6. Brain volume from baseline to month 6 |
6 months |
| Kappos 2010 | 1. Annualised relapse rate 2. Time to confirmed disability progression (confirmed after 3 months ) 3. Time to a first relapse 4. Time to disability progression (confirmed after 6 months) 5. Changes in the EDSS score 6. Changes in the MSFC z score between baseline and 24 months |
24 months | 1. Number of gadolinium‐enhancing lesions 2. Proportion of participants free from gadolinium‐enhancing lesions 3. Number of new or enlarged lesions on T2‐weighted MRI scans 4. Proportion of participants free from new or enlarged lesions on T2‐weighted scan 5. Volumes of hyperintense lesions on T2‐weighted scan 6. Volumes of hypointense lesions on T1‐weighted scans 7. Change in brain volume between baseline and 24 months 8. Safety and tolerability measures |
24 months |
| Saida 2012 | Percentage of participants free from relapse | 6 months | Participants free from gadolinium‐enhancing lesions | 6 months |
EDSS: Expanded Disability Status Scale; MSFC: Multiple Sclerosis Functional Composite
The primary outcome of each study is underlined
Confirmed relapse was defined as the occurrence of new symptoms, or worsening of previously stable or improving symptoms, and signs not associated with fever, lasting more than 24 hours. Symptoms had to appear at least 30 days after the onset of a preceding relapse (Cohen 2010; Saida 2012), and had to be accompanied by an increase of at least half a point in the EDSS score (Calabresi 2014; Cohen 2010; Kappos 2006; Kappos 2010) or one point in at least one of the functions in the Kurtzke Functional System score (excluding bowel‐bladder and mental systems) (Kappos 2006) or one point in each of two functions in the Kurtzke Functional Systems score, or two points in one of the functions in the Kurtzke Functional System score (excluding bowel–bladder or cerebral systems) (Calabresi 2014; Cohen 2010; Kappos 2010; Saida 2012).
Confirmed disability progression was defined as an increase of one point in the EDSS score (or 0.50 points if the baseline EDSS score was > 5·0), that was confirmed three months later in the absence of relapse (Calabresi 2014; Cohen 2010; Kappos 2010). Disability progression confirmed at six months was available in two studies at 24 months (Calabresi 2014; Kappos 2010). Assessment of disability was performed using the Multiple Sclerosis Functional Composite (MSFC) score change in one study (Kappos 2006). Disability progression was not considered by Fox 2014 and Saida 2012.
The treatment duration was six months in three trials (Fox 2014; Kappos 2006; Saida 2012), 12 months in one trial (Cohen 2010), and 24 months in two trials (Calabresi 2014; Kappos 2010).
The Characteristics of included studies tables provide further details.
All studies were sponsored by Novartis Pharma.
Description of participants
The overall population included in the six trials was 5152 participants with 3531 treated with fingolimod; 2061 with 0.5 mg daily, 1376 with 1.25 mg daily, and 94 with 5.0 mg daily. The comparison population included 1621 participants; 923 treated with placebo and 698 with intramuscular interferon beta‐1a or other DMDs. Enrolled participants were Caucasian, except in Saida 2012, which included Japanese participants. Participants were affected by relapsing‐remitting multiple sclerosis (RRMS) in all studies, and secondary progressive multiple sclerosis (SPMS) in a small percentage in two studies; 11% in Kappos 2006 and 2.3% in Saida 2012 (Table 4).
2. Baseline characteristics of the population included in the RCTs.
| Study name | Drugs | No. participants | Female (%) | Course of disease of RR‐SP (%) | Age, years, mean (SD) | Mean EDSS score (SD) | Disease duration, mean (SD) | Pre‐1 year number of relapses, mean (SD) | Percentage of pre‐study treatment‐naive participants | Percentage of participants with MRI enhancing lesions | Mean lesion volume on T2‐weighted images (mm3 ) (SD) |
| Calabresi 2014 | Placebo | 355 | 81 | 100 ‐ 0 | 40·1 (8·4) | 2·2 (1·5) | 10·6 (7·9) | 1·5 (0·9) | 27 | 36 | 5553 (7841) |
| Fingolimod 0.5 mg | 358 | 77 | 100 ‐ 0 | 40·6 (8·4) | 2·2 (1·4) | 10·4 (8·0) | 1·4 (0·9) | 26 | 39 | 5484 (8000) | |
| Fingolimod 1.25 mg | 370 | 76 | 100 ‐ 0 | 40·9 (8·9) | 2·3 (2·0) | 10·8 (8·2) | 1·5 (1·0) | 22 | 31 | 4936 (7286) | |
| Cohen 2010 | Interferon beta‐1a (Avonex) | 435 | 67.8 | 100 ‐ 0 | 36.0 (8.3) | 2.19 (1.26) | 7.4 (6.3) | 1.5 (0.8) | 43.7 | 36.9 | 4924 (5711) |
| Fingolimod 0.5 mg | 431 | 65.4 | 100 ‐ 0 | 36.7 (8.8) | 2.24 (1.33) | 7.5 (6.2) | 1.5 (1.2) | 44.8 | 32.6 | 5170 (6642) | |
| Fingolimod 1.25 mg | 426 | 68.8 | 100 ‐ 0 | 35.8 (8.4) | 2.21 (1.31) | 7.3 (6.0) | 1.5 (0.9) | 41.5 | 34.5 | 5085 (5962) | |
| Fox 2014 | DMD§ | 263 | 79.1 | 100 ‐ 0 | 45.1 (9.82) | 2.4 (1.32) | 11.7 (8.44) | 0.8 (1.32) | 0 | NR | NR |
| Fingolimod 0.5 mg | 790 | 76.1 | 100 ‐ 0 | 46.0 (9.82) | 2.4 (1.32) | 12.1 (8.38) | 0.8 (1.20) | 0 | NR | NR | |
| Kappos 2006 | Placebo | 93 | 66 | 90 ‐ 10 | 37.1 (19‐56)* | 2.6 (0.0‐6.5)* | 8.4 (0.2‐28.2)* | 1.2 (0‐5)* | NR | 51 | 8805 (123‐62,218)* |
| Fingolimod 1.25 mg | 94 | 75 | 89 ‐ 11 | 38.0 (19‐60)* | 2.7 (0.0‐6.0)* | 8.6 (0.3‐50.2)* | 1.3 (0‐5)* | NR | 47 | 10,219 (293‐104,504)* | |
| Fingolimod 5.0 mg | 94 | 71 | 87 ‐ 13 | 38.3 (18‐59)* | 2.5 (0.0‐6.0)* | 9.5 (0.5‐42.2)* | 1.3 (0‐4)* | NR | 57 | 8722 (349‐70,218)* | |
| Kappos 2010 | Placebo | 418 | 71.3 | 100 ‐ 0 | 37.2 (8.6) | 2.5 (1.3) | 8.1 (6.4) | 1.4 (0.7) | 59.6 | 37 | 6162 (7085) |
| Fingolimod 0.5 mg | 425 | 69.6 | 100 ‐ 0 | 36.6 (8.8) | 2.3 (1.3) | 8.0 (6.6) | 1.5 (0.8) | 57.4 | 38 | 6128 (7623) | |
| Fingolimod 1.25 mg | 429 | 68.8 | 100 ‐ 0 | 37.4 (8.9) | 2.4 (1.4) | 8.4 (6.9) | 1.5 (0.8) | 60.4 | 39.4 | 6829 (8491) | |
| Saida 2012 | Placebo | 57 | 68.4 | 100 ‐ 0 | 35.0 (8.9) | 2.1 (1.7) | 8.2 (7.3) | 1.7 (1.6) | NR | 42.1 | 31.6 (22.6)** |
| Fingolimod 0.5 mg | 57 | 70.2 | 94.7 ‐ 5.3 | 35.0 (9.0) | 2.3 (1.9) | 8.2 (6.8) | 1.4 (1.0) | NR | 42.1 | 30.4 (22.7)** | |
| Fingolimod 1.25 mg | 57 | 68.4 | 98.2 ‐ 1.8 | 36.0 (9.3) | 1.8 (1.7) | 7.1 (5.3) | 1.5 (0.9) | NR | 49.1 | 31.7 (23.3)** |
DMD: disease‐modifying drug;EDSS: Expanded Disability Status Scale; MRI: magnetic resonance imaging; NR: not reported; RR: relapsing‐remitting; SD: standard deviation; SP: secondary progressive
* Range (SD was not provided) ** Number of T2 lesions (volume was not provided) § interferon beta‐1b (Extavia® or Betaseron®) 0.25 mg injected subcutaneously every other day (46 participants); interferon beta‐1a (Avonex®) 30 μg intramuscular injected once a week (60 participants); interferon beta‐1a (Rebif®) 22 μg or 44 μg injected subcutaneously three times a week (65 participants); or glatiramer acetate (Copaxone®) 20 mg injected subcutaneously once‐daily (92 participants)
The inclusion criteria were similar among studies. Previous treatment with immunomodulating agents for at least six months were required in Fox 2014, and accepted in all other studies if the suspension occurred more than three months before trial onset (Calabresi 2014; Kappos 2006; Kappos 2010; Saida 2012), not specified in Cohen 2010.
Exclusion criteria were similar and included clinically significant systemic diseases, macular oedema (not specified for Kappos 2006), and diabetes (not specified for Kappos 2006 and Cohen 2010). Participants treated with immunosuppressants were excluded (Kappos 2010; Fox 2014), they had to interrupt treatment in the six months prior to randomisation (Saida 2012), interrupt azathioprine or methotrexate within six months, cyclophosphamide within 12 months, mitoxantrone or cladribine within 24 months (Kappos 2006), and natalizumab at least six months before randomisation (Calabresi 2014). Two studies specified as exclusion criteria the presence of cardiac abnormalities or leukopenia or lymphopenia (Kappos 2006; Saida 2012). Participants with varicella zoster immunoglobulin G (IgG) antibody negative at screening were excluded in three studies (Calabresi 2014; Fox 2014; Saida 2012).
The baseline clinical characteristics were homogeneous in term of age (ranging from 35.0 to 38.3 years), disability score (EDSS 1.8 to 2.7), disease duration (7.1 to 9.5 years), and mean relapse number in the year before randomisation (1.2 to 1.7). Percentage of prestudy treatment‐naive participants were different among studies, ranging from 0% to 60.4%. MRI enhancing lesions were detected in 31% to 49% of the participants, while the volume of MRI T2‐weighted lesions at baseline ranged from 4924 mm3 to 10,219 mm3 (Table 4).
Excluded studies
We excluded 18 studies because type of participants (three studies), design (seven studies) or intervention (one study) did not meet our inclusion criteria, five studies were not original articles (reporting pooled data of trials), and two studies were overviews (Characteristics of excluded studies).
Risk of bias in included studies
We evaluated risk of bias separately for benefit estimate (Characteristics of included studies), and adverse events monitoring (Table 5).
3. Methods of adverse events monitoring.
| Study name | Risk of bias | Did the researchers actively monitor for adverse events (AEs) (low risk of bias) or did they simply provide spontaneous reporting of AEs that arose (high risk of bias)? | Risk of bias | Did the authors define serious AEs (SAEs) according to an accepted international classification and report the number of SAEs? |
| Calabresi 2014 | Low | "We did extensive safety and tolerability assessments, in part as a response to preclinical safety concerns raised by the FDA and additional safety areas of interest identified in previous phase 2 and earlier clinical studies. We also recorded adverse events, serious adverse events, serious adverse events of special interest, 24 h Holter electrocardiography (ECG) post first‐dose and at 3 months, first‐dose bradycardia events, infections, laboratory tests, vital signs, ECG, echocardiography, pulmonary function. tests, chest high‐resolution CT,chest radiographs, ophthalmic examinations, including serial optical coherence tomography, and dermatological assessments." Clinical assessments were performed at screening and at randomisation (baseline), and study visits, including safety assessments, were scheduled at 2 weeks and 1, 2, 3, 6, 9, 12, 15, 18, 21, and 24 months after randomisation" | Unclear | "We also recorded adverse events, serious adverse events, serious adverse events of special interest, 24 h Holter electrocardiography (ECG) post fist‐dose and at 3 months, first‐dose bradycardia events, infections, laboratory tests, vital signs, ECG, echocardiography, pulmonary function tests, chest high‐resolution CT,chest radiographs, ophthalmic examinations, including serial optical coherence tomography, and dermatological assessments" |
| Cohen 2010 | Low | "An independent data and safety monitoring board evaluated overall safety in the fingolimod phase 3 program" and "Safety assessments were conducted during screening, at baseline, and at months 1, 2, 3, 6, 9, and 12" (pg 404) | Low | SAEs were predefined per standard criteria (death, life‐threatening event, persistent disability, congenital defect, unplanned hospitalisation, or otherwise medically significant) (FDA 2010 Clinical review of safety pg 151) |
| Fox 2014 | Low | "Safety and tolerability (secondary study objectives) were assessed via reporting of adverse events (AEs) and through physical examinations (ophthalmologist examinations, and evaluations of vital signs, chest x‐rays, and electrocardiograms [ECGs]), laboratory evaluations (measurement of hematology parameters, chemistry, urinalysis, serology, and lymphocyte counts)" | Unclear | Not specified |
| Kappos 2006 | Low | "An independent external data and safety monitoring board evaluated adverse events and other safety data" and "Adverse events were assessed and reported at each visit (scheduled and unscheduled) by the treating physicians. Laboratory evaluations were undertaken at a central laboratory". "Vital signs were obtained at each visit, and laboratory and hematologic measures were obtained at baseline, day 1, and months 1,3,6,9, and 12. Electrocardiograms were obtained at baseline, on days 1 and 7, and at months 1,3,6,12, and 24 hour Holter electrocardiographic monitoring was performed at selected sites at baseline, day 1, and month 3. Pulmonary function tests... were performed at screening and months 6 and 12" (pg 1126) | Low | Categorisation of SAEs conformed to ICH guidelines (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use) |
| Kappos 2010 | Low | "An independent data and safety monitoring board evaluated the safety" and "Study visits, including safety assessments, were scheduled at 2 weeks and 1, 2, 3, 6, 9, 12, 15, 18, 21, and 24 months after randomization" (pg 389) | Low | SAEs were predefined per standard criteria (death, life‐threatening event, persistent disability, congenital defect, unplanned hospitalisation, or otherwise medically significant) (FDA 2010 Clinical review of safety pg 151) |
| Saida 2012 | Low | "Adverse events, serious adverse events assessments were conducted at screening, baseline, days 1 and 15, and months 1,2,3,4,5 and 6" (pg 2) and "Safety assessment included recording of AEs, SAEs, hematology values, vital signs, results of dermatological and ophthalmological examinations and results of pulmonary and liver function tests" (Supplementary data online appendix) | Low | Categorisation of SAEs conformed to ICH guidelines (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use) |
AE: adverse event; CT: chest tomography; ECG: electrocardiography; FDA: Food and Drug Administration; SAE: serious adverse event
Allocation
We considered the method used to generate a random sequence and conceal allocation to be at low risk of bias in all trials except one; we judged Calabresi 2014 to be at unclear risk of bias.
Blinding
All included trials, except one open‐label trial were described as double‐blinded (Fox 2014). The drug in the treatment group was reported as identical in appearance to the drug in the control group. Assessors of clinical and radiological outcomes were reported as unaware of participants' assignment. We judged Fox 2014 to be at high risk of bias and the other trials to be at low risk of bias for blinding.
Incomplete outcome data
All trials provided a sufficient description of follow‐up and withdrawals. We judged three trials to be at high risk of attrition bias because significant differences in reasons of dropouts between treated and control groups were found ( Calabresi 2014 ; Fox 2014 ; Kappos 2010).
Selective reporting
We judged all trials to be at low risk of bias apart from Calabresi 2014, which we judged to be at unclear risk of bias as it did not report on clinically relevant results.
Other potential sources of bias
All trials were sponsored by Novartis Pharma. We judged four trials to be at high risk of bias because the study sponsor participated in conducting the study, or data analysis and some study co‐authors were affiliated to the pharmaceutical company (Calabresi 2014; Cohen 2010; Fox 2014; Kappos 2010) .
Further details are provided in relevant sections of the Characteristics of included studies tables and are presented as the 'Risk of bias' summary and 'Risk of bias' graph (Figure 2; Figure 3).
2.
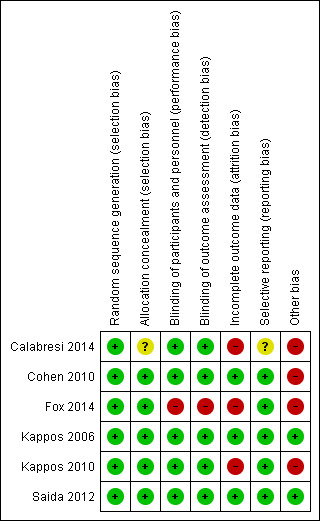
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
3.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Methods of adverse events monitoring
We judged the risk of bias regarding methods of adverse events monitoring to be low for all studies. We judged the risk of bias regarding methods of serious adverse events monitoring to be unclear for the Calabresi 2014 and for the Fox 2014 studies and low for four others (Table 5).
Effects of interventions
We defined two main comparisons, one evaluating the effect of fingolimod versus placebo and one evaluating the effect of fingolimod versus intramuscular IFN‐beta 1a or other DMDs. For each comparison, we considered the effect of fingolimod separately for the approved dose (0.5 mg) and the other treatment schedules (1.25 mg and 5.0 mg).
We reported the main results concerning benefit and withdrawals due to adverse events of fingolimod at the approved dose of 0.5 mg compared to placebo at 24 months in Table 1 and compared to intramuscular interferon beta‐1a at 12 months in Table 2.
Fingolimod compared to placebo
Primary Outcomes
The number of participants who were free from relapses during treatment with fingolimod 0.5 mg compared to placebo were retrieved from one trial (114 participants = 2.2%) at six months (Saida 2012), and two trials (1556 participants =30%) at 24 months (Calabresi 2014; Kappos 2010). At six months, there was a slight non‐significant benefit (risk ratio (RR) 1.22, 95% confidence interval (CI) 0.96 to 1.54) (Analysis 1.1). At 24 months the overall results indicated a benefit of fingolimod (RR 1.44, 95% CI 1.28 to 1.63; moderate quality evidence) (Table 1; Analysis 1.3). The higher doses were effective at six months at 1.25 mg (RR 1.27, 95% CI 1.11 to 1.45) and 5 mg (RR 1.30, 95% CI 1.10 to 1.53) (Analysis 1.1), and at 24 months at 1.25 mg (RR 1.51, 95% CI 1.29 to 1.76) (Analysis 1.3).
1.1. Analysis.

Comparison 1 Participants free from relapse, Outcome 1 At 6 months.
1.3. Analysis.

Comparison 1 Participants free from relapse, Outcome 3 At 24 months.
Data from two trials were available to calculate the number of participants without disability worsening during the first 24 months of treatment with fingolimod compared to placebo (Calabresi 2014; Kappos 2010). The results indicated little or no difference of fingolimod at 0.5 mg (RR 1.07, 95% CI 1.02 to 1.11; low quality evidence) (Table 1). Similar results were found when fingolimod was used at 1.25 mg (RR 1.08, 95% CI 1.03 to 1.12) (Analysis 2.2).
2.2. Analysis.

Comparison 2 Participants free from disability worsening, Outcome 2 At 24 months.
The number of participants who withdrew from the study because of adverse events with 0.5 mg of fingolimod compared to placebo was retrieved from two trials during the first six months (RR 2.00, 95% CI 0.53 to 7.61) (Kappos 2006; Saida 2012: Analysis 3.1); and two trials during the first 24 months of treatment (RR 1.42, 95% CI 0.89 to 2.25; very low quality evidence) (Calabresi 2014; Kappos 2010; Table 1; Analysis 3.3). The risk of discontinuing 1.25 mg of fingolimod due to adverse events compared to placebo significantly increased at 24 months (RR 1.93, 95% CI 1.48 to 2,52) (Analysis 3.3).
3.3. Analysis.

Comparison 3 Number of withdrawals due to adverse events, Outcome 3 Withdrawals due to adverse events over 24 months.
Detailed descriptive data on the type of adverse events, as reported in the primary studies, are provided in Figure 4. It should be noted that infections, hypertension, and abnormal liver tests were more frequent in the fingolimod group than in the placebo group.
4.

N, number of patients; n, number of events.
Significant differences (based on Fisher exact test) are reported in red.
The number of participants who withdrew because of serious adverse events were available from two trials at six months (Kappos 2006; Saida 2012), and two trials at 24 months (Calabresi 2014; Kappos 2010). No difference was found between placebo and fingolimod administered at 0.5 mg and 1.25 mg (Analysis 3.4; Analysis 3.6). A significant increased risk of discontinuation was found for fingolimod 5.0 mg versus placebo at six months (RR 2.77, 95% CI 1.04 to 7.38) (Analysis 3.4).
3.4. Analysis.

Comparison 3 Number of withdrawals due to adverse events, Outcome 4 Withdrawals due to serious adverse events over 6 months.
3.6. Analysis.

Comparison 3 Number of withdrawals due to adverse events, Outcome 6 Withdrawals due to serious adverse events over 24 months.
Detailed descriptive data on the type of serious adverse events, as reported in the primary studies, are provided in Figure 5. It should be noted that infections, basal‐cell carcinoma, and atrioventricular blocks were more frequent in the fingolimod group than in the placebo group.
5.

N, number of patients; n, number of events.
* One case of basal‐cell carcinoma was not reported as a serious adverse event by the site investigator (Calabresi 2014).
Significant differences (based on Fisher exact test) are reported in red.
The results regarding data analysed according to a likely scenario did not add any additional information (data not reported).
Secondary outcomes
The annualised relapse rate was evaluated by two trials at six months (Kappos 2006; Saida 2012), and two trials at 24 months (Calabresi 2014; Kappos 2010): at these time points the results favoured fingolimod 0.5 mg compared to placebo at six months (rate ratio 0.51, 95% CI 0.26 to 0.99; Analysis 4.1) and 24 months (rate ratio 0.50, 95% CI 0.40 to 0.62; moderate quality evidence) (Table 1; Analysis 4.3). Similar data were found for higher doses (Analysis 4.1, Analysis 4.3).
4.1. Analysis.

Comparison 4 Annualised relapse rate, Outcome 1 At 6 months.
4.3. Analysis.

Comparison 4 Annualised relapse rate, Outcome 3 At 24 months.
The number of participants free from MRI gadolinium‐enhancing lesions was evaluated by four trials at six months (Calabresi 2014; Kappos 2006; Kappos 2010; Saida 2012; Analysis 5.1), two trials at 12 months (Calabresi 2014; Kappos 2010; Analysis 5.2), and two trials at 24 months (Calabresi 2014; Kappos 2010; Analysis 5.3). We found better results for fingolimod doses compared to placebo at each time point: fingolimod 0.5 mg at six months (RR 1.42, 95% CI 1.33 to 1.51); 0.5 mg at 12 months (RR 1.39, 95% CI 1.30 to 1.48); and 0.5 mg at 24 months (RR 1.36, 95% CI 1.27 to 1.45; low quality evidence) (Table 1).
5.1. Analysis.

Comparison 5 Participants free from gadolinium‐enhancing lesions, Outcome 1 At 6 months.
5.2. Analysis.

Comparison 5 Participants free from gadolinium‐enhancing lesions, Outcome 2 At 12 months.
5.3. Analysis.

Comparison 5 Participants free from gadolinium‐enhancing lesions, Outcome 3 At 24 months.
The mean change of MRI T2‐weighted lesion load was evaluated by one trial at 12 months (Kappos 2010): the results favoured fingolimod 0.5 mg compared to placebo (mean difference (MD) ‐15.30, 95% CI ‐24.34 to ‐6.26); similar data were found when fingolimod was used at 1.25 mg for 12 months (Analysis 6.1). At 24 months the results also favoured fingolimod 0.5 mg compared to placebo (MD ‐20.43, 95% CI ‐34.03 to ‐6.83); similar data were found for fingolimod 1.25 mg (Analysis 6.2).
6.1. Analysis.

Comparison 6 Mean change of MRI T2‐weighted lesion load, Outcome 1 At 12 months.
6.2. Analysis.

Comparison 6 Mean change of MRI T2‐weighted lesion load, Outcome 2 At 24 months.
Quality of life was measured by the Hamburg Quality of Life Questionnarie in one trial at six months (Kappos 2006), and by the Euro quality of life scale (EQ‐5D) in one trial at 24 months (Calabresi 2014). No differences were found between fingolimod and placebo in either study (Analysis 7.1; Analysis 7.2).
7.1. Analysis.

Comparison 7 Quality of life, Outcome 1 At 6 months.
7.2. Analysis.

Comparison 7 Quality of life, Outcome 2 At 24 months.
Fingolimod compared to intramuscular interferon beta‐1a or other DMDs
Primary Outcomes
Data from one trial were available to evaluate the primary outcomes during the first 12 months of treatment with fingolimod 0.5 mg compared to intramuscular interferon beta‐1a (Cohen 2010).
The overall results (RR 1.18, 95% CI 1.09 to 1.27; moderate quality evidence) indicated a slight advantage for fingolimod 0.5 mg in favouring freedom from relapse (Table 2). Similar results were found when fingolimod was used at 1.25 mg (RR 1.15, 95% CI 1.06 to 1.24) (Analysis 1.2).
1.2. Analysis.

Comparison 1 Participants free from relapse, Outcome 2 At 12 months.
The results indicated no difference in favouring freedom from disability worsening at 12 months between fingolimod 0.5 mg and intramuscular interferon beta‐1a (RR 1.02, 95% CI 0.99 to 1.06; low quality evidence) (Table 2). Similar results were found when fingolimod was used at 1.25 mg (RR 1.01, 95% CI 0.98 to 1.05) (Analysis 2.1).
2.1. Analysis.

Comparison 2 Participants free from disability worsening, Outcome 1 At 12 months.
Compared to intramuscular interferon beta‐1a, the number of participants who withdrew due to adverse events was higher, but not significant for fingolimod 0.5 mg (RR 1.51, 95% CI 0.81 to 2.80; moderate quality evidence) (Table 2). Significant risk was found when used at 1.25 mg (RR 2.69, 95% CI 1.54 to 4.72) (Analysis 3.2).
3.2. Analysis.

Comparison 3 Number of withdrawals due to adverse events, Outcome 2 Withdrawals due to adverse events over 12 months.
Compared to intramuscular interferon beta‐1a, the number of participants who withdrew due to serious adverse events was higher, but not significant for fingolimod 0.5 mg (RR 1.21, 95% CI 0.72 to 2.02), and significantly higher for fingolimod 1.25 mg (RR 1.85, 95% CI 1.15 to 2.96) (Analysis 3.5).
3.5. Analysis.

Comparison 3 Number of withdrawals due to adverse events, Outcome 5 Withdrawals due to serious adverse events over 12 months.
Data from one trial were available to calculate the number of participants who withdrew due to adverse events during the first six months (Fox 2014): the RR was significantly higher for fingolimod 0.5 mg compared to other DMDs (RR 3.21, 95% CI 1.16 to 8.86) (Analysis 3.1).
3.1. Analysis.

Comparison 3 Number of withdrawals due to adverse events, Outcome 1 Withdrawals due to adverse events over 6 months.
Data from the same trial showed that the number of participants who withdrew due to serious adverse events were higher with fingolimod 0.5 mg compared to other DMDs (Fox 2014), but the result was not significant (RR 2.71, 95% CI 0.83 to 8.88) (Analysis 3.4).
Detailed descriptive data on the type of adverse and serious adverse events are provided in Figure 4 and Figure 5, respectively. The higher incidence of adverse events in fingolimod versus interferon beta‐1a, suggests lower tolerability for fingolimod. It should be noted that hypertension was more frequent in participants taking fingolimod, and depression was more frequent in the interferon beta‐1a group.
Secondary outcomes
The annualised relapse rate was evaluated by one trial at 12 months (Cohen 2010). A significant benefit for fingolimod 0.5 mg (RR 0.48, 95% CI 0.34 to 0.70) and fingolimod 1.25 mg (RR 0.61, 95% CI 0.47 to 0.78) doses compared to intramuscular interferon beta‐1a was observed (Analysis 4.2).
4.2. Analysis.

Comparison 4 Annualised relapse rate, Outcome 2 At 12 months.
The number of participants free from MRI gadolinium‐enhancing lesions at 12 months was evaluated by the same trial (Cohen 2010); a slight advantage for fingolimod 0.5 mg (RR 1.12, 95% CI 1.05 to 1.19) and fingolimod 1.25 mg (RR 1.13, 95% CI 1.06 to 1.20) compared to intramuscular interferon beta‐1a was observed (Analysis 5.2).
The mean change of MRI T2‐weighted lesion load at 12 months was evaluated by the same trial (Cohen 2010); we found no significant reduction for fingolimod 0.5 mg (MD ‐0.50, 95% CI ‐6.32 to 5.32) and fingolimod 1.25 mg (MD ‐3.70, 95% CI ‐9.18 to 1.78) doses compared to intramuscular interferon beta‐1a (Analysis 6.1).
Quality of life was measured at six months by the FS36 questionnaire in participants treated with fingolimod 0.5 mg compared to DMDs (Fox 2014). The results favoured fingolimod (Analysis 7.1).
Discussion
Summary of main results
Six RCTscontributed to this review. The overall population included 5152 participants; 3531 were randomly assigned to fingolimod (2061 of them treated with the approved dose of 0.5 mg daily) and 1621 controls, 923 treated with placebo and 698 with intramuscular interferon beta‐1a or other DMDs. The treatment duration was six months in three trials (Fox 2014; Kappos 2006; Saida 2012),12 months in one trial (Cohen 2010a), and 24 months in two trials (Calabresi 2014; Kappos 2010). All studies were sponsored by Novartis Pharma.
Fingolimod administered at the approved dose of 0.5 mg orally once‐daily is effective in increasing the number of people free from relapse (moderate quality of evidence) and from MRI gadolinium‐enhancing lesions (low quality of evidence), and in reducing the annualised relapse rate (moderate quality of evidence) at 24 months compared to placebo. A benefit on prevention of disability worsening was not observed (low quality of evidence). The risk of withdrawal due to adverse events was not significant (very low quality of evidence) (Table 1). Furthermore, MRI T2‐weighted lesion load changes at 12 and 24 months favour fingolimod versus placebo.
A slight advantage of fingolimod at a dose of 0.5 mg compared to intramuscular interferon beta‐1a in increasing freedom from relapse and from gadolinium‐enhancing lesions at 12 months was observed (moderate quality of evidence). A benefit was found for other measures of clinical activity (relapse rate: moderate quality of evidence). No difference between the two active treatments was found regarding prevention of disability worsening at 12 months (low quality of evidence). The risk of withdrawal due to adverse events was not significant (moderate quality of evidence) (Table 2) despite a worst tolerability due to the higher incidence of adverse events. Furthermore, no difference was observed regarding the mean change of MRI T2‐weighted lesion load at 12 months.
Overall completeness and applicability of evidence
All studies assessed the clinical and MRI outcomes selected for this review, except Fox 2014 who included only patient‐oriented outcomes. Participants were adults with relapsing‐remitting multiple sclerosis (RRMS), a high prestudy relapse frequency, low disability and disease duration over five years (Table 4).
The following four points, relevant for clinical practice, should be highlighted:
Measures of treatment response.
Worsening of disability.
Comparison with other active treatments.
Impact on quality of life.
The assessment of disease activity has been recently incorporated to redefine MS disease phenotypes (Lublin 2014). On the other hand, treatment expectations have evolved to include potential remission from MS symptoms to freedom from disease activity. We have chosen the number of participants without clinical (relapse and worsening of disability) and MRI (gadolinium‐enhancing lesions) events, as separate outcome measures. This is to underline the possible benefit from treatment. We have included a further MRI endpoint (total volume of abnormal T2‐hyperintensity) as a marker of disease severity (Lavery 2014). We have not included composite measures, such as, 'no evidence of disease activity' (NEDA), as they are not yet accepted as a primary outcome, and are of uncertain prognostic value (Giovannoni 2015; Rotstein 2015). Only post–hoc analysis concerning NEDA on pooled fingolimod trial populations has been published (Kappos 2015b; Nixon 2014), and useful data in the primary studies were lacking.
We did not find substantial evidence that fingolimod modified the risk of disability worsening, in spite of efficacy for inflammatory disease activity. We considered disability worsening as confirmed at six months follow‐up. Using less stringent criteria (three months), Calabresi 2014 and Kappos 2010 found that fingolimod was effective over the 24‐month period; however, this result may be related to reversible relapse‐related disability accrual. Dissociation between relapse and disability worsening is well documented during the natural course of the disease (Scalfari 2010; Scalfari 2013). On the other hand, meaningful changes might be difficult to detect using the outcomes currently approved to measure clinical disability, mainly in the early stages of the disease, and after a short treatment period (Cohen 2012; Lavery 2014). A divergent effect might also occur when different pathogenetic mechanisms occur (degenerative and inflammatory). In fact, no effect on progressive MS has been reported (Lublin 2016; Novartis 2016).
We only included one comparison concerning clinical and MRI outcomes in this review, i.e. fingolimod versus intramuscular interferon beta‐1a. We will evaluate ongoing trials of fingolimod compared to other approved DMDs in future updates of the review.
We did not find any benefit for fingolimod for quality of life in people with RRMS over two years of therapy, in comparison to placebo (Analysis 7.2). This result is relevant considering the expectation of oral therapy after the era of injectable drugs; following the introduction of fingolimod, patients with RRMS have switched to this drug rather than any other therapies, and they did so for convenience (Warrender‐Sparkes 2015). Type of scales and assessment methods used to measure quality of life in the included studies might have influenced the estimated effect of fingolimod for this outcome (Table 3).
The following limitations of the review need to be highlighted: .
The duration of the included trials was equal to or less than 24 months. Extension studies have been reported, but in most of them placebo‐treated participants switched to the fingolimod group. We excluded open‐label extension studies in this review.
The influence of patients' baseline characteristics on the effect of fingolimod was not explored due to lack of useful data in the included trials. Contradictory data are available in the literature. For example, one article reported that response to fingolimod was associated with patients' baseline high relapse frequency (Oh J 2013). A post‐hoc subgroup analysis of the FREEDOM study (Kappos 2010), reported lack of benefit of fingolimod on the relapse rate for people with MS older than 40 years and on disability worsening for those with baseline expanded disability status scale (EDSS) less than 3.5 score (Devonshire 2012). Fingolimod was found to be superior to intramuscular interferon beta‐1a in all subgroups defined by demographic factors or baseline disease characteristics (Cohen 2013). Another study reported that fingolimod demonstrated similar efficacy in patients with RRMS regardless of prior treatment history (Kremenchutzky 2014).
People with systemic disorders were excluded in all included trials, while in the real world comorbidity is frequent. Precautions and new recommendations need to be followed before and after starting treatment, or when restarting treatment with fingolimod (EMA 2011; EMA 2015).
-
We did not plan to explore safety outcomes beyond the duration of primary RCTs. The Food and Drug Administration (FDA) required planning of two studies (FDA 2010):
A post‐marketing observational prospective, parallel cohort study in relapsing multiple sclerosis patients to assess the potentially serious risk of: eye toxicity, cardiac and vascular toxicity, pulmonary toxicity, seizures, serious and opportunistic infections, malignancies, liver toxicity and atypical multiple sclerosis relapse. Specific outcomes examined should include, but not be limited to, macular oedema, symptomatic bradycardia, second and third degree atrioventricular block, and lymphoma. Final Report Submission: 15 December, 2020.
Develop and maintain a prospective, observational pregnancy exposure registry study conducted in the United States. Final Report Submission: 31 October, 2017.
At 31 October, 2011, 89 pregnancies were reported in completed or ongoing clinical studies, with 74 pregnancies in fingolimod treatment arms. Of 66 pregnancies with in utero exposure to fingolimod, there were 28 live births, nine spontaneous abortions, 24 elective abortions (one case each of tetralogy of Fallot, spontaneous intrauterine death, and failure of fetal development), two infants were born with malformations (one with congenital unilateral posteromedial bowing of the tibia and one with acrania). There were five cases (7.6%) of abnormal fetal development in the 66 pregnancies. Considering also the known risk of teratogenicity in animals, women of childbearing potential should use effective contraception during fingolimod therapy and for two months after treatment discontinuation (Karlsson 2014).
Quality of the evidence
We downgraded the quality of the evidence for all included outcomes at 24 months due to significant differences in reasons of incomplete outcome data between fingolimod 0.5 mg and placebo groups. We further downgraded the quality of evidence for disability worsening, withdrawals due to adverse events, and MRI gadolinium‐enhancing lesions due to insufficient information size and wide confidence intervals. We further downgraded the quality of evidence for withdrawals due to inconsistency. Overall we gave a GRADE rating of moderate for relapses, low for disability progression, very low for withdrawals due to adverse events, and low for MRI gadolinium‐enhancing lesions .
We downgraded the quality of the evidence for all included outcomes at 12 months due to insufficient information size between the fingolimod 0.5 mg and intramuscular interferon beta‐1a groups in Cohen 2010, resulting in moderate quality evidence. We further downgraded the quality of evidence for disability worsening because this study required only three months’ follow‐up to confirm sustained disability worsening. Although we had to accept the definition given in the original paper, we assessed this definition as unreliable in capturing unremitting disability worsening.
Potential biases in the review process
The search strategy for the trials, and contacts initiated with the main investigators, suggest the likelihood that we identified all relevant studies and obtained all relevant data.
Agreements and disagreements with other studies or reviews
Previous reviews on fingolimod as compared with placebo or other approved DMDs in RRMS have been published, but only descriptive data have been reported (Ali 2013; Doggrell 2010; Fox 2012; Gajofatto 2015; Gold 2011; Oh J 2013).
Other reviews have used a structured methodology, including network meta‐analysis, but they have considered a low number of RCTs, with different and limited endpoints. A systematic non‐Cochrane review assessed the efficacy and safety of fingolimod at different doses (Bao 2012); it was based on three RCTs published up to 2010. The results showed that fingolimod was safe and effective in treating RRMS, considering clinical and MRI (incidence rate of so‐called "intense lesion" on T2‐weighted) endpoints.
One review evaluated the comparative effects of fingolimod versus other DMDs using network meta‐analysis. It included ten studies, two of them assessing the effects of fingolimod (Del Santo 2011). The endpoint was limited to the relapse‐free rate at 12 months. The results suggested that fingolimod was superior to interferon beta‐1a (direct comparison) and glatiramer (indirect comparison) in terms of prevention of relapse at 12 months follow‐up.
The results of a Cochrane review evaluating the effect of immunomodulators and immunosuppressants in people with RRMS using network meta‐analysis, demonstrated that alemtuzumab, natalizumab, and fingolimod are the best choices against the recurrence of relapses, but the evidence was limited to the first 24 months of follow‐up (Tramacere 2015).
Authors' conclusions
Implications for practice.
The results of this review showed that fingolimod is a useful treatment of people with RRMS, because of its efficacy in the prevention of disease activity compared to placebo, although the benefit in terms of preventing disability worsening remains unclear. The direct comparison with other approved first‐line DMDs, in particular intramuscular interferon beta‐1a, indicates a higher benefit of fingolimod in terms of relapse prevention, but a significant risk of discontinuation in the first months of treatment. A higher incidence of adverse events was found, suggesting lower tolerability for fingolimod versus interferon beta‐1a, requiring careful monitoring over time.
However, the data were inadequate, for the low number of head‐to‐head RCTs and types of comparisons, with short follow‐up duration.
Implications for research.
There is a need to further explore the long‐term benefit and safety profile of fingolimod considering the risk of progressive multifocal leukoencephalopathy (FDA 2015), and basal cell carcinoma potentially associated with fingolimod therapy (EMA 2015). Head‐to‐head trials comparing fingolimod with other DMDs, and the evaluation of drug effects using other outcome measures, including disease activity freedom (Giovannoni 2015), are useful to guide clinicians to personalise patient treatment. The ongoing, and as yet unpublished trials, will possibly satisfy these issues.
What's new
| Date | Event | Description |
|---|---|---|
| 9 June 2015 | Amended | The author team has been amended |
Acknowledgements
We wish to thank: Dr. Anas Shaneh Saz, Rim Hasan and Suleiman Kojan for their contribution in writing the review protocol. We also thank Andrea Fittipaldo, Trials Search Co‐ordinator, for support provided in paper retrieval, and Sara Nuzzo for helping with data extraction of primary studies. We are grateful to Liliana Coco for valuable and helpful technical assistance provided, as well as for support in writing the review. We are grateful to Prof. Bianca Weinstock‐Guttman for valuable and helpful comments.
Appendices
Appendix 1. Keywords
{fingolimod} OR {FTY720} OR {FTY 720} OR {fingolimod hydrochloride} OR {FTY‐720} OR {2‐amino‐2‐(2‐(4‐octylphenyl)ethyl)‐1,3‐propanediol hydrochloride} OR {Gilenya} OR {sphingosine‐fosphate‐receptor antagonist} AND {relapsing remitting} OR {relapsing‐remitting }OR {remitting‐relapsing}OR {remitting relapsing}
Data and analyses
Comparison 1. Participants free from relapse.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 At 6 months | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Fingolimod 0.5 mg versus placebo | 1 | 114 | Risk Ratio (M‐H, Random, 95% CI) | 1.22 [0.96, 1.54] |
| 1.2 Fingolimod 1.25 mg versus placebo | 2 | 299 | Risk Ratio (M‐H, Random, 95% CI) | 1.27 [1.11, 1.45] |
| 1.3 Fingolimod 5.0 mg versus placebo | 1 | 184 | Risk Ratio (M‐H, Random, 95% CI) | 1.30 [1.10, 1.53] |
| 2 At 12 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Fingolimod 0.5 mg versus interferon beta‐1a | 1 | 860 | Risk Ratio (M‐H, Random, 95% CI) | 1.18 [1.09, 1.27] |
| 2.2 Fingolimod 1.25 mg versus interferon beta‐1a | 1 | 851 | Risk Ratio (M‐H, Random, 95% CI) | 1.15 [1.06, 1.24] |
| 3 At 24 months | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 Fingolimod 0.5 mg versus placebo | 2 | 1556 | Risk Ratio (M‐H, Random, 95% CI) | 1.44 [1.28, 1.63] |
| 3.2 Fingolimod 1.25 mg versus placebo | 2 | 1572 | Risk Ratio (M‐H, Random, 95% CI) | 1.51 [1.29, 1.76] |
Comparison 2. Participants free from disability worsening.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 At 12 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Fingolimod 0.5 mg versus interferon beta‐1a | 1 | 860 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.99, 1.06] |
| 1.2 Fingolimod 1.25 mg versus interferon beta‐1a | 1 | 851 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.98, 1.05] |
| 2 At 24 months | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Fingolimod 0.5 mg versus placebo | 2 | 1556 | Risk Ratio (M‐H, Random, 95% CI) | 1.07 [1.02, 1.11] |
| 2.2 Fingolimod 1.25 mg versus placebo | 2 | 1572 | Risk Ratio (M‐H, Random, 95% CI) | 1.08 [1.03, 1.12] |
Comparison 3. Number of withdrawals due to adverse events.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Withdrawals due to adverse events over 6 months | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Fingolimod 0.5 mg versus placebo | 1 | 114 | Risk Ratio (M‐H, Random, 95% CI) | 2.0 [0.53, 7.61] |
| 1.2 Fingolimod 0.5 mg versus DMDs | 1 | 1028 | Risk Ratio (M‐H, Random, 95% CI) | 3.21 [1.16, 8.86] |
| 1.3 Fingolimod 1.25 mg versus placebo | 2 | 298 | Risk Ratio (M‐H, Random, 95% CI) | 1.60 [0.63, 4.03] |
| 1.4 Fingolimod 5.0 mg versus placebo | 1 | 187 | Risk Ratio (M‐H, Random, 95% CI) | 1.98 [0.62, 6.35] |
| 2 Withdrawals due to adverse events over 12 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Fingolimod 0.5 mg versus interferon beta‐1a | 1 | 860 | Risk Ratio (M‐H, Random, 95% CI) | 1.51 [0.81, 2.80] |
| 2.2 Fingolimod 1.25 mg versus interferon beta‐1a | 1 | 851 | Risk Ratio (M‐H, Random, 95% CI) | 2.69 [1.54, 4.72] |
| 3 Withdrawals due to adverse events over 24 months | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 Fingolimod 0.5 mg versus placebo | 2 | 1556 | Risk Ratio (M‐H, Random, 95% CI) | 1.42 [0.89, 2.25] |
| 3.2 Fingolimod 1.25 mg versus placebo | 2 | 1572 | Risk Ratio (M‐H, Random, 95% CI) | 1.93 [1.48, 2.52] |
| 4 Withdrawals due to serious adverse events over 6 months | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 Fingolimod 0.5 mg versus placebo | 1 | 114 | Risk Ratio (M‐H, Random, 95% CI) | 1.67 [0.42, 6.65] |
| 4.2 Fingolimod 0.5 mg versus DMDs | 1 | 1028 | Risk Ratio (M‐H, Random, 95% CI) | 2.71 [0.83, 8.88] |
| 4.3 Fingolimod 1.25 mg versus placebo | 2 | 298 | Risk Ratio (M‐H, Random, 95% CI) | 2.36 [0.99, 5.66] |
| 4.4 Fingolimod 5.0 mg versus placebo | 1 | 187 | Risk Ratio (M‐H, Random, 95% CI) | 2.77 [1.04, 7.38] |
| 5 Withdrawals due to serious adverse events over 12 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 Fingolimod 0.5 mg versus interferon beta‐1a | 1 | 860 | Risk Ratio (M‐H, Random, 95% CI) | 1.21 [0.72, 2.02] |
| 5.2 Fingolimod 1.25 mg versus interferon beta‐1a | 1 | 851 | Risk Ratio (M‐H, Random, 95% CI) | 1.85 [1.15, 2.96] |
| 6 Withdrawals due to serious adverse events over 24 months | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 Fingolimod 0.5 mg versus placebo | 2 | 1556 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.55, 1.50] |
| 6.2 Fingolimod 1.25 mg versus placebo | 2 | 1572 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.74, 1.29] |
Comparison 4. Annualised relapse rate.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 At 6 months | 2 | Rate Ratio (Random, 95% CI) | Subtotals only | |
| 1.1 Fingolimod 0.5 mg versus placebo | 1 | Rate Ratio (Random, 95% CI) | 0.51 [0.26, 0.99] | |
| 1.2 Fingolimod 1.25 mg versus placebo | 2 | Rate Ratio (Random, 95% CI) | 0.44 [0.28, 0.70] | |
| 1.3 Fingolimod 5.0 mg versus placebo | 1 | Rate Ratio (Random, 95% CI) | 0.47 [0.26, 0.83] | |
| 2 At 12 months | 1 | Rate Ratio (Random, 95% CI) | 0.56 [0.46, 0.69] | |
| 2.1 Fingolimod 0.5 mg versus interferon beta‐1a | 1 | Rate Ratio (Random, 95% CI) | 0.48 [0.34, 0.70] | |
| 2.2 Fingolimod 1.25 versus interferon beta‐1a | 1 | Rate Ratio (Random, 95% CI) | 0.61 [0.47, 0.78] | |
| 3 At 24 months | 2 | Rate Ratio (Random, 95% CI) | Subtotals only | |
| 3.1 Fingolimod 0.5 mg versus placebo | 2 | Rate Ratio (Random, 95% CI) | 0.50 [0.40, 0.62] | |
| 3.2 Fingolimod 1.25 mg versus placebo | 2 | Rate Ratio (Random, 95% CI) | 0.47 [0.38, 0.59] |
Comparison 5. Participants free from gadolinium‐enhancing lesions.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 At 6 months | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Fingolimod 0.5 mg versus placebo | 3 | 1519 | Risk Ratio (M‐H, Random, 95% CI) | 1.42 [1.33, 1.51] |
| 1.2 Fingolimod 1.25 mg versus placebo | 4 | 1674 | Risk Ratio (M‐H, Random, 95% CI) | 1.43 [1.34, 1.53] |
| 1.3 Fingolimod 5 mg versus placebo | 1 | 158 | Risk Ratio (M‐H, Random, 95% CI) | 1.74 [1.35, 2.25] |
| 2 At 12 months | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Fingolimod 0.5 mg versus placebo | 2 | 1343 | Risk Ratio (M‐H, Random, 95% CI) | 1.39 [1.30, 1.48] |
| 2.2 Fingolimod 1.25 mg versus placebo | 2 | 1319 | Risk Ratio (M‐H, Random, 95% CI) | 1.39 [1.30, 1.48] |
| 2.3 Fingolimod 0.5 mg versus interferon beta‐1a | 1 | 728 | Risk Ratio (M‐H, Random, 95% CI) | 1.12 [1.05, 1.19] |
| 2.4 Fingolimod 1.25 mg versus interferon beta‐1a | 1 | 706 | Risk Ratio (M‐H, Random, 95% CI) | 1.13 [1.06, 1.20] |
| 3 At 24 months | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 Fingolimod 0.5 mg versus placebo | 2 | 1226 | Risk Ratio (M‐H, Random, 95% CI) | 1.36 [1.27, 1.45] |
| 3.2 Fingolimod 1.25 mg versus placebo | 2 | 1182 | Risk Ratio (M‐H, Random, 95% CI) | 1.43 [1.33, 1.52] |
Comparison 6. Mean change of MRI T2‐weighted lesion load.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 At 12 months | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.1 Fingolimod 0.5 mg versus placebo | 1 | 733 | Mean Difference (IV, Random, 95% CI) | ‐15.30 [‐24.34, ‐6.26] |
| 1.2 Fingolimod 1.25 mg versus placebo | 1 | 706 | Mean Difference (IV, Random, 95% CI) | ‐16.0 [‐25.23, ‐6.77] |
| 1.3 Fingolimod 0.5 mg versus interferon beta‐1a | 1 | 733 | Mean Difference (IV, Random, 95% CI) | ‐0.5 [‐6.32, 5.32] |
| 1.4 Fingolimod 1.25 mg versus interferon beta‐1a | 1 | 711 | Mean Difference (IV, Random, 95% CI) | ‐3.7 [‐9.18, 1.78] |
| 2 At 24 months | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2.1 Fingolimod 0.5 mg versus placebo | 2 | 1216 | Mean Difference (IV, Random, 95% CI) | ‐20.43 [‐34.03, ‐6.83] |
| 2.2 Fingolimod 1.25 mg versus placebo | 2 | 1171 | Mean Difference (IV, Random, 95% CI) | ‐32.51 [‐40.39, ‐24.62] |
Comparison 7. Quality of life.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 At 6 months | 2 | Mean Difference (Random, 95% CI) | Subtotals only | |
| 1.1 Fingolimod 1.25 mg versus placebo (Hamburg Quality of Life Questionnaire) | 1 | Mean Difference (Random, 95% CI) | ‐0.14 [‐9.13, 8.85] | |
| 1.2 Fingolimod 0.5 mg versus DMDs (Change in FS36 Mental component summary) | 1 | Mean Difference (Random, 95% CI) | 1.8 [0.42, 3.18] | |
| 1.3 Fingolimod 0.5 mg versus DMDs (Change in FS36 Physical component summary) | 1 | Mean Difference (Random, 95% CI) | 1.30 [0.30, 2.30] | |
| 2 At 24 months | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2.1 Fingolimod 0.5 mg versus placebo (Euro quality of life scale) | 1 | 713 | Mean Difference (IV, Random, 95% CI) | ‐0.01 [‐0.04, 0.02] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Calabresi 2014.
| Methods | Randomised, double‐blind, placebo‐controlled parallel groups, phase III trial Multicentre (117 centres) performed across eight countries, predominantly in USA (101), 1 centre in Australia, 1 centre in Austria, 2 centres in Canada, 3 centres in Poland, 3 centres in Romania, 5 centres in Turkey, 1 centre in United Kingdom Duration: 24 months Enrollment: from June 2006 to March 2009 Acronym: FREEDOMS II |
|
| Participants | 1083 participants with RRMS Inclusion criteria: 1. 18 to 55 years of age 2. Diagnosis of RRMS according to the 2005 revised McDonald criteria (Polman 2005) 3. One or more confirmed relapses during the preceding year (or two or more confirmed relapses during the previous two years) 4. EDSS score between 0 to 5.5 points (Kurtzke 1983) 5. No relapse or steroid treatment within 30 days before randomisation 6. Both treatment‐naive and previously treated people 7. Previously treated participants were eligible if interferon beta or glatiramer acetate therapy was stopped at least three months before randomisation and natalizumab treatment at least six months before randomisation Exclusion criteria: 1. Clinically significant systemic disease 2. Immune suppression (drug‐induced or disease induced) 3. Active infection or macular oedema, diabetes mellitus 4. History of malignancy (apart from successfully treated basal or squamous‐cell skin carcinoma) 5. Participants with specific cardiac, pulmonary, or hepatic disorders 6. Varicella ZV IgG antibody negative (Calabresi 2014, Supplementary web appendix) |
|
| Interventions | Participants were randomly allocated to one of the three groups: 1. Fingolimod 0.5 mg orally once‐daily (358 participants) 2. Fingolimod 1.25 mg orally once‐daily (370 participants) 3. Placebo orally once‐daily (355 participants) After review of data from the FREEDOMS and TRANSFORMS studies, on Nov 12, 2009, participants treated with the 1.25 mg dose, owing to the absence of clear added benefits and a higher risk for safety events such as infections and macular oedema (Calabresi 2014, Supplementary web appendix), were subsequently switched to the 0.5 mg dose in a blinded manner |
|
| Outcomes | Primary endpoint was the annualised relapse rate (defined as the number of confirmed relapses) at 24 month period A relapse was confirmed when it was accompanied by an increase of at least half a point on EDSS score, an increase of 1‐point in 2 different functional systems, or 2‐points in 1 functional system (excluding bowel, bladder, or cerebral functional systems) The clinical secondary objectives were as follows. 1. Time to disability progression confirmed at 3 months 2. Time to disability progression confirmed at 6 months Progression was defined as 1‐point EDSS change or 0.5‐point if baseline EDSS was > 5.0 3. Safety and tolerability 4. Time to first relapse 5. Proportion of relapse‐free patients 6. Change from baseline to the end of study on the MSFC score 7. Quality of life using the EQ‐5D and Patient Reported Indices in Multiple Sclerosis (PRIMUS) 8. Fatigue using the Modified Fatigue Impact Scale (MFIS) The MRI secondary objectives were: 1. Percent brain‐volume change from baseline at 24 months 2. Number and volume of gadolinium‐enhancing T1 lesions 3. Number of new or newly enlarged T2 lesions 4. Proportion of participants free of gadolinium‐enhancing T1 lesions 5. Proportion of participants free of new or newly enlarged T2 lesions 6. Proportion of participants free of new inflammatory activity (no gadolinium‐enhancing T1 lesions and no new or newly enlarged T2 lesions) 7. Percentage change from baseline in volume of gadolinium‐enhancing T1 lesions 8. Percentage change from baseline in volume of new or newly enlarged T2 lesions 9. Percentage change from baseline in brain volume |
|
| Notes | The trial was registered with clinicaltrials.gov, number NCT00355134 The study was sponsored by Novartis Pharma |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "randomly allocated patients (1:1:1; stratified by study centre). The randomisation sequence was generated with an automated system" pg 546 |
| Allocation concealment (selection bias) | Unclear risk | "the randomisation sequence was generated with an automated system" under the supervision of the Novartis Drug Supply Management team" pg 546 |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "both Fingolimod and placebo were dispensed in hard gelatin capsules of identical colour and size and packed in identical bottles" pg 546 |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "The efficacy assessments (i.e., confirmation of relapses, scheduled EDSS, and Multiple Sclerosis Functional Composite [MSFC] were done by an independent, specially trained, and certified assessor not otherwise involved in the treatment of patient"."In order to maintain the blind, efficacy assessments (i.e., scheduled EDSS and confirmation of relapses) were performed by an independent evaluating physician, not involved with any other aspects of subject care and management. Patients were instructed not to discuss adverse events with the independent evaluating physician" (Supplementary web appendix) All MRI scans were centrally reviewed by an independent radiologist (E‐WR) unaware of treatment allocation" |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Overall, 28.2% (305/1083) discontinued the study [32.2% (119/370) in fingolimod 1.25 mg, 24.0% (86/358) in fingolimod 0.5 mg, and 28.2% (100/ 355) in placebo], with some significant differences in reasons: unsatisfactory therapeutic effect (2.7% in fingolimod 1.25 mg, 1.7% in fingolimod 0.5 mg, and 4.8% in placebo) and adverse events or abnormal laboratory values (12.7% in fingolimod 1.25 mg, 10.1% in fingolimod 0.5 mg, and 5.1% in placebo) |
| Selective reporting (reporting bias) | Unclear risk | Differences between study design described in the article (protocol not available) and reported findings were not found. After 9 months from enrolment conclusion, participants treated with 1.25 mg were shifted to 0.5 mg. The number of participants shifted to 0.5 mg and the treatment duration were unknown |
| Other bias | High risk | The study was sponsored by Novartis Pharma, "The study sponsor participated in the design of the study, conduct of the study, data collection, data management, data analysis and interpretation, and preparation, review, and approval of the paper" pg 550, and four co‐authors of the published paper were affiliated to the pharmaceutical company |
Cohen 2010.
| Methods | Randomised, double‐blind, 3 parallel group phase III trial Multicentre (172 centres) performed in 18 countries (Argentina, Australia, Austria, Belgium, Brazil, Canada, Egypt, France, Germany, Greece, Hungary, Italy, Republic of Korea, Portugal, Spain, Switzerland, UK, USA). Duration: 12 months Enrollment: from May 2006 to September 2007 Acronym: TRANSFORMS |
|
| Participants | 1292 participants with RRMS Inclusion criteria: 1. 18 to 55 years of age 2. Relapsing‐remitting course 3. At least 1 documented relapse in the previous year or at least 2 documented relapses in the previous 2 years 4. EDSS score between 0 to 5.5 Exclusion criteria: 1. A documented relapse or corticosteroid treatment within 30 days before randomisation 2. Active infection 3. Macular edema 4. Immune‐suppression (either drug‐ or disease‐induced) 5. Clinically significant coexisting systemic disease Previous disease‐modifying therapy was not considered an exclusion criteria. The percentage of previously treated participants was 56.7%, in details 56.3% in interferon beta‐1a group, 55.2% in fingolimod 0.5 mg and 58.5% in fingolimod 1.25 mg. Most of them were treated with any interferon beta. Glatiramer acetate was previously administered in 15.7%, 13.1% and 15.4%, and natalizumab in 0.2%, 0.9% and 0.7% respectively |
|
| Interventions | Participants were randomly allocated to one of the three groups: 1. Fingolimod 1.25 mg orally once‐daily (426 participants) 2. Fingolimod 0.5 mg orally once‐daily (431 participants) 3. Interferon beta‐1a 30 ug intramuscularly once a week (435 participants) |
|
| Outcomes | Primary endpoint was the annualised relapse rate (defined as the number of confirmed relapses) at 12‐month period Relapse was defined as new, worsening, or recurrent neurologic symptoms that occurred at least 30 days after the onset of a preceding relapse, that lasted at least 24 hours without fever or infection, and that were accompanied by an increase of at least half a point on EDSS or an increase of at least 1‐point in 2 functional systems scores or of at least 2‐points in 1 functional system score (excluding changes in bowel or bladder function and cognition) Secondary endpoints: 1. Number of new or enlarged lesions on T2‐weighted MRI scans at 12 months 2. Time to confirmed disability progression Progression of disability was defined as a 1‐point increase in EDSS score (or a half point increase for participants with a baseline score 5.5) that was confirmed 3 months later in the absence of relapse |
|
| Notes | The trial was registered with clinicaltrials.gov, number: NCT00340834 The study was sponsored by Novartis Pharma |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Randomisation was performed in blocks of six within each site and was stratified according to site" pg 403 |
| Allocation concealment (selection bias) | Low risk | "Randomisation was performed centrally" and "Study‐group assignments were performed with the use of an interactive voice‐response system" pg 403 |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "During the trial, patients, study personnel, steering‐committee members, and the study statistician were unaware of study‐group assignments and leukocyte counts. Capsules, syringes, and packaging materials for active and placebo treatments were indistinguishable" pg 404 |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "At each site, a treating neurologist supervised medical management", "Patients were instructed to cover injection sites at visits, to not to discuss adverse events with clinical evaluators", and "Potential relapses triggered an unscheduled visit and were confirmed by the treating neurologist on the basis of blinded examination by the examining neurologist" pg 403‐404
The treating neurologist was possibly not blinded. Moreover, it is not clear how and when the examining neurologist evaluated the potential relapse MRI evaluators were unaware of study group assignment and leukocyte counts. An independent data and safety monitoring board evaluated overall safety in the fingolimod phase 3 program |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Overall, 10.8% (139/1292) discontinued the study [13.4% (57/426) in fingolimod 1.25 mg, 7.7% (33/43) in fingolimod 0.5 mg, and 11.3% (49/435) in interferon beta‐1a)], with some non‐significant differences in reasons: unsatisfactory therapeutic effect (0.7% in fingolimod 1.25 mg, 0.7% in fingolimod 0.5 mg, and 1.6% in interferon beta‐1a) adverse events (6.1% in fingolimod 1.25 mg, 2.1% in fingolimod 0.5 mg, and 2.1% in interferon beta‐1a) and abnormal laboratory values (0.9% in fingolimod 1.25 mg, 1.4% in fingolimod 0.5 mg, and 0.2% in interferon beta‐1a) |
| Selective reporting (reporting bias) | Low risk | Additional MRI data from the key endpoints were reported (protocol not available) |
| Other bias | High risk | The study was sponsored by Novartis Pharma, "data were analysed by the sponsor" pg 403, and five co‐authors of the published paper were affiliated to the pharmaceutical company |
Fox 2014.
| Methods | Randomised active comparator, parallel group, open‐label, phase IV trial
Multicentre study (157), 152 centres in the USA and 6 centres in Canada Duration: 6 months Enrollment: between August 2010 and August 2012. Acronymus: EPOC |
|
| Participants | 1053 participants with RRMS Inclusion criteria 1. 18 to 65 years of age 2. Diagnosis of relapsing MS in accordance with the 2005 McDonald criteria (Polman 2005) 3. EDSS score between 0 to 5.5 4. Treated with an injectable (DMD for at least 6 months before screening 5. Participants must be candidates for a change in therapy as determined by the treating physician 6. Treatment‐naïve to fingolimod Exclusion criteria 1. History of chronic disease of the immune system (except for MS) 2. Immunodeficiency 3. Malignancy other than localized basal cell carcinoma within the past 5 years 4. Cardiac arrest, myocardial infarction, ischaemic heart disease, or coronary spasm within 6 months 5. Mobitz type II second‐degree heart block, third‐degree atrioventricular block, or an increased QTc interval (4470 ms) 6. Uncontrolled diabetes mellitus (glycated haemoglobin > 7%) 7. Bone marrow transplant 8. Alcohol abuse within the past 5 years 9. Macular edema present at screening 10. Negative test for varicella zoster immunoglobulin G antibodies 11. Positive tests for hepatitis B, hepatitis C, or HIV 12. Active systemic bacterial, viral or fungal infections, tuberculosis 13. Pregnancy 14. Uncontrolled or poorly controlled cardiovascular and pulmonary disorders (hypertension or asthma; cardiac failure; severe respiratory disease or pulmonary fibrosis) 15. Chronic liver or biliary disease 16. Previous treatment with immunosuppressants, immunoglobulins, or monoclonal antibodies within 6 months before screening; any live or live attenuated vaccines within 1 month before screening; cladribine, cyclophosphamide, or mitoxantrone at any time; and class Ia or class III antiarrhythmic drugs at time of screening Participants randomly assigned to treatment with fingolimod changed from their pre‐randomisation DMD with no washout period. Participants randomised to the DMD group chose to either remain on the same therapy or, following a consultation with a physician, switch immediately to another approved DMD |
|
| Interventions | Participants were randomly allocated to one of the two groups: 1. Fingolimod 0.5 mg orally once‐daily (790 participants) 2. DMD (263 participants): interferon beta 1‐b (Extavia® or Betaseron®) 0.25 mg injected subcutaneously every other day (46 participants); interferon beta 1‐a (Avonex®)30 μg intramuscular injected once a week (60 participants); interferon beta‐1a (Rebif®) 22 μg or 44 μg injected subcutaneously three times a week (65 participants); or glatiramer acetate (Copaxone®) 20 mg injected subcutaneously once‐daily (92 participants) |
|
| Outcomes | Primary endpoint was to evaluate differences in satisfaction as measured by the Global Satisfaction subscale score on the Treatment Satisfaction Questionnaire for Medication (Atkinson 2004) Secondary objectives were 1, Effectiveness 2. Side effects 3. Fatigue 4. Depression 5. Activities of daily living 6. Health‐related QOL measured using the 36‐item Short‐Form Health Survey v2 (SF‐36 v2) (Jenkinson 1999) |
|
| Notes | The trial was registered with clinicaltrials.gov, number: NCT01216072 Four co‐authors of the published paper were affiliated to the pharmaceutical company (Novartis) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Patients were randomised using an interactive voice response system (IVRS) to either the once‐daily fingolimod (FTY720; GilenyaTM, Novartis Pharma AG, Basel, Switzerland) 0.5 mg arm or the injectable DMT arm, in a 3:1 ratio. A patient randomisation list was produced by an interactive voice response system using a validated system that automated the random assignment of patient numbers to the different treatment arms" pg 5 |
| Allocation concealment (selection bias) | Low risk | "A patient randomisation list was produced by the IVRS using a validated system that automated the random assignment of patient numbers to the different treatment arms" |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | "Fingolimod capsules (0.5 mg) were supplied, packaged, and labelled in accordance with the US Code of Federal Regulations governing the handling of investigational treatments. The capsules were dispensed by the study physician and supplied by Novartis Drug Supply Management. Patients randomised to the DMT group could choose to either remain on the same therapy" |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | The study was open‐ label |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Overall, 10.4% (110/1053) discontinued the study (9.6% ‐ 76/790 ‐ in fingolimod 0.5 mg, and 12.9% ‐ 34/263 ‐ in DMD), with some significant differences in reasons: unsatisfactory therapeutic effect (0.4% in fingolimod 0.5 mg, and 1.5% in DMD), adverse events (5.3% in fingolimod 0.5 mg, and 1.5% in DMD group) |
| Selective reporting (reporting bias) | Low risk | Inclusion and exclusion criteria were detailed in the protocol Cascione 2013 and summarised in the published primary study (Fox 2014). Missing data (SD of QOL values) request to the Authors (12 March 2015) were not provided |
| Other bias | High risk | The study was sponsored by Novartis Pharma, and 4 co‐authors of the published paper were affiliated to the pharmaceutical company. The criteria for changing treatment were undefined " Participants must be candidates for a change in therapy as determined by the treating physician. Participants randomised to the DMD group could have choose to either remain on the same therapy or, following a consultation with a physician, switch immediately to another approved DMD " |
Kappos 2006.
| Methods | Randomised, double‐blind, 3 parallel groups double‐dummy phase II (proof of concept) trial Multicentre (32 centres) performed in Canada and in 10 European countries (Denmark, Finland, France, Germany, Italy, Poland, Portugal, Spain, Switzerland, UK Duration: 6 months Enrollment: from May 2003 to April 2004 Acronym: FTY720 D2201 |
|
| Participants | 281 patients with MS, 246 with RRMS and 31 with SPMS Inclusion criteria: 1. Age 18‐60 years 2. Diagnosis of relapsing multiple sclerosis (Mc Donald 2001) 3. At least one of the following: two or more documented relapses during the previous 2 years, one or more documented relapses in the year before enrolment, and one or more gadolinium‐enhancing lesions detected on magnetic resonance imaging (MRI) at screening. 4. EDSS score between 0 to 6 5. Neurologically stable condition, with no evidence of relapse for at least 30 days before screening and during the screening and baseline phases Exclusion criteria: 1. Use of corticosteroids (within the previous 30 days) 2. Immunomodulatory therapy (within the previous 3 months) 3. Immunosuppressive treatment (azathioprine or methotrexate within 6 months, cyclophosphamide within 12 months, or mitoxantrone or cladribine within 24 months) 4. History of cardiac conditions that might increase the risk of a decrease in heart rate 5. White‐cell count less than 3500 per cubic millimetre 6. Lymphocyte count of less than 800 per cubic millimetre |
|
| Interventions | Participants were randomly allocated to one of the three groups: 1. Fingolimod 5.0 mg orally once‐ daily (94 patients) 2. Fingolimod 1.25 mg orally once‐ daily (94 patients) 3. Placebo orally once‐ daily (93 patients) |
|
| Outcomes | Primary endpoint was the total number of gadolinium‐enhancing lesions per patient recorded on T1‐weighted MRI at monthly intervals for 6 months. The clinical secondary objectives were: 1.Number of participants remaining free of relapse 2. Annualised relapse rate 3. Time to the first relapse Confirmed relapse was defined as the occurrence of new symptoms or worsening of previously stable or improving symptoms and signs not associated with fever, lasting more than 24 hours and accompanied by an increase of at least half a point in EDSS score or 1‐point in the score for at least 1 of the functional system (excluding the bowel and bladder and mental systems) The MRI secondary objectives were: 1. Total volume of gadolinium‐enhancing lesions per patient 2. Proportion of participants with gadolinium‐enhancing lesions 3. Total number of new lesion per patient on T‐weighted images 4. Changes in lesion volume on T2‐weighted images 5. Brain volume from baseline to month 6 |
|
| Notes | The study was sponsored by Novartis Pharma The trial was registered with clinicaltrials.gov,numbers: NCT00333138 (for core study) and NCT00235430 (for the extension) After the core study, participants could continue in the extension study; participants who had received active treatment in the core study continued with the same dose, and those who had received placebo were randomly assigned to receive 1.25 or 5.0 mg of fingolimod. The results have been reported for 227 out of 281 (81%) at 1 year (Kappos 2006), for 189 (67%) at 2 (O'Connor 2009), for 173 (62%) at 3 (Comi 2010), for 140 (49.8%) at 5 (Izquierdo 2013; Montalban 2011b,) for 122 (43.4%) at 7 years (Antel 2012; Montalban 2012) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "In the core study, patients were randomly assigned, in a 1:1:1 ratio, to 1.25 mg of fingolimod, 5.0 mg of fingolimod, or a matching placebo once daily" pg 1125 |
| Allocation concealment (selection bias) | Low risk | "Randomisation was stratified according to disease course (relapsing–remitting or secondary progressive) with the use of a centralized automated system that provided randomised packages of the study drug to each centre" pg 1125 |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "All drugs were given as identical capsules" and "The medication was prepackaged on the basis of a block size of 3; this information was not disclosed to investigators and monitors" pg 1125 and " Laboratory values that might have revealed the treatment assignment (e.g., lymphocyte counts) were not disclosed to treating physicians unless they exceeded prespecified safety limits" pg 1126 |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Information about participants assignments was not disclosed to investigators and monitors, "Relapses were confirmed by the treating physician on the basis of an examination by the EDSS rater who was not otherwise involved in patient care. When warranted, relapses were managed by the treating physician according to a standardized scheme" and "Neurologic assessments were performed by specially trained, independent neurologists who were unaware of the treatment assignments, were not involved in the everyday care of the patients, and had no access to their medical records" pg 1126 |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Overall, 1.4% (4/281) discontinued the study [2.1% (2/94) in fingolimod 5.0 mg, 1.1% (1/94) in fingolimod 1.25 mg, and 1.1% (1/93) in placebo] pg 1128 Reasons for lost‐to follow up were not reported |
| Selective reporting (reporting bias) | Low risk | Quality of life assessment was not registered either among the study endpoints of the published RCT or in the study registration at the clinicaltrials.gov. The data have been published Montalban 2011a |
| Other bias | Low risk | The study was supported by Novartis Pharma, Basel, Switzerland. There are no potential risks for other biases. The steering‐committee members and the sponsors designed the study. The authors had access to all data. An independent external data and safety monitoring board evaluated adverse events and other safety data as well as clinical and MRI efficacy data |
Kappos 2010.
| Methods | Randomised, double‐blind, 3 parallel group placebo controlled phase III trial Multicentre (138 centres) performed in 22 countries (Australia, Belgium, Canada, Czech Republic, Estonia, Finland, France, Germany, Greece, Hungary, Ireland, Israel, Netherlands, Poland, Romania, Russia, Slovakia, South Africa, Sweden, Switzerland, Turkey, UK) Duration: 2 years Enrollment: from January 2006 to August 2007 Acronym: FREEDOMS |
|
| Participants | 1272 participants with RRMS Inclusion criteria: 1. 18 to 55 years of age 2. Diagnosis of multiple sclerosis, according to the revised McDonald criteria (Polman 2005) 3. Relapsing–remitting course 4. One or more documented relapses in the previous year or two or more in the previous 2 years 5. EDSS score between 0 to 5.5 Exclusion criteria: 1. Relapse or corticosteroid treatment within 30 days before randomisation 2. Active infection 3. Macular edema 4. Diabetes mellitus 5. Immune suppression (drug‐ or disease‐induced) 6. Clinically significant systemic disease 7. Interferon beta or glatiramer acetate therapy within 3 months |
|
| Interventions | Participants were randomly allocated to one of the three groups: 1. Fingolimod 0.5 mg orally once‐daily (425) 2. Fingolimod 1.25 mg orally once‐daily (429) 3. Placebo orally once‐daily (418) |
|
| Outcomes | Primary endpoint was the annualised relapse rate (defined as the number of confirmed relapses per year) To constitute a confirmed relapse, the symptoms must have been accompanied by an increase of at least half a point in the EDSS score, of 1 point in each of 2 EDSS functional system scores, or of 2 points in 1 EDSS functional‐system score (excluding scores for the bowel–bladder or cerebral functional systems) The clinical secondary objectives were: 1. Time to the first relapse 2. Time to confirmed disability progression confirmed at 3 months 3. Time to confirmed disability progression confirmed at 6 months Time to confirmed disability progression was defined as an increase of 1‐point in the EDSS score (or half a point if the baseline EDSS score was equal to 5.5), confirmed after 3 months, with an absence of relapse at the time of assessment and with all EDSS scores measured during that time 4. Changes in the EDSS score and MSFC z score between baseline and 24 months 5. Safety and tolerability The MRI secondary objectives were: 1. Number of gadolinium‐enhancing lesions 2. Proportion of participants free from gadolinium‐enhancing lesions 3. Number of new or enlarged lesions on T2‐weighted MRI scans 4. Proportion of participants free from new or enlarged lesions on T2‐weighted scans 5. Volume of hyperintense lesions on T2‐weighted scans 6. Volume of hypointense lesions on T1‐weighted scans 7. Change in brain volume between baseline and 24 months |
|
| Notes | The trial was registered with clinicaltrials.gov number: NCT00289978 The study was sponsored by Novartis Pharma |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Randomization was performed centrally with the use of stratification according to site, with a block size of six within each site" pg 388 |
| Allocation concealment (selection bias) | Low risk | " Patients were randomly assigned, in a 1:1:1 ratio, to receive oral fingolimod capsules in a dose of 0.5 mg or 1.25 mg or matching placebo. Randomisation was performed centrally, with the use of a validated system and stratification according to site, with a block size of six within each site" pg 388 |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Double blind" (pg 388) procedure and methods "to ensure that all assessments remained unbiased regarding the study‐group assignments (i.e., unaffected by awareness of them) were adopted pg 388 |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk |
"To ensure that all assessments remained unbiased regarding the study‐group assignments (i.e., unaffected by awareness of them), an independent examining neurologist determined all the EDSS scores , this examining neurologist or a trained technician administered the Multiple Sclerosis Functional Composite pg 388. Another independent physician monitored patients for 6 or more hours after administration of the first dose of the study drug pg 389. Relapses were verified by the examining neurologist within 7 days after the onset of symptoms" MRI scans were analysed at a central MRI evaluation centre by radiologists who were unaware of the study group assignments and an independent data and safety monitoring board evaluated the safety and overall benefit–risk profiles |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Overall, 18.7% (238/1272) discontinued the study [22.4% (96/429) in fingolimod 1.25 mg, 13.2% (56/425) in fingolimod 0.5 mg, and 20.6% (86/418) in placebo], with some significant differences in reasons: unsatisfactory therapeutic effect (3.0% in fingolimod 1.25 mg, 1.4% in fingolimod 0.5 mg, and 6.0% in placebo) and abnormal laboratory values (4.7% in fingolimod 1.25 mg, 2.1% in fingolimod 0.5 mg, and 0.2% in placebo) |
| Selective reporting (reporting bias) | Low risk | Differences between study design described in the article (protocol not available) and reported findings were not found |
| Other bias | High risk | The study was sponsored by Novartis Pharma, "data were analysed by the sponsor" pg 388, and 4 co‐authors of the published paper were affiliated to the pharmaceutical company |
Saida 2012.
| Methods | Randomised, double‐blind, parallel group phase II trial Multicentre (43 centres) performed in Japan Duration: 6 months Enrollment: from October 2007 to February 2010 Acronym: none |
|
| Participants | 171 Japanese participants with MS, 167 with RRMS and 4 with SPMS Inclusion criteria: 1. 18–60 years of age 2. Diagnosis of MS according to the revised McDonald criteria (Polman 2005) 3. Relapsing course of the disease (relapsing–remitting or secondary progressive) 4. One or more relapses in the previous year or 2 or more relapses in the previous two years or at least one gadolinium enhanced T1‐weighted brain lesion within the 30 days prior to study commencement 5. EDSS score between 0 to 6 6. At least one T2‐weighted brain lesion Exclusion criteria 1. Long cord lesions of at least three vertebral segments on spinal MRI 2. Primary progressive MS 3. Relapse or corticosteroid treatment within 30 days before randomisation 4. Malignancy 5. Macular oedema 6. Diabetes mellitus 7. Active infection 8. Clinically significant systemic disease 9. Pregnancy 10. Received cladribine, cyclophosphamide, mitoxantrone, or other immunosuppressive or immunoglobulin medication in the six months prior to randomisation, or who had had plasmapheresis immunoadsorption or interferon beta therapy in the three months prior to randomisation 11. History of cardiac disorder including arrhythmia 12. Pulmonary condition including asthma 13. Leukopenia less than 3500 cell/mm3 or lymphocyte count of less than 800 14. Abnormal liver enzyme 15. Negative for varicella zoster at screening 16. Received any live or live attenuated vaccines |
|
| Interventions | Participants were randomly allocated to one of the three groups: 1. Fingolimod 0.5 mg orally once‐daily (57) 2. Fingolimod 1.25 mg orally once‐daily (57) 3. Placebo orally once‐daily (57) Relapses were treated with methylprednisolone up to 1000 mg/day for 3–5 days without an oral taper |
|
| Outcomes | Primary endpoint was the percentage of participants free from gadolinium‐enhancing lesions at 3 and 6 months Secondary endpoints were 1. Percentage of participants free from relapses over six months 2. Safety measures Confirmed relapse was defined as new, worsening, or recurrent neurological symptoms that occurred at least 30 days after the onset of a preceding relapse, lasted at least 24 hours without fever or infection and were accompanied by an increase of at least half a point in EDSS score or an increase of at least 1‐point in 2 functional systems scores or of at least 2‐points in 1 functional system (excluding changes in bowel‐bladder function and cognition) |
|
| Notes | The trial was registered with clinicaltrials.gov number: NCT00537082 The work was supported by Novartis Pharma KK and Mitsubishi Tanabe Pharma Corp., Tokyo, Japan |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Patients were randomly assigned, in a 1:1:1 ratio, to receive once‐daily fingolimod capsules, 0.5 mg or 1.25 mg, or matching placebo for six months" pg 2 |
| Allocation concealment (selection bias) | Low risk | Randomisation was performed by a central centre (Bellsystem 24 Inc., Tokyo), with the use of a validated system that assigned randomisation numbers to participants and automated the dynamic allocation of treatment arms to randomisation numbers. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | The identity of treatments was concealed by the use of study drugs that were identical in appearance, packaging, labelling and schedule of administration. "Patients, investigators, site personnel, first‐dose administrators remained blinded during the six‐month core study" pg 2 |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "MRI evaluators and data analysts remained blinded during the six‐month core study" pg 2 |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Overall, 14.0% (24/171) discontinued the study [15.8% (9/57) in fingolimod 1.25 mg, 15.8% (9/57) in fingolimod 0.5 mg, and 10.5% (6/57) in placebo] with some non‐significant differences in reasons: adverse events (10.5% in fingolimod 1.25 mg, 10.5% in fingolimod 0.5 mg, and 5.3% in placebo) unsatisfactory therapeutic effect (0% in fingolimod 1.25 mg, 0% in fingolimod 0.5 mg, and 3.5% in placebo) |
| Selective reporting (reporting bias) | Low risk | All relevant study endpoints were reported. Protocol was not available |
| Other bias | Low risk | The study was sponsored by Novartis Pharma, and 3 co‐authors of the published paper were affiliated to the pharmaceutical company. The inclusion criteria of participants are unclear (number at least one T2‐weighted brain lesion and diagnosis according to Polman 2005) |
DMD: disease‐modifying drug; EDSS: Expanded Disability Status Scale; EQ‐5D; Euro quality of life scale; IgG: immunoglobulin G; MRI: magnetic resonance imaging; MS: multiple sclerosis; MSFC: Multiple Sclerosis Functional Composite; RRMS: relapsing‐remitting multiple sclerosis; SPMS: secondary progressive multiple sclerosis
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Boulton 2013 | Type of participants: RCT evaluating the effect of fingolimod on pulmonary function in otherwise healthy people with moderate asthma |
| Chinea 2014 | Pooled data of RCTs: subgroup analysis on Hispanic population |
| Comi 2013 | Design: not randomised study (FIRST study) |
| Francis 2014 | Pooled data of RCTs on safety: relationship between lymphocyte counts and infections |
| Gold 2014 | Design: not randomised study on cardiac safety |
| Green 2013 | Type of participants: RCT evaluating acute optic neuritis |
| Havla 2013 | Design: not randomised study on people with relapsing‐remitting MS during the first year after switching from natalizumab to fingolimod |
| Kappos 2014 | Pooled data of RCTs on safety |
| Kappos 2014a | Pooled data of RCTs: subgroup analysis on participants previously treated with glatiramer acetate |
| Kappos 2015 | Type of intervention: RCT evaluating immune response to influenza vaccine administered to people with MS treated with fingolimod or placebo |
| Kappos 2015a | Design: cross‐over study evaluating the optimal timing for initiating fingolimod therapy following natalizumab discontinuation in RRMS. After baseline infusion of Natalizumab, patients were subsequently randomised to one of three treatment groups: a) 8‐week washout (8 weeks no treatment) followed by 24 weeks of treatment with fingolimod, b) 12‐week washout (8 weeks no treatment and 4 weeks placebo) followed by 20 weeks of treatment with fingolimod, or c) 16‐week washout (8 weeks no treatment and 8 weeks placebo) followed by 16 weeks of treatment with fingolimod |
| Karlsson 2014 | Overview of studies reporting outcomes of pregnancies that occurred during fingolimod treatment |
| laroni 2013 | Design: not randomised study on safety of the first dose of fingolimod in people with MS |
| Limmorth 2013 | Design: not randomised study of the cardiac safety profile of the initiation of fingolimod treatment (START study) |
| Lublin 2016 | Type of participants: primary progressive MS (INFORMS study) |
| Nolan 2013 | Design: not randomised study on ophthalmic findings (macular volume) in people with MS |
| Van Lokven 2013 | Design: not randomised study |
| Vollmer 2013 a | Pooled data of RCTs on disease outcome after fingolimod discontinuation |
| Zarbin 2013 | Overview reporting ophthalmic outcomes of people with MS receiving fingolimod |
MS: multiple sclerosis; RCT: randomised controlled trial
Characteristics of studies awaiting assessment [ordered by study ID]
NCT01317004.
| Methods | Randomised study evaluating the change in patient‐reported treatment satisfaction after 6 months of treatment with fingolimod 0.5 mg/day versus DMD standard of care, using the global satisfaction subscale of the Treatment Satisfaction Questionnaire for Medication (TSQM‐9) |
| Participants | RRMS |
| Interventions | Experimental: fingolimod Active comparator: standard therapy |
| Outcomes | Primary outcome measure: change in patient‐reported treatment satisfaction |
| Notes | This study has been completed (June 2014). Partial results on 61 patients of the EPOC study have been reported in the site clinicaltrials.gov (accessed 22 February 2016) |
NCT01333501.
| Methods | An 18‐month, open‐label, rater‐blinded, randomised, multicentre, active‐controlled, parallel group pilot study to assess efficacy and safety of fingolimod in comparison to interferon beta‐1b in treating the cognitive symptoms associated to RRMS and to assess possible relationship of these effects to regional brain atrophy |
| Participants | RRMS |
| Interventions | Experimental: fingolimod Active comparator: interferon beta‐1b |
| Outcomes | Cognitive dysfunction progression |
| Notes | The study has been completed (September 2015) (accessed 22 February 2016) |
NCT01534182.
| Methods | Phase IV, 6‐month, randomised, active comparator, open‐label, multicentre study to evaluate patient outcomes, safety and tolerability of fingolimod 0.5 mg/day in patients with relapsing‐remitting multiple sclerosis who are candidates for MS therapy change from previous DMD |
| Participants | RRMS |
| Interventions | Experimental: fingolimod Active comparator: standard DMD |
| Outcomes | Primary outcome measure: change in patient‐reported treatment satisfaction |
| Notes | This study has been completed (June 2013) and partial results on 298 participants of the EPOC study comparing fingolimod versus Interferon beta‐1a and glatiramer acetate have been reported in the site clinicaltrials.gov (accessed 22 February 2016) |
NCT01623596.
| Methods | Evaluation of patient retention of fingolimod versus currently approved DMD in patients with RRMS (PREFERMS). A 12‐month study where 1000 participants with RRMS will be randomised 1:1 to fingolimod or approved DMD. Participants will be in early stages of the disease and be treatment‐naive or have only been treated with one class of DMD (Interferon beta or glatiramer acetate) for no more than 5 years total exposure |
| Participants | RRMS |
| Interventions | Experimental: fingolimod Active comparator: disease modifying therapy |
| Outcomes | Primary outcome measures: retention on treatment |
| Notes | The study has been completed (August 2015) (accessed 22 February 2016) |
DMD: disease‐modifying drugs; MS: multiple sclerosis; RRMS: relapsing‐remitting multiple sclerosis;
Characteristics of ongoing studies [ordered by study ID]
EUCTR2013‐004622‐29‐IT.
| Trial name or title | A multicenter, randomised, open‐label study to assess the impact of natalizumab versus fingolimod on central nervous system tissue damage and recovery in active relapsing‐remitting multiple sclerosis subjects |
| Methods | Multicentre, randomised, open‐label study |
| Participants | Relapsing‐Remitting multiple sclerosis (RRMS) |
| Interventions | Experimental: fingolimod Active comparator: natalizumab |
| Outcomes | The effect of natalizumab compared to fingolimod on the evolution of new on‐treatment T1‐gadolinium‐enhancing lesions to persistent black holes over 52 weeks |
| Starting date | 10 November 2014 |
| Contact information | clinicaltrials@biogenidec.com |
| Notes | Authorised‐recruitment may be ongoing or finished (EUCTR) (access 6 August 2015) |
NCT01633112.
| Trial name or title | MS study evaluating safety and efficacy of two doses of fingolimod versus Copaxone |
| Methods | 12‐month, randomised, rater‐ and dose‐blinded study to compare the efficacy and safety of fingolimod 0.25 mg and 0.5 mg administered orally once‐daily with glatiramer acetate 20 mg administered subcutaneously once‐daily in patients with RRMS |
| Participants | RRMS |
| Interventions | Experimental: fingolimod Active comparator: glatiramer acetate |
| Outcomes | Primary outcome measure annualised relapse rate up to 12 months |
| Starting date | August 2012 |
| Contact information | Novartis Pharmaceuticals |
| Notes | This study is currently recruiting participants |
NCT01892722.
| Trial name or title | Two‐year, double‐blind, randomised multicenter, active‐controlled study to evaluate safety and efficacy of oral fingolimod versus intramuscular Interferon beta‐1a in paediatric patients with multiple sclerosis |
| Methods | Randomised controlled study |
| Participants | Paediatric patients with MS |
| Interventions | Oral fingolimod versus intramuscular Interferon beta‐1a |
| Outcomes | Primary outcome measure: frequency of relapses in patients treated for up to 24 months |
| Starting date | July 2013 |
| Contact information | Contact: Novartis Pharmaceuticals |
| Notes | This study is currently recruiting participants |
NCT02141022.
| Trial name or title | Computerised exercise training for cognitive remediation in adults with multiple sclerosis treated with Gilenya |
| Methods | RCT investigating the efficacy of computer‐based cognitive exercises as a means of cognitive remediation in patients with MS who are beginning Gileyna |
| Participants | MS |
| Interventions | Experimental: PACR program: plasticity based, adaptive cognitive remediation Active Comparator: Ordinary Computer Games |
| Outcomes | Primary outcome measure: change from baseline in neuropsychological test results at 12 weeks |
| Starting date | August 2013 |
| Contact information | Lauren Krupp, Stony Brook University |
| Notes | This study is ongoing, but not recruiting participants |
NCT02307838.
| Trial name or title | Long‐term follow‐up at 10 years of patients enrolled in the fingolimod phase II program in relapsing multiple sclerosis |
| Methods | Observational: to collect follow‐up data on patients who were randomised and received one dose of study drug (fingolimod) |
| Participants | RRMS |
| Interventions | None |
| Outcomes | Change from baseline in Expanded Disability Status Scale at 10 years |
| Starting date | June 2014 |
| Contact information | Novartis Pharmaceuticals |
| Notes | This study is currently recruiting participants |
NCT02342704.
| Trial name or title | The impact of natalizumab versus fingolimod on central nervous system tissue damage and recovery in active RRMS subjects (REVEAL) |
| Methods | To assess the effect of natalizumab compared to fingolimod on the evolution of new on‐treatment T1‐gadolinium‐enhancing (Gd+) lesions to persistent black holes over 52 weeks |
| Participants | RRMS |
| Interventions | Experimental: natalizumab Active Comparator: fingolimod |
| Outcomes | Cumulative number of ≥ 6‐months confirmed T1‐hypointense lesions arising from new on‐treatment T1 Gd+ |
| Starting date | November 2014 |
| Contact information | Contact: Biogen Idec |
| Notes | This study is currently recruiting participants |
MS: multiple sclerosis; RCT: randomised controlled trial; RRMS: relapsing‐remitting multiple sclerosis;
Differences between protocol and review
Background was amended.
Types of studies: cohort studies, case‐control studies, case reports or case‐series were not included in this review because an ad hoc review has been planned.
Participants: MS diagnostic criteria, which we accepted for inclusion of participants, have been added.
Primary outcome measures: 1) number of participants relapse‐free at six, 12 and 24 months, and number of participants free from disability progression at 12, 24 and 36 months were included as primary outcome measures more relevant to participants; 2) annualised relapse rate was moved to secondary outcomes; 3) number of participants who withdrew from the study because of serious adverse events was added as a primary outcome, and the rate of serious adverse events was excluded.
Secondary outcomes were amended.
Electronic searches were amended.
Dealing with missing data: a likely scenario was used as a sensitivity analysis to deal with missing data. An intention‐to‐treat analysis, using the last reported observed response ('carry forward'), previously reported in the review protocol, was not performed according to the Cochrane Handbook for Systematic Reviews of Interventions (Chapter 8.13.2.3) (Higgins 2011b).
'Summary of findings' tables were added.
Contributions of authors
| Roles and responsibilities | |
| Drafting the protocol | All review authors |
| Selecting which studies to include | LLM, RP, IP |
| Planning data sheet for study data extraction | LLM,IP |
| Extracting data from studies | LLM, IT, IP |
| Assessing risk of bias of studies | LLM, IP, IT |
| Entering data into RevMan | LLM, IP, IT |
| Carrying out data analysis and interpreting results | LLM, IT, GF |
| Drafting the manuscript and final review | LLM, GF |
| Editing | LLM |
| Approving the final version | All review authors |
Declarations of interest
Loredana La Mantia: none. Belal Firwana: none. Irene Tramacere: none. Ilaria Pacchetti: none. Roberto Palumbo: none. Graziella Filippini: none. As Co‐ordinating Editor, Dr. Filippini was excluded from the editorial process to ensure separation of the author and the editorial process. This includes all editorial decisions and related activities (e.g. Sign‐off for publication).
New
References
References to studies included in this review
Calabresi 2014 {published data only}
- Calabresi P, Radue EW, Goodin D, Jeffery D, Kottil R, Reder A, et al. Efficacy and safety of fingolimod in patients with relapsing‐remitting multiple sclerosis (RRMS): results from an additional 24‐month double‐blind, placebo‐controlled study (freedoms II study). Abstract meeting of the 64th American Academy of Neurology Annual Meeting, 2012, New Orleans, United States. Neurology. 2012.
- Calabresi PA, Goodin D, Jeffery D, Kappos L, Lublin FD, Rammohan K, et al. Efficacy and safety of fingolimod versus placebo: Primary outcomes from the phase 3 FREEDOMS II study in patients with relapsing‐remitting multiple sclerosis. Abstract meeting of the 28th Congress of the European Committee for Treatment and Research in Multiple Sclerosis, 2012, Lyon, France. Multiple Sclerosis. 2012; Vol. 18.
- Calabresi PA, Radue EW, Goodin D, Jeffery D, Rammohan KW, Reder AT, et al. Safety and efficacy of fingolimod in patients with relapsing‐remitting multiple sclerosis (FREEDOMS II): a double‐blind, randomised, placebo‐controlled, phase 3 trial. The Lancet Neurology 2014;13:545–56. [DOI] [PubMed] [Google Scholar]
- Coyle P, Cree B, Cabre P, Inglese M, Perumal J, Meng X, et al. Fingolimod efficacy and safety in an African‐American patient subgroup from freedoms II. Abstract meeting of the 66th American Academy of Neurology Annual Meeting, AAN 2014, Philadelphia, United States. Neurology. 2014; Vol. 82.
- Goodin D, Jeffery D, Kappos L, Lublin F, Radue EW, Rammohan K, et al. Fingolimod reduces annualized relapse rate in patients with relapsing‐remitting multiple sclerosis: Freedoms II study subgroup analysis. Abstract meeting of the 65th American Academy of Neurology Annual Meeting, 2013, San Diego, United States. Neurology. 2013; Vol. 80.
- Khan O, Cree B, Cabre P, Inglese M, Perumal J, Meng X, et al. The efficacy and safety of fingolimod in an African‐American patient subgroup from FREEDOMS II. Abtract meeting of The European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS); 2013 October 2‐5; Copenhagen, Denmark. Multiple Sclerosis. 2013.
- Radue E, Goodin D, Jeffery D, Kappos L, Lublin F, Rammohan K, et al. Fingolimod reduces magnetic resonance imaging inflammatory lesion activity versus placebo in patients with relapsing‐remitting multiple sclerosis: results from the phase 3. Multiple Sclerosis 2012;18(4):322‐3. [Google Scholar]
- Reder A, Jeffery D, Goodin D, Kappos L, Lublin F, Radue E, et al. Long‐term efficacy of fingolimod in patients with relapsing‐remitting multiple sclerosis: results from the phase 3 FREEDOMS II extension study. Multiple Sclerosis 2013;19:510‐1. [Google Scholar]
- Vollmer T, Goodin D, Jeffery D, Kappos L, Radue E, Rammohan K, et al. Effect of fingolimod on severe relapses, healthcare utilisation and relapse recovery in patients with relapsing‐remitting multiple sclerosis: results from the phase 3 FREEDOMS II study. Multiple Sclerosis 2012;18:438‐9. [Google Scholar]
- Vollmer T, Jeffery D, Goodin D, Kappos L, Lublin F, Radue EW, et al. Long‐term safety of fingolimod in patients with relapsing‐remitting multiple sclerosis: results from phase 3 freedoms II extension study. Neurology. 2013; Vol. 80.
- Winges KM, Werner JS, Harvey DJ, Cello KE, Durbin MK, Balcer LJ, et al. Baseline retinal nerve fiber layer thickness and macular volume quantified by OCT in the North American phase 3 fingolimod trial for relapsing–remitting multiple sclerosis. Journal of Neuro‐Ophthalmology 2013;33:322–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Cohen 2010 {published data only}
- Barkhof F, Cohen J, Montalban X, Comi G, Auberson L, Holdbrook F, et al. Fingolimod (FTY720) reduces brain volume loss over 12 months compared with intramuscular interferon beta‐1a: subgroup analyses of TRANSFORMS data based on inflammatory disease activity. Abstract meeting of the 5th Joint Triennal Congress of the European and Americas Committees for the treatment and research in Multiple Sclerosis; 2011 Oct 19–22, Amsterdam, The Netherlands. Multiple Sclerosis. 2011; Vol. 17.
- Barkhof F, Jong R, Sfikas N, Vera A, Francis G, Cohen J, TRANSFORMS study group. The influence of patient demographics, disease characteristics and treatment on brain volume loss in Trial Assessing Injectable Interferon vs FTY720 Oral in Relapsing‐Remitting Multiple Sclerosis (TRANSFORMS), a phase 3 study of fingolimod in multiple sclerosis. Multiple Sclerosis 2014;20(13):1704‐13. [DOI] [PubMed] [Google Scholar]
- Cohen JA, Barkhof F, Comi G, Hartung HP, Khatri BO, Montalban X, et al. Oral Fingolimod or intramuscular interferon for relapsing multiple sclerosis. New England Journal of Medicine 2010;362(5):402‐15. [DOI] [PubMed] [Google Scholar]
- Cohen JA, Barkhof F, Comi G, Izquierdo G, Khatri B, Montalban X, et al. Fingolimod versus intramuscular interferon in patient subgroups from TRANSFORMS. Journal of Neurology 2013;260(8):2023‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartung H, Barkhof F, Comi G, Kappos L, Khatri B, Montalban X, et al. Relationship between early disease activity and long‐term clinical outcome: results from the phase 3 TRANSFORMS study extension at 4.5 years in relapsing‐remitting multiple sclerosis. Abstract meeting of the twenty‐third meeting of the ENS; 2013 June 8‐11; Barcelona, Spain. Journal of Neurology. 2013; Vol. 260.
- Khatri B, Barkhof F, Comi G, Hartung H, Kappos L, Montalban X, et al. Long‐term efficacy data from the extension of the phase III TRANSFORMS study of fingolimod versus Interferon beta‐1a in relapsing‐remitting multiple sclerosis: 4.5 year follow‐up. Journal of Neurology 2012;259(1):S21. [Google Scholar]
- Khatri B, Barkhof F, Comi G, Hartung HP, Kappos L, Montalban X, et al. Comparison of fingolimod with Interferon beta‐1a in relapsing‐remitting multiple sclerosis: a randomised extension of the TRANSFORMS study. The Lancet Neurology 2011;10(6):520‐9. [DOI] [PubMed] [Google Scholar]
- Khatri B, Barkhof F, Comi G, Jin J, Francis G, Cohen J. Fingolimod treatment increases the proportion of patients who are free from disease activity in multiple sclerosis compared to IFN‐B1A: results from a phase 3, active‐controlled study (TRANSFORMS). Abstract meeting of the 64th American Academy of Neurology Annual Meeting, 2012, New Orleans, United States. Neurology. 2012; Vol. 78:1.
- Meng X, Cutter G, Chin P, Hashmonay R, Islam MZ. Effect of switching from intramuscular Interferon B‐1a to fingolimod on time to relapse in patients with relapsing‐remitting multiple sclerosis enrolled in a 1‐year extension of transforms. Abstract meeting of the 28th Congress of the European Committee for Treatment and Research in Multiple Sclerosis, 2013, Lyon, France. Neurology. 2013; Vol. 80.
- Montalban X, Barkhof F, Comi G, Hartung HP, Kappos L, Khatri B, et al. Long term efficacy of fingolimod in patients with relapsing‐remitting multiple sclerosis previously treated with interferon beta‐1a or disease modifying therapies: A post hoc analysis of the TRANSFORMS 4.5 year extension study. Journal of Neurology 2013;260:S124‐5. [Google Scholar]
Fox 2014 {published data only}
- Barbato L, Schofield L, McCague K, Pestreich L, Tobias K, Malhotra M. Randomized, open‐label study to evaluate patient‐reported outcomes (PRO) with fingolimod after changing from prior disease‐modifying therapy (DMT) for relapsing multiple sclerosis (MS): EPOC study rationale and design. Abstract meeting of the 136th Annual Meeting of the American Neurological Association; 2011 Sept 25‐27; San Diego, United States. Annals of Neurology. 2011.
- Calkwood J, Cree B, Crayton H, Kantor D, Brian Steingo B, Barbato L, et al. Impact of a switch to fingolimod versus staying on glatiramer acetate or beta interferons on patient‐ and physician‐reported outcomes in relapsing multiple sclerosis: post hoc analyses of the EPOC trial. BMC Neurology 2014;14(220):1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascione M, Wynn D, Agashivala N, McCague K, Pestreich L, Schofield L, et al. Summary scores for patient‐reported outcome measures in multiple sclerosis. Baseline data from the trial to Evaluate Patient OutComes, safety and tolerability of fingolimod (EPOC). Multiple Sclerosis 2012;18:488‐9. [Google Scholar]
- Cascione M, Wynn D, Barbato LM, Pestreich L, Schofield L, McCague K. Randomized, open‐label study to evaluate patient‐reported outcomes with fingolimod after changing from prior disease‐modifying therapy for relapsing multiple sclerosis: EPOC study rationale and design. Journal of Medical Economics 2013;16(7):859–65. [DOI] [PubMed] [Google Scholar]
- Crayton H, Hunter S, Huffman C, Agashivala N, Schofield L, McCague K, et al. Improved quality of life after therapy change to fingolimod. Journal of Neurology 2013;260:S127. [Google Scholar]
- Cree B, Kantor D, Steingo M, Agashivala N, Li S, McCague K, et al. Patient and physician reported outcomes after therapy switch from glatiramer acetate to fingolimod versus staying on glatiramer acetate. Multiple Sclerosis 2013;19:464‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiBernardo A, Agashivala N, Meng X, Hashmonay R, Barbato M, Chin P. Effect of fingolimod on two sub domains of the Beck depression inventory‐II In patients with relapsing multiple sclerosis. Abstract meeting of the 18th Annual Conference of Rehabilitation in MS; 2013 Oct 2‐5; Copenhagen, Denmark. Multiple Sclerosis. 2013.
- Edwards K, Crayton H, Calkwood J, Agashivala N, Li S, Chin P, et al. Patient‐and physician‐reported outcomes after therapy switch from interferon (beta) to fingolimod versus staying on interferon (beta) therapy. Abstract meeting of the 29th Congress of European Committee for Treatment and Research in MS; 2013 Oct 2‐5; Copenhagen, Denmark. Multiple Sclerosis. 2013; Vol. 19:231‐2.
- Fox E, Edwards K, Burch JG, Wynn DR, LaGanke C, Crayton H, et al. Outcomes of switching directly to oral fingolimod from injectable therapies: results of the Evaluate Patient OutComes (EPOC) study in relapsing multiple sclerosis. Multiple Sclerosis and Related Disorders 2014;3(5):607–19. [DOI] [PubMed] [Google Scholar]
- Gudesblatt M, Agashivala N, Randhaw S, Li S, Barbato L, Singer B. Outcomes of a switch to fingolimod to treat relapsing multiple sclerosis: A patient subgroup post hoc analysis. Journal of Multiple Sclerosis 2014; Vol. 2, issue 1:Open Access.
- Hughes B, Cascione M, Freedman M, Agius M, Kantor D, Gudesblatt M, et al. First‐dose effects of fingolimod after switching from injectable therapies in the randomized, open‐label, multicenter, Evaluate Patient OutComes (EPOC) study in relapsing multiple sclerosis. Multiple Sclerosis 2014;3(5):620‐8. [DOI] [PubMed] [Google Scholar]
- Singer B, Gudesblatt M, Agashivala N, Li S, Randhawa S, McCague K, et al. Patient‐reported outcomes after therapy switch to fingolimod: post‐hoc subgroup analysis of the EPOC study. Abstract meeting of the 66th American Academy of Neurology Annual Meeting, April 26–May 3, 2014, Philadelphia, United States. Neurology. 2014; Vol. 82.
Kappos 2006 {published data only}
- Antel J, Montalban X, O'Connor P, Vera A, Cremer M, Sfikas N, et al. Long‐term (7‐year) data from a phase 2 extension study of fingolimod in relapsing multiple sclerosis. Abstract meeting of The American Academy of Neurology, 64th AAN Annual Meeting; April 21 ‐ 28, 2012; New Orleans, United States. Neurology. 2012; Vol. 78.
- Cohen JA, Khatri B, Barkhof F, Comi G, Hartung HP, Montalban X, et al. Long‐term (up to 4.5 years) treatment with fingolimod in multiple sclerosis: results from the extension of the randomised TRANSFORMS study. Journal of Neurology, Neurosurgery, and Psychiatry 2015 June 25 [Epub ahead of print]. [DOI] [PMC free article] [PubMed]
- Comi G, O'Connor P, Montalban X, Antel J, Radue EW, Karlsson G, et al. Phase II study of oral fingolimod (FTY720) in multiple sclerosis: 3‐year results. Multiple Sclerosis 2009;16:197‐207. [DOI] [PubMed] [Google Scholar]
- Izquierdo G, O'Connor P, Montalban X, Rosenstiel P, Cremer M, Vera A, et al. Five‐year results from a phase 2 study of oral fingolimod in relapsing multiple sclerosis. Multiple Sclerosis 2014;20(7):877‐81. [DOI] [PubMed] [Google Scholar]
- Kappos L, Antel J, Comi G, Montalban X, O'Connor P, Polman CH, et al. Oral fingolimod (FTY720) for relapsing multiple sclerosis. New England Journal of Medicine 2006;355(11):1124‐40. [DOI] [PubMed] [Google Scholar]
- Montalban X, Comi G, Antel J, O'Connor P, Vera A, Cremer M, et al. Long‐term (>7‐year) efficacy and safety data from a phase II extension study of fingolimod in relapsing multiple sclerosis. Journal of Neurology 2012;259(1):S69‐70. [Google Scholar]
- Montalban X, Comi G, O'Connor P, Gold S, Vera A, Eckert B, et al. Oral fingolimod (FTY720) in relapsing multiple sclerosis: impact on health‐related quality of life in a phase II study. Multiple Sclerosis 2011;17(11):1341‐50. [DOI] [PubMed] [Google Scholar]
- Montalban X, O'Connor P, Antel J. Oral fingolimod (FTY720) shows sustained low rates of clinical and MRI disease activity in patients with relapsing multiple sclerosis: four‐year results from a phase II extension. Neurology. 2009; Vol. 72:A313.
- Montalban X, O'Connor P, Izquierdo G, Rosenstiel P, Cremer M, Prut L, et al. Long‐term fingolimod (FTY720) in relapsing MS: 5‐year results from an extension of a phase II, multicentre study show a sustained low level of disease activity. Multiple Sclerosis 2011;17(10):S442‐3. [Google Scholar]
- O'Connor P, Comi G, Montalban X, Antel J, Radue EW, Vera A, et al. Oral fingolimod (FTY720) in multiple sclerosis: two‐year results of a phase II extension study. Neurology 2009;72(1):73‐9. [DOI] [PubMed] [Google Scholar]
Kappos 2010 {published data only}
- Bergvall N, Sfikas N, Alsop J, Chin P, Rosensteil P, Kappos L. Consequences of different definitions of confirmed disability progression across randomised trials of MS therapies. Multiple Sclerosis 2012;18(4):473‐4. [Google Scholar]
- Camu W, Thouvenot E, Meinel M, Sfikas N, Chin P, Piani‐Meier D, et al. Influence of baseline clinical and demographic characteristics on disease evolution in the phase 3 FREEDOMS study in patients with relapsing‐remitting multiple sclerosis. Abtract meetings of the ECTRIMS 18th Annual Conference on Rehabilitation, 2013, Copenhagen, Denmark. Multiple Sclerosis. 2013; Vol. 19.
- Chin P, Rosenstiel P, Haering D, Francis G, Kappos L. Fingolimod leads to early clinical and MRI benefits in relapsing‐remitting multiple sclerosis. Abstract meeting of the twenty‐third ENS, 2013, Spain. Journal of Neurology. 2013.
- Cutter G, Chin P, Francis G, Meng X, Hashmonay R, Lublin F. Relapse is associated with residual deficits in relapsing‐remitting multiple sclerosis: Analysis of freedoms data. Abstract meeting, The American Academy of Neurology's 65th AAN Annual Meeting, 2013, San Diego, United States. Neurology. 2013; Vol. 80.
- Devonshire V, Havrdova E, Radue EW, O'Connor P, Zhang‐Auberson L, Agoropoulou C, et al. Relapse and disability outcomes in patients with multiple sclerosis treated with Fingolimod: subgroup analyses of the double‐blind, randomised, placebo‐controlled FREEDOMS study. The Lancet Neurology 2012;11:420–8. [DOI] [PubMed] [Google Scholar]
- Hohlfeld R, Calabresi PA, O'Connor P. Oral fingolimod (FTY720) reduces relapse rate in patients previously treated with disease‐modifying therapies for multiple sclerosis and in patients who are treatment naive: subgroup analysis of data from a 24‐month phase III study (FREEDOMS). Journal of Neurology. 2010; Vol. 257.
- Kappos L, De SN, Freedman MS, Cree BA, Radue EW, Sprenger T, et al. Inclusion of brain volume loss in a revised measure of 'no evidence of disease activity' (NEDA‐4) in relapsing‐remitting multiple sclerosis. Multiple Sclerosis 2015 Nov 19 [Epub ahead of print]. [DOI] [PMC free article] [PubMed]
- Kappos L, O'Connor P, Radue E, Polman C, Hohlfeld R, Selmaj K, et al. Long‐term effects of fingolimod in multiple sclerosis: The randomized FREEDOMS extension trial. Neurology 2015;84(15):1582‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappos L, Radue EW, O'Connor P, Polman C, Hohlfeld R, Calabresi P, et al. A placebo‐controlled trial of oral fingolimod in relapsing multiple sclerosis. New England Journal of Medicine 2010;362(5):387‐401. [DOI] [PubMed] [Google Scholar]
- Kappos L, Radue EW, O'Connor P, Polman C, Hohlfeld R, Calabresi P, et al. Long‐term efficacy and safety of fingolimod (FTY720) in relapsing‐remitting multiple sclerosis (RRMS): Results from the extension of the phase III FREEDOMS study. Neurology. 2012; Vol. 78:1.
- Kremenchutzky M, O'Connor P, Hohlfeld R, Zhang‐Auberson L, Rosenstiel P, Meng X, et al. Impact of prior treatment status and reasons for discontinuation on the efficacy and safety of fingolimod: Subgroup analyses of the Fingolimod Research Evaluating Effects of Daily Oral Therapy in Multiple Sclerosis (FREEDOMS) study. Multiple Sclerosis and Related Disorders 2013;3:341–9. [DOI] [PubMed] [Google Scholar]
- O'Connor P, Polman C, Hohlfeld R, Selmaj K, Olsson T, Agoropoulou C, et al. Phase III FREEDOMS study extension: Long‐term safety of fingolimod (FTY720) in relapsing‐remitting multiple sclerosis. Multiple Sclerosis 2012;18(4):223. [Google Scholar]
- Radue E, Kappos L, O'Connor P, Polman C, Hohlfeld R, Calabresi P, et al. Fingolimod significantly reduced brain volume loss in patients with relapsing‐remitting multiple sclerosis: 4‐year data from FREEDOMS extension study. Journal of Neurology. 2012; Vol. 259, issue 1:S21‐2.
- Radue E, Sprenger T, Vera A, Francis G, Rochotte E, Tomic D, et al. Effect of fingolimod on evolution of baseline enhancing MRI lesions into persistent T1 hypointense lesions: Post hoc analysis of the FREEDOMS study. Multiple Sclerosis 2014;20:112‐3. [Google Scholar]
- Radue EW, O'Connor P, Polman CH, Hohlfeld R, Calabresi P, Selmaj K, et al. Impact of fingolimod therapy on magnetic resonance imaging outcomes in patients with multiple sclerosis. Archives of Neurology 2012;69(10):1259‐69. [DOI] [PubMed] [Google Scholar]
Saida 2012 {published data only}
- Kira J, Itoyama Y, Kikuchi S, Hao Q, Kurosawa T, Nagato K, et al. Fingolimod (FTY720) therapy in Japanese patients with relapsing multiple sclerosis over 12 months: results of a phase 2 observational extension. BMC Neurology 2014;14:1‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kira J, Itoyama Y, Kikuchi S, Hao Q, Kurosawa T, Nagato K, et al. Oral fingolimod (FTY720) in Japanese patients with relapsing multiple sclerosis: Results of a 12‐month, phase 2 extension study. Multiple Sclerosis 2011;17(10):S193. [Google Scholar]
- Saida T, Kikuchi S, Itoyama Y, Hao Q, Kurosawa T, Nagato K, et al. A randomized, controlled trial of fingolimod (FTY720) in Japanese patients with multiple sclerosis. Multiple Sclerosis 2012;18:1269–77. [PUBMED: 22354739] [DOI] [PubMed] [Google Scholar]
- Saida T, Kikuchi S, Itoyama Y, Hao Q, Kurosawa T, Nagato K, et al. Oral fingolimod (FTY720) in Japanese patients with relapsing multiple sclerosis: Results of a 6‐month, randomised, double‐blind, placebo‐controlled, phase 2 study. Multiple Sclerosis 2011;17:S418‐9. [Google Scholar]
References to studies excluded from this review
Boulton 2013 {published data only}
- Boulton C, David OJ, Meiser K, Schmouder R. Tolerability and pulmonary pharmacodynamic effects during treatment initiation of once‐daily oral fingolimod in subjects with moderate asthma. Clinical Pharmacology in Drug Development 2013;2(1):2‐10. [DOI] [PubMed] [Google Scholar]
Chinea 2014 {published data only}
- Chinea A, Alvarenga R, Tomic D, DiBernardo A, Meng X, Hawker K. Efficacy and safety of fingolimod in hispanic patients: Pooled data from three phase 3 clinical trials. Neurology 2014;82:10. [Google Scholar]
Comi 2013 {published data only}
- Comi G, Gold R, Kappos L, Rosenstiel P, Sinha A, Tomic D. Relapse and safety outcomes in patients who transitioned from glatiramer acetate or interferon (beta) to fingolimod in the open‐label FIRST study. Multiple Sclerosis 2013;19:205. [Google Scholar]
Francis 2014 {published data only}
- Francis G, Kappos L, O’Connor P, Collins W, Tang D, Mercier F, et al. Temporal profile of lymphocyte counts and relationship with infections with fingolimod therapy. Multiple Sclerosis 2014;20(4):471–80. [DOI] [PubMed] [Google Scholar]
Gold 2014 {published data only}
- Gold R, Comi G, Palace J, Siever A, Gottschalk R, Bijarnia M, et al. Assessment of cardiac safety during fingolimod treatment initiation in a real‐world relapsing multiple sclerosis population: a phase 3b, open‐label study. Journal of Neurology 2014;261(2):267‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
Green 2013 {published data only}
- Green A, Sergott R, Bennett L, Hamilton S, Costello F, Dahlke F, et al. Fingolimod for the treatment of acute optic neuritis: design of a phase 2 study. Multiple Sclerosis 2013;19:238‐9. [Google Scholar]
Havla 2013 {published data only}
- Havla J, Tackenberg B, Hellwig K, Meinl I, Krumbholz M, Seitz F, et al. Fingolimod reduces recurrence of disease activity after natalizumab withdrawal in multiple sclerosis. Journal of Neurology 2013;260(5):1382‐7. [DOI] [PubMed] [Google Scholar]
Kappos 2014 {published data only}
- Kappos L, Zhang L, Francis AG, Cohen J. Fingolimod in relapsing multiple sclerosis: An integrated analysis of safety findings. Multiple Sclerosis and Related Disorders 2014;3:494–504. [DOI] [PubMed] [Google Scholar]
Kappos 2014a {published data only}
- Kappos L, Radue E, Karlsson G, Zheng H, Rosenstiel P, Jeffery D. Efficacy benefits of fingolimod 0.5 mg once daily in patients previously treated with glatiramer acetate: Pooled analysis of phase 3 FREEDOMS and FREEDOMS II studies. Neurology 2014;82(10 Suppl):193. [Google Scholar]
Kappos 2015 {published data only}
- Kappos L, Mehling M, Arroyo R, Izquierdo G, Selmaj K, Curovic‐Perisic V, et al. Randomized trial of vaccination in fingolimod‐treated patients with multiple sclerosis. Neurology 2015;84(9):872‐9. [DOI] [PubMed] [Google Scholar]
Kappos 2015a {published data only}
- Kappos L, Radue EW, Comi G, Montalban X, Butzkueven H, Wiendl H, et al. Switching from natalizumab to fingolimod. A randomized, placebo‐controlled study in RRMS. Neurology 2015;85(1):29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
Karlsson 2014 {published data only}
- Karlsson G, Francis G, Koren G, Heining P, Zhang X, Cohen J, et al. Pregnancy outcomes in the clinical development program of fingolimod in multiple sclerosis. Neurology 2014;82:674–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
laroni 2013 {published data only}
- Laroni A, Brogi D, Morra V, Guidi L, Pozzilli C, Comi G, et al. Safety of the first dose of fingolimod for multiple sclerosis: Results of an open‐label clinical trial. BMC Neurology 2014;14(65):1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Limmorth 2013 {published data only}
- Limmroth V, Hoyer S, Schuh K, Lang M, Hoffmann O, Ziemssen T. Good cardiac safety profile after fingolimod (Gilenya registered trademark) treatment initiation in patients with relapsing remitting multiple sclerosis: First interim analysis of the START study. Multiple Sclerosis. 2013; Vol. 19.
Lublin 2016 {published data only}
Nolan 2013 {published data only}
- Nolan R, Gelfand JM, Green AJ. Fingolimod treatment in multiple sclerosis leads to increased macular volume. Neurology 2013;80:139‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
Van Lokven 2013 {published data only}
- Lokven T, Ortler S, Moser S, Vollmar P, Ziemssen T. Comparison of therapy efficacy and satisfaction of German relapsing remitting multiple sclerosis (RRMS) patients on baseline therapy with fingolimod‐treated patients; results of an interim analysis of two non‐interventional studies (PANGAEA and PEARL). Multiple Sclerosis. 2013; Vol. 19:252.
Vollmer 2013 a {published data only}
- Vollmer T, Radue E, Vermersch P, Rosenstiel P, Putzki N, Meinel M, et al. Clinical and magnetic resonance imaging (MRI) disease activity after fingolimod discontinuation. Multiple Sclerosis 2013;19:227‐8. [Google Scholar]
Zarbin 2013 {published data only}
- Zarbin M, Jampol L, Jager R, Reder A, Francis G, Collins W, et al. Ophthalmic evaluations in clinical studies of fingolimod (FTY720) in multiple sclerosis. Ophthalmology 2013;120(7):1432‐9. [DOI] [PubMed] [Google Scholar]
References to studies awaiting assessment
NCT01317004 {published data only}
- NCT01317004. A 6‐month, randomized, active comparator, open‐label, multi‐center study to evaluate patient outcomes, safety and tolerability of fingolimod (FTY720) 0.5 mg/day in patients with relapsing remitting multiple sclerosis who are candidates for ms therapy change from previous disease modifying therapy (EPOC). clinicaltrials.gov/ct2/show/NCT01317004?term=NCT01317004&rank=1 (accessed 22 February 2016).
NCT01333501 {published data only}
- NCT01333501. An 18‐month, open‐label, Rater‐blinded, randomized, multi‐center, active‐controlled, parallel‐group pilot study to assess efficacy and safety of fingolimod in comparison to interferon beta 1b in treating the cognitive symptoms associated with relapsing‐remitting multiple sclerosis and to assess possible relationship of these effects to regional brain atrophy. clinicaltrials.gov/ct2/show/study/NCT01333501?term=NCT01333501&rank=1 (accessed 22 February 2016).
NCT01534182 {published data only}
- NCT01534182. A 6‐month, randomized, active comparator, open‐label, multi‐center study to evaluate patient outcomes, safety and tolerability of (Fingolimod) 0.5 mg/day in patients with relapsing remitting multiple sclerosis who are candidates for multiple sclerosis (MS) therapy change from previous disease modifying therapy (DMT). clinicaltrials.gov/ct2/show/NCT01534182?term=NCT01534182&rank=1 (accessed 10 December 2015).
NCT01623596 {published data only}
- NCT01623596. A 12‐month, Prospective, Randomized, Active‐controlled, Open‐label Study to Evaluate the Patient Retention of Fingolimod vs. Approved First‐line Disease Modifying Therapies in Adults With Relapsing Remitting Multiple Sclerosis (PREFERMS). https://clinicaltrials.gov/ct2/show/study/NCT01623596?term=NCT01623596&rank=1 22 February 2016.
References to ongoing studies
EUCTR2013‐004622‐29‐IT {published data only}
- EUCTR2013‐004622‐29‐IT. A multicenter, randomized, open‐label study to assess the impact of natalizumab versus fingolimod on central nervous system tissue damage and recovery in active relapsing‐remitting multiple sclerosis. https://www.clinicaltrialsregister.eu/ctr‐search/search?query=2013‐004622‐29 (accessed 10 December 2015).
NCT01633112 {published data only}
- NCT01633112. A 12‐month, randomized, rater‐ and dose‐blinded study to compare the efficacy and safety of fingolimod 0.25 mg and 0.5 mg administered orally once daily with glatiramer acetate 20 mg administered subcutaneously once daily in patients with relapsing‐remitting multiple sclerosis. https://clinicaltrials.gov/ct2/show/study/NCT01633112?term=NCT01633112&rank=1 (accessed 10 December 2015).
NCT01892722 {published data only}
- NCT01892722. Two‐year, double‐blind, randomised, multicenter, active‐controlled study to evaluate safety and efficacy of oral fingolimod versus interferon beta‐1a i.m. In pediatric patients with multiple sclerosis. https://clinicaltrials.gov/ct2/show/study/NCT01892722?term=NCT01892722&rank=1 (accessed 10 December 2015).
NCT02141022 {published data only}
- NCT02141022. A pilot study of plasticity‐based and adaptive cognitive remediation in adults with multiple sclerosis treated with Gilenya. https://clinicaltrials.gov/ct2/show/study/NCT02141022?term=NCT02141022&rank=1 (accessed 26 June 2015).
NCT02307838 {published data only}
- NCT02307838. Long‐term follow‐up at 10 years of patients enrolled in the fingolimod phase ii program in relapsing multiple sclerosis. https://clinicaltrials.gov/ct2/show/NCT02307838?term=NCT02307838&rank=1 (accessed 10 December 2015).
NCT02342704 {published data only}
- NCT02342704. A multicenter, randomized, open‐label study to assess the impact of Natalizumab versus fingolimod on central nervous system tissue damage and recovery in active relapsing‐remitting multiple sclerosis subjects. https://clinicaltrials.gov/ct2/show/study/NCT02342704?term=NCT02342704&rank=1 (accessed 10 December 2015).
Additional references
AIFA 2015
- Agenzia Italiana del Farmaco (AIFA). [Nota Informativa Importante sul primo caso di Leucoencefalopatia Multifocale Progressiva (PML) in un paziente con sclerosi multipla in trattamento con fingolimod]. http://www.agenziafarmaco.gov.it/it/content/nota‐informativa‐importante‐sul‐primo‐caso‐di‐leucoencefalopatia‐multifocale‐progressiva‐pml (accessed 10 December 2015).
Ali 2013
- Ali R, Nicholas RS, Muraro PA. Drugs in development for relapsing multiple sclerosis. Drugs 2013;73(7):623‐50. [DOI] [PubMed] [Google Scholar]
Alonso 2008
- Alonso A, Hernan MA. Temporal trends in the incidence of multiple sclerosis: a systematic review. Neurology 2008;71(2):129‐35. [PUBMED: 18606967] [DOI] [PMC free article] [PubMed] [Google Scholar]
Antel 2012
- Antel J, Montalban X, O'Connor P, Vera A, Cremer M, Sfikas N, et al. Long‐term (7‐year) data from a phase 2 extension study of fingolimod in relapsing multiple sclerosis. Meeting abstracts of The American Academy of Neurology's 64th AAN Annual Meeting; 2012 April 23; New Orleans. Neurology. 2012.
Atkinson 2004
- Atkinson MJ, Sinha A, Hass SL, Colman SS, Kumar RN, Brod M, et al. Validation of a general measure of treatment satisfaction, the Treatment Satisfaction Questionnaire for Medication (TSQM), using a national panel study of chronic disease. Health and Quality of Life Outcomes 2004;2:1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bao 2012
- Bao C, Zhou JH. Efficacy and safety of FTY720 in the treatment of relapsing‐remitting multiple sclerosis: A systematic review. Chinese Journal of Evidence‐Based Medicine 2012;12(4):445‐50. [Google Scholar]
Brinkmann 2002
- Brinkmann V, Davis MD, Heise CE, Albert R, Cottens S, Hof R. The immune modulator FTY720 targets sphingosine 1‐phosphate receptors. Journal of Biological Chemistry 2002;277:21453‐7. [DOI] [PubMed] [Google Scholar]
Cascione 2013
- Cascione M, Wynn D, Barbato LM, Pestreich L, Schofield L, McCague K. Randomized, open‐label study to evaluate patient‐reported outcomes with fingolimod after changing from prior disease‐modifying therapy for relapsing multiple sclerosis: EPOC study rationale and design. Journal of Medical Economics 2013;16(7):859–65. [DOI] [PubMed] [Google Scholar]
Chun 2010
- Chun J, Hartung H. Mechanism of action of oral Fingolimod (FTY720) in multiple sclerosis. Clinical Neuropharmacology 2010;33(2):91‐101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Cohen 2012
- Cohen JA, Reingold SC, Polman CH, Wolinsky JW, International Advisory Committee on Clinical Trials in Multiple Sclerosis. Disability outcome measures in multiple sclerosis clinicaltrials: current status and future prospects. Lancet Neurology 2012;11(5):467–76. [DOI] [PubMed] [Google Scholar]
Cohen 2013
- Cohen J, Barkhof F, Comi G, Izquierdo G, Khatri B, Montalban X, et al. Fingolimod versus intramuscular interferon in patient subgroups from TRANSFORMS. Journal of Neurology 2013;260(8):2023‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
Comi 2010
- Comi G, O'Connor P, Montalban X, Antel J, Radue EW, Karlsson G, et al. Phase II study of oral fingolimod (FTY720) in multiple sclerosis: 3‐year results. Multiple Sclerosis 2010;16(2):197‐207. [DOI] [PubMed] [Google Scholar]
Compston 2002
- Compston A, Coles A. Multiple sclerosis. Lancet 2002;359:1221‐31. [DOI] [PubMed] [Google Scholar]
Compston 2008
- Compston A, Coles A. Multiple sclerosis. Lancet 2008;372:1502‐17. [DOI] [PubMed] [Google Scholar]
David 2012
- David OJ, Kovaric GM, Schmouder RL. Clinical pharmacokinetics of fingolimod. Clinical Pharmacokinetics 2012;51(1):15‐28. [DOI] [PubMed] [Google Scholar]
Deeks 2011
- Deeks JJ, Higgins JPT, Altman DG (editors). Chapter 9: Analysing data and undertaking meta‐analyses. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Verion 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Del Santo 2011
- Santo F, Maratea D, Fadda V, Trippoli S, Messori A. Treatments for relapsing‐remitting multiple sclerosis: summarising current information by network meta‐analysis. European Journal of Clinical Pharmacology 2011;68:441‐8. [DOI] [PubMed] [Google Scholar]
Devonshire 2012
- Devonshire V, Havrdova E, Radue EW, O'Connor P, Zhang‐Auberson L, Agoropoulou C, et al. Relapse and disability outcomes in patients with multiple sclerosis treated with fingolimod: subgroup analyses of the double‐blind, randomised, placebo‐controlled FREEDOMS study. The Lancet Neurology 2012;11:420‐8. [DOI] [PubMed] [Google Scholar]
Doggrell 2010
- Doggrell SA. Oral fingolimod for relapsing‐remitting multiple sclerosis. Expert Opinion on Pharmacotherapy 2010;11(10):1777‐81. [DOI] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta‐analysis detected by a simple, graphical test. BMJ (Clinical Research Ed.) 1997;315:629‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Elhami 2011
- Elhami SR, Mohammad K, Sahraian MA, Eftekhar H. A 20‐year incidence trend (1989‐2008) and point prevalence (March 20, 2009) of multiple sclerosis in Tehran, Iran: a population‐based study. Neuroepidemiology 2011;36(3):141‐7. [DOI] [PubMed] [Google Scholar]
EMA 2015
- European Medicines Agency. Gilenya : EPAR ‐ Summary for the public. http://www.ema.europa.eu/ema/index.jsp?curl=pages/news_and_events/news/2015/12/news_detail_002447.jsp&mid=WC0b01ac058004d5c1 (accessed Decembre 2015). [ema.europa.eu/Find medicine/Humanmedicines/European Public Assessment Reports]
EMA 2011
- European Medicines Agency. Committee for medicinal products for human use (CHMP). Gylenia summary of product characteristics. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Public_assessment_report/human/002202/WC500104529.pdf (accessed December 2015).
FDA 2013
- US Food, Drug Administration. Safety. http://www.fda.gov/safety/medwatch/howtoreport/ucm053087.htm (accessed December 2015).
FDA 2010
- US Food and Drug Administration. Gilenya (Fingolimod) Product Approval Information 2010. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.Set_Current_Drug&ApplNo=022527&DrugName=GILENYA&ActiveIngred=FINGOLIMOD&SponsorApplicant=NOVARTIS&ProductMktStatus=1&goto=Search.DrugDetails 2010; Vol. (accessed December 2015).
FDA 2015
- Drug Safety Communication. Gilenya (Fingolimod): Drug Safety Communication ‐ FDA Warns About Cases of Rare Brain Infection. http://www.fda.gov/safety/medwatch/safetyinformation/safetyalertsforhumanmedicalproducts/ucm457183.htm (accessed December 2015).
Fox 2012
- Fox RJ, Rudick RA. Risk stratification and patient counselling for natalizumab in multiple sclerosis. Neurology 2012;78:436‐7. [DOI] [PubMed] [Google Scholar]
Freedman 2013
- Freedman MS. Treatment options for patients with multiple sclerosis who have a suboptimal response to Interferon‐ß therapy. European Journal of Neurology 2014;21:377‐87. [DOI] [PubMed] [Google Scholar]
Gajofatto 2015
- Gajofatto A, Turatti M, Monaco S, Benedetti MD. Clinical efficacy, safety, and tolerability of fingolimod for the treatment of relapsing‐remitting multiple sclerosis. Drug Healthcare and Patient Safety 2015;7:157‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
Giovannoni 2015
- Giovannoni G, Turnera B, Gnanapavana S, Offiahc C, Schmierera K, Marta M. Is it time to target no evident disease activity(NEDA) in multiple sclerosis?. Multiple Sclerosis and Related Disorders 215;4:329–33. [DOI] [PubMed] [Google Scholar]
Gold 2011
- Gold R. Oral therapies for multiple sclerosis: A review of agents in phase III development or recently approved. CNS Drugs 2011;25(1):37‐52. [DOI] [PubMed] [Google Scholar]
Higgins 2011a
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Higgins 2011b
- Higgins JPT, Altman DG, Sterne JAC (editors). Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Hillert 2012
- Hillert J. In the coming year we should abandon Interferons and glatiramer acetate as first line therapy for MS: no. Multiple Sclerosis 2013;19(1):26‐8. [DOI] [PubMed] [Google Scholar]
Hutchinson 2014
- Hutchinson M, Fox R, Havrdova E, Kurukulasuriya N, Sarda S, Agarwal S, et al. Efficacy and safety of BG‐12 (dimethyl fumarate) and other disease‐modifying therapies for the treatment of relapsing‐remitting multiple sclerosis: a systematic review and mixed treatment comparison. Current Medical Research and Opinion 2014;30(4):613‐27. [DOI] [PubMed] [Google Scholar]
Izquierdo 2013
- Izquierdo G, O'Connor P, Montalban X, Rosenstiel P, Cremer M, Vera A, et al. Five‐year results from a phase 2 study or oral fingolimod in relapsing multiple sclerosis. Multiple Sclerosis 2014;20:877‐81. [DOI] [PubMed] [Google Scholar]
Jenkinson 1999
- Jenkinson C, Stewart‐Brown S, Petersen S, Paice C. Assessment of the SF‐36 version 2 in the United Kingdom. Journal of Epidemiology and Community Health 1999;53:46‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kappos 2015b
- Kappos L, De SN, Freedman MS, Cree BA, Radue EW, Sprenger T, et al. Inclusion of brain volume loss in a revised measure of 'no evidence of disease activity' (NEDA‐4) in relapsing‐remitting multiple sclerosis. Multiple Sclerosis 2015 Nov 19 [Epub ahead of print]. [DOI] [PMC free article] [PubMed]
Koch‐Henriksen 2010
- Koch‐Henriksen N, Sørensen PS. The changing demographic pattern of multiple sclerosis epidemiology. Lancet Neurology 2010;9:520–32. [DOI] [PubMed] [Google Scholar]
Koutsouraki 2010
- Koutsouraki E, Costa V, Baloyannis S. Epidemiology of multiple sclerosis in Europe: a review. International Review of Psychiatry 2010;22(1):2‐13. [DOI] [PubMed] [Google Scholar]
Kremenchutzky 2014
- Kremenchutzky M, O'Connor P, Hohlfeld R, Zhang‐Auberson L, Rosenstiel P, Meng X, et al. Impact of prior treatment status and reasons for discontinuation on the efficacy and safety of fingolimod: Subgroup analyses of the Fingolimod Research Evaluating Effects of Daily Oral Therapy in Multiple Sclerosis (FREEDOMS) study. Multiple Sclerosis and Related Disorders. 2014; Vol. 3:341–9. [DOI] [PubMed]
Kurtzke 1983
- Kurtzke J. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS). Neurology 1983;33:1444‐52. [DOI] [PubMed] [Google Scholar]
Lavery 2014
- Lavery AM, Verhey LH, Waldman AT. Outcome measures in relapsing‐remitting multiple sclerosis: capturing disability and disease progression in clinical trials. Multiple Sclerosis International 2014;2014:1‐13. [ID 262350] [DOI] [PMC free article] [PubMed] [Google Scholar]
Liu 2013
- Liu J, Zhang C, Tao W, Liu M. Systematic review and meta‐analysis of the efficacy of sphingosine‐1‐phosphate (S1P) receptor agonist FTY720 (fingolimod) in animal models of stroke. The International Journal of Neuroscience 2013;123(3):163‐9. [DOI] [PubMed] [Google Scholar]
Lu 2013
- Lu E, Wang BW, Alwan S, Synnes A, Dahlgreen L, Sadovnick A, et al. A review of safety‐related pregnancy data surrounding the oral disease‐modifying drugs for multiple sclerosis. CNS Drugs 2014;28(2):89‐94. [DOI] [PubMed] [Google Scholar]
Lublin 2014
- Lublin FD, Reingold SC, Cohen JA, Cutter GR, Sørensen PS, Thompson AJ, et al. Defining the clinical course of multiple sclerosis. The 2013 revisions. Neurology 2014;83:278–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
Matloubian 2004
- Matloubian M, Lo CG, Cinamon G, Lesneski MJ, Xu Y, Brinkmann V, et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature 2004;427:355‐60. [DOI] [PubMed] [Google Scholar]
Mc Donald 2001
- McDonald WI, Compston A, Edan G, Goodkin D, Hartung HP, Lublin FD, et al. Recommended diagnosis criteria for multiple sclerosis: guidelines from the International Panel on the diagnosis of multiple sclerosis. Annals of Neurology 2001;50(1):121‐7. [DOI] [PubMed] [Google Scholar]
Miron 2008
- Miron VE, Jung CG, Kim HJ, Kennedy TE, Soliven B, Antel GP. FTY720 modulates human oligodendrocyte progenitor process extension and survival. Annals of Neurology 2008;63(1):61‐71. [DOI] [PubMed] [Google Scholar]
Moher 2009
- Moher D, Liberati A, Tetzlaff J, Altman DG, The PRISMA Group. Preferred reporting items for systematic reviews and meta‐analyses: The PRISMA Statement. BMJ 2009;339:2535. [PMC free article] [PubMed] [Google Scholar]
Montalban 2011a
- Montalban X, Comi G, O'Connor P, Gold S, Vera A, Eckert B, et al. Oral fingolimod (FTY720) in relapsing multiple sclerosis: impact on health‐related quality of life in a phase II study. Multiple Sclerosis 2011;17(11):1341‐50. [DOI] [PubMed] [Google Scholar]
Montalban 2011b
- Montalban X, O'Connor P, Izquierdo G, Rosenstiel P, Cremer M, Prut L, et al. Long‐term Fingolimod (FTY720) in relapsing MS: 5‐year results from an extension of a phase II, multicentre study show a sustained low level of disease activity. Abstract meeting of the 5th Joint Triennal Congress of The European and Americas Committees for Treatment and Research in Multiple Sclerosis, 2011, Amsterdam, The Netherlands. Multiple Sclerosis. 2011; Vol. 17.
Montalban 2012
- Montalban X, Comi G, Antel J, O'Connor P, Vera A, Cremer M, et al. Long‐term (>7‐year) efficacy and safety data from a phase II extension study of fingolimod in relapsing multiple sclerosis. Journal of Neurology 2012;259(1):S69‐70. [Google Scholar]
Nixon 2014
- Nixon R, Bergvall N, Tomic D, Sfikas N, Cutter G, Giovannoni G. No evidence of disease activity: indirect comparisons of oral therapies for the treatment of relapsing–remitting multiple sclerosis. Advances in Therapy 2014;31:1134–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
Noseworthy 2000
- Noseworthy JH, Lucchinetti CF, Rodriguez M, Weinshenker BG. Multiple Sclerosis. New England Journal of Medicine 2000;343:938‐52. [DOI] [PubMed] [Google Scholar]
Novartis 2016
- Novartis. Novartis provides on fingolimod phase III trial in primary progressive MS (PPMS). https://www.novartis.com/news/media‐releases/novartis‐provides‐update‐fingolimod‐phase‐iii‐trial‐primary‐progressive‐ms‐ppms (accessed 18 January 2016).
O'Connor 2009
- O'Connor P, Comi G, Montalban X, Antel J, Radue EW, Vera A, et al. FTY720 D2201 Study Group. Oral fingolimod (FTY720) in multiple sclerosis: two‐year results of a phase II extension study. Neurology 2009;72(1):73‐9. [DOI] [PubMed] [Google Scholar]
Oh J 2013
- Oh J, O'Connor PW. Safety, tolerability and efficacy of oral therapies for relapsing‐remitting multiple sclerosis. CNS Drugs 2013;27:591‐609. [DOI] [PubMed] [Google Scholar]
Parfenov 2013
- Parfenov V, Schluep M, Du Pasquier R. Assessing risk of multiple sclerosis therapies. Journal of the Neurological Sciences 2013;15:59‐65. [DOI] [PubMed] [Google Scholar]
Paugh 2006
- Paugh S, Cassidy M, He H, Milstien S, Sim‐Selley L, Spiegel S, et al. Sphingosine and its analog, the immunosuppressant 2‐Amino‐2‐(2‐[4‐octylphenyl]ethyl)‐1,3‐propanediol, interact with the CB1 cannabinoid receptor. Molecular Pharmacology 2006;70:41–50. [DOI] [PubMed] [Google Scholar]
Pinschewer 2000
- Pinschewer DD, Ochsenbein AF, Odermatt B, Brinkmann V, Hengartner H, Zinkernagel RM. FTY720 immunosuppression impairs effector T cell peripheral homing without affecting induction, expansion, and memory. Journal of Immunology 2000;164:5761‐70. [DOI] [PubMed] [Google Scholar]
Pittock 2004
- Pittock SJ, McClelland RL, Mayr WT, Jorgensen NW, Weinshenker BG, Noseworthy J, et al. Clinical implications of benign multiple sclerosis: a 20‐year population‐based follow‐up study. Annals of Neurology 2004;56:303‐6. [DOI] [PubMed] [Google Scholar]
Polman 2005
- Polman C, Reingold S, Edan G, Filippi M, Hartung H, Kappos L, et al. Diagnostic criteria for multiple sclerosis: 2005 revisions to the “McDonald Criteria”. Annals of Neurology 2005;58:840–6. [DOI] [PubMed] [Google Scholar]
Polman 2011
- Polman C, Reingold S, Banwell B, Clanet M, Cohen J, Filippi M, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the MacDonald criteria. Annals of Neurology 2011;69(2):292‐302. [DOI] [PMC free article] [PubMed] [Google Scholar]
RevMan 2015 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2015.
Richards 2002
- Richards RG, Sampson FC, Beard SM, Tappenden P. A review of the natural history and epidemiology of multiple sclerosis: implications for resource allocation and health economic models. Health Technology Assessment 2002;6(10):1‐73. [PUBMED: 12022938] [DOI] [PubMed] [Google Scholar]
Rotstein 2015
- Rotstein DL, Healy BC, Malik MT, Chitnis T, Weiner HL. Evaluation of no evidence of disease activity in a 7‐year longitudinal multiple sclerosis cohort. JAMA 2015;72(2):152‐8. [DOI] [PubMed] [Google Scholar]
Scalfari 2010
- Scalfari A, Neuhaus A, Degenhardt A, Rice P, Muraro P, Daumer M, et al. The natural history of multiple sclerosis, a geographically based study 10: relapses and long‐term disability. Brain 2010;133:1914–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
Scalfari 2013
- Scalfari A, Neuhaus A, Daumer M, Deluca GC, Muraro PA, Ebers GC. Early relapses, onset of progression, and late outcome in multiple sclerosis. JAMA 2013;70(2):214‐22. [DOI] [PubMed] [Google Scholar]
Tramacere 2015
- Tramacere I, Giovane C, Salanti G, D'Amico R, Filippini G. Immunomodulators and immunosuppressants for relapsing‐remitting multiple sclerosis: a network meta‐analysis. Cochrane Database of Systematic Reviews 2015, Issue 9. [DOI: 10.1002/14651858.CD011381.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Vickrey 1995
- Vickrey BG, Hays RD, Harooni R, Harooni R, Ellison GW. A health‐related quality of life measure for multiple sclerosis. Quality of Life Research 1995;4(3):187‐206. [DOI] [PubMed] [Google Scholar]
Warrender‐Sparkes 2015
References to other published versions of this review
Shaneh 2011
- Shaneh Saz A, Firwana BM, Hasan R, Kojan S, Mantia L, Filippini G. Fingolimod for relapsing remitting multiple sclerosis. Cochrane Database of Systematic Reviews 2011, Issue 10. [DOI: 10.1002/14651858.CD009371] [DOI] [PMC free article] [PubMed] [Google Scholar]