

Abstract
The CRISPR-associated (Cas) Cas12a is the effector protein for type V-A CRISPR systems. Cas12a is a sequence-specific endonuclease that targets and cleaves DNA containing a cognate short signature motif, called the protospacer adjacent motif (PAM), flanked by a 20 nucleotide (nt) segment that is complementary to the “guide” region of its CRISPR RNA (crRNA). The guide sequence of the crRNA can be programmed to target any DNA with a cognate PAM and is the basis for Cas12a’s current use for gene editing in numerous organisms and for medical diagnostics. While Cas9 (type II effector protein) is widely used for gene editing, Cas12a possesses favorable features such as its smaller size and creation of staggered double-stranded DNA ends after cleavage that enhances cellular recombination events. Collected here are protocols for the recombinant purification of Cas12a and the transcription of its corresponding programmable crRNA that are used in a variety of Cas12a-specific in vitro activity assays such as the cis, the trans and the guide-RNA independent DNA cleavage activities with multiple substrates. Correspondingly, protocols are included for the quantification of the activity assay data using ImageJ and the use of MATLAB for rate constant calculations. These procedures can be used for further structural and mechanistic studies of Cas12a orthologs and other Cas proteins.
1. Introduction
Cas12a is the signature endonuclease protein of the type V-A CRISPR-Cas (clustered regularly interspaced short palindromic repeats-CRISPR associated) adaptive immune system (Zetsche et al., 2015). Cas12a binds to a cognate RNA called the CRISPR RNA (crRNA), specifically by recognizing the 5′ end of the crRNA that possesses a pseudoknot structure to form a binary complex (Yamano et al., 2016). The binary complex surveils DNA and binds and cleaves it sequence-specifically if it possesses two specific features: (i) a protospacer adjacent motif (PAM), which is a short DNA motif that is specific for each Cas12a ortholog [for example, cognate PAM for Cas12a from Francisella novicida Cas12a (FnoCas12a) is 5′-TTN-3′]; and (ii) complementarity with the 20-nt “guide” region present at the 3′ end of the crRNA (Zetsche et al., 2015). After locating the PAM, the guide region of crRNA triggers formation of an R-loop with the DNA, forming the ternary complex. The ternary complex facilitates sequence specific cleavage of DNA [double stranded DNA (dsDNA) or single stranded DNA (ssDNA) that base pairs with the crRNA] using its endonuclease domain, RuvC (Chen et al., 2018; Gao, Yang, Rajashankar, Huang, & Patel, 2016; Zetsche et al., 2015).
Cas12a has a bilobed structure made of the recognition (REC) and nuclease (NUC) lobes (Fig. 1) (Dong et al., 2016; Gao et al., 2016; Yamano et al., 2016). The REC lobe comprises REC1 and REC2 domains, and the NUC lobe contains the RuvC, the PAM-interacting (PI) and the wedge (WED) domains, and the bridge helix (BH) (Yamano et al., 2016). The BH is an arginine-rich helix that bridges the REC and NUC lobes and is a conserved feature of type II and type V CRISPR systems (Yamano et al., 2016). When the Cas12a effector transitions from the binary to the ternary complex, the BH undergoes a conformational change that aids in target cleavage (Parameshwaran et al., 2021; Wörle, Jakob, Schmidbauer, Zinner, & Grohmann, 2021). Recent studies have shown the essential role that the BH plays in the Cas12a cleavage mechanism and mismatch tolerance (Parameshwaran et al., 2021; Wörle et al., 2021). The WED and REC1 domains recognize the dsDNA target through non-specific interactions with the sugar-phosphate backbone, while the PI domain sequence-specifically interacts with both strands of the PAM sequence to promote the unwinding of the dsDNA helix (Yamano et al., 2016). While Cas12a orthologs usually have a T-rich cognate PAM sequence (5′-TTN-3′ for FnoCas12a, and 5′-TTTN-3′ for Lachnospiraceae bacterium Cas12a (LbCas12a) and Acidaminococcus sp. Cas12a (AsCas12a) (Fonfara, Richter, Bratovič, Le Rhun, & Charpentier, 2016; Yamano et al., 2016; Zetsche et al., 2015), they can also recognize C-containing PAM sequences through altered interactions with the target DNA (Yamano et al., 2017). As the DNA unwinds upon PAM recognition, the crRNA can hybridize to its complementary sequence, the target DNA strand (TS), leaving a single stranded DNA, the non-target DNA strand (NTS), leading to the formation of the R-loop. The NTS is conducted by the PI domain to the RuvC active site for cleavage (Stella, Alcón, & Montoya, 2017; Swarts, van der Oost, & Jinek, 2017; Yamano et al., 2016). As the R-loop starts to form, Cas12a searches for complementarity with a 3–5-nt seed region near the PAM, which is very crucial in propagating the R-loop formation by further hybridization with the remaining nucleotides of the crRNA guide and the TS. While mismatches between the crRNA guide and the target DNA are tolerated to varying degrees based on the position of the mismatch, mismatches in the seed sequence drastically inhibit R-loop propagation, and by default, the cleavage activity of Cas12a (Stella et al., 2017). While cleaving a dsDNA, Cas12a exhibits a strict order in strand cleavage, NTS followed by TS cleavage (Swarts et al., 2017). After the NTS is cleaved, the REC and NUC lobes undergo conformational changes to position the TS in a single stranded form, with the correct polarity, in the catalytic pocket created by RuvC and a nearby domain called the “Nuc” domain (Cofsky et al., 2020; Swarts et al., 2017; Swarts & Jinek, 2019). The extra distance needed to accommodate the TS in a single stranded form in the catalytic site creates a staggered double stranded cut in the PAM distal end of the R-loop (Stella et al., 2018). This type of DNA cleavage triggered by PAM recognition, base pairing with the crRNA, and sequence-specific dsDNA cleavage is called “cis” cleavage in Cas12a terminology (Swarts & Jinek, 2019).
Fig. 1.

Structure of FnoCas12a-crRNA in complex with a dsDNA target. (A) Schematic representation of FnoCas12a domain organization. (B) Overall structure of the FnoCas12a-crRNA-target DNA complex. Domains are colored according to the scheme in panel A. (BH: bridge helix; PDB: 6I1K) (Swarts & Jinek, 2019).
Another interesting feature of Cas12a proteins is its ability to promote collateral ssDNA cleavage following cis DNA cleavage (Chen et al., 2018; S.-Y. Li et al., 2018). Based on available mechanisms, it is proposed that the release of the PAM-distal end of the DNA from the complex after cis cleavage, leaves the RuvC active site open for indiscriminate ssDNA cleavage (Swarts & Jinek, 2019). This activity is termed trans cleavage (Chen et al., 2018; Nguyen, Smith, & Jain, 2020) and has formed the basis of several molecular diagnostic tools where a ssDNA with a measurable probe is cleaved upon recognition of a specific DNA sequence for cis cleavage (Nguyen et al., 2020). Cis cleavage can be activated by a dsDNA possessing a PAM and a complementary sequence to the guide region of the crRNA or by a ssDNA that is complementary to the guide region of the crRNA (Chen et al., 2018).
Certain Cas proteins (Cas9 and Cas12a) have been shown to possess non-specific DNA cleavage in the absence of cognate crRNAs which is enabled by specific divalent metals, named as RNA-independent or guide-RNA free DNA cleavage (Saha et al., 2020; Sundaresan, Parameshwaran, Yogesha, Keilbarth, & Rajan, 2017). FnoCas12a can nonspecifically nick dsDNA plasmids and degrade ssDNA substrates in the presence of divalent metals such as Mn2+ and Co2+ (Sundaresan et al., 2017). Interestingly, AsCas12a is very active for RNA-independent DNA cleavage with the activity being triggered by Mg2+ as well and dsDNA being degraded following the initial nicking at higher metal concentrations (Li et al., 2020).
Cas12a is also unique since it possesses the ability to process its own crRNA through a unique active site that is different than the RuvC domain (Fonfara et al., 2016; Swarts et al., 2017). The CRISPR array of CRISPR systems (made up of alternating spacer and repeat sequences) is transcribed into one long transcript called the pre-crRNA (Fonfara et al., 2016). The repeat-derived region of the crRNA adopts a pseudoknot conformation that is recognized by the Cas12a protein while binding to the pre-crRNA (Dong et al., 2016; Fonfara et al., 2016). Cas12a then cleaves the repeat sequence upstream of the pseudoknot followed by further trimming to create a mature crRNA (Fonfara et al., 2016). The catalytic site for crRNA processing is located in the WED domain and contains two highly-conserved lysines and a histidine (Swarts et al., 2017). The mature crRNA consists of ~19-nt derived from the repeat sequence at the 5′ end, and ~24 nt from the spacer sequence at the 3′ end (Dong et al., 2016; Safari, Zare, Negahdaripour, Barekati-Mowahed, & Ghasemi, 2019; Swarts et al., 2017; Zetsche et al., 2015). The mechanism for trimming of the 3′ end is unknown currently. This property makes Cas12a unique in being self-sufficient for processing individual crRNAs from a tandem crRNA cassette that can be provided as a continuous stretch of DNA. This has enabled multiplexing, where different sites can be targeted simultaneously, a favorable feature for Cas12a genome applications (Paul & Montoya, 2020).
CRISPR-Cas systems have been engineered to use as genome editing and medical diagnostic tools because of their abilities to specifically target sites in the genome and perform a double-stranded cut which can then be repaired by the host’s DNA repair machinery (Bayat, Modarressi, & Rahimpour, 2018). Currently, Cas9 is the main CRISPR system used for gene editing and gene knockouts, but its large size and off-target activity make it a less-desirable tool (Shmakov et al., 2017). Cas12a is increasing in popularity as a genome tool because of its smaller size and multiplexing ability that helps in delivery using viral vectors and the creation of staggered DNA ends following cis cleavage that enhances recombinational events (Paul & Montoya, 2020; Zetsche et al., 2017). In addition, trans activity is specifically being used as molecular diagnostic tools, including specific detection of viral samples (Broughton et al., 2020; Mahas et al., 2021). The processive nature of trans activity amplifies signal enabling detection as low as 100 aM (Chen et al., 2018; L. Li et al., 2019; S.-Y. Li et al., 2018; Mahas et al., 2021). Catalytically dead Cas12a which is unable to induce dsDNA breaks is being used for gene repression and studies have shown that Cas12a possesses higher gene repression than catalytically dead Cas9 (Kim et al., 2017).
The properties of the Cas12a enzyme make it an excellent candidate for biomedical applications. Because of this, structural and mechanistic studies of Cas12a are very valuable to further enhance the biomedical applicability of Cas12a. The following protocols can be used for the structural and mechanistic studies of FnoCas12a and other Cas12a orthologs (Fig. 2).
Fig. 2.

A flowchart depicting the sequence of events from the combined protocols in this chapter.
2. Recombinant protein purification
Cas12a from several orthologs have been recombinantly expressed in Escherichia coli and biochemically and structurally characterized (Dong et al., 2016; Gao et al., 2016; Saha et al., 2020). In this report, protocols for FnoCas12a protein (UniProt ID: A0Q7Q2) will be presented. To recombinantly purify FnoCas12a, it can be expressed with a histidine (His) tag for Nickel-Nitriloacetic Acid (NTA) affinity purification and a maltose-binding protein (MBP) tag to increase protein solubility. The tags can be cleaved off after Ni-NTA column by introducing a protease cut site such as the tobacco etch virus (TEV) protease. The protocol presented below utilizes sonication for lysis, two affinity chromatography columns, nickel-NTA and cation exchange, and a size exclusion chromatography (SEC) (Parameshwaran et al., 2021). All these steps are variable. Alternative protocols use a HiTrap Heparin column in place of a cation exchange column (Mohanraju, Van Der Oost, Jinek, & Swarts, 2018) or use only a nickel-NTA column and an SEC column (Swarts et al., 2017).
2.1. Plasmids and bacterial strains for bacterial expression and purification of FnoCas12a
This protocol uses a pET28-based plasmid holding the Fno cas12a gene in the following pattern: His8-3C protease recognition site-MBP-TEV recognition site-FnoCas12a (Sundaresan et al., 2017). His8 tag (eight histidines) increases the affinity of the protein for the immobilized metal affinity chromatography (IMAC) column and the N-terminal Maltose Binding Protein (MBP) tag enhances folding and solubility of the target protein. 3C and TEV are sequence-specific proteases. To remove both tags from the purified protein, TEV protease can be used since it will produce FnoCas12a with minimal extra non-native amino acids. The expression plasmid is transformed into E. coli Rosetta strain 2 (DE3) cells for optimizing protein expression following established protocols. This plasmid should be propagated in medium containing both chloramphenicol and kanamycin to maintain the plasmids needed for tRNAs for rare codons and pET28 respectively. Bacterial expression plasmids are also available from the Addgene repository [FnoCas12a, (Addgene 113432), AsCas12a (Addgene 113430), LbCas12a (Addgene 113431)] (Chen et al., 2018), and can be used with the corresponding protocols for each plasmid type, which uses a similar overall procedure for protein purification.
2.2. FnoCas12a recombinant overexpression and purification
Grow an overnight primary culture of E. coli Rosetta strain 2 (DE3) transformed with the FnoCas12a plasmid in 50mL of 2xYT broth containing kanamycin (50 μg/mL) and chloramphenicol (34 μg/mL).
Prepare secondary cultures in two autoclaved 2-L Erlenmeyer flasks each containing 1 L of autoclaved 2xYT broth containing kanamycin and chloramphenicol with the final concentration of 50 and 34 μg/mL respectively. Use 10 mL of the overnight culture to inoculate 1 L 2xYT broth medium.
Grow secondary cultures for ~4 h (until optical density (OD) 600 nm is ~0.6–0.8). Take out 1 mL as an uninduced sample for analysis on a gel. (Pellet the uninduced sample, resuspend in 150 μL water and add 50 μL of 4× gel loading dye (50mM Tris-HCl pH 6.8, 2% SDS, 10% glycerol, 1% β-mercaptoethanol, 12.5mM EDTA, 0.02% bromophenol blue). Induce the rest of the culture with 0.2mM Isopropyl β-D-1-thiogalactopyranoside (IPTG) and let the culture grow overnight (~16 h) at 18°C. All cell culture is done with constant shaking at 180 RPM (revolutions per minute).
Centrifuge the overnight grown culture at 4°C for 20 min at 5000 relative centrifugal force (RCF). Resuspend the pellet in lysis buffer/Ni-NTA buffer A (20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), pH 7.5, 1M NaCl, 20mM Imidazole) and store it at −80°C until further use. A ratio of 1 g of cell pellet to 7mL of buffer can be used for optimal results.
On the day of protein purification, thaw the pellets on ice/water mixture. Pool the thawed pellets into a beaker. Once the pellet is completely thawed, add the protease inhibitors:phenylmethylsulfonyl fluoride (PMSF) and Benzamidine to a final concentration of 1 mM and Pepstatin and Leupeptin to a final concentration of 1 μg/mL.
Sonicate the cell suspension for a total of 3 min at 35 Amp with a 3 s ON and 20 s OFF cycle. Take out some sample as the post-sonication lysate.
Transfer the lysate into appropriate centrifuge tubes and spin at 30,000 RCF for 45 min. Take a sample of the pellet and the supernatant to run on the gel.
Load the supernatant onto a Cytiva HisTrap FF 5mL column (Ni-NTA column). If using a previously used column, it is ideal to strip the Ni2+ off the column and recharge the column with fresh Ni2+ as needed. Collect the flow through from the sample application step for analysis on a gel.
After loading the supernatant onto the Ni-NTA column, wash with 10 column volumes of Ni-NTA buffer A to remove non-specifically bound proteins. It is a good practice to monitor and ensure that the UV 280 nm is brought to the baseline (i.e., like the column equilibration step) by the end of this step.
Elute the bound protein off the Ni-NTA column using a continuous gradient to reach 100% B (Ni-NTA buffer B: 1× concentration is 20mM HEPES pH 7.5, 1M NaCl, 500mM Imidazole) in 15 column volumes. Usually, FnoCas12a elutes as a single major peak at around 175 mM imidazole. Once the run is completed, check every alternate fraction from the major peak, representative fractions from any minor peak, and other samples that were collected during the different steps in the protocol on a 7.5% denaturing sodium dodecyl sulfate-polyacrylamide (SDS) gel (Maizel & Jacob, 2000). The expected size of FnoCas12a with an attached MBP tag is ~196kDa.
Based on the protein banding pattern on the gel (Fig. 3A), pool the Ni-NTA fractions containing FnoCas12a into a beaker and take a pre-dialysis protein sample for gel analysis. Based on the intensity of the FnoCas12a band, add TEV protease to cleave off the MBP tag (~1 mg of TEV from a preparation starting with 2L cell culture; suggested ratio of TEV: protein of interest is 1:100, Raran-Kurussi, Cherry, Zhang, & Waugh, 2017). Transfer this sample to a 30kDa cut-off dialysis membrane and dialyze it against 1 L of dialysis buffer/ion exchange buffer A (20mM HEPES pH7.5, 150mM KCl, 2mM ethylenediaminetetraacetic acid (EDTA), 1mM dithiothreitol (DTT)) overnight with slow stirring at 4°C. We have experienced that diluting the pooled fractions with the Ni-NTA buffer B (~50–75mL for a 2 × 1 L preparation; done before the addition of TEV protease) before dialyzing prevents precipitation of the protein after the overnight dialysis.
Spin the overnight dialyzed protein (e.g., at 4°C for 15min at 4000 RCF) to remove any trace amounts of precipitates. Load the dialyzed protein onto a Cytiva HiTrapTM SP HP cation exchange column that has been equilibrated with dialysis buffer/ion exchange buffer A. Wash the column with 10 column volumes of ion exchange buffer A and elute with a continuous gradient of 0–100% using ion exchange buffer B (1× concentration: 20mM HEPES pH7.5, 1M KCl, 2mM EDTA, 1 mM DTT) over 20 column volumes. FnoCas12a elutes from the SP HP column at ~350mM KCl (Fig. 3B).
Check the fractions along with the shoulders of the peaks on a 7.5% SDS gel. The pre-dialysis and post dialysis samples should also be checked on the gel to ensure completeness of the TEV cleavage step. The expected molecular weight of FnoCas12a after TEV cleavage is 152 kDa.
Pool the fractions containing pure FnoCas12a and concentrate to ~1.7mL by centrifugation using a 30kDa cut off concentrator at 4°C. Load the sample on to an SEC (HiPrep™ 16/60 Sephacryl® S-300 HR or HiPrep™ 16/60 Sephacryl® S-200 HR) that has been equilibrated with SECbuffer (1× concentration is 20 mM HEPES pH7.5, 150mM KCl, 2mM EDTA and 1 mM TCEP). Elute the protein by isocratic elution using the SEC buffer. FnoCas12a usually elutes at ~60mL while using an S300 column that has a total column volume of 120 mL (Fig. 3C).
Check the SEC fractions on a 7.5% SDS PAGE gel. Also check the Abs (260/280) of each of the fractions to monitor nucleic acid contamination in the protein. Pool the fractions that are devoid of additional bands when visualized on the protein gel and based on the absorbance ratio. (Fractions with an Abs (260/280) ratio between 0.5 and 0.65 will have minimal nucleic acid contamination and are of good quality to be pooled together).
Concentrate the fractions using a 30 kDa cut-off spin column at 4°C. Keep monitoring the concentration of the protein and once it reaches an ideal concentration, aliquot and flash freeze the protein using liquid nitrogen. Store the aliquots at −80°C. It is a good practice to prepare smaller aliquots such that one frozen sample will be used for one activity assay. This prevents multiple freeze-thaw cycles of the purified FnoCas12a protein, which will maintain high quality active protein for biochemical assays. The molar extinction coefficient of FnoCas12a that has 1305 amino acids (5 amino acids are left after the TEV cleavage) is 144,330 M−1 cm−1.
Fig. 3.
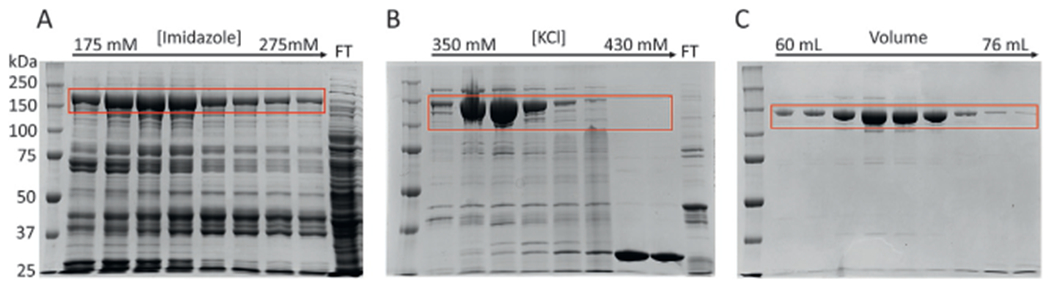
Purification of FnoCas12a from E. coli Rosetta cells by nickel-NTA (A), ion exchange (B), and size exclusion chromatography (C). The band corresponding to Cas12a is shown in red box in each gel. The molecular weight of FnoCas12a is 196kDa in Ni-NTA elution due to the attached MBP tag. For ion exchange and SEC columns, the expected molecular weight is 152kDa, after cleavage of the MBP tag. Flow Through (FT).
3. Preparation of crRNA
3.1. crRNA transcription
Cas proteins are guided by their crRNA to a specific DNA target that possesses a cognate PAM and a complementary sequence to the guide region of the crRNA. Even between Cas orthologs, the crRNA sequence can differ, and the overall secondary structures can be variable (Mir, Edraki, Lee, & Sontheimer, 2018; Yan et al., 2019). The FnocrRNA is usually around ~45 nucleotides (nt) long and contains ~19-nt of a repeat derived sequence and ~24-nt of the spacer sequence in the 5′–3′ direction (Fonfara et al., 2016; Safari et al., 2019; Zetsche et al., 2015). The repeat-derived sequence is specific for each ortholog, while the spacer sequence (i.e., the guide region) can be designed to target any specific DNA of interest possessing a flanking PAM sequence. The easiest option to produce crRNA is by in vitro transcription using oligo DNA templates. This requires two oligo DNAs: (i) a template strand (non-coding strand) which is used to transcribe crRNA; (ii) a T7 promoter strand that is needed for the T7 RNA Polymerase to transcribe the template strand.
Template strand: In the 5′–3′ direction, the template strand contains the reverse complement sequences of the following in the specific order as shown: the 24 to 25-nt targeting region of the DNA [i.e., the sequence of the target strand (TS) of the DNA; Fig. 4], the repeat-derived crRNA sequence, and the T7 promoter sequence.
T7 promoter strand: This oligo can be shorter possessing only the T7 polymerase promoter sequence in the 5′–3′ direction (if using T7 RNA Polymerase for in vitro transcription). Having only the promoter sequence enables the use of this strand for transcribing different crRNAs with unique sequences.
Fig. 4.

Sequences related to crRNA-DNA complex. (A) Schematic of the R-loop formed with the matched DNA substrate where the PAM sequence is gold, spacer sequence of the crRNA and the TS of the DNA that are complementary to each other are in red, and restriction enzyme sites are underlined. Arrows above the NTS point to nucleotide positions away from the PAM illustrating the mismatch (MM) positions between the DNA substrate and crRNA. Black triangles indicate Cas12a cleavage sites. (B) Schematic of the T7 promoter strand and template strand oligos for crRNA transcription. Bolded sequence is the T7 promoter sequence and its complement. Blue sequences are extra nucleotides added to increase transcription efficiency.
There are also modifications that can be added to the DNA template to improve RNA transcription. For example, the addition of three guanines to the 5′ terminus of the crRNA sequence can increase the efficiency of in vitro transcription by T7 RNA Polymerase. A step-by-step description of crRNA transcription is provided below. The reaction volume can be changed based on the amount of RNA required. A 200 μL transcription reaction following the protocol mentioned below will yield approximately 20–50 μg of RNA.
Obtain synthetic oligonucleotides for the template and T7 promoter strands from a vendor (e.g., Integrated DNA Technology, Sigma Aldrich, ThermoFisher Scientific). Prepare working stock solutions of the template strand at 10 μM and the T7 promoter strand at 20 μM concentrations. (Note: based on the scale of transcription required, the concentration of the strands can be varied.)
Reconstitute these oligos in molecular-grade water to produce the transcription duplex. For this, the template and T7 promoter strands can be annealed in a 1:1.5M ratio in an annealing buffer (e.g., 1× annealing buffer: 10mM Tris-HCl, pH 8, 50mM NaCl). Heat this mixture to 95°C for 5 min and allow to cool slowly to room temperature. Measure the concentration of the annealed DNA using extinction coefficient for double stranded DNA. The annealed DNA can be stored at −20°C and used as needed to perform transcription reactions.
Prepare 200 μL in vitro transcription reaction by mixing 1× transcription buffer (40 mM Tris-HCl, pH8, 1 mM spermidine, 50 μg bovine serum albumin, 20 mM MgCl2, 5mM DTT), 400 ng annealed transcription template, 8mM ATP, 8mM CTP, 8mM UTP, 9mM GTP, 50 μg RNasin (New England BioLabs), 1 μg inorganic phosphatase (New England BioLabs), and 40 μg T7 RNA polymerase. Incubate the reaction at 37°C for 4h. T7 RNA polymerase can be purified in house (Rio, 2013) or can be purchased from a vendor (use amounts specified by the vendor in this case). Adding T7 RNA polymerase in two steps can increase the efficiency of transcription (for example, 30 μg at the start of the reaction and an additional 10 μg after 3h).
Since the DNA template for transcription and the expected RNA product are of similar size, removing the DNA template will improve the quality of the RNA product. This can be achieved by DNase treatment at 37°C for 30 min [add 0.01 mg/mL DNase I (New England Biolabs) and 1× DNase buffer (10mM Tris-HCl, pH7.5, 2.5mM MgCl2, 0.5mM CaCl2) to the 200 μL transcription reaction].
The RNA in the transcription reaction can be precipitated for further purification. For this, add 3-volumes of cold 100% ethanol to the above reaction. Mix the tube by gentle inversions and store the mixture overnight (16–18h) at −20°C to precipitate the RNA out of solution. In 100% ethanol, the RNA can be stored at −20°C for longer periods of time. As needed, a small reaction volume can be tested on a 12% denaturing urea-formamide acrylamide gel along with an appropriate ladder to assess the success of transcription and the size of the RNA band (Beckert & Masquida, 2011).
3.2. crRNA purification
To ensure a good quality RNA product (i.e., a single band of the right size), it is a good practice to extract the in vitro transcribed RNA out of a denaturing urea acrylamide gel. A suggested procedure is below. (Note: while for quality analysis of RNA a urea-formamide acrylamide gel can be used, for RNA extraction and purification, urea acrylamide gels should be used. The pelleted RNA should be dissolved in an 8 M urea-containing buffer to ensure proper unfolding of RNA that is devoid of secondary structures.)
Centrifuge the RNA sample to pellet the RNA precipitate (for example, 21,130 RCF at 4°C for 30min or as appropriate). The precipitated RNA will form a small white pellet. Wash the pellet with two successive cold 80% ethanol washes. Flick the tube to dislodge the pellet while performing 80% ethanol washes. After the second wash, remove as much of the supernatant as possible without disturbing the RNA pellet. Completely evaporate all liquid from the pellet. This can be achieved by air drying for an appropriate time or by assisted drying using a vacufuge [e.g., Eppendorf Vacufuge plus 5305 V-AL (vacuum alcoholic solutions) set at 30°C]. During the washes and vacufuging, the RNA pellet will change its appearance from bright white to a translucent white indicating that salts are being washed out of the RNA pellet. Resuspend the RNA pellet by adding 200 μL of 1× gel loading solution [8 M urea, 1× TBE (89mM Tris-HCl, 89mM boric acid, 2mM EDTA), 0.02% bromophenol blue, 0.02% xylene cyanol] to the dried RNA pellet. Resuspension can be enhanced by frequent mixing of RNA in the gel loading solution and by slightly warming the solution. Ensure that the RNA pellet is completely dissolved before loading on the gel. (Note: to visualize RNA dissolution, it is a common practice to resuspend the RNA pellet in the gel loading dye devoid of the dyes, followed by adding the required amounts of dyes before loading the sample on the gel).
Prepare a 12% polyacrylamide gel containing 8 M urea. The length of the gel depends on the size of the RNA product and the number of products after transcription. For crRNA of 45-nt size, gels with the dimensions 320mm × 165mm × 1.5mm (h × w × d) are effective. Preheat the gel to 50°C and clean settled urea out of the wells before loading sample. Load the sample and run at 35 V for ~50–65 min until the RNA bands resolve. The bromophenol blue dye front can be used to track the gel running time (for 45-nt RNA product, it is advisable to stop the gel once the bromophenol blue band reaches the bottom of the gel. In a 12% polyacrylamide gel, the bromophenol blue dye migrates comparative to 12-nt, and the xylene cyanol dye migrates comparative to 55-nt (Ausubel et al., 1987). Running the bromophenol blue dye to the bottom of the gel ensures maximum separation between the 45-nt RNA band and the xylene cyanol band without the RNA band running out of the gel.
Wrap the gel in plastic wrap and visualize the RNA band by UV shadowing (for example, using a hand-held UV imager like the VWR UV-AC Dual Hand Lamp at wavelength 254 nm). It is very important to use a long wavelength UV and perform the visualization quickly to prevent RNA damage from UV. Use a marker to outline the RNA band (on the plastic wrap) and cut it around the marker. Transfer the gel piece into a 50 mL conical tube, crush the gel piece, and add enough volume of RNA elution buffer (50mM potassium acetate, 20mM KCl, adjust to pH 7) to cover the gel piece. Rotate the sample at 4°C overnight (16–18 h) to elute RNA into the solution. (Note: electroelution (for example using the CBS Scientific Electro-Eluter Concentrator ECU-040) from the RNA band is another option for this process).
Briefly centrifuge the 50mL conical tube at a low centrifugal force to collect all the liquid off the sides. Filter the liquid with a 0.22 μm filter to separate the gel pieces from the solution (e.g., using the 50 mL disposable Steriflip Vacuum-driven Filtration System with a 0.22 μm membrane). Add 2.5–3 volumes of cold 100% ethanol to precipitate RNA and store at −20°C, which will provide elution-1 of the RNA sample. Note: the gel pieces left behind after elution-1 can be returned to the 50 mL conical tube for another one or two elutions based on the amount of RNA present in the transcription reaction (estimate this based on the band intensity during ultraviolet (UV) shadowing). The elutions are typically stored in 100% ethanol at −20°C until further use to prevent RNA degradation.
To extract RNA from 100% ethanol, centrifuge the sample [for example, 3220 RCF at 4°C for 30 min (variable based on the centrifuge being used)]. Remove the supernatant and wash the pellet successively with two, 80% cold ethanol washes. After the second 80% ethanol wash, dry the RNA pellet and resuspend in RNase-free water or an appropriate buffer (1× annealing buffer, 10mM Tris-HCl, pH8, 50mM NaCl). Anneal the RNA by incubating it at 95°C for 2 min and slowly cooling to room temperature to allow the crRNA to attain its secondary fold. Quantify the folded RNA by measuring the absorbance at 260 nm. The folded FnoCas12a crRNA can be stored at 4°C for up to a week without significant activity reduction. The stability of crRNA should be closely monitored for specific Cas12a orthologs. Addition of divalent metal ions, (usually Mg2+ at 1 mM concentration) in the annealing reaction buffer is also an option for aiding proper crRNA folding (Babu et al., 2019). Inclusion of Mg2+ in the annealing reaction will require slight modifications in the annealing process, for example heating to a lower temperature (<60°C) or adding Mg2+ only during the slow cooling process when the temperature reaches around 60°C (Babu et al., 2019).
In addition to in vitro transcription using oligo DNA templates as mentioned in this protocol, other methods such as using a vendor to synthesize RNA (e.g., Integrated DNA Technologies, ThermoFisher Scientific, Sigma Aldrich) (Swarts & Jinek, 2019), in vitro transcription using a linearized plasmid holding the crRNA gene as the template using run-off transcription (Zoephel, Dwarakanath, Richter, Plagens, & Randau, 2012), and modification of oligo DNA template lengths for in vitro transcription can be adopted as needed (Karvelis et al., 2013).
4. Activity assays to characterize Cas12a properties
Previous studies have shown that FnoCas12a is capable of three different forms of DNA cleavage: (i) cis cleavage, (ii) trans cleavage, and (iii) RNA-independent or guide-free DNA cleavage (Fig. 5) (Chen et al., 2018; Sundaresan et al., 2017; Swarts & Jinek, 2019). In addition, Cas12a is able to process its crRNA to mature forms needed for DNA cleavage (Swarts et al., 2017).
Fig. 5.

Illustration of the components, substrates and products produced from the cis, trans, and guide-RNA independent cleavage assays. (Note: the products for the guide RNA-independent activity assay depend on the Cas12a ortholog and the divalent metal ion present (Li et al., 2020;Parameshwaran et al., 2021)) For assays with labels, only visible products are shown.
Cis cleavage is the guide RNA-dependent, sequence specific double stranded cleavage of a DNA target that possesses a complementary sequence as the guide region of the crRNA and a cognate PAM (Fig. 4). NTS is cleaved by the RuvC domain, while TS is cleaved by RuvC along with the assistance of Nuc domain (Yamano et al., 2016). There is a sequential order to cleave DNA by Cas12a, with NTS preceding TS cleavage, even though studies have shown that nicks or other destabilization of the NTS strand will allow TS cleavage without the prerequisite of NTS cleavage (Swarts & Jinek, 2019). Cis-DNA cleavage is the basis for gene editing applications using Cas12a.
Trans cleavage is the non-specific cleavage of ssDNA by the RuvC domain, which occurs as an after-effect of cis DNA cleavage. This cleavage needs crRNA and can be activated under the following conditions: (i) after cleavage of a dsDNA target by Cas12a followed by release of the PAM-distal DNA product that exposes RuvC active site for ssDNA binding and cleavage; (ii) Cas12a-crRNA bound to a TS strand representing a post-cleavage product (i.e., a 23-mer ssDNA bearing at least 15-nt complementarity with the guide-region of the crRNA and a cognate PAM) (Chen et al., 2018).
RNA-independent cleavage is the non-specific degradation of ssDNA or nicking of dsDNA in the presence of certain divalent metal ions such as Mn2+ and Co2+. This activity is crRNA-independent (Sundaresan et al., 2017). Recent literature shows that RNA-independent DNA cleavage activity can be promoted by Mg as well in other Cas12a orthologs such as AsCas12a (Li et al., 2020). Guide RNA free DNA cleavage by Cas9 was shown to induce DNA damage in human cells transfected with Cas9 devoid of guide RNA (Saha et al., 2020), and hence must be assessed for Cas protein orthologs while developing genome tools.
All the three in vitro DNA cleavage assays require purified Cas protein, but both cis and trans activity assays also require purified crRNA. The different cleavage mechanisms require the preparation of different DNA substrates. Protocols for each specific activity are mentioned in the following sections.
4.1. Cis DNA cleavage assay
Cis DNA cleavage is usually performed using a plasmid or a linear dsDNA substrate possessing a complementary sequence as the guide-region of the crRNA and flanked on the upstream by a cognate PAM (Fig. 4). In addition to analyzing cleavage of a target DNA possessing a completely complementary region as the crRNA guide (called matched DNA), sensitivity to cleave DNA possessing mismatches with the guide-region is also routinely performed to characterize new variants and orthologs of Cas12a. We describe the protocol for plasmid based cis cleavage assays below.
4.1.1. Preparation of matched and mismatched plasmid DNA substrates
Order NTS and TS oligos from IDT that consists of a sequence comprising a 24-nt region complementary to the crRNA-guide region and a 3-nt cognate PAM sequence. Since the protospacer sequence needs to be inserted into a plasmid, such as pUC19, the oligo ends should hold restriction site sequences that will be used to ligate the oligo into the plasmid in the correct direction (e.g., BamHI and EcoRI sites) (Fig. 4). (Note: It is advisable to order this oligo to resemble the post cleavage sequence left after restriction digestion, along with a 5′-phosphate on both ends to enable easy ligation into the plasmid.)
Anneal the TS and NTS oligos by mixing equal molars (for example: 10 μM each) of each of the strands in 1× annealing buffer (10 mM Tris-HCl, pH 8, 50mM NaCl). Heat the reaction to 95°C for 2min and then allow it to slowly cool to room temperature.
Phosphorylate the newly annealed DNA target insert following manufacture recommendation for the kinase enzyme. An example reaction includes mixing up to 300pmol of 5′ ends with 1× T4 polynucleotide kinase (PNK) buffer, 1 mM ATP and 10 units ofT4 PNK enzyme (New England BioLabs) to a final volume of 50 μL. Incubate the mixture at 37°C for 30 min. Inactivate the enzyme by incubating at 65°C for 20 min. (Note: If the oligos were ordered from IDT with phosphorylated 5′-ends, this step can be skipped.)
Digest 1 μg of pUC19 plasmid containing the ampicillin selection marker by adding 20 units of EcoRI (New England BioLabs), 20 units of BamHI (New England BioLabs) and NEBuffer r3.1 in a total reaction volume of 50 μL. Incubate at 37°C for 1 h.
To prevent self-ligation of the linearized pUC19, dephosphorylate the product after restriction digestion by adding 5 μL of Antarctic Phosphatase Reaction Buffer 10× (New England Biolabs) and 5 units of antarctic phosphatase (New England BioLabs) directly to the 1 μg of digested plasmid. Incubate the mixture at 37°C for 30min. Deactivate the enzyme by incubating at 80°C for 2 min.
To remove the presence of trace amounts of uncut plasmid that can produce negative colonies after ligation, extract the linearized pUC19 by resolving the digested products on a 0.8% agarose gel pre-stained with ethidium bromide. Cut out the digested band of the right size and use a gel extraction kit (Omega Biotek E.Z.N.A. Gel Extraction Kit, QIAGEN QIAquick Gel Extraction Kit) to extract the linearized plasmid from the gel.
Ligate the phosphorylated insert and the dephosphorylated plasmid vector following the recommendations for the specific ligase being used. For example, T4 ligase (New England BioLabs) recommends mixing 50 ng of vector with 37.5 ng of insert, 1× T4 Ligase Buffer (New England BioLabs) and 1 μL T4 DNA ligase (New England BioLabs) in a total reaction volume of 20 μL.
Transform 5–7 μL of the ligation reaction into 50 μL of E. coli DH5α cells following manufacture recommendations and plate on a Luria-Bertani-agar (LB-agar) culture plate containing 0.1mg/mL ampicillin and grow overnight at 37°C. Choose a few single colonies from this plate and confirm insertion of the protospacer by Sanger sequencing. Maintain glycerol and plasmid stocks of the positive clone.
The positive clone for the “matched/wild-type” plasmid substrate can be used as a template for site-directed mutagenesis (SDM) (Bachman, 2013) to develop the mismatched DNA substrates using a primer that introduces a single/multiple nucleotide “mismatch(es)” in the protospacer region (i.e., changing the DNA sequence to introduce non-complementarity with the guide-region of the crRNA). The mismatch positions are numbered according to the nucleotide’s position downstream of the PAM on the non-target strand (NTS). Alternatively, mismatches can be introduced by changing the nt of the guide region of the crRNA and keeping the protospacer sequence constant (Zetsche et al., 2015). In this case, in vitro transcriptions with different crRNA template strands need to be performed to create mismatch containing crRNA.
4.1.2. Cis DNA cleavage reaction
Cas12a is a single turn over enzyme under in vitro conditions for cis DNA cleavage. Most of the protocols assessing DNA cleavage properties of Cas12a orthologs and variants uses an excess of protein compared to the DNA substrate. Recombinantly purified Cas12a protein is bound with purified and annealed crRNA and incubated with DNA substrate in an appropriate buffer containing a divalent metal ion such as Mg2+ for a required amount of time. A protocol for cis DNA cleavage is as follows.
A reaction volume of 10 μL will contain the following: 1× cleavage buffer (20mM HEPES, pH7.5, 150mM KCl, 5% glycerol, 0.5mM DTT), 5mM MgCl2, 25nM Cas12a, 30nM crRNA, (1:1.2M ratio of Cas12a and crRNA), and 100ng of a plasmid DNA substrate (~5nM for pUC19-based substrate) containing the protospacer and PAM as mentioned in Section 4.1.1. Omit Cas protein in the control sample.
Incubate all the reaction components except the DNA substrate at 37°C for 10 min to form the ribonucleoprotein (RNP) complex.
Following this, add the DNA substrate and incubate at 37°C for 30min or another desired time.
Stop the reaction by adding the stop dye (1× composition: 50mM EDTA, 1% SDS, 10% glycerol, 0.08% orange G).
Resolve the reaction products on a 1% agarose gel and visualize the DNA bands by post-staining with 100 mL of 1 μg/mL ethidium bromide solution (Fig. 6). Post-staining is recommended to resolve linear, nicked, and supercoiled DNA bands, which need to be quantified separately to characterize the DNA cleavage properties of Cas proteins. Destaining with 100mL water following ethidium bromide staining is advisable to reduce background staining and saturating DNA band signal. The band intensities can be quantified as mentioned in Section 5 to assess the DNA cleavage properties of Cas12a protein. (Note: a user can optimize the staining and destaining protocol for ideal DNA band qualities as needed.)
Fig. 6.

Agarose gel showing cis DNA cleavage using a ds plasmid DNA substrate. Cis activity assay using a wild-type FnoCas12a (FnoCas12aWT) and a variant FnoCas12a (FnoCas12aVar; FnoCas12a-K969P/D970P) on a substrate with 100% complementarity with the guide sequence of the crRNA (WTS) or a substrate with a mismatch with the guide sequence of the crRNA and DNA target (MM2, MM3, MM8, numbers represent mismatch position downstream of PAM) are shown. See Fig. 4 for more details about mismatch positions. A 10 μL reaction with 100 ng of substrate, as mentioned in Section 4.1.2, was resolved on a 1% agarose gel. The control reaction was done in the absence of FnoCas12a protein. Arrows on the right show to where the nicked (N), linear (L) and supercoiled (SC) products migrated. FnoCas12aVar shows lower DNA linearization compared to FnoCas12aWT, which is more pronounced in mismatch containing DNA indicating the sensitivity of the variant in cleaving mismatch containing DNA.
Alternative methods to characterize Cas12a’s DNA cleavage activity includes use of linearized plasmid or use of annealed target strand (TS) and non-target strand (NTS) which can be labeled with 32P or fluorescein amidite (FAM) at both or one of the 5′ ends (Parameshwaran et al., 2021). Linearized substrate is created by restriction digestion of the plasmid substrate containing the protospacer matching the guide region of the crRNA flanked by a cognate PAM. Ideally, the restriction enzyme should be selected such that double stranded cleavage by Cas12a will create two linear bands with distinct sizes that can be resolved by an agarose gel. Similar rationale should be used while designing TS and NTS strands for oligo DNA cleavage assay. For oligo cleavage assays, the products should be resolved on a urea-formamide gel of appropriate percentage needed to resolve the different DNA bands (Parameshwaran et al., 2021). Another modification for oligo cleavage is labeling only one of the strands or using two distinct labels for each of the two DNA oligo strands to enable monitoring of NTS and TS cleavages separately.
4.2. Trans DNA cleavage assay
The trans activity assay requires ssDNA substrates. Both single stranded M13mp18 plasmid (New England BioLabs) and a ssDNA oligo (length can vary) can be used as substrates for trans cleavage. In addition to the ss DNA substrate, the trans activity assay also requires a ssDNA or dsDNA activator. The activator has a complementary sequence to the guide-region of the crRNA, and when they pair, a conformational change is induced in the protein which opens the RuvC catalytic pocket to allow ssDNA to enter and to be processively cleaved (Swarts & Jinek, 2019). (Note: Though trans activity is robust with ssDNA substrates, it has been reported to occur on dsDNA and RNA substrates with different Cas12a orthologs with appropriate reaction conditions (Fuchs, Curcuru, Mabuchi, Yourik, & Robb, 2019).)
4.2.1. Substrates
Procure single stranded M13mp18 plasmid (New England BioLabs) to use as a single-stranded, circular substrate.
Order a 54-nt oligo from IDT to be used as a single-stranded, linear substrate. This DNA can be labeled with 32P or FAM to specifically monitor the cleavage of this DNA since the reaction mix contains several other DNA strands (e.g., activator strands). (Note: the length of the DNA substrate can vary.)
4.2.2. Activators
For the single stranded activator, order an oligo (IDT) that contains a PAM sequence and at least 15-nt of complementarity with the guide region of the crRNA (Chen et al., 2018).
For the double stranded activator, order forward and reverse activator strands that contain Cas12a’s cognate PAM sequence and at least 15-nt of complementarity with the crRNA (Chen et al., 2018). Anneal the strands by mixing them in a 1:1 M ratio in 1× annealing buffer (10 mM Tris-HCl, pH 8, 50mM NaCl). Incubate at 95°C for 2min and allow to slowly cool to room temperature.
4.2.3. Reaction
A reaction volume of 10 μL will contain the following: 1× cleavage buffer (20mM HEPES, pH7.5, 150mM KCl, 5% glycerol, 0.5mM DTT), 5mM MgCl2, 25nM Cas12a, 30nM crRNA, (1:1.2M ratio of Cas12a and crRNA), 30 nM of activator, and 100ng of DNA substrate. Omit Cas protein in the control sample.
Incubate all the reaction components except the activator and the DNA substrate at 37°C for 10 min to form the ribonucleoprotein (RNP) complex.
Following this, add the activator and DNA substrate, and incubate at 37°C for 60min.
Stop reaction by adding the stop dye (1× composition: 50mM EDTA, 1% SDS, 10% glycerol, 0.08% orange G).
Resolve the reaction products on a 1% agarose gel (if using M13mp18 substrate) and visualize the DNA bands by post-staining with ethidium bromide as mentioned in Section 4.1.2 (Fig. 7). If using oligo DNA, resolve the products on an acrylamide gel appropriate for the size of the ssDNA substrate and visualize product formation by monitoring 32P or FAM or another appropriate label’s signal intensity. The band intensities can be quantified as mentioned in Section 5 to characterize the trans DNA cleavage of Cas12a.
Fig. 7.

A trans activity assay performed with increasing concentrations of wild-type FnoCas12a (FnoCas12aWT) or a variant FnoCas12a (FnoCas12aVar; FnoCas12a-K969P/D970P) complexed with crRNA (RNP). Activator was 24bp of dsDNA. A 10 μL reaction with 100ng of single-stranded substrate, as mentioned in Section 4.2.3, was resolved on a 1% agarose gel. Arrows on the right show to where the circular (Cr), linear (L), and degraded (D) products migrated. In the example shown, FnoCas12aWT has robust trans cleavage activity, while it is reduced in the variant (FnoCas12aVar).
4.3. RNA-independent DNA cleavage assay
Several Cas proteins can perform non-specific DNA cleavage through the RNA-independent DNA cleavage in the absence of a cognate crRNA/tracrRNA (trans activating crRNA). The types of DNA substrates that can be cleaved (e.g., ssDNA, dsDNA, plasmid, linear DNA) varies between different Cas proteins and the divalent metal ions that can promote the RNA-independent DNA cleavage also varies between different Cas proteins and orthologs (Li et al., 2020; Sundaresan et al., 2017). So, it is advisable to test different DNA substrates and different divalent ions while testing this activity in a Cas protein.
4.3.1. Substrates
Procure M13mp18 single-stranded circular plasmid (New England BioLabs), double-stranded plasmid (e.g., pUC19), linearized ss and ds plasmids using appropriate restriction enzymes, PCR amplified dsDNA of appropriate lengths that need to be tested, or oligos of appropriate length from a vendor that can be used for ssDNA or annealed to create short dsDNA substrates.
4.3.2. Reaction
A reaction volume of 10 μL will contain the following: 1× buffer (20mM HEPES, pH7.5, 150mM KCl, 2mM TCEP), 100nM purified Cas12a, 100 ng DNA substrate and 10 mM divalent metal.
Omit Cas12a in control sample to obtain the intensity of the uncleaved substrate. While using ds plasmid substrate (e.g., pUC19), create nicked and linear markers by digesting pUC19 with Nt.BspQI (one nicking site in pUC19) and EcoRI (one linearization site in pUC19) restriction enzymes respectively.
Incubate the reaction mix at 37°C for 30min.
Stop reaction with 1× stop dye (50mM EDTA, 1% SDS, 10% glycerol, 0.08% w/v Orange G).
Resolve the reaction products on a 1% agarose gel and post stain with ethidium bromide (Fig. 8).
Fig. 8.

RNA-independent activity assay performed with wild-type FnoCas12a (FnoCas12aWT) with no added divalent metal or in the presence of EDTA, Mg2+, or Mn2+ using a dsDNA plasmid [pUC19, (A)] or a ss DNA plasmid [M13mp18, (B)]. A 10 μL reaction with 100ng of substrate, as mentioned in Section 4.3.2, was resolved on a 1.2% agarose gel. The control reaction was done in the absence of FnoCas12a protein. The nicked and linear controls were made by digesting pUC19 plasmid with Nt. BspQI and EcoRI respectively. Arrows on the sides show to where the nicked (N), linear (L), supercoiled (SC), circular (Cr), and degraded (D) products migrated. The gels show that in FnoCas12a, Mn2+ induces nicking of dsDNA plasmid and degradation of ssDNA when guide-RNA is absent.
4.4. crRNA processing assay
Cas12a proteins have the intrinsic ability to process their crRNAs with an active site in the WED domain that is independent of the RuvC active site (Fonfara et al., 2016; Zetsche et al., 2017). Since crRNA processing does not involve the RuvC active site, this reaction can be performed with a “dead” Cas12a (dCas12a) that has an inactive RuvC site or a Cas12a protein with a functional RuvC active site. The following protocol is from the publication by Swarts et al. (2017) in Molecular Cell.
4.4.1. RNA substrates
Cas12a processes pre-crRNA into mature crRNAs and there is direct experimental evidence of Cas12a trimming the 5′ end of pre-crRNA (Safari et al., 2019). While the catalytic mechanism by which Cas12a processes pre-crRNA is metal-independent, it has been shown that divalent metal ions increase Cas12a’s affinity for crRNA by associating with the crRNA pseudoknot (Dong et al., 2016; Fonfara et al., 2016; Swarts et al., 2017). Possible RNA substrates include a wild-type pre-crRNA of appropriate length and sequence for the specific Cas12a ortholog, and a mature crRNA as a control for the size of processed crRNA. Other forms such as a pre-crRNA where the 2′-hydroxyl group upstream of the trimming site is disrupted to prevent its nucleophilic attack on the scissile phosphate can be used for mechanistic characterization of RNA cleavage. These RNA substrates can be produced using the protocol from Section 2 or through custom synthesis by vendors for modified RNAs, for example without the 2′-hydroxyl group.
4.4.2. Reaction
A reaction volume of 20 μL will contain the following: 2.5 μM purified Cas12a, 0.5× SEC buffer (1× SEC buffer, 20mM HEPES-KOH, pH7.5, 500mM KCl, 1mM DTT), 1 μM crRNA substrate, and 5mM of a chelating agent [e.g., EDTA, EGTA (ethylene glycol-bis (β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid)] or divalent cation (e.g., CaCl2, MgCl2, MnCl2, NiCl2). Incubate at 37°C for 10min.
Stop the reaction by adding 80mM EDTA and 0.8 mg/mL proteinase K, and incubate at 37°C for 30min. (Note: EGTA is better at chelating Ca2+ compared to EDTA.)
Add 20 μL of a 2× RNA loading dye, incubate at 95°C for 10 min and then resolve on a 7 M urea, 20% polyacrylamide gel.
Alternatively, fluorophores can be attached to the 5′ or 3′ end of the pre-crRNA substrate, and the reaction products can be resolved on a denaturing gel and fluorescence detected with a gel imager (Nguyen et al., 2020).
4.5. Time course activity assay
An activity assay where the reaction is stopped at progressive time points results in data that can be used to calculate rate constants of the reactions. The setup is the same as in the cleavage assays mentioned above except for stopping the reaction at the required time points.
Prepare a 10 μL reaction for each time point of interest (an example for time points is 0 s to 1 h, with a few time points in seconds (15 and 30 s) and the rest in minutes that are appropriate to capture the reaction kinetics) (Parameshwaran et al., 2021).
Stop the reaction at the desired time point with 1× stop dye.
Resolve the reaction products on an appropriate gel, and image the gels with an appropriate method (Fig. 9).
Fig. 9.

Time course cis DNA cleavage assay performed with FnoCas12aWT and a double-stranded substrate with a mismatch in position 8 (MM8). A 10 μL reaction with 100 ng of substrate, as mentioned in Section 4.5, was resolved on a 1% agarose gel. Arrows on the right show to where the nicked (N), linear (L) and supercoiled (SC) products migrated. Nicked DNA is the intermediate in the reaction and linear DNA is the final product. Note that the amount of nicked DNA raises first and then decreases when it is converted to linear product at longer time points.
4.6. Concentration titration assay
While characterizing a new Cas protein ortholog or a variant, it is advisable to test different concentrations of the protein-RNA complex to analyze differences in reaction saturation conditions when compared to a well characterized ortholog or the wild-type protein. The setup is the same as in the cleavage assays mentioned above except for using different concentrations of RNP or other reaction components (e.g., concentration of divalent metal, activator for trans cleavage).
Prepare a 10 μL reaction for each concentration point of interest (examples include protein titration from 0 to 1 μM; divalent metal analysis at 1, 5, and 10mM) (Parameshwaran et al., 2021).
Stop the reaction at the desired time point with 1× stop dye.
Resolve the reaction products on a gel and image appropriately (Fig. 7).
5. Quantification of Cas12a activity
Visual identification of the activity assay gels is not accurate enough to form conclusions. Quantification of DNA bands corresponding to substrates and products can provide accurate measurements of Cas12a activities. The DNA bands can be quantified using the ImageJ program (Rasband, 1997) and then used in calculations with normalization against the no protein control lane to obtain quantitative measurement of activity. To avoid experimental errors, it is important to include control lanes for each reaction to account for any variation in reaction components. Similarly, to account for differences in active protein fractions, it is recommended that different replications be performed for each experiment using at least two independent protein preparations, for example, three reaction replicates using two independent protein preparations. The following protocol and calculations refer to the possible quantification products produced by the activity assays.
For cis cleavage using a plasmid substrate, there are three different measurements that need to be taken for quantification: nicked (N) product [refers to a double-stranded circular DNA substrate that has undergone cleavage of only one strand, which is an intermediate during the linearization of supercoiled DNA], linear (L) product [refers to a double-stranded DNA substrate that has undergone cleavage of both strands], leftover super-coiled (SC) product (refers to uncut dsDNA substrate that was unused in the reaction).
For activity assays measuring trans cleavage using single-stranded circular DNA substrates, the gel will have the following forms of DNA: uncut circular (Cr, leftover unused substrate), linear (L, intermediate where DNA is linearized, but not progressively degraded) or degraded product (D, DNA substrate that has undergone progressive degradation). For trans cleavage quantification, following the intensity reduction of circular band is ideal for quantification.
5.1. Cis cleavage assay with dsDNA plasmid
For a double-stranded plasmid substrate with multiple cleavage products, measure intensities (I) of nicked (IN), linear (IL) and supercoiled (ISC) bands using ImageJ.
Perform a background correction as recommended by the ImageJ software.
- Subtract the intensities of nicked and linear DNA bands from the control lane (i.e., no protein lane) to account for the presence of nicked and linear bands in the supercoiled substrate by using the following equations:
(1)
where the subscript “C” represents the intensities of the control lane containing no Cas protein.(2) - Calculate total cleavage as:
(3) - Calculate the fraction of remaining uncleaved (supercoiled) substrate (Frac[SC]) as:
where the subscript “C” represents the intensities of the control lane containing no Cas protein. An alternate method to calculate remaining substrate is [1-(nicked + linear)] for each lane (see reference Parameshwaran et al., 2021 for details).(4) - Calculate the standard deviation (SD) as:
where R is the value from each replication, RAV is the average of data values from all the replications, and n is the number of replications. Note that R depends on what is being plotted (e.g., total cleavage (Eq. 3), linear product formation (Eq. 2), or monitoring the usage of supercoiled substrate (Eq. 4)).(5) - Calculate the standard error of mean (SEM) as:
(6) Plot appropriate graphs (bar or line) for visual representation of the data. Bar graph is used for single time or concentration points, while line graphs are used for time or concentration gradients.
Nicked, linear, or total cleavage can be used for plotting as required to analyze the results from activity assays. This can be useful while characterizing Cas12a variants or orthologs that shows differences in linearization and nicking compared to already characterized Cas12a proteins. For off-target analysis, reduction in linearization, while maintaining efficient nicking, is considered to have better mismatch sensitivity since nicked products are repaired during genome editing applications.
For modified cis cleavage assays that use labeled oligo DNA or linearized plasmid DNA as substrates, there are slight variations in the products being quantified. Details of these calculations can be referred from our Cas12a publication that tested different types of DNA substrates for cis cleavage (Parameshwaran et al., 2021).
5.2. Trans cleavage assay with ssDNA
For single stranded DNA where the DNA is degraded, calculate the fraction of remaining uncleaved (circular) substrate (Frac[CIR]) as:
| (7) |
where ICIR is the intensity of the uncleaved (circular) band left in the reaction with protein, and the subscript “C” represents the intensity of the uncleaved band in the control lane containing no Cas protein. SD and SEM can be calculated as mentioned in Eqs. (5) and (6).
6. Kinetic analysis
The data from time course assays can be fit with appropriate equations to obtain reaction rate constants. The choice of single exponential or double exponential fit depends on the properties of each protein being tested and can also vary based on experimental parameters. Details of different equations that can be used for kinetic analysis of Cas protein assays have been recently reported (Babu et al., 2019; Parameshwaran et al., 2021). Other kinetic analysis methods have been reported using oligo assays to monitor conformational changes along the reaction pathway by careful manipulation of the reaction conditions (Liu et al., 2020; Raper, Stephenson, & Suo, 2018; Strohkendl, Saifuddin, Rybarski, Finkelstein, & Russell, 2018). Equations for single and double exponential fit of the time course data is as follows:
Single-exponential decay equation:
| (8) |
y is the measured parameter (for example: disappearance of precursor), kobs is the reaction rate constant, t is time and a is the total active fraction (i.e., the amount of substrate being used up in the reaction).
Double-exponential decay equation:
| (9) |
Frac[SC] is specified by Eq. (4), k1 and k2 are the reaction rate constants, t is time and a1 and a2 are the fraction of supercoiled substrate that reacted respectively with the k1 and k2 rate constants. The total active fraction a = a1 + a2.
Time-course measurements can be fit with OriginLab (Origin(Pro) (Version 2022b), 2021) or MATLAB (MATLAB and Statistics Toolbox (Version 2021b), 2021) graphing and analysis software.
7. Conclusions
Cas12a is a versatile protein that possesses diverse activities: both DNA and RNA cleavages. DNA cleavage itself can occur by diverse reaction mechanisms that have led to unique biotechnology tool developments using Cas12a. Thus Cas12a provides a unique system for not only improved biotechnology and biomedical applications, but also characterization of unique reaction mechanisms from a fundamental mechanistic perspective. The protocols mentioned in this report can be easily adapted to different Cas12a orthologs and variants for their biochemical characterization.
Acknowledgments
The kinetic analysis methods were developed through our collaborative work with Dr. Peter Z. Qin at the University of Southern California. We thank Dr. Qin and his laboratory for kinetic analysis methods that are being reported in this methods manuscript. We thank the use of OU Protein Production and Characterization Core (PPC Core) for protein purification and instrument support. This core is supported by an IDeA grant from the NIGMS (grant number P20GM103640).
Funding
The work reported here was partially supported by the Oklahoma Center for the Advancement of Science and Technology (OCAST) award [grant number HR20–103, awarded to RR] and partially by a grant from the Research Council and the Office of the Vice President for Research and Partnerships of the University of Oklahoma Norman Campus to RR.
Footnotes
Conflicts of interest
Two US patents have been filed for a bridge helix variant of FnoCas12a protein with the patent numbers, US20200332275A1 and US20220213459A1. The authors declare no other competing financial interest.
References
- Ausubel FM, Brent R, Kingston RE, Struhl K, Smith JA, Moore DD, et al. (1987). Current protocols in molecular biology. Greene Pub. Associates, J. Wiley; xs. order fulfillment. [Google Scholar]
- Babu K, Amrani N, Jiang W, Yogesha SD, Nguyen R, Qin PZ, et al. (2019). Bridge helix of Cas9 modulates target DNA cleavage and mismatch tolerance. Biochemistry, 58(14), 1905–1917. 10.1021/acs.biochem.8b01241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachman J (2013). Site-directed mutagenesis. Methods in Enzymology, 529, 241–248. 10.1016/B978-0-12-418687-3.00019-7. [DOI] [PubMed] [Google Scholar]
- Bayat H, Modarressi MH, & Rahimpour A (2018). The conspicuity of CRISPR-Cpf1 system as a significant breakthrough in genome editing. Current Microbiology, 75(1), 107–115. 10.1007/s00284-017-1406-8. [DOI] [PubMed] [Google Scholar]
- Beckert B, & Masquida B (2011). Synthesis of RNA by in vitro transcription. Methods in Molecular Biology (Clifton, N.J.), 703, 29–41. 10.1007/978-1-59745-248-9_3. [DOI] [PubMed] [Google Scholar]
- Broughton JP, Deng X, Yu G, Fasching CL, Servellita V, Singh J, et al. (2020). CRISPR–Cas12-based detection of SARS-CoV-2. Nature Biotechnology, 38(7), 870–874. 10.1038/s41587-020-0513-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JS, Ma E, Harrington LB, Da Costa M, Tian X, Palefsky JM, et al. (2018). CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science, 360(6387), 436. 10.1126/science.aar6245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cofsky JC, Karandur D, Huang CJ, Witte IP, Kuriyan J, & Doudna JA (2020). CRISPR-Cas12a exploits R-loop asymmetry to form double-strand breaks. eLife, 9. 10.7554/eLife.55143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong D, Ren K, Qiu X, Zheng J, Guo M, Guan X, et al. (2016). The crystal structure of Cpf1 in complex with CRISPR RNA. Nature, 532(7600), 522–526. 10.1038/nature17944. [DOI] [PubMed] [Google Scholar]
- Fonfara I, Richter H, Bratovič M, Le Rhun A, & Charpentier E (2016). The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA. Nature, 532(7600), 517–521. 10.1038/nature17945. [DOI] [PubMed] [Google Scholar]
- Fuchs RT, Curcuru J, Mabuchi M, Yourik P, & Robb GB (2019). Cas12a trans-cleavage can be modulated in vitro and is active on ssDNA, dsDNA, and RNA. bioRxiv, 600890. 10.1101/600890. [DOI] [Google Scholar]
- Gao P, Yang H, Rajashankar KR, Huang Z, & Patel DJ (2016). Type V CRISPR-Cas Cpf1 endonuclease employs a unique mechanism for crRNA-mediated target DNA recognition. Cell Research, 26(8), 901–913. 10.1038/cr.2016.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karvelis T, Gasiunas G, Miksys A, Barrangou R, Horvath P, & Siksnys V (2013). CrRNA and tracrRNA guide Cas9-mediated DNA interference in Streptococcus thermophilus. RNA Biology, 10(5), 841–851. 10.4161/rna.24203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SK, Kim H, Ahn W-C, Park K-H, Woo E-J, Lee D-H, et al. (2017). Efficient transcriptional gene repression by type V-A CRISPR-Cpf1 from Eubacterium eligens. ACS Synthetic Biology, 6(7), 1273–1282. 10.1021/acssynbio.6b00368. [DOI] [PubMed] [Google Scholar]
- Li S-Y, Cheng Q-X, Liu J-K, Nie X-Q, Zhao G-P, & Wang J (2018). CRISPR-Cas12a has both cis- and trans-cleavage activities on single-stranded DNA. Cell Research, 28(4), 491–493. 10.1038/s41422-018-0022-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S-Y, Cheng Q-X, Wang J-M, Li X-Y, Zhang Z-L, Gao S, et al. (2018). CRISPR-Cas12a-assisted nucleic acid detection. Cell Discovery, 4(1), 1–4. 10.1038/s41421-018-0028-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Li S, Wu N, Wu J, Wang G, Zhao G, et al. (2019). HOLMESv2: A CRISPR-Cas12b-assisted platform for nucleic acid detection and DNA methylation quantitation. ACS Synthetic Biology, 8(10), 2228–2237. 10.1021/acssynbio.9b00209. [DOI] [PubMed] [Google Scholar]
- Li B, Yan J, Zhang Y, Li W, Zeng C, Zhao W, et al. (2020). CRISPR-Cas12a possesses unconventional DNase activity that can be inactivated by synthetic oligonucleotides. Molecular Therapy- -Nucleic Acids, 19, 1043–1052. 10.1016/j.omtn.2019.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M-S, Gong S, Yu H-H, Jung K, Johnson KA, & Taylor DW (2020). Engineered CRISPR/Cas9 enzymes improve discrimination by slowing DNA cleavage to allow release of off-target DNA. Nature Communications, 11(1), 3576. 10.1038/s41467-020-17411-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahas A, Hassan N, Aman R, Marsic T, Wang Q, Ali Z, et al. (2021). LAMP-coupled CRISPR–Cas12a module for rapid and sensitive detection of plant DNA viruses. Viruses, 13(3), 466. 10.3390/v13030466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maizel J, & Jacob V (2000). SDS polyacrylamide gel electrophoresis. Trends in Biochemical Sciences, 25(12), 590–592. 10.1016/S0968-0004(00)01693-5. [DOI] [PubMed] [Google Scholar]
- MATLAB and Statistics Toolbox (Version 2021b). (2021). The MathWorks, Inc. [Google Scholar]
- Mir A, Edraki A, Lee J, & Sontheimer EJ (2018). Type II-C CRISPR-Cas9 biology, mechanism and application. ACS Chemical Biology, 13(2), 357–365. 10.1021/acschembio.7b00855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohanraju P,VanDerOost J,Jinek M, & Swarts DC (2018). Heterologous expression and purification of the CRISPR-Cas12a/Cpf1 protein. Bio-Protocol, 8(9), e2842–e2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen LT, Smith BM, & Jain PK (2020). Enhancement of trans-cleavage activity of Cas12a with engineered crRNA enables amplified nucleic acid detection. Nature Communications, 11(1), 4906. 10.1038/s41467-020-18615-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Origin(Pro) (Version 2022b). (2021). OriginLab Corporation. [Google Scholar]
- Parameshwaran HP, Babu K, Tran C, Guan K, Allen A, Kathiresan V, et al. (2021). The bridge helix of Cas12a imparts selectivity in cis-DNA cleavage and regulates trans-DNA cleavage. FEBS Letters, 595(7), 892–912. 10.1002/1873-3468.14051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul B, & Montoya G (2020). CRISPR-Cas12a: Functional overview and applications. Biomedical Journal, 43(1), 8–17. 10.1016/j.bj.2019.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raper AT, Stephenson AA, & Suo Z (2018). Functional insights revealed by the kinetic mechanism of CRISPR/Cas9. Journal of the American Chemical Society, 140(8), 2971–2984. 10.1021/jacs.7b13047. [DOI] [PubMed] [Google Scholar]
- Raran-Kurussi S, Cherry S, Zhang D, & Waugh DS (2017). Removal of affinity tags with TEV protease. Methods in Molecular Biology, 1586, 221–230. 10.1007/978-1-4939-6887-9_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband WS (1997). ImageJ. U.S. National Institutes of Health. https://imagej.nih.gov/ij/ [Google Scholar]
- Rio DC (2013). Expression and purification of active recombinant T7 RNA polymerase from E. coli. Cold Spring Harbor Protocols, 2013(11), pdb.prot078527. 10.1101/pdb.prot078527. [DOI] [PubMed] [Google Scholar]
- Safari F, Zare K, Negahdaripour M, Barekati-Mowahed M, & Ghasemi Y (2019). CRISPR Cpf1 proteins: Structure, function and implications for genome editing. Cell & Bioscience, 9(1), 36. 10.1186/s13578-019-0298-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha C, Mohanraju P, Stubbs A, Dugar G, Hoogstrate Y, Kremers G-J, et al. (2020). Guide-free Cas9 from pathogenic Campylobacter jejuni bacteria causes severe damage to DNA. Science Advances, 6(25), eaaz4849. 10.1126/sciadv.aaz4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shmakov S, Smargon A, Scott D, Cox D, Pyzocha N, Yan W, et al. (2017). Diversity and evolution of class 2 CRISPR-Cas systems. Nature Reviews. Microbiology, 15(3), 169–182. 10.1038/nrmicro.2016.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stella S, Alcón P, & Montoya G (2017). Structure of the Cpf1 endonuclease R-loop complex after target DNA cleavage. Nature, 546(7659), 559–563. 10.1038/nature22398. [DOI] [PubMed] [Google Scholar]
- Stella S, Mesa P, Thomsen J, Paul B, Alcón P, Jensen SB, et al. (2018). Conformational activation promotes CRISPR-Cas12a catalysis and resetting of the endonuclease activity. Cell, 175(7), 1856–1871. e21 10.1016/j.cell.2018.10.045. [DOI] [PubMed] [Google Scholar]
- Strohkendl I, Saifuddin FA, Rybarski JR, Finkelstein IJ, & Russell R (2018). Kinetic basis for DNA target specificity of CRISPR-Cas12a. Molecular Cell, 71(5), 816–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaresan R, Parameshwaran HP, Yogesha SD, Keilbarth MW, & Rajan R (2017). RNA-independent DNA cleavage activities of Cas9 and Cas12a. Cell Reports, 21(13), 3728–3739. 10.1016/jxelrep.2017.11.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swarts DC, & Jinek M (2019). Mechanistic insights into the cis- and trans-acting DNase activities of Cas12a. Molecular Cell, 73(3), 589–600.e4. 10.1016/j.molcel.2018.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swarts DC, van derOost J, & Jinek M (2017). Structural basis for guide RNA processing and seed-dependent DNA targeting by CRISPR-Cas12a. Molecular Cell, 66(2), 221–233.e4. 10.1016/j.molcel.2017.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wörle E, Jakob L, Schmidbauer A, Zinner G, & Grohmann D (2021). Decoupling the bridge helix of Cas12a results in a reduced trimming activity, increased mismatch sensitivity and impaired conformational transitions. Nucleic Acids Research, 49(9), 5278–5293. 10.1093/nar/gkab286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamano T, Nishimasu H, Zetsche B, Hirano H, Slaymaker IM, Li Y, et al. (2016). Crystal structure of Cpf1 in complex with guide RNA and target DNA. Cell, 165(4), 949–962. 10.1016/jxell.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamano T, Zetsche B, Ishitani R, Zhang F, Nishimasu H, & Nureki O (2017). Structural basis for the canonical and non-canonical PAM recognition by CRISPR-Cpf1. Molecular Cell, 67(4), 633–645. e3. 10.1016/j.molcel.2017.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan WX, Hunnewell P, Alfonse LE, Carte JM, Keston-Smith E, Sothiselvam S, et al. (2019). Functionally diverse type V CRISPR-Cas systems. Science, 363(6422), 88–91. 10.1126/science.aav7271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetsche B, Gootenberg JS, Abudayyeh OO, Slaymaker IM, Makarova KS, Essletzbichler P, et al. (2015). Cpf1 is a single RNA-guided endonuclease of a Class 2 CRISPR-Cas system. Cell, 163(3), 759–771. 10.1016/j.cell.2015.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetsche B, Heidenreich M, Mohanraju P, Fedorova I, Kneppers J, DeGennaro EM, et al. (2017). Multiplex gene editing by CRISPR–Cpf1 using a single crRNA array. Nature Biotechnology, 35(1), 31–34. 10.1038/nbt.3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoephel J, Dwarakanath S, Richter H, Plagens A, & Randau L (2012). Substrate generation for endonucleases of CRISPR/Cas systems. Journal of Visualized Experiments: JoVE, 67, 4277. 10.3791/4277. [DOI] [PMC free article] [PubMed] [Google Scholar]