

Abstract
Gal et al address the issues raised by Gerber et al and reiterate that patients in their study showed decreased Misato homolog 1 (MSTO1) mRNA and protein levels, but also confirm finding of Gerber et al that the mutation is in MSTO2p pseudogene. Whether MSTO2p variant contributes to the observed decrease in MSTO1 levels in patients remains unclear.

The correspondence article by Gerber et al. “Autosomal recessive pathogenic MSTO1 variants in hereditary optic atrophy” (Gerber et al, 2023) is questioning the disease relevance of the c.22G>A p. (p.(Val8Met) or V8M) variant in MSTO1 we reported in 2017. They identified the same rare variant in 16/49 patients having optic neuropathy in heterozygous form. Gerber et al argue that the identified variant is a relatively common variant of MSTO2p lncRNA n.83G>A (rs11264409) which shows very strong homology with the reported MSTO1 V8M (rs762798018) missense mutation. The minor allele frequency of the MSTO2p pseudogenic alteration is 0.326 compared to the reported mutation which is a rare variant of MSTO1 with a 0.000202 allele frequency.
We resequenced both Patient 1 (I/1) and 2 (II/1) samples with our original primer that does not discriminate between MSTO1 and MSTO2p, and also with the MSTO1 and MSTO2p‐specific primers introduced by Gerber et al (2023). Using the original primers, the patients again presented heterozygous MSTO1 mutation but using the new primers they showed no mutation in MSTO1 and homozygous mutation in MSTO2p (Fig 1).
Figure 1. Sequencing results of exon 1 of MSTO1 and MSTO2P using Sanger methodology.
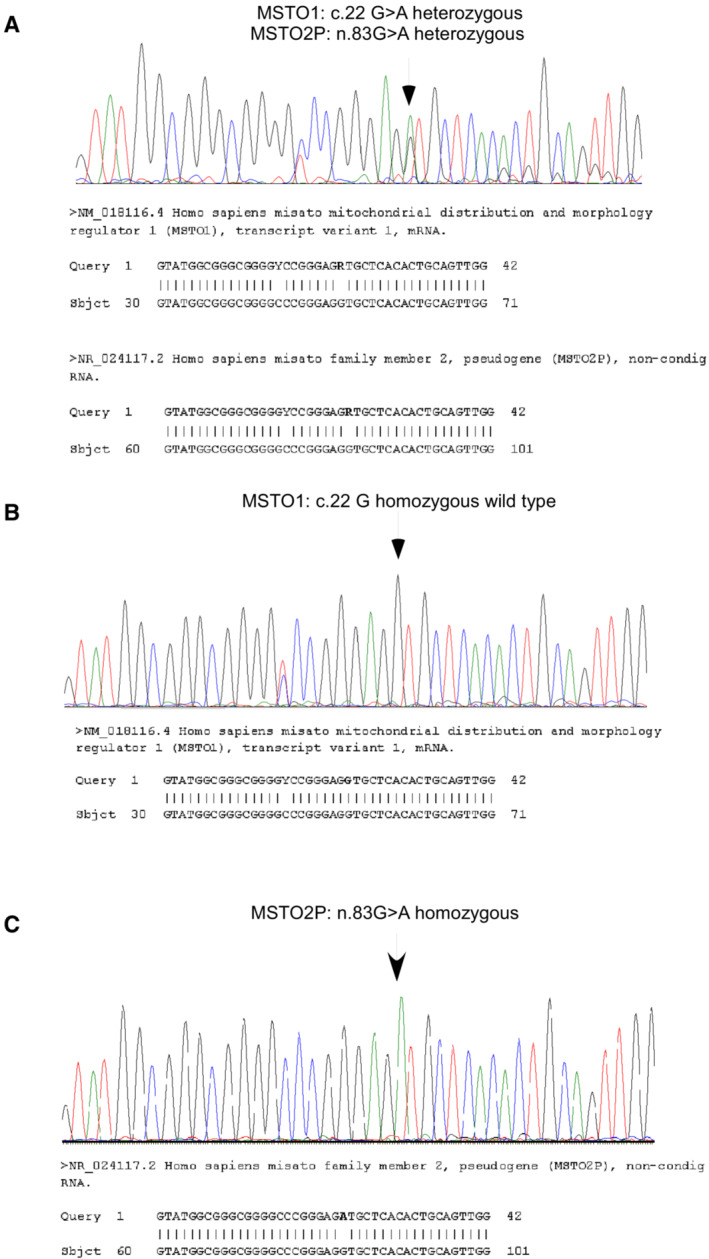
(A) The results of the repeated Sanger sequencing with the original primers (Gal et al, 2017). As a result of the sequencing, a heterozygous c.22G>A (MSTO1) and a heterozygous n.83G>A (MSTO2P) variant was present in the investigated region. Due to a high homology the MSTO1 and MSTO2P genes could not be separated. (B) MSTO1 sequencing with the newly designed section‐specific primers. As a result of the new sequencing, in the 22nd nucleotide position of the coding section of the. MSTO1 gene the guanine is present on both alleles. (C) MSTO2P sequencing with the newly designed section‐specific primers. In the MSTO2P gene the n.83G>A variant is present in homozygous form.
Database information (https://www.ncbi.nlm.nih.gov/snp/rs11264409) and results by Gerber et al (2023) indicated that the present mutation in MSTO2p is common and likely benign. However, it is worth noting that both patients 1 (I/1) and 2 (II/1) in our original paper Gal et al (2017) showed decreased MSTO1 mRNA and protein levels (fig 1E and F) and a mitochondrial phenotype (figs 2 and 3). Importantly, we showed that genetic rescue of MSTO1 in the patient cells reversed the mitochondrial changes (fig 4) and that silencing of MSTO1 in HeLa cells recapitulated the fusion defects observed in the patient cells (fig 5). Thus, changes in MSTO1 protein levels seem to contribute to the mitochondrial dynamics phenotype of the family.
The MSTO2p gene is transcribed but believed to be non‐coding. While it remains unclear which mRNA isoform (MSTO1 or MSTO2p) is decreased in our patients, the diminished band in our western blots is reflective of loss of MSTO1 protein in the patient cells (Fig 1F; Gal et al, 2017).
The new analysis suggests that in our patients, mutations in MSTO1 are not responsible for the decreased expression of the gene and protein, raising questions of how MSTO1 levels are regulated. One possibility would be that the homozygous mutation identified in MSTO2p make it act as a lncRNA that can regulate expression of MSTO1. In cancers, MSTO2p has been shown to work as a lncRNA that can regulate gene expression, including EZH2 (Wei et al, 2017; Wang et al, 2019; Guo & Zhang, 2022). Notably, heterozygous c.553G>C p.(Asp185His) (rs2302427) variation of the EZH2 gene was also detected in our Patients (I/1 and II/1). Based on this, the question arises as to whether the interaction of MSTO2p with another gene affects MSTO1 or mutations in a yet unidentified gene that can regulate MSTO1 levels is responsible for the phenotype in the family we reported previously (Gal et al, 2017).
Further studies will be necessary to validate if the homozygous MSTO2p mutation contributes or not to the decrease in MSTO1 mRNA and protein abundance identified in our patients. For example, quantification of MSTO1 and MSTO2p mRNA levels using primers specific for each isoform can define if the decrease in MSTO1 mRNA in patient fibroblasts reflects changes in one or both isoforms. Similarly, selective perturbations of MSTO1 and MSTO2p expression using siRNA or CRISPR can determine if MSTO2p might exert an effect on MSTO1 RNA expression. Finally, characterization of the mitochondrial phenotype in patient cells upon decreases of MSTO2p or overexpression of wild‐type or the homozygous mutant will further shed light onto the role of this pseudogene in patients carrying MSTO1 defects.
In summary, recent progress in discriminating of MSTO2p from MSTO1 made it possible to reinterpret our original WES results as causative to the disease phenotypes of our patient population. However, the pathway we described – from decreased MSTO1 to mitochondrial dynamics and function in the patient fibroblasts – remains well supported and, as in our original conclusion, likely contributes to the patients' disease. Effectively, these new data now allows to extend our original results in trying to understand whether and how changes in MSTO2p might contribute to disease phenotypes associated with MSTO1 loss.
EMBO Mol Med (2023) 15: e16251
Reply to: Gerber et al (August 2023)
See also: A Gai et al (July 2017) and A Gai et al (August 2023)
References
- Gal A, Balicza P, Weaver D, Naghdi S, Joseph SK, Varnai P, Gyuris T, Horvath A, Nagy L, Seifert EL et al (2017) MSTO1 is a cytoplasmic pro‐mitochondrial fusion protein, whose mutation induces myopathy and ataxia in humans. EMBO Mol Med 9: 967–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber S, Lessard L, Rouzier C, Ait-el-Mkadem Saadi S, Ameli R, Thobois S, Abouaf L, Bouhour F, Kaplan J et al (2023) Autosomal recessive pathogenic MSTO1 variants in hereditary optic atrophy. EMBO Mol Med 15: e16090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo M, Zhang X (2022) LncRNA MSTO2P promotes colorectal cancer progression through epigenetically silencing CDKN1A mediated by EZH2. World J Surg Oncol 20: 95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LJ, Sun GZ, Chen YF (2019) LncRNA MSTO2P promotes proliferation and autophagy of lung cancer cells by up‐regulating EZH2 expression. Eur Rev Med Pharmacol Sci 23: 3375–3382 [DOI] [PubMed] [Google Scholar]
- Wei Y, Chang Z, Wu C, Zhu Y, Li K, Xu Y (2017) Identification of potential cancer‐related pseudogenes in lung adenocarcinoma based on ceRNA hypothesis. Oncotarget 8: 59036–59047 [DOI] [PMC free article] [PubMed] [Google Scholar]