

Abstract
INTRODUCTION:
Sleep-wake disturbances are a prominent feature of Alzheimer’s Disease (AD). Atypical (non-amnestic) AD syndromes have different patterns of cortical vulnerability to AD. We hypothesized that atypical AD also shows differential vulnerability in subcortical nuclei that will manifest as different patterns of sleep dysfunction.
METHODS:
Overnight-EEG monitoring on forty-eight subjects, including 15 amnestic, 19 atypical AD and 14 controls. AD was defined based on neuropathological or biomarker-confirmation. We compared sleep architecture by visual scoring and spectral power analysis in each group.
RESULTS:
Overall, AD cases showed increased sleep fragmentation and N1 sleep than controls. Compared to atypical AD groups, typical AD showed worse N3 sleep dysfunction and relatively preserved REM sleep.
DISCUSSION:
Results suggest differing effects of amnestic and atypical AD variants on slow wave versus REM sleep, respectively, corroborating the hypothesis of differential selective vulnerability patterns of the subcortical nuclei within variants. Optimal symptomatic treatment for sleep dysfunction in clinical phenotypes may differ.
Keywords: Sleep, Alzheimer’s Disease, Selective vulnerability, Neuromodulatory Subcortical Systems, Locus Coeruleus
1. Introduction
Sleep alterations such as nighttime awakenings or daytime sleepiness are widespread among Alzheimer’s Disease (AD) patients[1]. Sleep dysfunction negatively affects the patient’s wellbeing and likely leads to greater cognitive decline[2], making sleep disturbances a leading cause of caregiver burden and early institutionalization in AD[3]. Despite their significant negative impact, sleep disorders in AD are still often misdiagnosed and undertreated[4]. Furthermore, the most used pharmacological treatments are non-specific, frequently causing adverse effects, warranting research to understand the neuronal basis of sleep dysfunction and improve treatment strategies[5].
AD is a biologically-based diagnosis requiring deposits of beta-amyloid plaques and neurofibrillary tangles in a particular pattern of pathological deposition [6]. In most cases, individuals with AD initially manifest short-term memory deficits that later evolve to include other cognitive domains (amnestic or typical phenotype). However, AD neuropathology can display a predominance of language, visuospatial or behavioral impairment instead of memory at early clinical stages (non-amnestic or atypical variants)[7]. A more profound knowledge of the diversity of behavioral features (e.g., sleep), biomarker specificities, and other underpinnings of AD heterogeneity would allow the development of tailored diagnostic-therapeutic strategies to address the particular needs of patients with atypical AD earlier and more appropriately. The brain control of the sleep cycle includes a series of subcortical structures specializing in wake-promoting, REM and non-REM sleep-promoting, and circadian clock regulating functions8. Most of these nuclei develop AD neuropathological changes[8]. In particular, key components of the arousal system: locus coeruleus (the primary source of brain noradrenaline), orexinergic neurons of the lateral hypothalamic areas, and histaminergic neurons of the tuberomammillary nucleus, develop AD-type tau pathology even before it is observed in the cortex [9–11]. In a study combining unbiased quantitative pathology with polysomnography, we recently showed that AD-related neurodegeneration of wake-promoting nuclei correlated with sleep dysfunction[12]. In vivo studies also show a link among locus coeruleus integrity, noradrenergic dysfunction, and disruption of the sleep-wake cycles[13–15].
Although the neuropathological patterns of typical, amnestic and atypical, non-amnestic AD show similarities, the cortical tau spread and atrophy patterns are presentation-specific[7,16,17]. We recently described a distinct pattern of neuropsychiatric features between amnestic and atypical AD variants, which suggests that nuclei of the neuromodulatory subcortical system (NSS) also show a different pattern of degeneration in AD clinical variants[5,18]. To date, there has been little focus to identify distinct sleep features among AD clinical variants, another manifestation of a likely specific pattern of NSS degeneration. However, optimal symptomatic treatment of sleep disturbances requires a deep understanding of its specific neurobiological basis to modulate the correct combination of neurotransmission systems. Uncovering the differences in sleep alterations across AD variants is of utmost importance. Biologically, polysomnography is a tool that can be used to discover sleep clinical features associated with different patterns of neuronal vulnerability in AD phenotypes that can ultimately provide insights into the disease pathogenesis[5]. Clinically, it will inform customized treatment.
We aimed to shed light on possible differences in sleep architecture between AD clinical phenotypes (amnestic/typical vs. non-amnestic/atypical) by investigating, in an exploratory approach, objective sleep metrics obtained via overnight video-electroencephalography (video-EEG) in a cohort of longitudinally followed, well-characterized AD cases. Cohort strengths include enrichment for atypical/non-amnestic variants, biomarker/autopsy-proven AD cases, and clinical data allowing us to account for potential effects of sleep modifiers such as anticholinesterase inhibitors, Selective Serotonin Reuptake Inhibitors and silent epileptiform activity. We hypothesized that sleep architecture is different between amnestic and non-amnestic AD, where N3-SWS, the sleep stage more strongly related to memory consolidation, is more impaired in amnestic AD than in non-amnestic AD.
2. Materials and methods
2.1. Participants:
Forty-eight subjects, including 15 individuals with amnestic AD, 19 individuals with non-amnestic AD (nine logopenic variant of primary progressive aphasia (lvPPA), ten posterior cortical atrophy (PCA) and 14 healthy controls were recruited at the Memory and Aging Center, UCSF, from 2008 to 2017. All data for this cross-sectional study were collected from a preexisting dataset designed to study epileptiform activity in AD, partially overlapping with Vossel et al. 2015 consisting of 33 AD and 19 controls [19]. Participant recruitment continued until 2017, reaching a cohort of 47 AD and 19 controls. Starting from this extended cohort, we included those participants meeting the following inclusion criteria (Suppl. Fig.1):
Confirmed postmortem diagnosis of AD (n=12) or highest likelihood of AD biomarker pathology by the National Institute on Aging and Alzheimer’s Association (NIA-AA) 2011 (n=22), therefore having at least one biomarker evidencing amyloid pathology (amyloid-PET or amyloid in CSF) and at least one biomarker evidencing neurodegeneration (tau in CSF, FDG-PET, tau-PET or MRI). Participants with a low or intermediate likelihood of AD pathology were excluded.
Optimal overnight video-EEG recording. Participants with suboptimal video-EEG (time gaps, technical issues) were excluded.
The study was approved by the UCSF Institutional Review Board, and all participants gave their written, informed consent.
Based on the clinical presentation and the neuropsychological profile, participants were classified as amnestic/typical or non-amnestic/atypical variants based on the predominant memory or non-memory cognitive domains impairment. Atypical phenotypes included predominant impairments in visuospatial (PCA) and language (lvPPA). Most of the included participants had a sporadic (non-genetic) early-onset AD (before 65, EOAD, n=30).
Controls were required to have a Mini-Mental State Examination (MMSE) score of ≥28, a Clinical Dementia Rating (CDR) Sum of Boxes (CDR-SOB) score of 0, no cognitive concerns reported by themselves or their informants, age-appropriate pattern on brain magnetic resonance imaging (MRI), and no neurological disorders.
2.2. Neuropathological evaluation
Neuropathological diagnoses (n=12) were based on an extensive dementia-oriented postmortem assessment at the UCSF/ Neurodegenerative Disease Brain Bank[21]. Twenty-six tissue blocks covering dementia-related regions of interest were dissected from the fixed slabs, and hematoxylin and eosin and immunohistochemical stains were applied following standard diagnostic procedures developed for patients with dementia. Neuropathological diagnosis followed currently accepted guidelines[21]. Overall severity of AD Neuropathologic Change (ADNC) was assigned using the National Institute on Aging (NIA)–Reagan criteria and NIA–Alzheimer Association criteria for AD[6].
2.3. Overnight long-term monitoring by video-electroencephalography
The methods for clinical sleep assessments are previously described[12]. All participants were evaluated at the Clinical and Translational Science Institute Clinical Research Center at Moffitt Hospital. The monitoring included long-term recording using silver cup electrodes in the standard international 10–20 electrode array and additional leads to record electrocardiography. EEGs included video telemetry recordings. After initial setup, participants were asked to hyperventilate for 3 minutes then rest and breath normally for 7 minutes with eyes closed. EEG recordings then continued overnight. Throughout the assessment, participants continued their usual medication regimen.
The long-term EEG monitoring data were exported to European Data Format (EDF) using Persyst 14 software, and sleep staging was performed in PRANA Production Suite 15.2 (Phi Tools, Strasbourg, France) following the American Academy of Sleep Medicine (AASM) criteria[22]. All studies were manually scored and reviewed by an experienced polysomnographic technologist (LY) and a trained neurologist (NF).
Electrode sites A1 and A2 were not available for all recordings. Therefore, the sleep staging montage was modified and re-referenced to contralateral temporal sites (T3, T4) post-recording to achieve standardization across all participants using six scalp electrodes (F3-T4, F4-T3, C3-T4, C4-T3, O1-T4, O2-T3) and two modified EOG (Fp1-T4, Fp2-T3). High-pass filters were set at 0.3 Hz and low-pass filters at 35 Hz for EEG and EOG. Raw data were visually scored in 30-s epochs into Wake, Stage N1, Stage N2, Stage N3, and REM sleep guided by AASM criteria. To overcome the lack of EMG, a visual spectrogram was created to compare against the staging hypnogram using RemLogic 3.4 (Natus Medical, Inc.San Carlos, CA) as a confirmation of wake and sleep periods as shown in (Supplementary Figure 2).
Measures of interest were total sleep time (TST, min), wake after sleep onset (WASO, min), REM latency (min), sleep maintenance (%), and percent time in Non-REM N1 sleep (%), N2 sleep (%), N3 sleep (%), and REM sleep (%), during sleep period time (SPT) and TST. Note that SPT refers to the duration of time from sleep onset to final awakening (including WASO), while TST refers to the total amount of sleep time scored, excluding awake time (WASO).
2.4. Electroencephalography data processing
Over-night video-EEG data were preprocessed using custom python scripts and functions from the YASA toolbox[23]. Artifacts were rejected by comparing over-night EEG data to a reference covariance matrix of clean data. Riemannian geometry was used to calculate the distance of each 5 second epoch of data from the reference covariance matrix. An epoch that exceeded a distance greater than 3 z-scores was flagged as an artifact. The efficacy of this approach was confirmed via visual inspection. The absolute power spectrum of the EEG data was computed using Welch’s method in 0.25 Hz bins. Since elevated delta during sleep in AD has been observed we focused the spectral analysis on the delta frequency band (1–4 Hz) [24]. Additionally, spindles were detected using automatic detection algorithms from the YASA toolbox (Supplementary Material 1). All analyses were performed separately for NREM stages and REM stages, and data from frontal (F3 and F4) and central (C3 and C4) were averaged before doing statistical analyses. Note that the terms ‘frontal’ and ‘central’ refer to the difference between frontal and central channels with contralateral temporal electrodes.
2.5. Statistical analyses
Statistical analyses were conducted using Stata/IC 16.1 (College Station, Texas, USA). Baseline characteristics by diagnostic groups are presented as means or frequencies. Kruskal-Wallis non-parametric test was used to test overall significance between populations followed by Dunn’s test to identify specific differences between groups. Additional regression models were adjusted by the presence of silent epileptiform activity, prescription of anticholinesterase inhibitors treatment and antidepressants or amyloid burden. This is an exploratory study and no correction for multiple comparisons has been applied. We expect that any pattern of statistically significant results will build upon each other and for this study to be a foundation for future studies with a larger sample size.
3. Results
3.1. Sample characteristics
Details on demographic and clinical data are described in Table 1. Amnestic and typical AD groups showed similar age at EEG, gender, age at disease onset, disease duration, cognitive (MMSE), and functional status (CDR SoB) at EEG. The presence of silent epileptiform activity and the prescription of anticholinesterase treatments was also similar between AD groups. The control group was slightly older than the amnestic AD at EEG time. As expected, controls showed higher MMSE and lower CDR SoB scores than AD groups. In addition, controls had no silent epileptiform activity and were not using anticholinesterase inhibitors.
Table 1.
Demographics and clinical data
| Amnestic AD (n=15) | Non-amnestic AD (n=19) | Controls (n=14) | Amnestic vs controls | Non-amnestic vs controls | Amnestic vs non-amnestic | |
|---|---|---|---|---|---|---|
| Age at onset (years) | 56.1 ± 9.0 | 55.7 ± 7.5 | N/A | N/A | N/A | ns |
| Age at EEG (years) | 60.6 ± 9.7 | 60.5 ± 6.7 | 64.5 ± 5.6 | * | ns | ns |
| Disease duration (years) | 5.1 ± 2.4 | 5.3 ± 1.9 | N/A | N/A | N/A | ns |
| Female (%) | 64.3 | 57.9 | 64.3 | ns | ns | ns |
| Education (years) | 15.9 ± 2.5 | 16.5 ± 2.8 | 17.4 ± 1.9 | ns | ns | ns |
| CDR at EEG time (%) | 0.86 ± 0.4 | 0.84 ± 0.4 | 0 ± 0 | ** | ** | ns |
| Normal | 0 | 0 | 100 | N/A | N/A | N/A |
| MCI | 43 | 36.9 | 0 | N/A | N/A | N/A |
| Mild dementia | 50 | 57.9 | 0 | N/A | N/A | N/A |
| Moderate dementia | 7 | 5.2 | 0 | N/A | N/A | N/A |
| CDR SoB at EEG time | 5.4 ± 2.3 | 4.6 ± 2.3 | 0 ± 0 | ** | ** | ns |
| MMSE at EEG time | 21.1 ± 5.7 | 22. ± 4.5 | 29.5 ± 0.8 | ** | ** | ns |
| Anticholinesterase Inhibitors (%) | 80 | 89.5 | 0 | ** | ** | ns |
| SSRI Antidepressants (%) | 46.7 | 42.1 | 7.1 | * | * | ns |
| Epileptiform activity (%) | 20 | 26.3 | 0 | P=0.07 | * | ns |
Data are shown as mean ± SD
Significance
P<0.05
P<0.01
ns, non-significant; N/A, not applicable
Abbreviations: CDR, Clinical Dementia rating; CDR-SoB, CDR Sum of Boxes; MMSE, Mini Mental State Examination; SSRI, Selective serotonin reuptake inhibitors
3.2. Sleep parameters over Sleep Period Time
Detailed results on sleep parameters over SPT are described in Table 2 and Fig. 1. Total Sleep Time (TST) was similar among amnestic AD, atypical AD, and controls. Despite similar TST, when compared to controls, amnestic AD showed higher WASO(%) (24.1 ± 12.6 vs. 16.6 ± 10.0, p<0.05) and N1(%) (9.2 ± 4.1 vs. 5.9 ± 2.8, p>0.01) and lower sleep maintenance (%) (75.9 ± 12.6 vs. 83.3 ± 10.0, p<0.05) and N3(%) (8.5 ± 5.4 vs. 13.7 ± 6.8). In the same line, atypical AD showed a higher rate of N1(%) (8.9 ± 5 vs 5.9 ± 2.8, p<0.05). In contrast to amnestic AD, atypical AD had less REM(%) (12.6 ± 5.6 vs 19.5 ± 6.5, p<0.01), longer REM latency (min) (133.7 ± 69 vs. 88.2 ± 32.8, p<0.05) and no differences in sleep maintenance, WASO, or N3 compared to controls. N3(%) was significantly lower in amnestic than in atypical AD (8.5 ± 5.4 vs. 15.1 ± 8.3, p<0.01). The distribution of sleep stages over SPT showed no difference between atypical non-amnestic/atypical variants (lvPPA, PCA) (Supplementary Fig. 3).
Table 2.
Sleep parameters over SPT
| Amnestic AD | Non-amnestic AD | Controls | Amnestic vs controls | Non-amnestic vs controls | Amnestic vs non-amnestic | |
|---|---|---|---|---|---|---|
| TST (min) | 372.8 ± 80.4 | 392.7 ± 79 | 390.6 ± 81.1 | ns | ns | ns |
| WASO (min) | 118.8 ± 67 | 109.7 ± 57.4 | 79.93 ± 54.1 | * | * | ns |
| N1 (min) | 45.2 ± 20.9 | 44.8 ± 26.4 | 28.11 ± 13.9 | ** | * | ns |
| N2 (min) | 202.3 ± 57.9 | 201.9 ± 53.9 | 202 ± 49.7 | ns | ns | ns |
| N3 (min) | 41.2 ± 24 | 74.9 ± 39.1 | 63.4 ± 32.6 | * | ns | ** |
| REM (min) | 78.87 ± 31.5 | 65 ± 31 | 91.3 ± 35.7 | ns | * | ns |
| Sleep Maintenance (%) | 75.9 ± 12.6 | 78 ± 12 | 83.3 ± 10.0 | * | ns | ns |
| WASO (%) | 24.1 ± 12.6 | 22.1 ± 12.1 | 16.6 ± 10.0 | * | ns | ns |
| N1 (%) | 9.2 ± 4.1 | 8.9 ± 5 | 5.9 ± 2.8 | ** | * | ns |
| N2 (%) | 41 ± 10 | 40.2 ± 9.2 | 43.1 ± 8.2 | ns | ns | ns |
| N3 (%) | 8.5 ± 5.4 | 15.1 ± 8.3 | 13.7 ± 6.8 | * | ns | ** |
| REM (%) | 16.1 ± 6 | 12.6 ± 5.6 | 19.5 ± 6.5 | ns | ** | ns |
| REM Latency (min) | 101.5 ± 58.1 | 133.7 ± 69 | 88.2 ± 32.8 | ns | * | ns |
Significance
P<0.05
P<0.01
ns, non-significant
Abbreviations: TST, Total Sleep Time; WASO, Wake after sleep onset
Figure 1.
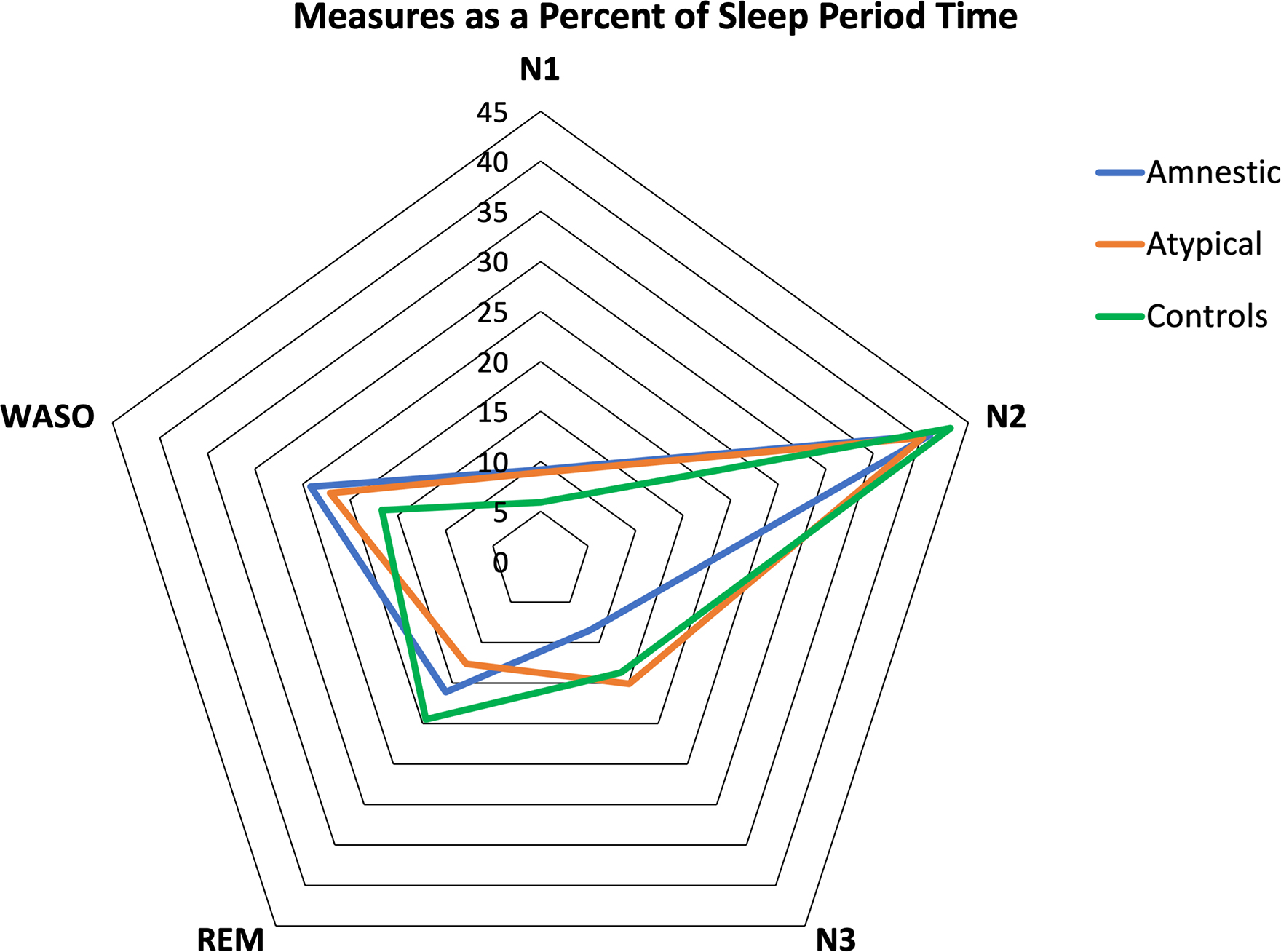
Distribution of sleep stages over sleep period time (SPT) in amnestic, atypical Alzheimer’s Disease and controls. Fig. 1 Shows the amount of each sleep stage (percentages, %) across groups. Note that the percentage of N3 is reduced in amnestics and REM sleep is reduced in atypical Alzheimer’s Disease. WASO and N1 are increased in Alzheimer’s Disease groups.
3.4. Sleep parameters over Total Sleep Time
Detailed results on sleep parameters over TST are described in Table 3, Figure 2. Amnestic AD showed higher N1(%) compared to controls (12.1 ± 5.2 vs. 7.4 ± 4.1, p<0.05). Amnestic AD showed lower N3(%) compared to controls (10.9 ± 5.8 vs 16.5 ± 7.8, p<0.05) and non-amnestic/atypical (10.9 ± 5.8 vs. 19.1 ± 10.1, p<0.01). Non-amnestic/atypical AD showed higher N1(%)(11.7 ± 6.8 vs. 7.4 ± 4.1, p<0.05) compared to controls and lower REM(%) compared to controls (15.9 ± 7.2 vs. 23.4 ± 6.1, p<0.01) and amnestics (15.9 ± 7.2 vs. 21.5 ± 7.4, p<0.01). Therefore, findings using TST and SPT were similar, aside from REM sleep, where non-amnestic AD had significantly less REM sleep as a percent of TST (excluding time awake after sleep onset) but not as a percent of SPT as compared to amnestics. The distribution of sleep stages over TPT showed no difference between atypical non-amnestic variants (lvPPA, PCA) (Supplementary Fig. 4). Linear regression models adjusting by potential confounders (epileptiform activity, the prescription of anticholinesterase inhibitors and antidepressants) were not significant and did not modify the effect of AD phenotypes over sleep parameters (Supplementary Table 1).’
Table 3.
Sleep parameters over TST
| Amnestic AD | Non-amnestic AD | Controls | Amnestic vs controls | Non-amnestic vs controls | Amnestic vs non-amnestic | |
|---|---|---|---|---|---|---|
| N1 (%) | 12.1 ± 5.2 | 11.7 ± 6.8 | 7.4 ± 4.1 | ** | * | ns |
| N2 (%) | 54.0 ± 9.0 | 52.0 ± 12.7 | 52.2 ± 9.4 | ns | ns | ns |
| N3 (%) | 10.9 ± 5.8 | 19.1 ± 10.1 | 16.5 ± 7.8 | * | ns | ** |
| REM (%) | 21.5 ± 7.4 | 15.9 ± 7.2 | 23.4 ± 6.1 | ns | ** | ** |
Significance
P<0.05
P<0.01
ns, non-significant
Figure 2.
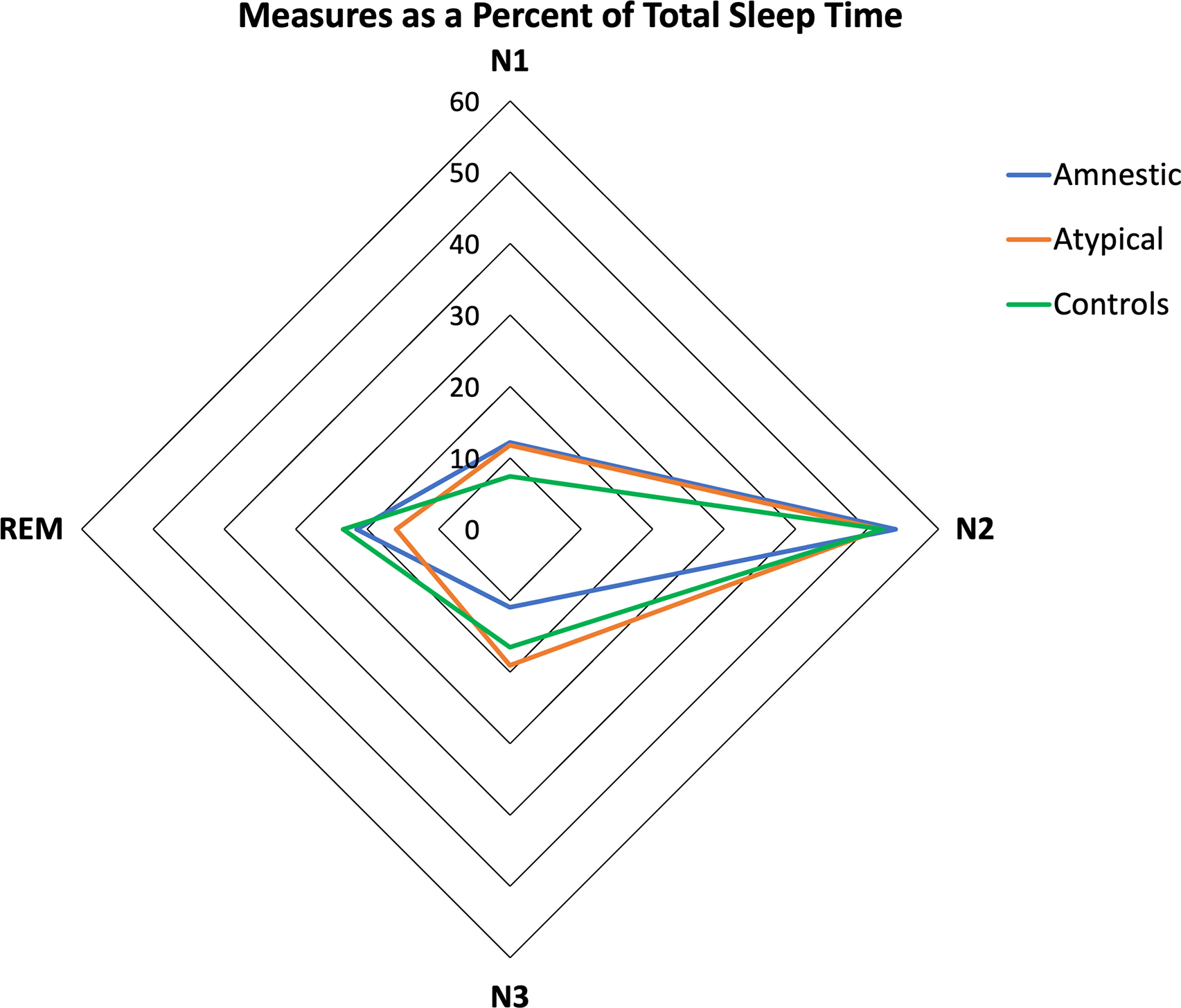
Distribution of sleep stages over TST in amnestic, atypical Alzheimer’s Disease and controls. Fig 2. Shows the amount of each sleep stage across groups. Note that the percentage of N3 is reduced in amnestics and, REM sleep is reduced in atypical Alzheimer’s Disease. N1 is increased in both Alzheimer’s Disease groups.
3.5. Effect of the global amyloid burden over the sleep parameters
Linear regression models adjusting by amyloid burden in amyloid-PET (Centiloids) were not significant (Supplementary Table 2). The amyloid burden was measured by Centiloids on a subsample of 27 PET scans (11 amnestic, 16 atypical). PET tracers used were (11)C-labeled Pittsburgh Compound-B (11C-PIB) (n=22) and 18F-AV-45 (florbetapir) (n=5).
3.6. Spectral analyses
Atypical AD had higher delta power during NREM stages 2 and 3 sleep as compared to both amnestic (Frontal SO P=0.02; Central SO P=0.01) and controls (Frontal SO P<0.01, Central SO P=0.03). There were no significant differences between amnestic AD and controls. Spindle measures did now show any differences between groups (Supplementary Table 3 and 4).
4. Discussion
This cross-sectional overnight video-EEG study demonstrates that biomarker/postmortem-validated AD patients with amnestic and atypical phenotypes show different profiles for sleep architecture and power spectral analysis (i.e., the percentage of sleep stages, delta power). Despite having similar total sleep time to controls, amnestic AD shows a reduced N3 (slow-wave sleep -SWS) phase and atypical AD shows reduced REM stage and increased REM latency, but similar N3 to controls. Delta power during N2 and N3 was higher in atypical than in amnestic AD. The discrepancy between N3 and REM is also significant when comparing amnestic to atypical AD. These differences remained significant after correcting for silent epileptiform activity, and the use of anticholinesterase inhibitors or antidepressants. In addition, the study confirmed a sleep fragmentation pattern consisting of more time spent awake (WASO) and also greater time in N1 sleep in AD overall compared to healthy controls.
Sleep fragmentation and difficulty reaching deeper sleep stages such as N3/SWS have been previously documented in AD[25]. However, these studies mostly included late-onset amnestic cohorts[1,26–28]. Our study confirms that N3/SWS deficits are also a feature of sporadic EOAD, when the clinical manifestation is amnestic. However, the finding that atypical AD had a relatively preserved N3/SWS stage but significant deficits in REM is novel. Although amyloid pathology has previously been associated with sleep alterations in healthy elders at risk for AD, it is unlikely that it is driving differential sleep patterns within AD variants due to a lack of differences in the load or distribution of beta-amyloid between AD typical and atypical forms [7, 17, 38]. Even within our cohort, we found amyloid burden to be similar between the two groups and unrelated to sleep parameters. Therefore overall, in light of previous studies, our current findings could suggest a different pattern of subcortical degeneration of nuclei regulating REM versus NREM sleep between AD subtypes. These findings are intriguing, marrying models of AD/memory models and sleep/memory models. The prevailing AD /memory model attributes hippocampal formation degeneration as the main contributor to early short-term memory disturbances in AD[7,29,30]. Patients with amnestic AD show a disproportionately high burden of neurofibrillary tangles in the hippocampus than patients with atypical phenotypes[7,17]. In relation to models of sleep and memory: N3/SWS has been classically related to memory processing and consolidation in physiological conditions[31–33]. Prior studies showed that reduced NREM N3/SWS leads to an impaired overnight sleep-dependent memory retention in otherwise healthy elders. In contrast, naps, including NREM sleep periods, could increase memory performance[31]. Our results raise the question of whether a lower vulnerability of SWS-promoting neurons and subsequently less impaired N3 stage contributes to the relatively better memory scores seen in atypical AD as well as the broader question of how the SWS-promoting generators interact with the hippocampal formation in affecting short-term memory function.
It is undisputed that impairment of sleep-wake cycles in AD is associated with greater cognitive decline, negatively impacting the quality of life, increasing caregiver burden, and causing early institutionalization[2,4,28]. Growing evidence has suggested that early tau accumulation within the neuromodulatory subcortical systems could play a central role in AD-related sleep disruption[34,35]. Comparison of night time sleep metrics and wake promoting nuclei in humans (orexinergic lateral hypothalamic area (LHA), the histaminergic tuberomammillary nucleus (TMN) and the noradrenergic locus coeruleus (LC)), found that wake promoting nuclei were associated with increased sleep disruption[12]. LC degeneration is associated with higher odds for presenting with symptomatic sleep disruption as well as other neuropsychiatric symptoms in early stages, even before tau spread reaches cortical areas (i.e., hippocampus) and leads to cognitive dysfunction[10,37]. Further, the LC is one of the first sites of tau-related neurodegeneration in AD (from Braak I-II already) as compared to other tauopathies, suggesting a selective vulnerability pattern of the LC wake-promoting neurons[9,12,36]. Our results showing a pattern of sleep disruption (higher awakenings, N1 and lower sleep maintenance) in a cohort enriched with early-onset presentations, suggests a dysfunctional arousal system driven by LC wake-promoting neurons. Indeed, the fact that EOAD manifests with a higher burden of behavioral and sleep disturbances in addition to the higher LC degeneration found on postmortem AD brain tissue, could indicate that vulnerability of the LC is even higher in early- than late-onset presentations[9,18,37]. Greater neuronal count within TMN is associated with less REM sleep and greater neuronal population in LC is associated with prolonged latency to REM sleep[12]. Together with this current work, it suggests further augmented function of the LC in non-amnestic/atypical EOAD with diminished REM sleep compared to amnestic EOAD. Further work is necessary on both the wake and sleep promoting nuclei in these cohorts to confirm these hypotheses
The specific areas driving such NREM impairment in AD (and particularly in amnestic AD) are unclear, especially since the physiological sleep-wake circuitry in the human brain remains under discussion given significant differences with the rodent model[8]. The classical N3/SWS regulation model suggested a reciprocal relationship between the sleep-promoting neurons on the intermediate nucleus and a network of the brainstem and hypothalamic neurons being attributed to an overall wake-promoting role. In this model these sleep-wake interconnected networks would interact in an ‘on/off switch’ manner, modulated by the circadian pacemaker in the suprachiasmatic nucleus. Nevertheless, recent animal investigations challenged this idea/model by highlighting brainstem areas such as the nucleus accumbens (NAc), the lateral hypothalamic area (LHA), and the medullary parafacial zone (PZ) as an N3-SWS generator[39–41]. In this line, the elevated adenosine levels on the NAc, the activation of neurons containing melanin-concentrating hormone in the LHA, and the GABAergic control of the PZ induce N3/SWS, whereas their inhibition suppresses sleep[39,41,42]. These interconnected areas hierarchically modulate thalamocortical synchronization and oscillation, resulting in the sleep/wake cycle. Though mainly based on animal studies, the current sleep/wake regulation conceptual network is complex, with several sleep-wake promoting centers[8,12]. From our current study findings of mostly NREM-N3/SWS sleep dysfunction in amnestic AD, we could infer that amnestics may present a specific pattern of tau-driven degeneration predominantly affecting these NREM sleep-wake interplaying areas.
In contrast, the decreased amount of REM sleep and delayed REM latency in non-amnestic/atypical AD suggest a predominance of REM sleep dysfunction in atypical phenotypes. The neural network that generates REM sleep consists of a large number of anatomic structures located in the brainstem, limbic system, thalamus, hypothalamus and cortex[43,44]. The critical REM-controlling areas are located in the mesopontine tegmentum and in the ventral medial medulla. Several monoaminergic and non-cholinergic structures in the mesopontine tegmentum act as REM-on and REM-off structures with reciprocal inhibitory projections acting as a flip-flop switch, and therefore transitioning from REM sleep to NREM sleep and back[45]. For instance, the noradrenergic locus coeruleus, orexinergic LHA, cholinergic neurons of the pedunculopontine nucleus (PPT), and the nucleus basalis of Meynert or GABAergic sublaterodorsal nucleus seem to promote REM sleep. In contrast, others, like the serotonergic dorsal raphe nucleus (DRN), inhibit REM sleep states[8]. These regions send projections inhibiting or activating nearby nuclei in the brainstem or other distant brain regions. Acting as REM sleep modulators, the decreased activity of noradrenergic LC, serotonergic DRN, histaminergic TMN and orexinergic LHA, allow the final REM sleep cortical activation. Previous findings from our group working with Progressive Supranuclear Palsy, a tauopathy neurodegenerative disease associated with executive dysfunction, which serves as a naturalistic model for extensive degeneration/vulnerability of the brainstem, found REM sleep deficits and profound sleep disruption[46]. In light of this and our current results, we could hypothesize that the interplay between these REM sleep-regulating areas might be more affected in neurodegenerative diseases associated with cognitive deficits other than memory earlier in disease progression, such as non-amnestic AD. Thus, this study suggests a selective vulnerability pattern of tau-derived neurodegeneration between AD variants with amnestics associated with NREM generators and non-amnestics with REM sleep generators. Changes in the LC have been particularly associated with REM dysfunction[12]. A more severe degeneration of REM-promoting neurons (and preservations on REM-off structures such as LC) in non-amnestic AD could underly the more significant REM dysfunction in this group. It could be argued that the lower REM sleep in non-amnestic vs amnestic AD could be affected through typical treatment of anticholinesterase inhibitors for AD which may be prescribed more readily for amnestic than non-amnestic patients. Though we did control for the use of these medications in our analyses, further investigation to the effect of a prolonged altered cholinergic system in AD and REM sleep is warranted. In addition, further research assessing tau degeneration in these nuclei within the AD spectrum is needed to confirm this hypothesis.
There is a possibility that co-pathologies might also contribute to defining these differences in sleep architecture between amnestic and non-amnestic AD. Our cohort is enriched with EOAD presentations, theoretically reducing the odds for overall co-pathologies. However, a particular non- AD pathological change, Lewy Body Disease (LBD), co-exists frequently in EOAD[21], worsens the clinical severity and modifies clinical presentations[4–50]. Some studies report up to 27% of REM sleep Behavior Disorder (RBD) prevalence in patients with AD -type dementia[51,52], which is likely a consequence of LBD co-pathology. Considering the high prevalence of LBD comorbidity in AD, it is probable that LBD is accentuating LBD -like symptoms in AD such as RBD, which could explain the REM sleep impairment found in non-amnestic/atypical EOAD in our study. However, this hypothesis remains untested, and further studies evaluating the association between RBD in AD and underlying LBD co-pathology are warranted.
This study’s strengths include the inclusion of autopsy-proven or biomarker-based AD cases –which had clinical and sleep data obtained using standardized methods by the same experts, a predominant early onset AD (EOAD) cohort, which reduces the odds of severe co-pathologies, enrichment of non-amnestic patients, and the inclusion of known sleep modifiers (i.e: anticholinesterase inhibitors, antidepressants, silent epileptiform activity) in the analytical models.
Nevertheless, our study has limitations. The use of overnight EEG telemetry recordings prevented us from characterizing specific sleep conditions such as obstructive sleep apnea, RBD, or periodic limb movements syndrome. Further studies using polysomnography are needed to clarify the influence of these factors on AD phenotype-associated differences in sleep patterns. To address the lack of some PSG parameters necessary to follow AASM scoring criteria strictly, we compared the hypnograms to spectrograms to inform reliability and improve scoring [54]. Also, we do not have EEG measures of the wake-period, which is a limitation as the wake-promoting nuclei degenerate early in AD, alpha waves correlate with cortical tau burden, and atypical AD cases have a disproportionately higher cortical tau burden than amnestics [7,17,52]. In addition, the lack of statistical differences in sleep/wake staging found between the AD atypical variants assessed (PCA, lvPPA) may be due to the small sample size of these subgroups, and requires further study with larger cohorts. Lastly, though our study attempted to recruit from a broad population, those who agreed to participate in our study were predominantly Caucasian.
In conclusion, sleep architecture in AD is not merely an exacerbation of age-related sleep changes but shows specific features across clinical variants. In line with prior literature, amnestic AD shows decreased N3 sleep. However, non-amnestic variants show a specific pattern consisting of predominant REM sleep dysfunction. The existence of specific sleep-wake profiles between variants suggests differential degenerative patterns of the sleep and wake-promoting systems within AD phenotypes, which would open the door to uncover the selective vulnerability on subcortical structures. Better understanding of the neurobiological changes underlying sleep differences between variants would promote tailored treatment avenues for sleep disorders in AD.
Supplementary Material
Highlights.
Alzheimer’s Disease (AD) variants show distinct patterns of sleep impairment.
Amnestic/typical AD has worse N3 - Slow Wave Sleep (SWS) impairment than atypical AD.
Atypical AD shows more REM deficits than typical AD.
Selective vulnerability patterns in subcortical areas may underly sleep differences.
Relatively preserved SWS may explain better memory scores in atypical vs. typical AD.
Research in context:
Systematic Review:
The authors reviewed the literature using PubMed and cited appropriate articles. Polysomnography studies previously analyzed the sleep pattern in Alzheimer’s Disease; however, most of the prior literature focuses on late-onset typical/amnestic AD. Very little is known about early-onset presentations with a predominance of atypical/non-amnestic syndromes.
Interpretation:
Our study provides compelling evidence that sleep patterns differ across variants. Beyond sleep fragmentation, amnestic AD shows a worse NREM N3/Slow-wave sleep impairment; conversely, the atypical/non-amnestic variants show a higher REM sleep dysfunction. Our study corroborates the hypothesis of differential selective vulnerability patterns of the subcortical nuclei within variants.
Future Directions:
Development of targeted treatments for sleep dysfunction may be needed across variants. Studies investigating the neurobiological basis of sleep dysfunction in AD spectrum may provide insight for deciphering the selective vulnerability of the neuromodulatory subcortical system. Further understanding of preserved N3/SWS as a mechanism of preserved memory is warranted.
Acknowledgements
The authors thank Raphael Vallat for sharing the YASA code, research team members, as well as the patients for their generous contribution to science.
Funding
This work was supported by Global Brain Health Institute, Tau Consortium/Rainwater Charity Foundation National Institute on Aging grants NIA R01 AG060477, NIA R01 AG064314, K24AG053435, K23-AG031861, K23AG038357, R01-AG027859, P01-AG1972403, and P30-AG062422, State of California Department of Health Services Alzheimer’s Disease Research Center of California grant 04–33516, Grant PCTRB-13–288476 from the Alzheimer’s Association, made possible by Part the Cloud Funding from the John Douglas French Alzheimer’s Foundation Funding from the SD. Bechtel, Jr. Foundation. NF recipient of GBHIALZUK-21–723831, AACSF_21_723056 and Rio Hortega grant CM21/00024.
Footnotes
Competing interests
The authors report no competing interests.
References
- [1].Liguori C, Placidi F, Izzi F, Spanetta M, Mercuri NB, Di Pucchio A. Sleep dysregulation, memory impairment, and CSF biomarkers during different levels of neurocognitive functioning in Alzheimer’s disease course. Alzheimers Res Ther 2020;12:5. 10.1186/s13195-019-0571-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Targa ADS, Benítez ID, Dakterzada F, Carnes A, Pujol M, Jorge C, et al. Sleep profile predicts the cognitive decline of mild-moderate Alzheimer’s disease patients. Sleep 2021;44:zsab117. 10.1093/sleep/zsab117. [DOI] [PubMed] [Google Scholar]
- [3].Okuda S, Tetsuka J, Takahashi K, Toda Y, Kubo T, Tokita S. Association between sleep disturbance in Alzheimer’s disease patients and burden on and health status of their caregivers. J Neurol 2019;266:1490–500. 10.1007/s00415-019-09286-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].McCleery J, Sharpley AL. Pharmacotherapies for sleep disturbances in dementia. Cochrane Database Syst Rev 2020;11:CD009178. 10.1002/14651858.CD009178.pub4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Falgàs N, Walsh CM, Neylan TC, Grinberg LT. Deepen into sleep and wake patterns across Alzheimer’s disease phenotypes. Alzheimers Dement 2021;17:1403–6. 10.1002/alz.12304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement 2012;8:1–13. 10.1016/j.jalz.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Murray ME, Graff-Radford NR, Ross OA, Petersen RC, Duara R, Dickson DW. Neuropathologically defined subtypes of Alzheimer’s disease with distinct clinical characteristics: a retrospective study. Lancet Neurol 2011;10:785–96. 10.1016/S1474-4422(11)70156-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lew CH, Petersen C, Neylan TC, Grinberg LT. Tau-driven degeneration of sleep- and wake-regulating neurons in Alzheimer’s disease. Sleep Med Rev 2021;60:101541. 10.1016/j.smrv.2021.101541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Oh J, Eser RA, Ehrenberg AJ, Morales D, Petersen C, Kudlacek J, et al. Profound degeneration of wake-promoting neurons in Alzheimer’s disease. Alzheimers Dement 2019;15:1253–63. 10.1016/j.jalz.2019.06.3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ehrenberg AJ, Suemoto CK, França Resende E de P, Petersen C, Leite REP, Rodriguez RD, et al. Neuropathologic Correlates of Psychiatric Symptoms in Alzheimer’s Disease. JAD 2018;66:115–26. 10.3233/JAD-180688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ehrenberg AJ, Nguy AK, Theofilas P, Dunlop S, Suemoto CK, Di Lorenzo Alho AT, et al. Quantifying the accretion of hyperphosphorylated tau in the locus coeruleus and dorsal raphe nucleus: the pathological building blocks of early Alzheimer’s disease. Neuropathol Appl Neurobiol 2017;43:393–408. 10.1111/nan.12387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Oh JY, Walsh CM, Ranasinghe K, Mladinov M, Pereira FL, Petersen C, et al. Subcortical Neuronal Correlates of Sleep in Neurodegenerative Diseases. JAMA Neurology 2022. 10.1001/jamaneurol.2022.0429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Van Egroo M, van Hooren RWE, Jacobs HIL. Associations between locus coeruleus integrity and nocturnal awakenings in the context of Alzheimer’s disease plasma biomarkers: a 7T MRI study. Alzheimers Res Ther 2021;13:159. 10.1186/s13195-021-00902-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Jacobs HIL, Riphagen JM, Ramakers IHGB, Verhey FRJ. Alzheimer’s disease pathology: pathways between central norepinephrine activity, memory, and neuropsychiatric symptoms. Mol Psychiatry 2021;26:897–906. 10.1038/s41380-019-0437-x. [DOI] [PubMed] [Google Scholar]
- [15].Dahl MJ, Mather M, Düzel S, Bodammer NC, Lindenberger U, Kühn S, et al. Rostral locus coeruleus integrity is associated with better memory performance in older adults. Nat Hum Behav 2019;3:1203–14. 10.1038/s41562-019-0715-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Graff-Radford J, Yong KXX, Apostolova LG, Bouwman FH, Carrillo M, Dickerson BC, et al. New insights into atypical Alzheimer’s disease in the era of biomarkers. Lancet Neurol 2021;20:222–34. 10.1016/S1474-4422(20)30440-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Petersen C, Nolan AL, de Paula França Resende E, Miller Z, Ehrenberg AJ, Gorno-Tempini ML, et al. Alzheimer’s disease clinical variants show distinct regional patterns of neurofibrillary tangle accumulation. Acta Neuropathol 2019;138:597–612. 10.1007/s00401-019-02036-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Falgàs N, Allen IE, Spina S, Grant H, Piña Escudero SD, Merrilees J, et al. The severity of neuropsychiatric symptoms is higher in early-onset than late-onset Alzheimer’s disease. Eur J Neurol 2021. 10.1111/ene.15203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Vossel KA, Ranasinghe KG, Beagle AJ, Mizuiri D, Honma SM, Dowling AF, et al. Incidence and impact of subclinical epileptiform activity in Alzheimer’s disease. Ann Neurol 2016;80:858–70. 10.1002/ana.24794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Jack CR. Preclinical Alzheimer’s disease: a valid concept. Lancet Neurol 2020;19:31. 10.1016/S1474-4422(19)30440-5. [DOI] [PubMed] [Google Scholar]
- [21].Spina S, La Joie R, Petersen C, Nolan AL, Cuevas D, Cosme C, et al. Comorbid neuropathological diagnoses in early versus late-onset Alzheimer’s disease. Brain 2021. 10.1093/brain/awab099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Berry RB, Gamaldo CE, Harding SM, Brooks R, Lloyd RM, Vaughn BV, et al. AASM Scoring Manual Version 2.2 Updates: New Chapters for Scoring Infant Sleep Staging and Home Sleep Apnea Testing. J Clin Sleep Med 2015;11:1253–4. 10.5664/jcsm.5176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Vallat R, Walker MP. An open-source, high-performance tool for automated sleep staging. ELife 2021;10:e70092. 10.7554/eLife.70092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Mander BA. Local Sleep and Alzheimer’s Disease Pathophysiology. Front Neurosci. 2020. Sep 23;14:525970. doi: 10.3389/fnins.2020.525970. PMID: 33071726; PMCID: PMC7538792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].D’Atri A, Scarpelli S, Gorgoni M, Truglia I, Lauri G, Cordone S, Ferrara M, Marra C, Rossini PM, De Gennaro L. EEG alterations during wake and sleep in mild cognitive impairment and Alzheimer’s disease. iScience. 2021. Apr 1;24(4):102386. doi: 10.1016/j.isci.2021.102386. PMID: 33981973; PMCID: PMC8086022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Liguori C, Spanetta M, Izzi F, Franchini F, Nuccetelli M, Sancesario GM, et al. Sleep-Wake Cycle in Alzheimer’s Disease Is Associated with Tau Pathology and Orexin Dysregulation. J Alzheimers Dis 2020;74:501–8. 10.3233/JAD-191124. [DOI] [PubMed] [Google Scholar]
- [27].Liguori C, Romigi A, Nuccetelli M, Zannino S, Sancesario G, Martorana A, et al. Orexinergic system dysregulation, sleep impairment, and cognitive decline in Alzheimer disease. JAMA Neurol 2014;71:1498–505. 10.1001/jamaneurol.2014.2510. [DOI] [PubMed] [Google Scholar]
- [28].Lucey BP, Wisch J, Boerwinkle AH, Landsness EC, Toedebusch CD, McLeland JS, et al. Sleep and longitudinal cognitive performance in preclinical and early symptomatic Alzheimer’s disease. Brain 2021;144:2852–62. 10.1093/brain/awab272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Sintini I, Graff-Radford J, Senjem ML, Schwarz CG, Machulda MM, Martin PR, et al. Longitudinal neuroimaging biomarkers differ across Alzheimer’s disease phenotypes. Brain 2020;143:2281–94. 10.1093/brain/awaa155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Ossenkoppele R, Schonhaut DR, Schöll M, Lockhart SN, Ayakta N, Baker SL, et al. Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer’s disease. Brain 2016;139:1551–67. 10.1093/brain/aww027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Stepan ME, Altmann EM, Fenn KM. Slow-wave sleep during a brief nap is related to reduced cognitive deficits during sleep deprivation. Sleep 2021;44:zsab152. 10.1093/sleep/zsab152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Walker MP. The role of slow wave sleep in memory processing. J Clin Sleep Med 2009;5:S20–26. [PMC free article] [PubMed] [Google Scholar]
- [33].Mander BA, Rao V, Lu B, Saletin JM, Lindquist JR, Ancoli-Israel S, et al. Prefrontal atrophy, disrupted NREM slow waves and impaired hippocampal-dependent memory in aging. Nat Neurosci 2013;16:357–64. 10.1038/nn.3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Braak H, Braak E. Neurofibrillary changes confined to the entorhinal region and an abundance of cortical amyloid in cases of presenile and senile dementia. Acta Neuropathol 1990;80:479–86. 10.1007/BF00294607. [DOI] [PubMed] [Google Scholar]
- [35].Eser RA, Ehrenberg AJ, Petersen C, Dunlop S, Mejia MB, Suemoto CK, et al. Selective Vulnerability of Brainstem Nuclei in Distinct Tauopathies: A Postmortem Study. J Neuropathol Exp Neurol 2018;77:149–61. 10.1093/jnen/nlx113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Cassidy CM, Therriault J, Pascoal TA, Cheung V, Savard M, Tuominen L, et al. Association of locus coeruleus integrity with Braak stage and neuropsychiatric symptom severity in Alzheimer’s disease. Neuropsychopharmacol 2022;47:1128–36. 10.1038/s41386-022-01293-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Bolton CJ, Tam JW. Differential Involvement of the Locus Coeruleus in Early- and Late-Onset Alzheimer’s Disease: A Potential Mechanism of Clinical Differences? MedRxiv 2020. 10.1101/2020.11.01.20224139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].La Joie R, Visani AV, Lesman-Segev OH, Baker SL, Edwards L, Iaccarino L, Soleimani-Meigooni DN, Mellinger T, Janabi M, Miller ZA, Perry DC, Pham J, Strom A, Gorno-Tempini ML, Rosen HJ, Miller, BL, Jagust WJ, Rabinovici GD. Association of APOE4 and Clinical Variability in Alzheimer Disease With The Pattern of Tau- and Amyloid-PET. Neurology. 2021. Feb 2;96(5):e650–e661. Doi: 10.1212/WNL.0000000000011270. Epub 2020 Dec 1. PMID: 33262228; PMCID: PMC7884991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Anaclet C, Ferrari L, Arrigoni E, Bass CE, Saper CB, Lu J, et al. The GABAergic parafacial zone is a medullary slow wave sleep-promoting center. Nat Neurosci 2014;17:1217–24. 10.1038/nn.3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Oishi Y, Xu Q, Wang L, Zhang B-J, Takahashi K, Takata Y, et al. Slow-wave sleep is controlled by a subset of nucleus accumbens core neurons in mice. Nat Commun 2017;8:734. 10.1038/s41467-017-00781-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Yamagata T, Kahn MC, Prius-Mengual J, Meijer E, Šabanović M, Guillaumin MCC, et al. The hypothalamic link between arousal and sleep homeostasis in mice. Proc Natl Acad Sci U S A 2021;118:e2101580118. 10.1073/pnas.2101580118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Schwartz MD, Kilduff TS. The Neurobiology of Sleep and Wakefulness. Psychiatr Clin North Am 2015;38:615–44. 10.1016/j.psc.2015.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Park S-H, Weber F. Neural and Homeostatic Regulation of REM Sleep. Front Psychol 2020;11:1662. 10.3389/fpsyg.2020.01662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Fraigne JJ, Torontali ZA, Snow MB, Peever JH. REM Sleep at its Core - Circuits, Neurotransmitters, and Pathophysiology. Front Neurol 2015;6:123. 10.3389/fneur.2015.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Iranzo A The REM sleep circuit and how its impairment leads to REM sleep behavior disorder. Cell Tissue Res 2018;373:245–66. 10.1007/s00441-018-2852-8. [DOI] [PubMed] [Google Scholar]
- [46].Walsh CM, Ruoff L, Walker K, Emery A, Varbel J, Karageorgiou E, et al. Sleepless Night and Day, the Plight of Progressive Supranuclear Palsy. Sleep 2017;40. 10.1093/sleep/zsx154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Zhang W, Zhang Q, Yang Q, Liu P, Sun T, Xu Y, et al. Contribution of Alzheimer’s disease neuropathologic change to the cognitive dysfunction in human brains with Lewy body-related pathology. Neurobiol Aging 2020;91:56–65. 10.1016/j.neurobiolaging.2020.02.022. [DOI] [PubMed] [Google Scholar]
- [48].Malek-Ahmadi M, Beach TG, Zamrini E, Adler CH, Sabbagh MN, Shill HA, et al. Faster cognitive decline in dementia due to Alzheimer disease with clinically undiagnosed Lewy body disease. PLoS One 2019;14:e0217566. 10.1371/journal.pone.0217566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Swirski M, Miners JS, de Silva R, Lashley T, Ling H, Holton J, et al. Evaluating the relationship between amyloid-β and α-synuclein phosphorylated at Ser129 in dementia with Lewy bodies and Parkinson’s disease. Alzheimers Res Ther 2014;6:77. 10.1186/s13195-014-0077-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Naasan G, Shdo SM, Rodriguez EM, Spina S, Grinberg L, Lopez L, et al. Psychosis in neurodegenerative disease: differential patterns of hallucination and delusion symptoms. Brain 2021;144:999–1012. 10.1093/brain/awaa413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Wang P, Wing YK, Xing J, Liu Y, Zhou B, Zhang Z, et al. Rapid eye movement sleep behavior disorder in patients with probable Alzheimer’s disease. Aging Clin Exp Res 2016;28:951–7. 10.1007/s40520-015-0382-8. [DOI] [PubMed] [Google Scholar]
- [52].Gagnon J-F, Petit D, Fantini ML, Rompré S, Gauthier S, Panisset M, et al. REM sleep behavior disorder and REM sleep without atonia in probable Alzheimer disease. Sleep 2006;29:1321–5. 10.1093/sleep/29.10.1321. [DOI] [PubMed] [Google Scholar]
- [53].Ranasinghe KG, Kudo K, Hinkley L, Beagle A, Lerner H, Mizuiri D, et al. Neuronal synchrony abnormalities associated with subclinical epileptiform activity in early onset Alzheimer’s disease. Brain 2021:awab442. 10.1093/brain/awab442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Prerau MJ, Brown RE, Bianchi MT, Ellenbogen JM, Purdon PL. Sleep Neurophysiological Dynamics Through the Lens of Multitaper Spectral Analysis. Physiology (Bethesda). 2017. Jan;32(1):60–92. doi: 10.1152/physiol.00062.2015. PMID: 27927806; PMCID: PMC5343535. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.