

Abstract
Ferroptosis has emerged as an attractive strategy in cancer therapy. Understanding the operational networks regulating ferroptosis may unravel vulnerabilities that could be harnessed for therapeutic benefit. Using CRISPR‐activation screens in ferroptosis hypersensitive cells, we identify the selenoprotein P (SELENOP) receptor, LRP8, as a key determinant protecting MYCN‐amplified neuroblastoma cells from ferroptosis. Genetic deletion of LRP8 leads to ferroptosis as a result of an insufficient supply of selenocysteine, which is required for the translation of the antiferroptotic selenoprotein GPX4. This dependency is caused by low expression of alternative selenium uptake pathways such as system Xc−. The identification of LRP8 as a specific vulnerability of MYCN‐amplified neuroblastoma cells was confirmed in constitutive and inducible LRP8 knockout orthotopic xenografts. These findings disclose a yet‐unaccounted mechanism of selective ferroptosis induction that might be explored as a therapeutic strategy for high‐risk neuroblastoma and potentially other MYCN‐amplified entities.
Keywords: ferroptosis, neuroblastoma, selenocysteine, selenoprotein, synthetic lethality
Subject Categories: Autophagy & Cell Death, Cancer, Neuroscience
The low‐density lipoprotein receptor (LRP8) was identified as a critical suppressor of ferroptosis in MYCN‐amplified neuroblastoma. Blocking selenium/selenocysteine uptake mechanisms via LRP8 offers a selective strategy to induce ferroptosis and disrupt GPX4 function.

The paper explained.
Problem
Ferroptosis, a distinct form of cell death, has emerged as a potential approach for eliminating solid tumors that are refractory to treatment. However, inhibiting the selenoprotein glutathione peroxidase 4 (GPX4), which suppresses ferroptosis, remains challenging due to the absence of suitable inhibitors and to the potential systemic toxicity. Our previous findings have demonstrated a notable dependence on GPX4 in high‐risk MYCN‐amplified neuroblastomas, yet the precise factors contributing to this phenomenon remain incompletely understood.
Results
Our study reveals that high‐risk MYCN‐amplified neuroblastomas exhibit a significant dependency on LRP8. Through genome‐wide and single‐cell transcriptomics CRISPR‐activation screens, we identify the low‐density lipoprotein receptor‐related protein 8 (LRP8) as a critical factor in selenium/selenocysteine metabolism in MYCN‐amplified cancers. We find that MYCN‐amplified cells ineficiently activate alternative selenium/selenocysteine pathways, resulting in vulnerability to LRP8 inhibition. These metabolic vulnerabilities provide a unique opportunity to target LRP8 to induce ferroptosis selectively and safely.
Impact
In light of the limited success in repurposing adult oncology drugs for neuroblastoma treatment, our research presents a significant advance by introducing innovative strategies based on ferroptosis. By specifically focusing on targeting LRP8, we have identified a potential breakthrough that not only offers novel therapeutic approaches for neuroblastoma but also holds promise for a range of pediatric malignancies and MYCN‐driven cancers.
Introduction
Ferroptosis is a cell death modality that is attracting increasing interest as a means to eradicate therapeutically challenging tumor entities (Viswanathan et al, 2017; Friedmann Angeli et al, 2019). Early studies reported that ferroptosis is triggered by cyst(e)ine starvation. Cysteine depletion can be initiated pharmacologically by small molecules such as Erastin that can specifically block cystine uptake via inhibiting the amino acid transporter system Xc−. System Xc− is a heterodimer consisting of SLC3A2 (also known as 4F2hc or CD98) and the specificity‐conferring subunit SLC7A11 (also known as xCT). Characterization of the molecular events leading to ferroptosis execution has shown that cysteine starvation promotes cell death by the acute depletion of the cysteine‐containing tripeptide glutathione (GSH), which in turn is essential to fuel the activity of the selenoprotein glutathione peroxidase 4 (GPX4; Yang et al, 2014). GPX4 utilizes reducing equivalents from glutathione (GSH) to suppress the accumulation of lipid hydroperoxides and, ultimately, the induction of ferroptotic cell death (Shah et al, 2018; Fig 1A).
Figure 1. Genome‐wide CRISPR activation screen identifies negative regulators of ferroptosis.
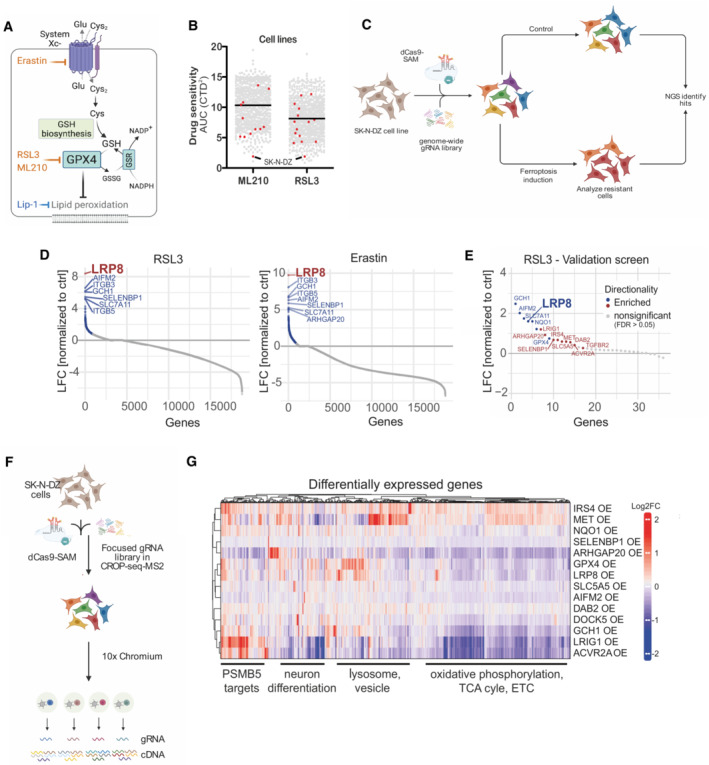
- Schematic depiction of ferroptosis regulators and small molecule modulators. Inhibitors are shown in light blue, and inducers in orange.
- Analyzis of the depmap portal (www.depmap.org) reveals MYCN‐amplified SK‐N‐DZ as hypersensitive cell lines to the GPX4 inhibitors RSL3 and ML210.
- Strategy of the genome‐wide CRISPR activation (CRISPRa) screen in MYCN‐amplified SK‐N‐DZ cells.
- Overexpression phenotypes conferring resistance to 300 nM RSL3 (left) or 1 μM Erastin (right) treatment. Significant hits are marked in blue (FDR ≤ 0.05), while the highest‐scoring hit, LRP8, is highlighted in red.
- Overexpression phenotypes conferring resistance to RSL3 (100 nM) induced ferroptosis in the pooled validation CRISPRa screen. Significantly enriched hits (FDR ≤ 0.05) are marked and labeled in blue (known ferroptosis regulators) or red (novel regulators identified in this study). The highest‐scoring hit from the primary screens, LRP8, is highlighted.
- Strategy of the single‐cell CRISPRa screen to characterize hits from the ferroptosis‐resistance screen. Guide RNA labels are recovered alongside the whole transcriptome readout for each cell.
- Transcriptomic consequences of CRISPRa of 14 scoring hits from the primary and the validation screens. Each row represents one CRISPRa cluster. For each cluster, the top 50 genes with the most significant differential expression (compared to the nontargeting control cluster) were selected and merged to a signature gene list represented by the columns. Columns and rows were hierarchically clustered based on Pearson's correlation.
Source data are available online for this figure.
Given that alternative pathways can provide cysteine and thus bypass the requirement for system Xc−, such as the transsulfuration pathway, there is an intense search for strategies and molecular tools to trigger ferroptosis at the level of GPX4. GPX4 inhibitors include the covalent inhibitors RSL3 and ML210, which perform in cultured cells but are inefficient in in vivo settings (Yang et al, 2014; Viswanathan et al, 2017; Liu et al, 2018; Eaton et al, 2019; Fig 1A). Thus, the lack of suitable in vivo GPX4 inhibitors and the predicted systemic toxicity of such compounds (Friedmann Angeli et al, 2014; Brutsch et al, 2015; Carlson et al, 2016) limit the translation of these discoveries into cancer therapies. Recently, we, and others, have uncovered that high‐risk MYCN‐amplified neuroblastomas are characterized by a striking GPX4 dependency (Floros et al, 2021; Lu et al, 2021; Alborzinia et al, 2022). The molecular determinants of this increased dependency remain elusive and untangling the mechanisms dictating ferroptosis hypersensitive states could potentially pave the way to exploit these vulnerabilities.
In this work, using genome‐wide and single‐cell CRISPR‐activation (CRISPRa) screens, we identify novel regulators of ferroptosis and detect shared transcriptional signatures and states regulating ferroptosis hypersensitivity. Specifically, we identify the low‐density lipoprotein receptor (LDLR)‐related protein 8 (LRP8, also known as APOER2) as a critical suppressor of ferroptosis in MYCN‐amplified neuroblastoma. We find that the LRP8 dependency in MYCN‐amplified neuroblastomas is partially due to the limited activity of alternative selenium transporters, specifically system Xc−. Therefore, our work demonstrates that the utilization of different selenium/selenocysteine acquisition pathways offers unanticipated opportunities to selectively and safely induce ferroptosis for therapeutic benefit.
Results
CRISPR activation screens identify novel regulators of ferroptosis
In order to gain additional insights into the process of ferroptosis, we reasoned that understanding the mechanisms that could increase the resistance of intrinsically sensitive cells would provide a means to identify yet uncharacterized regulators of ferroptosis (Fig 1A). Toward this end, we initially analyzed data from the depmap portal (www.depmap.org) in search of cell lines that are hypersensitive to the GPX4 inhibitors RSL3 and ML210 (Fig 1B). We selected the MYCN‐amplified neuroblastoma cell line SK‐N‐DZ, a representative of tumor entities that still defy current treatments and for which we and others have already reported a marked dependency on GPX4 (Lewerenz et al, 2018; Floros et al, 2021; Lu et al, 2021; Alborzinia et al, 2022), thus providing an ideal setting to interrogate the mechanisms underlying this hypersensitivity. Using this cell line, we performed genome‐wide CRISPR activation (CRISPRa) screens where we induced ferroptosis via two nonoverlapping mechanisms (Fig 1C), namely via inhibition of (i) GPX4 by RSL‐3 or (ii) system Xc− by Erastin (Fig 1A and C). Screen deconvolution based on the enrichment of gRNAs after ferroptosis induction allowed us to identify several known and novel ferroptosis regulators (Fig 1D, Dataset EV1, FDR ≤ 0.05). For instance, we found the overexpression of several known key ferroptosis regulators as hits including GCH1 (Kraft et al, 2020), AIFM2 (Bersuker et al, 2019; Mao et al, 2021), SLC7A11 (Dixon et al, 2014), GPX4 (Yang et al, 2014), and NQO1 (Sun et al, 2016) supporting the validity of our screening results. While the majority of hits were exclusive for one mechanism of ferroptosis induction, we found several overlapping hits (120 shared hits, Appendix Fig S1A). A secondary screen focusing on shared and functionally related hits (list provided in Dataset EV2) confirmed the resistance phenotype for most of the scoring hits when selected against the GPX4 inhibitor RSL3 (Fig 1E). To provide an unbiased understanding of the differential mechanism of cellular states that prevent ferroptosis, we coupled the focused screen to single‐cell RNA‐seq (scRNA‐seq, CROP‐seq) as a readout (Fig 1F). The focused library consisted of two guide RNAs (gRNAs) targeting each of the selected 36 candidate genes together with 10 nontargeting control gRNAs (NT‐ctrl). We obtained high‐quality data from ~11,000 CRISPRa‐assigned cells (mean of ~78,000 reads and median of ~6,600 genes per cell; Appendix Fig S1B). Selecting cells with a clear CRISPRa phenotype (see Materials and Methods) allowed us to retrieve 130 cells on average for each of the selected hits (32/36 target gene identities detected, Appendix Fig S1C). As expected, cells assigned to a particular perturbation cluster showed an increased expression of the targeted gene (Fig 1G, Appendix Fig S1E and F and Dataset EV3). Interestingly, the impact of several CRISPRa phenotypes converged on the expression of known ferroptosis regulators. For instance, overexpression phenotypes of IRS4 and MET shared the upregulation of known ferroptosis suppressors, including the transcription factor NFE2L2 that drives the expression of genes involved in redox signaling and oxidative protection (Yamamoto et al, 2018), the heat‐shock protein HSPB1, as well as the CoQ oxidoreductases AIFM2 (also known as FSP1 (Bersuker et al, 2019; Doll et al, 2019); Appendix Fig S1F and G). Gene Ontology analysis of differentially expressed genes upon CRISPRa showed significant enrichment in processes involved in cellular detoxification, energy metabolism, and proteasomal degradation that have been previously linked to ferroptosis (Dixon et al, 2012; Gao et al, 2019; Chen et al, 2021; Appendix Fig S1H). Interestingly, transcriptomic consequences of overexpressing the highest scoring hit in our primary screen, LRP8, clustered together with the GPX4 overexpression phenotype in our scCRISPRa screen, underscoring that the mechanism underlying ferroptosis resistance might overlap and be shared between multiple groups (Fig 1F and Appendix Fig S1D, F and I for the co‐expressed genes between GPX4 and LRP8 overexpression). Similarly, regulatory network inference and pathway activity analysis showed coclustering of the LRP8‐ and GPX4‐OE phenotypes further supporting an underlying shared transcriptional mechanism of ferroptosis resistance (Appendix Fig S1J). In sum, the scCRISPRa secondary screen faithfully recapitulated the gRNA identity in single‐cell assays and simultaneously provided transcriptional signatures of different ferroptosis‐resistant states derived from the genome‐wide screen.
SELENOP is a critical source of selenium to support the growth of a subset of MYCN‐amplified neuroblastomas
The identification of LRP8 as a critical regulator of ferroptosis sensitivity is consistent with data from the cancer therapeutic response portal (CTRP), showing that LRP8 expression strongly correlates with resistance to GPX4 inhibitors (ML210, RSL3, and ML162; Appendix Fig S2A; Basu et al, 2013). To functionally validate our findings, we first overexpressed LRP8 using a CRISPRa system in which SK‐N‐DZ cells became robustly resistant to RSL3 treatment (Extended Data Fig 2B). Similarly, stable overexpression of a Flag‐tagged hLRP8 in SK‐N‐DZ cells conveyed resistance to a panel of ferroptosis inducers covering differential modes of action (Fig 2A and B). In line with a specific inhibitory role for LRP8 in ferroptosis, an overall impact on sensitivity toward other cytotoxic compounds was not observed (Appendix Fig S2C). In accordance with the protective effect observed, LRP8 overexpression suppressed lipid peroxidation in cells treated with a GPX4 inhibitor (Fig 2C). Furthermore, overexpression of LRP8 was able to increase ferroptosis resistance in a larger panel of cell lines (Fig 2D). No other members of the LRP family showed similar effects when overexpressed, highlighting the specific role of LRP8 on ferroptosis resistance (Appendix Fig S2D and E).
Figure 2. LRP8 loss triggers ferroptosis in MYCN‐amplified neuroblastoma.
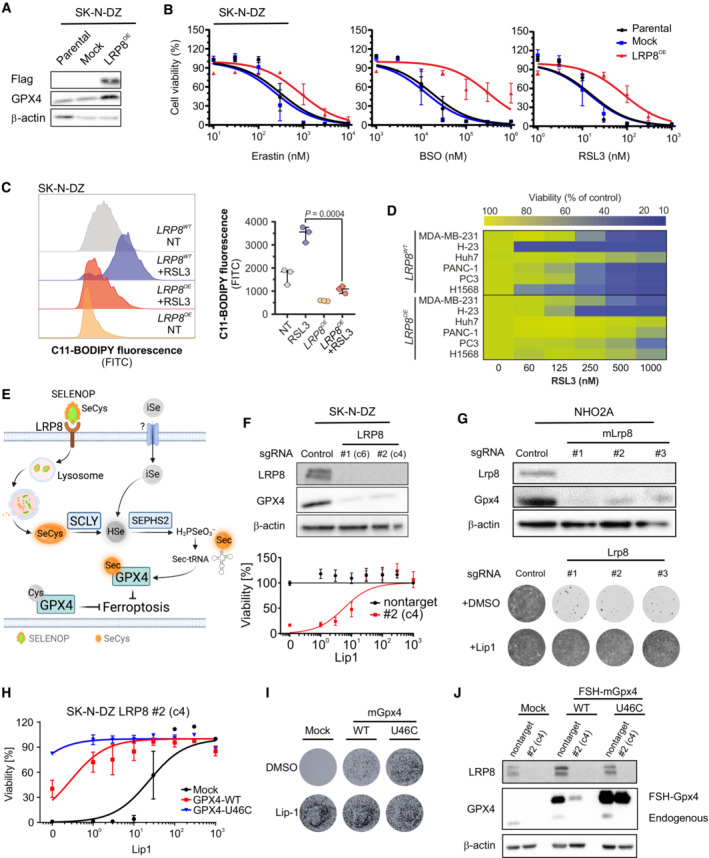
- Immunoblot analyzis of FLAG and GPX4 in SK‐N‐DZ cells overexpressing an empty vector or hLRP8‐Flag.
- Dose‐dependent toxicity of the ferroptosis inducers Erastin, BSO, and RSL3 in SK‐N‐DZ cell lines stably transduced with a vector expressing hLRP8‐Flag. Data are the mean ± SEM of n = 3 wells of a 96‐well plate from three independent experiments.
- Flow cytometry analyzis of BODIPY 581/591 C11 oxidation in SK‐N‐DZ overexpressing hLRP8‐Flag induced by RSL3 treatment (100 nM, 6 h) Data represent the mean ± SEM; n = 3 samples. Statistical analysis was performed using a two‐tailed Student's t‐test.
- Heat map depicting the dose‐dependent response of RSL3 in a panel of cell lines overexpressing hLRP8‐Flag.
- Schematic representation of selenium uptake mechanisms.
- Generation and characterization of LRP8 knockout cell lines using two independent gRNAs. Upper panel, immunoblot analyzis of LRP8 expression in cells transduced with gRNA targeting LRP8. Lower panel, the viability of cell lines, either wild‐type or knockout for LRP8 in the presence of increasing concentrations of Lip‐1. Data are the mean ± SEM of n = 3 wells of a 96‐well plate from two independent experiments.
- Recapitulation of LRP8 dependency in the NHO2A cell line derived from a murine model of MYCN amplification. The upper panel depicts the immunoblot of Lrp8 and Gpx4 in cells expressing three independent gRNAs targeting Lrp8. The lower panel shows the clonogenic capacity of Lrp8‐deficient cells and the protective effect of Lip‐1 (500 nM).
- Dose‐dependent response of SK‐N‐DZ LRP8 knockout clonal cell line (#2) expressing wild‐type and U46C‐GPX4 in the present of Lip1. Data are the mean ± SEM of n = 3 wells of a 96‐well plate from two independent experiments.
- The clonogenic capacity of an SK‐N‐DZ LRP8 knockout clonal cell line (#2) expressing wild‐type and U46C‐GPX4.
- Immunoblot analyzis of FLAG and GPX4 in SK‐N‐DZ cells overexpressing flag‐tagged WT or a U46C variant of GPX4 in wild‐type and knockout LRP8 background.
Source data are available online for this figure.
Next, we explored the mechanism by which LRP8 protects cells from ferroptosis. Previous studies have established that members of the low‐density lipoprotein (LDL) receptor‐related protein, including LRP2 and LRP8, are receptors for the selenium carrier protein SELENOP (Burk et al, 2006; Olson et al, 2007), indicating that modulation of selenocysteine metabolism would be the likely mechanism by which LRP8 protects cells from ferroptosis (Fig 2E). This premise agrees with the finding that overexpression of LRP8 leads to upregulation of GPX4 at the protein level (Fig 2A) without impacting RNA levels (Appendix Fig S2F), pointing toward an underlying post‐transcriptional regulation. Next, to address the requirement of LRP8, we generated LRP8‐deficient SK‐N‐DZ cells. Unexpectedly, without additional stressors, we observed that these cells readily underwent massive ferroptosis‐like cell death in the absence of ferroptosis‐inhibiting compounds (Fig 2F and Appendix Fig S2G). To further corroborate this observation and provide mechanical support for targeting LRP8 in neuroblastoma, we deleted Lrp8 in the murine TH‐MYCN neuroblastoma mouse model (Fig 2G) and a panel of neuroblastoma cell lines with and without MYCN amplification (Appendix Fig S2G–I). These additional models recapitulated the observation that LRP8 is essential to support the viability of human neuroblastoma cell lines and that it is also crucial in a cell line derived from a MYCN‐driven genetic neuroblastoma model (Appendix Fig S2H and I).
Furthermore, the loss of viability caused by LRP8 loss could be fully rescued by providing selenite that provides selenium by a mechanism independent of LRP8 and the ferroptosis inhibitor Liproxstatin‐1 (Lip‐1; Appendix Fig S2J). To validate the role of SELENOP, we used two distinct LRP8‐deficient models to show that SELENOP can only boost GPX4 expression in LRP8 proficient cells (Appendix Fig S3A and B). Lastly, the specificity of ferroptosis was confirmed by showing that only canonical inhibitors of ferroptosis can suppress cell death, whereas other cell death inhibitors cannot (Appendix Fig S3C).
Given the important role of selenocysteine metabolism in preventing ferroptosis, we next asked whether any correlation with the clinical outcome can be observed in cohorts of pediatric neuroblastoma patients. While LRP8 expression was not generally associated with event‐free survival, high expression of GPX4 and other members of the selenocysteine incorporation machinery (i.e., SEPHS2, PSTK, EEFSEC, and SECISBP2) showed a strong correlation with poor disease outcome. This finding is in line with the generally higher expression of members of the selenocysteine biosynthetic pathway in high‐risk neuroblastoma (Appendix Fig S3D). These data suggest that the efficient flux of selenium/selenocysteine through this pathway is a strong determinant for poor survival of neuroblastoma patients and may contribute to therapy resistance. This reasoning agrees with the observation that SK‐N‐DZ cells could not grow in selected batches of fetal bovine serum (FBS), which lack detectable SELENOP unless supplemented with exogenous sources of selenium or ferroptosis‐inhibiting compounds (Appendix Fig S4A and B). Accordingly, the protective effect conferred by the overexpression of LRP8 was dramatically reduced in low SELENOP conditions (Appendix Fig S4C).
Strong evidence for a direct link between LRP8, selenocysteine, GPX4, and cell survival we provide by showing that overexpression of wild‐type GPX4 was unable to fully rescue cell viability upon loss of LRP8. Importantly, the expression of GPX4 (U46C), a mutant that is approximately 100× catalytically less efficient but bypasses the requirement of selenocysteine loaded tRNAs for efficient translation (Ingold et al, 2018), could fully restore cell viability (Fig 2H–J). Consequently, only the levels of ectopic wild‐type (WT) and endogenous GPX4 were decreased by LRP8 deficiency, while the ectopic U46C GPX4 level remained unchanged (Fig 2H). In support of this, the knockdown of selenophosphate synthetase 2 (SEPHS2), a central enzyme in the metabolization of selenium, significantly reduced ferroptosis resistance of cells overexpressing LRP8 (Appendix Fig S4D). Additional evidence that ferroptosis is induced in LRP8‐deficient cells was provided by demonstrating that lipid peroxidation can be triggered in the absence of LRP8 and that this is prevented by the addition of ferrostatin‐1 (Fer‐1) or by the overexpression of the GPX4‐independent ferroptosis suppressor AIFM2 (FSP1; Appendix Fig S4E and F; Doll et al, 2019). Our data demonstrate that targeting SELENOP uptake via LRP8 is a valuable strategy to efficiently disrupt GPX4 maintenance and trigger ferroptosis in cultured MYCN‐amplified neuroblastoma cells.
Limited activity of system Xc− mediates the dependency on LRP8
Our observation that MYCN‐amplified neuroblastoma cells are highly dependent on LRP8 for selenium uptake is unanticipated, given that alternative forms and uptake mechanisms exist (Burk & Hill, 2015). Moreover, the specification analysis of FBS showed that selenite and selenate were present at approximately 1–2 nM. Still, these forms of selenium could not sufficiently support selenocysteine biosynthesis and consequently could not prevent ferroptosis in neuroblastoma cell lines (Appendix Fig S4B). Our rescue experiments suggested that additional 20‐fold excess of selenite was required to rescue viability (Appendix Fig S4B). These findings led us to hypothesize that alternative mechanisms of selenium provision must be inefficient or inactive in neuroblastoma cells. In order to gain insights into these alternative mechanisms, we performed a series of CRISPR‐based screens focusing on solute carriers (SLCs), as these transporters are the major mediators of soluble metabolite uptake into cells (Superti‐Furga et al, 2020). Aiming to identify SLC synthetic lethal interactions with LRP8 loss, we used cells in which the deletion of LRP8 did not lead to the loss of viability (HT1080 and A375; Appendix Fig S5A–D). Despite being viable upon genetic loss of LRP8, these cell lines display a high sensitivity toward several ferroptosis inducers (Appendix Fig S5B and D). Interestingly, screens in both cell lines retrieved the system Xc− subunits SLC7A11 and SLC3A2 as genes generating synthetic lethality with LRP8 deficiency (Fig 3A and B).
Figure 3. SLC‐focused CRISPR screen identifies alternative selenium uptake mechanisms.
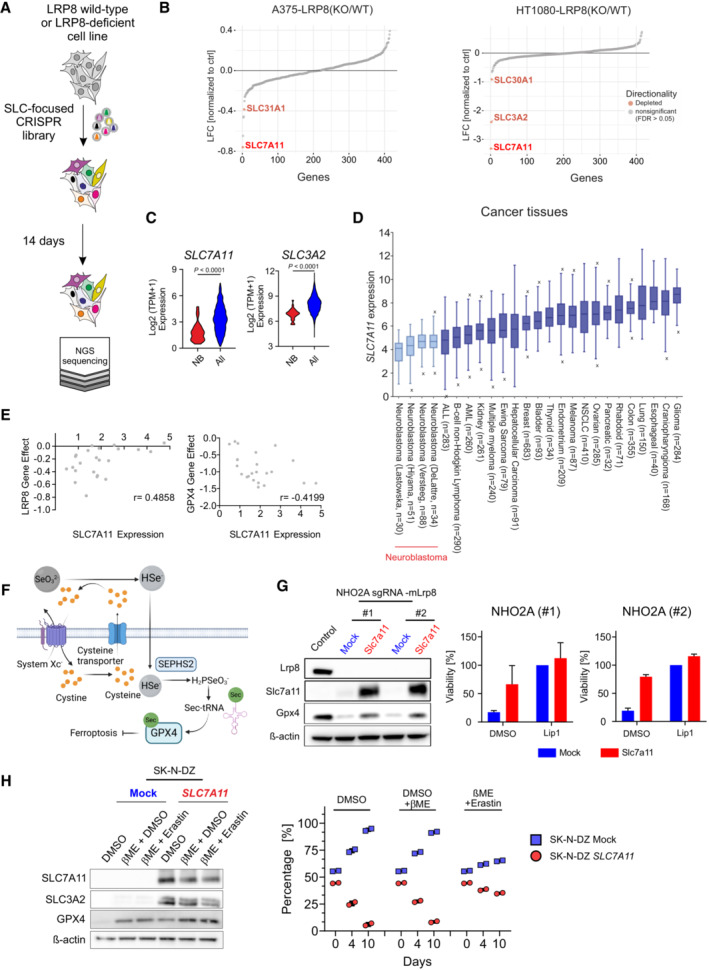
- Schematic representation of the SLC‐focused CRISPR knockout screen in the LRP8 wild‐type and LRP8‐deficient cell lines.
- Results of a CRISPR deletion screen conducted in H1080 and A375 cell lines displayed a log2 fold change between LRP8‐deficient and wild‐type cells.
- Comparison of SLC7A11 expression in a panel of 23 neuroblastoma cell lines against 1,349 non‐neuroblastoma cell lines demonstrating the lineage‐specific lower expression of SLC7A11 and SLC3A2 (http://www.depmap.org). Statistical analysis was performed using an Unpaired t‐test. ****P < 0.0001.
- SLC7A11 expression in cancer tissues of different entities. Data retrieved from the R2database. The center line in the box plots indicates the median value. Lower and upper hinges represent the 25th and 75th quantiles, the whiskers denote the 1.5× interquartile range, and outliers are marked in the plot.
- Dot plot depicting the correlation of the dependency of neuroblastoma cell lines on LRP8 and GPX4 (CERES score of −1 means full dependency based on CRISPR–Cas9 knockout screening data) and the expression levels of SLC7A11 in a panel of 27 neuroblastoma cell lines (depmap portal; https://depmap.org/portal/). Cell lines with low expression of SLC7A11 were found to be dependent on LRP8 (Pearson correlation r: 0.4858).
- Schematic representation of system Xc− of selenium uptake mechanisms.
- The immunoblot of Lrp8, Slc7a11 and Gpx4 in NHO2A stably expressing Cas9 and two independent sgRNAs targeting Lrp8 in an SLC7A11 or mock expressing background and viability of the cell lines in the presence of the DMSO, Lip1 (500 nM), or a combination of both. Cell viability assays depicted are mean ± SD of two independently performed experiments (n = 4 wells of a 96‐well plate).
- Generation and characterization of cells overexpressing SLC7A11. Immunoblot analysis of SLC7A11 and SLC3A2 from SK‐N‐DZ cells overexpressing SLC7A11 or mock. Cell competition assay of SK‐N‐DZ cell line overexpressing SLC7A11 or mock controls. For the experiment, cells were seeded at a ratio of 50/50 and 50,000 events were measured via flow cytometry at the depicted time points. Rescue experiments were performed in the presence of 50 μM beta‐Mercaptoethanol (beta‐ME) and 2 μM Erastin. Bars display percentage of eGFP‐ (blue) and Scarlet‐cells (red) with the means ± SD of two independent experiments. Statistical analysis was performed using an Unpaired t‐test comparing the abundance of SK‐N‐DZ cells overexpressing SLC7A11 (Red).
Source data are available online for this figure.
This indicates that system Xc−, in addition to its previously reported role in indirect uptake (Olm et al, 2009; Carlisle et al, 2020), is a major contributing factor for the direct uptake of inorganic (SeO3 2−) and organic forms (SeCys) of selenium (Fig 3B). In line with these findings, we noted that neuroblastoma show a generally low expression level of both system Xc− subunits (SLC7A11 and SLC3A2) compared with cancer tissues and cell lines of other entities (Fig 3C and D, and Appendix Fig S4H). These data suggest that the sensitivity and therefore dependency of neuroblastoma to LRP8 deletion could be at least partly due to the reduced activity of alternative selenium transporters. Publicly available data (depmap portal) indicate that in neuroblastomas, LRP8 dependency is inversely correlated with SLC7A11 expression with LRP8 knockout sensitive cell lines exhibiting lower SLC7A11 expression levels, while this is not observed for GPX4 (Fig 3E and Appendix Fig S4G). Lastly, providing support for our hypothesis that low SLC7A11 contributes to the increased LRP8 dependency, our data show that overexpression of SLC7A11 was able to restore GPX4 levels and partially rescue the viability of LRP8‐deficient cells (Fig 3F and G). However, despite these observations, we noted that SLC7A11 overexpression resulted in a profound growth defect of neuroblastoma cells. This effect was specific to SLC7A11 activity, as it was rescued by the addition of the system Xc− inhibitor Erastin (Fig 3H). Our initial characterization further demonstrated that in an isogenic model where MYCN is inducibly activated, the process was both aggravated by MYCN activity and cystine lelels (Appendix Fig S6A–D). Altogether, our data suggest that low expression of system Xc− components that limit intracellular uptake of inorganic selenium is the likely cause for the remarkable dependence of neuroblastoma cells on LRP8.
LRP8 is required for the initiation and maintenance of neuroblastoma in vivo
Given the critical role played by LRP8 and the selenocysteine metabolism in preventing ferroptosis in highly aggressive neuroblastoma subtypes with MYCN amplifications, we addressed whether LRP8 elimination could target neuroblastoma growth by inducing ferroptosis in vivo. We took advantage of our reported orthotopic animal model in which SK‐N‐DZ cells were implanted in the adrenal gland of NOD.Cg‐PrkdcscidIl2rgtm1Wjl/SzJ (NSG) mice (Alborzinia et al, 2022). Briefly, all cells were grown in the presence of liproxstatin‐1 (Lip‐1) before implantation to prevent ferroptotic cell death of LRP8‐deficient neuroblastoma cells. Subsequently, SK‐N‐DZ cells were implanted, and tumor growth was monitored using in vivo bioluminescence imaging (Fig 4A). LRP8‐deficient tumors showed a marked decrease in tumor growth and, compared with LRP8 proficient cells, showed a significantly increased overall survival of mice (76d ± vs. 31d ±, defined by reaching termination criteria) with a median survival of 29 days vs. 40 days (Fig 4B–D). More clinically relevant, we explored the therapeutic potential of targeting LRP8 in established SK‐N‐DZ neuroblastoma. For this, LRP8 proficient (LRP8WT) and deficient (LRP8KO) tumors were both allowed to grow in the presence of in vivo active Lip‐1 (Fig 4E), with the former ones serving as controls (Fig 4E, top, red). After LRP8‐deficient neuroblastomas were established (seven days of postimplantation with five days of Lip‐1 treatment), mice were randomized into two groups. Lip‐1 treatment was continued in the yellow group, while in the blue group, Lip‐1 injections, and thus ferroptosis inhibition, were stopped (Fig 4E, bottom). Analysis of the three cohorts after 14 days of postimplantation showed that both LRP8‐deficient xenograft groups (blue, yellow) showed impaired tumor growth compared with WT controls (red) despite Lip‐1‐mediated ferroptosis inhibition (Fig 4F and G). Importantly, Lip‐1 withdrawal in the randomized LRP8KO group for only 9 days (the maximum time approved by the animal protocol) already significantly reduced neuroblastoma growth also in established tumors, mimicking the clinical situation (Fig 4F and G). The data above support the requirement of LRP8 for the establishment and maintenance of MYCN‐amplified SK‐N‐DZ neuroblastoma by preventing ferroptosis. This is further supported by the robust loss of GPX4 protein in LRP8KO tumors (Fig 4H). Collectively, our data indicate that LRP8 is required to prevent ferroptosis by maintaining high levels of GPX4 in MYCN‐amplified orthotopic neuroblastoma models and suggest that inhibition of the SELENOP/LRP8 axis as a novel and selective strategy to trigger ferroptosis and thereby limit tumor growth in highly aggressive and hard to treat MYCN‐amplified neuroblastoma cells (Fig 4I).
Figure 4. LRP8 is essential for orthotopic neuroblastoma growth.
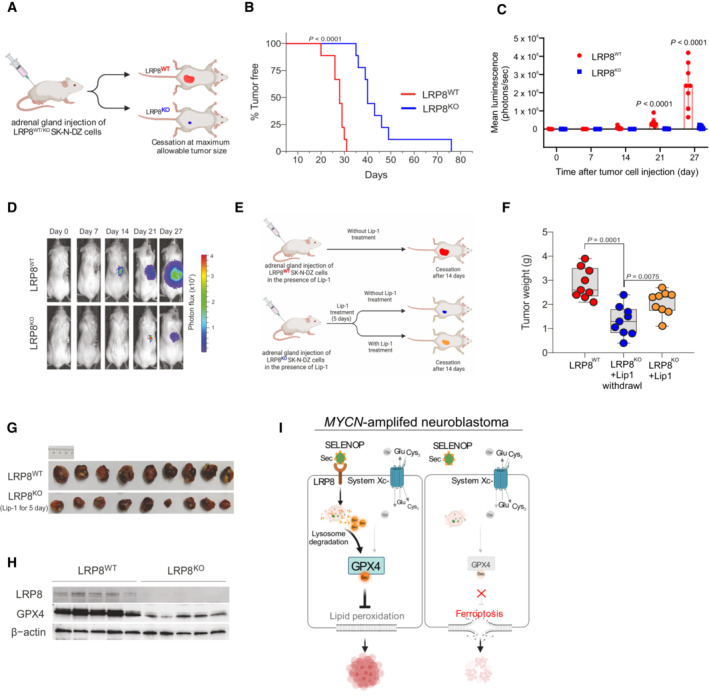
- Schematic representation of the orthotopic implantation of control (LRP8WT) or LRP8‐deficient (LRP8KO) SK‐N‐DZ cell lines.
- Kaplan–Meier plot displaying tumor‐free survival (TFS) for mice injected orthotopically with LRP8WT (red, n = 9) or LRP8KO (blue, n = 9) SK‐N‐DZ cells. A Gehan–Breslow–Wilcoxon test was conducted for statistical analyzis.
- Tumor growth upon orthotopic implantation of LRP8WT (red, n = 9) or LRP8KO (blue, n = 9) of SK‐N‐DZ cell line. Data represent the mean ± SEM; Mann–Whitney test one‐tailed P‐values are indicated.
- Representative luminescence images from each group are sown in (C).
- Outline of the orthotopic implantation and treatment scheme with Liproxstatin‐1 (Lip‐1) of LRP8WT and LRP8KO SK‐N‐DZ cells.
- Tumor weight of orthotopically implanted LRP8WT (red, n = 9) or LRP8KO (blue and yellow, n = 9 each) SK‐N‐DZ cells. All mice were treated with Lip‐1 for 5 days. After this, treatment was ceased for LRP8WT (red) and LRP8KO cohorts (blue) or maintained for an additional 14 days (yellow); see Materials and Methods for details. Tumors were analyzed at the endpoint. The center line in the box plots indicates the median value. Lower and upper hinges represent the 25th and 75th quantiles, the whiskers denote the Max and Min value. Mann–Whitney test one‐tailed P values are indicated.
- Representative images of tumors in the control (red) and LRP8KO groups (blue) treated with Lip‐1 for 5 days after implantation.
- Immunoblot analyzis of LRP8 and GPX4 levels from orthotopic tumors of LRP8WT or LRP8KO SK‐N‐DZ cells, treated with Lip‐1 for 5 days after implantation, followed by 14 days after randomization.
- Schematic representation for the proposed model of LRP8 inhibition essentiality. Comparison of selenium /selenocysteine uptake mechanisms via LRP8 and system Xc− in proliferating MYCN‐amplified cells, depicting the contribution of primarily LRP8/SELENOP supporting selenoprotein translation (left panel). Inhibition of LRP8 in system Xc− low cell selectively triggers ferroptosis in MYCN‐amplified neuroblastoma (right panel). SELENOP, Selenoprotein P; iSe, inorganic selenium; SeCys, Selenocysteine.
Source data are available online for this figure.
Discussion
The present study demonstrates that blocking selenium/selenocysteine uptake mechanisms could be an attractive indirect strategy to disrupt GPX4 function specifically and selectively to induce ferroptotic cell death in MYCN‐amplified neuroblastoma. We show here that selenium/selenocysteine can be obtained by cancer cells via multiple routes: First by the SELENOP/LRP8 axis and second, via the activity of system Xc− (SLC7A11/SLC3A2). Our data also imply that the striking LRP8 dependency may have emerged during tumor evolution as a result of the growth advantage of MYCN‐amplified neuroblastoma displaying low expression/activity of system Xc−. Of note, our study shows that forced expression of SLC7A11 prevents cell death of LRP8‐deficient neuroblastoma, but it also demonstrates its negative impact on the survival of these cells. While the mechanism is currently not clear, a possibility is that cystine might evoke dissulfide stress in a manner similar to a process dubbed disulfidptosis and described in a recent publication (Liu et al, 2023).
Thus, LRP8 blockade represents a rational strategy to indirectly deplete GPX4 and selectively trigger ferroptosis in MYCN‐amplified neuroblastoma and potentially other entities with low system Xc− expression/activity such as AML and lymphoma, while sparring normal cells. Of notice, recent reports have also identified LRP8 as a strong predictor of ferroptosis resistance both in cancer and in neurons (Greenough et al, 2022; Li et al, 2022). In both studies, the loss of LRP8 was not sufficient to spontaneously trigger ferroptosis neither in vitro nor in vivo. This echoes our observation that these dependencies are specific to defined contexts related to the fueling of the selenocysteine pathways. Overall, these discoveries could have broader implications. A recently published cancer dependency map of pediatric tumors identified LRP8 as an essential gene in pediatric Ewing sarcoma and medulloblastoma, all entities associated with MYCN amplifications (Dharia et al, 2021). Moreover, given the largely unsuccessful repurposing of adult oncology drugs for treating neuroblastoma and other childhood malignancies, these discoveries could provide an attractive framework to target ferroptosis in this entity. Targeting LRP8, for example, by developing LRP8/SELENOP neutralizing antibodies in MYCN‐amplified settings may overcome potential limitations of strategies that aim to inhibit GPX4 directly, expected to cause widespread organ toxicity as indicated by mouse genetic studies (Friedmann Angeli & Conrad, 2018).
One aspect that deserves attention is that all our studies are carried out in immunocompromised settings, and given that recent reports have suggested that CD8+ T‐Cells can drive tumor regression partially via ferroptosis (Matsushita et al, 2015; Liao et al, 2022) the effects observed here might be to some extent underestimated. Nevertheless, direct targeting of GPX4 in an immunocompetent setting might be a double‐edged sword as it could impair T‐cell function (Matsushita et al, 2015). Yet, targeting LRP8 might bypass such an unwanted off‐target effect since CD8+ T‐cells lack LRP8 expression. Therefore, additional studies on the inhibition of LRP8 in immunocompetent models are warranted to fully understand the therapeutic potential of targeting the LRP8/SELENOP axis for therapeutic benefit in neuroblastoma and other malignancies in particular the ones with amplified MYCN.
Materials ans Methods
Cell culture
The human neuroblastoma cell line SK‐N‐DZ (purchased from ATCC) was maintained in DMEM medium (Gibco) supplemented with 10% FCS (Gibco), 1× HEPES (Gibco), and 100 U/ml penicillin/streptomycin (Gibco) at 37°C with 5% CO2. Cell lines were tested for identity by SNP genotyping and mycoplasma contamination by the respective Multiplexion service (Heidelberg, Germany).
The human neuroblastoma cell line SH‐EP MYCN‐ER (Kindly provided by Prof. Dr. Martin Eilers; Herold et al, 2019), the murine neuroblastoma cell line NHO2A (derived from TH‐MYCN murine neuroblastoma model, kindly provided by Prof. Per Dr. Kogner), the human melanoma cell line A375 (purchased from ATCC), the human fibrosarcoma cell line HT1080 (purchased from ATCC) and the human hepatocellular carcinoma HepG2 (purchased from ATCC) were maintained in DMEM medium (Gibco) supplemented with 10% FCS (Gibco) and 100 U/ml penicillin/streptomycin (Gibco) at 37°C with 5% CO2. Cell lines were tested for mycoplasma contamination by the respective Multiplexion service (Heidelberg, Germany).
Immuoblotting
Cells were lysed in RIPA Buffer containing protease (Roche) inhibitor cocktail. Proteins were separated by SDS–polyacrylamide gel electrophoresis and electro transferred onto polyvinylidene difluoride membranes that were subsequently blocked in 5% defatted milk in Tris‐buffered saline (TBS) for 1 h room temperature (RT). Primary antibodies were diluted in a solution of 5% bovine serum albumin (BSA) in TBS and incubated overnight at 4°C. Membranes were washed and then incubated at RT for 2 h with horseradish peroxidase‐labeled secondary (1:3,000, anti‐rabbit #7074, anti‐mouse #7076 or anti‐rat #7077, Cell Signaling) diluted in a solution of 5% milk in TBS. Proteins were detected using enhanced chemiluminescence substrate (107‐5061, BioRad). Images were acquired using detection systems Amersham ImageQuant 800 (Cytiva, EUA) or Azure 400, (Biozym, Germany).
Following antibodies were used against β‐actin (1:5,000; A5441, Sigma‐Aldrich), GPX4 (1:1,000; no. ab125066, abcam), LRP8 (1:1,000; ab108208, abcam), SLC3A2 (CD98, 1:1,000; sc‐376815, Santa Cruz), SLC7A11 (1:10; antibody raised against a N‐terminal peptide of SLC7A11, clone 15C2‐111, developed in Munich + NaN3), SCLY (1:50; IG‐P1097, ImmunoGlobe GmbH), Flag (1:3,000; F3165, Sigma‐Aldrich), GPX1 (1:1,000; #3286, Cell Signaling), mouse Slc7a11 (1:10; monoclonal antibody generated at the HMGU, clone 4B3‐1113, developed in Munich + NaN3).
Molecular cloning
The human CRISPR activation pooled library Set A (Addgene plasmid #92379 was a gift from David Root and John Doench) was amplified as described previously (Joung et al, 2017). Briefly, Endura electrocompetent cells (Lucigen, Cat. No. 60242) were used to perform six electroporation reactions on a MicroPulserTM II (BioRad) following the manufacturer's instructions (pre‐set EC1 setting with V: 1.8 kV). Subsequently, cells were pooled and recovered at 37°C for 45 min on a shaking incubator. The cell suspension was diluted with LB‐medium, and 2 ml of bacterial suspension was spread evenly on prewarmed LB‐agar plates containing carbenicillin (100 μg/ml, 1× 245 mm square dish per 10,000 gRNAs in library) and incubated for 12 h at 37°C. The electroporation efficiency was assessed, and cells were harvested when library representation was > 100 bacterial colonies per gRNA. According to the manufacturer's instructions, the plasmid DNA was purified using NucleoBond® Xtra Maxi EF (Macherey‐Nagel, Cat. No. 740424). Sequencing libraries were prepared using the NEBNext 2× High‐Fidelity Master Mix (NEB, Cat. No. M0541L) with amplification primers partially complementary to the lentiviral gRNA backbone with overhangs introducing the Illumina P5/P7 adapter sequences. A pool of staggered P5 primer and 5% PhiX spike‐in were used to increase sequence diversity. Maintenance of gRNA distribution and library complexity was confirmed via next‐generation sequencing (NGS) on a Hi‐Seq2000 with the following read configuration: 125 cycles Read 1, 8 cycles index 1 (i7). Sequencing was performed by the High‐Throughput Sequencing Unit of the DKFZ Genomics and Proteomics Core Facility.
For the validation and the scCRISPRa screens, a custom gRNA library was constructed with selected hits from the primary screen. A pool of 82 oligonucleotides containing two gRNAs for each target and 10 NT control gRNAs were ordered from Twist Bioscience (San Francisco, USA) and cloned into a modified CROP‐seq‐MS2 vector via Golden Gate Assembly (NEB, Cat. No. E5520). The modified CROP‐seq‐MS2 plasmid was obtained as follows: the lentiviral CROP‐seq‐opti vector (Addgene plasmid #106280 was a gift from Jay Shendure) was sub‐cloned via restriction digest of the plasmid with NsiI‐HF (NEB, Cat. No. R3127) and SnaBI (NEB, Cat. No. R0130) and the insertion of a synthetic dsDNA fragment coding for a gRNA scaffold sequence with MS2 stem‐loop motifs (manufactured by Synbio Technologies, South Brunswick Township, USA; Sequence: 5′‐ATGCATGCTCTTCAACCTCAATAACGTTATATCCTGATTCACGTAATATTTTTGGGGTAAATTTAGTTCCTGTTCCATTAACTGCGCTAAAAATAATTTTTAAATCTTTTTTAGCTTCTTGCTCTTTTTTGTACGTCTCTGTTTTAGAGCTAGGCCAACATGAGGATCACCCATGTCTGCAGGGCCTAGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGGCCAACATGAGGATCACCCATGTCTGCAGGGCCAAGTGGCACCGAGTCGGTGCTTTTTTTAAGCTTGGCGTAACTAGATCTTGAGACACTGCTTTTTGCTTGTACTGGGTCTCTCTGGTTAGACCAGATCTGAGCCTGGGAGCTCTCTGGCTAACTAGGGAACCCACTGCTTAAGCCTCAATAAAGCTTGCCTTGAGTGCTTCAAGTAGTGTGTGCCCGTCTGTTGTGTGACTCTGGTAACTAGAGATCCCTCAGACCCTTTTAGTCAGTGTGGAAAATCTCTAGCAGTACGTA‐3′). The MS2 loops in the gRNA scaffold allow the recruitment of the p65‐HSF1 transactivator complex (expressed from pLentiMPH2). Next, the puromycin resistance gene was extended with a sequence coding for a viral p2A self‐cleaving motif and a tagBFP, thereby removing the stop codon from the puromycin resistance gene. For this, the plasmid was linearized with BstEII‐HF (NEB, Cat. No. R3162) and MluI‐HF (NEB, Cat. No. R3198), and the synthetic dsDNA fragment (manufactured by Twist Bioscience, Sequence: 5′‐GGTCACCGAGCTGCAAGAACTCTTCCTCACGCGCGTCGGGCTCGACATCGGCAAGGTGTGGGTCGCGGACGACGGCGCCGCCGTGGCGGTCTGGACCACGCCGGAGAGCGTCGAAGCGGGGGCGGTGTTCGCCGAGATCGGCCCGCGCATGGCCGAGTTGAGCGGTTCCCGGCTGGCCGCGCAGCAACAGATGGAAGGCCTCCTGGCGCCGCACCGGCCCAAGGAGCCCGCGTGGTTCCTGGCCACCGTCGGAGTCTCGCCCGACCACCAGGGCAAGGGTCTGGGCAGCGCCGTCGTGCTCCCCGGAGTGGAGGCGGCCGAGCGCGCCGGGGTGCCCGCCTTCCTGGAGACCTCCGCGCCCCGCAACCTCCCCTTCTACGAGCGGCTCGGCTTCACCGTCACCGCCGACGTCGAGGTGCCCGAAGGACCGCGCACCTGGTGCATGACCCGCAAGCCCGGTGCCGGAAGCGGAGCTACTAACTTCAGCCTGCTGAAGCAGGCTGGCGACGTGGAGGAGAACCCTGGACCTGGAGGGTCAGGGGGAGCCACCATGGTGTCTAAGGGCGAAGAGCTGATTAAGGAGAACATGCACATGAAGCTGTACATGGAGGGCACCGTGGACAACCATCACTTCAAGTGCACATCCGAGGGCGAAGGCAAGCCCTACGAGGGCACCCAGACCATGAGAATCAAGGTGGTCGAGGGCGGCCCTCTCCCCTTCGCCTTCGACATCCTGGCTACTAGCTTCCTCTACGGCAGCAAGACCTTCATCAACCACACCCAGGGCATCCCCGACTTCTTCAAGCAGTCCTTCCCTGAGGGCTTCACATGGGAGAGAGTCACCACATACGAAGACGGGGGCGTGCTGACCGCTACCCAGGACACCAGCCTCCAGGACGGCTGCCTCATCTACAACGTCAAGATCAGAGGGGTGAACTTCACATCCAACGGCCCTGTGATGCAGAAGAAAACACTCGGCTGGGAGGCCTTCACCGAAACTCTGTACCCCGCTGACGGCGGCCTGGAAGGCAGAAACGACATGGCCCTGAAGCTCGTGGGCGGGAGCCATCTGATCGCAAACGCCAAGACCACATATAGATCCAAGAAACCCGCTAAGAACCTCAAGATGCCTGGCGTCTACTATGTGGACTACAGACTGGAAAGAATCAAGGAGGCCAACAACGAGACCTACGTCGAGCAGCACGAGGTGGCAGTGGCCAGATACTGCGACCTCCCTAGCAAACTGGGGCACAAGCTTAATTAAACGCGT‐3′) was inserted to the plasmid backbone thus generating CROP‐seq‐MS2. The custom gRNA library was amplified in electrocompetent Endura bacteria (Lucigen, Cat. No. 60242), and plasmid DNA was purified as described. Library complexity was verified via NGS using the MiSeq V3 kit (Read configuration: 167 cycles Read 1, 8 cycles index 1 [i7]).
Individual gRNAs were cloned into the modified CROP‐seq‐MS2 or the pXPR_502 (Addgene #96923 was a gift from John Doench and David Root) lentivectors for CRISPRa experiments, and the pLKO5_RFP657 backbone for knockout (CRISPR‐KO) experiments via restriction digest of the respective lentivector with BsmBI (NEB, Cat. No. R0739). Oligonucleotides (Sigma‐Aldrich) with the gRNA sequences and complementary overhangs were phosphorylated, annealed, and inserted into the respective lentiviral delivery vector (Table 1).
Table 1.
Oligonucleotides used for cloning of individual gRNAs.
| Name | Sequence (5′‐3′) |
|---|---|
| Sense LRP8‐KO #1 | CACCGGCCACTGCATCCACGAACGG |
| Antisense LRP8‐KO #1 | AAACCCGTTCGTGGATGCAGTGGCC |
| Sense LRP8‐KO #2 | CACCGCTGCTTAGACCACAGCGACG |
| Antisense LRP8‐KO #2 | AAACCGTCGCTGTGGTCTAAGCAGC |
| Sense LRP8‐OE #1 | CACCGGGGCGGAGGCGGCAGCGGGA |
| Antisense LRP8‐OE #1 | AAACTCCCGCTGCCGCCTCCGCCCC |
| Sense LRP8‐OE #2 | CACCGGGCAGAGCCGAGTCAGAGAC |
| Antisense LRP8‐OE #2 | AAACGTCTCTGACTCGGCTCTGCCC |
| Sense AIFM2‐OE #1 | CACCGTAACCTTGACCCTGAGCGAA |
| Antisense AIFM2‐OE #1 | AAACTTCGCTCAGGGTCAAGGTTAC |
| Sense LRP8‐VBC #2 | CACCGTACGGCTGAAGAGAGTGCGT |
| Antisense LRP8‐VBC #2 | AAACACGCACTCTCTTCAGCCGTAC |
| Sense LRP8‐VBC #3 | CACCGGCAATAAGACCATCTCAG |
| Antisense LRP8‐VBC #3 | AAACCTGAGATGGTCTTATTGCC |
| Sense LRP8‐exon9 #3 | CACCGTACCATTACCTAGCCATGGA |
| Antisense LRP8‐exon9 #3 | AAACTCCATGGCTAGGTAATGGTAC |
| Sense LRP8‐exon9 #4 | CACCGGAACCTGGGGACTTAGACC |
| Antisense LRP8‐exon9 #4 | AAACGGTCTAAGTCCCCAGGTTCC |
| Sense mLrp8 #1 | CACCGATCTTCACGAACCGACACG |
| Antisense mLrp8 #1 | AAACCGTGTCGGTTCGTGAAGATC |
| Sense mLrp8 #2 | CACCGAGGTGCGGAGGATAGACC |
| Antisense mLrp8 #2 | AAACGGTCTATCCTCCGCACCTC |
Lentivirus production
Large‐scale lentivirus production was performed using a second‐generation lentivirus system and a calcium phosphate transfection kit (Invitrogen, Cat. No. K278001) in HEK293T cells. Briefly, early passaged HEK293T cells were cotransfected with the lentiviral transfer plasmid, a packaging plasmid (psPAX2, Addgene plasmid #12260 was a gift from Didier Trono), as well as with a plasmid encoding the VSV‐G envelope (pMD2.G, Addgene plasmid #12259 was a gift from Didier Trono). Viral supernatant was collected 48‐h post‐transfection, snap frozen, and stored at −80°C until use. Alternatively, for the generation of the cell expressing the following constructs; p442‐LRP8‐Flag, p442‐Mock, p442‐hSLC7A11, p442‐mSlc7a11, p442‐GFP and p442‐mScarlet, HEK293T cells were used to produce replication‐incompetent lentiviral particles pseudotyped with the ecotropic envelope protein of the murine leukemia virus (MLV) or the pantropic envelope protein VSV‐G. The third‐generation packaging plasmids (MDLg_pRRE and pRSV_Rev) and transfer plasmids were co‐transfected into HEK293T cells. Cell cultural supernatant containing viral particles was harvested 48‐h post‐transfection and used to transduce cell lines of interest. All experimental procedures for lentivirus production and transduction were performed in a biosafety level 2 laboratory.
Cell line generation for CRISPRa and CRISPR‐KO experiments
Polyclonal SK‐N‐DZ cells constitutively expressing the CRISPR activation machinery were engineered by transducing wild‐type cells with lentiviral particles carrying a dCas9‐VP64 (lenti dCas9VP64_Blast, Addgene plasmid #61425 was a gift from Feng Zhang) at a multiplicity of infection (MOI) of ~0.5. After recovery, cells were selected via Blasticidin treatment (20 μg/ml).
For the validation and scCRISPRa screens, SK‐N‐DZ cells expressing dCas9‐VP64 were transduced with lentiviral particles encoding the p65‐HSF1 transactivator complex (pLentiMPH2, Addgene plasmid #89308 was a gift from Feng Zhang). After recovery and selection with hygromycin B (200 μg/ml, Gibco, Cat. No. 10687010), cells were individualized by fluorescence‐activated cell sorting based on physical parameters (forward scatter and side scatter) and exclusion of dead cells via DAPI staining. Clonal cell lines were established and tested for their CRISPRa potential. Two independent clonal SK‐N‐DZ cell lines were selected for the validation screen, while the best‐scoring clonal cell line was used for the scCRISPRa screen.
For the CRISPR‐KO experiments, cells were transduced with lentivirus carrying a transgene that encodes a doxycycline‐inducible wild‐type Cas9 nuclease (pCW‐Cas9‐EGFP). After recovery, Cas9‐inducible cells were transduced with the gRNA lentivirus (pLKO5‐RFP657 backbone) at an MOI of ~0.3. Double‐positive cells (EGFP/RFP657) were individualized via FACS with the exclusion of dead cells (DAPI negative). Cas9 expression was induced by adding doxycycline (1 μg/ml, Sigma‐Aldrich, Cat. No. D9891) into the cell culture medium.
Genome‐wide CRISPRa screen to identify negative regulators of ferroptosis
Polyclonal SK‐N‐DZ cells expressing dCas9‐VP64 were transduced with the genome‐wide Calabrese CRISPRa library in triplicate at an MOI of ~0.3. For each replicate, 190 million cells were transduced, achieving a representation of ~1,000 cells per gRNA. After initial recovery for 4 days, cells were selected with puromycin (0.65 μg/ml) for 7 days. For each replicate, cells were split into four groups, of which one was harvested to determine baseline gRNA representation, another one maintained as the control group, and two others were treated with one of the ferroptosis‐inducing agents, RSL3 or Erastin, respectively, at a concentration lethal for wild‐type SK‐N‐DZ (300 nM/1 μM). Cells were maintained either with the addition of puromycin (control group) or puromycin and the respective drug for additional 2 weeks. The control cells were passaged, maintaining the gRNA representation of 1,000 cells per gRNA throughout the screen. Cells were harvested once the treatment groups reached approximately the same number as the control groups.
The genomic DNA was extracted using a commercial kit (Quick‐DNA Midirep Plus kit, Zymo Research, Cat. No. D4075) following the manufacturer's instructions and NGS libraries prepared as described. Libraries were sequenced as a multiplexed pool on a single lane of a HiSeq2000 chip with the following read configuration (125 cycles Read 1, 8 cycles index 1 [i7]).
Genome‐wide CRISPRa screen analysis and selection of candidate hits
Raw sequencing reads were processed as previously described (Spahn et al, 2017). Briefly, reads were trimmed to remove the constant vector sequence upstream of the gRNA sequences with cutadapt and then mapped to the reference library with Bowtie2. For each gRNA, the assigned reads were counted and normalized (cpm, counts per million reads). Using the control samples, a negative binomial count distribution was estimated and used to determine fold changes of individual gRNAs in the treatment samples. P‐values were computed and corrected for multiple testing (FDR). Finally, a gene‐level score was calculated from the mean log fold‐change of all gRNAs targeting the same gene. Candidate hits were selected using the top 100 enriched genes and examining protein interactions via StringDB (Szklarczyk et al, 2019) network analysis (R package version 2.4.0). Those hits were selected that showed a predicted interaction in the network (total of 36 genes, 162 predicted interactions, P‐value: 0.0055).
Secondary validation CRISPRa screen
Two monoclonal SK‐N‐DZ cell lines constitutively expressing dCas9‐VP64 and the p65‐HSF1 transactivator complex were transduced with the custom gRNA library targeting the transcription start sites of 36 selected hits from the primary screen (gRNA representation > 2,000×). After recovery and selection with puromycin (1 μg/ml), cells were split into one control group and a treatment group with RSL‐3 (100 nM). Cells were harvested after 2 weeks, and sequencing libraries were prepared as described. Libraries were sequenced as a multiplexed pool on a MiSeq V3 (Read 1: 167 cycles, index 1 [i7]: 8 cycles).
Secondary validation screen analysis
A count matrix of gRNA abundances was determined as described for the primary screen while mapping the reads to the custom reference library. To compute enrichment scores and P‐values, the robust rank aggregation workflow in the MaGeCK (Li et al, 2014) pipeline was used.
Single‐cell CRISPRa screen
A monoclonal SK‐N‐DZ expressing dCas9‐VP64 and p65‐HSF1 was transduced in duplicate with the custom gRNA library in the CROP‐seq‐MS2 backbone at a low MOI of ~0.07 to minimize the integration of multiple gRNAs. Transduction efficiency was confirmed via flow cytometric analysis of tagBFP expression. Cells were selected with puromycin at 1 μg/ml 24 h post‐transduction. Puromycin selection was continued for additional 4 days at 1.5 μg/ml. Ten days after transduction, cells were collected, and ~20,000 individual cells per lane were partitioned by the 10× Chromium Controller. Single‐cell RNA‐seq libraries were constructed using the Chromium Single Cell Gene Expression v3.1 kit (10× Genomics, Cat. No. 1000128) following the manufacturer's protocol, thereby retaining 40 ng of full‐length cDNA for the enrichment of gRNAs. Guide RNA sequences were selectively amplified by adapting a hemi‐nested PCR approach that was previously described (Hill et al, 2018) using the gRNA_enrichment1_fw (5′‐GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTCTTGTGGAAAGGACGAAACACCG‐3′) and gRNA_enrichment1_rv (5′‐CTACACGACGCTCTTCCGATCT‐3′) primers and the 2× KAPA HiFi HotStart Ready Mix (Roche, Cat. No. KK2601). In a subsequent PCR, 1 ng of purified PCR product was used as input to construct sequencing‐ready libraries with the gRNA‐enrichment2_fw (5′‐CAAGCAGAAGACGGCATACGAGAT[index1]GTGACTGGAGTTCAG‐3′) and gRNA_enrichment2_rv (5′‐AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTC‐3′) primers. The whole‐transcriptome single‐cell libraries were sequenced on a NovaSeq6000 with 28 cycles Read 1, 8 cycles index 1 (i7), and 91 cycles Read 2. The gRNA enrichment libraries were sequenced on a NextSeq550 high with the following read configuration: 32 cycles Read 1, 8 cycles index 1 (i7), and 120 cycles Read 2.
Single‐cell CRISPRa screen analysis
Whole‐transcriptome single‐cell reads were mapped to the reference genome (GRCh 38), and count matrices were generated using Cell Ranger (10× Genomics, version 6.0.1) with default parameters. Single‐cell sequencing data from two individual 10× Chromium lanes were combined using the aggregation function (cellranger aggr). Data normalization and downstream analysis were performed with the R package Seurat (version 4.0.1). Based on the inflection point per cell barcode, a threshold of 1,000 detected features per cell was set to filter out empty droplets and cells with low complexity. The count matrix was filtered for genes detected in at least 10 cells. For each cell, the UMI counts of each gene were divided by the total number of counts and multiplied by a scaling factor (10,000), after which they were log‐transformed. Guide RNA enrichment libraries were processed via fba (Duan & Hon, 2021; version 0.0.11) and mapped to the custom gRNA library, allowing two mismatches. The Seurat object was filtered for cells assigned to a particular gRNA, and cells with a strong CRISPRa phenotype were determined using Seurat's Mixscape function. Differential gene expression analysis was performed for each CRISPRa group with DESeq2 (version 1.32.0) using cells assigned to the NT group as control. The top 50 differentially expressed genes were selected (ranked by the adjusted P‐value) from each overexpression group and merged together to create the gene expression signature list. For Fig 1F, functionally related differentially expressed genes were labeled based on overrepresented terms retrieved from the Gene Ontology, the Reactome, the KEGG, and the Molecular Signatures databases and based on enrichment analysis performed with the R package clusterProfiler (version 4.0.2).
SLC‐focused CRISPR‐based screens for selenium‐relevant transporters
SK‐N‐DZ were transduced with the SLC‐focused CRISPR‐based library in duplicate at an MOI of 0.5. A total of 10 million cells were transduced for each replicate to achieve a representation of > 1,000. After recovery for 3 days, the cells were selected with 0.5 μg/ml puromycin and were maintained with 500 nM Lip‐1 for 5 days. After selection, for each replicate, the cells were split into five groups, which were treated with 20 nM Na2SeO3, 20 nM Selenocystiene, 200 nM Selenomethione, 50 μM ßME, and 500 nM Lip‐1, respectively. The cells were passaged, representing > 1,000 cells per gRNA throughout the screen. Cell pellets were harvested on Day 14 of treatment. The genomic DNA was extracted using a commercial kit (QIAGEN‐DNeasy) following the manufacturer's instructions. Libraries were sequenced as a multiplexed pool on a Nextseq500 (8 cycles index 2 [i5], 8 cycles index 1 [i7], 75 cycles Read 1, 75 cycles Read 2).
Generation of LRP8‐knockout cell lines
SK‐N‐DZ cells were transduced with lentiviral particles encoding LRP8 gRNAs (lentiCRISPRv2 blast, Addgene plasmid #98293 was a gift from Brett Stringer). After recovery and selection with blasticidin (10 μg/ml), cells were subcloned via limiting dilution. Clonal cell lines were established and tested for LRP8 expression by Western blot. Two clonal cell lines were selected for further experiments.
NHO2A cells were transduced with lentiviral particles encoding murine Lrp8 gRNAs (lentiCRISPRv2 blast, Addgene plasmid #98293 was a gift from Brett Stringer). After selection with blasticidin (10 μg/ml) and recovery, the cell lines were used for further experiments.
Clonal HT1080 and A375 cells constitutively expressing Cas9 were transfected with plasmids encoding gRNAs (pKLV2‐U6gRNA5(BbsI)‐PGKpuro2AmAG‐W, Addgene plasmid #67976 was a gift from Kosuke Yusa) targeting introns flanking LRP8 exon 9. After selection with puromycin (1 μg/ml), cells were grown by limiting dilution. Clonal cell lines were established and tested by genotyping PCR and Western blot. Two clonal cell lines were selected for further experiments and subsequent CRISPR‐based screens.
SLC‐focused CRISPR‐based screens in LRP8‐KO and wild‐type
LRP8‐KO and WT cell lines were transduced with the SLC‐focused CRISPR‐based library at an MOI of 0.5. A total of 10 million cells for each cell line were transduced, achieving > 1,000 cells per gRNA. After recovery and selection with 1 μg/ml puromycin, the LRP8‐KO and ‐WT cell lines were split and maintained at the gRNA representation of > 1,000. Cell pellets were harvested after 2 weeks of passaging. The genomic DNA was extracted with a commercial kit (QIAGEN‐DNeasy) following the manufacturer's instructions. Libraries were multiplexed and sequenced on a Nextseq 500 (8 cycles index 2 [i5], 8 cycles index 1 [i7], 75 cycles Read 1, 75 cycles Read 2). The mapping of raw sequencing reads to the reference library, and computing of enrichment scores and P‐values were processed using the MaGeCK pipeline. The MaGeCKFlute was used for the identification of gene hits and associated pathways.
Cell lines generation for a competition assay
Human neuroblastoma cell lines (SK‐N‐DZ and SH‐EP MYCN‐ER) expressing a lentiviral construct carrying SLC7A11 cDNA or an empty vector were generated using the methods described above. Briefly, lentiviral constructs were transduced into the corresponding cell line labeled with mScarlet or eGFP. The combination of construct/fluorophore is illustrated in the figures present in the main text. Established cell lines were counted and mixed at a 1:1 ratio, and the distribution was monitored during the period indicated in the main text. Rescue experiments were carried out using a combination of ßMe (50 μM) and Erastin (2 μM).
Analysis of cell viability
The impact of various compounds on cell viability was analyzed using the CellTiter‐Glo (CTG) assay (Promega). To determine changes in cell viability, 3,000 cells were seeded in the complete medium in 96‐well plates (Greiner Bio One) 24 h before the treatment. Cells were then treated for 72 h with the indicated concentration of compounds. Cell viability was analyzed using the CTG assay following the manufacturer's instructions. For in vitro clonogenic assays, 200 cells were seeded in a 12‐well plate for 14 days with each particular experimental condition, and colonies were stained with 1 ml 0.01% (w/v) crystal violet.
Analysis of lipid peroxidation
Approximately 105 cells were seeded in six‐well plates. Before lipid peroxidation was analyzed, the medium was removed, C11‐BODIPY (wavelength: 581/591 nm) diluted in Hank's Balanced Salt Solution (HBSS; Gibco), and added to wells at a final concentration of 4 μM. After 15‐min staining at 37°C inside the tissue culture incubator, cells were harvested gently, and lipid peroxidation levels were immediately analyzed using a BD FACS Aria™ III cell sorter.
Transient siRNA‐mediated gene knockdown
SK‐N‐DZ cells were seeded in 12‐well plates (200,000 cells/well) and 24 h later transiently transfected with a mix of RNAiMax (0.04 μl/well; Thermo Fisher Scientific) and 0.01 μM/well of siRNA following the manufacturers' instructions.
Selenium speciation analyses
We measured total selenium by inductively coupled plasma sectorfield mass spectrometry (ICP‐of‐MS), and the selenium species selenite (Se‐IV), selenate (Se‐VI), selenomethionine‐bound selenium (Se‐MET), selenocystine‐bound selenium (Se‐Cys), thioredoxin reductase‐bound selenium (Se‐TrxR), glutathione‐peroxidase‐bound selenium (Se‐GPx), selenoprotein‐P‐bound selenium (SELENOP), and albumin‐bound selenium (Se‐HSA) using ion exchange chromatography (IEC) coupled with ICP‐dynamic reaction cell mass spectrometry (ICP‐DRC‐MS) in analogy to methodologies previously established (Solovyev et al, 2013). The experimental settings for ICP‐sf‐MS (ELEMENT II, Thermo Scientific, Bremen Germany) were: radio frequency power: 1260 W, plasma gas flow: 16 l Ar/min auxiliary gas flow: 0.85 l Ar/min, nebulizer gas flow: 1.085 l Ar/min, daily optimized, dwell time 300 ms, ions monitored: 77Se, 78Se, high‐resolution mode. For the speciation of selenium compounds, we used the hyphenated system from Perkin Elmer (Rodgau, Germany) comprising a NexSAR gradient HPLC pump, autosampler, and NexION 300 D ICP‐DRC‐MS, completely controlled by Clarity software. The separation column for species separation was an ion exchange pre‐ and analytical column AG‐11 + AS‐11 (250 × 4 mm I.D.) from Thermo Dionex (Idstein, Germany). The sample volume was 50 μl. The mobile phases and chromatographic gradient were previously published (Solovyev et al, 2013). Briefly, the flow rate was 0.80 ml/min. The experimental settings for ICP‐DRC‐MS were: radio frequency power: 1250 W, plasma gas flow: 15 l Ar/min auxiliary gas flow: 1.05 l Ar/min, nebulizer gas flow: 0.92 l Ar/min, daily optimized, dwell time 300 ms, ions monitored: 77Se, 78Se, 80Se, DRC reaction gas: CH4 reaction at 0.58 ml/min, DRC rejection parameter q: 0.6. Five‐point calibration curves from 0 to 5,000 ng/l were linear with r 2 for the three Se isotopes being better than 0.999881. Data files from selenium chromatograms were processed with Clarity software for peak area integration.
Generation of HepG2 conditional medium and SELENOP enrichment
HepG2 cells were seeded to a 15‐cm2‐round dish with standard DMEM medium (10% FBS+ 1% Pen/Strep). The medium was changed to FBS‐free medium supplemented with 50 or 200 nM Na2SeO3 when HepG2 cells achieved ~70–80% confluent. After 48 h incubation, the medium was collected and briefly spined down to clarify the supernatant. The supernatant was transferred to the centrifugal filter (Amicon Ultra‐15, 30K. UFC903024) and centrifuged for 10 min at 3,000 g. After the supernatant was centrifuged through the centrifugal filter, 2 ml of FBS‐free medium was used to resuspend the concentratred fraction from the filter. HT1080‐Cas9 LRP8 WT/KO and SK‐N‐DZ cells were plated and treated with 10 nM Na2SeO3 or HepG2 concentrated medium in six‐well plates in the absence of FBS. After 24‐h incubation, the cell lysates were collected.
Analyses of pediatric cohorts of neuroblastoma samples
RNA sequencing of 498 neuroblastoma cases was performed as described previously (Zhang et al, 2015). In short, mRNA purification was performed using the Dynabeads mRNA Purification Kit (Invitrogen), and library construction was completed according to the standard TruSeq protocol. Clusters were generated according to the TruSeq PE cluster Kit version 3 reagent preparation guide (for cBot‐HiSeq/HiScanSQ). Paired‐end sequencing with 100 bp read length was performed on the Illumina HiSeq 2000 platform. Raw data processing, read mapping, and gene expression quantification were done using the Magic‐AceView analysis pipeline and AceView transcriptome reference (http://www.aceview.org) as described previously (Zhang et al, 2015). Genes with generally low read counts were removed using R (v4.1.1) and the function “filterByExpr” in R‐package edgeR (v3.34.1). Differential gene expression analysis was done using the empirical Bayes method implemented in the R‐package limma (v3.48.3).
In vivo orthotopic mouse experiments
All studies involving mice and experimental protocols were conducted in compliance with the German Cancer Center Institute guidelines and approved by the governmental review board of the state of Baden‐Wuerttemberg, Regierungspraesidium Karlsruhe, under the authorization number G‐176/19, followed the German legal regulations. Mouse strains used in the study: NOD.Cg‐PrkdcscidIl2rgtm1Wjl/SzJ (NSG, JAX stock #005557). Female mice, 3–4 months of age, were used for experiments. Mice have been housed in individually ventilated cages under temperature and humidity control. Cages contained an enriched environment with bedding material. To generate orthotopic mouse models for neuroblastoma, 2 × 105 SK‐N‐DZ cells were transplanted into the right adrenal gland after the surgical site was prepared. Cells were resuspended in a 1:1 (vol/vol) mix of growth factor‐reduced matrigel (Corning) and PBS. Overall, 20 μl of this cell suspension was injected into the right adrenal gland of anesthetized mice. After tumor cell transplantation, we monitored the mice for evidence of tumor development by bioluminescent signal using an IVIS Spectrum Xenogen device (Caliper Life Sciences). We observed a clear signal from the tumors 1 week after injecting 2 × 105 SK‐N‐DZ cells. For liproxstatin (Lip‐1), we used 10 mg/kg/day for the first 5 days. Lip‐1 treatment (every second day) continued in one group for another 2 weeks. Animals' health was monitored daily, and mice were euthanized as soon as they reached the abortion criteria defined in the procedure. The sample size was calculated with the help of a biostatistician using R version 3.4.0. Assumptions for power analysis were as follows: α error, 5%; β error, 20%. Mice were randomized into treatment groups before treatment. If animals had to be sacrificed before the predefined endpoint (due to weight loss or other termination criteria), they were excluded from any downstream analyses. All animal experiments were blinded during experiments and follow‐up assessments.
Author contributions
Hamed Alborzinia: Conceptualization; supervision; funding acquisition; writing – original draft; writing – review and editing. Zhiyi Chen: Data curation; formal analysis; investigation; writing – original draft; writing – review and editing. Umut Yildiz: Formal analysis; investigation; data curation; writing – original draft. Florencio Porto Freitas: Investigation. Felix C E Vogel: Investigation. Julianna Patricia Varga: Investigation. Jasmin Batani: Investigation. Christoph Bartenhagen: Data curation; formal analysis; visualization. Werner Schmitz: Data curation; formal analysis; methodology. Gabriele Büchel: Resources. Bernhard Michalke: Resources; methodology. Jiashuo Zheng: Investigation. Svenja Meierjohann: Resources; methodology. Enrico Girardi: Resources. Elisa Espinet: Investigation. Andrés F Flórez: Investigation. Ancely Ferreira dos Santos: Investigation. Nesrine Aroua: Investigation. Tasneem Cheytan: Investigation. Julie Haenlin: Methodology. Lisa Schlicker: Investigation. Thamara N Xavier da Silva: Investigation. Adriana Przybylla: Methodology. Petra Zeisberger: Methodology. Giulio Supert‐Furga: Resources. Martin Eilers: Resources. Marcus Conrad: Methodology. Marietta Fabiano: Investigation; methodology. Ulrich Schweizer: Resources; methodology. Matthias Fischer: Data curation; formal analysis. Almut Schulze: Supervision; funding acquisition. Andreas Trumpp: Conceptualization; funding acquisition; writing – review and editing. José Pedro Friedmann Angeli: Conceptualization; supervision; funding acquisition; writing – original draft; project administration; writing – review and editing.
Disclosure and competing interests statement
The authors declare the following financial interests/personal relationships, which may be considered as potential competing interests: MC is the cofounder of ROSCUE THERAPEUTICS GmbH and author of a patent application related to ferroptosis. GS‐F is coauthor of patent applications related to SLCs, cofounder of Solgate GmbH, and the Academic Project Coordinator of the IMI grants RESOLUTE and Resolution in partnership with Pfizer, Novartis, Bayer, Sanofi, Boehringer‐Ingelheim and Vifor Pharma. The GS‐F laboratory receives funds from Pfizer. All other authors declare no other relevant conflicts of interest.
Supporting information
Appendix S1
Dataset EV1
Dataset EV2
Dataset EV3
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Acknowledgments
JPFA acknowledges the support of the Junior Group Leader program of the Rudolf Virchow Center, University of Würzburg, the Deutsche Forschungsgemeinschaft (DFG), FR 3746/3‐1, SPP2306 (FR 3746/6‐1) and the CRC205 (INST 269/886‐1). This work was also supported by the SPP2306 (AT and HA), FOR2674, and SFB873 funded by the Deutsche Forschungsgemeinschaft (AT); the DKTK joint funding project “RiskY‐AML”; the “Integrate‐TN” Consortium funded by the Deutsche Krebshilfe, the Dietmar Hopp Foundation (AT). MC is supported by the DFG CO 291/7‐1 and the SPP2306 (CO 297/9‐1 and CO 297/10‐1) as well as the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement No. GA 884754). JZ is supported by a Humboldt Postdoctoral Fellowship. AS, LS, and FCEV are supported by SPP2306 (SCHU 2670/4‐1) and FOR2314 (SCHU 2670/2‐2). MF and CB were supported by the Deutsche Forschungsgemeinschaft (DFG; grant no. BA 6984/1‐1 and grant no. FI 1926/2‐1 to MF, and as part of the SFB 1399 to MF), the Förderverein für krebskranke Kinder e.V. Köln (endowed chair to MF), and the German Ministry of Science and Education (BMBF) as part of the e:Med initiative (grant no. 01ZX1603 and 01ZX1607 to MF). GS‐F acknowledges funding from the Austrian Academy of Sciences and European Research Council Advanced Grant 695214 GameofGates. EG was supported by a Marie Sklodowska‐Curie fellowship (MSCA‐IF‐2014‐661491). UY and HA thank the DKFZ Single‐Cell Open Lab (scOpenLab) for assistance with the single‐cell CRISPR‐activation screen. Illustrations Figs 1A, C, F, 2E, 4A, E, and I were created with BioRender.com. This publication was supported by the Open Access Publication Fund of the University of Würzburg. Open Access funding enabled and organized by Projekt DEAL.
EMBO Mol Med (2023) 15: e18014
Contributor Information
Hamed Alborzinia, Email: hamed.alborzy@gmail.com.
Andreas Trumpp, Email: a.trumpp@dkfz-heidelberg.de.
José Pedro Friedmann Angeli, Email: pedro.angeli@vichow.uni-wuerzburg.de.
Data availability
Raw screening data generated from the genome‐wide and validation CRISPRa screens are uploaded to the Sequence Read Archive under the accession code PRJNA866989 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA866989). Raw and processed data from the CROP‐seq experiment is deposited on the Gene Expression Omnibus (accession number GSE210762; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE210762).
References
- Alborzinia H, Florez AF, Kreth S, Bruckner LM, Yildiz U, Gartlgruber M, Odoni DI, Poschet G, Garbowicz K, Shao C et al (2022) MYCN mediates cysteine addiction and sensitizes neuroblastoma to ferroptosis. Nat Cancer 3: 471–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu A, Bodycombe NE, Cheah JH, Price EV, Liu K, Schaefer GI, Ebright RY, Stewart ML, Ito D, Wang S et al (2013) An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell 154: 1151–1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, Roberts MA, Tong B, Maimone TJ, Zoncu R et al (2019) The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575: 688–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brutsch SH, Wang CC, Li L, Stender H, Neziroglu N, Richter C, Kuhn H, Borchert A (2015) Expression of inactive glutathione peroxidase 4 leads to embryonic lethality, and inactivation of the Alox15 gene does not rescue such knock‐in mice. Antioxid Redox Signal 22: 281–293 [DOI] [PubMed] [Google Scholar]
- Burk RF, Hill KE (2015) Regulation of selenium metabolism and transport. Annu Rev Nutr 35: 109–134 [DOI] [PubMed] [Google Scholar]
- Burk RF, Hill KE, Motley AK, Austin LM, Norsworthy BK (2006) Deletion of selenoprotein P upregulates urinary selenium excretion and depresses whole‐body selenium content. Biochim Biophys Acta 1760: 1789–1793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlisle AE, Lee N, Matthew‐Onabanjo AN, Spears ME, Park SJ, Youkana D, Doshi MB, Peppers A, Li R, Joseph AB et al (2020) Selenium detoxification is required for cancer‐cell survival. Nat Metab 2: 603–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson BA, Tobe R, Yefremova E, Tsuji PA, Hoffmann VJ, Schweizer U, Gladyshev VN, Hatfield DL, Conrad M (2016) Glutathione peroxidase 4 and vitamin E cooperatively prevent hepatocellular degeneration. Redox Biol 9: 22–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Yu C, Kang R, Kroemer G, Tang D (2021) Cellular degradation systems in ferroptosis. Cell Death Differ 28: 1135–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dharia NV, Kugener G, Guenther LM, Malone CF, Durbin AD, Hong AL, Howard TP, Bandopadhayay P, Wechsler CS, Fung I et al (2021) A first‐generation pediatric cancer dependency map. Nat Genet 53: 529–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS et al (2012) Ferroptosis: an iron‐dependent form of nonapoptotic cell death. Cell 149: 1060–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, Thomas AG, Gleason CE, Tatonetti NP, Slusher BS et al (2014) Pharmacological inhibition of cystine‐glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 3: e02523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, Grocin AG, Xavier da Silva TN, Panzilius E, Scheel CH et al (2019) FSP1 is a glutathione‐independent ferroptosis suppressor. Nature 575: 693–698 [DOI] [PubMed] [Google Scholar]
- Duan J, Hon GC (2021) FBA: feature barcoding analysis for single cell RNA‐Seq. Bioinformatics 37: 4266–4268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton JK, Ruberto RA, Kramm A, Viswanathan VS, Schreiber SL (2019) Diacylfuroxans are masked nitrile oxides that inhibit GPX4 covalently. J Am Chem Soc 141: 20407–20415 [DOI] [PubMed] [Google Scholar]
- Floros KV, Cai J, Jacob S, Kurupi R, Fairchild CK, Shende M, Coon CM, Powell KM, Belvin BR, Hu B et al (2021) MYCN‐amplified neuroblastoma is addicted to iron and vulnerable to inhibition of the system Xc−/glutathione axis. Cancer Res 81: 1896–1908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedmann Angeli JP, Conrad M (2018) Selenium and GPX4, a vital symbiosis. Free Radic Biol Med 127: 153–159 [DOI] [PubMed] [Google Scholar]
- Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, Herbach N, Aichler M, Walch A, Eggenhofer E et al (2014) Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol 16: 1180–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedmann Angeli JP, Krysko DV, Conrad M (2019) Ferroptosis at the crossroads of cancer‐acquired drug resistance and immune evasion. Nat Rev Cancer 19: 405–414 [DOI] [PubMed] [Google Scholar]
- Gao M, Yi J, Zhu J, Minikes AM, Monian P, Thompson CB, Jiang X (2019) Role of mitochondria in ferroptosis. Mol Cell 73: 354–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenough MA, Lane DJR, Balez R, Anastacio HTD, Zeng Z, Ganio K, McDevitt CA, Acevedo K, Belaidi AA, Koistinaho J et al (2022) Selective ferroptosis vulnerability due to familial Alzheimer's disease presenilin mutations. Cell Death Differ 29: 2123–2136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herold S, Kalb J, Buchel G, Ade CP, Baluapuri A, Xu J, Koster J, Solvie D, Carstensen A, Klotz C et al (2019) Recruitment of BRCA1 limits MYCN‐driven accumulation of stalled RNA polymerase. Nature 567: 545–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill AJ, McFaline‐Figueroa JL, Starita LM, Gasperini MJ, Matreyek KA, Packer J, Jackson D, Shendure J, Trapnell C (2018) On the design of CRISPR‐based single‐cell molecular screens. Nat Methods 15: 271–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingold I, Berndt C, Schmitt S, Doll S, Poschmann G, Buday K, Roveri A, Peng X, Porto Freitas F, Seibt T et al (2018) Selenium utilization by GPX4 is required to prevent hydroperoxide‐induced ferroptosis. Cell 172: 409–422 [DOI] [PubMed] [Google Scholar]
- Joung J, Konermann S, Gootenberg JS, Abudayyeh OO, Platt RJ, Brigham MD, Sanjana NE, Zhang F (2017) Genome‐scale CRISPR‐Cas9 knockout and transcriptional activation screening. Nat Protoc 12: 828–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft VAN, Bezjian CT, Pfeiffer S, Ringelstetter L, Muller C, Zandkarimi F, Merl‐Pham J, Bao X, Anastasov N, Kossl J et al (2020) GTP Cyclohydrolase 1/tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Cent Sci 6: 41–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewerenz J, Ates G, Methner A, Conrad M, Maher P (2018) Oxytosis/ferroptosis‐(Re‐) emerging roles for oxidative stress‐dependent non‐apoptotic cell death in diseases of the central nervous system. Front Neurosci 12: 214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, Irizarry RA, Liu JS, Brown M, Liu XS (2014) MAGeCK enables robust identification of essential genes from genome‐scale CRISPR/Cas9 knockout screens. Genome Biol 15: 554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Ferguson L, Deol KK, Roberts MA, Magtanong L, Hendricks JM, Mousa GA, Kilinc S, Schaefer K, Wells JA et al (2022) Ribosome stalling during selenoprotein translation exposes a ferroptosis vulnerability. Nat Chem Biol 18: 751–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao P, Wang W, Wang W, Kryczek I, Li X, Bian Y, Sell A, Wei S, Grove S, Johnson JK et al (2022) CD8(+) T cells and fatty acids orchestrate tumor ferroptosis and immunity via ACSL4. Cancer Cell 40: 365–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Schreiber SL, Stockwell BR (2018) Targeting dependency on the GPX4 lipid peroxide repair pathway for cancer therapy. Biochemistry 57: 2059–2060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Nie L, Zhang Y, Yan Y, Wang C, Colic M, Olszewski K, Horbath A, Chen X, Lei G et al (2023) Actin cytoskeleton vulnerability to disulfide stress mediates disulfidptosis. Nat Cell Biol 25: 404–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Yang Q, Su Y, Ji Y, Li G, Yang X, Xu L, Lu Z, Dong J, Wu Y et al (2021) MYCN mediates TFRC‐dependent ferroptosis and reveals vulnerabilities in neuroblastoma. Cell Death Dis 12: 511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H, Koppula P, Wu S, Zhuang L, Fang B et al (2021) DHODH‐mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 593: 586–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushita M, Freigang S, Schneider C, Conrad M, Bornkamm GW, Kopf M (2015) T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J Exp Med 212: 555–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olm E, Fernandes AP, Hebert C, Rundlof AK, Larsen EH, Danielsson O, Bjornstedt M (2009) Extracellular thiol‐assisted selenium uptake dependent on the x(c)‐ cystine transporter explains the cancer‐specific cytotoxicity of selenite. Proc Natl Acad Sci U S A 106: 11400–11405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson GE, Winfrey VP, Nagdas SK, Hill KE, Burk RF (2007) Apolipoprotein E receptor‐2 (ApoER2) mediates selenium uptake from selenoprotein P by the mouse testis. J Biol Chem 282: 12290–12297 [DOI] [PubMed] [Google Scholar]
- Shah R, Shchepinov MS, Pratt DA (2018) Resolving the role of lipoxygenases in the initiation and execution of ferroptosis. ACS Cent Sci 4: 387–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solovyev N, Berthele A, Michalke B (2013) Selenium speciation in paired serum and cerebrospinal fluid samples. Anal Bioanal Chem 405: 1875–1884 [DOI] [PubMed] [Google Scholar]
- Spahn PN, Bath T, Weiss RJ, Kim J, Esko JD, Lewis NE, Harismendy O (2017) PinAPL‐Py: a comprehensive web‐application for the analysis of CRISPR/Cas9 screens. Sci Rep 7: 15854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, Tang D (2016) Activation of the p62‐Keap1‐NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 63: 173–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Superti‐Furga G, Lackner D, Wiedmer T, Ingles‐Prieto A, Barbosa B, Girardi E, Goldmann U, Gurtl B, Klavins K, Klimek C et al (2020) The RESOLUTE consortium: unlocking SLC transporters for drug discovery. Nat Rev Drug Discov 19: 429–430 [DOI] [PubMed] [Google Scholar]
- Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta‐Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P et al (2019) STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome‐wide experimental datasets. Nucleic Acids Res 47: D607–D613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore‐Ludlow B, Kaffenberger SD, Eaton JK, Shimada K, Aguirre AJ et al (2017) Dependency of a therapy‐resistant state of cancer cells on a lipid peroxidase pathway. Nature 547: 453–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Kensler TW, Motohashi H (2018) The KEAP1‐NRF2 system: a thiol‐based sensor‐effector apparatus for maintaining redox homeostasis. Physiol Rev 98: 1169–1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB et al (2014) Regulation of ferroptotic cancer cell death by GPX4. Cell 156: 317–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Yu Y, Hertwig F, Thierry‐Mieg J, Zhang W, Thierry‐Mieg D, Wang J, Furlanello C, Devanarayan V, Cheng J et al (2015) Comparison of RNA‐seq and microarray‐based models for clinical endpoint prediction. Genome Biol 16: 133 [DOI] [PMC free article] [PubMed] [Google Scholar]