

Abstract
Objective:
Pancreatic ductal adenocarcinoma (PDAC) displays a remarkable propensity towards therapy resistance. However, molecular epigenetic and transcriptional mechanisms enabling this are poorly understood. In this study we aimed to identify novel mechanistic approaches to overcome or prevent resistance in PDAC.
Design:
We utilized in vitro and in vivo models of resistant PDAC and integrated epigenomic, transcriptomic, nascent RNA and chromatin topology data. We identified a JunD-driven subgroup of enhancers, called interactive hubs (iHUBs), that mediate transcriptional reprogramming and chemoresistance in PDAC.
Results:
iHUBs display characteristics typical for active enhancers (H3K27ac enrichment) in both therapy sensitive and resistant states but exhibit increased interactions and production of enhancer RNA (eRNA) in the resistant state. Notably, deletion of individual iHUBs was sufficient to decrease transcription of target genes and sensitize resistant cells to chemotherapy. Overlapping motif analysis and transcriptional profiling identified the Activator Protein 1 (AP1) transcription factor JunD as a master transcription factor of these enhancers. JunD depletion decreased iHUB interaction frequency and transcription of target genes. Moreover, targeting either eRNA production or signaling pathways upstream of iHUB activation using clinically tested small molecule inhibitors decreased eRNA production and interaction frequency, and restored chemotherapy responsiveness in vitro and in vivo. Representative iHUB target genes were found to be more expressed in patients with poor response to chemotherapy compared to responsive patients.
Conclusion:
Our findings identify an important role for a subgroup of highly connected enhancers (iHUBs) in regulating chemotherapy response and demonstrate targetability in sensitization to chemotherapy.
Keywords: Enhancers, PDAC, resistance, enhancer promoter interaction, enhancer RNA, Transcription, JunD
INTRODUCTION
Less than half of patients with advanced pancreatic ductal adenocarcinoma (PDAC) survive longer than 12 months after diagnosis [1]. This dismal survival is accompanied by a steady rise in PDAC incidence across the world [2]. Two-thirds of PDAC tumors are resistant to current standard therapies [3]. Nab-Paclitaxel is a chemotherapy that targets microtubules and used in combination with gemcitabine as first-line palliative therapy in PDAC patients with poor performance status [4]. Paclitaxel significantly increases long-term survival [5]. Mechanisms of resistance to gemcitabine in PDAC have been extensively studied [6]. In contrast, fewer studies investigated resistance mechanisms for paclitaxel. Resistance to paclitaxel was linked to aberrant gene activation [7]. A transcriptionally permissive state perpetuates high adaptability in PDAC, enabling cells to thrive in chemotoxic environments and ultimately rendering most therapies ineffective. Thus, therapeutic targeting of transcriptional and epigenetic regulators may hinder adaptability and block evasion mechanisms.
Enhancers are distal regulatory elements that physically loop in 3D to interact with promoter regions and activate target genes [8]. Deregulated enhancers mediate transcriptional reprogramming in resistant leukemia [9], breast [10] and ovarian cancer [11]. Acetylation of histone 3 at lysine 27 (H3K27ac) is tightly linked to enhancer activation with a positive correlation between H3K27ac intensity and enhancer activity [12]. Enhancer-promoter interactions (EPI) determine enhancer-regulated gene activation and are highly tissue specific [13]. Rewiring of enhancer interactions occurs in various contexts including metastatic PDAC [14]. Architectural proteins such as CCCTC-binding factor (CTCF) and cohesin regulate genome compartmentalization, where interactions are enriched within topologically associating domains (TADs) [15]. Loss of CTCF has limited effects on TADs but leads to unfettered activation of gene transcription [16, 17]. On the other hand, cohesin perturbation leads to a loss of all 3D looping [18]. Thus, to date, there is no known mechanism to specifically disrupt enhancer-promoter interactions without inducing catastrophic events.
Super enhancers (SEs) are a subgroup of enhancers containing clusters of active elements that promote transcription of lineage-specific and oncogenic gene programs [19]. SEs play a crucial role in driving aggressiveness in PDAC [20, 21]. SEs are enriched for transcriptional activators such as bromodomain protein containing 4 (BRD4) and Mediator [19]. BRD4 promotes transcription at enhancers leading to enhancer RNA (eRNA) production [22]. While eRNA function has not yet been fully characterized, eRNAs are reported to strengthen enhancer-promoter interactions (EPIs) in estrogen-mediated transcription [23]. Additionally, eRNAs can interact with various structural proteins such as Mediator and cohesin [24,25] and have been reported to promote activation of transcriptional elongation by binding to Cyclin T1, thereby activating the Positive Transcription Elongation Factor-b complex [26]. However, specific targeting of eRNAs is challenging due their extremely short half-lives, making them essentially impervious to induction of premature termination by antisense oligonucleotide-mediated RNAseH-dependent degradation [27, 28].
To evaluate the role of enhancer reprogramming in PDAC, we established a highly resistant derivative of the L3.6pl pancreatic cancer cell line. This model allowed us to uncover a subgroup of enhancers called interactive hubs (iHUBs) that gain EPIs without necessarily displaying significant changes in H3K27ac enrichment. iHUBs produced higher levels of eRNA in resistant cells and patient-derived xenografts (PDXs) treated with chemotherapy. Deletion of a single iHUB using CRISPR/Cas9-mediated genome editing was sufficient to partially restore chemosensitivity. Additionally, we identified the Activator Protein 1 (AP1) transcription factor JunD as a major driver of iHUB-mediated transcriptional reprogramming and were able to sensitize a paclitaxel-resistant PDX by targeting iHUBs using low concentrations of a Cyclin-Dependent Kinase-9 inhibitor (CDK9i) or the MEK inhibitor trametinib. In conclusion, we uncovered that altered H3K27ac poorly correlates with resistance-associated enhancer rewiring. Instead, enhancers required for resistance-specific gene expression were associated with increased chromosomal interactions and targeting these iHUBs may provide a promising new approach to attenuate transcriptional reprogramming and promote or maintain sensitivity to chemotherapy.
RESULTS
Aberrant transcriptional activation in chemoresistant PDAC is not mediated by de novo activation of enhancers.
In order to establish an experimental model of chemotherapy-resistant PDAC, we maintained L3.6pl cells in increasing concentrations of paclitaxel for 100 days (Fig. 1A). While the parental L3.6pl cells (Par) had a half-maximal inhibitory concentration (IC50) of 1.6nM [95% CI 0.91–2.7nM] (Fig.S1A), paclitaxel-resistant cells (PacR) thrived in 64nM paclitaxel and exhibited a 100-fold increase in IC50: 158.6nM [95% CI 83.16–305nM] (Fig. 1B, Fig.S1B). Subsequently, we injected parental and resistant cells subcutaneously in NOD/SCID mice and after one week treated mice with 30mg/kg paclitaxel i.p. twice weekly for a total of three weeks (Fig. 1C). The weights and volumes of Par-derived xenograft tumors significantly decreased while there was no significant effect of paclitaxel treatment on PacR-derived tumors, confirming that this cell system represents a relevant model system for studying chemotherapy resistance in vivo (Fig. 1D, Fig.S1C–E).
Fig. 1. Aberrant transcriptional activation in chemoresistant PDAC is largely independent of de novo enhancer activation.
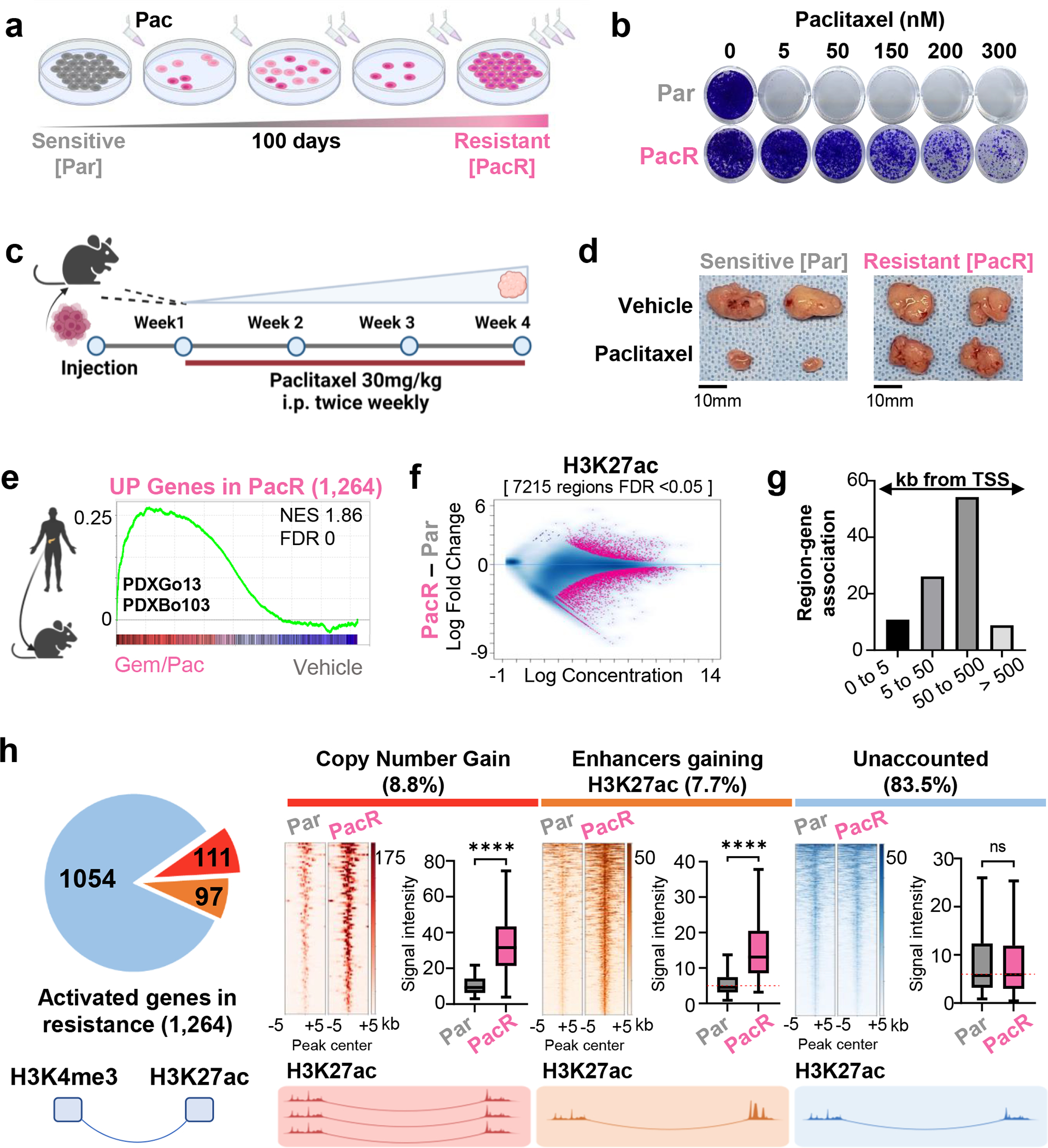
(a) A scheme depicting the establishment of paclitaxel resistant L3.6pl cells. Sensitive cells were treated with increasing concentrations of paclitaxel for 100 days. (b) Crystal Violet staining for proliferation assay where sensitive (Par) and paclitaxel resistant (PacR) cells are treated with increased concentrations of paclitaxel every 48 hours for 6 days. (PacR) are 100 times more resistant than sensitive cells (Par) (c) A scheme depicting experiment where xenografts are derived from Par and PacR. Cells were injected into immune-deficient mice and allowed to grow for 1 week before beginning treatment with paclitaxel or vehicle twice/weekly for 4 weeks. (d) Representative tumor sizes from Par and PacR derived xenografts showing that the PacR-derived xenografts are resistant in vivo. (e) Gene set enrichment analysis for activated genes in PacR (1,264 genes, FDR <0.05, log2 fold change >0.7) in 2 patient-derived xenografts treated with Gem/Pac for multiple cycles compared to the same patients-derived xenografts treated with vehicle. Plot shows a significant upregulation of activated PacR genes in treated patient-derived xenografts compared to vehicle. (f) MA plot shows differentially enriched regions for H3K27ac in PacR compared to Par. Significant regions are colored in pink. (g) GREAT analysis of regions gaining H3K27ac in PacR showing the distance to TSS for the group of regions that gain H3K27ac. Most of these regions are enriched at distal regions. (h) Pie chart showing that 111 out of 1264 PacR activated genes exhibit copy number gain either at TSS or interacting enhancer [red]. Only 97 genes out of 1264 activated genes interact with enhancers that significantly gain H3K27ac [orange]. The majority of genes (1054; 83.4%) interact with enhancers that display no significant change in H3K27ac enrichment [blue]. Heatmaps are showing H3K27ac binding profile at amplified regions [red], activated enhancers [orange], enhancers interacting with 1054 upregulated genes in blue. Box plots show intensity of H3K27ac at enhancers in RPKM.
To understand the mechanisms involved in resistance, we characterized the transcriptional changes in PacR by performing mRNA-seq for Par and PacR cells. We identified 1,887 genes that were significantly differentially regulated in PacR compared to Par [FDR < 0.05, Log2 Fold Change > 0.7 or =<−0.7] (Fig.S1F). Of these genes 1,264 were significantly upregulated and 623 were significantly downregulated. To validate that the transcriptional changes observed in PacR are representative of transcriptional reprogramming in patients, we utilized PDXs derived from two PDAC patients (PDXGo13, PDXBo103) and treated tumor-bearing mice with gemcitabine/paclitaxel or vehicle for multiple cycles before performing mRNA-seq. Genes identified to be activated in PacR were significantly upregulated upon chemotherapy treatment in both PDXs, supporting the existence and relevance of a common paclitaxel-induced transcriptional gene signature (Fig. 1E). The enrichment was observed when comparing the treated and control PDXs together and separately with common genes upregulated in both treated PDXs (Fig.S1G,H). Given that the PDXs were treated with gemcitabine/paclitaxel, we compared the PacR signature to the previously published GemR signature [56] and observed minimal overlap (~7.5% of PacR Up genes) and ~20% of the changes shared between PacR and the treated PDX (Fig.S2A–B, Table S1). This suggests that the transcriptional changes underlying the overlap between treated PDX and PacR cells are mainly mediated by the effects of paclitaxel.
Given the major transcriptional reprogramming observed in resistance, in addition to the significant role that epigenetic mechanisms play in regulating transcription, we investigated the epigenetic mechanisms driving aberrant transcriptional activation in resistance. Thus, we linked transcriptomic data with the genome-wide localization of the active epigenetic mark H3K27ac, which denotes active transcriptional start sites and enhancer regions. Interestingly, differential binding analysis identified more than 7,000 H3K27ac-enriched regions that were significantly changed in PacR [7,215 regions, FDR < 0.05] (Fig. 1F). GREAT analysis showed that most of these regions are distal from transcriptional starting sites (TSS) and thereby represent putative enhancers (Fig. 1G).
In order to evaluate the effect of these putative enhancers on transcription in resistant cells, we identified the target genes interacting in 3D with these enhancers. Enhancers are usually marked by H3K27ac but devoid of histone 3 trimethylated at lysine 4 (H3K4me3), while active TSS are marked by both [H3K27ac+, H3K4me3+]. Thus, we performed TSS-centric HiChIP for H3K4me3 as we observed minimal changes in H3K4me3 enrichment in Par and PacR (Fig.S2C). We expected that many upregulated genes would interact with activated enhancers that gain H3K27ac (evaluated by H3K27ac ChIP-seq). Alternatively, we expected that other upregulated genes will exhibit copy number variation either directly (TSS of gene) or indirectly (enhancer interacting with gene). To evaluate copy number variation (CNV), we performed CNV and differential binding analysis using input DNA (Fig.S2D,E). Surprisingly, only 16.5% of genes upregulated in resistance displayed either copy number gains at TSS of the gene, interacted with an enhancer with copy number gain, or interacted with an enhancer that gained H3K27ac in PacR compared to Par (Fig. 1H). This is in contrast to significantly downregulated genes where 39% of the genes interacted with regions losing H3K27ac (Fig.S2F). As enhancer activation is identified by a gain in H3K27ac enrichment, we evaluated the differential intensity of H3K27ac enrichment at the enhancers interacting with the activated genes with an unaccounted regulatory mechanism (the 83.5% of upregulated genes in blue in Fig. 1H). These enhancers displayed no significant changes in H3K27ac, meaning that they would not be defined as activated when using the standard paradigm of differential enrichment of H3K27ac to define de novo activated enhancers. Together, we established a representative system for studying enhancer reprogramming in chemotherapy-resistant PDAC. This is supported by the convergent gene programs activated in vitro and in PDXs treated with chemotherapy. Chromatin topology studies allowing for the identification of interacting distal enhancers and their associated target genes revealed that the majority of genes activated in PacR do not interact with an enhancer that gained H3K27ac.
Highly interactive enhancer hubs (iHUBs) mediate aberrant transcriptional activation in chemoresistant PDAC.
To investigate potential mechanisms underlying the unaccounted aberrant gene activation in resistance, we focused on the enhancers interacting with upregulated genes in resistant cells, but not displaying a gain of H3K27ac enrichment. Due to the established role of BRD4 as a crucial mediator of enhancer activity [19], we performed ChIP-seq for BRD4 in Par and PacR and evaluated the enrichment of BRD4 at these enhancers. Notably, despite the lack of change in H3K27ac enrichment at enhancers interacting with upregulated genes, BRD4 enrichment was significantly increased at these enhancers in resistant cells (Fig. 2A). Indeed, differential binding analysis identified a significant gain of BRD4 at 891 enhancer regions (H3K27ac+, H3K4me3−) and differentially separated Par and PacR in PCA analysis (Fig.S3A–B). Importantly, the gain of BRD4 is not due to a global increase in BRD4 protein levels (Fig.S3C). Interestingly, consistent with its potential role in chromatin organization [29], we observed a strong correlation between BRD4 signal at enhancers and enhancer interaction frequency (Fig. 2B, Fig.S3D).
Fig. 2. Highly interactive enhancer hubs (iHUBs) mediate aberrant transcriptional activation in chemoresistant PDAC.
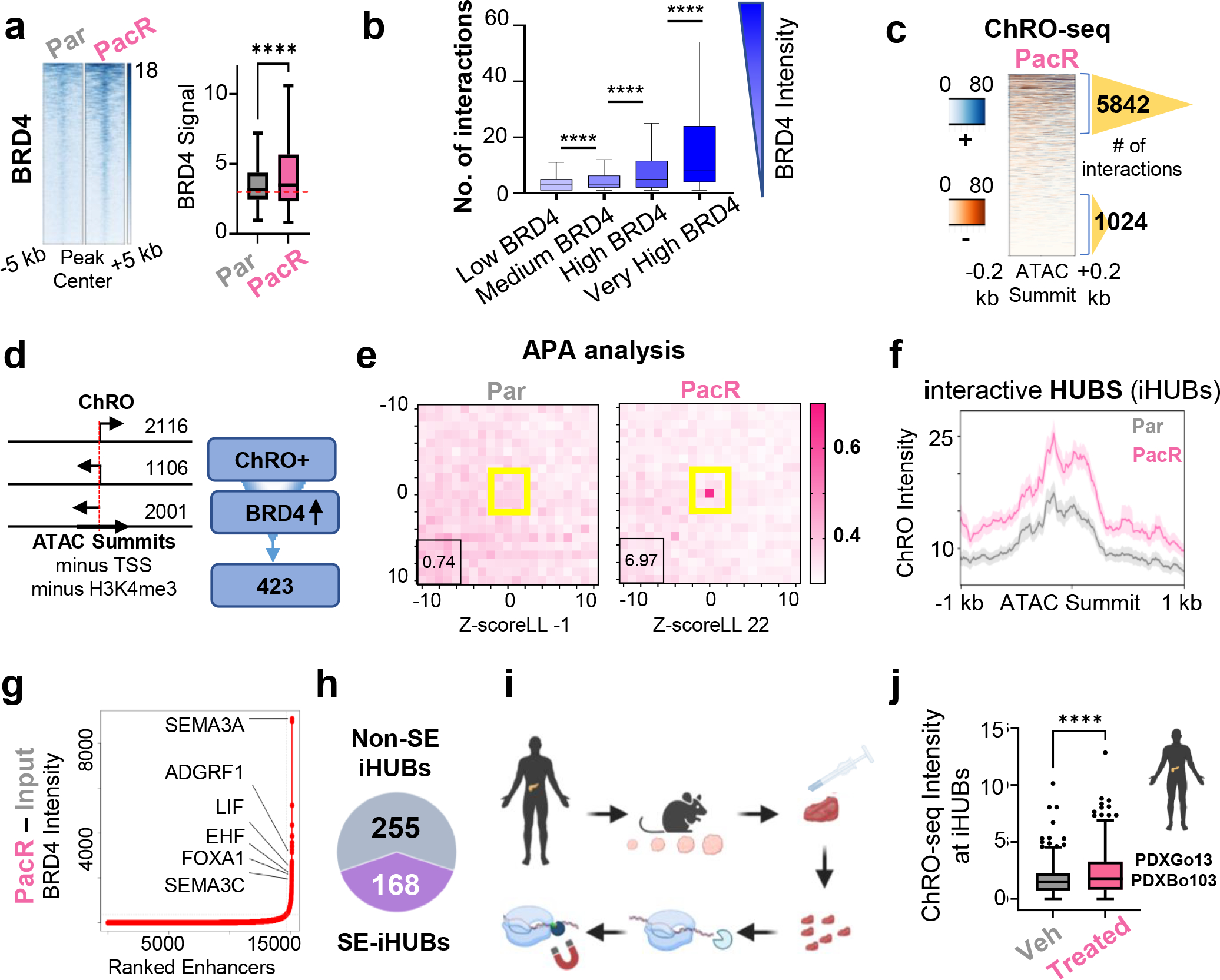
(a) Heatmap and box plot for intensity of BRD4 occupancy [RPKM] at enhancers interacting with upregulated genes of unaccounted mechanism. (b) Box plot showing the number of interactions for regions classified into 4 groups based on their BRD4 intensity. Darker blue represents more BRD4 intensity. The regions that are enriched by higher BRD4 intensity significantly exhibit more interaction frequency. (c) Heatmap of leChRO-seq signal at enhancers in PacR cells. Number of interactions are shown for the enhancers that produce more eRNA compared to enhancers that do not produce eRNA (blue, plus strand signal; orange, minus strand signal). Number of interactions are higher by 5.7 folds when comparing eRNA rich enhancers vs eRNA poor enhancers. (d) Schematic diagram depicting the identification of enhancers that gain eRNA and BRD4 in resistance. Regions are centered at ATAC summits and H3K27ac regions that are not defined as TSS and are not enriched for H3K4me3 are picked if they produce eRNA (sense, antisense or the opposite strand of a gene direction) and gain BRD4 in PacR. (e) APA analysis for regions identified in 2d to evaluate interaction frequency for these regions. Plots show more focal enrichment (higher EPIs) in PacR at enhancers that interact with activated genes in resistance and do not gain H3K27ac. A higher P2LL indicates higher enrichment. These enhancers are termed interactive hubs (iHUBs). (f) Plot profile of leChRO-seq signal in RPKM showing that iHUBs are more transcribed in PacR compared to Par. (g) ROSE algorithm plot identifying SEs in PacR. (h) Pie chart indicating that only a portion of the identified iHUBs (168 out 423) overlap with super enhancers. (i) Scheme depicting leChRO-seq from patient-derived xenografts. (j) Box plot showing significant gain of leChRO-seq signal in RPKM in two independent patient-derived xenografts that were treated with multiple cycles of chemotherapy compared to vehicle-treated xenografts. iHUBs identified from cell lines were used.
Given the role of BRD4 in driving enhancer RNA (eRNA) transcription [22] and the reported link between eRNA and enhancer-promoter interaction [23], we examined nascent RNA production by length extension chromatin run-on sequencing (leChRO-seq) in Par and PacR. leChRO-seq allows for the detection of short-lived RNAs including eRNA. Interestingly, we observed that the regions that gain BRD4 in PacR and produce high levels of enhancer RNA interact 5.6 times more frequently than non-transcribed enhancers (Fig. 2C). To identify enhancer regions that produce eRNA, we performed ATAC-seq in Par and PacR cells and focused on regions that produce one-sided transcripts or transcripts in the opposite strand of mRNA transcription. We further overlapped these enhancers with significantly gained BRD4 regions in PacR (Fig. 2D). This focused approach identified 423 enhancers of interest. We evaluated the frequency of interactions for these regions using aggregate plot analysis (APA) in our H3K4me3 HiChIP data. Notably, we observed focal enrichment only in PacR, indicating that these 423 enhancers are highly interactive in PacR compared to Par, where they are not as interactive [P2LL 6.97:0.74, Z score LL 22: −1] (Fig. 2E). Therefore, based on their high interaction frequency, we termed this set of enhancers “interactive hubs” (iHUBs). Indeed, iHUB target genes showed significant enrichment in PacR with 332 genes significantly enriched in PacR compared to Par (Fig.S3E). We observed that these iHUBs are specific to paclitaxel as their target genes were not significantly enriched in gemcitabine-resistant cells (Fig.S3F). Consistent with the gain of BRD4, we observed a significant gain of the BRD4-bound epigenetic mark, H4K5ac [30], at iHUBs (Fig.S3G). To rule out that these regions are an artifact of the HiChIP method, we ensured that there was no significant change in H3K4me3 enrichment at the associated TSS of the genes interacting with these 423 enhancers (Fig.S3H). Additionally, we investigated the intensity of leChRO-seq at iHUBs. Interestingly, we observed a significant gain of eRNA production in resistance (Fig. 2F, Fig.S3I).
Based on the established role of SE in controlling cell fate-determining genes, we investigated whether iHUBs are variants of SE. To this end, we identified SE in PacR using the ROSE algorithm (Fig. 2G) and found that only 168 of the 423 iHUBs intersected with SE, while 73 SE intersected with iHUBs (Fig. 2H, Fig.S3J). The smaller number of SE overlapping with iHUBs suggests that some SE contained multiple iHUBs within them. Importantly, it also suggests that iHUBs represent a potentially meaningful subgroup of enhancers largely independent of SE. We further confirmed the clinical and in vivo relevance of our findings related to iHUB transcription by performing leChRO-seq in vehicle and Gem/Pac-treated PDX samples (PDXBo103 and PDXGo13; Fig. 2I). By inferring iHUBs from our in vitro findings, we observed increased eRNA transcription at iHUBs in treated PDX compared to vehicle (Fig. 2J).
In conclusion, we identified a subgroup of highly interactive enhancers termed iHUBs that mediate transcriptional activation in resistance and are characterized by high BRD4 localization and eRNA production that is not necessarily coupled to changes in the enrichment of H3K27ac.
iHUBs mediate chemoresistance in PDAC.
After identifying a crucial role of iHUBs in mediating transcriptional activation, we sought to determine the role of iHUBs in mediating resistance. In order to identify iHUBs potentially relevant to patients, we integrated gene expression data from PDAC patients from the TCGA database with CancerRxTissue [31] to predict PDAC patient tumors that are more sensitive to paclitaxel (Fig.S4A). We focused on patient samples that cluster based on their expected IC50 and performed differential expression analysis (Fig.S4B). Subsequently, we plotted genes based on their differential enrichment in resistant patient tumors and identified LIF, IGFBP6, CLDN1, GPX4, and PON2 as expressed genes that have preferential enrichment in less responsive patient tumors (Fig.S4C).
Leukemia inhibitory factor (LIF) has previously been reported to play an oncogenic role in KRAS-induced PDAC. Moreover, LIF is upregulated in paclitaxel-resistant endometrial cancer [32]. Insulin-Like Growth Factor Binding Protein 6 (IGFBP6) was reported to be upregulated in PDAC tissue [33]. Additionally, Glutathione Peroxidase 4 (GPX4) has a crucial protective role in oxidative stress and its inhibition is associated with overcoming resistance [34]. On the other hand, Paraoxonase 2 (PON2) was reported to promote growth and metastasis of PDAC [35]. Less is known about the role of Dehydrogenase/Reductase 3 (DHRS3) in PDAC, but it has been linked to lymphocyte infiltration of the pancreas [36]. Claudin 1 (CLDN1) promotes proliferation of PDAC cells in response to tumor necrosis factor-alpha [37]. Based on their reported roles in cancer [38] and therapeutic response [39], as well as their amenability to pharmacological targeting, we specifically focused on LIF and GPX4. Both LIF- and GPX4-interacting iHUBs showed increased interaction in PacR compared to Par (Fig. 3A–B). They also showed a significant gain in BRD4 occupancy (Fig.S4D). This is in contrast to enhancers that are not iHUBs, which show comparable interaction frequency in Par and PacR (Fig.S4E–G). To validate this in a group of comparable size, we used bedtools to identify random enhancers that are active in PacR and are similar in size to iHUBs. In contrast to iHUBs, no significant change in interaction frequency was found in two independent sets of random enhancers active in PacR (Fig.S5A).
Fig. 3. iHUBs mediate chemoresistance in PDAC.
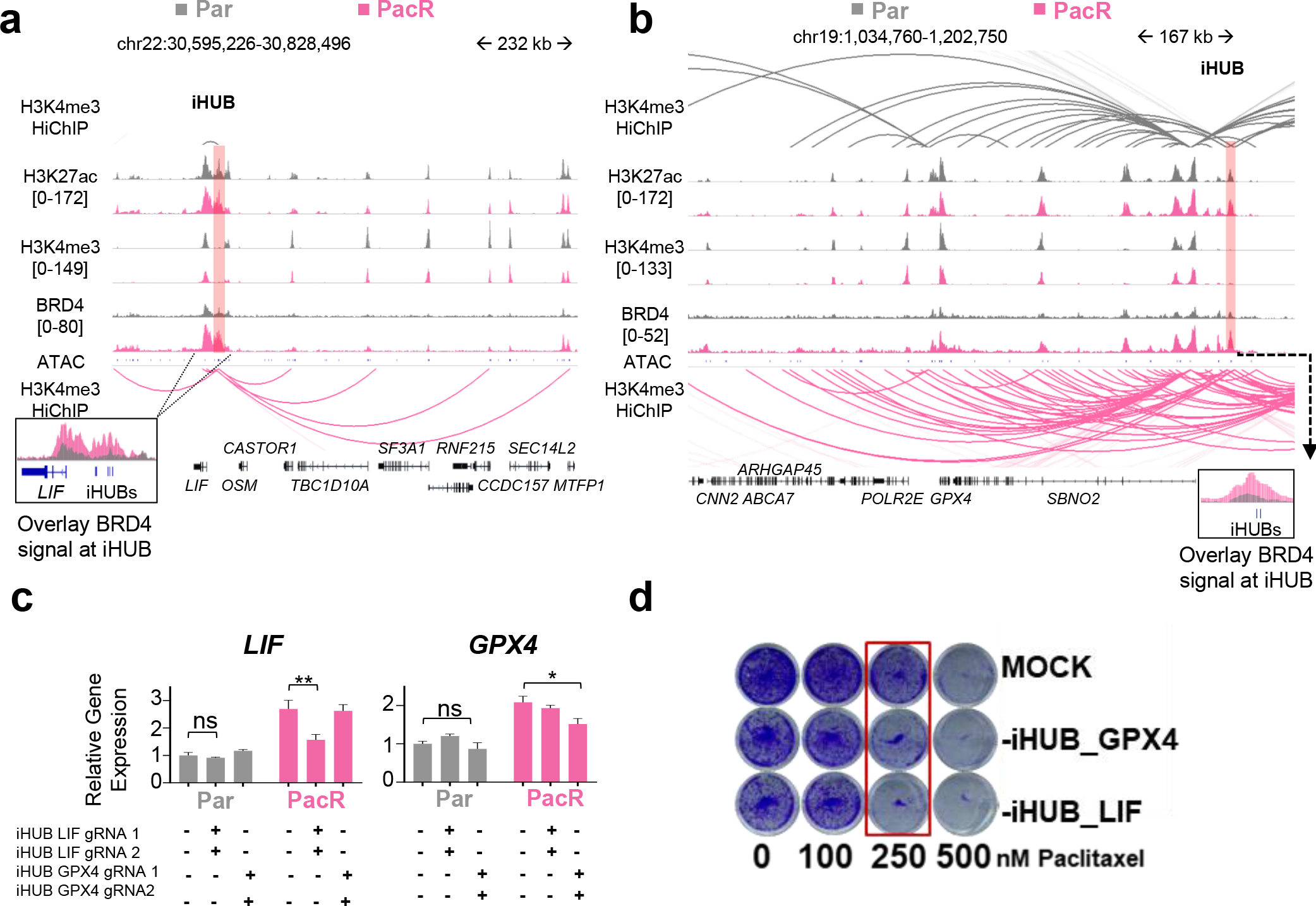
(a,b) Occupancy profiles of H3K27ac, H3K4me3, ATAC at LIF and GPX4 iHUBs in Par and PacR cells showing minimal changes in H3K27ac and H3K4me3, but significant gain of BRD4 occupancy [RPKM]. Arcs in the HiChIP data show an increase in interaction frequency in PacR (pink) compared to Par cells (gray). (c) Quantitative PCR for LIF and GPX4 mRNAs in control cells (MOCK) or following deletion of LIF or GPX4 iHUBs. Both LIF and GPX4 mRNAs were significantly downregulated upon the deletion of their respective iHUBs in PacR. Deletion of iHUBs in Par did not affect LIF and GPX4 mRNA levels, supporting a specific role of iHUBs in resistance. (d Crystal violet staining for proliferation assay upon deletion of LIF or GPX4 iHUBs compared to MOCK showing sensitization of PacR cells when they lose one iHUB.
To evaluate the functional contribution of iHUBs to resistance, we employed CRISPR/Cas9-mediated genome editing to specifically delete individual enhancers. In each, we deleted regions encompassing the ATAC peak (~180bp for LIF iHUB and ~450bp for GPX4 iHUB) and validated the loss of the deleted region (Fig.S5B–E). Consistent with an essential role in transcriptional activation of target gene expression, the deletion of either the LIF- or GPX4-iHUBs specifically decreased the expression of the respective target gene (Fig. 3C, Fig.S5F). Notably, the effect of deleting individual iHUBs on target gene expression was specific to the PacR cells where LIF and GPX4 expression was only dependent on the iHUB in the resistant state. The deletion did not cause downregulation of other nearby genes (Fig.S5G). Remarkably, the deletion of either the LIF- or GPX4-iHUB alone was sufficient to partially re-sensitize PacR to paclitaxel (Fig. 3D, Fig.S6A). Consistently, the LIF-LIFR signaling inhibitor EC330 significantly sensitized PacR cells (Fig.S6B). In addition, knockdown of GPX4 sensitized PacR cells (Fig.S6C). Low concentration of the GPX4 inhibitor ML-162 also sensitized PacR and Capan-1 cells (Fig.S6D–E).
Together, we confirmed the role of iHUBs in mediating transcriptional activation and paclitaxel resistance in pancreatic cancer whereby deletion of a single iHUB was sufficient to elicit an appreciable chemosensitizing effect.
iHUB function requires AP-1 transcription factor activity.
After identifying a crucial role of iHUBs in mediating resistance, we sought to identify transcription factors responsible for directing iHUB function. Therefore, we used HOMER to identify the top transcription factor binding motifs enriched at iHUBs and integrated this information with gene expression and regulation patterns in PacR vs. Par (Fig. 4A). Using this approach, we identified the AP-1 transcription factor JunD as a putative iHUB-enriched transcription factor. Accordingly, we performed ChIP seq for JunD in Par and PacR and observed that JunD binds at iHUB regions (Fig. 4B). We also observed a significant increase in JunD binding in PacR compared to Par (Fig.S7A–B). Given the essential role of upstream MEK-MAPK signaling in activating AP-1 transcription factor activity via direct phosphorylation [40], we investigated whether inhibition of MAPK signaling with the clinical MEK inhibitor trametinib can sensitize PacR cells. Indeed, when combined with paclitaxel, trametinib significantly sensitized PacR cells to paclitaxel treatment (Fig. 4C). To study the effects of JunD depletion on the expression of iHUB targets, we knocked down JunD for 24 h and performed RNA-seq (Fig.S7C). Thereby, we observed that JunD depletion significantly downregulated both iHUB-associated eRNA and mRNA in PacR (Fig. 4D–E; Fig.S7D). To confirm that the decrease in expression is due to loss of enhancer interactions at iHUBs, we performed H3K4me3 HiChIP in PacR and observed a significant decrease in interaction frequency at iHUBs upon JunD depletion (Fig. 4F, Fig.S8A–C). We hypothesized that JunD promotes interactions at iHUBs by recruiting Mediator to increase interaction with target genes. Therefore, we performed ChIP seq for the Mediator Complex Subunit 1 (MED1). Consistently, we observed that MED1 occupancy is significantly upregulated at iHUBs in PacR compared to Par and this increase is reversed by JunD depletion in PacR cells (Fig. 4G).
Fig. 4. JunD drives iHUB formation in chemoresistance in PDAC.
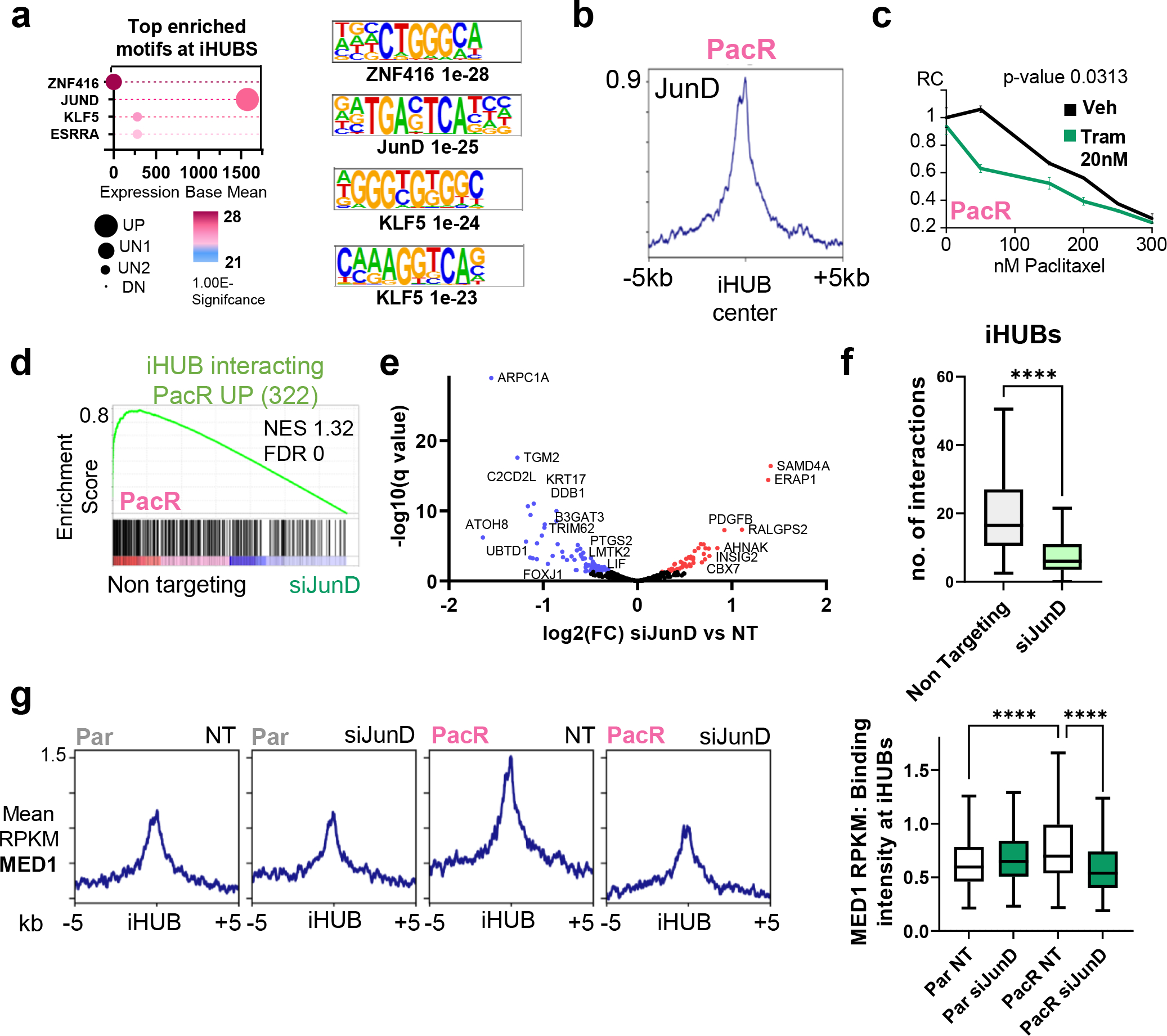
(a) Cleveland plot depicting the top motifs enriched at iHUBs (defined by HOMER) coupled with expression base mean and regulation patterns in PacR compared to Par. JunD is the top hit for transcription factors that are enriched at iHUBs. JunD is expressed and upregulated in PacR compared to Par. (b) Binding profile for JunD in PacR cells at iHUB regions [peaks center is the ATAC summit; extended for −/+5kb]. (c) Relative showing significant sensitization upon treatment with low concentrations of the trametinib when combined with paclitaxel. (d) Gene set enrichment analysis for iHUB interacting activated genes in PacR (322 genes) in PacR with JunD knockdown (24 hours) compared to Non targeting. Plot shows a significant downregulation of iHUB activated PacR genes in JunD knockdown. (e) Volcano plot for differentially regulated genes upon JunD knockdown in PacR cells. [Blue: significantly downregulated, Red: significantly upregulated]. Notably, GPX4 is not significantly downregulated. (f) Box plot showing number of interactions at IHUBs in PacR when comparing non-targeting to JunD knockdown. It shows a significant decrease in the number of iHUB interactions upon JunD depletion. (g) Plot Profile and box plots showing localization intensity of Med1 at iHUB regions. Plots show a significant upregulation of Med1 enrichment in PacR cells compared to Par cells and a significant downregulation of Med1 enrichment upon JunD knockdown.
In conclusion, our data reveal a central importance of JunD in driving iHUB activity and that JunD depletion impairs iHUB target gene expression by leading to decreased MED1 occupancy and interaction frequency.
Perturbation of iHUB transcription decreases interaction frequency and reverses resistance.
As eRNA production is a main characteristic of iHUBs, we investigated whether transcription of iHUBs is necessary for the high interaction frequency or is a bystander effect. To further validate the gain of eRNA production at iHUBs, we performed ChIP-seq for RNA Polymerase II (Pol II) in Par and PacR. Consistent with our leChRO-seq data, Pol II occupancy at iHUBs was increased in PacR compared to Par (Fig 5A–B). Consistently, we were able to detect higher levels of eRNA produced by iHUB-LIF and iHUB-GPX4 in PacR compared to Par (Fig.S9A). Inhibition of CDK9 activity was previously shown to particularly affect eRNA synthesis [41]. Thus, we used low concentrations of the specific CDK9 inhibitor and clinical candidate, Enitociclib [formerly known as VIP152 and BAY1251152] to perturb transcriptional elongation at iHUB enhancers. Indeed, Enitociclib led to a significant decrease in iHUB eRNA and target gene expression (Fig. 5C). Interestingly, this concentration (100nM) was specific since it had no effect on the expression of eRNA and mRNA in Par cells (Fig.S9B). Moreover, we found that Enitociclib is synergistic with paclitaxel in PacR [ZIP synergy score of 6.8] (Fig. 5D). Additionally, low concentrations of Enitociclib were able to sensitize other pancreatic cancer cells such as MIAPaCa-2 to paclitaxel treatment (Fig.S9C).
Fig. 5. Perturbation of iHUB transcription decreases interaction frequency and reverses resistance.
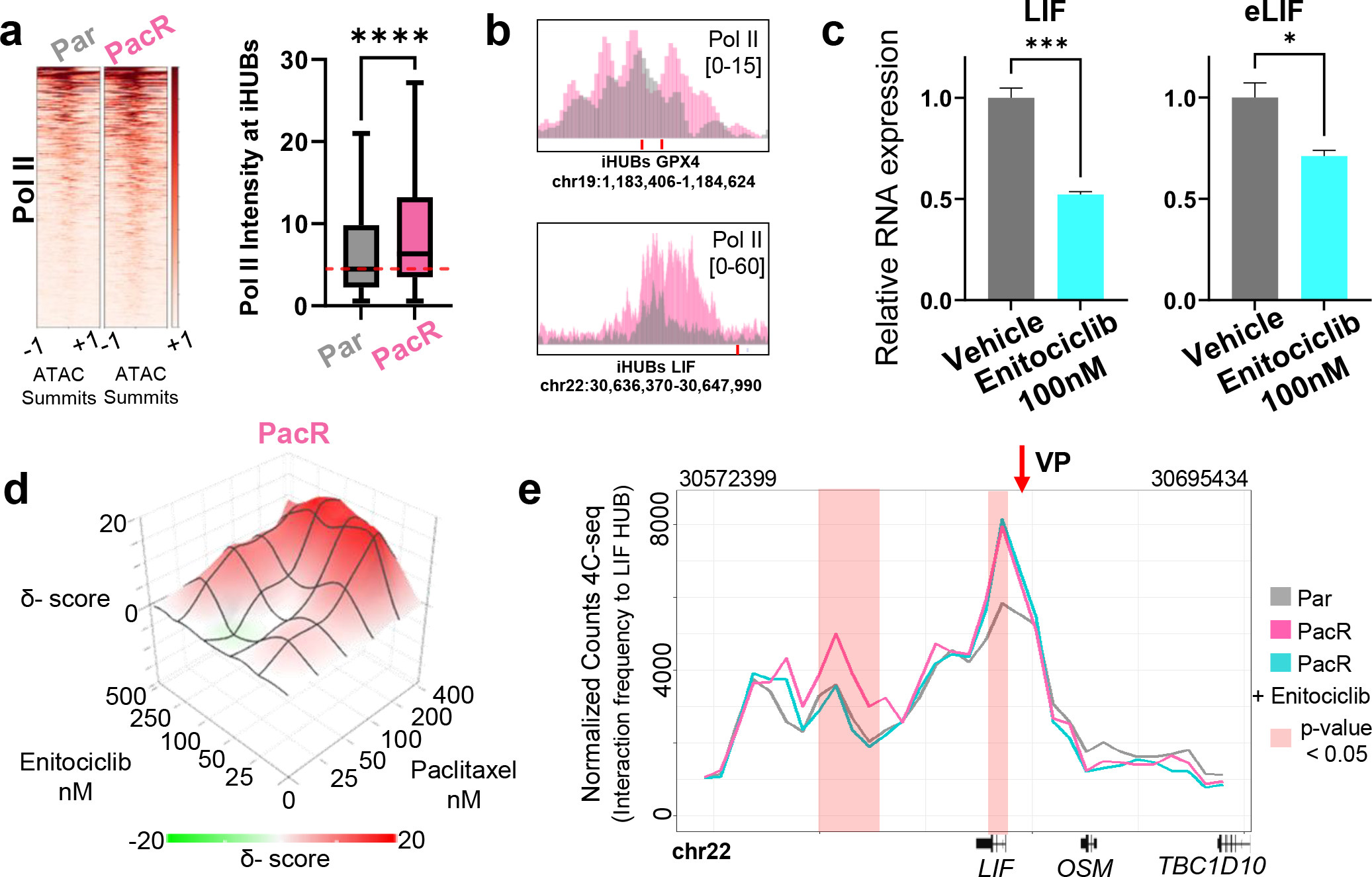
(a) Heatmap and box plot showing a significant increase in Pol II enrichment at iHUBs in PacR. Regions are centered at ATAC summits and extended −/+1kb (b) Binding profile of Pol II at the iHUBs associated with the LIF and GPX4 genes showing increased enrichment in PacR. (c) Quantitative PCR data showing significant decrease of LIF mRNA and eRNA upon treatment with 100nM of the CDK9i Enitociclib for 4 hours. (d) Plot showing the inhibition of cell growth upon treatment with different concentrations of paclitaxel and Enitociclib displaying significant synergy [ZIP synergy score of 6.8]. (e) Plot showing interaction frequencies detected by 4C-seq with the LIF iHUB as a focal viewpoint. Increased normalized 4C-seq counts represents higher interaction of the regions to the LIF iHUB. Interaction frequency of the LIF TSS to the LIF iHUB region is significantly increased in PacR compared to Par. These gained interactions are significantly decreased upon treatment with Enitociclib (100nM for 4 h).
Based on these results, we next examined the potential role of CDK9-dependent eRNA production in controlling enhancer-gene interactions by performing 4C-Seq for the LIF- and GPX4-iHUBs in PacR treated with Enitociclib and compared to PacR and Par. Remarkably, the low concentration of Enitociclib led to a decrease of interaction frequency between the iHUB (the viewpoint in the 4C-Seq experiment) and the TSS of the target gene (Fig. 5E, Fig.S9D).
Thus, our results show that low concentrations of a clinical candidate CDK9 inhibitor, Enitociclib, specifically inhibit resistance-associated eRNA expression thereby decreasing enhancer-target gene interaction frequency and reversing chemotherapeutic resistance in vitro. This supports a general model where iHUBs activate specific detrimental pathways in resistance.
Targeting iHUBs reverses resistance in vivo.
While our in vitro results suggested that CDK9 inhibition may be sufficient to induce chemotherapeutic sensitivity, we next sought to determine the potential clinical implications of our findings using a relevant translational model. For this purpose, we utilized a PDX derived from a Mayo Clinic PDAC patient tumor that was not responsive to paclitaxel treatment (PDX298). We performed a four-arm treatment study in which mice were either treated with vehicle, Enitociclib, paclitaxel, or a combination of Enitociclib and paclitaxel (Fig. 6A). As expected, paclitaxel and Enitociclib did not significantly decrease tumor size compared to vehicle (Fig. 6B–C). In contrast, the combination of Enitociclib and paclitaxel not only significantly decreased tumor size but also led to tumor shrinkage compared to pretreatment size. Additionally, Enitociclib treatment led to a significant decrease in the expression of iHUB eRNAs such as eLIF, eGPX4, and eTRIM62 (Fig.S10A). Thus, combinatorial Enitociclib and paclitaxel therapy was effective in sensitizing a resistant patient-derived xenograft in vivo.
Fig. 6. Targeting iHUBs reverses resistance in vivo.
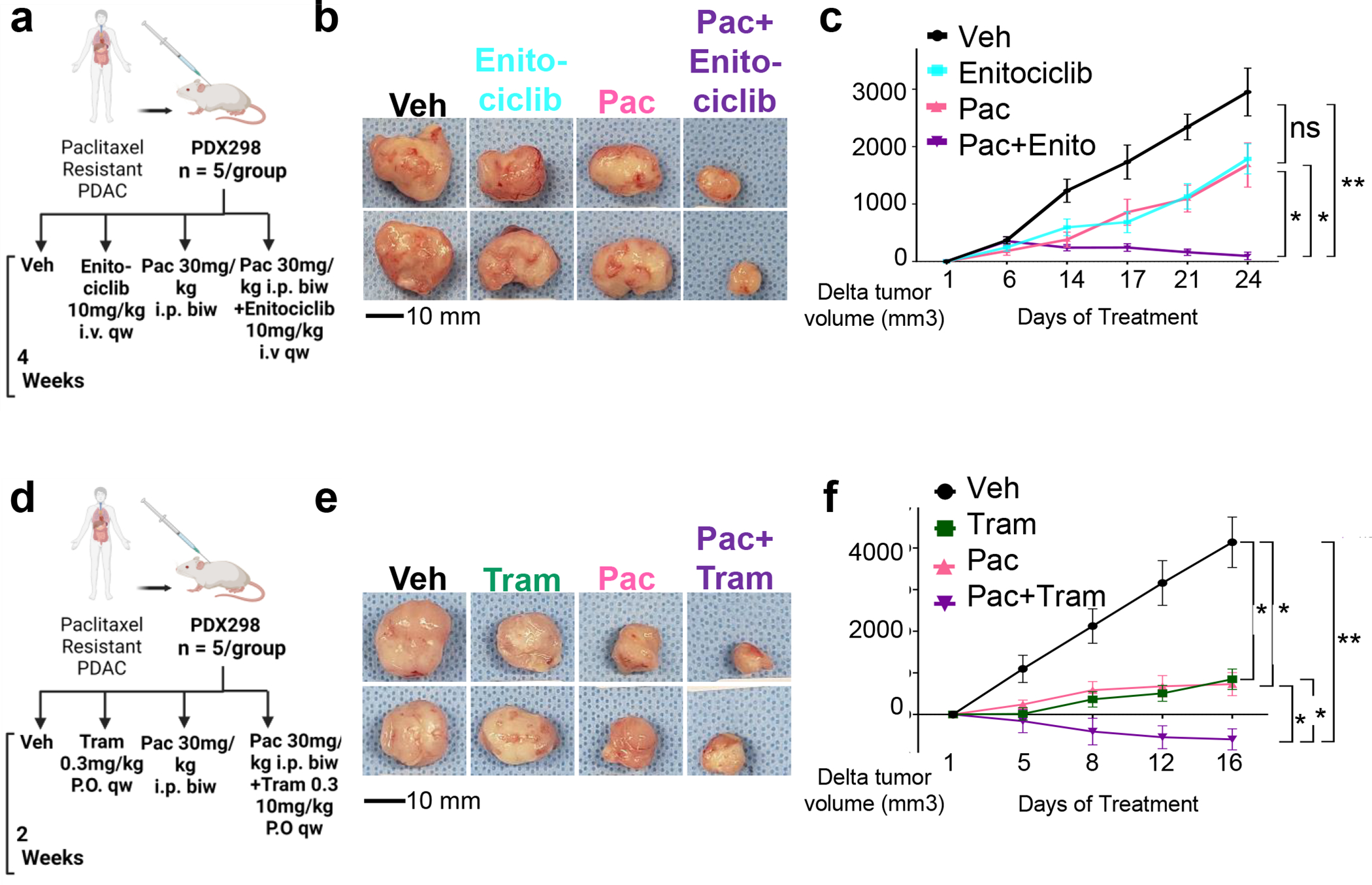
(a) To test the utility of Enitociclib in vivo, a patient-derived xenograft was established from a patient that progressed under paclitaxel treatment (PDX298). Tumor-bearing mice were treated with vehicle, Enitociclib (10mg/kg i.v. once weekly), paclitaxel (30mg/kg i.p. twice weekly) or a combination of paclitaxel (30mg/kg i.p. twice weekly) and Enitociclib (10mg/kg i.v. once weekly). (b) Representative tumor sizes after treatment with paclitaxel, Enitociclib or combination therapy leading to decreased tumor size. (c) Delta tumor size plot for the four arms of therapy for the resistant PDX298 showing no significant decrease of tumor size in both monotherapies compared to tumor shrinkage in the arm with combination. (d-f) An experiment analogous to (a-c) performed with the resistant PDX298, but with trametinib (0.3mg/kg) instead of Enitociclib.
Given the essential role of the MEK/MAPK-activated AP1 transcription factor in controlling resistance-associated gene expression and enhancer activation, we also tested the ability of trametinib to reverse resistance in vivo. Using the same therapy-resistant PDX model system described above (Fig. 6D), we observed that combining trametinib treatment with paclitaxel dramatically decreased tumor size compared to individual therapies (Fig. 6E–F). Thus, inhibition of iHUB transcription factor activity by trametinib treatment sensitizes resistant PDAC PDX tumors via targeting iHUB function.
To evaluate if expression of iHUB targets is increased in patients with poor response to chemotherapy, we performed immunohistochemistry (IHC) for GPX4 and LIF in nine pancreatic cancer samples from tumors with variable response. Response was defined based on clinical and radiologic information or pathologic criteria (Table.S2). Patients were stratified in three groups (three patients with poor response, three with partial response and three with good response). We observed a general tendency where tumors from patients with poor response highly express LIF and GPX4 (Fig. 7A, Fig.S10B).
Fig. 7. LIF and GPX4 expression in pancreatic tumors with variable response to chemotherapy [Pac/Gem].

(a) H&E staining and immunohistochemistry for LIF and GPX4 in 9 tumor sections from pancreatic cancer patients. Patients were stratified based on their response to treatment based on clinical, radiologic or pathologic criteria. Samples from patients with poorer response to therapy show higher staining intensity for GPX4 and LIF. (b) Graphical scheme showing enhancers that gain enhancer-promoter interaction frequency in resistance, thereby forming interactive hubs (iHUBs). iHUBs can be targeted by preventing the release of promoter proximal pausing (CDK9i) or directly targeting the activation of JunD by MEKi.
In conclusion, our results identify a novel mechanism of chemoresistance in PDAC whereby genes required for therapy resistance are activated by enhancers that gain interaction frequency which we refer to as iHUBs (Fig. 7B). These iHUBs are dependent on JunD and induce transcriptional changes due to their highly interactive state. Notably, targeting of either eRNA production (by the CDK9 inhibitor Enitociclib) or transcription factor activity essential for iHUB function (by inhibiting MEK/MAPK signaling upstream of the AP1 transcription factor JunD with trametinib) provide two promising mechanistically distinct, but functionally related, approaches to overcome or prevent therapeutic resistance in PDAC.
DISCUSSION
SEs are functionally important drivers of aberrant transcription in disease [19]. Similarly, we identified a subgroup of enhancers that gain interaction frequency (iHUBs) and act as major mediators of chemoresistance in PDAC. This is in concordance with Huang et al. who identified highly interactive subgroups of SEs, as well as other reports identifying highly interactive enhancers in other systems [42, 43, 44]. In contrast, iHUBs are not exclusively located in SEs. iHUBs are activated by gaining EPIs and driven by JunD. Notably, the deletion of a single iHUB is sufficient to sensitize resistant cells to chemotherapy. Moreover, targeting of iHUBs by decreasing EPI was sufficient to sensitize a PDX derived from a clinically chemoresistant PDAC patient tumor to chemotherapy.
It is important to note that the definition of iHUBs is not necessarily linked to enhancer regions that are the most interactive in resistance, but rather the differential interaction frequency that promotes resistance-associated gene programs. In this study, BRD4 occupancy and eRNA production showed more correlation to gained interaction frequency compared to H3K27ac. Thus, our results demonstrate that using increased H3K27ac as the only proxy for enhancer activity is not sufficient for identifying many functionally relevant enhancers. This is consistent with recent reports showing that while active histone modifications are highly correlative and predictive of transcription activity, particular open chromatin regions do not necessarily follow this rule. It is likely that iHUBs belong to these distinct regions [45].
Notably, not all regions which gained BRD4 were iHUBs as seen by the fact that super enhancers called based on BRD4 signal did not encompass all iHUBs. Given that BRD4 was reported to be dispensable for retaining EPIs [46], it is most probable that sequence-specific transcription factors, such as JunD, are necessary to promote interaction. Based on this, incorporation of 3D dimensional data when studying enhancers will be important in order to identify functionally relevant regulatory elements that can be targeted in disease.
Our findings confirm that eRNAs have stabilizing effects on EPI, which was previously suggested for estrogen-dependent transcription [23]. Indeed, as shown in our 4C-seq data, perturbation of eRNA production leads to decreased EPIs at iHUBs. We defined the CDK9 perturbation as specific because it did not affect eRNA synthesis at these enhancers in parental cells. This is also consistent with the reported role of high levels of eRNA promoting local EPIs in the brain [47]. However, it contrasts with Rahman et al. reporting that eRNA production is not important in sustaining target gene transcription [48]. Given the strong link between eRNA and EPI, we suggest that eRNA can be used as a proxy for interaction frequency in samples where interrogating 3D chromatin looping is technically challenging. Consistently, we can detect increased eRNA production at iHUBs in PDX tissue from PDAC patients. Thus, detection of certain eRNAs in tumor samples by semi-quantitative PCR or RNA in situ hybridization is a feasible approach that can be used to evaluate the EPI status of certain enhancers in patients. This will allow for the prediction of acquired or intrinsic resistance in patients and identify the optimal adjunct therapies that can prevent or attenuate resistance. Additionally, targeting of eRNA can also be a promising therapeutic approach. It is possible that the sensitizing effects of CDK9 inhibition may include non-iHUB targets and it will be necessary to validate if their use will lead to higher toxicity and side effects in clinical applications.
In this study, the AP1 transcription factor JunD was observed to be highly enriched at iHUBs. As motif analysis cannot differentiate between different AP1 transcription factors we focused on JunD as it was one of the most highly expressed transcription factors in our system. Indeed, JunD was reported to promote LPS-induced DNA looping in macrophages [49]. Consistently, AP1 factors were reported to regulate CTCF-independent DNA looping [50]. AP1 also recruits the SWI/SNF chromatin remodeling complex and is thereby tightly linked to enhancer activation and DNA looping [51]. This may explain the high adaptability at AP1-enriched enhancers, priming their conversion to highly interactive iHUBs in disease, thereby mediating aberrant transcriptional activation.
Promising therapies to overcome PDAC resistance are currently under investigation in clinical trials. However, these are limited to specific resistance-promoting pathways in PDAC, such as GSK3β [NCT03678883] and EGFR [NCT01505413]. Our findings that iHUB formation via gained EPI can be a major mechanism of treatment evasion in PDAC, suggests that effective therapeutic iHUB targeting can be a very promising approach to increase the efficacy, not only of other chemotherapy regimens, but also of targeted therapy approaches in PDAC. Due to the high tissue specificity of EPIs, which exceeds that of differential enhancer markings [13], targeting iHUBs is expected to spare non-tumor cells due to the lack of effects on gene expression programs where iHUBs are in their minimally interactive configuration. In this study, we demonstrate that the transcriptional effects of iHUB deletion are exclusive to cells where these are in the highly interactive configuration. Additionally, we suggest that altered patterns of enhancer interactions activate genes that promote resistance, metastasis, or aggressiveness in different cancer types. Thus, iHUB targeting has the potential to widely prevent transcription-mediated resistance in other cancer types.
We focused our efforts on therapeutic agents that are already in clinical phase trials to increase the translational potential of our work. Both trametinib and Enitociclib are currently in clinical trials including two studies with Enitociclib currently recruiting cancer patients [NCT02635672, NCT04978779] and 11 studies in PDAC examining the effects of trametinib. A previous Phase II trial using trametinib showed promising results in locally recurrent pancreatic cancer after surgery [52]. Conversely, trametinib did not show improvement in untreated metastatic PDAC when combined with gemcitabine [53]. This can be due to inadequate combination partners (paclitaxel vs gemcitabine) or inappropriate dosing. Given the good safety profile of these agents, a wider evaluation of their use in resistant PDAC patients may provide a promising outlook.
It is important to note that slight changes of H3K4me3 intensity at promoters could potentially introduce bias in interaction detected by HiChIP in this study. However, the finding that a ChIP-independent approach (4C-Seq) confirmed these data suggests that that the observed gained 3D chromatin conformational changes are robust. In conclusion, our results reveal the fundamental importance of EPI in enhancer-mediated activation of resistance-associated gene expression. This new paradigm of enhancer activation was seen irrespective of when resistance was attained and was seen in acquired resistance in cell lines, in xenografts, or directly in the patient (prior to PDX generation). Thereby, we identify an important subgroup of enhancers which control the transcription of a set of important genes that allows tumor cells to withstand chemotoxicity. Importantly, treatment with low concentrations of clinical candidates targeting the involved mechanisms specifically attenuates EPI at these enhancers and reverses resistance, thereby providing viable and promising new approaches to increase PDAC patient survival.
METHODS
Establishment of Resistant Cells
L3.6pl pancreatic cancer cells were maintained in minimum essential media (MEM; Thermo Fisher Scientific) supplemented with 10% FBS (Thermo Fisher Scientific), 1% penicillin/streptomycin (Sigma-Aldrich), and 1% L-Glutamine (Sigma-Aldrich). For establishment of resistant cells, paclitaxel (T7191; Sigma-Aldrich) was diluted in dimethyl sulfoxide (DMSO) (Roth) and added the cells starting from 2 nM (the estimated IC50). Once the cells started to thrive, the concentration of paclitaxel was doubled until the cells were able to grow in 64 nM after 100 days. Subsequently, resistant cells were maintained in 64 nM of paclitaxel in MEM media at all times. Detailed protocols are available in supplementary materials and methods.
HiChIP and ChRO-seq
H3K4me3 HiChIP was performed using the protocol for absolute quantification of architecture (AQuA-HiChIP) [54]. Nascent RNA was detected using length extension ChRO-seq (leChRO-seq) [55]. Please refer to supplementary materials and methods for more details.
Establishment of Patient-derived Xenografts
All details are in supplementary materials and methods.
Statistical Analysis
Significance was evaluated using the unpaired t-test if samples passed the Shapiro–Wilk test. Otherwise, the Mann-Whitney statistical test was used. For paired comparisons that failed the Shapiro-Wilk test, we used wilcoxon matched pairs signed rank test. p-values are indicated by **** for =< 0.0001, *** for =< 0.001, ** for =< 0.01 and * for =< 0.05.
Supplementary Material
What is already known on this topic
Changes in cell fate are usually accompanied by de novo activation of distal enhancer regions characterized by changes in H3K27ac enrichment. Enhancer reprogramming mediates transcriptional changes in metastatic PDAC.
What this study adds
We identified a subgroup of enhancers called iHUBs that do not necessarily exhibit a change in H3K27ac enrichment, but rather become transcriptionally active and display increased interaction frequency with target genes required for therapy resistance in PDAC. These enhancers are driven by the AP1 transcription factor JunD.
How this study might affect research, practice or policy
Changes in enhancer activity may not necessarily be coupled to changes in enrichment of H3K27ac. Since iHUB activity can be inhibited to restore therapeutic responsiveness using clinically relevant small molecule inhibitors, targeting iHUBs may represent a promising new approach to reverse and prevent resistance in PDAC.
ACKNOWLEDGMENTS
We thank G. Salinas, F. Ludewig, and S. Lutz from the Transcriptome and Genome Analysis Laboratory of the University Medical Center Göttingen, Germany for performing some of the next generation sequencing. We would like to thank Davide Povero, John R. Hawse, William A. Faubion and their lab members. Model figures were generated using BioRender.com.
FUNDING
Research reported in this publication was supported by the Robert Bosch Stiftung [RBMF/RBCT; no grant/award number] to S.A.J., the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health under Award Number P30DK084567 (Pilot and feasibility award to F.H.H), CTSA Grant Number KL2 TR002379 from the National Center for Advancing Translational Science to N.J.S, Deutsche Krebshilfe (PiPAC Consortium) [70112505] to Z.N, J.T.S, E.H and S.A.J., and the Deutsche Forschungsgemeinschaft (DFG) [JO 815/3–2 to S.A.J., KFO5002 to E.H.]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations
- PDAC
Pancreatic Ductal Adenocarcinoma
- CDK9i
Cyclin-dependent Kinase-9 Inhibitor
- CNV
Copy Number Variation
- EPI
Enhancer-Promoter Interaction
- eRNA
Enhancer RNA
- iHUB
Interactive Hub
- PacR
Paclitaxel-Resistant Cells
- Par
Parental Cells
- PDX
Patient-Derived Xenograft
- SE
Super Enhancer
- TAD
Topologically Associating Domain
Footnotes
ETHICS DECLARATIONS
The authors declare no competing interests. Resistant PDX was established under Mayo Clinic IRBs (66–06, 354–06, 19–012104). All PDXGo13-related experiments in this study are approved by the UMG IRB (Göttingen, Germany; 70112108). Samples for IHC were collected by the Clinical Core of the Cell signaling Center in the Gastroenterology department (21–004887).
DATA AVAILABILITY
All raw and processed data are available at www.ebi.ac.uk/arrayexpress under accession number E-MTAB-11730–33, E-MTAB-11737–40, E-MTAB-11744, E-MTAB-12705,7–8. All samples and their respective accession numbers are available in Tables S3–S5[56].
References
- 1.Huang L, Jansen L, Balavarca Y, Babaei M, van der Geest L, Lemmens V, et al. Stratified survival of resected and overall pancreatic cancer patients in Europe and the USA in the early twenty-first century: a large, international population-based study. BMC Med 2018;16:125-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rawla P, Sunkara T, Gaduputi V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J Oncol 2019;10:10–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Juiz NA, Iovanna J, Dusetti N. Pancreatic Cancer Heterogeneity Can Be Explained Beyond the Genome. Front Oncol 2019;9:246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sohal DPS, Kennedy EB, Cinar P, Conroy T, Copur MS, Crane CH, et al. Metastatic Pancreatic Cancer: ASCO Guideline Update. J Clin Oncol 2020:Jco2001364. [DOI] [PubMed] [Google Scholar]
- 5.Goldstein D, El-Maraghi RH, Hammel P, Heinemann V, Kunzmann V, Sastre J, et al. nab-Paclitaxel plus gemcitabine for metastatic pancreatic cancer: long-term survival from a phase III trial. J Natl Cancer Inst 2015;107. [DOI] [PubMed] [Google Scholar]
- 6.Sarvepalli D, Rashid MU, Rahman AU, Ullah W, Hussain I, Hasan B, et al. Gemcitabine: A Review of Chemoresistance in Pancreatic Cancer. Crit Rev Oncog 2019;24:199–212. [DOI] [PubMed] [Google Scholar]
- 7.Yao X, Hu JF, Li T, Yang Y, Sun Z, Ulaner GA, et al. Epigenetic regulation of the taxol resistance-associated gene TRAG-3 in human tumors. Cancer Genet Cytogenet 2004;151:1–13. [DOI] [PubMed] [Google Scholar]
- 8.Spitz F, Furlong EE. Transcription factors: from enhancer binding to developmental control. Nat Rev Genet 2012;13:613–26. [DOI] [PubMed] [Google Scholar]
- 9.Bell CC, Fennell KA, Chan Y-C, Rambow F, Yeung MM, Vassiliadis D, et al. Targeting enhancer switching overcomes non-genetic drug resistance in acute myeloid leukaemia. Nature Communications 2019;10:2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fu X, Pereira R, De Angelis C, Veeraraghavan J, Nanda S, Qin L, et al. FOXA1 upregulation promotes enhancer and transcriptional reprogramming in endocrine-resistant breast cancer. Proceedings of the National Academy of Sciences 2019;116:26823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shang S, Yang J, Jazaeri AA, Duval AJ, Tufan T, Lopes Fischer N, et al. Chemotherapy-Induced Distal Enhancers Drive Transcriptional Programs to Maintain the Chemoresistant State in Ovarian Cancer. Cancer Res 2019;79:4599–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A 2010;107:21931–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He B, Chen C, Teng L, Tan K. Global view of enhancer-promoter interactome in human cells. Proc Natl Acad Sci U S A 2014;111:E2191–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roe J-S, Hwang C-I, Somerville TDD, Milazzo JP, Lee EJ, Da Silva B, et al. Enhancer Reprogramming Promotes Pancreatic Cancer Metastasis. Cell 2017;170:875–88.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pope BD, Ryba T, Dileep V, Yue F, Wu W, Denas O, et al. Topologically associating domains are stable units of replication-timing regulation. Nature 2014;515:402–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaushal A, Mohana G, Dorier J, Özdemir I, Omer A, Cousin P, et al. CTCF loss has limited effects on global genome architecture in Drosophila despite critical regulatory functions. Nature Communications 2021;12:1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nora EP, Goloborodko A, Valton A-L, Gibcus JH, Uebersohn A, Abdennur N, et al. Targeted Degradation of CTCF Decouples Local Insulation of Chromosome Domains from Genomic Compartmentalization. Cell 2017;169:930–44.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rao SSP, Huang SC, Glenn St Hilaire B, Engreitz JM, Perez EM, Kieffer-Kwon KR, et al. Cohesin Loss Eliminates All Loop Domains. Cell 2017;171:305–20.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lovén J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013;153:320–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hamdan FH, Johnsen SA. DeltaNp63-dependent super enhancers define molecular identity in pancreatic cancer by an interconnected transcription factor network. Proc Natl Acad Sci U S A 2018;115:E12343–e52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Somerville TDD, Xu Y, Miyabayashi K, Tiriac H, Cleary CR, Maia-Silva D, et al. TP63-Mediated Enhancer Reprogramming Drives the Squamous Subtype of Pancreatic Ductal Adenocarcinoma. Cell reports 2018;25:1741–55.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nagarajan S, Hossan T, Alawi M, Najafova Z, Indenbirken D, Bedi U, et al. Bromodomain protein BRD4 is required for estrogen receptor-dependent enhancer activation and gene transcription. Cell Rep 2014;8:460–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li W, Notani D, Ma Q, Tanasa B, Nunez E, Chen AY, et al. Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature 2013;498:516–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lai F, Orom UA, Cesaroni M, Beringer M, Taatjes DJ, Blobel GA, et al. Activating RNAs associate with Mediator to enhance chromatin architecture and transcription. Nature 2013;494:497–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsai PF, Dell’Orso S, Rodriguez J, Vivanco KO, Ko KD, Jiang K, et al. A Muscle-Specific Enhancer RNA Mediates Cohesin Recruitment and Regulates Transcription In trans. Mol Cell 2018;71:129–41.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao Y, Wang L, Ren S, Wang L, Blackburn PR, McNulty MS, et al. Activation of P-TEFb by Androgen Receptor-Regulated Enhancer RNAs in Castration-Resistant Prostate Cancer. Cell Rep 2016;15:599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lai F, Damle SS, Ling KK, Rigo F. Directed RNase H Cleavage of Nascent Transcripts Causes Transcription Termination. Mol Cell 2020;77:1032–43.e4. [DOI] [PubMed] [Google Scholar]
- 28.Lee JS, Mendell JT. Antisense-Mediated Transcript Knockdown Triggers Premature Transcription Termination. Mol Cell 2020;77:1044–54.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Linares-Saldana R, Kim W, Bolar NA, Zhang H, Koch-Bojalad BA, Yoon S, et al. BRD4 orchestrates genome folding to promote neural crest differentiation. Nat Genet 2021;53:1480–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jung M, Philpott M, Müller S, Schulze J, Badock V, Eberspächer U, et al. Affinity Map of Bromodomain Protein 4 (BRD4) Interactions with the Histone H4 Tail and the Small Molecule Inhibitor JQ1 *. Journal of Biological Chemistry 2014;289:9304–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Y, Umbach DM, Krahn JM, Shats I, Li X, Li L. Predicting tumor response to drugs based on gene-expression biomarkers of sensitivity learned from cancer cell lines. BMC Genomics 2021;22:272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tang W, Ramasamy K, Pillai SMA, Santhamma B, Konda S, Pitta Venkata P, et al. LIF/LIFR oncogenic signaling is a novel therapeutic target in endometrial cancer. Cell Death Discovery 2021;7:216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu XF, Guo CY, Liu J, Yang WJ, Xia YJ, Xu L, et al. Gli1 maintains cell survival by up-regulating IGFBP6 and Bcl-2 through promoter regions in parallel manner in pancreatic cancer cells. J Carcinog 2009;8:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li B, Yang L, Peng X, Fan Q, Wei S, Yang S, et al. Emerging mechanisms and applications of ferroptosis in the treatment of resistant cancers. Biomedicine & Pharmacotherapy 2020;130:110710. [DOI] [PubMed] [Google Scholar]
- 35.Nagarajan A, Dogra SK, Sun L, Gandotra N, Ho T, Cai G, et al. Paraoxonase 2 Facilitates Pancreatic Cancer Growth and Metastasis by Stimulating GLUT1-Mediated Glucose Transport. Mol Cell 2017;67:685–701.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Widjaja-Adhi MAK, Palczewski G, Dale K, Knauss EA, Kelly ME, Golczak M, et al. Transcription factor ISX mediates the cross talk between diet and immunity. Proc Natl Acad Sci U S A 2017;114:11530–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kondo J, Sato F, Kusumi T, Liu Y, Motonari O, Sato T, et al. Claudin-1 expression is induced by tumor necrosis factor-alpha in human pancreatic cancer cells. Int J Mol Med 2008;22:645–9. [PubMed] [Google Scholar]
- 38.Corcoran RB, Contino G, Deshpande V, Tzatsos A, Conrad C, Benes CH, et al. STAT3 plays a critical role in KRAS-induced pancreatic tumorigenesis. Cancer Res 2011;71:5020–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A, et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 2017;551:247–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vinciguerra M, Vivacqua A, Fasanella G, Gallo A, Cuozzo C, Morano A, et al. Differential phosphorylation of c-Jun and JunD in response to the epidermal growth factor is determined by the structure of MAPK targeting sequences. J Biol Chem 2004;279:9634–41. [DOI] [PubMed] [Google Scholar]
- 41.Hah N, Murakami S, Nagari A, Danko CG, Kraus WL. Enhancer transcripts mark active estrogen receptor binding sites. Genome Res 2013;23:1210–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang J, Li K, Cai W, Liu X, Zhang Y, Orkin SH, et al. Dissecting super-enhancer hierarchy based on chromatin interactions. Nature Communications 2018;9:943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miguel-Escalada I, Bonàs-Guarch S, Cebola I, Ponsa-Cobas J, Mendieta-Esteban J, Atla G, et al. Human pancreatic islet three-dimensional chromatin architecture provides insights into the genetics of type 2 diabetes. Nat Genet 2019;51:1137–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Madsen JGS, Madsen MS, Rauch A, Traynor S, Van Hauwaert EL, Haakonsson AK, et al. Highly interconnected enhancer communities control lineage-determining genes in human mesenchymal stem cells. Nat Genet 2020;52:1227–38. [DOI] [PubMed] [Google Scholar]
- 45.Wang Z, Chivu AG, Choate LA, Rice EJ, Miller DC, Chu T, et al. Prediction of histone post-translational modification patterns based on nascent transcription data. Nat Genet 2022;54:295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Crump NT, Ballabio E, Godfrey L, Thorne R, Repapi E, Kerry J, et al. BET inhibition disrupts transcription but retains enhancer-promoter contact. Nature Communications 2021;12:223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhou Y, Xu S, Zhang M, Wu Q. Systematic functional characterization of antisense eRNA of protocadherin α composite enhancer. Genes Dev 2021;35:1383–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rahman S, Zorca CE, Traboulsi T, Noutahi E, Krause MR, Mader S, et al. Single-cell profiling reveals that eRNA accumulation at enhancer–promoter loops is not required to sustain transcription. Nucleic Acids Research 2017;45:3017–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhao W, Wang L, Zhang M, Wang P, Zhang L, Yuan C, et al. NF-κB– and AP-1–Mediated DNA Looping Regulates Osteopontin Transcription in Endotoxin-Stimulated Murine Macrophages. The Journal of Immunology 2011;186:3173–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Seo J, Koçak DD, Bartelt LC, Williams CA, Barrera A, Gersbach CA, et al. AP-1 subunits converge promiscuously at enhancers to potentiate transcription. Genome Res 2021;31:538–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sen M, Wang X, Hamdan FH, Rapp J, Eggert J, Kosinsky RL, et al. ARID1A facilitates KRAS signaling-regulated enhancer activity in an AP1-dependent manner in colorectal cancer cells. Clin Epigenetics 2019;11:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhu X, Cao Y, Liu W, Ju X, Zhao X, Jiang L, et al. Stereotactic body radiotherapy plus pembrolizumab and trametinib versus stereotactic body radiotherapy plus gemcitabine for locally recurrent pancreatic cancer after surgical resection: an open-label, randomised, controlled, phase 2 trial. Lancet Oncol 2022;23:e105–e15. [DOI] [PubMed] [Google Scholar]
- 53.Infante JR, Somer BG, Park JO, Li CP, Scheulen ME, Kasubhai SM, et al. A randomised, double-blind, placebo-controlled trial of trametinib, an oral MEK inhibitor, in combination with gemcitabine for patients with untreated metastatic adenocarcinoma of the pancreas. Eur J Cancer 2014;50:2072–81. [DOI] [PubMed] [Google Scholar]
- 54.Gryder BE, Khan J, Stanton BZ. Measurement of differential chromatin interactions with absolute quantification of architecture (AQuA-HiChIP). Nat Protoc 2020;15:1209–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chu T, Rice EJ, Booth GT, Salamanca HH, Wang Z, Core LJ, et al. Chromatin run-on and sequencing maps the transcriptional regulatory landscape of glioblastoma multiforme. Nature Genetics 2018;50:1553–64. [dataset] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kutschat AP, Hamdan FH, Wang X, Wixom AQ, Najafova Z, Gibhardt CS, et al. STIM1 Mediates Calcium-dependent Epigenetic Reprogramming in Pancreatic Cancer. Cancer Res 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw and processed data are available at www.ebi.ac.uk/arrayexpress under accession number E-MTAB-11730–33, E-MTAB-11737–40, E-MTAB-11744, E-MTAB-12705,7–8. All samples and their respective accession numbers are available in Tables S3–S5[56].