

Graphical abstract

Keywords: Nanopore, Structural variant, Third-generation sequencing, SERPINC1
Highlights
-
•
Structural variants (SVs) calling tools are compared using real data.
-
•
We compare at the genome level the SVs detected by nanopore sequencing and aCGH.
-
•
We use coverage data for polishing calls and improve consensus.
-
•
Our method contributes to identify SVs and filtering out erroneous SV calls.
Abstract
Introduction
Whole-genome sequencing using nanopore technologies can uncover structural variants, which are DNA rearrangements larger than 50 base pairs. Nanopore technologies can also characterize their boundaries with single-base accuracy, owing to the kilobase-long reads that encompass either full variants or their junctions. Other methods, such as next-generation short read sequencing or PCR assays, are limited in their capabilities to detect or characterize structural variants. However, the existing software for nanopore sequencing data analysis still reports incomplete variant sets, which also contain erroneous calls, a considerable obstacle for the molecular diagnosis or accurate genotyping of populations.
Methods
We compared multiple factors affecting variant calling, such as reference genome version, aligner (minimap2, NGMLR, and lra) choice, and variant caller combinations (Sniffles, CuteSV, SVIM, and NanoVar), to find the optimal group of tools for calling large (>50 kb) deletions and duplications, using data from seven patients exhibiting gross gene defects on SERPINC1 and from a reference variant set as the control. The goal was to obtain the most complete, yet reasonably specific group of large variants using a single cell of PromethION sequencing, which yielded lower depth coverage than short-read sequencing. We also used a custom method for the statistical analysis of the coverage value to refine the resulting datasets.
Results
We found that for large deletions and duplications (>50 kb), the existing software performed worse than for smaller ones, in terms of both sensitivity and specificity, and newer tools had not improved this. Our novel software, disCoverage, could polish variant callers’ results, improving specificity by up to 62% and sensitivity by 15%, the latter requiring other data or samples.
Conclusion
We analyzed the current situation of >50-kb copy number variants with nanopore sequencing, which could be improved. The methods presented in this work could help to identify the known deletions and duplications in a set of patients, while also helping to filter out erroneous calls for these variants, which might aid the efforts to characterize a not-yet well-known fraction of genetic variability in the human genome.
Introduction
Structural variants (SVs) are heterogeneous gross genetic defects spanning more than 50 base pairs (bp) that include copy number variations (CNVs), which can be either duplications or deletions; and other rearrangements of DNA: insertions, inversions, and translocations [1]. Despite the number of SVs in the genome being smaller than that of single nucleotide variations (SNVs) and small insertions or deletions (indels), SVs account for a larger number of variable bases [2] and are more likely to be pathogenic than SNVs or indels [3]. Furthermore, the importance of SVs could still be underestimated because of the limitations of the current molecular detection methods [4]. Thus, there is an increasing interest to identify and characterize SVs, particularly in biomedicine.
Current molecular algorithms used to identify pathogenic variants first recommend to rule out SNVs and indels by using short-read sequencing [5]. SVs are usually screened in cases with no SNVs or indels found. Nowadays, there is a relatively large range of methods used for SV detection, both specific to a gene/region (targeted scan) and genome-wide. Most of the specific methods are based on PCR, such as a) long-range PCR, in which a high-fidelity, high-processivity polymerase amplifies a region in the order of kilobases (kb), and the fragments are then sequenced [6]; b) real-time qPCR, where the results of a case are compared to those of the control or to standard curves to determine the number of copies of a sequence [7]; c) multiple ligand probe amplification (MLPA), which uses labeled probes, so multiple regions can be amplified, and after amplification, the product is proportional to the gene dosage, so again a control is needed for comparison [7]; and d) fluorescence in situ hybridization (FISH), which uses labeled probes that hybridize against their target, without any amplification. For resolutions in the range of kilobases, however, probes must be used on stretched chromosome fibers, and experiments on multiple targets are costly and time-consuming [7]. Non-specific, genome-wide techniques include methods such as a) Giemsa-banded karyotyping, for chromosomal alterations [8]; b) comparative genome hybridization (CGH), which uses fluorescent labels to quantify a sample against a control, and was adapted to microarrays from [9] single nucleotide polymorphism (SNP) arrays, used in a similar manner [7]; and c) optical genome mapping (OGM), which measures the distance between labeled probes to detect CNVs [10].
All of these methods present limitations to detect and characterize SVs, such as scope, resolution, or the ability to discover novel mutations, and none of them reaches nucleotide resolution. Next-generation sequencing (NGS) also detects SVs; however, it lacks sensitivity and fails to detect 30%–90% of the SVs, while still producing high false positive rates [11], [12], despite recent applications reporting 86% sensitivity for CNVs and 100% for > 10-kb CNVs [12].
The development of third-generation sequencing technologies, such as PacBio and Nanopore, has improved SV detection by means of their kilobase-long reads, at the cost of a lower per-base accuracy [1]. PacBio uses a DNA polymerase, a circular template with the sequence of interest inserted, and fluorescently labeled deoxyribonucleotide triphosphates (dNTPs) to produce reads in the range of tens of kilobases [13]. In each polymerization step, the fluorescent probe is excited, its emission recorded, and finally cleaved before the next dNTP is added. In contrast, nanopore sequencing does not rely on DNA polymerization; instead, linear molecules of DNA are attached to an adapter and a motor protein. These motor proteins unwind the DNA molecules, which go through pores embedded on a membrane, disrupting the ionic current in the pores. These disruptions are measured and analyzed to determine the nucleotide sequence in real time [13]. The resulting reads can reach several hundreds of kilobases in length, even megabases (Mb). A key advantage of these sequencing technologies is the ability to locate the boundaries of SVs with single base resolution, which is valuable information for running confirmation experiments.
Numerous efforts have been made to develop analysis tools for SV detection using data from Nanopore and PacBio [14]. Unfortunately, the limitations of these methods still cause a fraction of SVs to go undiscovered, while reporting false positives [1], [15], [16]. As we demonstrate, this seems to be worse for larger (>50 kb) SVs. While these variants may be less common, they are linked to neurological and rare diseases [17], are associated to blood serum levels of biological compounds [18] and may be linked to adaptations to different environments of human populations [19]; therefore, detecting them accurately is of scientific interest.
In this work, we focused on improving the identification of large CNVs by using one of the abovementioned technologies, nanopore sequencing. To achieve this, we explored different pipelines varying in software and reference data to examine which one generated the best set of SV calls. Then, we developed a new tool that analyzed the coverage data, allowing us to further polish calling and improve consensus with other techniques. We applied this method to a group of patients carrying different pathogenic SVs. The results were compared with those obtained by applying CGH.
Our case study considered seven patients with antithrombin (AT) deficiency, which is an autosomal, monogenic, dominant disorder that significantly increases the risk of thrombosis. AT deficiency is mainly caused by defects in the coding gene (SERPINC1), mostly SNVs or small indels [5]. However, up to 5% of the AT deficiency cases with a molecular diagnosis are caused by SVs. Thus, AT deficiency, as a monogenic disease already linked to SVs, is an interesting disease as a starting point for an SV detection study, as it presents a clear genotype and the genetic cause is located at a particular gene. Most SVs causing AT deficiency are deletions with variable length, but duplications and retrotransposon insertions have also been found [20].
SVs causing AT deficiency are usually detected by MLPA, although this method fails to detect some of the SVs causing AT deficiency [21]. Furthermore, the length of the defect is not precisely defined, and no nucleotide resolution is obtained. Thus, in order to cover this limitation, the samples were evaluated using CGH. CGH was initially introduced as a genome-wide method to detect copy number gains and losses larger than 10 Mb [22], but it has evolved to an array format (aCGH) that uses probes covering the human genome [23].
In this study, by comparing at the genome level, CNVs detected by aCGH and nanopore sequencing in patients with the AT deficiency caused by a CNV affecting SERPINC1, we found that the performance for detecting variants with a size greater than 50 kb was worse. We tested this on a total of three aligners and four variant callers, resulting in 11 variant sets per patient. To check these results, we used a publicly available and extensive dataset as a benchmark for our pipeline. With these results in mind, here, we present a novel coverage analysis tool, disCoverage, which might help in detecting large CNVs.
Materials and methods
Patient data
The study was conducted on seven patients with thrombosis that had AT quantitative type I deficiency caused by a CNV affecting SERPINC1. These SVs were detected by MLPA after negative results of sequencing the whole gene [24]. The clinical, genetic, and biochemical information of these patients is presented in Table 1. P1-4 and P6-7 were described previously [21], with corresponding identifiers P3, P4, P7, P1, P6, and P8; and P1-7 also were described previously [25], with corresponding identifiers P6, P3, P24, P25, P16, P35, and P20, respectively.
Table 1.
Descriptive information of the study participants. Column 5 indicates whether the mutation affecting SERPINC1 was found with aCGH. The last three columns indicate whether CuteSV, NanoVar, Sniffles, and SVIM, in this order, found the mutation affecting SERPINC1 after the indicated aligner. Abbreviations: DVT: deep venous thromboembolism, CSVT: central sinovenous thrombosis WGD: whole SERPINC1 gene deletion, ex: exon, DEL: deletion, DUP: duplication. Column 5 indicates whether the mutation affecting SERPINC1 was found with aCGH. NA19240 is external to the study and does not suffer, to the best of our knowledge, AT-deficiency. Anti-FXa is “anti-factor Xa activity”, a measure of antithrombin activity expressed as a percentage of the reference value obtained in a pool of 100 healthy blood donors.
| lra | Minimap2 | NGMLR | ||||||
|---|---|---|---|---|---|---|---|---|
| Patient | Thrombosis | Anti-FXa (%) | MLPA | Detected by aCGH | CNVs detected by aCGH | CuteSV/Sniffles/SVIM | CuteSV/NanoVar/Sniffles/SVIM | CuteSV/NanoVar/Sniffles/SVIM |
| P1 | DVT (21 y.o.) | 42 | WGD | N | 11 | N, N, N | Y, Y, Y, Y | Y, Y, Y, Y |
| P2 | ND | 45 | WGD | Y | 10 | N, N, N | N, N, N, Y | N, N, Y, Y |
| P3 | ND | 45 | DEL ex. 1–5 | Y | 14 | N, N, N | N, N, Y, Y | Y, N, Y, Y |
| P4 | CSVT | 30 | DEL ex. 1 | N | 11 | Y, Y, N | Y, Y, Y, Y | Y, Y, Y, Y |
| P5 | Arterial | 57 | WGD | N | 7 | N, N, N | Y, N, Y, Y | N, N, Y, Y |
| P6 | DVT | 61 | DUP ex. 1–5 | N | 17 | N, N, N | N, N, N, N | N, N, N, N |
| P7 | DVT | 52 | DEL ex. 2–5 | N | 14 | Y, Y, Y | N, Y, N, Y | Y, Y, Y, Y |
Ethics statement
All included subjects gave their informed consent to enter the study, which was approved by the Ethics Committee (EST 31/18) of Morales Meseguer Hospital, and performed in accordance with the 1964 Declaration of Helsinki and their later amendments.
Genetic analysis
Long-read whole-genome nanopore sequencing (LR WGS) was performed using a PromethION device. Basecalling was performed with Guppy (3.0.4 e7dbc23 to 3.2.8 bd67289). P3 and P6 runs were inefficient because of the low pore number during sequencing. For P3, a second run was conducted, and both sets of results were combined.
Array comparative genome hybridization (aCGH) was performed with high-density CytoScan® HD Array (Thermo Fisher Scientific, Inc.), which includes 2.67 million markers for copy number analysis, 750,000 SNP probes, and 1.9 million non-polymorphic probes. Following hybridization, a laser scanner (GeneChip® 3000 Scanner) was used for scanning the arrays, and the images were extracted and analyzed using the Affymetrix Gene Chip Command Console software (version 4.0) and the Chromosome analysis software (ChAS v.1.2/na33.2) (Fisher Scientific, Inc.), and interpreted with the aid of the UCSC genome browser [26].
Nanopore data processing
The data processing pipeline for this study consisted of the following steps: (1) alignment against a reference human genome, (2) variant calling, and (3) variant filtering and merging. After these steps, (4) the obtained variants were compared with those obtained using aCGH and (5) a coverage analysis was performed. We will describe these steps next, and their connection is shown in Fig. 1. The relevant tools are summarized in Supplementary Table 2.
Fig. 1.

Flowchart describing the steps for SV analysis done in this work. Nanopore reads from our cases, and from a control dataset from an individual whose SVs have been extensively characterized by Chaisson et al., 2019, were aligned using three different programs. The alignments generated, three per sample, were analyzed by four variant callers (except the combination of lra and NanoVar, which do not seem to be compatible). Then, the four (or three, for lra) SV sets based on each aligner were merged using SURVIVOR. This set could then be compared against aCGH, for our samples, or against the truth set, for NA19240. We then checked whether the coverage could be used to support CNVs, particularly the ones that were not found by callers. To deal with aCGH coordinates, which were only available for the hg19 reference, the alignment and variant calling were repeated for our samples.
Table 2.
Individual sequencing statistics for the study cases, and NA19240.
| Patient | Mean read length |
Percentage of genome covered (MAPQ ≥ 20) (lra/minimap2/NGMLR) |
Median and (Q3-Q1) coverage (MAPQ ≥ 20) (lra/minimap2/NGMLR) |
|---|---|---|---|
| P1 | 5387.96 | 92/92/92 | 17 (20–13)/18 (21–14)/17 (21–13) |
| P2 | 7508.66 | 93/92/92 | 25 (29–20)/26 (30–21)/25 (30–20) |
| P3 | 3955.37 | 93/92/92 | 15 (18–12)/16 (20–13)/16 (19–12) |
| P4 | 7331.80 | 93/93/92 | 24 (29–19)/25 (29–20)/25 (29–19) |
| P5 | 6369.04 | 93/92/92 | 18 (21–14)/18 (22–14)/18 (21–14) |
| P6 | 3795.30 | 92/91/91 | 5 (6–3)/5 (7–3)/5 (7–3) |
| P7 | 6166.71 | 93/92/91 | 13 (16–10)/14 (17–11)/14 (16–10) |
| NA19240 | 14,565 | 93/92/91 | 16 (19–13)/17 (20–13)/15 (18–11) |
In this work, three different programs were used for alignment in combination with up to four variant callers. Regarding the aligners, three were used: NGMLR [27] 0.2.7, build from July 2, 2018; minimap2 [28] 2.17-r941; and lra [29]. The first two are recommendations for sensitivity and speed [1], and the latter is included in the latest Oxford Nanopore Technologies’ (ONT) recommendations.
Alignments were performed against hg38 [30] (which includes some corrections [31], also explained here [1]) for the three aligners, and hg19 [32] for minimap2 and NGMLR, to test specific variants. For sorting and indexing the alignment files, samtools [33] 1.9 was used for minimap2 and NGMLR (to maintain compatibility with NGMLR), and 1.12 for lra.
SVs were called by Sniffles [27] 1.0.12 (github build cloned on February 10, 2021), SVIM [34] 1.4.0, CuteSV [35], and NanoVar [36]. Sniffles and SVIM were chosen to maximize sensitivity [1], CuteSV from the ONT latest recommendations and independent benchmarks [15], and NanoVar for its promising performance [36]. Sniffles, SVIM, and CuteSV only consider reads with a mapping quality of > 20, but as NanoVar does not apply such a filter, it was run on filtered BAM files to emulate this option. For hg19, only Sniffles and SVIM after minimap2 and NGMLR were used, as they performed the best with the SVs of interest in our study.
Call sets from Sniffles, CuteSV, and NanoVar were filtered using a minimum supporting reads threshold of 5, and those from SVIM using a quality value of 5. All were sorted and indexed by bcftools [33] 1.9. They were later merged using SURVIVOR [37]. The SVs were then limited to > 50-kb CNVs. The aCGH coordinates were translated from hg19 to hg38 with USCS liftOver [38]. Coordinate lifting’s limitations were taken into consideration; to account for them, the repetition of the workflow on hg19 allowed to compare the results and assess whether the differences truly came from the lifting. To compare the considered techniques, as aCGH was less precise, a difference in size of 33% and a difference in coordinates of 3 Mb were allowed, although types had to match: a “Gain” from aCGH was called “DUP” (duplication) by nanopore sequencing, and a “Loss”, a “DEL” (deletion).
Sequencing statistics were calculated using NanoStat [39]. Coverage was measured using mosdepth [40] 0.3.1 by chromosome and on regions of interest with a mapping quality threshold of 20.
Data analysis was performed in R [41], with a series of packages for different purposes: for data preparation, data.table [42] and tidyverse [43]; for plotting, circlize [44] and ggplot2 [45]; and stringi [46] for text operations.
Statistical analysis of coverage using disCoverage
The tool presented in this work, disCoverage, acted as a wrapper for the coverage calculation and then processed the coverage data. It relied on SV’s coordinates retrieved from the variant callers listed in the previous section, or from other techniques, such as aCGH, which was used in this work.
The per-base coverage of SVs and their surroundings (1 Mb in each direction, to account for coordinates’ error and coverage variability) were measured by mosdepth [40] with the input encoded in the BED format. For this study, these files were generated manually, using Sniffles’ results, when possible, or from aCGH (the Sniffles coordinates are presented in Supplementary Table 1). With the latter, the coordinates for the seven SVs were adjusted manually.
The mean coverage of each putative SV was compared with its surroundings, using a threshold of 40% shift in coverage. Statistical significance was checked with the implementation of Student’s t-test (Welch’s approximation) in R. As each base had a coverage value, the group size (one group being both the surrounding regions, and the other, the CNV’s coordinates or tentative ones) was sufficiently large to generate values too small to be normally processable in R, which allowed precise calculations only of decimal numbers greater than 2.225074e − 308. To circumvent this, the t parameter and the degrees of freedom returned by R were used to calculate the natural logarithm of the p-value for a two-tailed test (2 ∗ cdf (−|t|, degrees of freedom), where cdf denotes the cumulative distribution function of the t-Student distribution). Retrieving the cdf value as a logarithm avoided the generation of very low numeric values (which otherwise would have been turned into zero, or calculated with precision errors). Then, the p-values were calculated from their logarithms by using the exponential function and the Rmpfr [47] package with 100 bits of precision. These two steps avoided working with low values in R’s statistics function and then allowed us to retrieve and work normally with p-values below the precision threshold.
Additionally, disCoverage generated plots as those presented in the subfigures of Fig. 5, Fig. 6, which allowed us to visually examine the coverage differences in the regions of interest.
Fig. 5.

Representative examples of cases with SVs detected by aGCH and nanopore sequencing. A) Example of a deletion (loss), B) example of an insertion (gain), and C) examples of a deletion detected by callers after NGMLR alignment, D) but not after minimap2, although the coverage analysis supported the deletion with both the aligners. Segments in the putative SV region are colored green, segments with coverage greater than the chromosome mean (brown continuous line) are colored blue, and red for lower. The mean coverage of the SV is represented by the red horizontal segment, and the surroundings’ mean, by the orange dotted line. ***: p-value ≤ 1e − 13482, **: p-value ≤ 1e − 3117, and for Student’s t-test, see Statistical coverage analysis for details. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Fig. 6.
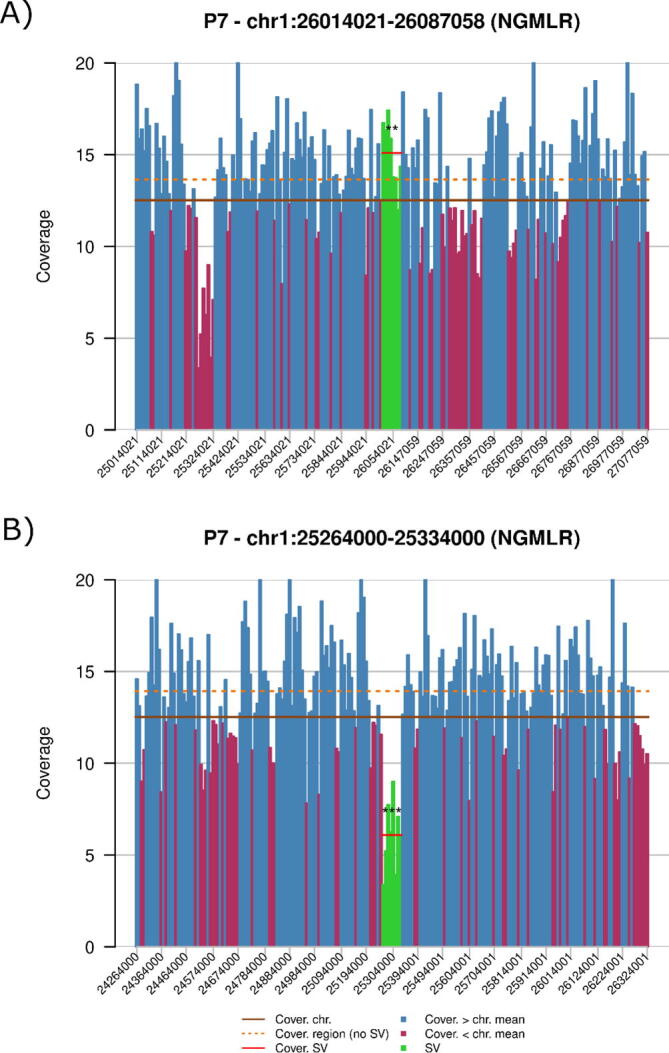
Representative example of a SV detected by aGCH not detected by Sniffles or SVIM analysis of nanopore sequencing, but showing significantly different coverage and being compatible with the type of SV identified by aCGH in a close chromosomal region. A) Nanopore data on the region pointed by the SV (loss) detected in aCGH (chr1:26059700, green area) do not support a deletion, but nearby, around chr1:25265000, red area, a potential deletion is suggested. B) Nanopore data on this new coordinate (green area) support a coverage significantly lower than in the surrounding regions. ***: p-value ≤ 1e − 13482, **: p-value ≤ 1e − 3117, and for Student’s t-test, see Statistical coverage analysis for details. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
For this study, a p-value threshold was selected using an ROC curve from pRoc [48]. For this purpose, we used the CNVs from P1 to P7, considering as positive cases those with a coefficient between the SV coverage and its surroundings of>0.4. The threshold of p-value < 1e − 13482 provided a specificity of 100% and was selected by using two criteria: Youden’s and closest to the top-left corner of the graph. A less stringent threshold, p < 1e − 3117, provided a sensitivity of 100%. These values might not work universally, and disCoverage allows inputting different ones.
Results
Sequencing metrics
The mean read length was 5.83 kb ± 4.49 kb, and the mean number of sequenced and aligned bases reported by NanoStat and determined by the three aligners (lra, minimap2, and NGMLR) was 57.89 ± 22.39/60.29 ± 22.93/65.14 ± 25.11 Gb and 52.96 ± 20.80/56.51 ± 21.88/52.44 ± 20.53 Gb, respectively (these statistics varied by aligner because only the mapped reads were considered for their computing). Although NGMLR obtained more sequenced bases than minimap2 and lra in all the patients, the number of aligned bases was always higher for minimap2. Only patient 6, who showed shorter reads, had a reduced number of sequenced bases and aligned bases as compared to the rest of the patients (Figs. S1, S2, and S3).
Accordingly, the percentage of the genome covered (with mapping quality values of at least 20) by nanopore sequencing was 92.71% when using lra, 93.86% when using minimap2, and 92.86% with NGMLR (Fig. S4). As expected, patients with the highest number of aligned bases (P2 and P4) showed the highest coverage, having 50% of their genome above 20 × coverage, while patient 6 showed the lowest genome coverage: no positions in the patient’s genome reached a coverage depth of 20.
Cnvs identified by aCGH and nanopore sequencing
With aCGH, we could identify 84 CNVs in all the patients, with a mean of 12 ± 3.27 CNVs/patient. All the patients had a similar distribution of SVs involving a gain or a loss, and the global distribution of these two types of SVs in the whole cohort was similar (40 losses and 44 gains). Fig. 2 shows the number and type of SVs identified by aCGH in each patient. Because of the limitations of this method regarding SV size, the gross gene defects affecting SERPINC1 were only found by aCGH in two patients: P2 and P3.
Fig. 2.

Number and type of SVs found by aCGH in each patient. For each case, this is split into copy gains and losses, as reported by aCGH. The green sections indicate the amount of “gains” or duplications, and the yellow ones, “losses” or deletions. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
For nanopore data, we reported the results from the union of variant callers (three for lra, and four for minimap2/NGMLR), for each aligner. Nanopore sequencing showed a remarkably high number of total SV calls. For aligners lra/minimap2/NGMLR, the mean number of total SV calls was 23,874.86 ± 9,139.74/38,462.14 ± 12,207.19/35,392 ± 11,696.86, respectively. Most of the SVs detected by the nanopore sequencing were deletions (48%/43%/42% of the total SVs for lra/minimap2/NGMLR) and insertions (47%/34%/33%), although this method also detected other types of SVs, such as duplications (1%/10%/12%), inversions (3%/2%/3%), and translocations (1%/10%/10%). It was interesting that the proportion of these less frequent SV types was higher with NGMLR and minimap2 than with lra. Fig. 3 shows the number and type of SVs detected in each patient. Again, P6 was the patient with the least number of SVs detected. Note that the SVs affecting SERPINC1 were detected in all the cases by nanopore sequencing.
Fig. 3.

Number and type of SVs found by nanopore sequencing per patient and aligner. Deletions are shown in green, duplications in yellow, insertions in purple, and translocations in blue. N1 refers to NA19240, the control dataset. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Concordance and discrepancies of SVs detected by aCGH and nanopore sequencing
This analysis only considered > 50-kb CNVs occurring in chromosomes 1–22, X, and Y. The results differed depending on the variant caller program used and aligner. Moreover, the results differed when using hg38 or hg19 as the reference. Only 11/23/31 CNVs detected by aCGH (13.10%/27.38%/36.90%) were also detected by nanopore sequencing with variant callers when using lra, minimap2, or NGMLR (Table 4 for deletions and Table 5 for duplications). Supplementary Table 1 shows the CNVs identified by both the methods in each patient, focusing on the hg38 and hg19 coordinates.
Table 4.
Consensus between aCGH and nanopore sequencing, and disCoverage effect for deletions (DEL). N1 refers to NA19240. *For NA19240, the values in these columns are > 50-kb deletions in the truth set and > 50-kb deletions present in the truth set also found with nanopore sequencing by our pipeline, respectively.
| Patient | DEL found with aCGH* | DEL found by aCGH and nanopore sequencing* |
>50-kb DEL after disCoverage |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| lra |
minimap2 |
NGMLR |
lra |
minimap2 |
NGMLR |
|||||
| CuteSV, Sniffles, SVIM | Any caller | CuteSV, NanoVar, Sniffles, SVIM | Any caller | CuteSV, NanoVar, Sniffles, SVIM | Any caller | CuteSV, Sniffles, SVIM | CuteSV, NanoVar, Sniffles, SVIM | |||
| P1 | 4 | 1,0,0 | 1 | 0,1,1,0 | 2 | 2,1,3,0 | 3 | 1,0,10 | 1,11,3,8 | 7,12,9,15 |
| P2 | 7 | 2,0,1 | 2 | 0,1,4,0 | 4 | 1,1,4,1 | 4 | 4,26,24 | 4,17,3,16 | 9,19,7,37 |
| P3 | 4 | 2,0,0 | 2 | 1,1,3,0 | 3 | 3,1,3,0 | 3 | 0,10,8 | 2,10,1,5 | 7,11,9,14 |
| P4 | 6 | 2,2,0 | 2 | 0,2,3,2 | 3 | 3,2,5,2 | 4 | 4,32,24 | 2,21,6,14 | 13,18,16,30 |
| P5 | 2 | 0,0,0 | 0 | 1,0,1,0 | 2 | 0,0,1,0 | 1 | 3,19,16 | 3,20,9,12 | 9,19,4,28 |
| P6 | 6 | 0,0,0 | 0 | 0,1,1,0 | 1 | 1,1,1,0 | 1 | 0,0,0 | 0,6,0,1 | 1,3,1,1 |
| P7 | 9 | 2,0,0 | 2 | 1,2,3,0 | 3 | 1,2,4,0 | 0 | 0,13,9 | 2,15,2,7 | 6,13,8,21 |
| N1 | 94 | 0,0,0 | 0 | 0,0,0,1 | 1 | 0,0,0,0 | 0 | 0,4,4 | 4,22,15,20 | 5,24,14,30 |
Table 5.
Consensus between aCGH and nanopore sequencing, and disCoverage effect for duplications (DUP). N1 refers to NA19240. *For NA19240, the values in these columns are > 50-kb duplications in the truth set and > 50-kb duplications present in the truth set also found with nanopore sequencing by our pipeline, respectively.
| Patient | SVs found with aCGH* | DUP found by aCGH and nanopore sequencing* |
>50-kb DUP after disCoverage |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| lra |
minimap2 |
NGMLR |
lra |
minimap2 |
NGMLR |
|||||
| CuteSV, Sniffles, SVIM | Any caller | CuteSV, NanoVar, Sniffles, SVIM | Any caller | CuteSV, NanoVar, Sniffles, SVIM | Any caller | CuteSV, Sniffles, SVIM | CuteSV, NanoVar, Sniffles, SVIM | |||
| P1 | 7 | 0,0,0 | 0 | 0,1,1,0 | 1 | 1,1,1,0 | 1 | 1,1,1 | 3,1,4,3 | 2,1,5,7 |
| P2 | 3 | 0,0,0 | 0 | 0,0,0,0 | 0 | 1,0,2,0 | 2 | 2,3,4 | 0,0,5,1 | 4,0,7,8 |
| P3 | 9 | 1,1,1 | 1 | 1,2,2,1 | 2 | 2,2,2,1 | 1 | 1,2,2 | 2,2,3,2 | 1,1,3,2 |
| P4 | 4 | 1,0,0 | 1 | 1,0,1,0 | 1 | 2,0,2,0 | 2 | 6,6,8 | 2,1,4,2 | 2,0,8,6 |
| P5 | 5 | 1,0,0 | 1 | 1,0,1,1 | 1 | 1,0,1,1 | 1 | 5,6,2 | 1,3,3,2 | 9,2,6,11 |
| P6 | 7 | 0,0,0 | 0 | 1,0,1,1 | 1 | 1,0,1,1 | 1 | 0,0,0 | 1,0,1,1 | 2,0,3,3 |
| P7 | 3 | 0,0,0 | 0 | 0,0,0,0 | 0 | 0,0,1,0 | 1 | 1,1,1 | 0,1,1,0 | 0,0,0,1 |
| N1 | 28 | 0,0,0 | 0 | 0,0,0,0 | 0 | 0,0,0,0 | 0 | 1,1,0 | 4,22,15,20 | 5,24,14,30 |
We found a total of 1930, 683, and 1432 > 50-kb CNVs for the lra, minimap2, and NGMLR alignments. The mean number of SVs per patient was 275.71 ± 136.06, 97.57 ± 40.93, and 204.57 ± 94.70 for lra, minimap2, and NGMLR, respectively. Fig. 4 shows the number and type of CNVs identified by nanopore sequencing in each patient. Note that NGMLR and lra detected a considerably higher number of SVs with these features than minimap2. We also observed that these two types of SVs had a similar distribution with NGMLR’s alignments (50.28% deletions and 49.72% duplications), while the percentage of deletions found with lra’s and minimap2′s alignments was significantly higher than that of duplications (70% and 30%, 65.59% and 34.41%).
Fig. 4.

Number and type of CNVs > 50 kb found by nanopore sequencing per patient. Green is for deletions, and yellow for duplications. N1 refers to NA19240, the control dataset. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Interestingly, the coordinates obtained for each concordant SV detected by both the methods, namely aCGH and nanopore sequencing, did not match for any SV, but the differences ranged from 11 bp to 2,956,260 bp, and might be different depending on the version of the reference genome used (hg38 or hg19) (Supplementary Table 1).
A factor to consider in the comparison was the allele dose of the selected variants. Array hybridization reported both hetero- and homozygous CNVs (43.42% of the deletions and 40.79% of the duplications were marked as heterozygous). It was possible to find 27.3%/42.4%/54.5% of the heterozygous deletions with the lra/minimap2/NGMLR alignments, and 0%/40.0%/40.0% of the homozygous ones. For duplications, the agreement was 6.5%/16.1%/25.8% and 14.3%/14.3%/42.9% for homozygous and heterozygous ones, respectively.
Aiming to explain the discrepant results between these two methods, we first analyzed the nanopore coverage data for the SVs detected by aCGH that were not detected by nanopore sequencing. The coverage was measured by using mosdepth inside SVs’ coordinates and on their surroundings. The results revealed a significant (p-value < 1e − 13482 with Welch’s approximation of Student’s t-test) difference in coverage compatible with the type of SV detected by aCGH. Thus, an aCGH “loss” corresponded with a lower coverage by the nanopore sequencing, while a “gain” had higher coverage than the surroundings areas. Representative examples of the coverage obtained by nanopore sequencing for the SVs detected by aCGH, including concordances and discrepancies that might be solved by this approach, are shown in Fig. 5.
We also observed that for several SVs detected by aCGH not called by using nanopore sequencing, a compatible difference in coverage was not found in the region indicated by the array, but it was observed nearby (<1 Mb) (Fig. 6). After adjusting to these new coordinates, we found nine regions compatible with the CNVs identified by aCGH (Supplementary Table 1).
All the CNVs identified by aCGH that were also found with nanopore sequencing (using Sniffles on the NGMLR alignment, which generated the most complete set of calls) except two were also supported by the coverage analysis. The two CNVs for which the coverage analysis failed were a duplication on chromosome 11 in P5 and a duplication on chr22 in P2 (see Supplementary Table 1 for the aCGH coordinates). For the latter, it was not considered supported by the coverage because it did not reach the required 40% difference in coverage. Additionally, P1′s deletion in chr1:146174166–146231540 was supported by the coverage in the coordinates reported by aCGH, although the NGMLR + Sniffles pipeline called it at chr1:143226638–143275280, where there was no coverage difference.
The rest of the CNVs found by aCGH were not found by the callers on minimap2′s (N not found = 49) or NGMLR’s (N not found = 40) alignments and were not supported by the coverage. For seven of these CNVs, there was supporting coverage data from nanopore sequencing, but these data showed irregular coverage across the surroundings or the presence of specific features, such as adjacent centromeres, which prevented the consideration of a potential SV with nanopore sequencing. Finally, for the five SVs detected by aCGH, the coordinates could not be translated from hg19 to hg38, because the region was split. One of these SVs, the duplication in chr10 in P7, was also identified by minimap2 and NGMLR when aligning against hg19.
Fig. 7 summarizes the status of the SVs detected by aCGH after the nanopore sequencing analysis in each chromosome and in each patient.
Fig. 7.

Structural variants identified by aCGH and their status after nanopore analysis. Concentric semi-dashed circumferences represent P1–7 from outermost to innermost, while the most external one shows genome coordinates and karyotype. Each dot represents an SV call from aCGH, and its color indicates whether it was found by our nanopore analysis and how. A) Results obtained using hg38 as a reference. B) Results obtained using hg19 as a reference. Coordinates are taken from aCGH results. Variants that would obscure each other are shifted away from the semi-dashed lines to avoid this; check Supplementary Table 1 for the full list of coordinates. SVs found by variant callers are colored green; those only found by our coverage analysis, blue; and those not found, red. Nearby dots have been shifted across the radius so as to not obscure the close SVs. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
SVs only found with nanopore sequencing
Even after discarding all the SVs that the aCGH could not detect, the variant callers detected 1930/683/1432 > 50-kb CNVs (lra/minimap2/NGMLR alignments) across the study from the nanopore data not reported by aCGH. A manual inspection of these SVs revealed that 980/185/603 were very large (>10 Mb), some encompassing other SVs (such as the ones confirmed by MLPA) or centromeres, which suggested that these > 10-Mb SVs were false positive artifacts.
SV calling for control dataset
To evaluate the variant calling in our pipeline, we used a publicly available set of nanopore reads [49] of the well-characterized NA19240 as the control dataset and a previously reported set of variants [50] as the gold standard. The variants were from an exhaustive study that yielded a high-confidence set by combining multiple techniques [51] for applications such as the one considered in this study. One flow cell from PromethIon sequencing was used as the reference; this resulted in a coverage similar to the rest of the cases presented (a mean of 16.2×, which fell within our range, see Table 2). The distribution of SVs for lra/minimap2/NGMLR was 68.8/66.0%/64.0% deletions, 0.3%/4.9%/6.1% duplications, 29.9%/24.1%/24.9% insertions, 1.0%/1.0%/1.4% inversions, and 0.1%/3.9%/3.7% translocations. The CNV call data are shown in Table 3.
Table 3.
SVs detected by using the different bioinformatic tools reported in the study participants and control. The presented numbers are in the format total deletions; total duplications/>50-kb deletions; >50-kb duplications. N1 refers to NA19240.
| Aligner | Caller | P1 | P2 | P3 | P4 | P5 | P6 | P7 | N1 |
|---|---|---|---|---|---|---|---|---|---|
| lra | CuteSV | 4 941;63/ 16;31 |
13 304;204/ 98;78 |
10 186;82/ 47;35 |
13 130;212/ 103;76 |
11 941;210/ 91;73 |
2 195;26/ 19;11 |
9 468;82/ 42;35 |
22 618;43/ 13;13 |
| Sniffles | 11 599;57/ 119;28 |
12 375;150/ 194;54 |
8 878;81/ 104;43 |
12 307;157/ 208;59 |
11 059;187/ 207;65 |
2 120;36/ 51;13 |
8 136;63/ 95;28 |
18 316;61/ 50;32 |
|
| SVIM | 8 827;29/ 11;11 |
10 242;97/ 16;28 |
7 395;38/ 5;14 |
10 259;105/ 18;33 |
9 248;122/ 19;31 |
1 755;18/ 0;5 |
7 240;32/ 6;10 |
13 209;24/ 0;13 |
|
| minimap2 | CuteSV | 7 007;826/ 14;17 |
6 213;950/ 19;20 |
5 280;735/ 12;14 |
11 718;1 822/ 27;34 |
14 524;2 192/ 33;27 |
3 441;280/ 6;7 |
4 774;707/ 8;9 |
29 874;1 699/ 29;30 |
| NanoVar | 18 807;4 542/ 27;23 |
15 724;4 969/ 37;27 |
15 321;4 433/ 17;16 |
15 730;3 980/ 39;32 |
14 773;3 203/ 33;23 |
4 140;939/ 7;5 |
14 039;4 333/ 27;15 |
28 547;2 690/ 32;27 |
|
| Sniffles | 15 547;2 628/ 38;10 |
14 129;3 436/ 47;16 |
11 816;2 483/ 30;5 |
14 167;2 452/ 45;17 |
12 526;1 375/ 33;7 |
3 001;89/ 10;1 |
10 417;2 340/ 26;2 |
24 548;1 004/ 48;18 |
|
| SVIM | 10 629;3 480/ 2;8 |
11 187;3 941/ 6;8 |
8 989;3 275/ 2;4 |
11 315;2 991/ 5;8 |
10 191;2 114/ 4;5 |
2 514;302/ 0;1 |
8 564;3 085/ 3;1 |
16 808;1 674/ 4;10 |
|
| NGMLR | CuteSV | 15 617;3 009/ 31;87 |
5 499;1 542/ 21;84 |
12 391;2 664/ 28;66 |
14 943;4 014/ 53;122 |
4 957;1 370/ 13;47 |
2 897;459/ 15;22 |
4 134;976/ 18;31 |
21 455;1 805/ 24;51 |
| NanoVar | 16 985;3 211/ 26;15 |
14 890;5 236/ 44;16 |
14 070;2 971/ 25;7 |
14 932;3 627/ 41;21 |
13 904;4 835/ 30;14 |
3 586;821/ 6;2 |
12 933;4 233/ 31;5 |
24 417;2 488/ 41;8 |
|
| Sniffles | 13 590;2 051/ 69;69 |
12 817;4 689/ 111;104 |
10 507;1 743/ 61;53 |
12 955;3 049/ 121;100 |
11 523;4 039/ 81;67 |
2 464;267/ 19;15 |
9 457;2 948/ 51;41 |
19 847;967/ 63;37 |
|
| SVIM | 9 743;2 450/ 23;38 |
10 380;4 935/ 23;46 |
8 268;2 048/ 18;28 |
10 510;3 388/ 34;55 |
9 552;4 316/ 18;47 |
2 110;319/ 3;13 |
7 952;3 269/ 14;21 |
12 276;1 430/ 9;20 |
We managed to find 37.93%/42.71%/40.05% (lra/minimap2/NGMLR) of the deletions in Chaisson et al.’s dataset, 3.70%/13.81%/16.43% duplications, and 36.24%/37.56%/34.85% insertions (these results are split by size range in Supplementary Table 3). The median difference in the coordinates between Chaisson et al.’s truth and call sets was 12.5 bp/13.0 bp/14.0 bp for each aligner. However, the agreement was considerably lower for > 50-kb CNVs, with just 0/1/0 SV (which was a deletion) out of 122 (94 deletions and 28 duplications), which were different for each aligner used. Despite this, more > 50-kb CNVs were called by the nanopore analysis: a total of 91/153/168 for lra/minimap2/NGMLR, so the agreement was lower than that observed in the study cases.
Despite the poor consensus, disCoverage allowed us to discard 83/115/114 SVs not present in the gold standard from the lra/minimap2/NGMLR datasets, reducing the number of false positives considerably.
Discussion
Third-generation sequencing is an emerging technology particularly useful for the identification and characterization of SVs [13]. However, it is still necessary to improve bioinformatic tools used to detect SVs by using long-read sequencing data. Therefore, we compared the data obtained by aCGH and nanopore sequencing from seven patients with AT deficiency caused by SVs affecting SERPINC1, which were first detected by MLPA. While nanopore sequencing was sufficient to detect the gene defects carried by these patients, the comparison of SVs larger than 50 kb detected by these methods allowed us to identify the weak points of current programs used to identify SVs from the nanopore data, as well as to propose new approaches that might provide a better identification of SVs by using this promising technology.
As showcased in this work, obtaining a complete list of SVs requires compromising specificity at the least. In addition, many factors play an important role in variant calling, ranging from the quality of the experimental results to the selection of software tools. Next, we will discuss these aspects.
Non-bioinformatic factors affecting SV discovery
Regarding the in vitro part of the study, two variables seem responsible for the completeness of the results: coverage depth and read length. For coverage depth, a higher value allows for a more stringent threshold for the variants’ read support. P6 is a good example; the few nanopore reads explained why only 3 out of 17 SVs detected by aCGH were found by nanopore sequencing. However, after the examination of the unfiltered call sets, two additional SVs were found with three supporting reads each (Supplementary Table 1). In contrast, our results did not seem to point read length as a decisive parameter for the successful identification of large SVs, as the mean read length for NA19240 was 2.4 fold of that of the study cases, and the detection of > 50-kb CNVs was lower than 1% (Table 3), against the 37% of the most successful combination of tools applied in this work. This was likely because the > 50-kb CNVs were detected by split reads instead of reads containing the whole variant. For general use, 20-kb length reads have been reported to produce optimal results [16].
Furthermore, the allele dose might be another factor to consider as heterozygous SVs usually require more coverage to be detected [16]. As shown in our results, while the recall percentage was similar for homozygous and heterozygous deletions, the nanopore results were more complete for homozygous duplications.
Finally, the particular nature of the target SVs in this work might have limited the existing programs from finding them. Variant callers rely on algorithms that examine the alignment against the reference genome. Thus, SVs are detected when supplementary alignments are found. In our study, 13.10%/27.38%/36.90% (lra/ minimap2/NGMLR) of the SVs detected by aCGH were also detected by nanopore sequencing using these variant callers. The SVs that were not found were explained by the absence of supplementary alignments supporting the variants. One reason why there were no such alignments in these cases might be the relatively low coverage, which reduced the likelihood of reads covering the boundaries of the SV. There might also be the possibility that the mutant allele had not been sequenced, but this seemed unlikely to have occurred for 70% of our variants. A different explanation for these missing reads might be that DNA breaking during library preparation occurred more frequently in these regions. Indeed, regions with SVs could be somehow unstable, facilitating both fracturing during experiments and in-cell DNA recombination, the latter being an SV genesis mechanism [52], [53].
Evaluation of variant calling
The rate of detection of SVs by nanopore sequencing using the aCGH results as a reference (∼13%–36%) seemed low, considering the capabilities of nanopores. To further explore this, we used the available data from the well-characterized individual NA19240 to study the agreement with this case.
While the correct calling of the totality of SV was ∼ 40%, it was lower for duplications than for deletions and insertions (Supplementary Table 3).
Despite the large number of SV calls, only one > 50-kb CNV with minimap2 was found to be in common with this truth set. This was certainly lower than the ∼40% for all the SVs, which pointed to the variant callers underperforming with large SVs. It was also lower than the agreement between nanopore sequencing and aCGH in our study; this difference might be attributed to the reference SV set obtained using different techniques. The kit that we used for genomic hybridization detected SVs in a specific size range and was limited to CNVs, while the NA19240 combined several techniques, which resulted in a broader scope. Other studies, however, have reported similar performance across SV lengths [16]. As we used the same tools with similar settings, the nature of the variant set used for the comparison might be the cause of this discrepancy; moreover, >50-kb variants seemed to be usually grouped with > 10-kb ones [12], [16]. Supplementary Table 3 includes the recall data for NA19240 split by SV type and length and reveals a steep decrease in > 50-kb SVs.
Despite the low agreement with aCGH, nanopore sequencing remains a valuable tool that detects all the pathogenic SVs in SERPINC1 [20], in addition to being able to call mutations in a considerably wider size range for all types of variants.
Comparison between different alignments and SV calling tools
In this work, three different programs were used for alignment in combination with up to four variant callers, and we found that not every workflow led to the same variants being detected. Sniffles produced the best results in calling the SVs detected by aCGH (N = 31, with NGMLR). SVIM, CuteSV, and NanoVar only managed to report a fraction of these SVs. Only NanoVar found a single SV that Sniffles did not. Furthermore, SVIM characterized some of them worse, reporting them as a pair of breakpoints (BND).
As for the alignment process, the ones produced by NGMLR allowed Sniffles to find nine more SVs also present in the aCGH results, and the coverage analysis supported an additional one when run with NGMLR-produced alignments. Although NGMLR had limitations, such as being slower [1] and having some bugs at the time of writing this manuscript, it was the approach that generated the largest consensus of SVs between aCGH and nanopore sequencing. Its results contained all of minimap2′s, except for the deletion on chr8 in P7, which was detected with the read support below the filtering threshold (Supplementary Table 1).
While the previous statements applied to > 500-kb CNVs, newer tools might offer better results when checking for a broader range of variants. In fact, minimap2, CuteSV, NanoVar, and SVIM yielded results that shared more variants with the NA19240 truth set (15,112, 14,475, and 14,163) than Sniffles (13,150), although the first two called more variants (43,467 and 44,954) than Sniffles (35,077), while SVIM reported fewer (27,684). The results for the other were worse, particularly for lra, and are presented in Supplementary Table 2.
Coverage analysis complements SV discovery of nanopore data
Coverage analysis has already been used for searching for CNVs with NGS data, although with limited success [54]. A comparison of the data obtained by aCGH and nanopore sequencing revealed that a significant number of SVs detected by the array but not detected by variant callers with nanopore data (∼15%) might be identified by a statistical analysis of the coverage. Thus, this approach improved variant calling with nanopore applications. However, disCoverage is not a variant caller and needs putative SV positions, which could be obtained from an additional technique, or maybe more interestingly, by a study of multiple species samples from the same species. The variants could be merged to generate a set of SVs, which could be then re-checked on every sample, which might help at polishing calls on a population.
Unfortunately, coverage analysis has some limitations. Coverage is not uniform in a sample, and local peaks and valleys are frequently observed without necessarily pointing to an SV; furthermore, unresolved regions in the genome insert drastic shifts in coverage. Moreover, even when studying per-base coverage, it is not clear where the shift happens.
Coverage analysis may also be used for filtering out some SVs that, despite being supported by variant callers, do not seem to imply changes in the DNA material. This can only be applied to some types, namely deletions and duplications, which has been proven useful in our study.
NA19240 data were used for assessing the performance of variant callers on our samples and for testing disCoverage. We applied both approaches, filtering the called SVs and checking the coverage support for truth set events, to our cases and the data from NA19240 and increased the agreement between techniques. The results from P1–7 were discussed in detail in the Results section. Similarly, for NA19240, we could discard 29/62 out of the 100 called SVs when using minimap2 and NGMLR, respectively. Out of the 122 SVs of interest in the truth set, 55/61 SVs were supported by the coverage. This represented an increase in specificity when discarding ∼56%/62% of the erroneous calls for each aligner. Considering that SV detection is still in development [14], a fraction of the remaining “false” positives could be present in NA19240’s genome, considering that they were supported by alignment and coverage. The number of SVs recovered by disCoverage was one order of magnitude larger than that of variant callers for the truth set, although this was certainly influenced by the callers that only found one SV from the gold standard. For the > 50-kb CNVs in our study cases, disCoverage performed similarly to callers, and it was able to rescue a few more variants (Fig. 7), although it needed the coordinates as the input.
Therefore, the coverage analysis implemented in our tool was useful to study large SVs from the nanopore sequencing data. The decision to compare against a wide (2 Mb) surrounding region seemed to have allowed to overcome the coverage irregularity, and the selected p-values were useful to detect SVs with confidence.
In this work, we used a threshold for p-values smaller than 0.05. This has been used for particular applications in the scope of biological studies [55], [56], and general corrections for p-values are available, as 0.05 may not be suitable for all applications [57]. As a matter of fact, recent works have proposed that the 0.05 threshold should be lowered [58]. With this in mind, we combined the t-test with the coverage difference as a criterion to report any SV as supported by the coverage. Our p-value threshold was meaningful for our cases and was useful to polish the variant sets. However, further studies are required to confirm its optimality.
As shown in this work, coverage analysis could be used to find additional support for possible SVs with another technology’s results as the input. This process could be extended for application to situations in which a set of genes or loci are suspected to be affected, which could be helpful for molecular diagnosis. Therefore, the other side of our analysis is also of interest: variant callers generated large sets of variants, and reducing the number of SVs could help in finding the mutation responsible for a given disease. High confidence sets could be generated from the intersection of several callers’ results [1], and disCoverage could be an alternative (for deletions and duplications) that would not require several callers to report the same difficult variant.
Nanopore sequencing and aCGH differences
The variants’ coordinates were another major difference between aCGH and nanopore sequencing that contributed to the explanation of the discrepant results. The coordinates for the SVs discovered with nanopore sequencing were more accurate because the alignments allowed for placing them with a 1-base precision, which is one of the strengths of third-generation sequencing [13]. These differences were measured and compared (Fig. 8) for the CNVs found with both the reference versions. The variability of accuracy for the aCGH coordinates was considerable, as there were coordinates of SVs determined by these two methods with almost complete concordance (11 bp) to a difference that almost reach 3 Mb (2,956,260 bp). Interestingly, the discrepancy of coordinates was more evident when using hg38 as the reference, which suggested that this difference was an artifact from the coordinate translation between references. Although for a deletion in chr17:43229084–43303788 (hg38 coordinates), the difference was greater than 2 Mb in both the cases, and the other three SVs found in hg19 but not in hg38 had a difference greater than 1 Mb. We considered these calls to be the same CNVs as their aCGH counterparts. This was based on the premise that, as they had the same nature (e.g., “Loss” and “DEL”), similar size, and relatively close location, it was likely that they were the same variant. We found this more likely than such similar variants existing this closely, but only one being detected by callers or having a significant impact of coverage. Lifting variants from one reference to the other introduces errors in coordinates, which makes the discrepancies between techniques more noticeable (Fig. 8). Consequently, converted sets of coordinates should be used carefully.
Fig. 8.

Boxplot of the absolute value of aCGH and nanopore coordinate difference (start and end mean) per patient, for hg19 (green) and hg38 (orange). Coordinates are taken from NGMLR and Sniffles, which yielded the best results for > 50-kb CNVs. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
The majority of > 50-kb CNVs identified by nanopore sequencing (99.43%/96.63%/97.84% of them for lra, minimap2, and NGMLR) were not detected by aCGH. Very large SVs (>10 Mb), particularly those encompassing other SVs or centromeres, were probably artifacts and were not considered true, despite being supported by several reads and supplementary alignments.
Finally, note that while CGH and nanopore sequencing reported a considerably different number of variants (∼20 CNVs against ∼30,000 SVs of all types, respectively), the difference was lower for > 50-kb CNVs, of which nanopore sequencing reported ∼ 100. Additionally, after running disCoverage, most of them were filtered out (Table 2, Table 3), and the number of variants called was more similar between the two. The use of CGH was interesting because it provided a clear example of how large variants were harder to detect. While new tools were developed, the results for this particular subset did not improve. Despite being difficult to study, large SVs can be pathogenic [20], and therefore, its characterization was important.
Conclusion
Nanopore sequencing is a very useful third-generation sequencing technology to find variants that cannot be detected by applying short-read sequencing technologies, and they are useful for other studies, for example, haplotyping [59]. Nevertheless, this technology and its related software tools are at different levels of maturity and require further development.
Our results led to the conclusion that, for SV calling, the hg38 version of the reference human was better, which was in agreement with the literature on smaller variants [60], although SVs whose coordinates could not be translated (8 in our study) were impossible to find in hg38. Regarding software, the alignment produced by NGMLR generated more complete sets of SV calls when using Sniffles, with more SVs as a trade-off. SVIM, with only one flow cell per patient, called fewer SVs and characterized them worse. However, using the NA19240 dataset, we assessed that this was specific to large CNVs that yielded satisfactory results, although Sniffles and CuteSV obtained the best results.
Future applications may yield higher depth coverage alongside more accurate reads. Additionally, software applications will be developed to better parse anomalies in alignments and characterize SVs more accurately. Meanwhile, newer tools seem to bring improvements in the field [15], [16]. However, the variant sets reported by third-generation sequencing are still incomplete and contain false positives [1], [15], [16]. As reported, this issue seemed to be worse for larger CNVs, and more recent tools have not improved this, and the problem does not seem to be observed or addressed. Large CNVs, and SVs in general, are still of interest for the impact they have [17]–[19]. We presented that the coverage analysis from the same sequencing experiment that was used to call variants could alleviate the problem.
Regarding disCoverage, we found that it could support many more aCGH > 50 kb from Chaisson et al.’s truth set than variant callers. Thus, in addition to filtering out CNV calls, it could be used for detecting the known (previously or discovered in the same studio) polymorphic CNVs in populations, which is another current use of nanopore sequencing [61].
We believe that this study, which we have extended from AT to genomic scale, and the methodology applied to compare nanopore sequencing with aCGH at the whole-genome level will contribute to the creation of a knowledge foundation that may allow future projects to tackle other pathologies.
Code and data availability
The code used for analysis is available in the following GitHub repository: https://github.com/javiercguard/nanopore_pipeline_21. The tool disCoverage is available at https://github.com/javiercguard/disCoverage. The data that support the findings (P1–7) of this study are available from NIHR BioResource, but restrictions apply to the availability of these data, and so, these data are not publicly accessible. Data from NA19240 were downloaded from a public dataset [49], [50].
CRediT authorship contribution statement
Javier Cuenca-Guardiola: Conceptualization, Methodology, Formal Analysis, Validation, Investigation, Writing. Belén de la Morena-Barrio: Conceptualization, Methodology, Formal Analysis, Validation, Investigation, Writing. Juan L. García: Investigation, Writing. Alba Sanchis-Juan: Investigation, Writing. Javier Corral: Conceptualization, Methodology, Formal Analysis, Validation, Supervision, Writing. Jesualdo T. Fernández-Breis: Conceptualization, Methodology, Formal Analysis, Validation, Supervision, Writing.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
We gratefully acknowledge the NIHR BioResource centers and staff for their contribution. We thank the National Institute for Health Research and NHS Blood and Transplant. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR, or the Department of Health and Social Care. Javier Cuenca-Guardiola is funded by the Ministerio de Universidades through grant FPU19/03662. Belén DMB has a postdoctoral contract from CIBERER. This work has been partially possible thanks to the funding of the Instituto de Salud Carlos III and the European Regional Development Fund through grant PI21/00174, and the Instituto de Salud Carlos III a nd the Next Generation EU grant PMP21/00052.
Footnotes
Peer review under responsibility of Cairo University.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jare.2022.10.012.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.De Coster W., et al. Structural variants identified by Oxford Nanopore PromethION sequencing of the human genome. Genome Res. Jul. 2019;29(7):1178–1187. doi: 10.1101/gr.244939.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sudmant P.H., et al. An integrated map of structural variation in 2,504 human genomes. Nature. Oct. 2015;526(7571):75–81. doi: 10.1038/nature15394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eichler E.E. Genetic Variation, Comparative Genomics, and the Diagnosis of Disease. N Engl J Med. Jul. 2019;381(1):64–74. doi: 10.1056/NEJMra1809315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bowden R., et al. Sequencing of human genomes with nanopore technology. Nat Commun. Dec. 2019;10(1):1869. doi: 10.1038/s41467-019-09637-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Corral J., de la Morena-Barrio M.E., Vicente V. The genetics of antithrombin. Thromb Res. Sep. 2018;169:23–29. doi: 10.1016/j.thromres.2018.07.008. [DOI] [PubMed] [Google Scholar]
- 6.Davies PA, Gray G. Long-Range PCR. In: PCR Mutation Detection Protocols, vol. 187, New Jersey: Humana Press, 2002, pp. 051–055. doi: https://doi.org/10.1385/1-59259-273-2:051.
- 7.Ceulemans S, van der Ven K, Del-Favero J. Targeted Screening and Validation of Copy Number Variations. In: Genomic Structural Variants, vol. 838, L. Feuk, Ed. New York, NY: Springer New York, 2012, pp. 311–328. doi: https://doi.org/10.1007/978-1-61779-507-7_15. [DOI] [PubMed]
- 8.Hu Q., Maurais E.G., Ly P. Cellular and genomic approaches for exploring structural chromosomal rearrangements. Chromosome Res Int J Mol Supramol Evol Asp Chromosome Biol. Mar. 2020;28(1):19–30. doi: 10.1007/s10577-020-09626-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pinkel D., et al. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat Genet. Oct. 1998;20(2):207–211. doi: 10.1038/2524. [DOI] [PubMed] [Google Scholar]
- 10.Chan S. et al., Structural Variation Detection and Analysis Using Bionano Optical Mapping. In: Copy Number Variants, vol. 1833, D. M. Bickhart, Ed. New York, NY: Springer New York, 2018, pp. 193–203. doi: https://doi.org/10.1007/978-1-4939-8666-8_16. [DOI] [PubMed]
- 11.Mahmoud M., Gobet N., Cruz-Dávalos D.I., Mounier N., Dessimoz C., Sedlazeck F.J. Structural variant calling: the long and the short of it. Genome Biol. Nov. 2019;20(1):246. doi: 10.1186/s13059-019-1828-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Minoche A.E., et al. ClinSV: clinical grade structural and copy number variant detection from whole genome sequencing data. Genome Med. Feb. 2021;13(1):32. doi: 10.1186/s13073-021-00841-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Logsdon G.A., Vollger M.R., Eichler E.E. Long-read human genome sequencing and its applications. Nat Rev Genet. Oct. 2020;21(10):597–614. doi: 10.1038/s41576-020-0236-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Amarasinghe S.L., Su S., Dong X., Zappia L., Ritchie M.E., Gouil Q. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. Diciembre 2020,;21(1):30. doi: 10.1186/s13059-020-1935-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dierckxsens N., Li T., Vermeesch J.R., Xie Z. A benchmark of structural variation detection by long reads through a realistic simulated model. Genome Biol. Dec. 2021;22(1):342. doi: 10.1186/s13059-021-02551-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang T., et al. Long-read sequencing settings for efficient structural variation detection based on comprehensive evaluation. BMC Bioinf. Nov. 2021;22(1):552. doi: 10.1186/s12859-021-04422-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Z., Roberts R., Mercer T.R., Xu J., Sedlazeck F.J., Tong W. Towards accurate and reliable resolution of structural variants for clinical diagnosis. Genome Biol. Mar. 2022;23(1):68. doi: 10.1186/s13059-022-02636-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen L., et al. Association of structural variation with cardiometabolic traits in Finns. Am J Hum Genet. Apr. 2021;108(4):583–596. doi: 10.1016/j.ajhg.2021.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deng L., et al. Analysis of five deep-sequenced trio-genomes of the Peninsular Malaysia Orang Asli and North Borneo populations. BMC Genomics. Nov. 2019;20:842. doi: 10.1186/s12864-019-6226-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de la Morena-Barrio B, Stephens J, de la Morena-Barrio ME, Stefanucci L, Padilla J, Miñano A, Gleadall N, García JL, López-Fernández MF, Morange PE, Puurunen M, Undas A, Vidal F, Raymond FL, Vicente V, Ouwehand WH, Corral J, Sanchis-Juan A; NIHR BioResource. Long-Read Sequencing Identifies the First Retrotransposon Insertion and Resolves Structural Variants Causing Antithrombin Deficiency. Thromb Haemost. 2022 Aug;122(8):1369-1378. doi: 10.1055/s-0042-1749345. Epub 2022 Jun 28. PMID: 35764313; PMCID: PMC9393088. [DOI] [PMC free article] [PubMed]
- 21.De la Morena-Barrio B.et al., Identification of the first large intronic deletion responsible of type I antithrombin deficiency not detected by routine molecular diagnostic methods, Br. J. Haematol., vol. 186, no. 4, pp. e82–e86, Aug. 2019, doi: https://doi.org/10.1111/bjh.15913 [DOI] [PubMed]
- 22.Kallioniemi A., et al. Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science. Oct. 1992;258(5083):818–821. doi: 10.1126/science.1359641. [DOI] [PubMed] [Google Scholar]
- 23.Barrett M.T., et al. Comparative genomic hybridization using oligonucleotide microarrays and total genomic DNA. Proc Natl Acad Sci U S A. Dec. 2004;101(51):17765–17770. doi: 10.1073/pnas.0407979101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de la Morena-Barrio M.E., et al. Hypoglycosylation is a common finding in antithrombin deficiency in the absence of a SERPINC1 gene defect. J Thromb Haemost. 2016;14(8):1549–1560. doi: 10.1111/jth.13372. [DOI] [PubMed] [Google Scholar]
- 25.de la Morena-Barrio B. et al., Molecular Dissection of Structural Variations Involved in Antithrombin Deficiency, J. Mol. Diagn. JMD, pp. S1525-1578(22)00042–3, Feb. 2022, doi: https://doi.org/10.1016/j.jmoldx.2022.01.009. [DOI] [PubMed]
- 26.Kent W.J., et al. The Human Genome Browser at UCSC. Genome Res. Jun. 2002;12(6):996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sedlazeck F.J., et al. Accurate detection of complex structural variations using single-molecule sequencing. Nat Methods. Jun. 2018;15(6):461–468. doi: 10.1038/s41592-018-0001-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. Sep. 2018;34(18):3094–3100. doi: 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ren J., Chaisson M.J.P. lra: A long read aligner for sequences and contigs. PLOS Comput Biol. Jun. 2021;17(6):e1009078. doi: 10.1371/journal.pcbi.1009078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Genome Reference Consortium. Genome Reference Consortium Human Build 38 (GRCh38). NCBI, 2013. Accessed: Feb. 07, 2022. [Online]. Available: ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/000/001/405/GCA_000001405.15_GRCh38/seqs_for_alignment_pipelines.ucsc_ids/GCA_000001405.15_GRCh38_no_alt_analysis_set.fna.gz
- 31.Li H. Which human reference genome to use?. Which human reference genome to use? https://lh3.github.io/2017/11/13/which-human-reference-genome-to-use (accessed Jul. 12, 2020).
- 32.Genome Reference Consortium. Genome Reference Consortium Human Build 37 (GRCh37). NCBI, 2013. Accessed: Feb. 07, 2022. [Online]. Available: ftp://ftp-trace.ncbi.nih.gov/1000genomes/ftp/technical/reference/human_g1k_v37.fasta.gz
- 33.Danecek P., et al. Twelve years of SAMtools and BCFtools. GigaScience. Feb. 2021;vol. 10, no. 2:p. giab008. doi: 10.1093/gigascience/giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heller D., Vingron M. SVIM: structural variant identification using mapped long reads. Bioinformatics. Sep. 2019;35(17):2907–2915. doi: 10.1093/bioinformatics/btz041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang T., et al. Long-read-based human genomic structural variation detection with cuteSV. Genome Biol. Aug. 2020;21(1):189. doi: 10.1186/s13059-020-02107-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tham C.Y., et al. NanoVar: accurate characterization of patients’ genomic structural variants using low-depth nanopore sequencing. Genome Biol. Mar. 2020;21(1):56. doi: 10.1186/s13059-020-01968-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jeffares D.C., et al. Transient structural variations have strong effects on quantitative traits and reproductive isolation in fission yeast. Nat Commun. Jan. 2017;8(1):14061. doi: 10.1038/ncomms14061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuhn R.M., Haussler D., Kent W.J. The UCSC genome browser and associated tools. Brief Bioinform. Mar. 2013;14(2):144–161. doi: 10.1093/bib/bbs038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.De Coster W., D’Hert S., Schultz D.T., Cruts M., Van Broeckhoven C. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics. Agosto 2018,;34(15):2666–2669. doi: 10.1093/bioinformatics/bty149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pedersen B.S., Quinlan A.R. Mosdepth: quick coverage calculation for genomes and exomes. Bioinformatics. Mar. 2018;34(5):867–868. doi: 10.1093/bioinformatics/btx699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.R Core Team . R Foundation for Statistical Computing; Vienna, Austria: 2020. R: A Language and Environment for Statistical Computing. [Online]. Available: [Google Scholar]
- 42.Dowle M, Srinivasan A. data.table: Extension of ‘data.frame‘. 2021. [Online]. Available: https://CRAN.R-project.org/package=data.table
- 43.Wickham H., et al. Welcome to the tidyverse. J Open Source Softw. 2019;4(43):1686. doi: 10.21105/joss.01686. [DOI] [Google Scholar]
- 44.Gu Z., Gu L., Eils R., Schlesner M., Brors B. circlize implements and enhances circular visualization in R. Bioinformatics. 2014;30(19):2811–2812. doi: 10.1093/bioinformatics/btu393. [DOI] [PubMed] [Google Scholar]
- 45.Wickham H. Springer-Verlag; New York: 2016. ggplot2: Elegant Graphics for Data Analysis. [Online]. Available: [Google Scholar]
- 46.Gagolewski M. stringi: Fast and portable character string processing in R. 2021. [Online]. Available: https://stringi.gagolewski.com/
- 47.Maechler M. Rmpfr: R MPFR - Multiple Precision Floating-Point Reliable. 2020. [Online]. Available: https://CRAN.R-project.org/package=Rmpfr
- 48.Robin X., et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinf. 2011;12:77. doi: 10.1186/1471-2105-12-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.De Coster W., et al. PromethION WGS data of NA19240 (run 1) European Nucleotide Archive. 2019 ftp://ftp.sra.ebi.ac.uk/vol1/run/ERR258/ERR2585112/NA19240_run1.fastq.gz [Online]. Available: [Google Scholar]
- 50.Chaisson MJP, Sanders AD, Zhao X, other, “nstd152 (Chaisson et al. 2019).” dbVar, 2019. Accessed: Feb. 07, 2022. [Online]. Available: https://www.ncbi.nlm.nih.gov/dbvar/studies/nstd152/
- 51.Chaisson M.J.P., et al. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat Commun. Apr. 2019;10(1):1784. doi: 10.1038/s41467-018-08148-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carvalho C.M.B., Lupski J.R. Mechanisms underlying structural variant formation in genomic disorders. Nat Rev Genet. Apr. 2016;17(4):224–238. doi: 10.1038/nrg.2015.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shaikh T.H., et al. Low copy repeats mediate distal chromosome 22q11.2 deletions: Sequence analysis predicts breakpoint mechanisms. Genome Res. Apr. 2007;17(4):482–491. doi: 10.1101/gr.5986507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Klambauer G., et al. cn.MOPS: mixture of Poissons for discovering copy number variations in next-generation sequencing data with a low false discovery rate. Nucleic Acids Res. May 2012;40(9):e69–e. doi: 10.1093/nar/gks003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fadista J., Manning A.K., Florez J.C., Groop L. The (in)famous GWAS P -value threshold revisited and updated for low-frequency variants. Eur J Hum Genet. Aug. 2016;24(8):1202–1205. doi: 10.1038/ejhg.2015.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kaler A.S., Purcell L.C. Estimation of a significance threshold for genome-wide association studies. BMC Genomics. Jul. 2019;20(1):618. doi: 10.1186/s12864-019-5992-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jafari M., Ansari-Pour N. Why, When and How to Adjust Your P Values? Cell J Yakhteh. 2019;20(4):604–607. doi: 10.22074/cellj.2019.5992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Di Leo G., Sardanelli F. Statistical significance: p value, 0.05 threshold, and applications to radiomics—reasons for a conservative approach. Eur Radiol Exp. Mar. 2020;4(1):18. doi: 10.1186/s41747-020-0145-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Martin M. et al.. WhatsHap: fast and accurate read-based phasing. bioRxiv, p. 085050, Nov. 2016, doi: https://doi.org/10.1101/085050.
- 60.Pan B., et al. Similarities and differences between variants called with human reference genome HG19 or HG38. BMC Bioinf. Mar. 2019;20(2):101. doi: 10.1186/s12859-019-2620-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Beyter D., et al. Long-read sequencing of 3,622 Icelanders provides insight into the role of structural variants in human diseases and other traits. Nat Genet. Jun. 2021;53(6):779–786. doi: 10.1038/s41588-021-00865-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The code used for analysis is available in the following GitHub repository: https://github.com/javiercguard/nanopore_pipeline_21. The tool disCoverage is available at https://github.com/javiercguard/disCoverage. The data that support the findings (P1–7) of this study are available from NIHR BioResource, but restrictions apply to the availability of these data, and so, these data are not publicly accessible. Data from NA19240 were downloaded from a public dataset [49], [50].