

Abstract
Purpose of review:
We recently localized a new KCC3 transporter to the apical membrane of type-B intercalated cells. This gives us an opportunity to revisit the roles of the K-Cl cotransporters-3 in kidney and integrate the new findings to our current knowledge of the biology of the bicarbonate secreting cells.
Recent findings:
Here we review the basic properties of the K-Cl cotransporter with a particular attention to the responsiveness of the transporter to cell swelling. We summarize what is already known about KCC3b and discuss new information gained from our localizing of KCC3a in type-B intercalated cells. We integrate the physiology of KCC3a with the main function of the type-B cell, i.e., bicarbonate secretion through the well-characterized apical Cl−/HCO3− exchanger and the basolateral Na-HCO3 cotransporter.
Summary:
Both KCC3b and KCC3a seem to be needed for maintaining cell volume during enhanced inward cotransport of Na-glucose in proximal tubule and Na-HCO3 in intercalated cells. In addition, apical KCC3a might couple to pendrin function to recycle Cl−, particularly in conditions of low salt diet and therefore low Cl− delivery to the distal tubule. This function is critical in alkalemia, and KCC3a function in the pendrin-expressing cells may contribute to the K+ loss which is observed in alkalemia.
Keywords: K+ transport, Cell volume, Distal nephron, Type-B intercalated cells, bicarbonate
Introduction
The K-Cl cotransporter mediates the electroneutral, Cl− coupled, movement of K+ ions across plasma membranes. The transporter belongs to the SLC12 family of solute carriers, itself a member of a superfamily of amino acid/polyamine/organocation transporters (1). The gene encoding K-Cl cotransport originated in protists and duplicated twice during evolution, resulting into four distinct genes in higher vertebrates: SLC12A4 (KCC1), A5 (KCC2), A6 (KCC3), and A7 (KCC4).
The K-Cl cotransporter was originally described as a functional transport unit in sheep and human red blood cells (2, 3). The transporter was shown to be activated by N-ethylmaleimide (2), cell swelling (3), and inhibitable by high doses of furosemide (4). Like the Na-K-2Cl cotransporter (5), the K-Cl cotransporter functional unit is a dimer (6). The recent cryo-electron microscopy (cryo-EM) structures of NKCC1 (7**, 8*), KCC1-KCC4 (9**–11) confirms the dimeric nature of the transporters. As cells express multiple K-Cl cotransporters (12–14), it is believed that they likely express heterodimers. Work performed in heterologous expression systems has provided evidence for cotransporter heterodimerization (6, 15).
Swelling activation is a key property of K-Cl cotransporters
K-Cl cotransport activity is activated by dephosphorylation and inhibited by phosphorylation (16, 17). A simple two-state model fits nicely the data of swelling activation of K-Cl cotransport (18) with swelling principally inhibiting the kinase that shuts off KCC function. The current consensus in the field is that individual transporters at the membrane are either on (dephosphorylated) or off (phosphorylated), and the overall transport measured in a cell, depends upon the number of transporters expressed at the membrane that are in the on state. K-Cl cotransporters possess at their amino terminus a binding site for Ste20p-like kinases (19) and it is the phosphorylation of two key threonine residues, located in the COOH tail of the cotransporter, that inactivates KCC (20). Despite the recent resolution of the structure of the transporters, their activation/de-activation by phosphorylation are still poorly understood.
Expression of KCC3 in kidney
The cloning of human KCC3 revealed the existence of two alternative start sites or alternative promoters (21–23). Mount and Race independently cloned KCC3 from human muscle (21) and placenta (22), respectively. Hiki and colleagues also isolated KCC3 from human umbilical vein endothelial cells (23). As shown in Figure 1, the 109 kb hKCC3 gene starts with exon 1a and finishes with exon 25. Exon 1b is located some 17.6 kb downstream of exon 1a and 43 kb upstream of exon 2. Exon 2 is a small 45 bp alternatively spliced cassette exon encoding a short (15 amino acid long) peptide. Detailed transcript analysis revealed that KCC3a, the isoform containing exon 1a, is highly expressed in muscle and brain, whereas KCC3b, the isoform starting at exon 1b, is predominantly found in the kidney (14). KCC3 is expressed at the basolateral of proximal tubule epithelial cells (24). However, until recently, no antibodies could distinguish between the KCC3a and KCC3b isoforms. After generating and validating a KCC3a-specific antibody, we examined KCC3a localization along the nephron (25**). Except for specific cells labeled in a discontinuous pattern in the cortex, the KCC3a signal was extremely low in kidney sections from wild-type mice. We localized KCC3a in intercalated cells of type-B or type-nonA/nonB using co-staining with pendrin antibody, an intercalated cell marker (26). While the majority of the signal was detected at the apical membrane alongside pendrin, a few cells showed weaker signal at the basolateral membrane (25**). Additional co-labeling experiments revealed that the KCC3a signal was only present in V-ATPase positive cells, distinct from cells expressing the sodium chloride cotransporter (NCC), the epithelial sodium channel (ENaC), and calbindin-28, indicating that KCC3a expression was absent in the distal convoluted tubule (DCT, identified with antibody reacting with phosphorylated NCC or pNCC), and CNT principal cells (identified with ENaC and calbindin-28). The absence of KCC3a signal in the proximal tubule also indicated to us that the KCC3 signal observed in the proximal tubule must be originating from KCC3b.
Figure 1. Structure of the SLC12A6 gene.

The human SLC12A6 gene is located on Chr. 15: 34,229,784-34,338,060 (minus strand) between ER membrane protein complex subunit 4 (EMC4 – 34,225013-34,230156) for which the two genes overlap their 3’ end, and ribonucleoprotein, NOP10 (34,339,159-34,343,180). SLC12A6 is composed of 26-27 exons, depending on whether exon 1a is a single exon (as in mouse) or broken up into 2 exons. Transcription starts at either exon 1a (for KCC3a) or exon 1b (for KCC3b). Exon 2 is a small 45 bp alternatively spliced cassette exon encoding a 15 amino acid peptide. There is evidence that this exon is alternatively spliced in tissues expressing KCC3a or KCC3b.
Function of KCC3 in the kidney
Creatinine, urea, Na+, and H+ serum concentrations in KCC3-KO mice do not differ from controls. However, hourly diuresis and increased water consumption were observed in KCC3 knockout mice, although with no variation in urine osmolality or electrolyte concentration (27). In a separate study, Gerardo Gamba’s group showed that while Na+ or K+ depletion had no effect on KCC3 abundance, hyperglycemia caused a 2-fold increase in KCC3 expression in the rodent kidney (28). On this observation, they inferred that KCC3 function in the proximal tubule was to either maintain sodium-glucose cotransporter-2 (SGLT2) activity by promoting the basolateral outflow of Na+ through the Na+ pump, or to prevent SGLT2 activity from causing the epithelial cells to swell (28). The cell swelling hypothesis is attractive as mathematical models of active epithelial ion transport, e.g., in DCT, indicate the importance of adding a K-Cl cotransporter for the cells to maintain their volume (29).
In two recent papers, we addressed the role of KCC3a in the connecting tubule. In the first manuscript, we demonstrated that KCC3a expression was increased by a variety of manipulations that led to alkalosis. For instance, we observed increased KCC3a expression following 24-h water restriction or salt deficient diet (25**). This observed increase can be accounted for by contraction alkalosis, as for a defined amount of HCO3− in the blood, a decrease in plasma volume (contraction) will result in increased HCO3− concentration or alkalosis. Furthermore, the condition that resulted in the largest increase in KCC3a expression was feeding the mice with 280 mM bicarbonate in the drinking water (25**). As this treatment resulted in increased pendrin and KCC3a abundance and KCC3 deletion resulted in decreased pendrin abundance, we proposed a functional relationship between pendrin and KCC3a (25**). In conditions of alkalemia, the cortical collecting duct secretes HCO3− via pendrin, an apical, electroneutral Cl−/HCO3− exchanger. This function is mediated by type-B intercalated cells. Pendrin secretes HCO3− in exchange of Cl− in the filtrate, thus contributing to acid-base balance (30–34). Many studies have shown pendrin stimulated by metabolic alkalosis (35–40).
K+ loss is typically observed in alkalemia (41). It is well known that the intracellular storage of K+ and the capacity of principal cells to secrete K+, which harbor renal outer medullary potassium channels (ROMK), and large conductance calcium-activated potassium channels known as Maxi-K or BK channels, can be influenced by acid-base balance (42). Increased luminal or systemic levels of HCO3−, pH, distal Na+ and fluid flow all stimulate ROMK, and BK channel, which in turn causes an increase in distal K+ secretion (42–45). Further studies are needed to dissect the contributions of ROMK, the BK channel, and KCC3a to the K+ loss observed in alkalemia.
Although, as just discussed, the K+ loss is often associated with K+ channel activity like ROMK in principal cells, a recent study demonstrated that pendrin was required for K+ excretion under a high KHCO3 diet (46). In our recent study, we hypothesized that pendrin and KCC3a are functionally coupled (Figure 2A), thereby accounting for the electroneutral K+ loss that occurs through KCC3a when Cl− is recycled to sustain pendrin activity (25**). Note that an alternative model involves cell volume regulation mediated by KCC3. Indeed, bicarbonate transport across the intercalated cell may cause cell swelling (47, 48). Therefore, instead of Cl− recycling, water may be recycled via KCC3 activity, again to the detriment of K+ (Figure 2B). This hypothesis would also be consistent with the fact that KCC3 activity is otherwise silent under isosmotic conditions. Once again, this KCC function was mathematically modeled in the rat DCT; the transporter was needed to maintain cell volume during various stages of epithelial transport (29).
Figure 2. Working model of KCC3a roles in type-B (or nonA/nonB) intercalated cells.
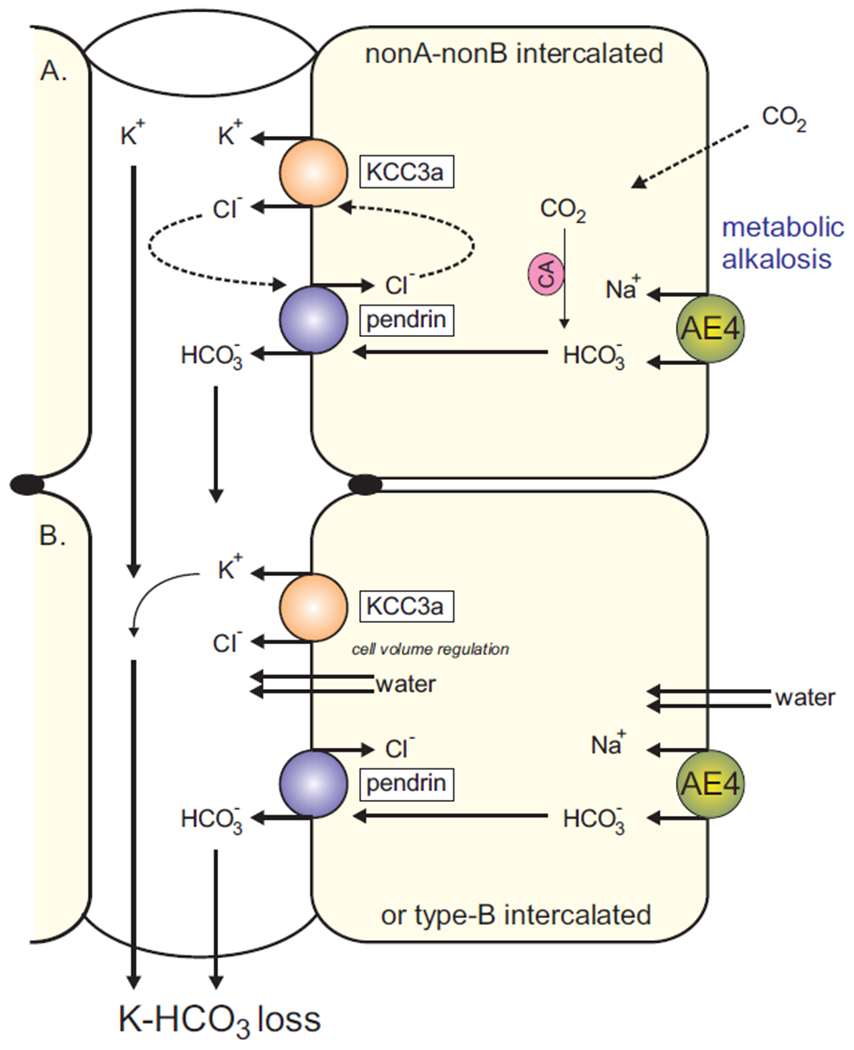
KCC3 is expressed at the apical membrane where it mediates the efflux on K+ and Cl−. A: Under conditions of bicarbonate excretion, the Cl− ion that enters the cell through the Cl−/HCO3− exchanger recycles through KCC3. This activity results into the excretion (loss) of K+. The Na-HCO3 cotransporter AE4 is a major pathway HCO3− entry in the cell. B: AE4 activity (import of ions) leads to cell swelling, while pendrin activity (exchange of ions) would be volume neutral. A role for KCC3a would be to maintain cell volume during bicarbonate transport. Cl− recycling and volume regulation are not mutually exclusive but complementary.
Our two models of Figure 2 assumed that the gradients support the efflux of K+ and Cl− (and water) into the lumen. According to microperfusion studies in the distal nephron, the luminal concentrations of K+ and Cl− in the distal nephron are 2 mM and 30-60 mM, respectively (referenced in (29)). Since the outside product of [K+] x [Cl−] is 20–30 times smaller than the inner product, it would require an excess of 40–50 mM luminal K+ to reverse the gradients. Therefore, transport through apical KCC3 is poised in the direction of secretion or K+ loss. If KCC3a facilitates the loss of K+, one would anticipate a decreased expression of KCC3a under low K+ diet. Against this expectation, we observed increased KCC3a abundance in mice fed with a K+ deficient diet. While this increase in KCC3a can be explained by the K+ deficient diet causing alkalosis (49, 50), it is surprising that under low K+ availability, the kidney would upregulate a transporter that facilitates the loss of K+. One could postulate that under these conditions the transporters are mostly silent. However, this would be inconsistent with recent data showing that when pendrin knockout mice are placed on a diet low in Na+, Cl− and K+, they excrete a far greater amount of K+ than wild-type mice (51*). Under normal settings, pendrin-KO mice can maintain a normal acid-base balance (52), but when they are maintained on a NaCl + K+ deficient diet, they experience severe metabolic alkalosis. The enhanced K+ excretion seen in pendrin-KO mice fed a diet deficient in both K+ and NaCl would therefore be consistent with additive or synergistic overexpression of the KCC3a.
Other transporters affecting pendrin function
Several studies have shown that the cystic fibrosis transmembrane conductance regulator (CFTR) interacts with pendrin possibly to stabilize the exchanger in the membrane (53–55). Under baseline conditions, deletion of CFTR has no impact on pendrin abundance (56), in contrast to deletion of KCC3 having a significant impact on pendrin (25**). Evidence suggests, however, that under base loading “stimulated” conditions, CFTR is required for proper pendrin function (56). Moreover, another study reported decreased apical expression and enhanced intracellular localization of pendrin in CFTR-KO mice compared with wild-type mice during bicarbonate loading (57). Thus, there seems to also be a functional link between the exchanger and the Cl− channel, maybe like the relationship between ROMK and NKCC2 in the thick ascending limb. Note that while CFTR might very well interact to facilitate pendrin function, the Cl− channel cannot explain the K+ loss observed in alkalemia.
A similar function had been assigned to the Na+ driven Cl−/HCO3− exchanger (NDCBE), which makes recycling of Cl− and HCO3− possible. NDCBE is necessary to preserve intravascular volume and Na+ balance when the body is depleted of salts (58). However, nonA and nonB intercalated cells do not express NDCBE and have higher pendrin abundance and activity (26, 59). As the abundance of KCC3a overlaps with pendrin in the cortex, it suggests expression of KCC3a in nonA/nonB intercalated cells; thus, the cotransporter is more likely to recycle Cl− than NDCBE. A net result of this operation would be KHCO3 secretion in a state of alkalemia. It appears, however, that pendrin can couple with a variety of transporters and, thus, a particular physiological need may dictate which transporter will functionally pair with pendrin. Further studies are required to identify these different physiological conditions and the corresponding transporters that pendrin couples with.
The entry pathway(s) of K+ and HCO3− into the type-B intercalated cells are still unclear. According to textbooks, proton pumps instead of Na+/K+ pumps power renal intercalated cells. However, the Na+/K+-ATPase is in fact expressed in intercalated cells (60) and thus, like in many other cells, K+ enters intercalated cells through the Na+/K+ pump. In contrast to type-A intercalated cells which express NKCC1 at the basolateral membrane (61, 62), type-B cells lack the cotransporter. Thus, aside from the pump, it is unclear what other transport pathways participate to the entry of K+ at the basolateral membrane of type-B intercalated cells. As far as bicarbonate is concerned, it can enter the cell as CO2 diffusion followed by hydration to HCO3 by carbonate anhydrases or enter through the Na+ dependent Cl−/HCO3− exchanger - AE4 (SLC4A9) (63). According to recent study, AE4 serves as both a HCO3− entry channel into type-B intercalated cells and a HCO3− sensor at the basolateral membrane (64**). It is possible that AE4 is as critical for the KCC3a function as it is for the pendrin function.
Regulation of KCC3a in the distal nephron
In our 2022 paper, we showed that deletion of KCC3 decreased the abundance of pendrin in mice (25**). This suggests that pendrin may be dependent on KCC3 function. Whether the reverse situation was true, i.e., deletion of pendrin affects KCC3 expression, had yet to be addressed. Furthermore, the mechanism by which alkalemia stimulates KCC3a expression was unknown. Are aldosterone and/or angiotensin-II involved in the process? Our most recent KCC3a study published in 2023 focused on these issues. In this paper, we showed that KCC3a abundance at the apical membrane of the intercalated cells increased after 24 hours and remained elevated after 7 days of NaHCO3 treatment (65*). Other study, however, has demonstrated that after 7 days of the HCO3− load, pendrin abundance returns to baseline, even though pendrin abundance initially increases with HCO3− (39). We also investigated whether KCC3a abundance changed when acute acidosis was induced via NH4Cl treatment. We observed that a 24-hour NH4Cl treatment had no effect on the abundance of KCC3a or pendrin, contrary to a previous observation that pendrin abundance decreased upon chronic NH4Cl treatment (66). This difference in pendrin behavior could be explained by differences in sample preparations: membrane protein preparation versus crude protein preparation, which contains both membrane and cytosolic proteins. Another possible explanation is that KCC3a abundance was already at its lowest in the baseline condition. However, we might observe the effect of NH4Cl on KCC3a when KCC3a expression is already upregulated.
Our work also demonstrated that KCC3a abundance was increased with NaHCO3, KHCO3 or K-citrate treatments (65*). K-citrate would function as KHCO3 because citrate enters the Krebs cycle, producing CO2 alongside ATP, leading to the formation of HCO3−. These results confirmed that it is the organic anion and not the cation that increases KCC3a abundance. As pendrin expression in intercalated cells is affected by aldosterone and angiotensin-II (67, 68), we addressed the role of these factors in KCC3a activation. We showed that KCl supplementation in the diet, which is known to stimulate aldosterone production, did not increase KCC3a abundance. KCC3a abundance is increased by 23 hours of water restriction, Na+ deficient diet, or otherwise known as contraction alkalosis (25**); however, angiotensin-II, a key and significant signaling molecule under these circumstances, may also contribute to the increase in KCC3a abundance (69). High salt diet suppresses angiotensin-II release (70), yet we found that HCO3− under either high salt diet or normal salt diets increased the abundance of KCC3a and pendrin, compared with high salt diet alone (65*). In addition, systemic suppression of angiotensin-II functions by blocking the angiotensin A1 receptor with losartan had no effect on KCC3a abundance on normal or Na+ deficient diets, thus ruling out any significant role for angiotensin-II in the increase in KCC3a abundance after 23 hours of water restriction or Na+ deficient diet (65*). Taken together, our findings indicate that the main signaling molecule is HCO3−, not aldosterone or angiotensin-II. Experiments performed in mouse cortical M-1 cells, which express the intercalated cell markers pendrin, V-ATPase, and Gpr116 (71, 72) showed that KCC3a expression in the cells was significantly increased in bicarbonate-containing culture media, compared to HEPES-containing culture media, whereas pH had no effect (65*). Thus, HCO3− seems to be the primary signaling molecule responsible for the increased KCC3a abundance.
In contrast to KCC3b, whose mRNA levels are increased in hyperglycemia (28), the increase in KCC3a expression observed in alkalemia does not involve transcription (65*). In fact, despite the significant increase in protein abundance, KCC3a mRNA levels remained unchanged in the kidney of mice fed with bicarbonate. This observation suggests alterations in protein translation or, more likely, changes in protein degradation. This discovery presents a new avenue for further investigation.
The lack of reliable antibodies against phosphorylated KCC3 poses a challenge in studying KCC3 function. The availability of a KCC3 knockout mouse model should in principle help confirming the involvement of KCC3 in K+ loss in alkalemia. However, due to the severe peripheral neuropathy disorder, we lost two cohorts of KCC3 knockout mice in metabolic cages. As we developed a conditional KCC3 knockout mouse model (73), we should be able to drive KCC3 deletion specifically in intercalated cells. One important confounding factor is ROMK, which constitutes a major pathway for K+ secretion (74). As mentioned above, whether the contribution of KCC3 to K+ loss will be significant enough to challenge the contribution of ROMK is an important question to answer. Maybe KCC3 function will be most relevant when ROMK participation in K+ loss is minimal, e.g., in conditions of low Na+ delivery to the distal nephron. This is consistent with the elegant study of Charles Wingo who provided evidence for cortical collecting tubule K+ and Cl− secretion in rabbits adapted to a KHCO3-rich and NaCl-deficient diet (75**).
In conclusion, two separate isoforms of KCC3 are expressed in kidney: KCC3a and KCC3b. Early work showed abundant KCC3b transcript, while barely detectable KCC3a mRNA in mouse kidney. Recent work by us identified the site of KCC3a expression: the apical membrane of type-B intercalated cells. Our work showed that KCC3a expression is stimulated by contraction alkalosis and metabolic alkalosis, thereby constituting a pathway for non-conductive K+ secretion in alkalemia. This discovery opens new avenues of research but also an opportunity to develop drugs that would minimize excessive loss of K+ in alkalemia.
Distinct KCC3 transporters are expressed in the kidney cortex: KCC3b in proximal tubule and KCC3a in distal tubule.
KCC3a is observed alongside pendrin at the apical membrane of type-B (or nonA/nonB) intercalated cells.
KCC3a expression is stimulated by contraction alkalosis and metabolic alkalosis.
KCC3a mediates K+ secretion (or K+ loss) in alkalemia.
Bicarbonate itself seems to be the major factor leading to increased KCC3a protein expression.
Financial support and sponsorship
This work was supported by NIH grant: DK093501, Leducq foundation grant: 17CVD05, and the B.H. Robbins Endowed Directorship from the Department of Anesthesiology at Vanderbilt University to E.D.
Footnotes
Conflicts of interest
The authors declare no conflicts of interest.
References
- 1.Wong FH, Chen JS, Reddy V, et al. The amino acid-polyamine-organocation superfamily. J Mol Microbiol Biotechnol. 2012;22(2):105–13. [DOI] [PubMed] [Google Scholar]
- 2.Lauf PK, Theg BE. A chloride dependent K+ flux induced by N-ethylmaleimide in genetically low K+ sheep and goat erythrocytes. Biochem Biophys Res Commun. 1980;70:221–42. [DOI] [PubMed] [Google Scholar]
- 3.Dunham PB, Steward GW, Ellory JC. Chloride-activated passive potassium transport in human erythrocytes. Proc Natl Acad Sci USA 1980;77:1711–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lauf PK. Thiol-dependent passive K/Cl transport in sheep red cells. IV. Furosemide inhibition as a function of external Rb+, Na+, and Cl−. J Membrane Biol. 1984;77:57–62. [DOI] [PubMed] [Google Scholar]
- 5.Moore-Hoon ML, Turner RJ. The structural unit of the secretory Na+-K+-2Cl− cotransporter (NKCC1) is a homodimer. Biochemistry. 2000;39(13):3718–24. [DOI] [PubMed] [Google Scholar]
- 6.Ding J, Ponce-Coria J, Delpire E. A trafficking-deficient mutant of KCC3 reveals dominant-negative effects on K-Cl cotransport function. PLoS One. 2013;8(4):e61112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chew TA, Orlando BJ, Zhang J, et al. Structure and mechanism of the cation-chloride cotransporter NKCC1. Nature. 2019;572(7770):488–92. [DOI] [PMC free article] [PubMed] [Google Scholar]; (**) First cryo-EM structure of a member of the cation-chloride (SLC12) cotransporter family. The overall structure and ion binding sites of zebrafish NKCC1 is revealed.
- 8.Moseng MA, Su CC, Rios K, et al. Inhibition mechanism of NKCC1 involves the carboxyl terminus and long-range conformational coupling. Sci Adv. 2022;8(43):eabq0952. [DOI] [PMC free article] [PubMed] [Google Scholar]; (*) Cryo-EM strcuture of human NKCC1 in complex with furosemide or bumetanide.
- 9.Liu S, Chang S, Han B, et al. Cryo-EM Structures of the Human Cation-Chloride Cotransporter KCC1. Science. 2019;366:505–8. [DOI] [PubMed] [Google Scholar]; (**) First cryo-EM structure of a K-Cl cotransporter revealing the existence (conservation) of two chloride binding sites. As K-Cl cotransport mediates the strict movement of 1 K+:1 Cl− per transport cycle (see referencve 16), this key observation indicates that one site is occupied but does not participate in transport. This observation is consistent with kinetics analyses of red blood cell K-Cl cotransport performed in the early 1990s.
- 10.Chi X, Li X, Chen Y, et al. Cryo-EM structures of the full-length human KCC2 and KCC3 cation-chloride cotransporters. Cell Res. 2020;31(4):482–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xie Y, Chang S, Zhao C, et al. Structures and an activation mechanism of human potassium-chloride cotransporters. Science Adv. 2020;6:eabc5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pan D, Kalfa TA, Wang D, et al. K-Cl cotransporter gene expression during human and murine erythroid differentiation. J Biol Chem. 2011;286(35):30492–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu J, Karadsheh M, Delpire E. Developmental regulation of the neuronal-specific isoform of K-Cl cotransporter KCC2 in postnatal rat brains. J Neurobiol. 1999;39:558–68. [PubMed] [Google Scholar]
- 14.Pearson M, Lu J, Mount DB, Delpire E. Localization of the K-Cl cotransporter, KCC3, in the central and peripheral nervous systems: expression in choroid plexus, large neurons, and white matter tracts. Neuroscience. 2001;103(2):483–93. [DOI] [PubMed] [Google Scholar]
- 15.Casula S, Shmukler BE, Wilhelm S, et al. A dominant negative mutant of the KCC1 K-Cl cotransporter: both N- and C-terminal cytoplasmic domains are required for K-Cl cotransport activity. J Biol Chem. 2001;276(45):41870–8. [DOI] [PubMed] [Google Scholar]
- 16.Jennings ML, Adame MF. Direct estimate of 1:1 stoichiometry of K(+)-Cl(−) cotransport in rabbit erythrocytes. Am J Physiol (Cell Physiol). 2001;281(3):C825–C32. [DOI] [PubMed] [Google Scholar]
- 17.Jennings ML, Schultz RK. Okadaic acid inhibition of KCl cotransport. Evidence that protein dephosphorylation is necessary for activation of transport by either swelling or N-ethylmaleimide. J Gen Physiol 1991;97:799–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jennings ML, al-Rohil N. Kinetics of activation and inactivation of swelling-stimulated K+/Cl− transport. The volume-sensitive parameter is the rate constant for inactivation. J Gen Physiol. 1990;95:1021–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Piechotta K, Lu J, Delpire E. Cation-chloride cotransporters interact with the stress-related kinases SPAK and OSR1. J Biol Chem. 2002;277(52):50812–9. [DOI] [PubMed] [Google Scholar]
- 20.Rinehart J, Maksimova YD, Tanis JE, et al. Sites of regulated phosphorylation that control K-Cl cotransporter activity. Cell. 2009;138(3):525–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mount DB, Mercado A, Song L, et al. Cloning and Characterization of KCC3 and KCC4, new members of the cation-chloride cotransporter gene family. J Biol Chem. 1999;274:16355–62. [DOI] [PubMed] [Google Scholar]
- 22.Race JE, Makhlouf FN, Logue PJ, et al. Molecular cloning and functional characterization of KCC3, a new K-Cl cotransporter. Am J Physiol (Cell Physiol). 1999;277:C1210–C9. [DOI] [PubMed] [Google Scholar]
- 23.Hiki K, D’Andrea RJ, Furze J, et al. Cloning, characterization, and chromosomal location of a novel human K+-Cl− cotransporter. J Biol Chem. 1999;274:10661–7. [DOI] [PubMed] [Google Scholar]
- 24.Mercado A, Vazquez N, Song L, et al. NH2-terminal heterogeneity in the KCC3 K+-Cl− cotransporter. Am J Physiol Renal Physiol. 2005;289(6):F1246–F61. [DOI] [PubMed] [Google Scholar]
- 25.Ferdaus MZ, Terker AS, Koumangoye R, Delpire E. KCC3a, a strong candidate pathway for K+ loss in alkalemia. Front Cell Dev Biol. 2022;10:931326. [DOI] [PMC free article] [PubMed] [Google Scholar]; (**) Original observation of KCC3a expression at the apical membrane of type-B intercalated cells and stimulation by metabolic alkalosis.
- 26.Kim YH, Kwon TH, Frische S, et al. Immunocytochemical localization of pendrin in intercalated cell subtypes in rat and mouse kidney. Am J Physiol Renal Physiol. 2002;283(4):F744–F54. [DOI] [PubMed] [Google Scholar]
- 27.Garneau AP, Marcoux AA, Noël M, et al. Ablation of Potassium-Chloride Cotransporter Type 3 (Kcc3) in Mouse Causes Multiple Cardiovascular Defects and Isosmotic Polyuria. PLoS One. 2016;11(5):e0154398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Melo Z, Cruz-Rangel S, Bautista R, et al. Molecular evidence for a role for K(+)-Cl(−) cotransporters in the kidney. Am J Physiol Renal Physiol. 2013;305(10):F1402–F11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weinstein AM. A mathematical model of rat distal convoluted tubule. I. Cotransporter function in early DCT. Am J Physiol Renal Physiol. 2005;289(4):F699–F720. [DOI] [PubMed] [Google Scholar]
- 30.Star RA, Burg MB, Knepper MA. Bicarbonate secretion and chloride absorption by rabbit cortical collecting ducts. Role of chloride/bicarbonate exchange. J Clin Invest. 1985;76(3):1123–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weiner ID, Weill AE, New AR. Distribution of Cl−/HCO3− exchange and intercalated cells in rabbit cortical collecting duct. Am J Physiol. 1994;267(6 Pt 2):F952–F64. [DOI] [PubMed] [Google Scholar]
- 32.Weiner ID, Hamm LL. Regulation of intracellular pH in the rabbit cortical collecting tubule. J Clin Invest. 1990;85(1):274–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Emmons C Transport characteristics of the apical anion exchanger of rabbit cortical collecting duct beta-cells. Am J Physiol. 1999;276(4):F635–F43. [DOI] [PubMed] [Google Scholar]
- 34.Emmons C, Kurtz I. Functional characterization of three intercalated cell subtypes in the rabbit outer cortical collecting duct. J Clin Invest. 1994;93(1):417–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Verlander JW, Hassell KA, Royaux IE, et al. Deoxycorticosterone upregulates PDS (Slc26a4) in mouse kidney: role of pendrin in mineralocorticoid-induced hypertension. Hypertension. 2003;42(3):356–62. [DOI] [PubMed] [Google Scholar]
- 36.Verlander JW, Kim YH, Shin W, et al. Dietary Cl(−) restriction upregulates pendrin expression within the apical plasma membrane of type B intercalated cells. Am J Physiol Renal Physiol. 2006;291(4):F833–F9. [DOI] [PubMed] [Google Scholar]
- 37.Mohebbi N, Perna A, van der Wijst J, et al. Regulation of two renal chloride transporters, AE1 and pendrin, by electrolytes and aldosterone. PLoS One. 2013;8(1):e55286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Frische S, Kwon TH, Frøkiaer J, et al. Regulated expression of pendrin in rat kidney in response to chronic NH4Cl or NaHCO3 loading. Am J Physiol Renal Physiol. 2003;284(3):F584–F93. [DOI] [PubMed] [Google Scholar]
- 39.Wagner CA, Finberg KE, Stehberger PA, et al. Regulation of the expression of the Cl−/anion exchanger pendrin in mouse kidney by acid-base status. Kidney Int. 2002;62(6):2109–17. [DOI] [PubMed] [Google Scholar]
- 40.Atkins JL, Burg MB. Bicarbonate transport by isolated perfused rat collecting ducts. Am J Physiol. 1985;249(4 Pt 2):F485–F9. [DOI] [PubMed] [Google Scholar]
- 41.Engelking LR. Metabolic Alkalosis. Textbook of Veterinary Physiological Chemistry: Academic Press; 2015. p. 576–83. [Google Scholar]
- 42.Aronson PS, Giebisch G. Effects of pH on Potassium: New Explanations for Old Observations. J Am Assoc Nephrol. 2011;22:1981–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pech V, Pham TD, Hong S, et al. Pendrin modulates ENaC function by changing luminal HCO3. J Am Soc Nephrol. 2010;21(11):1928–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Khuri RN, Strieder WN, Giebisch G. Effects of flow rate and potassium intake on distal tubular potassium transfer. Am J Physiol. 1975;228(4):1249–61. [DOI] [PubMed] [Google Scholar]
- 45.Satlin LM, Carattino MD, Liu W, Kleyman TR. Regulation of cation transport in the distal nephron by mechanical forces. Am J Physiol Renal Physiol. 2006;291(5):F923–F31. [DOI] [PubMed] [Google Scholar]
- 46.Al-Qusairi L, Grimm R, Zapf AM, et al. Dietary Anion Controls Potassium Excretion: It’s More Than a Poorly Absorbable Anion Effect. J Am Assoc Nephrol. 2022;33:FR-OR42. [Google Scholar]
- 47.Procino G, Milano S, Tamma G, et al. Co-regulated pendrin and aquaporin 5 expression and trafficking in Type-B intercalated cells under potassium depletion. Cell Physiol Biochem. 2013;32(7):184–99. [DOI] [PubMed] [Google Scholar]
- 48.Rodighiero S, Bottà G, Bazzini C, Meyer G. Pendrin overexpression affects cell volume recovery, intracellular pH and chloride concentration after hypotonicity-induced cell swelling. Cell Physiol Biochem. 2011;28(3):559–70. [DOI] [PubMed] [Google Scholar]
- 49.Lee Hamm L, Hering-Smith KS, Nakhoul NL. Acid-base and potassium homeostasis. Semin Nephrol. 2013;33(3):257–64. [DOI] [PubMed] [Google Scholar]
- 50.Han KH. Mechanisms of the effects of acidosis and hypokalemia on renal ammonia metabolism. Electrolyte Blood Press. 2011;9(2):45–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pham TD, Elengickal AJ, Verlander JW, et al. Pendrin-null mice develop severe hypokalemia following dietary Na(+) and K(+) restriction: role of ENaC. Am J Physiol Renal Physiol. 2022;322(5):F486–F97. [DOI] [PMC free article] [PubMed] [Google Scholar]; (*) One likely mechanism for the severe hypokalemia is KCC3a. If true, this would indicate that the function of the K-Cl cotransporter does not require pendrin function. The low salt conditions, or rather low Cl− conditions, would favor the driving force for K-Cl efflux (K+ loss) at the luminal membrane.
- 52.Kim YH, Verlander JW, Matthews SW, et al. Intercalated cell H+/OH− transporter expression is reduced in Slc26a4 null mice. Am J Physiol Renal Physiol. 2005;289(6):F1262–F72. [DOI] [PubMed] [Google Scholar]
- 53.Tamma G, Dossena S. Functional interplay between CFTR and pendrin: physiological and pathophysiological relevance. Front Biosci (Landmark Ed). 2022;27(2):75. [DOI] [PubMed] [Google Scholar]
- 54.Ko SB, Zeng W, Dorwart MR, et al. Gating of CFTR by the STAS domain of SLC26 transporters. Nat Cell Biol. 2004;6(4):343–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chang MH, Plata C, Sindic A, et al. Slc26a9 is inhibited by the R-region of the cystic fibrosis transmembrane conductance regulator via the STAS domain. J Biol Chem. 2009;284(41):28306–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Berg P, Svendsen SL, Hoang TTL, et al. Impaired renal HCO(3) (−) secretion in CFTR deficient mice causes metabolic alkalosis during chronic base-loading. Acta Physiol (Oxf). 2021;231(3):e13591. [DOI] [PubMed] [Google Scholar]
- 57.Varasteh Kia M, Barone S, McDonough AA, et al. Downregulation of the Cl−/HCO3− Exchanger Pendrin in Kidneys of Mice with Cystic Fibrosis: Role in the Pathogenesis of Metabolic Alkalosis. Cell Physiol Biochem. 2018;45(4):1551–65. [DOI] [PubMed] [Google Scholar]
- 58.Sinning A, Radionov N, Trepiccione F, et al. Double Knockout of the Na+-Driven Cl−/HCO3− Exchanger and Na+/Cl− Cotransporter Induces Hypokalemia and Volume Depletion. J Am Soc Nephrol. 2017;28(1):130–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Roy A, Al-bataineh MM, Pastor-Soler NM. Collecting duct intercalated cell function and regulation. Clin J Am Soc Nephrol. 2015;10(2):305–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sabolić I, Herak-Kramberger CM, Breton S, Brown D. Na/K-ATPase in intercalated cells along the rat nephron revealed by antigen retrieval. J Am Soc Nephrol. 1999;10(5):913–22. [DOI] [PubMed] [Google Scholar]
- 61.Ginns SM, Knepper MA, Ecelbarger CA, et al. Immunolocalization of the secretory isoform of Na-K-Cl cotransporter in rat renal intercalated cells. J Am Soc Nephrol. 1996;7(12):2533–42. [DOI] [PubMed] [Google Scholar]
- 62.Wall SM, Fischer MP, Mehta P, et al. Contribution of the Na+-K+-2Cl− cotransporter NKCC1 to Cl− secretion in rat OMCD. Am J Physiol Renal Physiol. 2001;280(5):F913–F21. [DOI] [PubMed] [Google Scholar]
- 63.Hentschke M, Hentschke S, Borgmeyer U, et al. The murine AE4 promoter predominantly drives type B intercalated cell specific transcription. Histochem Cell Biol. 2009;132(4):405–12. [DOI] [PubMed] [Google Scholar]
- 64.Vitzthum H, Koch M, Eckermann L, et al. The AE4 transporter mediates kidney acid-base sensing. Nat Commun. 2023;14(1):3051. [DOI] [PMC free article] [PubMed] [Google Scholar]; (**) This key study reveals that contrary to previously published models, the Na-HCO3 cotransporter AE4 functions as an importer of Na+ and HCO3− at the base of type-B intercalated cells. This has enormous consequence as its activity is now key to HCO3− secretion, and its activity likely results into swelling of the cell which then.
- 65.Ferdaus MZ, Terker AS, Koumangoye R, et al. Bicarbonate is the primary inducer of KCC3a expression in renal cortical B-type intercalated cells. Am J Physiol Cell Physiol. 2023;324(5):C1171–C8. [DOI] [PMC free article] [PubMed] [Google Scholar]; (*) As the title of the paper states, in this study, we demonstrated that KCC3a expression was not affected by factors like aldosterone or angiotensin-II but was solely responsive to changes in bicarbonate itself. The mechanism of stimulation is unknown but does not involves transcription.
- 66.Hafner P, Grimaldi R, Capuano P, et al. Pendrin in the mouse kidney is primarily regulated by Cl− excretion but also by systemic metabolic acidosis. Am J Physiol Cell Physiol. 2008;295(6):C1658–C67. [DOI] [PubMed] [Google Scholar]
- 67.Pham TD, Verlander JW, Wang Y, et al. Aldosterone Regulates Pendrin and Epithelial Sodium Channel Activity through Intercalated Cell Mineralocorticoid Receptor-Dependent and -Independent Mechanisms over a Wide Range in Serum Potassium. J Am Soc Nephrol. 2020;31(3):483–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Verlander JW, Hong S, Pech V, et al. Angiotensin II acts through the angiotensin 1a receptor to upregulate pendrin. Am J Physiol Renal Physiol. 2011;301(6):F1314–F25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Harrison-Bernard LM. The renal renin-angiotensin system. Adv Physiol Educ. 2009;33(4):270–4. [DOI] [PubMed] [Google Scholar]
- 70.Drenjančević-Perić I, Jelaković B, Lombard JH, et al. High-salt diet and hypertension: focus on the renin-angiotensin system. Kidney Blood Press Res. 2011;34(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Adler L, Efrati E, Zelikovic I. Molecular mechanisms of epithelial cell-specific expression and regulation of the human anion exchanger (pendrin) gene. Am J Physiol Cell Physiol. 2008;294(5):C1261–C76. [DOI] [PubMed] [Google Scholar]
- 72.Kui M, Pluznick JL, Zaidman NA. The transcription factor Foxi1 promotes expression of V-ATPase and Gpr116 in M-1 cells. Am J Physiol Renal Physiol. 2023;324(3):F267–F73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ding J, Delpire E. Deletion of KCC3 in parvalbumin neurons leads to locomotor deficit in a conditional mouse model of peripheral neuropathy associated with agenesis of the corpus callosum. Behav Brain Res. 2014;274C:128–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Welling PA, Ho K. A comprehensive guide to the ROMK potassium channel: form and function in health and disease. Am J Physiol Renal Physiol. 2009;297(4):F849–F63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wingo CS. Potassium secretion by the cortical collecting tubule: effect of Cl gradients and ouabain. Am J Physiol Renal Physiol. 1989;256:F303–13. [DOI] [PubMed] [Google Scholar]; (**) While this paper was published a long time ago, it is truly remarkable in the context of this review. This study clearly provides early evidence of an electroneutral K+ transport mechanism at the apical membrane of cortical collecting duct cells, a transporter that we have now identified as KCC3a. In addition, it independently confirms (as we were not aware of it at the time of our study) the physiological conditions under which KCC3a is likely most relevant: alkalemic and low salt conditions.