

Abstract
An accurate and cost-efficient methodology for the estimation of the enthalpies of formation for closed-shell compounds composed of C, H, O, and N atoms is presented and validated against critically-evaluated experimental data. The computational efficiency is achieved through the use of the Resolution-of-Identity (RI) and Domain-Based Local Pair-Natural Orbital Coupled Cluster (DLPNO-CCSD(T)) approximations, which results in drastic reduction in both the computational cost and the number of necessary steps for a composite quantum chemical method. The expanded uncertainty for the proposed methodology evaluated using a data set of 45 thoroughly vetted experimental values for molecules containing up to 12 heavy atoms is about 3 kJ·mol−1, competitive with those of typical calorimetric measurements. For the compounds within the stated scope, the methodology is shown to be superior to a representative, more general, and widely-used composite quantum chemical method, G4.
Graphical Abstract
Introduction
The importance of reliable and readily-accessible values for the gas-phase enthalpies of formation (ΔfH○) of organic compounds is well recognized. Reliable experimental data are available only for a limited number of cases, resulting in a long history of estimation method development. The oldest and most widely used approach is the application of empirical group-contribution schemes.1–3 Group-contribution methods are very accessible as they require almost no computational effort. However, they are limited both in scope (group value availability) and in accuracy (additivity approximation within the chosen group decomposition scheme). The alternative is the use of quantum chemical methods which are free of these limitations. Unlike the group-contribution approaches, quantum chemical methods do not yield ΔfH○ directly; it is derived from either the enthalpy of atomization or the enthalpy of suitable, preferably isodesmic, reaction (subject to availability of reliable experimental enthalpies of formation for all participants except for the one under consideration). The former approach can be formally viewed as a direct method, given tabulated experimental and precomputed data for all atoms involved. However, strong multireference nature of atomic configurations necessitates advanced quantum chemical calculations to obtain atomization enthalpies of acceptable accuracy. Practical application of quantum chemistry for prediction of ΔfH○ requires at least two steps: one needs (1) to obtain the optimized model geometry and its electronic energy (E) and (2) to compute vibrational frequencies needed for evaluation of zero-point vibrational energy (ZPVE) and the enthalpy change from 0 K to the reference temperature of 298.15 K (). These two calculations are very computationally-expensive and one has to use relatively low levels of theory and small basis sets even for moderately-sized compounds. Consequently, the energies obtained after optimization are rarely of acceptable accuracy and additional steps, in the form of single-point energy calculations at higher levels of theory and with larger basis sets, need to be taken. As using both at the same time is often also computationally-prohibitive, several multiple-step procedures were proposed, including Gaussian (Gn, n=1,2,3,4),4–7 Complete Basis Set (CBS),8–11 HEAT,12,13 ATOMIC,14,15 and Weizmann (Wn, n=1,2,3,4)16–18 protocols; more sophisticated schemes are under active development.19,20 Most of these approaches involve multiple single-point energy calculations with balanced combinations of theory levels and basis set sizes, thus reducing the overall computational requirements. The final energy is derived from the results of these multiple steps. It should also be noted that routine use of high-accuracy HEAT or the Wn theories is extremely computationally-expensive on modern mainstream hardware, even for moderately-sized molecules. Consequently, the “budget” Gn and CBS procedures presently dominate the practical estimation of the enthalpies of formation.21–25 Most methods from these families approximate coupled-cluster with single, double, and perturbative triple excitations (CCSD(T)) level of theory with large (or extrapolated to infinite) basis set. Recent benchmarking for ΔfH○ derived from atomization enthalpies reported the best performance at the level of 2.5–3 kJ·mol−1 standard deviation (s) for C/H/O systems22 and nitrogen-containing organics,23 although reliable experimental data for nitrogen compounds are limited,24,25 making large-scale assessment problematic.
This brings another issue related to the performance assessment: the method performance is normally evaluated by comparing predictions with reliable experimental data. The majority of ΔfH○ for organic compounds are determined from their energies of combustion measured in bomb calorimeters. This requires a few grams of a sample of very high purity, preferably above 99.9 %. The high purity is important since the relative standard uncertainty in the energies of combustion is typically about 0.02 %. Accurate chemical analysis of the combustion products is also necessary to achieve this data quality. This analysis is especially important for large molecules and cases involving heteroatoms. Furthermore, since bomb experiments are usually conducted with compounds in the condensed state, the enthalpy of vaporization or sublimation is required to derive the gas-phase ΔfH○. While the standard uncertainty in gas-phase enthalpies of formation is about 1 kJ·mol−1 for state-of-the-art studies, uncertainties of a few kJ·mol−1 are typical for the majority of the competent measurements. This imposes a limitation on determination of the performance metrics as one cannot achieve accuracy better than that of the data themselves.
The focus of this work is the development of a method for efficient estimation of ΔfH○ suitable for practical applications with moderate computational resources. Of note is the fact that most of composite (multiple-step) methods rely on numerical solution of exact model equations. However, a number of methods based on efficient approximate solution exist, and some of them have evolved to a level of practical maturity. Specifically, the “Resolution-of-Identity” (RI) (also referred to as “Density-Fitted”, DF) methods can substantially accelerate Self-Consistent Field (SCF), Density Functional Theory (DFT), and the Møller-Plesset second-order perturbation theory (MP2) calculations.26–28 With the use of the RI approximations, the first step of a composite method, geometry optimization, can be conducted very efficiently and with significantly larger basis sets as compared to those that can be afforded in a course of the canonical solution. Furthermore, if a sufficiently accurate and efficient CCSD(T) approximation is available, one can bypass multiple steps and use this approximation with a large basis set as a final step of the method. The recently reported DLPNO-CCSD(T) approach29–32 offers a very efficient and accurate approximation of the canonical CCSD(T) with nearly linear scaling of the computational time with the system size. Provided that the approximations are sufficiently accurate for the application considered here, a drastic reduction in computational time and memory requirements can be achieved or, alternatively, the calculations for very large molecules become possible. This represents the basic idea of the proposed method. The specific implementation details are given below.
Methods
Computational methods
In this study, we focus on efficient estimation of the enthalpies of formation for closed-shell compounds with compositions restricted to C, H, O, and N elements. We consider two methods, B3LYP-D3(BJ)33 and RI-MP2, for geometry optimization and frequency calculations, and DLPNO-CCSD(T) for single-point electronic energy calculations. The balanced Karlsruhe “def2” triple- and quadruple-zeta basis sets34 were used in these calculations. Additional computations with the popular G4 method, representative of the current budget composite methods, were also done for comparison. The direct estimation of the enthalpy of formation is performed with the following equation:
| (1) |
The summation in the last term of Eq. (1) is performed over all chemical elements present in the compound (N equals to 4 in this study); ni is the ith element count, and hi is the element-specific constant. Eq. (1) is mathematically equivalent to the derivation of ΔfH○ using the enthalpy of atomization, and hi can be formally defined via computed atomic electronic energies, reference enthalpies of formation, and reference enthalpy changes for individual atomic species. The present implementation of DLPNO-CCSD(T) does not support open-shell systems, a restriction that does not apply to gas-phase atomic species in their ground states considered in this study. To circumvent this problem, we treat hi as empirical constants and determine them from the regression analysis against the experimental data. This approach resembles earlier semi-empirical “atom-equivalent” proposals35–37 to convert SCF and DFT energies to ΔfH○, but with the full rigor of explicitly accounting for ZPVE and terms. Our tests have shown that exclusion of these terms results in a nearly 4-fold increase in the standard deviation between the experiment and the model. It should also be noted that absolute DLPNO-CCSD(T) energies used in Eq. (1) are very sensitive to the DLPNO threshold parameters;31 consequentely, the hi regression constants depend on them as well. “TightPNO” settings31 were used in all cases and need to be applied if hi constants reported here are used to predict ΔfH○. The use of default, “NormalPNO” settings31 leads to larger data scatter, manifested in about 0.1–0.2 kJ·mol−1 increase in the standard deviation and wider ranges of deviations between the experimental and predicted values. Because hi constants are determined empirically using the experimental data, they are also expected to compensate, at least to some extent, for the deficiencies in the computed ZPVE and the lack of the post-CCSD(T)38 contributions.
The computational schemes tested using the present approach are listed in Table 1. They include four schemes based on RI-MP2 geometries (“small”, “small+”, “medium”, and “large”); the naming follows an increase in the basis sets used in the scheme. Additionally, the combination based on B3LYP-D3(BJ) geometry, “medium-DFT”, with the basis sets corresponding to the “medium” scheme was tested. Energies in all schemes, except for the “small+”, were obtained from a single-point DLPNO-CCSD(T) calculation. The “small+” scheme is a test of a more complex composite protocol that includes additional MP2 energy correction, similar to the Gaussian theory methodology.5 In comparison with the “small”, “small+” includes an additional single-point RI-MP2/def2-QZVP calculation, and the DLPNO-CCSD(T)/def2-TZVP energy is corrected by the energy increment between the corresponding RI-MP2/def2QZVP and RI-MP2/def2-TZVP values.
Table 1:
Computational schemes testeda
| scheme | E | ZPVE & |
|---|---|---|
| small | DLPNO-CCSD(T)/def2-TZVP//RI-MP2/def2-TZVP | B3LYP-D3(BJ)/def2-TZVP |
| medium | DLPNO-CCSD(T)/def2-QZVP//RI-MP2/def2-TZVP | B3LYP-D3(BJ)/def2-TZVP |
| large | DLPNO-CCSD(T)/def2-QZVP//RI-MP2/def2-QZVP | B3LYP-D3(BJ)/def2-TZVP |
| medium-DFT | DLPNO-CCSD(T)/def2-QZVP//B3LYP-D3(BJ)/def2-TZVP | B3LYP-D3(BJ)/def2-TZVP |
| small+ | E (small) + A E (RI-MP2)b | B3LYP-D3(BJ)/def2-TZVP |
| G4c | G4 | G4 |
| G4-E | G4 | B3LYP-D3(BJ)/def2-TZVP |
Finally, two schemes based on the popular G4 procedure were evaluated for comparison. The scheme labeled “G4” represents canonical G4 calculation of ΔfH○ via the enthalpy of atomization. The scheme “G4-E” is the case when only E was taken from the G4 results, while ΔfH○ was computed using Eq. (1) with ZPVE, , and the hi constants determined in the same manner as for the schemes based on DLPNO-CCSD(T).
From the initial tests, it was recognized that the vibrational frequency analysis needed for evaluation of ZPVE and terms posed a significant “bottleneck” in practical RI-MP2 calculations. To keep the computational costs down, the vibrational frequencies used in all proposed schemes (except for the canonical G4) were computed with B3LYP-D3(BJ)/def2-TZVP using the geometries optimized at the same level. For the schemes based on geometries other than those produced with B3LYP-D3(BJ)/def2-TZVP, this introduces an additional optimization step. However, the overall computational effort still remains lower as compared to the alternative of using RI-MP2 for frequency calculations. Prior to their use, the computed frequencies were scaled with the factors of 0.96 for hydrogen stretches and 0.985 for all other modes. The terms were evaluated using conventional rigid rotor - harmonic oscillator approximation,40 and anharmonicities due to internal rotations were ignored. The compounds for which anharmonicities can significantly affect were deliberately avoided during the data set selection.
The assignments for scaling factors used here are consistent with reported recommendations41–43 and the results do not show strong sensitivity to variations in their values. Their fidelity was also tested by including them in the optimization procedure along with hi constants. This did not lead to significant performance improvements (standard deviation reduction was within 0.1 kJ·mol−1) or variations in the scaling factor for the lower frequencies, but resulted in unrealistically low values of the scaling factor for hydrogen stretches, about 0.8.
All computations (except for G4) were performed with ORCA44 v.3.0.3 package. The G4 calculations were performed with Gaussian 09.45
Critically-evaluated data set
The data set of reliable, critically-evaluated experimental enthalpies of formation at 298.15 K for 45 compounds compiled from evaluated data reviews39,46,47 and from the original experimental works is given in Table 2. Only cases with at least two independent experimental verifications were considered. If the values in the reviews were consistent and no new data were available, the recommended values from the reviews were used. Otherwise, the experimental data and their uncertainties were evaluated to identify outliers. The uncertainties included contributions due to repeatability, calibration, auxiliary compounds, and chemical analysis, if this information was available. The most reliable combustion energies were weight-averaged and the condensed-state enthalpies of formation were derived using the enthalpies of formation for carbon dioxide and water recommended by CODATA.48 The uncertainties in the enthalpies of formation also included the uncertainties in ΔfH○ for reference compounds present in the combustion equations. Similar analysis for the enthalpies of vaporization and sublimation included both calorimetric results and temperature-dependent vapor pressures. An effort was made to achieve a balanced representation of different functional groups and to avoid cases exhibiting significant conformational ambiguity or, as mentioned earlier, vibrations with strong anharmonicity affecting evaluation of terms. The expanded uncertainties (0.95 confidence level) were below 2 kJ·mol−1 in all cases. We note that much larger data sets with the same restrictions on elemental composition and electronic structure as applied here were reported in the literature (e.g., Ref. 37). However, the imposed strict requirements of the confirmed experimental accuracy are critical for robust determination of parameters in Eq. (1) and meaningful assessment of the method performance, and they firmly constrain any significant extension of the present set of compounds. It should be emphasized that development of the present data set (inclusive of the associated critical evaluation procedures) is significant and absolutely critical part of the present effort.
Table 2:
List of compounds, experimental ΔfH○, and deviations between the experiment and the calculationsa
| experimentalb | c | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| name | formula | U | Ref. | S | M | L | M-DFT | S+ | G4 | G4-E | |
| methane | CH4 | −74.53 | 0.06 | 39 | 0.6 | −1.7 | −1.7 | −1.6 | −1.7 | 0.1 | 1.7 |
| ethane | C2H6 | −83.78 | 0.15 | 39 | 1.6 | −0.3 | −0.3 | −0.2 | −0.3 | −0.6 | 1.1 |
| propane | C3H8 | −104.6 | 0.2 | 39 | 1.8 | 0.2 | 0.2 | 0.3 | 0.2 | −1.2 | 0.8 |
| butane | C4H10 | −125.8 | 0.3 | 39 | 1.8 | 0.4 | 0.4 | 0.5 | 0.3 | −1.5 | 0.8 |
| isobutane | C4H10 | −135.1 | 0.5 | 49–51 | 0.2 | −1.5 | −1.5 | −1.4 | −1.6 | −2.5 | −0.2 |
| neopentane | C5H12 | −168.0 | 0.8 | 46 | 1.0 | −1.6 | −1.6 | −1.4 | −1.6 | −0.7 | 1.7 |
| cyclohexane | C6H12 | −123.3 | 0.8 | 46 | 1.1 | 0.2 | 0.2 | 0.3 | 0.1 | −3.7 | −1.9 |
| ethylene | C2H4 | 52.53 | 0.14 | 39 | 2.3 | 0.3 | 0.3 | 0.1 | 0.7 | 0.2 | 1.1 |
| propene | C3H6 | 20.3 | 0.3 | 39 | 1.8 | 0.3 | 0.3 | 0.2 | 0.6 | −0.4 | 0.9 |
| (E)-2-butene | C4H8 | −11.2 | 0.5 | 39 | 0.8 | −0.6 | −0.6 | −0.7 | −0.5 | −1.6 | 0.1 |
| (z)-2-butene | C4H8 | −7.3 | 0.5 | 39 | −1.1 | −1.8 | −1.9 | −1.9 | −1.7 | −3.2 | −1.7 |
| cyclohexene | C6H10 | −5.0 | 1.0 | 52–54 | 0.8 | 0.4 | 0.4 | 0.4 | 0.5 | −3.5 | −2.5 |
| norbornene | C7H10 | 81.9 | 1.7 | 55–57 | 3.7 | 2.7 | 2.6 | 3.0 | 2.8 | 1.0 | 1.7 |
| 1,3-butadiene | C4H6 | 110.0 | 1.0 | 46 | −0.1 | −1.7 | −1.7 | −1.9 | −1.1 | −1.8 | −0.8 |
| ethyne | C2H2 | 228.32 | 0.14 | 39 | 3.1 | 0.7 | 0.8 | 0.5 | 1.0 | −0.5 | −0.2 |
| propyne | C3H4 | 185.1 | 0.5 | 58–60 | 2.5 | 0.0 | 0.2 | −0.2 | 0.2 | −0.7 | −0.5 |
| 1-butyne | C4H6 | 165.4 | 0.9 | 39 | 1.4 | −0.8 | −0.7 | −0.9 | −0.7 | −2.2 | −1.7 |
| benzene | C6H6 | 82.9 | 0.9 | 47 | 0.1 | 0.9 | 1.0 | 0.9 | 1.1 | −1.8 | −2.0 |
| styrene | C8H8 | 148.0 | 1.4 | 46 | −2.0 | −1.4 | −1.4 | −1.7 | −1.3 | −1.1 | −1.4 |
| naphthalene | C10H8 | 150.6 | 1.5 | 47 | −0.6 | 1.0 | 1.0 | 1.2 | 0.6 | 3.6 | 2.0 |
| biphenyl | C12H10 | 180.2 | 1.8 | 61–68 | −0.8 | 0.8 | 0.9 | 0.8 | 0.6 | 3.2 | 1.6 |
| water | H2O | −241.83 | 0.04 | 48 | −13.5 | −0.4 | −0.3 | −0.5 | −0.6 | −1.8 | −1.2 |
| carbon dioxide | CO2 | −393.51 | 0.13 | 48 | 9.6 | 1.5 | 1.5 | 1.9 | 2.0 | 3.6 | 1.6 |
| methanol | CH 4 O | −200.7 | 0.2 | 39 | −3.8 | 1.0 | 1.0 | 0.9 | 0.9 | 0.2 | 0.9 |
| ethanol | C2H6O | −234.6 | 0.2 | 39 | −3.8 | 0.9 | 1.0 | 0.9 | 0.8 | −0.7 | 0.2 |
| 2-propanol | C3H8O | −272.8 | 0.4 | 39 | −3.5 | 0.8 | 0.8 | 0.8 | 0.6 | −0.9 | 0.3 |
| 2-methylpropan-2-ol | C4H10O | −312.5 | 0.8 | 46 | −3.5 | −0.2 | −0.2 | −0.1 | −0.4 | −0.2 | 1.2 |
| phenol | C6H6O | −95.7 | 1.1 | 69–74 | −7.8 | −2.8 | −2.7 | −2.8 | −2.8 | −4.9 | −5.6 |
| 1-naphthol | C10H8O | −27.5 | 1.7 | 75–77 | −7.2 | −1.7 | −1.7 | −1.4 | −2.2 | 2.0 | 0.1 |
| dimethyl ether | C2H6O | −184.0 | 0.4 | 39 | 2.6 | 0.7 | 0.7 | 0.5 | 0.9 | 0.7 | 1.6 |
| anisole | C7H8O | −69.9 | 1.0 | 78–82 | 0.3 | −0.3 | −0.4 | −0.6 | −0.2 | 0.0 | −0.7 |
| methanal | CH 2 O | −109.16 | 0.11 | 39 | 6.5 | 2.1 | 2.1 | 1.9 | 2.8 | 2.7 | 2.3 |
| ethanal | C2H4O | −165.5 | 0.3 | 39 | 4.6 | 0.8 | 0.8 | 0.8 | 1.2 | 0.6 | 0.6 |
| propanone | C3H6O | −216.1 | 0.4 | 39 | 3.5 | 0.2 | 0.2 | 0.3 | 0.4 | −0.3 | 0.0 |
| formic acid | CH2O2 | −378.5 | 0.2 | 39 | 1.9 | 1.3 | 1.3 | 1.2 | 1.2 | 0.2 | −0.6 |
| acetic acid | C2H4O2 | −432.8 | 0.6 | 39 | −1.2 | −1.4 | −1.4 | −1.4 | −1.8 | −2.8 | −3.4 |
| benzoic acid | C7H6O2 | −294.1 | 1.0 | 83, d | 0.8 | 1.5 | 1.6 | 1.5 | 0.9 | 1.2 | −0.9 |
| ammonia | H3N | −45.56 | 0.03 | 39 | −7.2 | −0.2 | −0.1 | −0.2 | −0.5 | −2.8 | −0.6 |
| acetonitrile | C2H3N | 74.0 | 0.3 | 46 | 11.0 | 2.2 | 2.4 | 2.4 | 2.8 | 1.2 | 2.0 |
| urea | CH4N2O | −237.8 | 0.5 | 84–92 | −5.5 | −2.0 | −2.0 | −2.1 | −2.8 | −5.6 | −3.6 |
| piperidine | C5H11N | −47.3 | 0.8 | 93–97 | 2.2 | 2.6 | 2.6 | 2.8 | 2.7 | −0.8 | 1.5 |
| pyridine | C5H5N | 140.4 | 0.7 | 46 | 5.5 | 2.0 | 2.0 | 2.0 | 2.6 | 0.6 | 0.8 |
| aniline | C6H7N | 87.1 | 1.2 | 98–102 | −3.7 | 0.6 | 0.7 | 0.4 | 0.6 | −1.5 | −0.5 |
| nitrobenzene | C6H5NO2 | 66.0 | 1.1 | 102–104,e | 5.6 | −1.7 | −1.8 | −1.5 | −0.4 | 8.5 | 6.6 |
| benzamide | C7H7NO | −99.8 | 1.0 | 98,99,105–109 | −2.6 | −1.7 | −1.7 | −1.7 | −2.2 | −2.1 | −2.5 |
| standard deviation | 4.6 | 1.4 | 1.4 | 1.4 | 1.5 | 2.5 | 2.0 | ||||
a energy units are kJ·mol−1;
b U represents expanded uncertainty (0.95 level of confidence);
c S, M, L, M-DFT, and S+ refer to “small”, “medum”, “large”, “medium-DFT”, and “small+” schemes, respectively (see Table 1);
d enthalpy of sublimation was evaluated using the NIST ThermoData Engine 10.1 software110 with full list of literature sources available;111
e the enthalpy of combustion reported in Ref. 103 was corrected to −(3086.7 ± 0.7) kJ·mol−1 because the term for adjustment to the standard pressure (estimated to be 6 J·g−1) had an incorrect sign; the resulting liquid-phase standard enthalpy of formation is 11.1 ± 1.0 kJ·mol−1
Performance metric and uncertainty of predictions
For the main model performance metric, the standard deviation was used:
| (2) |
where , exp and , calc are experimental and computed enthalpies of formation for the jth compound, respectively, M the total number of compounds in the data set, and np the number of optimized parameters (equals N for all schemes except for the canonical G4 method, for which it is zero).
For the model with optimized parameters, the standard uncertainty of the predicted value can be estimated as112
| (3) |
where n is the row-vector of the chemical element counts in the compound for which the prediction was made, and V is the covariance matrix:
| (4) |
In the above equation, N is the M × N design matrix of the linear least squares problem defined by Eq. (1) and composed of row-vectors of element counts for the compounds in the data set. Combining Eqs (3) and (4), one can obtain
| (5) |
As seen, the expression in the brackets of Eq. (5) has no dependency on the computational scheme used in the method; it depends only on compositions of the compounds in the data set and the compound for which the estimate is being made. It is, therefore, possible to precompute (NTN)−1 matrix for the data set adopted here and use it for a priori assessment of the relative contributions of the two terms in Eq. (5) to the standard uncertainty. The (NTN)−1 matrix computed for the present data set is given in Table 3. Clearly, some off-diagonal terms (e.g., carbon-hydrogen) are rather significant, indicative of the obvious fact that element counts in closed-shell organic compounds are correlated following the chemical bonding patterns. All off-diagonal terms are negative, suggesting the rate of uncertainty increase with compound size that is slower than what would be expected for uncorrelated element counts. For all compounds in the present data set (with sizes ranging from water to biphenyl), the term in Eq. (5) associated with (NTN)−1 can be neglected and u(ΔfH○) N is nearly equal to the corresponding standard deviation s.
Table 3:
| C | H | O | N | |
|---|---|---|---|---|
| C | 0.005073 | −0.003146 | −0.001 679 | −0.000443 |
| H | −0.003146 | 0.002 544 | −0.000692 | −0.001579 |
| O | −0.001679 | −0.000 692 | 0.037142 | −0.008760 |
| N | −0.000443 | −0.001579 | −0.008 760 | 0.103257 |
Finally, the commonly reported expanded uncertainty (coverage factor of 2, corresponding to 0.95 confidence for normal distribution) is twice of the standard uncertainty,
| (6) |
Results and Discussion
The results for all schemes are presented in Table 2 (deviations between the experiment and predictions for individual compounds and standard deviations for each scheme), Table 4 (regression constants hi), and Fig. 1 (box-and-whisker diagram of deviation distributions). As seen, the “small” scheme based on def2-TZVP basis set yields rather high standard deviation of 4.6 kJ·mol−1 and 3 outliers. Elevating the basis set for the singlepoint DLPNO-CCSD(T) energy calculations to def2-QZVP results in dramatic improvement over the “small” scheme. All three schemes using DLPNO-CCSD(T)/def2-QZVP energies, “medium”, ”large”, and ”medium-DFT”, exhibit very similar performance. They differ only in terms of the method used to generate the optimized model geometry; no noticeable effect on performance is observed for the choices tested (Table 2 and Fig. 1). The obtained regression constants hi are also very close for these three cases (Table 4). The standard deviations for “medium”, ”large”, and ”medium-DFT” schemes do not exceed 1.5 kJ·mol−1 (corresponding to about 3 kJ·mol−1 expanded uncertainty), and they feature no apparent outliers. ΔfH○ for all compounds in the set are predicted within 3 kJ·mol−1 for all three schemes. Among the three, “medium-DFT” is the most economical computationally and can be suggested as the first choice to consider. However, the other two schemes, “medium” and ”large”, do not pose significantly higher computational expenses and may be considered in situations that can benefit from the use of RI-MP2 geometries over those obtained with B3LYP-D3(BJ).
Table 4:
Regression constants −hi in Eq. (1) for the tested computational schemes
| scheme | C | H | O | N |
|---|---|---|---|---|
| small | 99 880.13 | 1516.17 | 197071.22 | 143562.88 |
| medium | 99 904.57 | 1525.81 | 197129.56 | 143 605.50 |
| large | 99 904.58 | 1525.78 | 197129.66 | 143605.53 |
| medium-DFT | 99 904.56 | 1525.80 | 197129.63 | 143605.41 |
| small+ | 99 907.55 | 1527.03 | 197131.91 | 143608.06 |
| G4-E | 100044.38 | 1528.35 | 197275.39 | 143749.04 |
Figure 1:
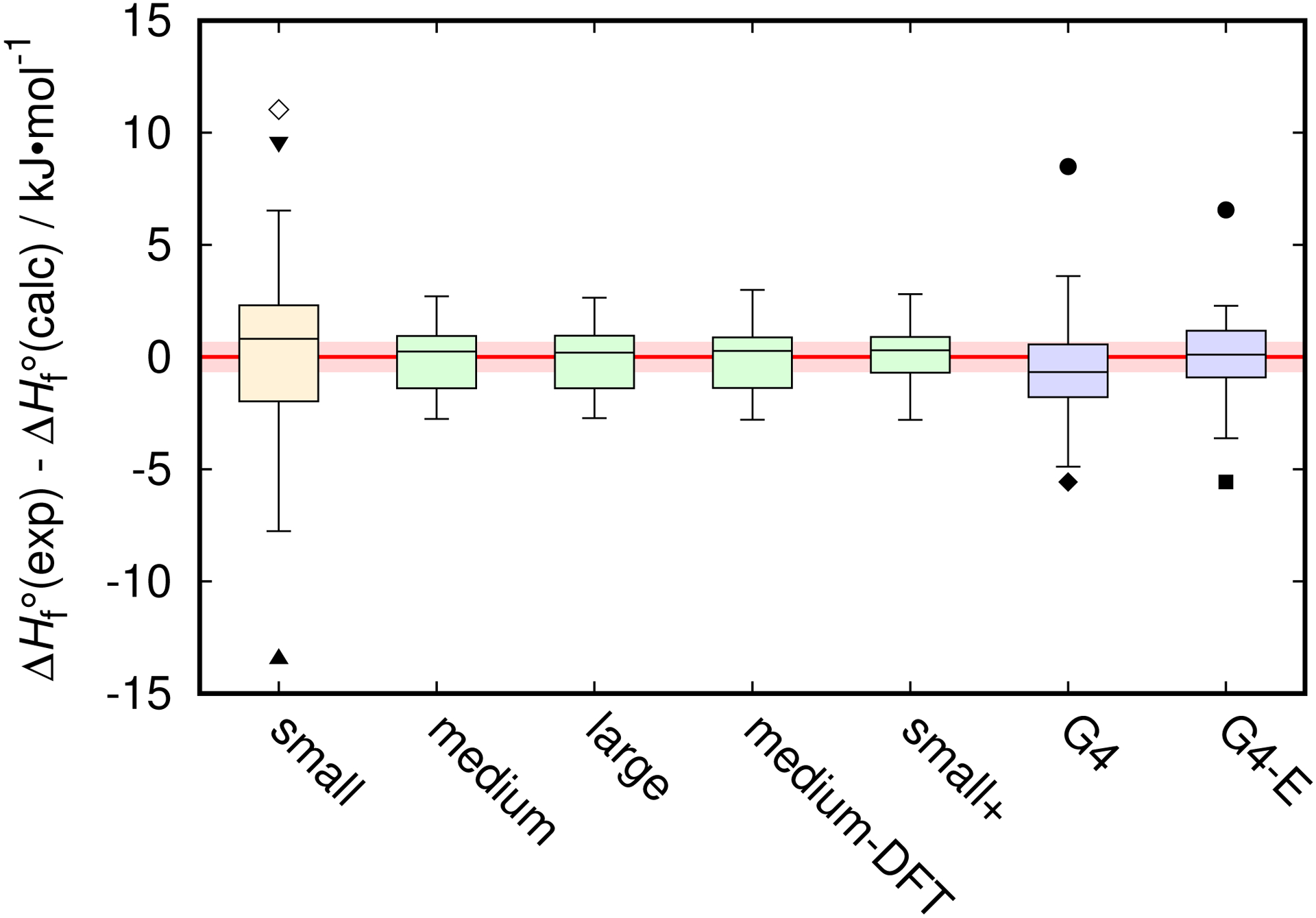
Box-and-whisker diagram of the deviations between the experimental and computed ΔfH○. Whiskers indicate data extrema within 1.5 of the interquartile range from the corresponding box edges.113 The outliers: ● - nitrobenzene, ◆ - urea, ■ - phenol, ▴ - water, ▾ - carbon dioxide, ◇ - acetonitrile. The shaded area represents a 50 % confidence interval (consistent with the box sizes) for the state-of-the-art calorimetric measurements corresponding to 0.95 confidence level of 2 kJ·mol−1.
The “small+” scheme presents an interesting dilemma. Formally, it can be viewed as an intermediate case between the “small” and the “‘medium” schemes: instead of performing full DLPNO-CCSD(T)/def2-QZVP calculation as in the case of the “medium” scheme, the DLPNO-CCSD(T)/def2-TZVP energy of “small” scheme is corrected with the RI-MP2 energy increment due to basis set increase from def2-TZVP to def2-QZVP. The resulting standard deviation for the “small+” scheme is nearly the same as those for the three DLPNO-CCSD(T)/def2-QZVP-based methods discussed above (only about 0.1 kJ·mol−1 increase). However, the distribution of deviations between the experimental and predicted values for this scheme appears noticeably different (Fig. 1). The deviations span over approximately the same range as for the DLPNO-CCSD(T)/def2-QZVP-based schemes, but the interquartile range (box size in Fig. 1) is smaller. This implies a narrower middle portion of the distribution with higher “tails”. More detailed analysis of the data in Table 2 indicates that the changes in the distribution are not uniform across the data set: while improvement is generally observed for the hydrocarbons (which represent the largest fraction of the data set), it is accompanied by some degradation of predictions for oxygenates and especially nitrogen-containing compounds, mainly responsible for the elevated “tails” of the distribution. This is not unexpected as oxygen and nitrogen contributions are more affected by the basis set size and the theory level. Therefore, although “small+” represents an attractive budget alternative, the DLPNO-CCSD(T)/def2-QZVP-based schemes are expected to be more reliable for the general use.
As seen in Fig. 1, the canonical G4 atomization scheme shows significant bias (systematically overpredicts ΔfH○) for the present data set, consistent with prior observations.22 Two outliers, urea and nitrobenzene, are present. “Parametrization” of G4 via Eq. (1), “G4-E” scheme, allows more objective comparison of G4 procedure with the present results by introducing the same set of adjustable parameters (it should be noted, however, that G4 energies already incorporate an empirical term, “the higher-level correction”7). “G4-E” does have significantly reduced bias as compared to the canonical G4. However, the outliers still persist (nitrobenzene and phenol), resulting in only moderate reduction in the standard deviation, from 2.5 to 2.0 kJ·mol−1 (Table 2). With the outliers excluded, the remaining compounds are predicted within 4.9 and 3.7 kJ·mol−1 for “G4” and “G4-E“ schemes, respectively. Comprehensive and scrupulous analysis of the original experimental data for the outliers exhibited by the G4-based methods (urea, phenol, and nitrobenzene) carried out in this study did not yield reasons to suspect significant experimental errors. Similar problem with the G4 method for nitrobenzene was reported previously, and the discrepancy was circumvented with an empirical correction.104 Recent extensive G4 investigation114 suggested revision to the experimental ΔfH○ of phenol based on their theoretical findings. The reported G4 results114 are consistent with the present G4-based predictions. On the other hand, the deviation of the present DLPNO-CCSD(T)/def2-QZVP-based results from our critically-evaluated experimental value is within 2.8 kJ·mol−1. Although it is one of the highest deviations for this data set, it remains within two standard deviations, giving no sufficient grounds for revision.
Finally, the computational performance of the presented procedures needs to be mentioned. The efficiency of the most expensive step, DLPNO-CCSD(T), was documented in detail by its developers.32 In the present study with the “TightPNO” settings,31 DLPNOCCSD(T)/def2-QZVP calculations took about 30 min for butane (4 heavy atoms) and 9.5 hours for biphenyl (12 heavy atoms) on 10 Intel Xeon E5–4617 CPU cores at 2.9 GHz with 100 Gb of RAM and 7200 rpm mechanical disk RAID storage. This performance opens possibility for large-scale applications of the presented methodology over a wide range of molecular sizes.
Conclusions
The proposed computational schemes provide simple and economical approach to estimate the enthalpies of formation of closed-shell organic compounds requiring only 3–5 steps performed using very cost-efficient approximations. The results obtained with the critically-evaluated experimental data set containing molecules with up to 12 heavy atoms suggest the expanded uncertainties of predicted values to be about 3 kJ·mol−1, well below 4 kJ·mol−1 of the target “chemical accuracy”19 and competitive with the typical experimental uncertainties. For the compounds within the stated scope, the proposed schemes were also found to be superior to the more general, budget composite method (G4) widely used at present. Due to their efficiency, the schemes can be used for large-scale validation of the existing data collections and emerging new data.115 Furthermore, as more accurate and efficient methods become available,116 the approach presented here can be easily upgraded via straightforward reparametrization of Eq. (1).
Acknowledgement
The authors thank anonymous reviewer who suggested testing of the “small+” scheme. Contribution of the U.S. National Institute of Standards and Technology and not subject to copyright in the United States. Trade names are provided only to specify procedures adequately and do not imply endorsement by the National Institute of Standards and Technology. Similar products by other manufacturers may be found to work as well or better. The authors declare no competing financial interest.
References
- (1).Benson SW Thermochemical kinetics: methods for the estimation of thermochemical data and rate parameters, 2nd ed.; Wiley-Interscience; Wiley, New York, 1976. [Google Scholar]
- (2).Cohen N Revised Group Additivity Values for Enthalpies of Formation (at 298 K) of Carbon-Hydrogen and Carbon-Hydrogen-Oxygen Compounds. J. Phys. Chem. Ref. Data 1996, 25, 1411–1481. [Google Scholar]
- (3).Holmes JL; Aubry C Group Additivity Values for Estimating the Enthalpy of Formation of Organic Compounds: An Update and Reappraisal. 1. C, H, and O. J. Phys. Chem. A 2011, 115, 10576–10586. [DOI] [PubMed] [Google Scholar]
- (4).Curtiss LA; Jones C; Trucks GW; Raghavachari K; Pople JA Gaussian-1 theory of molecular energies for second-row compounds. J. Chem. Phys 1990, 93, 2537–2545. [Google Scholar]
- (5).Curtiss LA; Raghavachari K; Trucks GW; Pople JA Gaussian-2 theory for molecular energies of first- and second-row compounds. J. Chem. Phys 1991, 94, 7221–7230. [Google Scholar]
- (6).Curtiss LA; Raghavachari K; Redfern PC; Rassolov V; Pople JA Gaussian-3 (G3) theory for molecules containing first and second-row atoms. J. Chem. Phys 1998, 109, 7764–7776. [Google Scholar]
- (7).Curtiss LA; Redfern PC; Raghavachari K Gaussian-4 theory. J. Chem. Phys 2007, 126, 084108. [DOI] [PubMed] [Google Scholar]
- (8).Petersson GA; Bennett A; Tensfeldt TG; Al-Laham MA; Shirley WA; Mantzaris J A complete basis set model chemistry. I. The total energies of closed-shell atoms and hydrides of the first-row elements. J. Chem. Phys 1988, 89, 2193–2218. [Google Scholar]
- (9).Ochterski JW; Petersson GA; Montgomery JA A complete basis set model chemistry. V. Extensions to six or more heavy atoms. J. Chem. Phys 1996, 104, 2598–2619. [Google Scholar]
- (10).Montgomery JA; Frisch MJ; Ochterski JW; Petersson GA A complete basis set model chemistry. VI. Use of density functional geometries and frequencies. J. Chem. Phys 1999, 110, 2822–2827. [Google Scholar]
- (11).Montgomery JA; Frisch MJ; Ochterski JW; Petersson GA A complete basis set model chemistry. VII. Use of the minimum population localization method. J. Chem. Phys 2000, 112, 6532–6542. [Google Scholar]
- (12).Tajti A; Szalay PG; Császár AG; Kállay M; Gauss J; Valeev EF; Flowers BA; Vázquez J; Stanton JF HEAT: High accuracy extrapolated ab initio thermochemistry. J. Chem. Phys 2004, 121, 11599–11613. [DOI] [PubMed] [Google Scholar]
- (13).Bomble YJ; Vázquez J; Kállay M; Michauk C; Szalay PG; Császár AG; Gauss J; Stanton JF High-accuracy extrapolated ab initio thermochemistry. II. Minor improvements to the protocol and a vital simplification. J. Chem. Phys 2006, 125, 064108. [DOI] [PubMed] [Google Scholar]
- (14).Bakowies D Ab initio thermochemistry using optimal-balance models with isodesmic corrections: The ATOMIC protocol. J. Chem. Phys 2009, 130, 144113. [DOI] [PubMed] [Google Scholar]
- (15).Bakowies D Ab initio thermochemistry with high-level isodesmic corrections: Validation of the ATOMIC protocol for a large set of compounds with first-row atoms (H, C, N, O, F). J. Phys. Chem. A 2009, 113, 11517–11534. [DOI] [PubMed] [Google Scholar]
- (16).Martin JML; de Oliveira G Towards standard methods for benchmark quality ab initio thermochemistry - W1 and W2 theory. J. Chem. Phys 1999, 111, 1843–1856. [Google Scholar]
- (17).Boese AD; Oren M; Atasoylu O; Martin JML; Kállay M; Gauss J W3 theory: Robust computational thermochemistry in the kJ/mol accuracy range. J. Chem. Phys 2004, 120, 4129–4141. [DOI] [PubMed] [Google Scholar]
- (18).Karton A; Rabinovich E; Martin JML; Ruscic B W4 theory for computational thermochemistry: In pursuit of confident sub-kJ/mol predictions. J. Chem. Phys 2006, 125, 144108. [DOI] [PubMed] [Google Scholar]
- (19).Peterson KA; Feller D; Dixon DA Chemical accuracy in ab initio thermochemistry: current strategies and future challenges. Theor. Chem. Acc 2012, 131, 1079. [Google Scholar]
- (20).Karton A A computational chemist’s guide to accurate thermochemistry for organic molecules. Wiley Interdiscip. Rev.: Comput. Mol. Sci 2016, 6, 292–310. [Google Scholar]
- (21).Rayne S; Forest K Estimated Gas-Phase Standard State Enthalpies of Formation for Organic Compounds Using the Gaussian-4 (G4) and W1BD Theoretical Methods. J. Chem. Eng. Data 2010, 55, 5359–5364. [Google Scholar]
- (22).Simmie JM; Somers KP Benchmarking Compound Methods (CBS-QB3, CBS-APNO, G3, G4, W1BD) against the Active Thermochemical Tables: A Litmus Test for Cost-Effective Molecular Formation Enthalpies. J. Phys. Chem. A 2015, 119, 7235–7246. [DOI] [PubMed] [Google Scholar]
- (23).Simmie JM A Database of Formation Enthalpies of Nitrogen Species by Compound Methods (CBS-QB3, CBS-APNO, G3, G4). J. Phys. Chem. A 2015, 119, 10511–10526. [DOI] [PubMed] [Google Scholar]
- (24).Suntsova MA; Dorofeeva OV Use of G4 Theory for the Assessment of Inaccuracies in Experimental Enthalpies of Formation of Aliphatic Nitro Compounds and Nitramines. J. Chem. Eng. Data 2014, 59, 2813–2826. [Google Scholar]
- (25).Suntsova MA; Dorofeeva OV Comment on “Use of G4 Theory for the Assessment of Inaccuracies in Experimental Enthalpies of Formation of Aliphatic Nitro Compounds and Nitramines”. J. Chem. Eng. Data 2015, 60, 1532–1533. [Google Scholar]
- (26).van Alsenoy C Ab initio calculations on large molecules: The multiplicative integral approximation. J. Comput. Chem 1988, 9, 620–626. [Google Scholar]
- (27).Feyereisen M; Fitzgerald G; Komornicki A Use of approximate integrals in ab initio theory. An application in MP2 energy calculations. Chem. Phys. Lett 1993, 208, 359–363. [Google Scholar]
- (28).Weigend F; Häser M RI-MP2: first derivatives and global consistency. Theor. Chem. Acc 1997, 97, 331–340. [Google Scholar]
- (29).Riplinger C; Neese F An efficient and near linear scaling pair natural orbital based local coupled cluster method. J Chem. Phys 2013, 138, 034106. [DOI] [PubMed] [Google Scholar]
- (30).Riplinger C; Sandhoefer B; Hansen A; Neese F Natural triple excitations in local coupled cluster calculations with pair natural orbitals. J. Chem. Phys 2013, 139, 134101. [DOI] [PubMed] [Google Scholar]
- (31).Liakos DG; Sparta M; Kesharwani MK; Martin JML; Neese F Exploring the Accuracy Limits of Local Pair Natural Orbital Coupled-Cluster Theory. J. Chem. Theory Comput 2015, 11, 1525–1539. [DOI] [PubMed] [Google Scholar]
- (32).Liakos DG; Neese F Is It Possible To Obtain Coupled Cluster Quality Energies at near Density Functional Theory Cost? Domain-Based Local Pair Natural Orbital Coupled Cluster vs Modern Density Functional Theory. J. Chem. Theory Comput 2015, 11, 4054–4063. [DOI] [PubMed] [Google Scholar]
- (33).Grimme S; Ehrlich S; Goerigk L Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem 2011, 32, 1456–1465. [DOI] [PubMed] [Google Scholar]
- (34).Weigend F; Ahlrichs R Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys 2005, 7, 3297–3305. [DOI] [PubMed] [Google Scholar]
- (35).Dewar MJS; Storch DM Comparative tests of theoretical procedures for studying chemical reactions. J. Am. Chem. Soc 1985, 107, 3898–3902. [Google Scholar]
- (36).Mole SJ; Zhou X; Liu R Density Functional Theory (DFT) Study of Enthalpy of Formation. 1. Consistency of DFT Energies and Atom Equivalents for Converting DFT Energies into Enthalpies of Formation. J. Phys. Chem 1996, 100, 14665–14671. [Google Scholar]
- (37).Tirado-Rives J; Jorgensen WL Performance of B3LYP Density Functional Methods for a Large Set of Organic Molecules. J. Chem. Theory Comput 2008, 4, 297–306. [DOI] [PubMed] [Google Scholar]
- (38).Karton A How large are post-CCSD(T) contributions to the total atomization energies of medium-sized alkanes? Chem. Phys. Lett 2016, 645, 118–122. [Google Scholar]
- (39).Ruscic B Active Thermochemical Tables (ATcT) values based on ver. 1.118 of the Thermochemical Network (2015). http://atct.anl.gov, accessed: July 5, 2016.
- (40).McQuarrie DA Statistical Mechanics; University Science Books, Sausalito, CA, 2000. [Google Scholar]
- (41).Merrick JP; Moran D; Radom L An Evaluation of Harmonic Vibrational Frequency Scale Factors. J. Phys. Chem. A 2007, 111, 11683–11700. [DOI] [PubMed] [Google Scholar]
- (42).Bao JL; Zheng J; Alecu I; Zhao Y; Truhlar DG Database of Frequency Scale Factors for Electronic Model Chemistries, Version 3, Beta 2. https://comp.chem.umn.edu/freqscale/version3b2.htm, accessed: February 10, 2017.
- (43).Kesharwani MK; Brauer B; Martin JML Vibrational Energy Scale Factors for Double-Hybrid Density Functionals (and Other Selected Methods): Can Anharmonic Force Fields Be Avoided? J. Phys. Chem. A 2015, 119, 1701–1714. [DOI] [PubMed] [Google Scholar]
- (44).Neese F The ORCA program system. Wiley Interdiscip. Rev.: Comput. Mol. Sci 2012, 2, 73–78. [Google Scholar]
- (45).Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Mennucci B; Petersson GA et al. Gaussian 09 Revision D.01. Gaussian Inc., Wallingford CT, 2013. [Google Scholar]
- (46).Pedley JB Thermochemical Data and Structures of Organic Compounds; TRC Data Series; Thermodynamics Research Center, College Station, TX, 1994. [Google Scholar]
- (47).Roux MV; Temprado M; Chickos JS; Nagano Y Critically evaluated thermochemical properties of polycyclic aromatic hydrocarbons. J. Phys. Chem. Ref. Data 2008, 37, 1855–1996. [Google Scholar]
- (48).Cox JD; Wagman DD; Medvedev VA CODATA Key Values for Thermodynamics; Hemisphere Publishing Corp., New York, 1989. [Google Scholar]
- (49).Pines H; Kvetinskas B; Kassel LS; Ipatieff VN Determination of Equilibrium Constants for Butanes and Pentanes. J. Am. Chem. Soc 1945, 67, 631–637. [Google Scholar]
- (50).Prosen EJ; Maron FW; Rossini FD Heats of Combustion, Formation, and Isomerization of Ten C4 Hydrocarbons. J. Res. Nat. Bur. Stand 1951, 46, 106–112. [Google Scholar]
- (51).Pittam DA; Pilcher G Measurements of heats of combustion by flame calorimetry. Part 8. Methane, ethane, propane, n-butane and 2-methylpropane. J. Chem. Soc., Faraday Trans.1 1972, 68, 2224–2229. [Google Scholar]
- (52).Labbauf A; Rossini FD Heats of combustion, formation, and hydrogenation of 14 selected cyclomonoölefin hydrocarbons. J. Phys. Chem 1961, 65, 476–480. [Google Scholar]
- (53).Good WD; Smith NK Enthalpies of combustion of toluene, benzene, cyclohexane, cyclohexene, methylcyclopentane, 1-methylcyclopentene, and n-hexane. J. Chem. Eng. Data 1969, 14, 102–106. [Google Scholar]
- (54).Steele WV; Chirico RD; Knipmeyer SE; Nguyen A; Smith NK; Tasker IR Thermodynamic properties and ideal-gas enthalpies of formation for cyclohexene, phthalan (2,5-dihydrobenzo-3,4-furan), isoxazole, octylamine, dioctylamine, trioctylamine, phenyl isocyanate, and 1,4,5,6-tetrahydropyrimidine. J. Chem. Eng. Data 1996, 41, 1269–1284. [Google Scholar]
- (55).Kozina MP; Bychikhina LV; Galchenko GL; Milvitskaya EM; Ordubadi M; Plate AF Enthalpies of Nortricyclene and Norbornene Formation. Doklady Akad. Nauk SSSR 1976, 226, 1105–1108. [Google Scholar]
- (56).Steele WV; Chirico RD; Knipmeyer SE; Nguyen A; Smith NK Thermodynamic properties and ideal-gas enthalpies of formation for butyl vinyl ether, 1,2-dimethoxyethane, methyl glycolate, bicyclo[2.2.1]hept-2-ene, 5-vinylbicyclo[2.2.1]hept-2-ene, trans-azobenzene, butyl acrylate, di-tert-butyl ether, and hexane-1,6-diol. J. Chem. Eng. Data 1996, 41, 1285–1302. [Google Scholar]
- (57).Jochems R; Dekker H; Mosselman C; Somsen G The use of the LKB 8721–3 vaporization calorimeter to measure enthalpies of sublimation. The enthalpies of sublimation of bicyclo[2.2.1]hept-2-ene (norbornene), bicyclo[2.2.1]heptane (norbornane), and tricyclo[3.3.1.13,7]decane (adamantane). J. Chem. Thermodyn 1982, 14, 395–398. [Google Scholar]
- (58).Wagman DD; Kilpatrick JE; Pitzer KS; Rossini FD Heats, equilibrium constants, and free energies of formation of the acetylene hydrocarbons through the pentynes, to 1,500° K. J. Res. Nat. Bur. Stand 1945, 35, 467–496. [Google Scholar]
- (59).Conn JB; Kistiakowsky GB; Smith EA Heats of organic reactions. VIII. Some further hydrogenations, including those of some acetylenes. J. Am. Chem. Soc 1939, 61, 1868–1876. [Google Scholar]
- (60).Cox JD; Pilcher G Thermochemistry of Organic and Organometallic Compounds; Academic Press, New York, 1970. [Google Scholar]
- (61).Montgomery RL; Rossini FD; Mansson M Enthalpies of combustion, vaporization, and formation of phenylbenzene, cyclohexylbenzene, and cyclohexylcyclohexane; enthalpy of hydrogenation of certain aromatic systems. J. Chem. Eng. Data 1978, 23, 125–129. [Google Scholar]
- (62).Coleman DJ; Pilcher G Heats of combustion of biphenyl, bibenzyl, naphthalene, anthracene and phenanthrene. Trans. Faraday Soc 1966, 62, 821–827. [Google Scholar]
- (63).Mackle H; O’Hare PAG A high-precision aneroid semi-micro combustion calorimeter. Trans. Faraday Soc 1963, 59, 2693–2701. [Google Scholar]
- (64).Parks GS; Vaughan LM The heat of combustion of biphenyl. J. Am. Chem. Soc 1951, 73, 2380–2381. [Google Scholar]
- (65).Chirico RD; Knipmeyer SE; Nguyen A; Steele WV The thermodynamic properties of biphenyl. J. Chem. Thermodyn 1989, 21, 1307–1331. [Google Scholar]
- (66).Clark T; Knox T; Mackle H; McKervey MA; Rooney JJ Heats of sublimation of some cage hydrocarbons by a temperature scanning technique. J. Chem. Soc. Faraday Trans.1 1975, 71, 2107–2110. [Google Scholar]
- (67).Morawetz E Enthalpies of vaporization for a number of aromatic compounds. J. Chem. Thermodyn 1972, 4, 455–460. [Google Scholar]
- (68).Bradley RS; Cleasby TG The vapour pressure and lattice energy of some aromatic ring compounds. J. Chem. Soc 1953, 1690–1692. [Google Scholar]
- (69).Parks GS; Manchester KE; Vaughan LM Heats of combustion and formation of some alcohols, phenols, and ketones. J. Chem. Phys 1954, 22, 2089–2090. [Google Scholar]
- (70).Andon RJL; Biddiscombe DP; Cox JD; Handley R; Harrop D; Herington EFG; Martin JF Thermodynamic properties of organic oxygen compounds. Part I. Preparation and physical properties of pure phenol, cresols, and xylenols. J. Chem. Soc 1960, 5246–5254. [Google Scholar]
- (71).Balson EW Studies in vapour pressure measurement, Part II. A new all-glass manometer sensitive to 0.001 mm. Trans. Faraday Soc 1947, 43, 48–53. [Google Scholar]
- (72).Nitta I; Seki S Vapor Pressure of Phenol. Nippon Kagaku Zasshi 1948, 69, 143–145. [Google Scholar]
- (73).Biddiscombe DP; Martin JF Vapour pressures of phenol and the cresols. Trans. Faraday Soc 1958, 54, 1316–1322. [Google Scholar]
- (74).Parsons GH; Rochester CH; Wood CEC Effect of 4-substitution on the thermodynamics of hydration of phenol and the phenoxide anion. J. Chem. Soc. B 1971, 533–536. [Google Scholar]
- (75).Colomina M; Roux MV; Turrion C Thermochemical properties of naphthalene compounds II. Enthalpies of combustion and formation of the 1-and 2-naphthols. J. Chem. Thermodyn 1974, 6, 571–576. [Google Scholar]
- (76).Ribeiro da Silva MAV; Ribeiro da Silva MDMC; Pilcher G Enthalpies of combustion of 1-hydroxynaphthalene, 2-hydroxynaphthalene, and 1,2-, 1,3-, 1,4-, and 2,3-dihydroxynaphthalenes. J. Chem. Thermodyn 1988, 20, 969–974. [Google Scholar]
- (77).Chirico RD; Steele WV; Kazakov AF Thermodynamic properties of 1-naphthol: Mutual validation of experimental and computational results. J. Chem. Thermodyn 2015, 86, 106–115. [Google Scholar]
- (78).Simões RG; Agapito F; Diogo HP; da Piedade MEM Enthalpy of Formation of Anisole: Implications for the Controversy on the O-H Bond Dissociation Enthalpy in Phenol. J. Phys. Chem. A 2014, 118, 11026–11032. [DOI] [PubMed] [Google Scholar]
- (79).Badoche MM Chaleurs de Combustion du Phénol, du m-Crésol et de leurs Éthers. Bull. Soc. Chim. Fr 1941, 8, 212–220. [Google Scholar]
- (80).Lebedeva ND; Katin Yu. A. Heats of Combustion of Monosubstituted Benzenes. Russ. J. Phys. Chem 1972, 46, 1088–1089. [Google Scholar]
- (81).Fenwick JO; Harrop D; Head AJ Thermodynamic properties of organic oxygen compounds 41. Enthalpies of formation of eight ethers. J. Chem. Thermodyn 1975, 7, 943–954. [Google Scholar]
- (82).Hales JL; Lees EB; Ruxton DJ Thermodynamic properties of organic oxygen compounds. Part 18. Vapour heat capacities and heats of vaporization of ethyl ketone, ethyl propyl ketone, methyl isopropy1 ketone, and methyl phenyl ether. Trans. Faraday Soc 1967, 63, 1876–1879. [Google Scholar]
- (83).Certificate of Analysis. Standard Reference Material 39j Benzoic Acid (Calorimetric Standard), NIST, Gaithersburg, MD, 2007. https://www-s.nist.gov/srmors/certificates/39j.pdf, accessed: July 5, 2016. [Google Scholar]
- (84).Huffman HM Thermal data. XII. The heats of combustion of urea and guanidine carbonate and their standard free energies of formation. J. Am. Chem. Soc 1940, 62, 1009–1011. [Google Scholar]
- (85).Johnson WH Enthalpies of combustion and formation of acetanilide and urea. J. Res. Nat. Bur. Stand 1975, 79, 487–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Månsson M; Sunner S Heat of formation of sulphuric acid. Acta Chem. Scand 1963, 17, 723–727. [Google Scholar]
- (87).Aleksandrov Yu. I.; Osipova TR; Yushkevich VF; Murashova SV; Oleinik BN Study of Urea as a Reference Compound for Combustion Calorimetry. Termodin. Org. Soedin 1979, 65–69. [Google Scholar]
- (88).Kabo G. Ya.; Miroshnichenko EA; Frenkel ML; Kozyro AA; Simirskii VV; Krasulin AP; Vorob’eva VP; Lebedev Yu. A. Thermochemistry of alkyl derivatives of urea. Doklady Akad. Nauk SSSR, Ser. Khim 1990, 750–755. [Google Scholar]
- (89).Diogo HP; de Piedade MEM A micro-combustion calorimeter suitable for samples of mass 10 mg to 50 mg. Application to solid compounds of C, H, and O, and of C, H, O, and N. J. Chem. Thermodyn 1995, 27, 197–206. [Google Scholar]
- (90).Ribeiro da Silva MDMC; Santos LMNBF; Silva ALR; Fernandes Ó; Acree WE Jr Energetics of 6-methoxyquinoline and 6-methoxyquinoline N-oxide: the dissociation enthalpy of the (N–O) bond. J. Chem. Thermodyn 2003, 35, 1093–1100. [Google Scholar]
- (91).Zaitsau Dz.; Kabo GJ; Kozyro AA; Sevruk VM The effect of the failure of isotropy of a gas in an effusion cell on the vapor pressure and enthalpy of sublimation for alkyl derivatives of carbamide. Thermochim. Acta 2003, 406, 17–28. [Google Scholar]
- (92).Emel’yanenko VN; Kabo GJ; Verevkin SP Measurement and prediction of thermochemical properties: improved increments for the estimation of enthalpies of sublimation and standard enthalpies of formation of alkyl derivatives of urea. J. Chem. Eng. Data 2006, 51, 79–87. [Google Scholar]
- (93).Bedford AF; Beezer AE; Mortimer CT Heats of formation and bond energies. Part X. 1,2,5,6-tetrahydropyridine, piperidine, and piperazine. J. Chem. Soc 1963, 2039–2043. [Google Scholar]
- (94).Good WD Enthalpies of combustion of nine organic nitrogen compounds related to petroleum. J. Chem. Eng. Data 1972, 17, 28–31. [Google Scholar]
- (95).Osborn AG; Douslin DR Vapor pressure relations of 13 nitrogen compounds related to petroleum. J. Chem. Eng. Data 1968, 13, 534–537. [Google Scholar]
- (96).Messerly JF; Todd SS; Finke HL; Good WD; Gammon BE Condensed-phase heat-capacity studies and derived thermodynamic properties for six cyclic nitrogen compounds. J. Chem. Thermodyn 1988, 20, 209–224. [Google Scholar]
- (97).Hossenlopp IA; Archer DG Enthalpies of vaporization of piperidine and 1,2-dimethylbenzene; gas-phase isobaric heat capacities of piperidine. J. Chem. Thermodyn 1988, 20, 1061–1068. [Google Scholar]
- (98).Anderson CM; Gilbert EC The Apparent Energy of the N-N Bond as Calculated from Heats of Combustion. J. Am. Chem. Soc 1942, 64, 2369–2372. [Google Scholar]
- (99).Cole LG; Gilbert EC The Heats of Combustion of Some Nitrogen Compounds and the Apparent Energy of the N-N Bond. J. Am. Chem. Soc 1951, 73, 5423–5427. [Google Scholar]
- (100).Hatton WE; Hildenbrand DL; Sinke GC; Stull DR Chemical Thermodynamic Properties of Aniline. J. Chem. Eng. Data 1962, 7, 229–231. [Google Scholar]
- (101).Steele WV; Chirico RD; Knipmeyer SE; Nguyen A Vapor Pressure, Heat Capacity, and Density along the Saturation Line: Measurements for Benzenamine, Butylbenzene, sec-Butylbenzene, tert-Butylbenzene, 2,2-Dimethylbutanoic Acid, Tridecafluoroheptanoic Acid, 2-Butyl-2-ethyl-1,3-propanediol, 2,2,4-Trimethyl-1,3-pentanediol, and 1-Chloro-2-propanol. J. Chem. Eng. Data 2002, 47, 648–666. [Google Scholar]
- (102).Kusano K; Wadsö I Enthalpy of vaporization of some organic substances at 25° C and test of calorimeter. Bull. Chem. Soc. Jpn 1971, 44, 1705–1707. [Google Scholar]
- (103).Lebedeva ND; Katin Yu. A.; Akhmedova G. Ya. Standard Enthalpy of Formation for Nitrobenzene. Dep. VINITI 2945–71, 1971. [Google Scholar]
- (104).Verevkin SP; Emel’yanenko VN; Diky V; Dorofeeva OV Enthalpies of formation of nitromethane and nitrobenzene: New experiments vs. quantum chemical calculations. J. Chem. Thermodyn 2014, 73, 163–170. [Google Scholar]
- (105).Kalugina TG; Kiparisova EG Enthalpies of Combustion and of Formation of Derivatives of Carbamide. Russ. J. Phys. Chem 1987, 61, 261–262. [Google Scholar]
- (106).Steele WV; Chirico RD; Nguyen A; Hossenlopp IA; Smith NK Determination of Ideal-gas Enthalpies of Formation for Key Compounds. AIChE Symp. Series No. 279 1990, 86, 138–154. [Google Scholar]
- (107).Gomez LAT; Sabbah R Thermodynamique de substances azotees. IX. Etude thermochimique de la benzamide. Comparaison des grandeurs energetiques liees a la structure de quelques amides et thioamides. Thermochim. Acta 1982, 58, 311–315. [Google Scholar]
- (108).Almeida ARRP; Monte MJS Thermodynamic study of benzamide, N-methylbenzamide, and N, N-dimethylbenzamide: vapor pressures, phase diagrams, and hydrogen bond enthalpy. J. Chem. Eng. Data 2010, 55, 3507–3512. [Google Scholar]
- (109).Verevkin SP; Emel’yanenko VN; Nagrimanov RN Nearest-Neighbor and Non-Nearest-Neighbor Interactions between Substituents in the Benzene Ring. Experimental and Theoretical Study of Functionally Substituted Benzamides. J. Phys. Chem. A 2016, 120, 9867–9877. [DOI] [PubMed] [Google Scholar]
- (110).NIST ThermoData Engine Version 10.0 - Pure Compounds, Binary Mixtures and Reactions. NIST Standard Reference Database 103b, Gaithersburg, MD, 2016. https://www.nist.gov/srd/nist-standard-reference-database-103b, accessed: January 13, 2017. [Google Scholar]
- (111).ThermoLit - NIST Literature Report Builder for Thermophysical and Thermochemical Property Measurements. NIST Standard Reference Database 171, Gaithersburg, MD, 2016. http://trc.nist.gov/thermolit, accessed: January 13, 2017. [Google Scholar]
- (112).Daniel C; Wood FS Fitting Equations to Data; Wiley, New York, 1980. [Google Scholar]
- (113).Benjamini Y Opening the Box of a Boxplot. Am. Stat 1988, 42, 257–262. [Google Scholar]
- (114).Dorofeeva OV; Ryzhova ON Enthalpy of Formation and O-H Bond Dissociation Enthalpy of Phenol: Inconsistency between Theory and Experiment. J. Phys. Chem. A 2016, 120, 2471–2479. [DOI] [PubMed] [Google Scholar]
- (115).Diky V; Chirico RD; Kazakov AF; Muzny CD; Frenkel M ThermoData Engine (TDE): Software Implementation of the Dynamic Data Evaluation Concept. 4. Chemical Reactions. J. Chem. Inf. Model 2009, 49, 2883–2896. [DOI] [PubMed] [Google Scholar]
- (116).Riplinger C; Pinski P; Becker U; Valeev EF; Neese F Sparse maps – A systematic infrastructure for reduced-scaling electronic structure methods. II. Linear scaling domain based pair natural orbital coupled cluster theory. J. Chem. Phys 2016, 144, 024109. [DOI] [PubMed] [Google Scholar]