

Abstract
Zika virus (ZIKV) is a re-emerging mosquito-borne flavivirus that can have devastating health consequences. The developmental and neurological effects from a ZIKV infection arise in part from the virus triggering cellular stress pathways and perturbing transcriptional programs. To date, the underlying mechanisms of transcriptional control directing viral restriction and virus-host interaction are understudied. Activating Transcription Factor 3 (ATF3) is a stress-induced transcriptional effector that modulates the expression of genes involved in a myriad of cellular processes, including inflammation and antiviral responses, to restore cellular homeostasis. While ATF3 is known to be upregulated during ZIKV infection, the mode by which ATF3 is activated and the specific role of ATF3 during ZIKV infection is unknown. In this study, we show via inhibitor and RNA interference approaches that ZIKV infection initiates the integrated stress response pathway to activate ATF4 which in turn induces ATF3 expression. Additionally, by using a CRISPR-Cas9 system to deplete ATF3, we found that ATF3 acts to limit ZIKV gene expression in A549 cells. In particular, the ATF3-dependent anti-ZIKV response occurred through regulation of innate immunity and autophagy pathways. We show that ATF3 differentially regulates the expression of innate immune response genes and suppresses the transcription of autophagy related genes to influence autophagic flux. Our study therefore highlights an important role for the integrated stress response pathway and ATF3 in establishing an antiviral effect during ZIKV infection.
Keywords: Zika virus, Flavivirus, Transcription Factor, Integrated Stress Response, Autophagy, Innate immune response
Introduction
Zika virus (ZIKV) is a flavivirus that is spread mainly by Aedes mosquitoes (1) and causes self-limiting infections characterized by mild symptoms such as fever, headache, and joint pain (2). The re-emergence of ZIKV from 2007 to 2016 produced large outbreaks in Yap Island, French Polynesia, and the American region (2-4). These outbreaks implicated the virus in intrauterine-linked complications termed congenital Zika syndrome which includes microcephaly, congenital malformations, and fetal demise (5, 6). Additionally, the recent surges in infection also revealed an association with Guillain-Barre syndrome, a neurological disease which results in paralysis and affects adults (7). Combined these damaging effects make re-emerging ZIKV a significant public health challenge (8),which is worsened in part due to the different transmission routes and the absence of antiviral drugs and vaccines. Improving our understanding of the core mechanisms of viral processes, virus-host interactions, and viral restriction may provide valuable clues to help offset this re-emerging public health challenge.
ZIKV has a single-stranded positive-sense RNA genome, approximately 11,000 nucleotides in length, that is translated into a single polyprotein upon viral entry into a host cell. Viral translation occurs on the endoplasmic reticulum (ER) membrane and is followed by proteolytic cleavage of the polyprotein. This process produces structural proteins (capsid [C], precursor membrane [prM], envelope [E]) involved in virus formation and non-structural proteins required for protein processing (NS2B and NS3), viral replication (NS1, NS2A, NS3. NS4A, NS4B, NS5, RNA dependent RNA polymerase [RdRp]), and immune evasion (NS1, NS5) (9, 10). After these viral proteins are made, the viral genome is replicated on the ER membrane. This process triggers extensive remodeling of the membrane as host proteins together with viral nonstructural (NS) proteins assemble to form the replication complex (11-13). The replicated genome subsequently associates with structural proteins to form the nascent virion on the ER membrane at sites juxtaposed to the replication complex (9). As a result of the immense structural changes induced, and the accumulation of misfolded proteins in the ER, cellular homeostasis is disrupted. In response, the cell activates two distinct but overlapping signaling networks namely the unfolded protein response (UPR) and the Integrated Stress Response (ISR) (14).
The ISR is a large network of signaling pathways in eukaryotic cells that is stimulated by external and internal stressors including viral infection, nutrient deprivation, and ER stress. These stressors activate a four-member family of eIF2α kinases, PERK (Protein Kinase R-like ER kinase), PKR (Protein Kinase R; a double-stranded RNA-dependent protein kinase), GCN2 (general control non-derepressible-2) and HRI (heme-regulated eIF2α kinase) (14). All four kinases share sequence similarity in their catalytic domains but have different regulatory domains (15). Therefore, each kinase responds to a distinct stress, but all target the translation initiation factor eIF2 and phosphorylate the serine 51 residue of the alpha subunit (15). This phosphorylation event inhibits the guanine nucleotide exchange factor for the eIF2 complex, eIF2B and prevents the assembly of translation pre-initiation complexes (16). Ultimately, eIF2α phosphorylation represses global cap-dependent translation but promotes the preferential translation of select mRNAs that play key roles in resolving the stress (17).
Activating transcription factor 4 (ATF4) is one of the best studied ISR-specific effector proteins that acts as a master regulator of stress and is selectively translated through a mechanism involving the activation of upstream open reading frames upon eIF2α phosphorylation (18). When induced, ATF4 controls the transcriptional programs of a cohort of genes involved in cell survival or cell death. The overall outcome of ATF4 expression is context specific and is influenced by the cell type, type of stressor and the duration of stress (19, 20). One target of ATF4 is Activating Transcription Factor 3 (ATF3), another ISR gene activated in response to stress. Depending on the cellular environment or nature of the stress, ATF3 can be activated by other effectors beside ATF4 (21). Like ATF4, ATF3 belongs to the ATF/CREB family of transcription factors and can function as either a transcriptional activator or repressor (22). It has a DNA binding domain as well as a basic leucine zipper (bZip) region that is important for dimer formation (23). When promoting transcription of target genes, ATF3 heterodimerizes with other bZip proteins like c-JUN, while in a repressive role, ATF3 forms homodimers or stabilizes inhibitory co-factors at promoter sites (23, 24). Generally, ATF3 modulates various cellular processes like autophagy, innate immune and inflammatory responses, DNA damage response, and cell cycle progression (21). During viral infection, activation of ATF3 produces paradoxical outcomes (25-28). Notably during Japanese encephalitis virus (JEV) infections, ATF3 putatively repressed the expression of select interferon stimulated and autophagy genes to enhanced viral protein and RNA levels (26). Like ZIKV, JEV is a neurotropic mosquito-borne flavivirus. In contrast however, JEV is phylogenetically grouped into a different clade within the flavivirus genus. Given that ATF3 has both pro- and viral functions (25-28), we wondered if ATF3 might exhibit similar or different activities during ZIKV infection.
Our recent global transcriptome analysis of human neuronal cells infected with ZIKV and Dengue virus (DENV) also revealed a connection with ATF3 (29). Specifically, RNA-seq and gene ontology analyses of human SH-SY5Y neuronal cells infected with two strains of ZIKV, Uganda (MR799) and Puerto Rico (PRVABC59), and DENV serotype 2 revealed an upregulation of immune response genes in both ZIKV strains but not in DENV. Additionally, genes involved in cellular responses were significantly upregulated particularly in PRVABC59 infected cells, including genes associated with both ER stress and the UPR pathway (ATF4, ATF3 and CHOP/DDIT3) (29). Elevated ATF4 expression suggested that the ISR pathway was putatively activated during ZIKV PRVABC59 infection, which in turn would stimulate ATF3 expression and downstream targets like CHOP for stress management. However, the functional significance of ATF3 in ZIKV infection and if it has pro- or anti-viral functions, had not been determined.
In this study, we used ISR-specific inhibitors and ATF4 gene silencing approaches to show that depletion of ATF4 decreased ZIKV gene expression and the ISR pathway stimulated ATF4 expression which directly activated ATF3 during ZIKV infection. We further demonstrated that in the absence of ATF3, ZIKV protein and RNA levels increased indicating that ATF3 functioned to restrict viral infection. Finally, we determined that knockout of ATF3 enhanced the expression of autophagy genes and differentially affected the expression of anti-viral innate immune genes during ZIKV infection. Our data reveal the overlapping effects of ATF3 regulation within the cell and highlight that ATF3-driven cross regulation of innate immunity and autophagy pathways collectively impedes ZIKV infection.
Results
ZIKV promotes strong ATF3 expression 24-hours post infection.
In a previous gene expression study, we observed that ZIKV PRVABC59 infection in a neuronal cell line (SH-SY5Y) stimulated immune and stress response genes such as ATF3 and CHOP (29). ZIKV is known to rearrange ER membranes and activate the UPR (11). To investigate ATF3 expression in uninfected A549 lung adenocarcinoma cells in response to ER stress, we first treated A549 cells with tunicamycin, and then examined protein expression at 0.5-, 2-, 4- and 6-hours post-treatment. Tunicamycin inhibits the first step of protein glycosylation to affect the folding of glycosylated proteins in the ER (30). The accumulation of these misfolded proteins in the ER lumen induces ER stress, activation of PERK, a UPR sensor, which phosphorylates eIF2α and enhances translation of ATF4 to induce ATF3 expression (31). By immunoblot analysis we found that ATF4 protein levels increased from 2-hours after tunicamycin treatment with ATF3 protein strongly expressed at 4- and 6-hours after treatment (Figure 1A). Additionally, at 4- and 6-hours post-treatment, RT-qPCR analysis revealed an increase in mRNA expression of ATF3, ATF4 and CHOP (Figure 1B-1D). The coincident upregulation of ATF4 and CHOP mRNA with ATF3 expression is consistent with the target and effector functions of ATF3 (21). Thus, in A549 cells, tunicamycin and the induction of ER stress activates ATF3 expression at 4 and 6 hours.
Figure 1. ATF3 expression is induced by chemical and viral induction of ER stress.

(A-D) A549 cells were treated with 2 nM tunicamycin (TU) and harvested at 0-, 0.5-, 2-, 4- and 6-hours after treatment. (A) Cellular proteins ATF3 and ATF4 were analyzed by western blot. GAPDH was used as the loading control. The western blot shown is representative of at least 3 independent experiments. (B-D) The fold change of ATF4, ATF3 and CHOP mRNA levels relative to β-actin mRNA were also determined by RT-qPCR. (E-J) A549 WT cells were infected with ZIKV PRVABC (moi of 10 PFU/cell) for 0-, 12-, 24- or 48-hours. (E) Cellular and viral proteins were assayed by western blot with GAPDH as the loading control. Protein levels are representative of at least 3 independent experiments. (F-I) RT-qPCR analyses were used to determine the fold change in expression of ATF4, ZIKV, ATF3 and CHOP mRNAs, where the specific mRNA was normalized to β-actin mRNA. (J) Viral titers from virions released into the media at each time point were determined by plaque assay. N=3, and error bars show ± SD. Statistical significance was determined by Student T-test. *p<0.01, **p<0.001, ***p<0.0005, ****p<0.0001, ns-not significant.
To determine when ATF3 was stimulated during ZIKV infection, we infected A549 cells with ZIKV PRVABC59 (moi=10 PFU/cell) and examined viral and cellular proteins and RNA levels at different timepoints following infection. The highest level of the ZIKV nonstructural protein NS1 was observed at 24 hours post-infection and correlated with peak ATF3 protein expression (Figure 1E). ATF4 expression increased from 12- to 24-hours following infection and remained steady until 48 hours. Consistent with this trend, viral, ATF4, ATF3 and CHOP mRNA significantly increased at 24 hours post-infection (Figure 1F-I). Since high viral protein and RNA production occurred at 24 hours post-infection, we reasoned that translation and replication peaked 24 hours after ZIKV infection and declined by 48 hours as virion packaging occurred. As predicted, a high titer of virions was released 48 hours after infection (Figure 1J). We similarly examined ATF3 expression following infection with MR766, the original ZIKV strain isolated in Uganda in 1947 (32, 33). MR766 also induced ATF3 mRNA and protein expression, albeit at 48 hours post-infection compared to 24 hours for PRVABC59 (data not shown). Together, these data indicated that peak viral protein and RNA expression strongly coincided with ATF3 RNA and protein expression.
ATF3 restricts ZIKV gene expression.
To determine the functional importance of ATF3 during ZIKV infection, we generated an ATF3 knock-out (KO) A549 cell line using CRISPR-Cas9 gene editing and a guide RNA targeting exon 2 (Figure S1A). We validated ATF3 KO by sequence analysis (data not shown) and by comparing ATF3 expression in WT and KO cell lines treated with DMSO or tunicamycin (Figure 1B). Indeed, in WT A549 cells ATF3 expression was induced by tunicamycin treatment, but ATF3 protein was absent in the KO cells (Figure S1B). Notably, RT-qPCR analysis showed that ATF3 mRNA was upregulated in the KO cells (Figure S1C). Because the gRNA used to generate the KO cells targets a region within exon 2 which contains the start codon, transcription of ATF3 was not ablated by INDELS introduced during editing but did affect translation of the ATF3 protein (Figure S1A-C). Hence, when the upstream effector of ATF3, which was unaffected in KO cells, was induced upon stress, the effector activated the transcription of ATF3, but downstream translation was impeded.
Next, WT and ATF3 KO cells were mock-infected or infected with ZIKV PRVABC59 at two different moi (1 and 10 PFU/cell). Cells were harvested at 24 hours post-infection, and virus and ATF3 expression examined by western blotting and RT-qPCR. Our data showed that ZIKV infection induced ATF3 protein expression in WT cells but not in ATF3 KO cells (Figure 2A). Interestingly, we found that in ATF3 deficient cells the levels of the ZIKV NS1 protein were notably increased compared to those in WT cells (Figure 2A). Consistent with the increase in ZIKV protein, viral RNA was significantly upregulated in ATF3 deficient cells compared to WT cells (Figure 2B). We additionally performed plaque assays to quantify virion titer produced in WT and ATF3 KO cells and determined that a greater number of infectious particles were produced in the absence of ATF3 (Figure 2D). To validate these data, we also examined ZIKV gene expression in WT and ATF3 KO HCT-116 colorectal cells (34), and observed a similar increase in ZIKV protein and RNA levels (Figure S1D-E). Taken together, these results indicate that ATF3 expression suppressed ZIKV gene expression, and this effect was not cell type specific.
Figure 2. ATF3 expression restricts ZIKV gene expression.

(A) A representative western blot showing ZIKV NS1 and ATF3 expression in both WT and ATF3 KO A549 cell lines. GAPDH was the loading control. (B-C) RT-qPCR analyses of ZIKV and ATF3 RNA levels normalized to β-actin mRNA in KO cells compared to WT cells. (D) Virions released during infection in WT and KO cells was quantified as the average viral titer (PFU/ml) using the plaque assay method. N=3, Error bars show ± SD. Statistical significance was determined by Student T-test. ****p<0.0001, ns-not significant.
ATF3 is activated through the ISR pathway during ZIKV infection.
A number of effector proteins (e.g., ATF4, p53, NF-kB and JNK) associated with different signaling pathways are known to induce ATF3 expression (21). Given that ZIKV induces changes in ER membrane morphology, activates ER stress sensors (IRE-1, ATF6 and PERK) and the presence of double-stranded viral RNA intermediates activate PKR, we reasoned that increased ATF3 expression was initiated through the ISR pathway. Specifically, activation of the ISR kinases during ZIKV infection would lead to a shutdown of cap-dependent translation, increase translation of ATF4, and subsequent activation of ATF3 (Figure 3A). To investigate if the ISR pathway was responsible for ATF3 activation during ZIKV infection, we inhibited the ISR pathway in mock- and ZIKV-infected cells using a general ISR inhibitor (ISRIB). ISRIB acts on eIF2B, a guanine nucleotide exchange factor involved in translation and renders the cells resistant to the effects of eIF2α phosphorylation (35, 36). ISRIB or DMSO (vehicle control) were added to cells 1-hour after the initial virus infection and maintained in the media until cells were harvested at 24 hours post-infection. ZIKV infection in DMSO treated cells elicited strong viral protein and RNA expression, high viral titers, and increased ATF4 levels - all consistent with ZIKV inducing the ISR pathway. However, in the presence of ISRIB, virus protein and RNA expression and virion production decreased (Figure 3B, 3F & 3G). The effects of ISRIB on ZIKV infection were not the result of inhibitor toxicity as a cell viability assay showed that treatment with 500 nM of ISRIB for 24 hours did not affect A549 cell growth (Figure S2A). Thus, the ISR pathway is an important modulator of ZIKV gene expression.
Figure 3. ZIKV activates ATF3 through the Integrated Stress Response (ISR) pathway.

(A) Schematic of the ISR pathway. Stress conditions like virus infections, ER stress, amino acid deprivation and oxidative stress induce stalling of most cap-dependent translation by the phosphorylation of eIF2α and induces the translation of ATF4. ATF4 in turn activates ATF3 to restore cellular homeostasis (11-14). A549 cells were mock-infected or infected with the ZIKV (PRVABC59, moi=10 PFU/cell) in the presence or absence of ISRIB, an ISR inhibitor. Cells were harvested 24-hours post-infection, and (B) cellular and viral proteins analyzed by western blot. The fold change in (C) ZIKV, (D) ATF4, (E) ATF3 and (F) ASNS mRNA levels relative to β-actin mRNA were determined by RT-qPCR. (G) Viral titers were measured by plaque assay. N=3 Error bars show ± SD. Statistical significance was determined by Student T-test. **p<0.001, ***p<0.0005, ****p<0.0001, ns-not significant.
We next examined the consequence of ISRIB on ATF4, the central integrator of the ISR pathway. In mock-infected cells treated without or with ISRIB, ATF4 protein and RNA levels remained unchanged (Figure 3B & 3C). However, in ZIKV-infected ISRIB-treated cells ATF4 protein levels decreased and mirrored the levels in mock-infected cells in the absence or presence of ISRIB. These data support the function of ISRIB as a pharmacological inhibitor of the ISR pathway (Figure 3B & 3C). We also verified the inhibitor activity by measuring the mRNA levels of asparagine synthetase (ASNS), a well characterized downstream target of ATF4 (37, 38). Specifically in the presence of ISRIB, cellular translation would progress and ATF4 protein expression, and that of the downstream targets such as ASNS, would be suppressed. Indeed, ASNS mRNA levels were reduced in both mock- and ZIKV-infected cells treated with ISRIB (Figure 3D). In contrast, ZIKV-infected cells treated with DMSO showed increased ATF4 protein and increased ASNS mRNA abundance (Figure 3C & 3D).
Last, we examined ATF3 protein and mRNA expression (Figure 3B & 3E). ATF3 expression was not activated in mock-infected cells treated with DMSO or ISRIB. As expected, during ZIKV infection ATF3 mRNA and protein were expressed, while in the presence of ISRIB the levels of ATF3 mRNA decreased (Figure 3E). Unexpectedly however, ATF3 protein levels notably increased with ISRIB treatment (Figure 3B). We speculated that during ZIKV infection and inhibitor treatment, the increased ATF3 protein levels might be a result of ATF3 remaining in the cytoplasm and thus being more soluble following cell lysis. To determine if the increase in ATF3 protein levels was the result of a redistribution of this transcription factor between the nucleus and cytoplasm, we performed subcellular fraction on mock- and ZIKV-infected cells treated with DMSO or ISRIB. We examined the nuclear and cytoplasmic fractions by western blot using fibrillarin and β-tubulin as cellular markers for the respective fractions. Subcellular fractionation showed that the increased levels of ATF3 protein in ZIKV-infected cells treated with ISRIB were present in the nuclear fraction (Figure S2B). These results show that following ZIKV infection and inhibition of the ISR pathway, consistent with the transcriptional function ATF3 predominantly localized to the nucleus.
Because ATF3 mRNA levels decreased in ZIKV-infected cells treated with ISRIB but the protein significantly increased (Figure 3E & 3A), we examined whether this response was specific to the broad ISR inhibitor or if an ISR kinase-specific inhibitor would have the same response. We therefore treated mock- and ZIKV-infected cells without or with GSK2606414, an inhibitor that blocks autophosphorylation of PERK (39) and downstream activation of the ISR pathway induced by ER stress (Figure 3A). Similar to the effect of ISRIB, viral protein and RNA were expressed with ZIKV-infection and were decreased with PERK inhibition (Figure S3A & S3B). ATF4 protein and mRNA levels on the other hand increased in ZIKV-infected cells treated with the PERK inhibitor (Figure S3A & S3D), which was likely the result of activation of the other ISR kinases (Figure 3A), such as PKR in response to ZIKV-infection (40, 41). Similar to ZIKV-infected cells in the presence of ISRIB, inhibition of PERK decreased ATF3 mRNA levels and notably increased ATF3 protein levels (Figure S3E & S3A). Overall, these results show that during ZIKV infection, ATF3 is activated through the ISR pathway, and is expected to modulate cellular stress by regulating transcription of specific genes. However, when the ISR pathway is inhibited, ATF3 protein expression may be upregulated, through either translation or inhibition of protein turnover, to control the cellular stress induced during viral infection.
ATF4 is the key activator of ATF3 during ZIKV infection.
Our data show that the ISR pathway is an important regulator of ZIKV gene expression and contributor to ATF3 activation. Thus, we next investigated if the master regulator of the ISR pathway i.e., ATF4 was the upstream activator of ATF3 during ZIKV infection. To this end, we depleted ATF4 with shRNAs stably transduced in A549 cells, and then either mock or ZIKV PRVABC59 infected A549 cells. As a control, we used A549 cells stably expressing a scramble non-targeting shRNA. Viral and cellular protein and RNA were analyzed 24 hours post-infection. To determine if depletion of ATF4 would affect ATF3 expression, we first treated cells with tunicamycin or DMSO (vehicle control) to induce ATF3 expression. In control non-targeting shRNA transduced cells treated with tunicamycin we observed an increase in ATF4 and ATF3 expression (Figure 4A). ZIKV infection upregulated ATF4 and ATF3 protein and RNA abundance (Figure 4A, 4B & Figure S4A-C). Conversely, knock-down of ATF4 significantly reduced ATF3 levels in tunicamycin-treated and ZIKV-infected cells (Figure 4A & 4B, and Figure S4A & S4C). Interestingly, and in contrast to the deletion of ATF3 in A549 cells (Figure 2), we found that depletion of ATF4 decreased ZIKV protein and RNA levels (Figure 4A & 4C). These data suggest that in ZIKV-infected cells, ATF4 is the key activator of ATF3, and ATF4 expression acts to promote ZIKV gene expression.
Figure 4. ATF4 induces ATF3 expression and promotes ZIKV protein and RNA expression.
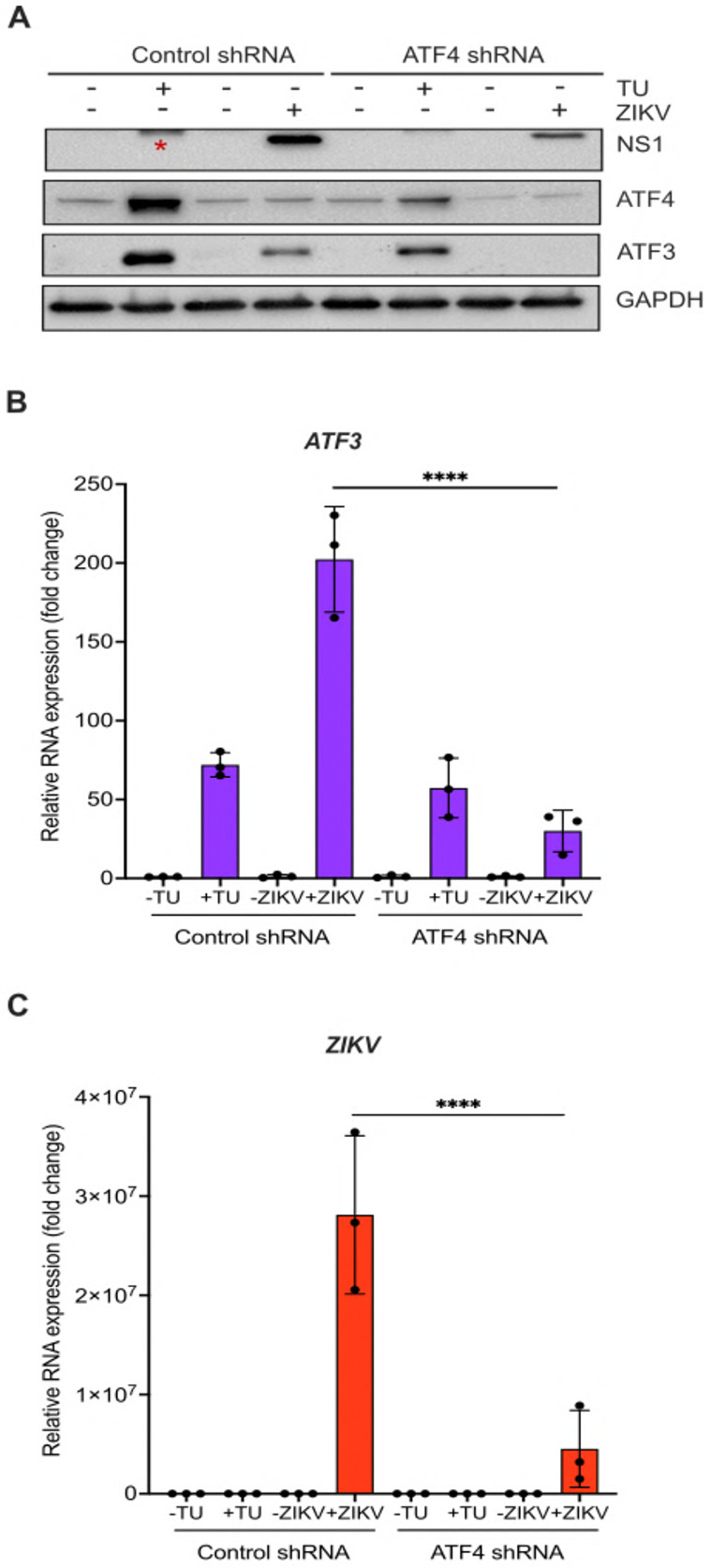
A549 cells expressing either control or ATF4 targeting shRNA were treated with tunicamycin (TU) or infected with ZIKV (moi=10 PFU/cell). (A) ATF4, ATF3 and ZIKV NS1 proteins were assayed via western blot. (B-C) Fold change in ZIKV and ATF3 RNA levels relative to β-actin mRNA were determined by RT-qPCR. N=3 Error bars show ± SD. Statistical significance was determined by Student T-test. **p<0.005, * non-specific band.
ATF3 and ATF4 have opposing effects during ZIKV infection.
Our data show that ATF3 expression has an antiviral role by reducing ZIKV gene expression, while the upstream effector protein ATF4 has a proviral role (Figure 2A, 2B, 4A & 4C). With these opposing functions, we hypothesized that if both ATF3 and ATF4 were depleted, viral expression would be restored to levels comparable with WT infected cells. To test this hypothesis, we transfected WT and ATF3 KO cell lines with either a control siRNA or siRNA targeting ATF4. These cells were then mock-infected or infected with ZIKV (moi=10 PFU/cell). By western blot and RT-qPCR we determined that ATF4 was successfully depleted in both WT and ATF3 KO cells (Figure 5A & 5C). Consistent with the data in Figure 4, depletion of ATF4 in WT cells decreased the abundance of ZIKV protein and RNA, and the activation of ATF3. In line with our prediction, we observed that ZIKV protein and RNA levels were rescued, albeit incomplete, in cells lacking ATF3 and depleted of ATF4 (Figure 5A & 5B). Therefore, ATF3 and ATF4 expression have opposing roles that together modulate the cellular response to ZIKV infection.
Figure 5. ATF3 restricts while ATF4 promotes ZIKV infection.
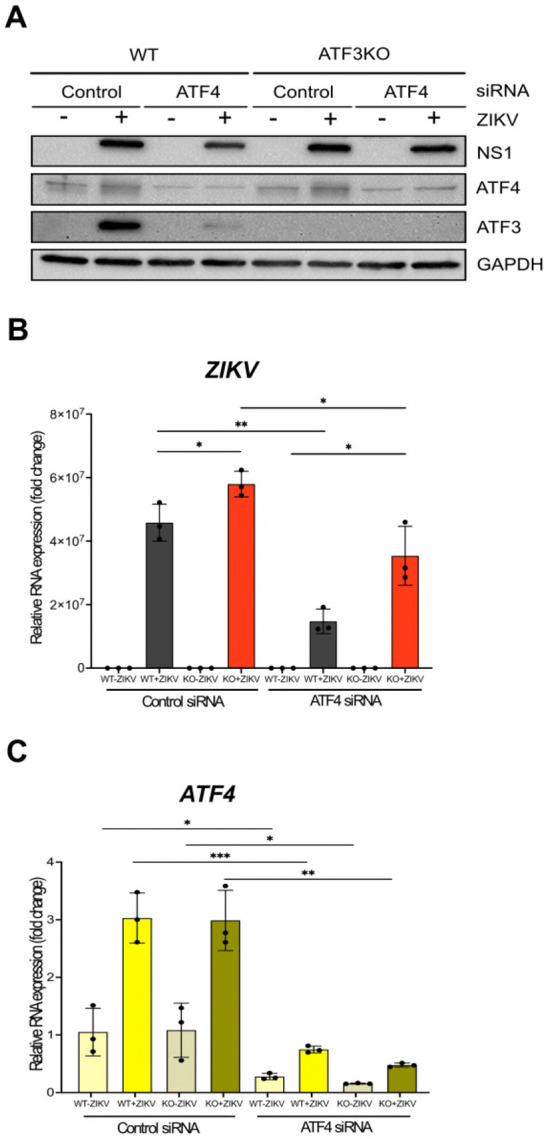
A549 WT and ATF3 KO cells expressing either control or ATF4 targeting siRNA were infected without or with ZIKV (moi=10; −/+Z). (A) ZIKV NS1, ATF4 and ATF3 proteins were analyzed by western blot with GAPDH as the loading control. (B-C) Fold change of ZIKV, ATF4, and ATF3 RNA levels relative to β-actin mRNA were assayed by RT-qPCR. N=3 Error bars show ± SD. Statistical significance was determined by Student t-test. *p < 0.05; **p < 0.01; ***p < 0.001, ns-not significant.
ATF3 regulates the antiviral immune response.
In the absence of ATF3, ZIKV protein, RNA and titers increase (Figure 2). One mode by which ATF3 might restrict ZIKV gene expression is by regulating the transcription of distinct genes that antagonize ZIKV. Indeed, ATF3 has been shown to both stimulate and dampen the immune response (21). In response to viral infection, the innate immune pathway is activated to restrict virus infection (42). In particular, the primary response is initiated by pattern recognition receptors which recognize different viral components and leads to expression of type 1 interferon (IFN β, Figure 6A). The release of interferon initiates the secondary innate immune response and expression of interferon stimulated genes (ISGs) that block different stages of infection (Figure 6A) (43). To determine if ATF3 promotes the expression of antiviral genes, we examined the abundance of select mRNA transcripts involved in either the primary or secondary phases (Figure 6A) of the innate immune response. Many of these genes (RIG-I, STAT1, STAT2, IRF9, ISG15 and IFIT2) were previously reported in murine cells to have predicted ATF3 binding sites in the promoter regions (26). We analyzed expression in WT and ATF3 KO cells that were mock- or ZIKV PRVABC59 (moi of 1 and 10 PFU/cell). The levels of IFN-β mRNA increased in response to ZIKV regardless of the presence or deletion of ATF3 in the A549 cells, with a more robust response in ATF3 KO cells (Figure 6C). Consistent with another report (26), the levels of IFN-α did not change (data not shown). The abundance of RIG-I, STAT1, IRF9 and ISG15 mRNAs decreased in ZIKV-infected ATF3 KO cells compared to WT cells (Figure 6B, 6D, 6E & 6F), suggesting that ATF3 affects the expression of these innate immune response genes. In contrast, the abundance of STAT2 mRNA did not change (data not shown), and the levels of IFIT2 increased in ATF3 KO ZIKV-infected cells (Figure 6G). ATF3 has previously been shown to function as an activator and repressor of transcription (21). Thus, the differential expression of the select mRNA transcripts associated with the innate immune response pathway are likely the consequence of this differential transcriptional regulation of the ATF3-directed host-response to ZIKV infection. Together, these data suggest that in the absence of ATF3, a dampened transcriptional response of select innate immune genes in part facilitates the increase in the abundance of ZIKV protein, RNA, and viral titers.
Figure 6. ATF3 negatively or positively regulates specific antiviral genes during ZIKV infection.

A549 cells WT and ATF3 KO cells were mock-infected or infected with ZIKV PRVABC (moi=1 and 10 PFU/cell) and antiviral gene expression examined 24-hours post infection. (A) Schematic of the antiviral innate immune response pathway. Key antiviral genes assayed at various steps of the pathway are highlighted in blue. (B-G) RT-qPCR analyses of immune response genes RIG-I, IFN-β, STAT1, IRF9, ISG15 and IFIT2. Target RNAs were normalized to β-actin mRNA and fold change determined. N=3, Error bars show ± SD. Statistical significance was determined by Student T-test. *p<0.01, **p<0.001, ***p<0.0005, ****p<0.0001, ns-not significant.
ATF3 limits ZIKV infection by suppressing autophagy.
In addition to modulating ER stress and the innate immune response, ZIKV has also been reported to subvert the autophagy pathway early during infection to promote viral replication (44, 45). Interestingly, in response to stress ATF3 has been shown to bind with the promoter sequences of two autophagy related genes namely Beclin-1 and ATG5 (26, 46). Given this interaction, we first investigated the effect of ATF3 on the expression of autophagy genes during ZIKV infection. These genes are associated with distinct steps in the autophagy pathway and were previously found to be upregulated in JEV-infected neuronal cells depleted of ATF3 (26). A549 WT and ATF3 KO cells were mock-infected or infected with ZIKV at moi of 1 and 10 PFU/cell. At 24-hours post-infection we examined by RT-qPCR the abundance of ATG3, ATG4, ATG5, ATG12, ATG13, ATG15, ATG101, ULK1 and ULK2 genes. In WT cells infected with ZIKV we observed a modest, albeit not significant, increase in ATG5, ATG12, ATG101 and ULK2 mRNAs (Figure 7A-D). In contrast however, in the ZIKV-infected ATF3 KO cells, the levels of these same transcripts were significantly increased (Figure 8A-D). Taken together, these data suggest that activation of ATF3 downregulates the expression of select autophagy genes.
Figure 7. ATF3 negatively regulates select autophagy genes to influence autophagic flux.

A549 cells WT and ATF3 KO cells were mock-infected or infected with ZIKV PRVABC (moi=1 and 10 PFU/cell) and the expression of select autophagy genes examined 24-hours post infection. (A-D) RT-qPCR analyses of autophagy related genes ATG5, ATG12, ATG101 and ULK2 normalized to β-actin mRNA. N=3, Error bars show ± SD. Statistical significance was determined by Student T-test. *p<0.01, **p<0.001, ***p<0.0005. (E-F) A549 WT and ATF3 deficient cells were exposed to starvation media for 1-, 2- and 4 hours. Autophagy markers LC3B and p62/SQSTM1 were examined by western blotting with GAPDH as the loading control. (G-H) A549 WT and ATF3 KO cells were mock-infected or infected with ZIKV PRVABC (moi=1 and 10 PFU/cell) and autophagy-associated proteins LC3B-I, LC3B-II and p62/ SQSTM1 were analyzed by western blot at 24-hours post infection. Immunoblots shown are representatives from 3 independent experiments. (I) p62/ SQSTM1 gene expression relative to β-actin was examined by RT-qPCR. In (G) cells were infected with ZIKV at moi=1 and 10 PFU/cell, while in (H-I) cells were infected with ZIKV at moi=10 PFU/cell. N=3, Error bars show ± SD. Statistical significance was determined by Student T-test. *p<0.01, **p<0.001.
To investigate if upregulation of select autophagy genes in ATF3 KO cells might influence autophagic flux during ZIKV infection we next examined the abundance of two autophagy markers LC3B, which is cleaved from LC3B-I to LC3B-II as autophagy proceeds (47, 48) and p62/SQSTM1, a cargo adapter that is degraded during autophagy (Figure 7) (47, 49, 50). We first examined the consequence on LC3B-II and p62/SQSTM1 under starvation conditions by growing A549 WT and ATF3 KO cells in starvation media for 1, 2 and 4 hours. In WT cells we observed that the levels of LC3B-II and p62/SQSTM1 levels declined with time compared to cells maintained in normal media (Figure 7E). In contrast, in the starved ATF3 KO cells the overall levels of LC3B-II and p62/SQSTM1 proteins appeared elevated compared to WT cells and the abundance of LC3B-II and p62/SQSTM1 modestly decreased after 4 hours of starvation conditions (Figure 7F). These data suggest that in the absence of ATF3, autophagic flux in response to starvation conditions is delayed. Next, we investigated if ATF3 also affected autophagy during ZIV infection. In WT cells, LC3B-II and p62/SQSTM1 protein levels increased during ZIKV infection compared to mock infection (Figure 7G & 7H). Meanwhile in both mock and ZIKV infected ATF3 KO cells, LC3B-II and p62/SQSTM1 protein levels were upregulated (Figure 7G & 7H). Thus, the possible delay of autophagic flux in the absence of ATF3 present in control cells, may be further impaired in virus infected cells. These data suggest that ATF3 may in part restrict ZIKV infection by regulating autophagy and thus also ZIKV replication (45, 46).
Discussion
ATF3 mediates adaptive responses via the positive or negative modulation of cellular processes including immune response, autophagy, and apoptosis (21, 22). For virus infections, ATF3 expression can produce anti-viral outcomes by regulating the transcription of host antiviral genes or benefit the virus by dampening the expression of genes necessary for virus restriction and/or resolution of virus-induced stress (25-28). We previously showed that ATF3 was upregulated during ZIKV infection of SH-SY5Y cells (51), however the upstream effector proteins inducing ATF3 expression and the consequence of ATF3 activation on ZIKV gene expression was unknown.
In this study we determined that peak ATF3 expression coincides with robust ZIKV protein and RNA expression at 24 hours after infection in A549 cells (Figure 1). We identified the ISR pathway as the upstream signaling cascade of ATF3 activation during ZIKV infection (Figure 3 & Figure S3) with ATF4 as the direct effector of ATF3 in this pathway (Figure 4). This observation is consistent with ZIKV activating the ISR through the ER sensor PERK and PKR. Upon stress induction, these kinases phosphorylate eIF2α which attenuate global protein synthesis and trigger ATF4 translation leading to ATF3 induction (52, 53). Finally, we show that ATF3 directs expression of innate immune response and autophagy-related genes to restrict ZIKV gene expression. Taken together these data highlights an important role for the integrated stress response pathway and ATF3 in establishing an antiviral effect during ZIKV infection.
Following activation during infection, the ISR either protects against viral infections or is subverted or blocked to promote viral replication. Evidence of these roles have been demonstrated in several studies involving viruses within the Flaviviridae family (54-59). For example, in hepatitis C virus (HCV) infection studies, PKR as well as PERK and ATF6 were co-opted to support viral replication through inhibiting the IFN pathway and inducing autophagy respectively (55, 56). Similarly, in a JEV infection model, the virus counteracted the antiviral effects of the ISR by specifically blocking PKR activation and eIF2α phosphorylation using viral protein NS2A thereby ensuring effective viral replication (57). In parallel, during DENV infections in Huh7 and A549, stimulation of PERK and IRE-1α signaling led to increased viral replication (58). However, in the case of West Nile virus (WNV), previous reports indicated that infection induced PERK and PKR kinases leading to apoptosis and repressed viral replication (54, 59). Like other flaviviruses, ZIKV infection activated the PERK arm of the ISR pathway in human neural stem cells, and in embryonic mouse cortices after intra-cerebroventricular injection with the virus (60). The resulting increase in ATF4, ATF3 and CHOP mRNA levels caused a neurogenic imbalance which notably however, co-treatment with the PERK inhibitor GSK2656157 attenuated (60). Consistent with these data, in A549 ZIKV-infected cells, we observed that GSK2656157 inhibited PERK activation, restricted translation of ATF4, reduced ATF3 and PERK mRNA accumulation and decreased ZIKV protein and RNA levels (Figure S3). Thus, activation and regulation of the ISR likely has a more significant role in viral infection than previously appreciated.
After ISR activation, eIF2α is phosphorylated leading to a global reduction in cellular protein synthesis. However, noncanonical mechanisms, such as the presence of an internal ribosomal entry sites or upstream open reading frame (uORF), allows for the translation of some cellular mRNAs such as ATF4 during this global reduction (61). In our study, ATF4 RNA and protein levels were upregulated by ZIKV infection accordingly. We determined that shRNA-depletion of ATF4 during tunicamycin- or ZIKV-induced ER-stress, downregulated ATF3 expression indicating that ATF4 activated ATF3. In depleting ATF4, ZIKV protein and RNA expression was also blunted suggesting that expression of ATF4 supports ZIKV infection (Figure 4). Consistent with our findings, other studies demonstrated that ATF4 drives proviral outcomes. Notably, ATF4 was described to promote human immunodeficiency virus 1 (HIV-1), human herpes virus 8 (HHV-8), and murine cytomegalovirus (MCMV) infections by directly controlling transcription (62-66). ATF4 was also found to positively affect porcine reproductive and respiratory syndrome virus (PRRSV), a single-stranded positive-sense RNA virus that replicates in cytoplasm (66). Our finding that ATF4 has proviral functions during ZIKV infection (Figure 4) could therefore be due to activation of ATF4-dependent genes like GADD34 (growth arrest and DNA damage-inducible protein 34) which downregulates the ISR in a negative feedback loop through the recruitment of protein phosphatase 1 (PP1) to dephosphorylate eIF2α (67, 68). In contrast to our results, DENV-2 infection enhanced ATF4 nuclear accumulation to confer an antiviral state (69). Therefore, depending on the virus, ATF4 may positively or negatively regulate viral fate through downstream events. Future studies will explore the mode by which ATF4 positively regulates ZIKV, either transcriptionally or by the protein affecting specific steps in the ZIKV infectious cycle.
When we inhibited the ISR pathway during ZIKV infection using ISRIB, a broad ISR inhibitor (Figure 3) or GSK2606414, a PERK inhibitor (Figure S3), ATF4 protein expression was reduced and ATF3 mRNA levels were negligible. These results align with ATF4 being the upstream effector protein of ATF3 in the ISR pathway. Unexpectedly however, ATF3 protein, but not RNA, levels dramatically increased (Figure 3 and Figure S3) following inhibition of the ISR and ZIKV infection, but not after tunicamycin treatment and inhibition of the PERK pathway (Figure S3 and data not shown). We postulated that this accumulation in ATF3 protein might be a result of this transcription factor not being imported into the nucleus, or being relocalized from the nucleus into the cytoplasm, where the cytosolic form was more soluble, and hence more abundant, than the nuclear form. However, consistent with the transcriptional role of ATF3, we observed that the protein is predominantly in the nucleus (Figure S2). We also considered that, like ATF4, ATF3 might be translationally regulated via an upstream open reading frame (70). Inspection of the 5’ UTR revealed a short UTR length and the absence of an upstream (or downstream) AUG codon that could direct this stress-induced translational control mechanism. We therefore speculate that under the appropriate stress conditions, ATF3 protein levels are regulated by either an alternate translational control mechanism such as via an internal ribosomal entry site and/or protein stability/turnover pathways (71, 72). Indeed, ATF3 protein stability has been shown to be regulated by UBR1/UBR2 and MDM2 ubiquitinases and the ubiquitin-specific peptidase 33 (USP33) protein (73, 74). It is therefore possible that differential expression of ubiquitinases and/or deubiquitinases following inhibition of the ISR pathway during ZIKV infection changes ATF3 protein levels. Additional experiments would need to be undertaken to investigate such regulation. It also remains to be determined if the accumulated ATF3 protein is transcriptionally functional either as an activator or repressor (22).
As a stress response factor, ATF3 is upregulated in response to different viral infections producing positive or negative effects depending on the virus (25-28). During HSV infection, neuronal stress induces ATF3 which binds the promoter region of the HSV LAT RNA and facilitates HSV latency (25). For RNA viruses, ATF3 indirectly affects viral gene expression by transcriptionally controlling the expression of cellular RNAs to promote LMCV, VSV*ΔG(Luc) replicon, MCMV and JEV infections (26, 27, 64, 75). In contrast we find that in ATF3 KO cells, the abundance of ZIKV protein, RNA and titers increase indicating that rather than a proviral role, ATF3 functions to restrict ZIKV infection. Notably, this function was not specific to cell type nor ZIKV isolate (Figure 2 and Figure S1). Despite viral studies indicating a pro- or antiviral function for ATF3, the mechanism by which ATF3 acts to affect the different viruses is poorly described.
ATF3 affects a host of systems, including cell cycle (76), apoptosis (77) neuron regeneration (78, 79), serine and nucleotide biosynthesis (80, 81) and the immune response (21). For the latter, ATF3 functions has been described as a rheostat that regulates the immune response (21). For instance, in ATF3-deficient bone marrow-derived macrophages (BMDM), the expression of IFN-β and other downstream components were upregulated compared to WT cells, and this attenuated LMCV and VSV*ΔG(Luc) replicon infections (28). Likewise in NK cells, ATF3 negatively regulated IFN-γ expression however, the reverse was observed in MCMV infected ATF3 knockout mice compared to WT mice (26). Similarly, interferon stimulated genes (ISGs) were upregulated in JEV infected Neuro2A and MEF cells depleted of ATF3, and chromatin immunoprecipitation studies showed that ATF3 bound to select promoter regions in STAT1, IRF9 and ISG15 (26). Given these prior studies showing ATF3 regulating the immune response, we hypothesized that by transcriptionally controlling genes involved in the innate immune response, ATF3 promotes ISG expression during ZIKV infection. From our data, the absence of ATF3 specifically led to a decrease in the transcription of immune response genes, IRF9, ISG15, RIG-I and STAT1 (Figure 6) which supports the role of ATF3 as a transcriptional activator of these genes during ZIKV infection. It is worth noting that, depletion of ATF3 did not suppress all innate immune effectors as IFIT2 and IFN-β (IFNB1) were upregulated in both WT and ATF3 KO cells, albeit the mRNA levels were further increased in the ATF3 KO cells (Figure 5). In BMDM, two ATF3 binding sites were identified in the promoter and upstream region of IFNB1, where the second binding site functioned to negatively regulate IFNB1 levels (28). It is possible that in our A549 KO system this second binding site is nonfunctional and thus IFNB1 expression is not subjected to feedback regulation. Alternatively, other studies predict that ATF3 potentially suppresses interferon expression by remodeling the nucleosome, keeping the chromosome in a transcriptionally inactive state through interacting with histone deacetylase 1 (28, 82). Future transcriptomic studies defining ATF3 promoter occupancy during ZIKV infection will elucidate how this stress induced transcription factor differentially directs the expression of IFNB1 and other ISGs.
In addition to modulating the immune response, ATF3 also affects autophagy (26, 46), a cellular pathway that is induced and usurped by flaviviruses (83). During JEV infection of cells depleted of ATF3, the levels of select autophagy genes, LC3-II (a marker of autophagy) and ATG5 proteins, were increased (26). Given that ATF3 was shown by others to negatively regulate autophagy and innate immune response for JEV infection (26), we also sought to elucidate the impact of ATF3 on autophagy during ZIKV infection. We determined that during ZIKV infection, ATF3 negatively regulates autophagy as transcript levels of selected autophagy genes, ATG5, ATG12, ATG101 and ULK2 were higher in ZIKV-infected ATF3 knockout cells compared to WT cells (Figure 7A-7D). Since ZIKV gene expression was increased in ATF3 KO cells, we reasoned that the increase in autophagy gene levels and putative autophagy membranes would support increased ZIKV replication compartments. However, for this process to be feasible, ZIKV and/or ATF3 activation would under wildtype conditions be expected to restrict autophagic flux. Indeed, ZIKV has been shown to antagonize selective autophagy which has been shown to have antiviral functions (41, 84). For example, DENV and ZIKV viral NS3 proteases cleaved FAM134B, an ER-localized receptor autophagy machinery component, to prevent ER turnover and increase flavivirus replication (85). Additionally, ZIKV NS5 protein interaction with Adjuba, an initiator of multiprotein complexes and mitotic kinase activator (84, 86, 87), prevented downstream signaling and the selective turnover of mitochondria (41). In WT A549 cells incubated with starvation media, we observed an initial increase in LC3-II and p62/SQSTM1 levels which decreased over time showing the autophagic flux in response to starvation (Figure 7D). In comparison, in A549 ATF3 KO cells the autophagic response was slower (Figure 7E). Notably, the levels of LC3-II were higher in both mock- and ZIKV-infected ATF3 KO cells compared to WT (Figure 7G). While the LC3-II levels in ZIKV-infected ATF3 KO cells raise the possibility that autophagy was induced and stalled, when we examined the levels of p62/SQSTM1 (Figure 7H), we observed a modest decrease in protein levels consistent with the delayed autophagic response to starvation (Figure 7E). Notably, p62/SQSTM1 mRNA transcript levels were increased in ZIKV-infected ATF3 KO cells (Figure 7I), which might compensate for protein turnover (Figure 7H). Thus, the delayed autophagic flux in ATF3 KO cells, might support the increased formation of replication sites on these membranes (88). It is also possible that the ATF3-directed regulation of autophagy might function to interface with the innate immune response to maintain cell homeostasis. Additional experiments are needed to elucidate whether ATF3 regulation of autophagy functions to modulate immune signaling pathways.
Sood and colleagues first showed that ATF3 was upregulated during JEV infection and that RNAi depletion of ATF3 decreased JEV protein and RNA abundances and viral titers (26). Moreover, during JEV infection, ATF3 was reported to negatively regulate antiviral response and autophagy genes, likely by controlling transcription (26). Our data showing a positive effect on immune response gene expression (Figure 6) and a negative effect on autophagy (Figure 7) in ATF3 KO cells contrast data from previous JEV experiments. These differences might be explained by differences in the cell types used in these experiments and effects of dimerization on ATF3 function. ATF3 can have both promoter and repressor functions (89) depending on whether this stress inducible transcription factor homodimerizes or forms a heterodimer with other transcription factors. The JEV studies were undertaken in mouse Neuro2A and mouse embryonic fibroblast cells (26), while we used human A549 lung adenocarcinoma and HCT-116 colorectal carcinoma cells. Differences in the abundance of interacting partners between mouse and human cell lines may influence ATF3 dimerization which in turn may influence the transcriptional responses. Alternatively, as JEV and ZIKV are part of different flavivirus clades the difference in ATF3 function may be related to a virus specific response. Future studies are needed to elucidate the virus genetic determinants that modulate ATF3 function.
In summary, here we show that during ZIKV infection the stress-induced transcription factor ATF3, which is activated through the ISR pathway and ATF4, differentially controls the transcription of select innate immune response and autophagy genes. Our work highlights that transcriptional control of cellular factors such as activating specific transcription factors can be pivotal in cellular response to virus infection.
Materials and Methods
Cell Lines and ZIKV
A549 (Human lung epithelial adenocarcinoma, ATCC CCL185) wild type (WT) and ATF3 knock-out (KO) cell lines were maintained in Dulbecco’s minimal essential medium (DMEM; Life Technologies) supplemented with 10% fetal bovine serum (FBS; Seradigm), 10 mM nonessential amino acids (NEAA; Life Technologies), 5 mM L-glutamine (Life Technologies) and 1% sodium pyruvate (0.055 g/liter; Life Technologies). HCT-116 WT and ATF3 KO cells were grown in McCoy’s 5A media (Corning, #10-050-CV) supplemented with 10% fetal bovine serum (FBS; Seradigm). The HCT-116 wild-type and ATF3 knockout cell lines were generously provided by Dr. Morgan Sammons, University at Albany-SUNY. These cells were maintained in McCoy’s 5A media (Corning) that was supplemented with 10% FBS and 1% penicillin and streptomycin (50,000 units/L penicillin, 0.05 g/L streptomycin; Life Technologies). Vero cells (ATCC CRL-81) were cultured in DMEM supplemented with 10% FBS, 1% penicillin and streptomycin and 10 mM HEPES (Life Technologies). HEK293FT cells (Life Technologies) were grown in DMEM with 10% FBS, 10 mM NEAA and 5 mM L-glutamine. All cell lines were cultured at 37 °C with 5% CO2 in a water-jacketed incubator. ZIKVPR (Puerto Rico PRVABC59) strain was a gift from Dr. Laura Kramer (Wadsworth Center NYDOH) with permission from the CDC. Viral stocks were prepared in C6/36 cells by infecting near confluent cells at an moi of 0.1 and incubating at 27°C. At 7 days post infection, media from infected cells were collected and aliquots were stored at −80°C. To validate infection, RNA was extracted and examine by RT-qPCR and viral titers were measured by plaque assay.
Creating the ATF3 Knock-out (KO) A549 Cell Line
We generated A549 ATF3 KO cells in our laboratory using the CRISPR/Cas9 system. The following gRNA sequence targeting ATF3 was cloned into plentiCRISPRv2 plasmid: 5’-CCACCGGATGTCCTCTGCGC-3’ (Genscript). HEK293FT cells were co-transfected with pLentiCRISPRv2-ATF3 CRISPR gRNA, and pMD2.G (Addgene) and psPAX2 (Addgene) packaging plasmids using JetOptimus DNA transfection reagent (Polyplus) according to the manufacturer’s protocol. Media containing lentivirus was collected 24- and 48-hours post transfection and pooled together. The pooled lentivirus media was filtered with a 0.45 mm pore filter and used to transduce A549 cells in the presence of 6 mg/ml polybrene. Twenty-four hours later, the lentivirus-containing media was removed, replaced with fresh media and cells were incubated at 37°C. After 24 hours of incubation, the transduced cells were transferred into new tissue culture dishes and puromycin (1 mg/ml) selection was carried out for 4 days by which time all A549 WT control cells were killed by the antibiotic. Individual clones were isolated by diluting, seeding in a 96-well plate, and incubating at 37°C. Following expansion, clones were screened by western blotting and RT-qPCR. DNA was then isolated from successful KO clones using DNAzol extraction. PCR was subsequently carried out with forward and reverse primers (5’-CTGCCTCGGAAGTGAGTGCT-3’ and 5’- AACAGCCCCCTGCCTAGAAC-3’) designed to exon 2. The PCR products were cloned into pCR2.1 Topo vector and sequence analyzed by Sanger sequencing to verify the KO.
ZIKV Infection
Cells were previously seeded to be near 80% confluency on day of infection. Control cells were trypsinized and counted to determine the multiplicity of infection (moi). Cells were infected at an moi of 10. An appropriate aliquot of viral stock was thawed at RT, diluted in PBS to a final volume of 1 ml and added to cells. For mock-infected plates, 1 ml of PBS was added. Cells were incubated at 37°C for 1 hour, rocking every 15 minutes. An hour later, 9 ml of media was added per plate and returned to the incubator for 24 hours.
siRNA and shRNA Transfections
Single stranded oligos synthesized by Integrated DNA Technologies (IDT) were used for transient transfections. Sense (5’-CGUACGCGGAAUACUUCGAUU-3’) and anti-sense (5’-UCGAAGUAUUCCGCGUACGUU-3’) oligos targeting the control gene GL2 (90), were prepared by incubating in annealing buffer (150mM Hepes [pH 7.4], 500 mM potassium acetate, and 10 mM magnesium acetate) for 1 minute at 90°C followed by a 1-hour incubation at 37°C. The duplex had a final concentration of 20 μM. Prior to transfection, A549 cells were seeded at 4x105 in 6-well plates for 24 hours. The cells were then transfected with 50 nM control and ATF4 SilencerSelect siRNA (ThermoFisher Scientific, Catalog no. s1702) using Lipofectamine RNAi Max transfection reagent (Invitrogen) based on the manufacturer’s protocol.
Stable transfections were performed following the lentivirus approach. HEK293FT cells were transfected with 1μg of TRC-pLKO.1-Puro plasmid containing either non-targeting shRNA (CAACAAGATGAAGAGCACCAA) or ATF4-targeted shRNA (GCCTAGGTCTCTTAGATGATT) (Sigma-Aldrich), together with 1 μg mixture of packaging plasmids (pMD2.G and psPAX2) prepared in JetPRIME reagent and buffer (Polyplus) as per manufacturer’s instructions. After 24 and 48 hours of transfection, media containing lentivirus was harvested, pooled together, and filtered through a 0.45 μm filter. Pre-seeded A549 cells were subsequently transduced with the lentivirus in the presence of 6ug/ml of polybrene. After 24 hours, the lentivirus-containing media was removed, replaced with fresh media and cells were incubated at 37°C for 24 hours. Following incubation, the transduced cells were transferred into new tissue culture dishes and puromycin (1 mg/ml) selection was carried out for 4 days. Finally, we screened the transfected cells by western blot and RT-qPCR to assess the efficiency of knockdown.
Chemical Treatments
Tunicamycin (Sigma) was dissolved in DMSO at a stock concentration of 2 mM. ER stress was induced by treating cells with 2 mM tunicamycin for 6 hours at 37°C. GSK2606414 (PERK inhibitor; Sigma) was dissolved in DMSO to achieve a 30 mM stock concentration. Cells that were mock and ZIKV infected were co-treated with PERK inhibitor for 24 hours at 37°C., was reconstituted at 5 mM stock concentration in DMSO and used at 500 nM on cells for 24 hours at 37°C.
Harvest of Chemically Treated and ZIKV-infected Cells
Virus infected and chemically treated cells were harvested as follows; first media was aspirated from the cell culture dishes. Cells were gently washed twice with 4 ml cold PBS and aspirated. A volume of 1 ml cold PBS was then added to plates, cells were scraped off the plate using a cell lifter and the cell suspension was thoroughly mixed. Equal volumes of 500 μl were aliquoted into two separate tubes. Cell suspensions were centrifuged at 14,000 rpm for 30 seconds to pellet the cells. The supernatant was aspirated off and cells in one tube were prepared for protein analysis while the other tube was prepared for RNA analysis.
Cell Viability Assay
A549 cells in a 96-well plate were seeded at 4x103 cells/well in 100 μl media and incubated at 37°C 2 days prior to cell viability measurements. The next day, cells were treated with the pharmacological inhibitor in 100 μl of media and incubated at 37°C. After 24 hours, 100 μl of CellTiter-Glo 2.0 reagent (Promega) was added to each well and allowed to equilibrate to room temperature for 30 minutes. The mixture was rocked on an orbital shaker for 2 minutes and incubated in the dark for 10 minutes. The luminescence was read using a Promega GloMax 96 Microplate Luminometer.
Amino Acid Starvation
A549 WT and ATF3 KO cells were washed once with pre-warmed PBS. The cells were then washed twice with pre-warmed starvation medium (140 mM NaCl, 1 mM CaCl2, 1 mM MgCl2, 5 mM glucose, and 20 mM Hepes, pH 7.4) and incubated with starvation medium supplemented with 1% BSA (91).
Western Blot Analysis
Cells were lysed with RIPA buffer (100 mM Tris-HCl pH 7.4, 0.1% sodium dodecyl sulphate (SDS), 1% Triton X-100, 1% deoxycholic acid, 150 mM NaCl) containing protease inhibitors (EDTA-free; ThermoScientific) and incubated on ice for 20 minutes. The lysates were centrifuged at 14,000 rpm for 20 minutes at 4°C and the clarified supernatant collected. Protein concentrations were quantified using the DC protein assay kit (Bio-Rad). Twenty-five micrograms (25 μg) of proteins were separated on 8%, 10% or 12% SDS-polyacrylamide (PAGE) gel at 100 V for 2 hours. Proteins from gels were transferred on to polyvinylidene difluoride membrane (Millipore) at 30V overnight, 100V for 1 hour or 70V for 45 minutes at 4°C, respectively. The blots were activated in absolute methanol and stained with PonceauS (Sigma) to determine transfer efficiency. Subsequently, blots were washed in PBS buffer with 0.1% Tween (PBS-T) and blocked in 5% milk or 5% BSA in PBS-T for 1 hour at room temperature. The blots were incubated with primary antibodies diluted in blocking buffer for 2 hours at room temperature or overnight at 4°C. This was followed with three 10-minute PBS-T washes after which the blots were incubated in secondary antibodies diluted with blocking buffer for 1 hour at room temperature. The blots were washed 3 times in PBS-T and the proteins were visualized using Clarity Western ECL blotting substrate (Bio-Rad) or SuperSignal West Femto (ThermoScientific). The following primary antibodies were used: rabbit anti-ZIKV NS1 (GeneTex; 1:10,000), mouse anti-GAPDH (ProteinTech; 1:10,000), rabbit anti-ATF3 (Abcam; 1:1,000), rabbit anti-ATF4 (D4B8) (Cell Signaling; 1:1,000), rabbit anti-PERK (D11A8) (Cell Signaling; 1:1,000), rabbit anti-eIF2α (Cell Signaling; 1:1,000), rabbit anti-p-eIF2α (D9G8) (Cell Signaling; 1:1,000), rabbit anti-LC3B (D11) (Cell Signaling; 1:1,000) and mouse anti-p62/SQSTM1 (Abnova; 1:4,000). Donkey anti-rabbit-IgG-HRP (Invitrogen), donkey anti-mouse-IgG-HRP (Santa Cruz Biotech) were used as secondary antibodies at a 1:10,000 dilution.
Plaque Assays
Vero cells were seeded in 6-well plates at a density of 7x105/well and incubated at 37°C with 5% CO2 overnight. The following day, ten-fold serial dilutions from 10−1 to 10−6 of media from infections were prepared in 1xPBS. The media on Vero cells seeded the previous day was aspirated, 150 μl of 1xPBS was added to mock well and 150 μl of each virus dilution was added to the remaining wells. The cells were incubated at 37°C with 5% CO2 for 1hour, with gentle rocking every 15 minutes. After incubation, the PBS or virus dilution in PBS was aspirated and 3 ml of overlay consisting of 1:1 2xDMEM (500 mL of RNase-free water, 84 mM of sodium bicarbonate, 5% FBS and 1% penicillin and streptomycin, at pH 7.4) and 1.2% avicel was added to each well and the plates were incubated at 37°C with 5% CO2. At day 5 post-infection, overlay was aspirated, cells were fixed with 1 ml of 7.4% formaldehyde for 10 minutes at room temperature, rinsed with water and plaques developed using 1% crystal violet (Sigma) in 20% methanol.
RT-qPCR Analysis
Total RNA was isolated from cells using TRIzol reagent (Ambion by Life Technologies) and the RNA Clean and Concentrator kit (Zymo Research). The RNA was DNase-treated using the TURBO DNA-free™ kit (Invitrogen) and reverse transcribed using the High-Capacity cDNA Reverse Transcription reagents (Applied Biosystems). The resulting cDNA was used for qPCR analysis with iTaq Universal SYBR Green Supermix reagents (Biorad) and CFX384 Touch Real-Time PCR system (Biorad). The RT-qPCR primer sequences are shown in Table 1.
Table 1:
Primers used for RT-qPCR
| Gene name | Forward (5’-to-3’) | Reverse (5’-to-3’) |
|---|---|---|
| ZIKV | CCTTGGATTCTTGAACGAGGA | AGAGCTTCATTCTCCAGATCAA |
| Beta-actin (ACTB) | GTCACCGGAGTCCATCACG | GACCCAGATCATGTTTGAGACC |
| ATF3 | TGTCAAGGAAGAGCTGAGGTTTG | GATTCCAGCGCAGAGGACAT |
| ATF4 | CAGACGGTGAACCCAATTGG | CAACCTGGTCGGGTTTTGTT |
| ASNS | GGTACATCCCGACAGTGATGATATT | CCTGGACACTATGAAGTTTTGGATT |
| CHOP | CCTGGTTCTCCCTTGGTCTTC | AGCCCTCACTCTCCAGATTCC |
| RIG-I | AGAGCACTTGTGGACGCTTT | ATACACTTCTGTGCCGGGAGG |
| IFN-β | GGCGTCCTCCTTCTGGAACT | GCCTCAAGGACAGGATGAACTT |
| STAT1 | TTCACCCTTCTAGACTTCAGACC | GGAACAGAGTAGCAGGAGGG |
| STAT2 | CGGGACATTCAGCCCTTTTC | TGGCTCTCCACAGGTGTTTC |
| IRF9 | AGCTCTCCTCCAGCCAAGACA | CCAGCAAGTATCGGGCAAAGG |
| IFIT2 | AAGCACCTCAAAGGGCAAAAC | TCGGCCCATGTGATAGTAGAC |
| ISG15 | GTACAGGAGCTTGTGCCGT | GCCTTCAGCTCTGACACCGA |
| ATG3 | GGCAATGGGCTACAGGGGAA | ACCGCTTATAGCACGGCACA |
| ATG5 | AGACCTTCTGCACTGTCCATCT | TGCAATCCCATCCAGAGTTGC |
| ATG12 | AAGTGGGCAGTAGAGCGAACA | TGGTCTGGGGAAGGAGCAAAG |
| ATG13 | CAGGTCCCGGCCTCCGTAAT | TTGTCCAGGTCCTTTCTGTCCT |
| ATG14 | GACCTGGTGGACTCCGTGGAC | GTCGATAAACCTCTCCCGGTCG |
| ATG101 | CGCTCCTCCAGCTTCCGAGT | AAGCTCGGCTCATGCCCTTC |
| ULK2 | CGCCAGAAAACTGATTGGGAGG | TCTGCGAGGTCTCCACCATT |
Statistical Analysis
The data shown is from at least 3 independent experiments. Data was analyzed using Prism 9.4.1 software (GraphPad, La Jolla, CA, USA) to establish statistical significance. We performed two-tailed student t-test for two groups and three-way ANOVA for multiple group comparisons. A P-value of <0.001, <0.01 or <0.05 was considered significant.
Supplementary Material
Figure S1. ATF3 expression is induced by tunicamycin and ZIKV in A549 and HCT-116 cells respectively. (A) A schematic showing ATF3 gene organization, and the target of the guide RNA used to create the ATF3 KO cell line. B) A549 WT and ATF3 KO cells were treated with tunicamycin for 6 hours. ATF3 proteins were analyzed after drug treatment by western blotting and GAPDH served as the loading control. The western blot shown is representative of 3 independent experiments. (B) The fold change of ATF3 mRNA relative to β-actin mRNA in WT and ATF3 KO cells was measured by RT-qPCR after tunicamycin treatment. (C) HCT-116 WT and ATF3 KO cells were infected with ZIKV PRVABC59 (moi=10 PFU/cell) for 48 hours. ATF3 and viral NS1 proteins were analyzed by western blot with GAPDH used as the loading control. ZIKV RNA levels were also determined by RT-qPCR. mRNA levels were determined from 3 independent experiments and error bars show ± SD. Statistical significance was determined by Student T-test. *p<0.05.
Figure S2. ISR inhibition does not affect cell viability or localization of ATF3 during ZIKV infection. (A) A549 WT cells were treated with DMSO or ISRIB or had no treatment, and viable cells were measured as luciferase unit using the cell viability assay. (B) A549 cells were either mock-infected or infected with ZIKV (PRVABC59, moi=10 PFU/cell) in the presence or absence of ISRIB. Cellular and nuclear fractions were prepared from cells harvested 24-hours post-infection. The resultant subcellular fractions were analyzed by immunoblotting and probed with NS1, ATF4, ATF3, fibrillarin and β-tubulin antibodies. Fibrillarin and β-tubulin were used as nuclear and cytoplasmic markers respectively. Results shown are from 3 independent experiments.
Figure S3. PERK inhibition enhances ATF3 protein levels but reduces ATF3 mRNA levels during ZIKV infection. PERK, one of four kinases central to the ISR pathway was targeted by treating A549 mock- or ZIKV-infected (PRVABC59, moi=10 PFU/cell) with or without an inhibitor (GSK2606414). (A) Cellular and viral proteins were analyzed by western blot. The fold change in (B) ZIKV, (C) PERK (D) ATF4 and (E) ATF3 levels relative to β-actin mRNA were determined by RT-qPCR. (F) Cell viability of non-treated cells and cells treated with DMSO, or PERK inhibitor were determined. N=3 Error bars show ± SD. Statistical significance was determined by Student T-test. **p<0.001, ****p<0.0001, ns-not significant.
Figure S4. ATF4 triggers ATF3 expression and positively regulates ZIKV replication and translation. A549 cells expressing either control or ATF4-specific shRNA were treated with tunicamycin (TU) or infected with ZIKV (moi=10 PFU/cell). (A) A representative western blot probed with ATF4, ATF3 and ZIKV NS1 antibodies. (B-C) Relative mRNA expression of ATF4 and ATF3 genes measured as fold change were determined by RT-qPCR. All experiments were done in triplicates. Statistical significance was determined by Student T-test. ***p<0.005, ns-not significant.
Importance.
ZIKV is a re-emerging mosquito-borne flavivirus associated with congenital Zika syndrome in infants and Guillain Barré syndrome in adults. As a cytoplasmic virus, ZIKV co-opts host cellular mechanisms to support viral processes and consequently, reprograms the host transcriptional profile. Such viral-directed transcriptional changes and their pro- or anti-viral significance remain understudied. We previously showed that ATF3, a stress-induced transcription factor, is significantly upregulated in ZIKV infected mammalian cells, along with other cellular and immune response genes. Here, we specifically define the intracellular pathway responsible for ATF3 activation and elucidate the impact of ATF3 expression on ZIKV infection. Our data provides novel insights into the role of the integrated stress response pathway in stimulating ATF3 which differentially regulates the innate immune response and autophagy at the transcript level to antagonize ZIKV gene expression. This study establishes a framework that links viral-induced stress response to transcriptional regulation of host defense pathways and thus expands the depth of knowledge on virus-mediated transcriptional mechanisms during ZIKV infection which in turn will inform future therapeutic strategies.
Acknowledgments
This work was supported by a grant from National Institutes of Health (R01GM123050) to CTP. PB is supported by a generous predoctoral fellowship from the American Heart Association (Award ID: 903514). The research in this manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the NIH or AHA. We thank Dr. Morgan Sammons (University at Albany-SUNY) for the helpful discussions on transcriptional control mechanisms. We also gratefully acknowledge members of the Pager lab, and Drs. Marlene Belfort and John Cleary at UAlbany and The RNA Institute for their thoughtful comments and suggestions on this manuscript.
Abbreviations
- ATF3
Activating transcription factor 3
- ATF4
Activating transcription factor 4
- BMDMs
Bone marrow-derived macrophages
- CHOP
C/EBP homologous protein
- DENV
Dengue virus
- DMSO
Dimethyl sulfoxide
- eIF2α
Eukaryotic initiation factor 2-alpha
- GCN2
General control non-derepressible-2
- HRI
Heme-regulated eIF2α kinase
- IFN
Interferon
- ISG
Interferon stimulated genes
- ISR
Integrated stress response
- ISRIB
Integrated stress response inhibitor
- JEV
Japanese encephalitis virus
- MCMV
murine cytomegalovirus
- NS
Nonstructural
- PKR
Protein kinase R; double-stranded RNA-dependent protein kinase
- PERK
Protein kinase R-like ER kinase
- UPR
Unfolded protein response
- ZIKV
Zika virus
- ZIKV PRVABC59
Zika virus Puerto Rico isolate
References
- 1.Dick GWA. 1952. Zika Virus (I). Isolations and serological specificity. Trans R Soc Trop Med Hyg 46:509–520. [DOI] [PubMed] [Google Scholar]
- 2.Duffy MR, Chen TH, Hancock WT, Powers AM, Kool JL, Lanciotti RS, Pretrick M, Marfel M, Holzbauer S, Dubray C, Guillaumot L, Griggs A, Bel M, Lambert AJ, Laven J, Kosoy O, Panella A, Biggerstaff BJ, Fischer M, Hayes EB. 2009. Zika virus outbreak on Yap Island, Federated States of Micronesia. New England Journal of Medicine 360:2536–2543. [DOI] [PubMed] [Google Scholar]
- 3.Musso D, Nilles EJ, Cao-Lormeau VM. 2014. Rapid spread of emerging Zika virus in the Pacific area. Clinical Microbiology and Infection 20:O595–O596. [DOI] [PubMed] [Google Scholar]
- 4.Baud D, Gubler DJ, Schaub B, Lanteri MC, Musso D. 2017. An update on Zika virus infection. The Lancet 390:2099–2109. [DOI] [PubMed] [Google Scholar]
- 5.Panchaud A, Stojanov M, Ammerdorffer A, Vouga M, Baud D. 2016. Emerging role of Zika virus in adverse fetal and neonatal outcomes. Clin Microbiol Rev. American Society for Microbiology 10.1128/CMR.00014-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoen B, Schaub B, Funk AL, Ardillon V, Boullard M, Cabié A, Callier C, Carles G, Cassadou S, Césaire R, Douine M, Herrmann-Storck C, Kadhel P, Laouénan C, Madec Y, Monthieux A, Nacher M, Najioullah F, Rousset D, Ryan C, Schepers K, Stegmann-Planchard S, Tressières B, Voluménie J-L, Yassinguezo S, Janky E, Fontanet A. 2018. Pregnancy Outcomes after ZIKV Infection in French Territories in the Americas. New England Journal of Medicine 378:985–994. [DOI] [PubMed] [Google Scholar]
- 7.Cao-Lormeau VM, Blake A , Mons S, Lastere S, Roche C, Vanhomwegen J, T Dub T, Baudouin L, A Teissier A, P Larre7, AL Vial8, C Decam9, V Choumet6, SK Halstead10, Prof HJ Willison10, L Musset11, JC Manuguerra5,6, Prof P Despres12, Prof E Fournier13, HP Mal and FG 1Unit. 2016. Guillain-Barré Syndrome outbreak caused by ZIKA virus infection in French Polynesia VM. Lancet 387:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.2016. WHO Statement on the first meeting of the International Health Regulations (2005) Emergency Committee on Zika Virus and observed increase in neurological disorders and neonatal malformations. Saudi Med J. [Google Scholar]
- 9.Sager G, Gabaglio S, Sztul E, Belov GA. 2018. Role of host cell secretory machinery in zika virus life cycle. Viruses 10:2013–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ye Q, Liu ZY, Han JF, Jiang T, Li XF, Qin CF. 2016. Genomic characterization and phylogenetic analysis of Zika virus circulating in the Americas. Infection, Genetics and Evolution 43:43–49. [DOI] [PubMed] [Google Scholar]
- 11.Mohd Ropidi MI, Khazali AS, Nor Rashid N, Yusof R. 2020. Endoplasmic reticulum: A focal point of Zika virus infection. J Biomed Sci 27:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Romero-Brey I, Bartenschlager R. 2016. Endoplasmic reticulum: The favorite intracellular niche for viral replication and assembly. Viruses 8:1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Welsch S, Miller S, Romero-Brey I, Merz A, Bleck CKE, Walther P, Fuller SD, Antony C, Krijnse-Locker J, Bartenschlager R. 2009. Composition and Three-Dimensional Architecture of the Dengue Virus Replication and Assembly Sites. Cell Host Microbe 5:365–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM. 2016. The integrated stress response. EMBO Rep 17:1374–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Donnelly N, Gorman AM, Gupta S, Samali A. 2013. The eIF2α kinases: Their structures and functions. Cellular and Molecular Life Sciences 70:3493–3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clemens M. 1996. Protein kinases that phosphorylate eIF2 and eIF2B, their role in eukaryotic cell translational control. In “Transla- tional Control,” 139–172. [Google Scholar]
- 17.Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, Stojdl DF, Bell JC, Hettmann T, Leiden JM, Ron D. 2003. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell 11:619–633. [DOI] [PubMed] [Google Scholar]
- 18.Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D. 2000. Regulated Translation Initiation Controls Stress-Induced Gene Expression in Mammalian Cells. Mol Cell 6:1099–1108. [DOI] [PubMed] [Google Scholar]
- 19.Wortel IMN, van der Meer LT, Kilberg MS, van Leeuwen FN. 2017. Surviving Stress: Modulation of ATF4-Mediated Stress Responses in Normal and Malignant Cells. Trends in Endocrinology and Metabolism 28:794–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang S, Chen XA, Hu J, Jiang JK, Li Y, Chan-Salis KY, Gu Y, Chen G, Thomas C, Pugh BF, Wang Y. 2015. ATF4 gene network mediates cellular response to the anticancer PAD inhibitor YW3-56 in triple-negative breast cancer cells. Mol Cancer Ther 14:877–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hai T, Wolford CC, Chang YS. 2010. ATF3, a hub of the cellular adaptive-response network, in the pathogenesis of diseases: Is modulation of inflammation a unifying component? Gene Expr 15:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rohini M, Haritha Menon A, Selvamurugan N. 2018. Role of activating transcription factor 3 and its interacting proteins under physiological and pathological conditions. Int J Biol Macromol 120:310–317. [DOI] [PubMed] [Google Scholar]
- 23.Liang G, Wolfgang CD, Chen BPC, Chen T, Hai T. 1996. Guosheng Liang‡, Curt D. Wolfgang‡, Benjamin P. C. Chen‡, Tsu-Hua Chen§ ¶ , and Tsonwin Hai‡§. Biochemistry 271:1695–1701. [Google Scholar]
- 24.Hashimoto Y, Zhang C, Kawauchi J, Imoto I, Adachi MT, Inazawa J, Amagasa T, Hai T, Kitajima S. 2002. An alternatively spliced isoform of transcriptional repressor ATF3 and its induction by stress stimuli. Nucleic Acids Res 30:2398–2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shu M, Du T, Zhou G, Roizman B. 2015. Role of activating transcription factor 3 in the synthesis of latency-associated transcript and maintenance of herpes simplex virus 1 in latent state in ganglia. Proc Natl Acad Sci U S A 112:E5420–E5426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sood V, Sharma KB, Gupta V, Saha D, Dhapola P, Sharma M, Sen U, Kitajima S, Chowdhury S, Kalia M, Vrati S. 2017. ATF3 negatively regulates cellular antiviral signaling and autophagy in the absence of type I interferons. Sci Rep 7:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rosenberger CM, Clark AE, Treuting PM, Johnson CD, Aderem A. 2008. ATF3 regulates MCMV infection in mice by modulating IFN-γ expression in natural killer cells. Proc Natl Acad Sci U S A 105:2544–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Labzin LI, Schmidt S v., Masters SL, Beyer M, Krebs W, Klee K, Stahl R, Lütjohann D, Schultze JL, Latz E, de Nardo D. 2015. ATF3 Is a Key Regulator of Macrophage IFN Responses. The Journal of Immunology 195:4446–4455. [DOI] [PubMed] [Google Scholar]
- 29.Bonenfant G, Meng R, Shotwell C, Badu P, Payne AF, Ciota AT, Sammons MA, Berglund JA, Pager CT. 2020. Asian Zika virus isolate significantly changes the transcriptional profile and alternative RNA splicing events in a neuroblastoma cell line. Viruses 12:510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takatsuki A, Tamura G. 1982. Inhibition of glycoconjugate biosynthesis by tunicamycin, p. 35–70. In Tunicamycin. Japan Scientific Societies Press Tokyo. [Google Scholar]
- 31.Hetz C, Zhang K, Kaufman RJ. 2020. Mechanisms, regulation and functions of the unfolded protein response. Nat Rev Mol Cell Biol. Nature Research 10.1038/s41580-020-0250-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dick GWA. 1952. Zika virus (II). Pathogenicity and physical properties. Trans R Soc Trop Med Hyg 46:521–534. [DOI] [PubMed] [Google Scholar]
- 33.Dick GWA. 1952. Zika Virus (I). Isolations and serological specificity. Trans R Soc Trop Med Hyg 46:509–520. [DOI] [PubMed] [Google Scholar]
- 34.Yan F, Ying L, Li X, Qiao B, Meng Q, Yu L, Yuan X, Ren ST, Chan DW, Shi L, Ni P, Wang X, Xu D, Hu Y. 2017. Overexpression of the transcription factor ATF3 with a regulatory molecular signature associates with the pathogenic development of colorectal cancer. Oncotarget 8:47020–47036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rabouw HH, Langereis MA, Anand AA, Visser LJ, de Groot RJ, Walter P, van Kuppeveld FJM. 2019. Small molecule ISRIB suppresses the integrated stress response within a defined window of activation. Proc Natl Acad Sci U S A 116:2097–2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zyryanova AF, Kashiwagi K, Rato C, Harding HP, Crespillo-Casado A, Perera LA, Sakamoto A, Nishimoto M, Yonemochi M, Shirouzu M, Ito T, Ron D. 2021. ISRIB Blunts the Integrated Stress Response by Allosterically Antagonising the Inhibitory Effect of Phosphorylated eIF2 on eIF2B. Mol Cell 81:88–103.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Linares JF, Cordes T, Duran A, Reina-Campos M, Valencia T, Ahn CS, Castilla EA, Moscat J, Metallo CM, Diaz-Meco MT. 2017. ATF4-Induced Metabolic Reprograming Is a Synthetic Vulnerability of the p62-Deficient Tumor Stroma. Cell Metab 26:817–829.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barbosa-Tessmann IP, Chen C, Zhong C, Schuster SM, Nick HS, Kilberg MS. 1999. Activation of the unfolded protein response pathway induces human asparagine synthetase gene expression. Journal of Biological Chemistry 274:31139–31144. [DOI] [PubMed] [Google Scholar]
- 39.Axten JM, Medina JR, Feng Y, Shu A, Romeril SP, Grant SW, Li WHH, Heerding DA, Minthorn E, Mencken T, Atkins C, Liu Q, Rabindran S, Kumar R, Hong X, Goetz A, Stanley T, Taylor JD, Sigethy SD, Tomberlin GH, Hassell AM, Kahler KM, Shewchuk LM, Gampe RT. 2012. Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}-2,3-dihydro- 1H-indol-5-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK). J Med Chem 55:7193–7207. [DOI] [PubMed] [Google Scholar]
- 40.García MA, Gil J, Ventoso I, Guerra S, Domingo E, Rivas C, Esteban M. 2006. Impact of Protein Kinase PKR in Cell Biology: from Antiviral to Antiproliferative Action. Microbiology and Molecular Biology Reviews 70:1032–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ponia SS, Robertson SJ, McNally KL, Subramanian G, Sturdevant GL, Lewis M, Jessop F, Kendall C, Gallegos D, Hay A, Schwartz C, Rosenke R, Saturday G, Bosio CM, Martens C, Best SM. 2021. Mitophagy antagonism by ZIKV reveals Ajuba as a regulator of PINK1 signaling, PKR-dependent inflammation, and viral invasion of tissues. Cell Rep 37. [DOI] [PubMed] [Google Scholar]
- 42.McFadden MJ, Gokhale NS, Horner SM. 2017. Protect this house: cytosolic sensing of viruses. Curr Opin Virol 10.1016/j.coviro.2016.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McFadden MJ, Gokhale NS, Horner SM. 2017. Protect this house: cytosolic sensing of viruses. Curr Opin Virol. Elsevier B.V. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gratton R, Agrelli A, Tricarico PM, Brandão L, Crovella S. 2019. Autophagy in zika virus infection: A possible therapeutic target to counteract viral replication. Int J Mol Sci 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Echavarria-Consuegra L, Smit JM, Reggiori F. 2019. Role of autophagy during the replication and pathogenesis of common mosquito-borne flavi- and alphaviruses. Open Biol 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lin H, Li HF, Chen HH, Lai PF, Juan SH, Chen JJ, Cheng CF. 2014. Activating transcription factor 3 protects against pressure-overload heart failure via the autophagy molecule Beclin-1 pathway. Mol Pharmacol 85:682–691. [DOI] [PubMed] [Google Scholar]
- 47.Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, Adhihetty PJ, Adler SG, Agam G, Agarwal R, Aghi MK, Agnello M, Agostinis P, Aguilar P V, Aguirre-Ghiso J, Airoldi EM, Ait-Si-Ali S, Akematsu T, Akporiaye ET, Al-Rubeai M, Albaiceta GM, Albanese C, Albani D, Albert ML, Aldudo J, Algül H, Alirezaei M, Alloza I, Almasan A, Almonte-Beceril M, Alnemri ES, Alonso C, Altan-Bonnet N, Altieri DC, Alvarez S, Alvarez-Erviti L, Alves S, Amadoro G, Amano A, Amantini C, Ambrosio S, Amelio I, Amer AO, Amessou M, Amon A, An Z, Anania FA, Andersen SU, Andley UP, Andreadi CK, Andrieu-Abadie N, Anel A, Ann DK, Anoopkumar-Dukie S, Antonioli M, Aoki H, Apostolova N, Aquila S, Aquilano K, Araki K, Arama E, Aranda A, Araya J, Arcaro A, Arias E, Arimoto H, Ariosa AR, Armstrong JL, Arnould T, Arsov I, Asanuma K, Askanas V, Asselin E, Atarashi R, Atherton SS, Atkin JD, Attardi LD, Auberger P, Auburger G, Aurelian L, Autelli R, Avagliano L, Avantaggiati ML, Avrahami L, Awale S, Azad N, Bachetti T, Backer JM, Bae D-H, Bae J, Bae O-N, Bae SH, Baehrecke EH, Baek S-H, Baghdiguian S, Bagniewska-Zadworna A, Bai H, Bai J, Bai X-Y, Bailly Y, Balaji KN, Balduini W, Ballabio A, Balzan R, Banerjee R, Bánhegyi G, Bao H, Barbeau B, Barrachina MD, Barreiro E, Bartel B, Bartolomé A, Bassham DC, Bassi MT, Bast RC, Basu A, Batista MT, Batoko H, Battino M, Bauckman K, Baumgarner BL, Bayer KU, Beale R, Beaulieu J-F, Beck GR, Becker C, Beckham JD, Bédard P-A, Bednarski PJ, Begley TJ, Behl C, Behrends C, Behrens GMN, Behrns KE, Bejarano E, Belaid A, Belleudi F, Bénard G, Berchem G, Bergamaschi D, Bergami M, Berkhout B, Berliocchi L, Bernard A, Bernard M, Bernassola F, Bertolotti A, Bess AS, Besteiro S, Bettuzzi S, Bhalla S, Bhattacharyya S, Bhutia SK, Biagosch C, Bianchi MW, Biard-Piechaczyk M, Billes V, Bincoletto C, Bingol B, Bird SW, Bitoun M, Bjedov I, Blackstone C, Blanc L, Blanco GA, Blomhoff HK, Boada-Romero E, Böckler S, Boes M, Boesze-Battaglia K, Boise LH, Bolino A, Boman A, Bonaldo P, Bordi M, Bosch J, Botana LM, Botti J, Bou G, Bouché M, Bouchecareilh M, Boucher M-J, Boulton ME, Bouret SG, Boya P Boyer-Guittaut M, Bozhkov P V, Brady N, Braga VMM, Brancolini C, Braus GH, Bravo-San Pedro JM, Brennan LA, Bresnick EH, Brest P, Bridges D, Bringer M-A, Brini M, Brito GC, Brodin B, Brookes PS, Brown EJ, Brown K, Broxmeyer HE, Bruhat A, Brum PC, Brumell JH, Brunetti-Pierri N, Bryson-Richardson RJ, Buch S, Buchan AM, Budak H, Bulavin D V, Bultman SJ, Bultynck G, Bumbasirevic V, Burelle Y, Burke RE, Burmeister M, Bütikofer P, Caberlotto L, Cadwell K, Cahova M, Cai D, Cai J, Cai Q, Calatayud S, Camougrand N, Campanella M, Campbell GR, Campbell M, Campello S, Candau R, Caniggia I, Cantoni L, Cao L, Caplan AB, Caraglia M, Cardinali C, Cardoso SM, Carew JS, Carleton LA, Carlin CR, Carloni S, Carlsson SR, Carmona-Gutierrez D, Carneiro LAM, Carnevali O, Carra S, Carrier A, Carroll B, Casas C, Casas J, Cassinelli G, Castets P, Castro-Obregon S, Cavallini G, Ceccherini I, Cecconi F, Cederbaum AI, Ceña V, Cenci S, Cerella C, Cervia D, Cetrullo S, Chaachouay H, Chae H-J, Chagin AS, Chai C-Y, Chakrabarti G, Chamilos G, Chan EYW, Chan MT V, Chandra D, Chandra P, Chang C-P, Chang RC-C, Chang TY, Chatham JC, Chatterjee S, Chauhan S, Che Y, Cheetham ME, Cheluvappa R, Chen C-J, Chen G, Chen G-C, Chen G, Chen H, Chen JW, Chen J-K, Chen M, Chen M, Chen P, Chen Q, Chen Q, Chen S-D, Chen S, Chen SS-L, Chen W, Chen W-J, Chen WQ, Chen W, Chen X, Chen Y-H, Chen Y-G, Chen Y, Chen Y, Chen Y, Chen Y-J, Chen Y-Q, Chen Y, Chen Z, Chen Z, Cheng A, Cheng CHK, Cheng H, Cheong H, Cherry S, Chesney J, Cheung CHA, Chevet E, Chi HC, Chi S-G, Chiacchiera F, Chiang H-L, Chiarelli R, Chiariello M, Chieppa M, Chin L-S, Chiong M, Chiu GNC, Cho D-H, Cho S-G Cho WC, Cho Y-Y, Cho Y-S, Choi AMK. Choi E-J, Choi E-K, Choi J, Choi ME, Choi S-I, Chou T-F, Chouaib S, Choubey D, Choubey V, Chow K-C, Chowdhury K, Chu CT, Chuang T-H, Chun T, Chung H, Chung T, Chung Y-L, Chwae Y-J, Cianfanelli V, Ciarcia R, Ciechomska IA, Ciriolo MR, Cirone M, Claerhout S, Clague MJ, Clària J, Clarke PGH, Clarke R, Clementi E, Cleyrat C, Cnop M, Coccia EM, Cocco T, Codogno P, Coers J, Cohen EEW, Colecchia D, Coletto L, Coll NS, Colucci-Guyon E, Comincini S, Condello M, Cook KL, Coombs GH, Cooper CD, Cooper JM, Coppens I, Corasaniti MT, Corazzari M, Corbalan R, Corcelle-Termeau E, Cordero MD, Corral-Ramos C, Corti O, Cossarizza A, Costelli P, Costes S, Cotman SL, Coto-Montes A, Cottet S, Couve E, Covey LR, Cowart LA, Cox JS, Coxon FP, Coyne CB, Cragg MS, Craven RJ, Crepaldi T, Crespo JL, Criollo A, Crippa V, Cruz MT, Cuervo AM, Cuezva JM, Cui T, Cutillas PR, Czaja MJ, Czyzyk-Krzeska MF, Dagda RK, Dahmen U, Dai C, Dai W, Dai Y, Dalby KN, Dalla Valle L, Dalmasso G, D’Amelio M, Damme M, Darfeuille-Michaud A, Dargemont C, Darley-Usmar VM, Dasarathy S, Dasgupta B, Dash S, Dass CR, Davey HM, Davids LM, Dávila D, Davis RJ, Dawson TM, Dawson VL, Daza P, de Belleroche J, de Figueiredo P, de Figueiredo RCBQ, de la Fuente J, De Martino L, De Matteis A, De Meyer GRY, De Milito A, De Santi M, de Souza W, De Tata V, De Zio D, Debnath J, Dechant R, Decuypere J-P, Deegan S, Dehay B, Del Bello B, Del Re DP, Delage-Mourroux R, Delbridge LMD, Deldicque L, Delorme-Axford E, Deng Y, Dengjel J, Denizot M, Dent P, Der CJ, Deretic V, Derrien B, Deutsch E, Devarenne TP, Devenish RJ, Di Bartolomeo S, Di Daniele N, Di Domenico F, Di Nardo A, Di Paola S, Di Pietro A, Di Renzo L, DiAntonio A, Díaz-Araya G, Díaz-Laviada I, Diaz-Meco MT, Diaz-Nido J, Dickey CA, Dickson RC, Diederich M, Digard P, Dikic I, Dinesh-Kumar SP, Ding C, Ding W-X, Ding Z, Dini L, Distler JHW, Diwan A, Djavaheri-Mergny M, Dmytruk K, Dobson RCJ, Doetsch V, Dokladny K, Dokudovskaya S, Donadelli M, Dong XC, Dong X, Dong Z, Donohue TM, Doran KS, D’Orazi G, Dorn GW, Dosenko V, Dridi S, Drucker L, Du J, Du L-L, Du L, du Toit A, Dua P, Duan L, Duann P, Dubey VK, Duchen MR, Duchosal MA, Duez H, Dugail I, Dumit VI, Duncan MC, Dunlop EA, Dunn WA, Dupont N, Dupuis L, Durán R V, Durcan TM, Duvezin-Caubet S, Duvvuri U, Eapen V, Ebrahimi-Fakhari D, Echard A, Eckhart L, Edelstein CL, Edinger AL, Eichinger L, Eisenberg T, Eisenberg-Lerner A, Eissa NT, El-Deiry WS, El-Khoury V, Elazar Z, Eldar-Finkelman H, Elliott CJH, Emanuele E, Emmenegger U, Engedal N, Engelbrecht A-M, Engelender S, Enserink JM, Erdmann R, Erenpreisa J, Eri R, Eriksen JL, Erman A, Escalante R, Eskelinen E-L, Espert L, Esteban-Martínez L, Evans TJ, Fabri M, Fabrias G, Fabrizi C, Facchiano A, Færgeman NJ, Faggioni A, Fairlie WD, Fan C, Fan D, Fan J, Fang S, Fanto M, Fanzani A, Farkas T, Faure M, Favier FB, Fearnhead H, Federici M, Fei E, Felizardo TC, Feng H, Feng Y, Feng Y, Ferguson TA, Fernández ÁF, Fernandez-Barrena MG, Fernandez-Checa JC, Fernández-López A, Fernandez-Zapico ME, Feron O, Ferraro E, Ferreira-Halder CV, Fesus L, Feuer R, Fiesel FC, Filippi-Chiela EC, Filomeni G, Fimia GM, Fingert JH, Finkbeiner S, Finkel T, Fiorito F, Fisher PB, Flajolet M, Flamigni F, Florey O, Florio S, Floto RA, Folini M, Follo C, Fon EA, Fornai F, Fortunato F, Fraldi A, Franco R, Francois A, François A, Frankel LB, Fraser IDC, Frey N, Freyssenet DG, Frezza C, Friedman SL, Frigo DE, Fu D, Fuentes JM, Fueyo J Fujitani Y, Fujiwara Y, Fujiya M, Fukuda M, Fulda S, Fusco C, Gabryel B, Gaestel M, Gailly P, Gajewska M, Galadari S, Galili G, Galindo I, Galindo MF, Galliciotti G, Galluzzi L, Galluzzi L, Galy V, Gammoh N, Gandy S, Ganesan AK, Ganesan S, Ganley IG, Gannagé M, Gao F-B, Gao F, Gao J-X, García Nannig L, García Véscovi E, Garcia-Macía M, Garcia-Ruiz C, Garg AD, Garg PK, Gargini R, Gassen NC, Gatica D, Gatti E, Gavard J, Gavathiotis E, Ge L, Ge P, Ge S, Gean P-W, Gelmetti V, Genazzani AA, Geng J, Genschik P, Gerner L, Gestwicki JE, Gewirtz DA, Ghavami S, Ghigo E, Ghosh D, Giammarioli AM, Giampieri F, Giampietri C, Giatromanolaki A, Gibbings DJ, Gibellini L, Gibson SB, Ginet V, Giordano A, Giorgini F, Giovannetti E, Girardin SE, Gispert S, Giuliano S, Gladson CL, Glavic A, Gleave M, Godefroy N, Gogal RM, Gokulan K, Goldman GH, Goletti D, Goligorsky MS, Gomes A V, Gomes LC, Gomez H, Gomez-Manzano C, Gómez-Sánchez R, Gonçalves DAP, Goncu E, Gong Q, Gongora C, Gonzalez CB, Gonzalez-Alegre P, Gonzalez-Cabo P, González-Polo RA, Goping IS, Gorbea C, Gorbunov N V, Goring DR, Gorman AM, Gorski SM, Goruppi S, Goto-Yamada S, Gotor C, Gottlieb RA, Gozes I, Gozuacik D, Graba Y, Graef M, Granato GE, Grant GD, Grant S, Gravina GL, Green DR, Greenhough A, Greenwood MT, Grimaldi B, Gros F, Grose C, Groulx J-F, Gruber F, Grumati P, Grune T, Guan J-L, Guan K-L, Guerra B, Guillen C, Gulshan K, Gunst J, Guo C, Guo L, Guo M, Guo W, Guo X-G, Gust AA, Gustafsson ÅB, Gutierrez E, Gutierrez MG, Gwak H-S, Haas A, Haber JE, Hadano S, Hagedorn M, Hahn DR, Halayko AJ, Hamacher-Brady A, Hamada K, Hamai A, Hamann A, Hamasaki M, Hamer I, Hamid Q, Hammond EM, Han F, Han W, Handa JT, Hanover JA, Hansen M, Harada M, Harhaji-Trajkovic L, Harper JW, Harrath AH, Harris AL, Harris J, Hasler U, Hasselblatt P, Hasui K, Hawley RG, Hawley TS, He C, He CY, He F, He G, He R-R, He X-H, He Y-W, He Y-Y, Heath JK, Hébert M-J, Heinzen RA, Helgason GV, Hensel M, Henske EP, Her C, Herman PK, Hernández A, Hernandez C, Hernández-Tiedra S, Hetz C, Hiesinger PR, Higaki K, Hilfiker S, Hill BG, Hill JA, Hill WD, Hino K, Hofius D, Hofman P, Höglinger GU, Höhfeld J, Holz MK, Hong Y, Hood DA, Hoozemans JJM, Hoppe T, Hsu C, Hsu C-Y, Hsu L-C, Hu D, Hu G, Hu H-M, Hu H, Hu MC, Hu Y-C, Hu Z-W, Hua F, Hua Y, Huang C, Huang H-L, Huang K-H, Huang K-Y, Huang S, Huang S, Huang W-P, Huang Y-R, Huang Y, Huang Y, Huber TB, Huebbe P, Huh W-K, Hulmi JJ, Hur GM, Hurley JH, Husak Z, Hussain SNA, Hussain S, Hwang JJ, Hwang S, Hwang TIS, Ichihara A, Imai Y, Imbriano C, Inomata M, Into T, Iovane V, Iovanna JL, Iozzo R V, Ip NY, Irazoqui JE, Iribarren P, Isaka Y, Isakovic AJ, Ischiropoulos H, Isenberg JS, Ishaq M, Ishida H, Ishii I, Ishmael JE, Isidoro C, Isobe K, Isono E, Issazadeh-Navikas S, Itahana K, Itakura E, Ivanov AI, Iyer AK V, Izquierdo JM, Izumi Y, Izzo V, Jäättelä M, Jaber N, Jackson DJ, Jackson WT, Jacob TG, Jacques TS, Jagannath C, Jain A, Jana NR, Jang BK, Jani A, Janji B, Jannig PR, Jansson PJ, Jean S, Jendrach M, Jeon J-H, Jessen N, Jeung E-B, Jia K, Jia L, Jiang H, Jiang H, Jiang L, Jiang T, Jiang X, Jiang X, Jiang X, Jiang Y, Jiang Y, Jiménez A, Jin C, Jin H, Jin L, Jin M, Jin S, Jinwal UK, Jo E-K, Johansen T, Johnson DE, Johnson GVW, Johnson JD, Jonasch E, Jones C, Joosten LAB, Jordan J, Joseph A-M, Joseph B, Joubert AM, Ju D, Ju J, Juan H-F, Juenemann K, Juhász G, Jung HS, Jung JU, Jung Y-K, Jungbluth H, Justice MJ, Jutten B, Kaakoush NO, Kaarniranta K, Kaasik A, Kabuta T, Kaeffer B, Kågedal K, Kahana A, Kajimura S, Kakhlon O, Kalia M, Kalvakolanu D V, Kamada Y, Kambas K, Kaminskyy VO, Kampinga HH, Kandouz M, Kang C, Kang R, Kang T-C, Kanki T, Kanneganti T-D, Kanno H, Kanthasamy AG, Kantorow M, Kaparakis-Liaskos M, Kapuy O, Karantza V, Karim MR, Karmakar P, Kaser A, Kaushik S, Kawula T, Kaynar AM, Ke P-Y, Ke Z-J, Kehrl JH, Keller KE, Kemper JK, Kenworthy AK, Kepp O, Kern A, Kesari S, Kessel D, Ketteler R, Kettelhut I do C, Khambu B, Khan MM, Khandelwal VKM, Khare S, Kiang JG, Kiger AA, Kihara A, Kim AL, Kim CH, Kim DR, Kim D-H, Kim EK, Kim HY, Kim H-R, Kim J-S, Kim JH, Kim JC, Kim JH, Kim KW, Kim MD, Kim M-M, Kim PK, Kim SW, Kim S-Y, Kim Y-S, Kim Y, Kimchi A, Kimmelman AC, Kimura T, King JS, Kirkegaard K, Kirkin V, Kirshenbaum LA, Kishi S, Kitajima Y, Kitamoto K, Kitaoka Y, Kitazato K, Kley RA, Klimecki WT, Klinkenberg M, Klucken J, Knævelsrud H, Knecht E, Knuppertz L, Ko J-L, Kobayashi S, Koch JC, Koechlin-Ramonatxo C, Koenig U, Koh YH, Köhler K, Kohlwein SD, Koike M, Komatsu M, Kominami E, Kong D, Kong HJ, Konstantakou EG, Kopp BT, Korcsmaros T, Korhonen L, Korolchuk VI, Koshkina N V, Kou Y, Koukourakis MI, Koumenis C, Kovács AL, Kovács T, Kovacs WJ, Koya D, Kraft C, Krainc D, Kramer H, Kravic-Stevovic T, Krek W, Kretz-Remy C, Krick R, Krishnamurthy M, Kriston-Vizi J, Kroemer G, Kruer MC, Kruger R, Ktistakis NT, Kuchitsu K, Kuhn C, Kumar AP, Kumar A, Kumar A, Kumar D, Kumar D, Kumar R, Kumar S, Kundu M, Kung H-J, Kuno A, Kuo S-H, Kuret J, Kurz T, Kwok T, Kwon TK, Kwon YT, Kyrmizi I, La Spada AR, Lafont F, Lahm T, Lakkaraju A, Lam T, Lamark T, Lancel S, Landowski TH, Lane DJR, Lane JD, Lanzi C, Lapaquette P, Lapierre LR, Laporte J, Laukkarinen J, Laurie GW, Lavandero S, Lavie L, LaVoie MJ, Law BYK, Law HK, Law KB, Layfield R, Lazo PA, Le Cam L, Le Roch KG, Le Stunff H, Leardkamolkarn V, Lecuit M, Lee B-H, Lee C-H, Lee EF, Lee GM, Lee H-J, Lee H, Lee JK, Lee J, Lee J, Lee JH, Lee M, Lee M-S, Lee PJ, Lee SW, Lee S-J, Lee S-J, Lee SY, Lee SH, Lee SS, Lee S-J, Lee S, Lee Y-R, Lee YJ, Lee YH, Leeuwenburgh C, Lefort S, Legouis R, Lei J, Lei Q-Y, Leib DA, Leibowitz G, Lekli I, Lemaire SD, Lemasters JJ, Lemberg MK, Lemoine A, Leng S, Lenz G, Lenzi P, Lerman LO, Lettieri Barbato D, Leu JI-J, Leung HY, Levine B, Lewis PA, Lezoualc’h F, Li C, Li F, Li F-J, Li J, Li K, Li L, Li M, Li M, Li Q, Li R, Li S, Li W, Li W, Li X, Li Y, Lian J, Liang C, Liang Q, Liao Y, Liberal J, Liberski PP, Lie P, Lieberman AP, Lim HJ, Lim K-L, Lim K, Lima RT, Lin C-S, Lin C-F, Lin F, Lin F, Lin F-C, Lin K, Lin K-H, Lin P-H, Lin T, Lin W-W, Lin Y-S, Lin Y, Linden R, Lindholm D, Lindqvist LM, Lingor P, Linkermann A, Liotta LA, Lipinski MM, Lira VA, Lisanti MP, Liton PB, Liu B, Liu C, Liu C-F, Liu F, Liu H-J, Liu J, Liu J-J, Liu J-L, Liu K, Liu L, Liu L, Liu Q, Liu R-Y, Liu S, Liu S, Liu W, Liu X-D, Liu X, Liu X-H, Liu X, Liu X, Liu X, Liu Y, Liu Y, Liu Z, Liu Z, Liuzzi JP, Lizard G, Ljujic M, Lodhi IJ, Logue SE, Lokeshwar BL, Long YC, Lonial S, Loos B, López-Otín C, López-Vicario C, Lorente M, Lorenzi PL, Lõrincz P, Los M, Lotze MT, Lovat PE, Lu B, Lu B, Lu J, Lu Q, Lu S-M, Lu S, Lu Y, Luciano F, Luckhart S, Lucocq JM, Ludovico P, Lugea A, Lukacs NW, Lum JJ, Lund AH, Luo H, Luo J, Luo S, Luparello C, Lyons T, Ma J, Ma Y, Ma Y, Ma Z, Machado J, Machado-Santelli GM, Macian F, MacIntosh GC, MacKeigan JP, Macleod KF, MacMicking JD, MacMillan-Crow LA, Madeo F, Madesh M, Madrigal-Matute J, Maeda A, Maeda T, Maegawa G, Maellaro E, Maes H, Magariños M, Maiese K, Maiti TK, Maiuri L, Maiuri MC, Maki CG, Malli R, Malorni W, Maloyan A, Mami-Chouaib F, Man N, Mancias JD, Mandelkow E-M, Mandell MA, Manfredi AA, Manié SN, Manzoni C, Mao K, Mao Z, Mao Z-W, Marambaud P, Marconi AM, Marelja Z, Marfe G, Margeta M, Margittai E, Mari M, Mariani F V, Marin C, Marinelli S, Mariño G, Markovic I, Marquez R, Martelli AM, Martens S, Martin KR, Martin SJ, Martin S, Martin-Acebes MA, Martín-Sanz P, Martinand-Mari C, Martinet W, Martinez J, Martinez-Lopez N, Martinez-Outschoorn U, Martínez-Velázquez M, Martinez-Vicente M, Martins WK, Mashima H, Mastrianni JA, Matarese G, Matarrese P, Mateo R, Matoba S, Matsumoto N, Matsushita T, Matsuura A, Matsuzawa T, Mattson MP, Matus S, Maugeri N, Mauvezin C, Mayer A, Maysinger D, Mazzolini GD, McBrayer MK, McCall K, McCormick C, McInerney GM, McIver SC, McKenna S, McMahon JJ, McNeish IA, Mechta-Grigoriou F, Medema JP, Medina DL, Megyeri K, Mehrpour M, Mehta JL, Mei Y, Meier U-C, Meijer AJ, Meléndez A, Melino G, Melino S, de Melo EJT, Mena MA, Meneghini MD, Menendez JA, Menezes R, Meng L, Meng L, Meng S, Menghini R, Menko AS, Menna-Barreto RFS, Menon MB, Meraz-Ríos MA, Merla G, Merlini L, Merlot AM, Meryk A, Meschini S, Meyer JN, Mi M, Miao C-Y, Micale L, Michaeli S, Michiels C, Migliaccio AR, Mihailidou AS, Mijaljica D, Mikoshiba K, Milan E, Miller-Fleming L, Mills GB, Mills IG, Minakaki G, Minassian BA, Ming X-F, Minibayeva F, Minina EA, Mintern JD, Minucci S, Miranda-Vizuete A, Mitchell CH, Miyamoto S, Miyazawa K, Mizushima N, Mnich K, Mograbi B, Mohseni S, Moita LF, Molinari M, Molinari M, Møller AB, Mollereau B, Mollinedo F, Mongillo M, Monick MM, Montagnaro S, Montell C Moore DJ, Moore MN, Mora-Rodriguez R, Moreira PI, Morel E, Morelli MB, Moreno S, Morgan MJ, Moris A, Moriyasu Y, Morrison JL, Morrison LA, Morselli E, Moscat J, Moseley PL, Mostowy S, Motori E, Mottet D, Mottram JC, Moussa CE-H, Mpakou VE, Mukhtar H, Mulcahy Levy JM, Muller S, Muñoz-Moreno R, Muñoz-Pinedo C, Münz C, Murphy ME, Murray JT, Murthy A, Mysorekar IU, Nabi IR, Nabissi M, Nader GA, Nagahara Y, Nagai Y, Nagata K, Nagelkerke A, Nagy P, Naidu SR, Nair S, Nakano H, Nakatogawa H, Nanjundan M, Napolitano G, Naqvi NI, Nardacci R, Narendra DP, Narita M, Nascimbeni AC, Natarajan R, Navegantes LC, Nawrocki ST, Nazarko TY, Nazarko VY, Neill T, Neri LM, Netea MG, Netea-Maier RT, Neves BM, Ney PA, Nezis IP, Nguyen HTT, Nguyen HP, Nicot A-S, Nilsen H, Nilsson P, Nishimura M, Nishino I, Niso-Santano M, Niu H, Nixon RA, Njar VCO, Noda T, Noegel AA, Nolte EM, Norberg E, Norga KK, Noureini SK, Notomi S, Notterpek L, Nowikovsky K, Nukina N, Nürnberger T, O’Donnell VB, O’Donovan T, O’Dwyer PJ, Oehme I, Oeste CL, Ogawa M, Ogretmen B, Ogura Y, Oh YJ, Ohmuraya M, Ohshima T, Ojha R, Okamoto K, Okazaki T, Oliver FJ, Ollinger K, Olsson S, Orban DP, Ordonez P, Orhon I, Orosz L, O’Rourke EJ, Orozco H, Ortega AL, Ortona E, Osellame LD, Oshima J, Oshima S, Osiewacz HD, Otomo T, Otsu K, Ou JJ, Outeiro TF, Ouyang D, Ouyang H, Overholtzer M, Ozbun MA, Ozdinler PH, Ozpolat B, Pacelli C, Paganetti P, Page G, Pages G, Pagnini U, Pajak B, Pak SC, Pakos-Zebrucka K, Pakpour N, Palková Z, Palladino F, Pallauf K, Pallet N, Palmieri M, Paludan SR, Palumbo C, Palumbo S, Pampliega O, Pan H, Pan W, Panaretakis T, Pandey A, Pantazopoulou A, Papackova Z, Papademetrio DL, Papassideri I, Papini A, Parajuli N, Pardo J, Parekh V V, Parenti G, Park J-I, Park J, Park OK, Parker R, Parlato R, Parys JB, Parzych KR, Pasquet J-M, Pasquier B, Pasumarthi KBS, Patschan D, Patterson C, Pattingre S, Pattison S, Pause A, Pavenstädt H, Pavone F, Pedrozo Z, Peña FJ, Peñalva MA, Pende M, Peng J, Penna F, Penninger JM, Pensalfini A, Pepe S, Pereira GJS, Pereira PC, Pérez-de la Cruz V, Pérez-Pérez ME, Pérez-Rodríguez D, Pérez-Sala D, Perier C, Perl A, Perlmutter DH, Perrotta I, Pervaiz S, Pesonen M, Pessin JE, Peters GJ, Petersen M, Petrache I, Petrof BJ, Petrovski G, Phang JM, Piacentini M, Pierdominici M, Pierre P, Pierrefite-Carle V, Pietrocola F, Pimentel-Muiños FX, Pinar M, Pineda B, Pinkas-Kramarski R, Pinti M, Pinton P, Piperdi B, Piret JM, Platanias LC, Platta HW, Plowey ED, Pöggeler S, Poirot M, Polčic P, Poletti A, Poon AH, Popelka H, Popova B, Poprawa I, Poulose SM, Poulton J, Powers SK, Powers T, Pozuelo-Rubio M, Prak K, Prange R, Prescott M, Priault M, Prince S, Proia RL, Proikas-Cezanne T, Prokisch H, Promponas VJ, Przyklenk K, Puertollano R, Pugazhenthi S, Puglielli L, Pujol A, Puyal J, Pyeon D, Qi X, Qian W, Qin Z-H, Qiu Y, Qu Z, Quadrilatero J, Quinn F, Raben N, Rabinowich H, Radogna F, Ragusa MJ, Rahmani M, Raina K, Ramanadham S, Ramesh R, Rami A, Randall-Demllo S, Randow F, Rao H, Rao VA, Rasmussen BB, Rasse TM, Ratovitski EA, Rautou P-E, Ray SK, Razani B, Reed BH, Reggiori F, Rehm M, Reichert AS, Rein T, Reiner DJ, Reits E, Ren J, Ren X, Renna M, Reusch JEB, Revuelta JL, Reyes L, Rezaie AR, Richards RI, Richardson DR, Richetta C, Riehle MA, Rihn BH, Rikihisa Y, Riley BE, Rimbach G, Rippo MR, Ritis K, Rizzi F, Rizzo E, Roach PJ, Robbins J, Roberge M, Roca G, Roccheri MC, Rocha S, Rodrigues CMP, Rodríguez CI, de Cordoba SR, Rodriguez-Muela N, Roelofs J, Rogov V V, Rohn TT, Rohrer B, Romanelli D, Romani L, Romano PS, Roncero MIG, Rosa JL, Rosello A, Rosen K V, Rosenstiel P, Rost-Roszkowska M, Roth KA, Roué G, Rouis M, Rouschop KM, Ruan DT, Ruano D, Rubinsztein DC, Rucker EB, Rudich A, Rudolf E, Rudolf R, Ruegg MA, Ruiz-Roldan C, Ruparelia AA, Rusmini P, Russ DW, Russo GL, Russo G, Russo R, Rusten TE, Ryabovol V, Ryan KM, Ryter SW, Sabatini DM, Sacher M, Sachse C, Sack MN, Sadoshima J, Saftig P, Sagi-Eisenberg R, Sahni S, Saikumar P, Saito T, Saitoh T, Sakakura K, Sakoh-Nakatogawa M, Sakuraba Y, Salazar-Roa M, Salomoni P, Saluja AK, Salvaterra PM, Salvioli R, Samali A, Sanchez AMJ, Sánchez-Alcázar JA, Sanchez-Prieto R, Sandri M, Sanjuan MA, Santaguida S, Santambrogio L, Santoni G, dos Santos CN, Saran S, Sardiello M, Sargent G, Sarkar P, Sarkar S, Sarrias MR, Sarwal MM, Sasakawa C, Sasaki M, Sass M, Sato K, Sato M, Satriano J, Savaraj N, Saveljeva S, Schaefer L, Schaible UE, Scharl M, Schatzl HM, Schekman R, Scheper W, Schiavi A, Schipper HM, Schmeisser H, Schmidt J, Schmitz I, Schneider BE, Schneider EM, Schneider JL, Schon EA, Schönenberger MJ, Schönthal AH, Schorderet DF, Schröder B, Schuck S, Schulze RJ, Schwarten M, Schwarz TL, Sciarretta S, Scotto K, Scovassi AI, Screaton RA, Screen M, Seca H, Sedej S, Segatori L, Segev N, Seglen PO, Seguí-Simarro JM, Segura-Aguilar J, Seki E, Seiliez I, Sell C, Semenkovich CF, Semenza GL, Sen U, Serra AL, Serrano-Puebla A, Sesaki H, Setoguchi T, Settembre C, Shacka JJ, Shajahan-Haq AN, Shapiro IM, Sharma S, She H, Shen C-KJ, Shen C-C, Shen H-M, Shen S, Shen W, Sheng R, Sheng X, Sheng Z-H, Shepherd TG, Shi J, Shi Q, Shi Q, Shi Y, Shibutani S, Shibuya K, Shidoji Y, Shieh J-J, Shih C-M, Shimada Y, Shimizu S, Shin DW, Shinohara ML, Shintani M, Shintani T, Shioi T, Shirabe K, Shiri-Sverdlov R, Shirihai O, Shore GC, Shu C-W, Shukla D, Sibirny AA, Sica V, Sigurdson CJ, Sigurdsson EM, Sijwali PS, Sikorska B, Silveira WA, Silvente-Poirot S, Silverman GA, Simak J, Simmet T, Simon AK, Simon H-U, Simone C, Simons M, Simonsen A, Singh R, Singh S V, Singh SK, Sinha D, Sinha S, Sinicrope FA, Sirko A, Sirohi K, Sishi BJN, Sittler A, Siu PM, Sivridis E, Skwarska A, Slack R, Slaninová I, Slavov N, Smaili SS, Smalley KSM, Smith DR, Soenen SJ, Soleimanpour SA, Solhaug A, Somasundaram K, Son JH, Sonawane A, Song C, Song F, Song HK, Song J-X, Song W, Soo KY, Sood AK, Soong TW, Soontornniyomkij V, Sorice M, Sotgia F, Soto-Pantoja DR, Sotthibundhu A, Sousa MJ, Spaink HP, Span PN, Spang A, Sparks JD, Speck PG, Spector SA, Spies CD, Springer W, Clair DS, Stacchiotti A, Staels B, Stang MT, Starczynowski DT, Starokadomskyy P, Steegborn C, Steele JW, Stefanis L, Steffan J, Stellrecht CM, Stenmark H, Stepkowski TM, Stern ST, Stevens C, Stockwell BR, Stoka V, Storchova Z, Stork B, Stratoulias V, Stravopodis DJ, Strnad P, Strohecker AM, Ström A-L, Stromhaug P, Stulik J, Su Y-X, Su Z, Subauste CS, Subramaniam S, Sue CM, Suh SW, Sui X, Sukseree S, Sulzer D, Sun F-L, Sun J, Sun J, Sun S-Y, Sun Y, Sun Y, Sun Y, Sundaramoorthy V, Sung J, Suzuki H, Suzuki K, Suzuki N, Suzuki T, Suzuki YJ, Swanson MS, Swanton C, Swärd K, Swarup G, Sweeney ST, Sylvester PW, Szatmari Z, Szegezdi E, Szlosarek PW, Taegtmeyer H, Tafani M, Taillebourg E, Tait SWG, Takacs-Vellai K, Takahashi Y, Takáts S, Takemura G, Takigawa N, Talbot NJ, Tamagno E, Tamburini J, Tan C-P, Tan L, Tan ML, Tan M, Tan Y-J, Tanaka K, Tanaka M, Tang D, Tang D, Tang G, Tanida I, Tanji K, Tannous BA, Tapia JA, Tasset-Cuevas I, Tatar M, Tavassoly I, Tavernarakis N, Taylor A, Taylor GS, Taylor GA, Taylor JP, Taylor MJ, Tchetina E V, Tee AR, Teixeira-Clerc F, Telang S, Tencomnao T, Teng B-B, Teng R-J, Terro F, Tettamanti G, Theiss AL, Theron AE, Thomas KJ, Thomé MP, Thomes PG, Thorburn A, Thorner J, Thum T, Thumm M, Thurston TLM, Tian L, Till A, Ting JP, Titorenko VI, Toker L, Toldo S, Tooze SA, Topisirovic I, Torgersen ML, Torosantucci L, Torriglia A, Torrisi MR, Tournier C, Towns R, Trajkovic V, Travassos LH, Triola G, Tripathi DN, Trisciuoglio D, Troncoso R, Trougakos IP, Truttmann AC, Tsai K-J, Tschan MP, Tseng Y-H, Tsukuba T, Tsung A, Tsvetkov AS, Tu S, Tuan H-Y, Tucci M, Tumbarello DA, Turk B, Turk V, Turner RFB, Tveita AA, Tyagi SC, Ubukata M, Uchiyama Y, Udelnow A, Ueno T, Umekawa M, Umemiya-Shirafuji R, Underwood BR, Ungermann C, Ureshino RP, Ushioda R, Uversky VN, Uzcátegui NL, Vaccari T, Vaccaro MI, Váchová L, Vakifahmetoglu-Norberg H, Valdor R, Valente EM, Vallette F, Valverde AM, Van den Berghe G, Van Den Bosch L, van den Brink GR, van der Goot FG, van der Klei IJ, van der Laan LJW, van Doorn WG, van Egmond M, van Golen KL, Van Kaer L, van Lookeren Campagne M, Vandenabeele P, Vandenberghe W, Vanhorebeek I, Varela-Nieto I, Vasconcelos MH, Vasko R, Vavvas DG, Vega-Naredo I, Velasco G, Velentzas AD, Velentzas PD, Vellai T, Vellenga E, Vendelbo MH, Venkatachalam K, Ventura N, Ventura S, Veras PST, Verdier M, Vertessy BG, Viale A, Vidal M, Vieira HLA, Vierstra RD, Vigneswaran N, Vij N, Vila M, Villar M, Villar VH, Villarroya J, Vindis C, Viola G, Viscomi MT, Vitale G, Vogl DT, Voitsekhovskaja O V, von Haefen C, von Schwarzenberg K, Voth DE, Vouret-Craviari V, Vuori K, Vyas JM, Waeber C, Walker CL, Walker MJ, Walter J, Wan L, Wan X, Wang B, Wang C, Wang C-Y, Wang C, Wang C, Wang C, Wang D, Wang F, Wang F, Wang G, Wang H, Wang H, Wang H-G, Wang H, Wang H-D, Wang J, Wang J, Wang M, Wang M-Q, Wang P-Y, Wang P, Wang RC, Wang S, Wang T-F, Wang X, Wang X, Wang X-W, Wang X, Wang X, Wang Y, Wang Y, Wang Y, Wang Y-J, Wang Y, Wang Y, Wang YT, Wang Y, Wang Z-N, Wappner P, Ward C, Ward DM, Warnes G, Watada H, Watanabe Y, Watase K, Weaver TE, Weekes CD, Wei J, Weide T, Weihl CC, Weindl G, Weis SN, Wen L, Wen X, Wen Y, Westermann B, Weyand CM, White AR, White E, Whitton JL, Whitworth AJ, Wiels J, Wild F, Wildenberg ME, Wileman T, Wilkinson DS, Wilkinson S, Willbold D, Williams C, Williams K, Williamson PR, Winklhofer KF, Witkin SS, Wohlgemuth SE, Wollert T, Wolvetang EJ, Wong E, Wong GW, Wong RW, Wong VKW, Woodcock EA, Wright KL, Wu C, Wu D, Wu GS, Wu J, Wu J, Wu M, Wu M, Wu S, Wu WKK, Wu Y, Wu Z, Xavier CPR, Xavier RJ, Xia G-X, Xia T, Xia W, Xia Y, Xiao H, Xiao J, Xiao S, Xiao W, Xie C-M, Xie Z, Xie Z, Xilouri M, Xiong Y, Xu C, Xu C, Xu F, Xu H, Xu H, Xu J, Xu J, Xu J, Xu L, Xu X, Xu Y, Xu Y, Xu Z-X, Xu Z, Xue Y, Yamada T, Yamamoto A, Yamanaka K, Yamashina S, Yamashiro S, Yan B, Yan B, Yan X, Yan Z, Yanagi Y, Yang D-S, Yang J-M, Yang L, Yang M, Yang P-M, Yang P, Yang Q, Yang W, Yang WY, Yang X, Yang Y, Yang Y, Yang Z, Yang Z, Yao M-C, Yao PJ, Yao X, Yao Z, Yao Z, Yasui LS, Ye M, Yedvobnick B, Yeganeh B, Yeh ES, Yeyati PL, Yi F, Yi L, Yin X-M, Yip CK, Yoo Y-M, Yoo YH, Yoon S-Y, Yoshida K-I, Yoshimori T, Young KH, Yu H, Yu JJ, Yu J-T, Yu J, Yu L, Yu WH, Yu X-F, Yu Z, Yuan J, Yuan Z-M, Yue BYJT, Yue J, Yue Z, Zacks DN, Zacksenhaus E, Zaffaroni N, Zaglia T, Zakeri Z, Zecchini V, Zeng J, Zeng M, Zeng Q, Zervos AS, Zhang DD, Zhang F, Zhang G, Zhang G-C, Zhang H, Zhang H, Zhang H, Zhang H, Zhang J, Zhang J, Zhang J, Zhang J, Zhang J, Zhang L, Zhang L, Zhang L, Zhang L, Zhang M-Y, Zhang X, Zhang XD, Zhang Y, Zhang Y, Zhang Y, Zhang Y, Zhang Y, Zhao M, Zhao W-L, Zhao X, Zhao YG, Zhao Y, Zhao Y, Zhao Y, Zhao Z, Zhao ZJ, Zheng D, Zheng X-L, Zheng X, Zhivotovsky B, Zhong Q, Zhou G-Z, Zhou G, Zhou H, Zhou S-F, Zhou X, Zhu H, Zhu H, Zhu W-G, Zhu W, Zhu X-F, Zhu Y, Zhuang S-M, Zhuang X, Ziparo E, Zois CE, Zoladek T, Zong W-X, Zorzano A, Zughaier SM. 2016. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12:1–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tanida I, Ueno T, Kominami E. 2008. LC3 and Autophagy BT - Autophagosome and Phagosome, p. 77–88. In Deretic V (ed.), Methods Mol Biol. Humana Press, Totowa, NJ. [DOI] [PubMed] [Google Scholar]
- 49.Rogov V, Dötsch V, Johansen T, Kirkin V. 2014. Interactions between Autophagy Receptors and Ubiquitin-like Proteins Form the Molecular Basis for Selective Autophagy. Mol Cell 10.10167/.molcel.2013.12.014. [DOI] [PubMed] [Google Scholar]
- 50.Larsen KB, Lamark T, Øvervatn A, Harneshaug I, Johansen T, Bjørkøy G. 2010. A reporter cell system to monitor autophagy based on p62/SQSTM1. Autophagy 6:784–793. [DOI] [PubMed] [Google Scholar]
- 51.Bonenfant G, Meng R, Shotwell C, Berglund JA, Pager CT. 2020. Asian Zika virus isolate significantly changes the transcriptional profile and alternative RNA splicing events in a neuroblastoma cell line. Viruses 660209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM. 2016. The integrated stress response. EMBO Rep 17:1374–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jiang H-Y, Wek SA, McGrath BC, Lu D, Hai T, Harding HP, Wang X, Ron D, Cavener DR, Wek RC. 2004. Activating Transcription Factor 3 Is Integral to the Eukaryotic Initiation Factor 2 Kinase Stress Response. Mol Cell Biol 24:1365–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Samuel MA, Whitby K, Keller BC, Marri A, Barchet W, Williams BRG, Silverman RH, Gale M, Diamond MS. 2006. PKR and RNase L Contribute to Protection against Lethal West Nile Virus Infection by Controlling Early Viral Spread in the Periphery and Replication in Neurons. J Virol 80:7009–7019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Arnaud N, Dabo S, Maillard P, Budkowska A, Kalliampakou KI, Mavromara P, Garcin D, Hugon J, Gatignol A, Akazawa D, Wakita T, Meurs EF. 2010. Hepatitis c virus controls interferon production through PKR activation. PLoS One 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang J, Kang R, Huang H, Xi X, Wang B, Wang J, Zhao Z. 2014. Hepatitis C virus core protein activates autophagy through EIF2AK3 and ATF6 UPR pathway-mediated MAP1LC3B and ATG12 expression. Autophagy 10:766–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tu Y-C, Yu C-Y, Liang J-J, Lin E, Liao C-L, Lin Y-L. 2012. Blocking Double-Stranded RNA-Activated Protein Kinase PKR by Japanese Encephalitis Virus Nonstructural Protein 2A. J Virol 86:10347–10358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee YR, Kuo SH, Lin CY, Fu PJ, Lin YS, Yeh TM, Liu HS. 2018. Dengue virus-induced ER stress is required for autophagy activation, viral replication, and pathogenesis both in vitro and in vivo. Sci Rep 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Medigeshi GR, Lancaster AM, Hirsch AJ, Briese T, Lipkin WI, DeFilippis V, Früh K, Mason PW, Nikolich-Zugich J, Nelson JA. 2007. West Nile Virus Infection Activates the Unfolded Protein Response, Leading to CHOP Induction and Apoptosis. J Virol 81:10849–10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gladwyn-Ng I, Cordón-Barris L, Alfano C, Creppe C, Couderc T, Morelli G, Thelen N, America M, Bessières B, Encha-Razavi F, Bonnière M, Suzuki IK, Flamand M, Vanderhaeghen P, Thiry M, Lecuit M, Nguyen L. 2018. Stress-induced unfolded protein response contributes to Zika virus-associated microcephaly. Nat Neurosci. Nature Publishing Group; 10.1038/s41593-017-0038-4. [DOI] [PubMed] [Google Scholar]
- 61.Chen HH, Tarn WY. 2019. uORF-mediated translational control: recently elucidated mechanisms and implications in cancer. RNA Biol. Taylor and Francis Inc. 10.1080/15476286.2019.1632634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Caselli E, Benedetti S, Gentili V, Grigolato J, Di Luca D. 2012. Short communication: Activating transcription factor 4 (ATF4) promotes HIV type 1 activation. AIDS Res Hum Retroviruses 28:907–912. [DOI] [PubMed] [Google Scholar]
- 63.Lee SD, Yu KL, Park SH, Jung YM, Kim MJ, You JC. 2018. Understanding of the functional role(s) of the Activating Transcription Factor 4(ATF4) in HIV regulation and production. BMB Rep 51:388–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Qian Z, Xuan B, Chapa TJ, Gualberto N, Yu D. 2012. Murine Cytomegalovirus Targets Transcription Factor ATF4 To Exploit the Unfolded-Protein Response. J Virol 86:6712–6723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Caselli E, Benedetti S, Grigolato J, Caruso A, Di Luca D. 2012. Activating transcription factor 4 (ATF4) is upregulated by human herpesvirus 8 infection, increases virus replication and promotes proangiogenic properties. Arch Virol 157:63–74. [DOI] [PubMed] [Google Scholar]
- 66.Gao P, Chai Y, Song J, Liu T, Chen P, Zhou L, Ge X, Guo X, Han J, Yang H. 2019. Reprogramming the unfolded protein response for replication by porcine reproductive and respiratory syndrome virus. PLoS Pathog 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Novoa I, Zeng H, Harding HP, Ron D, Neibert DW, Hollander MC, Luethy JD, Papathanasiou M, Fragoli J, Holbrook NJ, Hoffman-Lieberman B. 2001. Feedback Inhibition of the Unfolded Protein Response by GADD34-mediated Dephosphorylation of eIF2 The Journal of Cell Biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mufrrih M, Chen B, Chan S-W. 2021. Zika Virus Induces an Atypical Tripartite Unfolded Protein Response with Sustained Sensor and Transient Effector Activation and a Blunted BiP Response. mSphere 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fraser JE, Wang C, Chan KWK, Vasudevan SG, Jans DA. 2016. Novel dengue virus inhibitor 4-HPR activates ATF4 independent of protein kinase R-like Endoplasmic Reticulum Kinase and elevates levels of eIF2α phosphorylation in virus infected cells. Antiviral Res 130:1–6. [DOI] [PubMed] [Google Scholar]
- 70.Vattem KM, Wek RC. 2004. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lee JM, Hammarén HM, Savitski MM, Baek SH. 2023. Control of protein stability by post-translational modifications. Nat Commun. Nature Research; 10.1038/s41467-023-35795-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Thompson SR. 2012. So you want to know if your message has an IRES? Wiley Interdiscip Rev RNA 10.1002/wrna.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mishra R, Lahon A, Banerjea AC. 2020. Dengue Virus Degrades USP33–ATF3 Axis via Extracellular Vesicles to Activate Human Microglial Cells. The Journal of Immunology 205:1787–1798. [DOI] [PubMed] [Google Scholar]
- 74.Vu TTM, Varshavsky A. 2020. The ATF3 Transcription Factor Is a Short-Lived Substrate of the Arg/N-Degron Pathway. Biochemistry 59:2796–2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Labzin LI, Schmidt S V., Masters SL, Beyer M, Krebs W, Klee K, Stahl R, Lütjohann D, Schultze JL, Latz E, De Nardo D. 2015. ATF3 Is a Key Regulator of Macrophage IFN Responses. The Journal of Immunology 195:4446–4455. [DOI] [PubMed] [Google Scholar]
- 76.Akbarpour Arsanjani A, Abuei H, Behzad-Behbahani A, Bagheri Z, Arabsolghar R, Farhadi A. 2022. Activating transcription factor 3 inhibits NF-κB p65 signaling pathway and mediates apoptosis and cell cycle arrest in cervical cancer cells. Infect Agent Cancer 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kooti A, Abuei H, Farhadi A, Behzad-Behbahani A, Zarrabi M. 2022. Activating transcription factor 3 mediates apoptotic functions through a p53-independent pathway in human papillomavirus 18 infected HeLa cells. Virus Genes 58:88–97. [DOI] [PubMed] [Google Scholar]
- 78.Katz HR, Arcese AA, Bloom O, Morgan JR. 2022. Activating Transcription Factor 3 (ATF3) is a Highly Conserved Pro-regenerative Transcription Factor in the Vertebrate Nervous System. Front Cell Dev Biol. Frontiers Media S.A. 10.3389/fcell.2022.824036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hunt D, Raivich G, Anderson PN. 2012. Activating transcription factor 3 (ATF3) and the nervous system. Front Mol Neurosci 10.3389/fnmol.2012.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li X, Gracilla D, Cai L, Zhang M, Yu X, Chen X, Zhang J, Long X, Ding HF, Yan C. 2021. ATF3 promotes the serine synthesis pathway and tumor growth under dietary serine restriction. Cell Rep 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Di Marcantonio D, Martinez E, Kanefsky JS, Huhn JM, Gabbasov R, Gupta A, Krais JJ, Peri S, Tan YF, Skorski T, Dorrance A, Garzon R, Goldman AR, Tang HY, Johnson N, Sykes SM. 2021. ATF3 coordinates serine and nucleotide metabolism to drive cell cycle progression in acute myeloid leukemia. Mol Cell 81:2752–2764.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gilchrist M, Thorsson V, Li B, Rust AG, Korb M, Kennedy K, Hai T, Bolouri H, Aderem A. 2006. Systems biology approaches identify ATF3 as a negative regulator of Toll-like receptor 4. Nature 441:173–178. [DOI] [PubMed] [Google Scholar]
- 83.Chiramel AI, Best SM. 2018. Role of autophagy in Zika virus infection and pathogenesis. Virus Res 254:34–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chen X, Stauffer S, Chen Y, Dong J. 2016. Ajuba phosphorylation by CDK1 promotes cell proliferation and tumorigenesis. Journal of Biological Chemistry 291:14761–14772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lennemann NJ, Coyne CB. 2017. Dengue and Zika viruses subvert reticulophagy by NS2B3-mediated cleavage of FAM134B. Autophagy 13:322–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jia H, Peng H, Hou Z. 2020. Ajuba: An emerging signal transducer in oncogenesis. Pharmacol Res. Academic Press; 10.1016/j.phrs.2019.104546. [DOI] [PubMed] [Google Scholar]
- 87.Hirota Toru, Kunitoku Naoko, Sasayama Takashi, Marumoto Tomotoshi, Zhang Dongwei, Nitta Masayuki, Hatakeyama Katsuyoshi, Saya Hideyuki. 2003. Aurora-A and an Interacting Activator, the LIM Protein Ajuba, Are Required for Mitotic Commitment in Human Cells. Cell 114:585–598. [DOI] [PubMed] [Google Scholar]
- 88.Choi Y, Bowman JW, Jung JU. 2018. Autophagy during viral infection - A double-edged sword. Nat Rev Microbiol. Nature Publishing Group; 10.1038/s41579-018-0003-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Liang G, Wolfgang CD, Chen BPC, Chen T, Hai T. 1996. ATF3: Gene Genomic Organization, Promoter and Regulation. J Biol Chem 271:1695–1701. [DOI] [PubMed] [Google Scholar]
- 90.Elbashir Sayda M., Harborth Jens, Lendeckel Winfried, Yalcin Abdullah, Weber Klaus, Tuschl Thomas. 2001. Generation of target cells. Nature 411:494–498. [DOI] [PubMed] [Google Scholar]
- 91.Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, Griffiths G, Ktistakis NT. 2008. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. Journal of Cell Biology 182:685–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. ATF3 expression is induced by tunicamycin and ZIKV in A549 and HCT-116 cells respectively. (A) A schematic showing ATF3 gene organization, and the target of the guide RNA used to create the ATF3 KO cell line. B) A549 WT and ATF3 KO cells were treated with tunicamycin for 6 hours. ATF3 proteins were analyzed after drug treatment by western blotting and GAPDH served as the loading control. The western blot shown is representative of 3 independent experiments. (B) The fold change of ATF3 mRNA relative to β-actin mRNA in WT and ATF3 KO cells was measured by RT-qPCR after tunicamycin treatment. (C) HCT-116 WT and ATF3 KO cells were infected with ZIKV PRVABC59 (moi=10 PFU/cell) for 48 hours. ATF3 and viral NS1 proteins were analyzed by western blot with GAPDH used as the loading control. ZIKV RNA levels were also determined by RT-qPCR. mRNA levels were determined from 3 independent experiments and error bars show ± SD. Statistical significance was determined by Student T-test. *p<0.05.
Figure S2. ISR inhibition does not affect cell viability or localization of ATF3 during ZIKV infection. (A) A549 WT cells were treated with DMSO or ISRIB or had no treatment, and viable cells were measured as luciferase unit using the cell viability assay. (B) A549 cells were either mock-infected or infected with ZIKV (PRVABC59, moi=10 PFU/cell) in the presence or absence of ISRIB. Cellular and nuclear fractions were prepared from cells harvested 24-hours post-infection. The resultant subcellular fractions were analyzed by immunoblotting and probed with NS1, ATF4, ATF3, fibrillarin and β-tubulin antibodies. Fibrillarin and β-tubulin were used as nuclear and cytoplasmic markers respectively. Results shown are from 3 independent experiments.
Figure S3. PERK inhibition enhances ATF3 protein levels but reduces ATF3 mRNA levels during ZIKV infection. PERK, one of four kinases central to the ISR pathway was targeted by treating A549 mock- or ZIKV-infected (PRVABC59, moi=10 PFU/cell) with or without an inhibitor (GSK2606414). (A) Cellular and viral proteins were analyzed by western blot. The fold change in (B) ZIKV, (C) PERK (D) ATF4 and (E) ATF3 levels relative to β-actin mRNA were determined by RT-qPCR. (F) Cell viability of non-treated cells and cells treated with DMSO, or PERK inhibitor were determined. N=3 Error bars show ± SD. Statistical significance was determined by Student T-test. **p<0.001, ****p<0.0001, ns-not significant.
Figure S4. ATF4 triggers ATF3 expression and positively regulates ZIKV replication and translation. A549 cells expressing either control or ATF4-specific shRNA were treated with tunicamycin (TU) or infected with ZIKV (moi=10 PFU/cell). (A) A representative western blot probed with ATF4, ATF3 and ZIKV NS1 antibodies. (B-C) Relative mRNA expression of ATF4 and ATF3 genes measured as fold change were determined by RT-qPCR. All experiments were done in triplicates. Statistical significance was determined by Student T-test. ***p<0.005, ns-not significant.