

Abstract
Purpose:
Using next generation sequencing (NGS), The Cancer Genome Atlas (TCGA) found that endometrial carcinomas (ECs) fall under one of four molecular subtypes, and a POLE mutation status, mismatch repair (MMR) and p53 immunohistochemistry (IHC)-based surrogate has been developed. We sought to retrospectively classify and characterize a large series of unselected ECs that were prospectively subjected to clinical sequencing by utilizing clinical molecular and IHC data.
Experimental Design:
All patients with EC with clinical tumor-normal MSK-IMPACT NGS from 2014–2020 (n=2,115) were classified by integrating molecular data (i.e., POLE mutation, TP53 mutation, MSIsensor score) and MMR and p53 IHC results. Survival analysis was performed for primary EC patients with upfront surgery at our institution.
Results:
Utilizing our integrated approach, significantly more ECs were molecularly classified (1,834/2,115, 87%) as compared to the surrogate (1,387/2,115, 66%, p<0.001), with an almost perfect agreement for classifiable cases (Kappa 0.962, 95% CI 0.949–0.975). Discrepancies were primarily due to TP53 mutations in p53-IHC-normal ECs. Of the 1,834 ECs, most were of copy number (CN)-high molecular subtype (40%), followed by CN-low (32%), MSI-high (23%) and POLE (5%). Histologic and genomic variability was present among all molecular subtypes. Molecular classification was prognostic in early- and advanced-stage disease, including early-stage endometrioid EC.
Conclusions:
The integration of clinical NGS and IHC data allows for an algorithmic approach to molecularly classifying newly diagnosed EC, while overcoming issues of IHC-based genetic alteration detection. Such integrated approach will be important moving forward given the prognostic and potentially predictive information afforded by this classification.
Keywords: molecular classification, endometrial carcinoma, clinical sequencing, mutation, immunohistochemistry
1. INTRODUCTION
Endometrial cancer (EC) is the sixth most common cancer worldwide with an increasing incidence and disease-associated mortality in 2020, estimated at 417,000 and 97,000, respectively [1]. Similar to other solid tumors, EC is a clinically, histologically and molecularly heterogeneous disease [2]. Through comprehensive molecular analysis, The Cancer Genome Atlas (TCGA) identified four molecular EC subtypes: 1) POLE (ultramutated) harboring POLE exonuclease domain hotspot mutations, 2) microsatellite instability (MSI)-high (hypermutated, MSI-H, DNA mismatch repair [MMR]-deficient), 3) copy number-high (CN-H) harboring high levels of CN alterations and recurrent TP53 mutations, and 4) copy number-low (CN-L) lacking defining features of the other subtypes, also referred to as ECs of ‘non-specific molecular profile’ [3]. For implementation of molecular classification into the clinic, a surrogate, the Proactive Molecular Risk Classifier for Endometrial Cancer (ProMisE) algorithm, was developed [4]. This surrogate involves the mutational assessment of the POLE exonuclease domain and immunohistochemical (IHC) analysis of the MMR proteins (i.e., MLH1, MSH2, MSH6 and PMS2) and p53 [5], and has now been included in the NCCN guidelines and World Health Organization (WHO) classification of Female Genital Tumors [6, 7]. ProMisE identifies four molecular subtypes that are analogous but not identical to the genomic subtypes described in TCGA [8]. For example, in the original TCGA study [3], only 92% of ECs classified as CN-H molecular subtype harbored TP53 mutations, suggesting that the surrogate classifier misses a subset of non-TP53-altered CN-H ECs.
Integration of clinical next-generation sequencing (NGS) assays of tumor and germline DNA holds expanding diagnostic, prognostic, and therapeutic implications. The use of NGS also provides an orthogonal approach for the identification of MSI or TP53 mutations not captured by IHC. Since 2014, the FDA-authorized tumor-normal MSK-IMPACT (Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets) targeted sequencing assay has been utilized at our institution [9, 10], and is offered to all newly diagnosed EC patients presenting for care. In addition to a high-depth NGS-based detection of mutations and CN alterations, MSK-IMPACT provides information on MSI and tumor mutational burden [10]. In this study, we sought to retrospectively classify a large series of unselected ECs of all histologic types by integrating clinical molecular data with IHC results. We further aimed to demonstrate the utility of relevant alternative parameters for molecular classification, including MSIsensor score and TP53 mutation, define the concordance with the conventional molecular subtyping approach (ProMisE), and assess mutational profiles and oncologic outcomes of the defined molecular subtypes and specific histologic subsets of the disease.
2. MATERIALS AND METHODS
2.1. Case selection
This retrospective study was approved by the Institutional Review Board at MSK, and all patients provided written consent for genomic profiling. ECs subjected to clinical tumor-normal MSK-IMPACT sequencing from test inception in 2014 through 12/31/2020 were included (n=2,121; Fig. 1A). MSK-IMPACT sequencing was performed on tumor tissue from the primary diagnostic procedure (hysterectomy or pre-operative biopsy) or from recurrent tumor sample. For patients with multiple specimens that underwent MSK-IMPACT, sequencing data from the earliest occurrence (i.e., primary tumor or first recurrence specimen) were evaluated. Demographic and clinical data were extracted by review of electronic medical records. Cases were eligible for clinical outcomes analyses if MSK-IMPACT was performed prior to a documented recurrence with resultant unambiguous molecular classification, and if treatment planning with upfront surgical staging was performed at our institution (Supplementary Fig. S1).
Figure 1. Patient selection and molecular classification of endometrial cancers integrating clinical tumor-normal sequencing and immunohistochemistry data.

A. CONSORT diagram summarizing the endometrial cancer patients included in this study. B. Method employed for classification of endometrial cancers into the molecular subtypes using clinical MSK-IMPACT sequencing results and immunohistochemistry. C. Distribution of histologic subtypes and molecular subtypes for 1,834 endometrial cancers subjected to clinical sequencing. CN-H, copy number-high; CN-L, copy number-low; G1, grade 1; G2, grade 2; G3, grade 3; MSI-H, microsatellite instability-high; NOS, not otherwise specified.
2.2. Histologic typing, MMR and p53 IHC
Pathology reports authored by departmental gynecologic pathologists throughout the study timeframe were reviewed. These contain histopathologic data evaluated through a uniform diagnostic approach with biweekly diagnostic consensus conferences, as previously described [11]. Histologic type, FIGO 2009 stage, and endometrioid tumor grade were recorded based on the patient’s initial pathologic diagnosis. All histologic subtypes were included (i.e., endometrioid, serous, clear cell, carcinosarcoma, undifferentiated/dedifferentiated, and mixed/high-grade not otherwise specified (NOS)). ECs of unclassified histology were re-reviewed by gynecologic pathologists (L.H.E. or A.M.-B.) and, if unresolved, remained in ‘other’. Similarly, ECs of endometrioid histology without defined FIGO grading were re-reviewed and designated ‘N/A’ if unresolved. The highest histologic grade for endometrioid type ECs was recorded from either the pre-operative biopsy or hysterectomy specimen. MMR and p53 IHC were generally performed on the same specimen as DNA extraction for MSK-IMPACT sequencing. If either IHC staining was missing, it was obtained from the pre-operative or recurrence specimen, if available. ECs lacking MLH1 or PMS2 protein expression were typically subjected to MLH1 promoter methylation analysis [12].
2.3. Genomic data extraction
The genomic data extracted from the MSK-IMPACT assay, which targets between 341 (2014) and 505 (2020) cancer-related genes, included somatic mutation count, POLE mutational status, TP53 mutation status, MSIsensor score, tumor purity, fraction of genome altered (FGA), and tumor mutational burden (TMB) [10, 11]. POLE exonuclease domain hotspot mutations were defined based on Leon-Castillo et al. [13]. MSIsensor scores of ≥10 were considered MSI-H, between ≥3 and <10 as MSI-indeterminate, and <3 as stable (MSS), as described [14].
2.4. Molecular classification of EC integrating clinical NGS and IHC data
Given that mutation and CN alteration detection as well as MSIsensor score are affected by the tumor cell content of a given sample, we included only high-quality samples with a minimum tumor purity of 20%. In addition, samples lacking any somatic mutations were excluded (Fig. 1A). Molecular subtype assignment was performed hierarchically (Fig. 1B): i) POLE molecular subtype was defined by the presence of a known POLE hotspot exonuclease domain mutation [13]. If a somatic POLE mutation was present at a known hotspot position/residue with a different amino acid change (e.g., V411M rather than known hotspot V411L), it was deemed unclassifiable. ii) MSI-H molecular subtype was assigned if the MSIsensor score was ≥10 [14, 15] and/or if MMR-deficient (MMRd) based on IHC irrespective of MSIsensor score. MMRd by IHC was defined as absence of MLH1, MSH2, and/or PMS2 and MSH6 protein expression from all tumor cell nuclei, in the presence of an internal control [16]. IHC results with equivocal or inconclusive staining results were not considered MMRd if the MSIsensor score was <10. iii) CN-H molecular subtype was assigned based on the presence of a TP53 homozygous deletion or a pathogenic driver mutation defined by OncoKB, CIViC, and/or Cancer Hotspots in cBioPortal [17–19]. When available, p53 IHC results were used as a reference. We also assigned TP53 wild-type ECs to the CN-H molecular subtype if they harbored an MDM2 amplification paired with aberrant p53 IHC expression [20]. iv) CN-L molecular subtype was assigned if a tumor sample did not harbor any of the defining features of the other subtypes [21, 22]. ECs with ambiguous results were subjected to a central re-review of histologic type (see above), IHC results and/or molecular findings (E.R-D., A.M-B., L.H.E and B.W.); if unresolved, EC were deemed ‘unclassifiable’ and excluded from classification and downstream analyses. In addition, ECs were classified into the molecular subtypes according to the modified ProMisE algorithm as reported in the NCCN/ WHO guidelines [5–7], utilizing i) POLE hotspot mutation, ii) MMRd by IHC, and iii) p53 abnormal by IHC.
2.5. Statistical analysis
Continuous variables were assessed by Kruskal-Wallis rank sum test and categorical variables by Fisher’s exact test. Cohen’s κ coefficients and corresponding 95% confidence intervals were used to measure agreement between molecular subtype assignments [23]. Progression-free survival (PFS) was measured from the date of surgery to the date of last gynecologic assessment or recurrence or death, whichever first. Disease status and disease progression were extracted from medical oncology notes, in combination with follow-up imaging studies according to the PRISSMM data model approach [24]. Overall survival (OS) was measured from date of surgery to date of last known follow-up or date of death. PFS and OS related analyses were only performed including patients who were treated at MSK for their primary disease and using their primary tumor sample for MSK-IMPACT. Kaplan-Meier method was applied to estimate the median survival time and survival rate at a specific time. The associations between variables and survival outcomes were tested by using Log-rank test for categorical variables and Wald test based on Cox-proportional hazard (CoxPH) model for continuous variables. Landmark analyses, with one month landmark time point, was applied to examine time-dependent variable of adjuvant treatment [25]. Multivariate CoxPH models were built to determine the independent prognostic factors for PFS and OS. All analyses were performed using R 4.1.2 (https://www.R-project.org/). All p-values were two-sided. A p-value less than 0.05 was considered significant.
3. RESULTS
3.1. Molecular subtypes of ECs subjected to clinical NGS sequencing
Of the 2,115 ECs subjected to MSK-IMPACT sequencing between 1/1/2014 and 12/31/2020, 281 ECs (13%) were excluded due to low tumor purity, lack of somatic mutations, ambiguous results, and/or duplicates, leaving a final cohort of 1,834 ECs of all histologic subtypes for molecular classification (Fig. 1A). For most ECs (82%, 1,501/1,834), clinical sequencing was performed on the initial diagnosis specimen, 323 (18%) on the recurrence tumor specimen, and 10 (0.5%) on non-descriptive tumor specimens. In the primary diagnosis group, MSK-IMPACT sequencing was performed primarily on hysterectomy specimens (1,312/1,501, 87%) rather than pre-operative biopsies (189/1,501, 13%). In the recurrence tumor group, MSK-IMPACT sequencing was mostly performed on the recurrence biopsy itself (318/323, 98.5%), and rarely on the initial diagnostic tumor. MMR and p53 IHC were performed clinically for 1,470 (80%) and 1,143 (62%) ECs, respectively.
Integrating the clinical NGS and IHC data for molecular classification (Fig. 1B) revealed that the majority of ECs at our tertiary cancer center were of CN-H molecular subtype (734/1,834, 40%), followed by CN-L (575/1,834, 31.3%), MSI-H (428/1,834, 23.3%) and POLE (97/1,834, 5.3%) molecular subtypes (Fig. 1C). The CN-H ECs either harbored pathogenic driver TP53 mutations (n=729), TP53 homozygous deletions (n=4) or MDM2 amplification coupled with aberrant p53 IHC expression (n=1). For 63% (459/734) of CN-H ECs, p53 IHC was available; of the 733 CN-H ECs with TP53 alterations, 94% (431/458) had aberrant p53 protein expression (Fig. 2A). Pathogenic TP53 missense mutations were the most commonly identified in CN-H ECs (512/734); followed by frameshift deletions (64/734) and nonsense mutations (58/734; Fig. 3A). The 27 TP53-mutant CN-H ECs with p53 wild-type expression by IHC harbored hotspot missense mutations (15/27), frameshift deletions (5/27), nonsense mutations (4/27), hotspot splice-site mutations (2/27) or a homozygous deletion (1/27). As expected, the majority of CN-H ECs were of high-grade histologic subtypes (Table 1). In contrast, ECs of CN-L molecular subtype (575/1,834, 31%) were primarily of endometrioid histology (482/575, 84%), but also included unclassified (28/575, 5%), clear cell (20/575, 3%) and mixed/ high-grade NOS ECs (17/575, 3%; Fig. 1C, Table 1).
Figure 2. Endometrial cancer molecular classification according to the integrated clinical tumor-normal sequencing/immunohistochemistry-based and the conventional ProMisE approaches.

A. TP53 mutations and p53 expression patterns by immunohistochemistry in endometrial cancers classified as of copy number-high molecular subtype. B. Sequencing-derived MSI-sensor scores and DNA mismatch repair (MMR) deficiency/ proficiency by immunohistochemistry in endometrial cancers classified as of MSI-H molecular subtype. C. Agreement of molecular subtype assignments using our integrated molecular and immunohistochemistry-based approach (top) and the Proactive Molecular Risk Classifier for Endometrial Cancer (ProMisE; second row from top). Information on the TP53 mutation status, p53 immunohistochemistry, MSIsensor score and MMR immunohistochemistry are shown below.
Figure 3. Landscape of somatic mutations and gene copy number alterations of endometrial cancers by molecular subtype integrating clinical sequencing and immunohistochemistry.
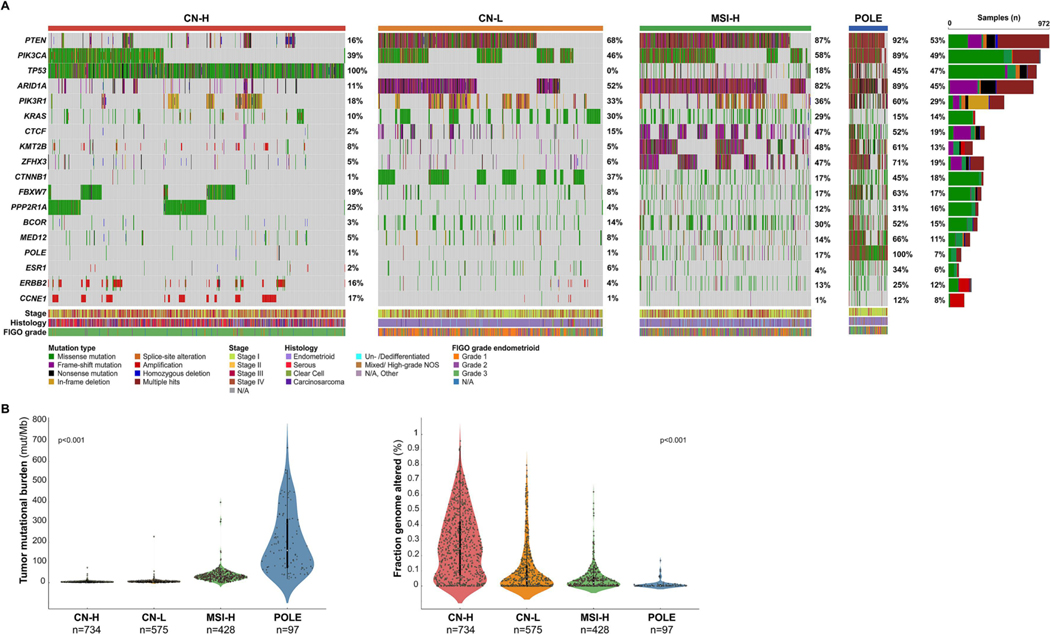
A. Oncoprints depicting the most recurrent somatic alterations in cancer-related genes in endometrial carcinomas by molecular subtype. Each column represents a tumor and the Oncoprint rows depict alterations for each gene. The bottom part of the graph shows the summary of histopathologic and clinical information for each case. The bar graph on the right of the panel shows the number and distribution of alterations for each gene across all subtypes. Mutation types and clinicopathologic features are color coded according to the legend. B. Tumor mutational burden in endometrial cancer by molecular subtype. C. Fraction of genome altered endometrial cancer by molecular subtype. P-value, Kruskal-Wallis rank sum test. CN-H, copy number-high; CN-L, copy number-low; MSI-H, microsatellite instability-high; N/A, not available; NOS, not otherwise specified.
Table 1.
Clinicopathologic and molecular characteristics of 1,834 endometrial cancers according to molecular subtype defined by integrating clinical tumor-normal sequencing and immunohistochemistry data.
| Total n=1,834 | POLE n=97 | MSI-H n=428 | CN-H n=734 | CN-L n=575 | p value* | |
|---|---|---|---|---|---|---|
| Age at diagnosis years, median (range) | ||||||
| BMI kg/m2, median (range) | 29.6 (14.9–67.6) | 26.3 (17.7–43.0) | 30.5 (16.6–59.3) | 29.1 (17.5–66.2) | 30.7 (14.9–67.6) | <0.001 |
| Histology, n (%) | <0.001 | |||||
| Endometrioid | 976 (53) | 75 (77) | 345 (81) | 74 (10) | 482 (84) | |
| Serous | 274 (15) | 0 | 3 (0.7) | 265 (36) | 6 (1) | |
| Clear Cell | 56 (3.1) | 1 (1) | 4 (0.9) | 31 (4.2) | 20 (3.5) | |
| Carcinosarcoma | 223 (12) | 0 | 14 (3.3) | 195 (27) | 14 (2.4) | |
| De-/ Undifferentiated | 39 (2.1) | 2 (2.1) | 25 (5.8) | 4 (0.5) | 8 (1.4) | |
| Mixed/ high-grade NOS | 170 (9.3) | 10 (10) | 28 (6.5) | 115 (16) | 17 (3) | |
| Other | 96 (5.2) | 9 (9.3) | 9 (2.1) | 50 (6.8) | 28 (4.9) | |
| Endometrioid grade, n (%) | <0.001 | |||||
| 1 | 473 (50) | 25 (35) | 128 (38) | 19 (28) | 301 (64) | |
| 2 | 287 (30) | 22 (31) | 131 (39) | 11 (16) | 123 (26) | |
| 3 | 183 (19) | 25 (35) | 74 (22) | 37 (55) | 47 (10) | |
| Stage, n (%) | <0.001 | |||||
| I | 956 (58) | 79 (85) | 256 (66) | 256 (39) | 365 (71) | |
| II | 72 (4.3) | 3 (3.2) | 18 (4.6) | 29 (4.4) | 22 (4.3) | |
| III | 341 (21) | 7 (7.5) | 82 (21) | 182 (28) | 70 (14) | |
| IV | 288 (17) | 4 (4.3) | 34 (8.7) | 190 (29) | 60 (12) | |
| Adjuvant treatment†, n (%) | <0.001 | |||||
| None | 371 (36) | 30 (36) | 95 (35) | 43 (14) | 203 (58) | |
| Chemo +/− RT | 452 (44) | 19 (23) | 108 (40) | 245 (77) | 80 (23) | |
| RT | 199 (19) | 35 (42) | 67 (25) | 30 (9.4) | 67 (19) | |
| Tissue type sequenced, n (%) | <0.001 | |||||
| Primary | 1501 (82) | 93 (97) | 355 (83) | 590 (81) | 463 (81) | |
| Recurrence/ metastasis | 323 (18) | 3 (3) | 73 (17) | 139 (19) | 108 (19) | |
| Tumor purity (%), median (range) | 40 (20–90) | 40 (20–85) | 40 (20–90) | 50 (20–90) | 40 (20–90) | <0.001 |
| TMB (mutations/Mb), median (range) | 6.1 (0.0–667.9) | 159.4 (19.3–667.9) | 30.1 (0.0–397.9) | 4.4 (0.8–74.6) | 6.1 (0.0–228.2) | <0.001 |
| Somatic mutations (n), median (range) | 7 (1–758) | 171 (22–758) | 34 (1–483) | 5 (1–85) | 7 (1–258) | <0.001 |
| Fraction genome altered (%), median (range) | 5.6 (0.0–95.7) | 0.1 (0.0–16.8) | 2.2 (0.0–62.2) | 24.7 (0.0–95.7) | 3.5 (0.0–79.7) | <0.001 |
| MSIsensor score, median (range) | 0.5 (0.0–47.2) | 0.2 (0.0–22.5) | 16.6 (0.0–47.2) | 0.6 (0.0–8.0) | 0.1 (0.0–8.5) | <0.001 |
Kruskal-Wallis rank sum test; Fisher’s exact test; Fisher’s Exact Test for Count Data with simulated p-value (based on 2000 replicates)
Adjuvant treatment: none, chemotherapy with or without radiation therapy, radiation therapy alone
BMI, body mass index; Chemo, chemotherapy; CN-H, copy number-high; CN-L, copy number-low; MSI-H, microsatellite instability-high; RT, radiotherapy; TMB, tumor mutational burden.
Of the 428 ECs of MSI-H molecular subtype, MMR IHC was available for 402 cases (94%). Of the 289 ECs with an MSIsensor score ≥10, MMR IHC results were concordant in 95% of ECs (250/289). In 39 ECs classified as MSI-H based on MSIsensor, 67% (26/39) did not have MMR IHC performed and 33% (13/39) had intact MMR protein expression (Fig. 2B). The ECs with intact MMR expression did not have methylation studies performed. Of note, the median tumor mutation burden (TMB) did not differ between MSI-H ECs with an MSIsensor score ≥10 and loss of MMR protein expression as compared to those with an MSIsensor score ≥10 and intact MMR protein expression (34.5 mt/Mb vs 36.0 mt/Mb, p=0.98). The remaining 139 ECs had an MSIsensor <10 and were classified as MSI-H molecular subtype based on loss MMR protein expression by IHC. The predominant histology of MSI-H ECs was endometrioid (345/428, 81%), and un-/dedifferentiated ECs were commonly of MSI-H molecular subtype (25/39, 64%).
Of the ECs subjected to clinical sequencing, 5% were of POLE molecular subtype (97/1,834). V411L and P286R were the most common POLE hotspot mutation identified (38/97, 39% and 33/97, 34%, respectively; Supplementary Fig. S2A). Like MSI-H or CN-L ECs, POLE ECs were primarily of endometrioid histology (75/97, 77%). Of note, mixed/high-grade NOS was found in 10 POLE ECs (10%) and another 9 (9.3%) POLE ECs were of unclassified/other histology (Figs. 1 and 3).
3.2. Comparison of integrated vs conventional surrogate molecular subtyping approach
We next sought to compare our integrated molecular and IHC-based with the conventional ProMisE molecular subtyping. As noted above, MMR IHC (80%) and p53 IHC (62%) were clinically performed only for a subset of ECs. With the addition of tumor-normal clinical sequencing data to IHC results we classified a significantly larger set of ECs from our initial 2,115 patient cohort into the molecular subtypes (1,834/2115, 86.7%) as compared to the ProMisE algorithm (1,387/2115, 65.5%, p<0.001), the latter of which requires both MMR and p53 IHC for all non-POLE cases.
Using the final study cohort (n=1,834), we next ascertained agreement between molecular subtype assignments by our integrated and the ProMisE classification. Overall, we found only a moderate agreement between our integrated molecular/ IHC and the ProMisE classification due to the large number of unclassified cases (Kappa 0.555, 95% confidence interval (CI) 0.531–0.579); Fig. 2C, Supplementary Table S1). When focusing on the ECs classifiable by the ProMisE algorithm (n=1,208), we recorded an almost perfect agreement (Kappa 0.962, 95% CI 0.949–0.975). Misclassifications were due to normal MMR or p53 expression patterns by IHC in the presence of high MSIsensor scores or TP53 mutations, respectively. A subset of CN-L ECs (n=25) by ProMisE harbored TP53 alterations but had normal p53 IHC-based expression, which were classified as CN-H using our integrated approach. In addition, a subset of CN-L (n=6) and CN-H (n=2) ECs by ProMisE had high MSIsensor scores but were MMR-proficient by IHC (Fig. 2C, Supplementary Table S1).
3.3. Clinicopathologic and somatic genomic features of EC molecular subtypes
There was a significant difference amongst the four molecular subtypes assigned by our integrated classification when comparing BMI, age at diagnosis, histology, stage, and adjuvant treatment type (p<0.001 each; Table 1). Surgical staging data were available for 90% of the ECs with molecular classification (1,657/1,834); the remaining either had clinical staging (advanced/metastatic disease) or no regional lymph node assessment at time of surgery. Patients with CN-H ECs had a greater association with stage III/IV disease (57% vs. 12–30%, p<0.001) and adjuvant chemotherapy with or without radiation treatment (77% vs 23–40%, p<0.001; Table 1). There was no difference in histology (p=0.24) or FIGO stage (p=0.09) between primary EC (n=1,501) and recurrent EC (n=323) tissue subjected to MSK-IMPACT sequencing (Supplementary Table S2).
Consistent with previous observations [3, 26], endometrioid cancers were preferentially of CN-L and MSI-H molecular subtypes (Supplementary Fig. S2B). In our series, 4% (30/760) of grade 1/2 and 20% (37/183) of grade 3 endometrioid ECs were of CN-H molecular subtype, as were almost all the serous ECs (265/274, 97%) and carcinosarcomas (195/223, 87%; Supplementary Fig. S2B).
Assessment of the somatic pathogenic mutations, amplifications and homozygous deletions affecting the cancer-related genes tested revealed that PIK3CA, PTEN, TP53 and ARID1A alterations were the most common across all molecular subtypes (Fig. 3A). ERBB2 and CCNE1 amplification were primarily restricted to CN-H ECs (ERBB2 amplification CN-H 13.4%, CN-L 1.7%; CCNE1 amplification CN-H 17.4%, CN-L 0.9%) and absent in MSI-H and POLE ECs (Fig. 3A). Consistent with the whole-exome sequencing-based data from TCGA [3], the tumor mutational burden (TMB) was highest for POLE ECs (median 159.4 mut/Mb, range 19.3–667.9) compared to ECs of MSI-H molecular subtype (median 30.1 mut/Mb, range 0.0–397.9) or CN-H (4.4 mut/Mb, range, 0.8–74.6) and CN-L ECs (median 6.1, range 0.0–228.2, p<0.001; Fig. 3B). Chromosomal instability was highest in ECs of CN-H molecular subtype (median FGA 24.7, range 0.0–95.7) compared to CN-L ECs (median FGA 3.5, range 0.0–79.7) or the hypermutated MSI-H (median FGA 2.2, range 0.0–62.2) and POLE ECs (median FGA 0.1, range 0.0–16.8; p<0.001; Fig. 3C).
3.4. Association with oncologic outcome
The median follow-up time for the 925 primary EC patients that met criteria for survival analysis was 22.3 months (range 0.5–214 months; CONSORT diagram, Supplementary Fig. S1). In this subset of patients there was a significant difference between ECs of the different molecular subtypes when comparing clinical characteristics (p<0.001 each; Supplementary Table S3).
When assessing the one-year progression-free (PFS) and overall survival (OS), we found that patients with ECs of POLE molecular subtype had the best outcomes, and those with ECs of CN-H molecular subtypes had the worst outcomes (one-year PFS: POLE 98.7% (95% CI 91.1–99.8%), CN-L 94.6% (95% CI 91.2–96.6%), MSI-H 91.3% (95% CI 86.7–94.4%), and CN-H 73.0% (95% CI 67.2–78%, p<0.001; one-year OS POLE 100%, MSI-H 98.1% (95% CI 95–99.3%), CN-L 99.3% (95% CI 97.3–99.8%), and CN-H 94.9 % (95% CI 91.4–97%), p<0.001; Fig. 4A). Multivariate analysis revealed that age at diagnosis (i.e., ≥ 60 years; HR 1.75, 95% C 1.22–2.50), FIGO stage (i.e., stage III HR 4.23, 95% CI 2.77–6.44; and stage IV HR 7.13, 95% CI 4.63–11.0) and non-endometrioid histology (HR 1.78, 95% CI 1.12–2.82) remained statistically significantly associated with poor PFS (Supplementary Table S4). The multivariate analysis for OS had significant associations with age at diagnosis (≥60 years; HR 2.57, 95% CI 1.32–5.02) and FIGO stage (i.e., stage III HR 4.25, 95% CI 1.99–9.08; and stage IV 9.43, 95% CI 4.45–20.0).
Figure 4. Survival outcomes of endometrial cancer patients by molecular subtype using an integrated clinical sequencing – immunohistochemistry-based classification approach.

Survival was assessed in 925 patients with endometrial cancer of all histologic types whose tumors were subjected to MSK-IMPACT prior to recurrence, had upfront surgery, were surgically staged, and had surgery performed at our institution (see Supplementary Fig. S1). A. Kaplan-Meier curve comparing progression-free survival (PFS) and overall survival (OS) in all 925 endometrial cancer patients by molecular subtype. B. PFS (n=679) and OS (n=502) of endometrial cancer patients with stage I/II disease. C. PFS and OS of endometrial cancer patients with stage III/IV disease (n=246). For stage III and stage IV analysis separately, see Supplementary Fig. S3. D. PFS and OS of patients with stage I/II endometrioid endometrial cancer (n=502). Survival compared with log-rank test. *p-value defined excluding the POLE molecular subtype due to the lack of events. CN-H, copy number-high; CN-L, copy number-low; MSI-H, microsatellite instability-high.
As a next step, we assessed the association of the molecular subtypes with outcomes separately for EC patients diagnosed with early-stage disease (stages I/II) and advanced-stage disease (stages III/IV). The molecular classification continued to be associated significantly with outcome, and POLE and CN-H ECs had the best and the worst outcomes in either early-stage or late-stage disease, respectively. We did observe, however, that there was a substantial decrease in advanced-stage survival outcomes for MSI-H (1-year PFS 82.8%, 95%CI 70.4–90.4%) and advanced-stage CN-L (1-year PFS 77.9%, 95%CI 61.8–87.8%) when compared to early-stage MSI-H (1-year PFS 94.2%, 95% CI 89.2–97%) and early stage CN-L (1-year PFS 97.2%, 95% CI 94.1–98.6%), respectively (Figs. 4B and 4C; Supplementary Fig. 3).
We further sought to assess whether the molecular subtype classification is prognostic in early-stage endometrioid ECs. While the majority of stage I/II endometrioid ECs were of CN-L (n=266, 53%) or MSI-H (n=152, 30%) molecular subtypes, stage III/IV ECs were most commonly MSI-H (n=48, 52%) or CN-L (n=29, 31%; p<0.001; Supplementary Table S5). The molecular subtypes were significantly associated with PFS in stage I/II endometrioid ECs, with POLE and CN-L ECs having the best (2-year PFS POLE: 96% (95% CI, 86–99%); CN-L: 98% (95% CI, 95–99%)), MSI-H intermediate (88%; 95% CI, 80–93%) and CN-H ECs having the worst outcomes (72%; 95% CI, 43–88%; p<0.001; Fig. 4D).
3.5. Serous ECs, endometrioid ECs and carcinosarcomas of CN-H molecular subtype
Analysis of the levels of chromosomal instability amongst CN-H serous ECs (n=265), CN-H endometrioid ECs (n=74) and CN-H carcinosarcomas (n=195) revealed that carcinosarcomas of CN-H molecular subtype had significantly higher FGAs (median 34.5, range 0.0–90.4) compared to CN-H serous ECs (median 22.3, range 0.0–75.4) and CN-H endometrioid ECs (median 12.9, range 0.0–95.7; p<0.001; Fig. 5A). Notably, PFS and OS was not statistically significantly different between CN-H serous, endometrioid and carcinosarcomas (survival cohort; n=192; p>0.1; Fig. 5B).
Figure 5. Survival outcomes in high-risk histologic types and in copy number-low endometrial cancer patients by chromosomal instability.

A. Fraction of genome altered (FGA, %) among copy number-high (CN-H) endometrial cancers of serous histology (n=265), endometrioid histology (n=74), and carcinosarcoma (n=195). B. Progression-free survival (PFS) and overall survival (OS) of patients meeting survival criteria with endometrial carcinomas of CN-H serous (n=82), CN-H endometrioid (n=32), and CN-H carcinosarcoma (n=78) histologic types. C. PFS and OS of patients with CN-low (CN-L) endometrial cancer (n=249; survival cohort) dichotomized by the median FGA of CN-H endometrial cancer (median 20%; Supplementary Fig. S4). Survival compared with log-rank test.
3.6. CN-L and MSI-H ECs with high chromosomal instability
Together with the observation that the majority but not all (92%) CN-H ECs in the TCGA study harbored a TP53 mutations [3], we hypothesized that TP53 mutation/ p53 IHC may be an incomplete surrogate of high levels of CN alterations and that analysis of chromosomal instability may be additive. In this exploratory analysis, in the survival cohort, 6% (21/324) of unselected primary CN-L ECs and 0.8% (2/249) of unselected primary MSI-H ECs had an FGA above the median of 20% found in CN-H ECs (Supplementary Fig. S4). In the CN-L group, these patients had worse outcomes than CN-L ECs with lower levels of chromosomal instability (PFS: CN-L FGA ≥20 HR 7.52, 95% CI 3.51–16.09 vs. FGA <20; p<0.001; OS FGA ≥20 HR 17.33, 95% CI 4.87–61.71; p<0.001; Figures 5C-D).
4. DISCUSSION
Here we demonstrate that through the utilization of prospective clinical tumor-normal sequencing data, we expand the number of ECs that can be classified into the molecular subtypes as compared to the ProMisE algorithm, which relies on the availability of clinical MMR and p53 IHC. Our integrated classification is mostly concordant with that of the ProMisE classification but also provides a refinement via the incorporation of the MSIsensor score as a validated proxy for MMRd/MSI status and TP53 driver mutations for p53 IHC [14, 27, 28]. More specifically, clinical sequencing provides TP53 alteration status, which in the absence of MMR/POLE mutations is the defining feature of CN-H ECs. By integrating NGS data, we found ECs that would have had a different or missing TCGA classification if based only on POLE mutation sequencing plus MMR/p53 IHC results.
Our data on CN-H ECs confirm that this group of tumors is associated with higher-risk, non-endometrioid histology and poor prognostic outcomes relative to other molecular subtypes. In the original description of the molecular classification by TCGA [3], a combination of TMB, MSI and somatic CN alterations was utilized for the molecular classification; all (60/60, 100%) ECs of CN-H molecular subtype harbored the highest levels of CN alterations (cluster 4), whereas none of the POLE (0/17) or CN-L ECs (0/90) and only 3% (2/65) of MSI-H ECs were of somatic CN alteration cluster 4 [3]. CN-L ECs are heterogenous tumors with varying outcomes [22, 29], and we observed that CN-L tumors with levels of chromosomal instability similar to those seen on average in CN-H ECs are associated with significantly worse prognosis. Additional studies are warranted to further refine the CN-L group and to validate approaches for the identification of CN-L ECs with high levels of CN alterations associated with poor outcomes.
Analysis of this large population of ECs allowed for the assessment of the molecular subtype distribution in the less common histologic types. We found that each molecular subtype group had varying percentages of non-endometrioid histology ECs, particularly carcinosarcoma, clear cell, un-/de-differentiated and mixed/ high-grade NOS. Our study confirms recent data evaluating 1,336 EC patients from 29 Canadian institutions [30], where the majority of the 83 POLE tumors were of endometrioid histology (88% Jamieson et al [30] vs 77% this study), but had also representation of other histologies including carcinosarcoma, serous, and dedifferentiated carcinoma.
In this study, we found our patient outcomes were consistent with the existing literature, including TCGA [3, 4, 8, 31–33]. Of note, however, our survival subgroup only included patients who underwent MSK-IMPACT sequencing prior to a documented recurrence requiring escalation of care, underwent upfront surgery at our institution, and had surgical staging including regional lymph node analysis to reduce further bias in the analysis. Multivariate analysis identified MSI-H molecular subtype, CN-H molecular subtype, age ≥60 years/ diagnosis, FIGO stages III/IV and non-endometrioid histology to be associated with worse PFS outcomes. Our survival analyses further demonstrate that molecular classification is associated with outcome when assessing patients with advanced stage disease (i.e., stages III/IV) at diagnosis, especially patients with MSI-H and CN-L ECs having worse PFS and OS compared to their early-stage (i.e., stages I/II) complement.
Other than POLE exonuclease domain hotspot mutations and TP53 mutations, genomic heterogeneity can be observed within each molecular subtype group. We further confirm that PTEN and PIK3CA are highly recurrently altered across subtypes, whereas FBXW7, PPP2R1A, ERBB2 and CCNE1 alterations are enriched in CN-H ECs. The addition of trastuzumab to carboplatin and paclitaxel is NCCN listed for patients with stage III-IV or recurrent HER2-positive EC based on a randomized phase 2 study [34]. This also remains an area of active clinical development with the recent FDA approval of trastuzumab deruxtecan for the treatment of HER2-positive breast [35] and gastric cancer [36], and ERBB2-mutant lung cancer [37]. Further refinement of the molecular subtypes in clinically meaningful ways is required, however, particularly for CN-L and MSI-H ECs, as these groups comprise patients with tumors that may behave poorly relative to their umbrella subtype group and harbor different targetable alterations. Recent studies also investigated the additional use of estrogen receptivity within the CN-L subgroup, which may lead to further stratify and identify higher-risk, subtype-specific ECs [38, 39].
Our study has several limitations, including bias inherent to any retrospective study. First, MMR and p53 IHC data were not available for all patients. For patients presenting for care at our institution at the time of a recurrence, MMR IHC analyses were often not repeated due to lack of sample availability and interpretation relied upon recorded documentation. In addition, IHC analyses performed on the biopsy were not typically repeated on the hysterectomy specimen given that high concordance has been reported in the literature [40]. For NGS data, we used a 20% tumor purity cut-off to ascertain high quality sequencing results; further studies would be required to further refine and define the optimal tumor cellularity cut-off from sequencing studies. We recognize the cost and availability of NGS may pose a barrier for obtaining POLE, MSIsensor, and TP53 results within and across institutions. Though the use of targeted sequencing in EC remains investigational, the potential to stratify patients for a tailored adjuvant treatment may ultimately lead to cost-effective alternatives. Lastly, to minimize survival bias, we applied a very stringent criteria for the inclusion of cases in the survival analyses.
In this study, we demonstrate that in a large, unselected, prospectively clinically sequenced series of ECs the integration of molecular and IHC data leads to the classification of a larger set of ECs into the TCGA molecular subtypes as compared to the ProMisE classifier. In addition, orthogonal sequencing-based information on TP53 mutation status and MSI refines the classification of cases with discordant IHC and mutation/ MSI status capturing a greater number of classifiable tumors. Given the insights gained from the molecular studies over the past decade, the NCCN has included the consideration of comprehensive genomic profiling for the initial evaluation of uterine neoplasms [6]. With the more widespread access of NGS, the integration of mutational data into the molecular stratification may assist in the further subclassification/refinement of some of the molecular subtypes and will be essential for prospective clinical trials to identify the best possible adjuvant approach for this heterogenous disease.
Supplementary Material
Supplementary Figure S1. CONSORT diagram summarizing the endometrial cancer patients included in the survival analyses.
Supplementary Figure S2. Distribution of POLE mutations and of molecular subtypes across endometrial cancer histologic types defined using an integrated clinical sequencing - immunohistochemistry approach.
Supplementary Figure S3. Survival outcomes of endometrial cancer patients with stage III and IV disease by molecular subtype.
Supplementary Figure S4. Fraction of genome altered in endometrial cancers of different molecular subtypes.
Supplementary Table S1. Agreement between our integrated clinical sequencing – immunohistochemistry-based approach and the ProMisE algorithm for molecular classification of endometrial cancers.
Supplementary Table S2. Distribution of histology, stage, and molecular subtype amongst primary and recurrent endometrial cancers subjected to MSK-IMPACT sequencing and molecular subtyping.
Supplementary Table S3. Clinico-pathologic and molecular characteristics of endometrial cancers according to molecular subtype for the survival cohort (n=925).
Supplementary Table S4. Univariate and multivariate analysis on progression-free survival for patients meeting survival criteria (n=925).
Supplementary Table S5. Clinico-pathologic characteristics by molecular subtypes for stage I/II endometrioid endometrial carcinoma.
Highlights.
Clinical sequencing and immunohistochemistry data integration allows for endometrial cancer (EC) molecular classification
Integrated classification is highly concordant with existing surrogates while allowing more ECs to be classified
Integrated classification overcomes issues of immunohistochemistry-based detection of genetic alterations
Integrated molecular classification has prognostic value across all stages and histologic types of EC
ACKNOWLEDGEMENTS
Research reported in this publication was supported in part by a Cancer Center Support Grant of the NIH/NCI (Grant No. P30CA008748). D. Zamarin is supported by the Ovarian Cancer Research Foundation Liz Tilberis Award, and the Department of Defense Ovarian Cancer Research Academy (OC150111). B. Weigelt is supported in part by a Breast Cancer Research Foundation grant. L.H. Ellenson and B. Weigelt are supported in part by a Cycle for Survival grant. D. Zamarin, C. Aghajanian and B. Weigelt are supported in part by BreakThrough Cancer Foundation grants.
N.R. Abu-Rustum reports Stryker/ Novadaq and GRAIL grants paid to the institution, outside the current study. C.F. Friedman reports institutional research support from Seagen, Merck, BMS, AstraZeneca, Mersana and Hotspot Therapeutics; consulting fees from BMS, Seagen and Aadi Biosciences; honoraria for lectures from Onclive; meeting/ travel support by Puma; participation on Data Safety Monitoring or Advisory Board of Merck, Genentech and Marengo (all uncompensated). D. Zamarin reports institutional research support from AstraZeneca, Merck, Plexxikon Synthekine and Genentech; consulting fees from AstraZeneca, Synthekine, Astellas, Tessa Therapeutics, Memgen, Celldex, Crown Biosciences, Hookipa Biotech, Kalivir, Xencor and GSK; royalties from Merck; and stock options from Accurius Therapeutics, ImmunOS Therapeutics and Calidi Biotherapeutics, all outside the submitted work. V. Makker reports meeting/ travel support by Eisai and Merck; participation on Data Safety Monitoring or Advisory Board of Duality, Merck, Karyopharm, Exelexis, Eisai, Karyopharm, BMS, Clovis, Faeth Immunocore, Morphosys, AstraZeneca, Novartis, GSK, Bayer (all unpaid), and study support to the institution by Merck, Eisai, AztraZeneca, Faeth, Karyopharm, Zymeworks, Duality, Clovis, Bayer and Takeda. Y. Liu reports institutional research funding from Repare Therapeutics, AstraZeneca and GSK; honoraria from Total Health and Sarah Lawrence College; and travel/ meeting support by AstraZeneca, outside the submitted work. B. Weigelt reports a research grant from REPARE Therapeutics paid to the institution, outside the submitted work. D.S. Chi reports personal fees from Apyx Medical, Verthermia Inc., Biom ‘Up, and AstraZeneca, as well as recent or current stock/options ownership of Apyx Medical, Verthemia, Intuitive Surgical, Inc., TransEnterix, Inc., Doximity, Moderna, and BioNTech SE. E.L. Jewell reports personal fees from Covidien/Medtronic. M. M. Leitao is an ad hoc speaker for Intuitive Surgical, Inc.; outside the submitted work, he is on the Advisory Board of Ethicon/Johnson & Johnson and Takeda; and reports grants paid to the institution by KCI/Acelity. C. Aghajanian reports clinical trial funding paid to the institution from AstraZeneca; consulting fees (advisory board) from Eisai/Merck, Roche/Genentech, Abbvie, AstraZeneca/Merck, and Repare Therapeutics; advisory board participation (no fee) for Blueprint Medicine; and leadership/fiduciary roles for the GOG Foundation Board of Directors (travel cost reimbursement) and NRG Oncology Board of Directors (unpaid).
Footnotes
CONFLICTS OF INTEREST
The remaining authors have no conflicts of interest to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021;71:209–49. [DOI] [PubMed] [Google Scholar]
- [2].Murali R, Soslow RA, Weigelt B. Classification of endometrial carcinoma: more than two types. Lancet Oncol. 2014;15:e268–78. [DOI] [PubMed] [Google Scholar]
- [3].Cancer Genome Atlas Research Network, Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Talhouk A, McConechy MK, Leung S, Li-Chang HH, Kwon JS, Melnyk N, et al. A clinically applicable molecular-based classification for endometrial cancers. Br J Cancer. 2015;113:299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Murali R, Delair DF, Bean SM, Abu-Rustum NR, Soslow RA Evolving Roles of Histologic Evaluation and Molecular/Genomic Profiling in the Management of Endometrial Cancer. J Natl Compr Canc Netw. 2018;16:201–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].National Comprehensive Cancer Network. NCCN Guidelines for Uterine Neoplasms V.1.2022. 2022. [Google Scholar]
- [7].WHO Classification of Tumours Editorial Board. Female Genital Tumours. 5th Edition ed. Lyon: IARC; 2020. [Google Scholar]
- [8].Kommoss S, McConechy MK, Kommoss F, Leung S, Bunz A, Magrill J, et al. Final validation of the ProMisE molecular classifier for endometrial carcinoma in a large population-based case series. Ann Oncol. 2018;29:1180–8. [DOI] [PubMed] [Google Scholar]
- [9].Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J Mol Diagn. 2015;17:251–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23:703–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Momeni-Boroujeni A, Dahoud W, Vanderbilt CM, Chiang S, Murali R, Rios-Doria EV, et al. Clinicopathologic and Genomic Analysis of TP53-Mutated Endometrial Carcinomas. Clin Cancer Res. 2021;27:2613–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Walsh MD, Cummings MC, Buchanan DD, Dambacher WM, Arnold S, McKeone D, et al. Molecular, pathologic, and clinical features of early-onset endometrial cancer: identifying presumptive Lynch syndrome patients. Clin Cancer Res. 2008;14:1692–700. [DOI] [PubMed] [Google Scholar]
- [13].Leon-Castillo A, Britton H, McConechy MK, McAlpine JN, Nout R, Kommoss S, et al. Interpretation of somatic POLE mutations in endometrial carcinoma. J Pathol. 2020;250:323–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Middha S, Zhang L, Nafa K, Jayakumaran G, Wong D, Kim HR, et al. Reliable Pan-Cancer Microsatellite Instability Assessment by Using Targeted Next-Generation Sequencing Data. JCO Precis Oncol. 2017;2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Manning-Geist BL, Liu YL, Devereaux KA, Da Cruz Paula A, Zhou QC, Ma W, et al. Microsatellite instability-high endometrial cancers with MLH1 promoter hypermethylation have distinct molecular and clinical profiles. Clin Cancer Res. 2022; 28:4302–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Modica I, Soslow RA, Black D, Tornos C, Kauff N, Shia J. Utility of immunohistochemistry in predicting microsatellite instability in endometrial carcinoma. Am J Surg Pathol. 2007;31:744–51. [DOI] [PubMed] [Google Scholar]
- [17].Chang MT, Bhattarai TS, Schram AM, Bielski CM, Donoghue MTA, Jonsson P, et al. Accelerating Discovery of Functional Mutant Alleles in Cancer. Cancer Discov. 2018;8:174–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Chakravarty D, Gao J, Phillips SM, Kundra R, Zhang H, Wang J, et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis Oncol. 2017;2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Griffith M, Spies NC, Krysiak K, McMichael JF, Coffman AC, Danos AM, et al. CIViC is a community knowledgebase for expert crowdsourcing the clinical interpretation of variants in cancer. Nat Genet. 2017;49:170–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Nakamura M, Obata T, Daikoku T, Fujiwara H. The Association and Significance of p53 in Gynecologic Cancers: The Potential of Targeted Therapy. Int J Mol Sci. 2019;20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Vermij L, Smit V, Nout R, Bosse T. Incorporation of molecular characteristics into endometrial cancer management. Histopathology. 2020;76:52–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Momeni-Boroujeni A, Nguyen B, Vanderbilt CM, Ladanyi M, Abu-Rustum NR, Aghajanian C, et al. Genomic landscape of endometrial carcinomas of no specific molecular profile. Mod Pathol. 2022;35:1269–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Landis JR, Koch GG The measurement of observer agreement for categorical data. Biometrics. 1977;33:159–74. [PubMed] [Google Scholar]
- [24].Kehl KL, Xu W, Lepisto E, Elmarakeby H, Hassett MJ, Van Allen EM, et al. Natural Language Processing to Ascertain Cancer Outcomes From Medical Oncologist Notes. JCO Clin Cancer Inform. 2020;4:680–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Levesque LE, Hanley JA, Kezouh A, Suissa S. Problem of immortal time bias in cohort studies: example using statins for preventing progression of diabetes. Brit Med J. 2010;340. [DOI] [PubMed] [Google Scholar]
- [26].Da Cruz Paula A, DeLair DF, Ferrando L, Fix DJ, Soslow RA, Park KJ, et al. Genetic and molecular subtype heterogeneity in newly diagnosed early- and advanced-stage endometrial cancer. Gynecol Oncol. 2021;161:535–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Singh N, Piskorz AM, Bosse T, Jimenez-Linan M, Rous B, Brenton JD, et al. p53 immunohistochemistry is an accurate surrogate for TP53 mutational analysis in endometrial carcinoma biopsies. J Pathol. 2020;250:336–45. [DOI] [PubMed] [Google Scholar]
- [28].Kobel M, Ronnett BM, Singh N, Soslow RA, Gilks CB, McCluggage WG Interpretation of P53 Immunohistochemistry in Endometrial Carcinomas: Toward Increased Reproducibility. Int J Gynecol Pathol. 2019;38 Suppl 1:S123–S31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Depreeuw J, Stelloo E, Osse EM, Creutzberg CL, Nout RA, Moisse M, et al. Amplification of 1q32.1 Refines the Molecular Classification of Endometrial Carcinoma. Clin Cancer Res. 2017;23:7232–41. [DOI] [PubMed] [Google Scholar]
- [30].Jamieson A, Huvila J, Thompson EF, Leung S, Chiu D, Lum A, et al. Variation in practice in endometrial cancer and potential for improved care and equity through molecular classification. Gynecol Oncol. 2022;165:201–14. [DOI] [PubMed] [Google Scholar]
- [31].Stelloo E, Nout RA, Osse EM, Jurgenliemk-Schulz IJ, Jobsen JJ, Lutgens LC, et al. Improved Risk Assessment by Integrating Molecular and Clinicopathological Factors in Early-stage Endometrial Cancer-Combined Analysis of the PORTEC Cohorts. Clin Cancer Res. 2016;22:4215–24. [DOI] [PubMed] [Google Scholar]
- [32].Cosgrove CM, Tritchler DL, Cohn DE, Mutch DG, Rush CM, Lankes HA, et al. An NRG Oncology/GOG study of molecular classification for risk prediction in endometrioid endometrial cancer. Gynecol Oncol. 2018;148:174–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Bosse T, Nout RA, McAlpine JN, McConechy MK, Britton H, Hussein YR, et al. Molecular Classification of Grade 3 Endometrioid Endometrial Cancers Identifies Distinct Prognostic Subgroups. Am J Surg Pathol. 2018;42:561–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Fader AN, Roque DM, Siegel E, Buza N, Hui P, Abdelghany O, et al. Randomized Phase II Trial of Carboplatin-Paclitaxel Versus Carboplatin-Paclitaxel-Trastuzumab in Uterine Serous Carcinomas That Overexpress Human Epidermal Growth Factor Receptor 2/neu. J Clin Oncol. 2018;36:2044–51. [DOI] [PubMed] [Google Scholar]
- [35].Cortés J, Kim S-B, Chung W-P, Im S-A, Park YH, Hegg R, et al. Trastuzumab Deruxtecan versus Trastuzumab Emtansine for Breast Cancer. New Engl J Med. 2022;386:1143–54. [DOI] [PubMed] [Google Scholar]
- [36].Shitara K, Bang Y-J, Iwasa S, Sugimoto N, Ryu M-H, Sakai D, et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Gastric Cancer. New Engl J Med. 2020;382:2419–30. [DOI] [PubMed] [Google Scholar]
- [37].Li BT, Smit EF, Goto Y, Nakagawa K, Udagawa H, Mazières J, et al. Trastuzumab Deruxtecan in HER2-Mutant Non–Small-Cell Lung Cancer. New Engl J Med. 2021;386:241–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Jamieson A, Huvila J, Chiu D, Thompson EF, Scott S, Salvador S, et al. Grade and Estrogen Receptor Expression Identify a Subset of No Specific Molecular Profile Endometrial Carcinomas at a Very Low Risk of Disease-Specific Death. Mod Pathol. 2023;36:100085. [DOI] [PubMed] [Google Scholar]
- [39].Vermij L, Jobsen JJ, Leon-Castillo A, Brinkhuis M, Roothaan S, Powell ME, et al. Prognostic refinement of NSMP high-risk endometrial cancers using oestrogen receptor immunohistochemistry. Br J Cancer. 2023;128:1360–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Talhouk A, Hoang LN, McConechy MK, Nakonechny Q, Leo J, Cheng A, et al. Molecular classification of endometrial carcinoma on diagnostic specimens is highly concordant with final hysterectomy: Earlier prognostic information to guide treatment. Gynecol Oncol. 2016;143:46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1. CONSORT diagram summarizing the endometrial cancer patients included in the survival analyses.
Supplementary Figure S2. Distribution of POLE mutations and of molecular subtypes across endometrial cancer histologic types defined using an integrated clinical sequencing - immunohistochemistry approach.
Supplementary Figure S3. Survival outcomes of endometrial cancer patients with stage III and IV disease by molecular subtype.
Supplementary Figure S4. Fraction of genome altered in endometrial cancers of different molecular subtypes.
Supplementary Table S1. Agreement between our integrated clinical sequencing – immunohistochemistry-based approach and the ProMisE algorithm for molecular classification of endometrial cancers.
Supplementary Table S2. Distribution of histology, stage, and molecular subtype amongst primary and recurrent endometrial cancers subjected to MSK-IMPACT sequencing and molecular subtyping.
Supplementary Table S3. Clinico-pathologic and molecular characteristics of endometrial cancers according to molecular subtype for the survival cohort (n=925).
Supplementary Table S4. Univariate and multivariate analysis on progression-free survival for patients meeting survival criteria (n=925).
Supplementary Table S5. Clinico-pathologic characteristics by molecular subtypes for stage I/II endometrioid endometrial carcinoma.